
Are transition metal phthalocyanines active for urea synthesis via electrocatalytic coupling of CO2 and N2?†
Yungan
Huang
a,
Ting
Fan
 b and
Yongfei
Ji
*a
b and
Yongfei
Ji
*a
aSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, Guangdong, P. R. China. E-mail: yongfeiji2018@gzhu.edu.cn
bSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, Guangdong, P. R. China
First published on 28th November 2024
Abstract
Electrocatalytic coupling of CO2 and N2 to synthesize urea presents a promising approach to address global energy and environmental challenges. Despite the potential, developing an efficient catalyst capable of activating both CO2 and N2 while suppressing side reactions remains a significant challenge. Recent studies have indicated that CuPc and CoPc exhibit notable activity in this process. Herein, we report a theoretical analysis of the catalytic performance of 3d–5d transition metal phthalocyanines (MPcs) in the electrocatalytic urea synthesis reaction. Our findings reveal that MPcs generally exhibit limited activity due to the poor competitiveness of N2 for adsorption sites and the high energy barrier associated with CO–N2 coupling, which hinders their ability to compete with CO reduction and/or N2 reduction pathways. Furthermore, the coupling between CO and NH2* is either insufficient for N2 reduction or is outcompeted by ammonia formation. We propose that enhancing N2 adsorption could facilitate C–N coupling, offering a potential strategy for the design of single-atom catalysts aimed at improving urea synthesis efficiency.
1 Introduction
Urea plays an essential role in industrial production as it serves not only as a primary nitrogen fertilizer for agriculture but also as a key chemical feedstock for the synthesis of various essential chemicals.1 Currently, urea is industrially produced through the reaction of ammonia with carbon dioxide (NH3 + CO2 → CO(NH2)2 + H2O) under high temperature (450–500 K) and pressure (150–250 bar).1 The ammonia utilized in this process is predominantly derived from the energy-intensive Haber–Bosch process,1 which requires significant energy input to break the triple bond in N2. The development of efficient and sustainable urea production methods presents a formidable challenge.2,3Recently, electrocatalytic coupling of CO2 with N2 for urea synthesis has been attracting increasing scientific interest.1,2 In 2016, Kayan et al. successfully demonstrated the coupling of CO2 and N2 to form urea on a Pt electrode at −0.16 V potential, albeit under 60 bar pressure and with low urea yield and selectivity. In 2020, Wang achieved the CO2–N2 coupling for urea synthesis under ambient conditions on a PdCu alloy at −0.4 V vs. a reversible hydrogen electrode (RHE),4 where CO was found to couple with surface-adsorbed N2 to form NCON intermediates, which can be readily converted to urea. Since these pioneering studies, numerous materials have been explored both experimentally and theoretically for CO2–N2 coupling in urea synthesis. Materials such as MoP,5 Sb doped BiOx,6 Cu3N with N vacancies,7 CuBi,8 and Bi2S39 have shown significant activity for the coupling of CO2 and N2. Because the coactivation of CO2 and N2 is very crucial for C–N coupling, many catalysts with two or more components to provide different types of active sites have been designed. For instance, Zhang et al. prepared the Mott–Schottky Bi–BiVO4 heterostructure,10 BiFeO3/BiVO4 heterostructure,11 and artificial frustrated Lewis pairs on a nickel borate12 catalyst for the co-activation of CO2 and N2. Luo et al. synthesized a SiW6Mo6@MIL-101(Cr) catalyst for the co-activation of CO2 at a Mo site and N2 at a W site.13 Pan et al. found that Pd1Cu1–TiO2 is more active than Pd1–TiO2 for urea synthesis due to a much lower C–N coupling barrier on the former catalyst.14 Recently, Wang et al. achieved a Faraday efficiency (FE) of 63.5% for urea synthesis with a Zn–Mn dual atom catalyst.15 CO is proposed to be inserted into the triple bond in N2 before it is reduced, which is the key to achieving a high FE for urea. From the theoretical aspect, a variety of materials, including single- and dual-atom catalysts,16,17 Mo2P, and so on, have been explored.18 However, efficient CO–N2 coupling remains challenging, and early transition metals or other reactive elements are required for this step. However, the performance of existing materials is still far from satisfactory.
Transition metal phthalocyanines (MPcs) are molecular catalysts characterized by a single transition metal atom embedded within a porphyrin ring, structurally analogous to single-atom catalysts supported on nitrogen-doped graphene. The catalytic properties of the metal site in MPcs closely resemble those of graphene,19 and MPcs have been applied in a wide range of catalytic research studies.20–22 For example, Duan et al. applied NiPc for CO2 reduction and found that the field effect may lower the barrier of the reaction.23 Hao Li et al. investigated the pH dependence of the M–NC type catalysts (including MPcs) for oxygen reduction and identified an “acid trap” that should be avoided in the design of M–NC type catalysts.24 For urea synthesis, Shibata et al. explored the performance of various MPcs in the electrocatalytic synthesis of urea from CO2 and nitrite25 in 2001. They found that the FE for urea is related to the catalyst's ability to simultaneously reduce CO2 and nitrite. More recently, Ghorai et al. demonstrated that CoPc supported on MoS2 can efficiently transform CO2 and N2 to urea with a urea yield of 175.6 mg h−1 mgcat−1 at −0.7 VRHE.26 Similarly, CuPc nanotubes were found to co-reduce CO2 and N2 to urea with a yield of 143.47 μg h−1 mgcat−1 and a faradaic efficiency of 12.99% at −0.6 VRHE.27 These studies suggest that even single-atom catalysts can facilitate the coupling of CO2 and N2 (or nitride) to urea, despite the presence of only one metal site for the adsorption and activation of intermediates. The MPc catalysts may function as tandem catalysts that can convert both the CO2 and N-containing molecules to the coupling intermediates, and the coupling reaction probably proceeds via the Eley–Rideal mechanism in which one of the C–N coupling intermediates desorb to react with the other adsorbed one.
However, the efficiency of CoPc and CuPc catalysts in urea synthesis remains suboptimal, highlighting the need for the exploration of more efficient MPc catalysts. In this study, we employ density functional theory (DFT) computations to evaluate the performance of 3d–5d transition metal phthalocyanines in the electrocatalytic coupling of CO2 and N2 for urea synthesis. Our findings identify several factors that limit the performance of MPc-based catalysts in this reaction, and we propose strategies to enhance their activity in the subsequent discussion.
2 Computational methods
All calculations were performed utilizing density functional theory implemented in the Vienna ab initio simulation package (VASP).28–30 The projector-augmented wave pseudopotential was employed for electron–ion interaction.31 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was adopted,32 with a plane-wave basis set cutoff energy of 460 eV. The metal phthalocyanines were modeled within a hexagonal simulation cell with the size of 22.20 Å × 22.20 Å × 14.00 Å to maintain a minimum distance of 7.00 Å between the atoms in adjacent periodic images. The Brillouin zone was sampled exclusively at the Gamma point. The dispersive interactions were accounted for using the DFT-D3 method.33,34 The solvent effects were included by applying corrections of −0.25 eV to COOH* species and −0.10 eV to oxygen-terminated species.35 Structural optimizations were performed until the maximum force on any atom was less than 0.02 eV Å−1. The transition state for C–N coupling is sought using the nudged elastic band method with climbing images.36,37 The Gibbs free energy changes for the reactions were calculated using the computational hydrogen electrode model, which relates the energy of the (H+ + e−) pair to that of H2(g).38 A systematic correction of −0.13 eV and 0.29 eV was applied to gas-phase CO2 and CO molecules to align reaction free energies with experimental values.39 Free energy corrections were made using the harmonic approximation with the gas-phase molecules treated as ideal gases under standard conditions.35 The adsorption energy (Eads) of gas-phase species at MPc were defined as Eads = E(mole/sub) − E(mole) − E(sub) where E(mole/sub) is the total electronic energy of the MPc with the molecule adsorbed, E(mole) is the electronic energy of the free molecule, and E(sub) is the electronic energy of the MPc. The free energy change for the adsorption (ΔGads) was determined by adding the free energy corrections to Eads.3 Results and discussion
3.1 The competitive adsorption between CO2/CO and N2
In the reaction, the gas-phase molecules may compete for the adsorption site. We first investigate the competitive adsorption of CO2, CO, and N2. The typical adsorption structures are depicted in Fig. 1. CO2 is found to be physically adsorbed in a linear configuration on MPcs when the metal M is a late transition metal, including group 9–12 transition metals such as Co, Ni, Cu, Zn, Rh, Pd, Ag, Cd, Ir, Pt, and Au, as well as Mn, Fe. In contrast, on early transition metal sites, CO2 is chemically adsorbed in a bent configuration with the carbon and one oxygen atom coordinated to the metal. This difference is attributed to the higher number of unoccupied d-orbitals in early transition metals, leading to a higher d-band center and thus stronger adsorption capabilities.16 On CrPc, we found that CO2 can be chemically adsorbed, but this state is 0.65 eV less stable than the physically adsorbed state. For N2, physical adsorption occurs at group 10–12 metal sites, while chemical adsorption in an end-on mode is observed on other transition metal sites. Ghorai et al. reported that on CuPc, N2 can be adsorbed at the N site on the MPc rings,27 but we found that adsorption at the transition metals site is consistently more favorable. For example, the adsorption of N2 at the Co site is more favorable than the N site by 0.22 eV. For CO, it is also physically adsorbed on group 10–12 transition metal elements and chemically on the early ones. When N2 and CO are chemically adsorbed, they adopt a vertical orientation, whereas physical adsorption results in a more tilted configuration. Compared to N2 and CO, the reluctance of CO2 to be chemically adsorbed on group 8–9 metals is likely due to its need to bend, which demands more reactive metal sites for effective chemical adsorption.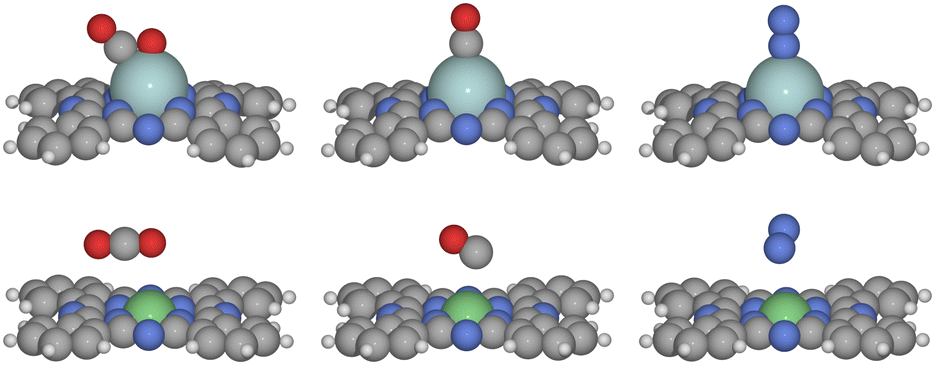 | ||
| Fig. 1 The optimized structure of chemically adsorbed CO2, CO and N2 on ZrPc and physically adsorbed CO2, CO and N2 on NiPc. | ||
The parity plot between ΔGads of CO2 and N2 is shown in Fig. 2(a). We found that ΔGads(CO2) is lower than ΔGads(N2) on nearly all MPcs except for VPc, RePc, OsPc, and RuPc. This observation contrasts somewhat with the results above, which demonstrated that N2 can be chemically adsorbed on a wider range of transition metal sites. In some cases, this is due to free energy corrections. Take FePc for example, Eads(CO2) and Eads(N2) on FePc in a vacuum are −0.30 eV and −0.47 eV, suggesting a stronger interaction between N2 and FePc. However, the sum of free energy correction and solvent correction to Eads(CO2) is 0.20 eV lower than that to Eads(N2), resulting in ΔGads(CO2) being 0.03 eV lower than ΔGads(N2). Therefore, when ΔGads(CO2) − ΔGads(N2) > 0.20 eV, for example, on HfPc and NbPc, the interaction of MPc with CO2 is stronger than with N2.
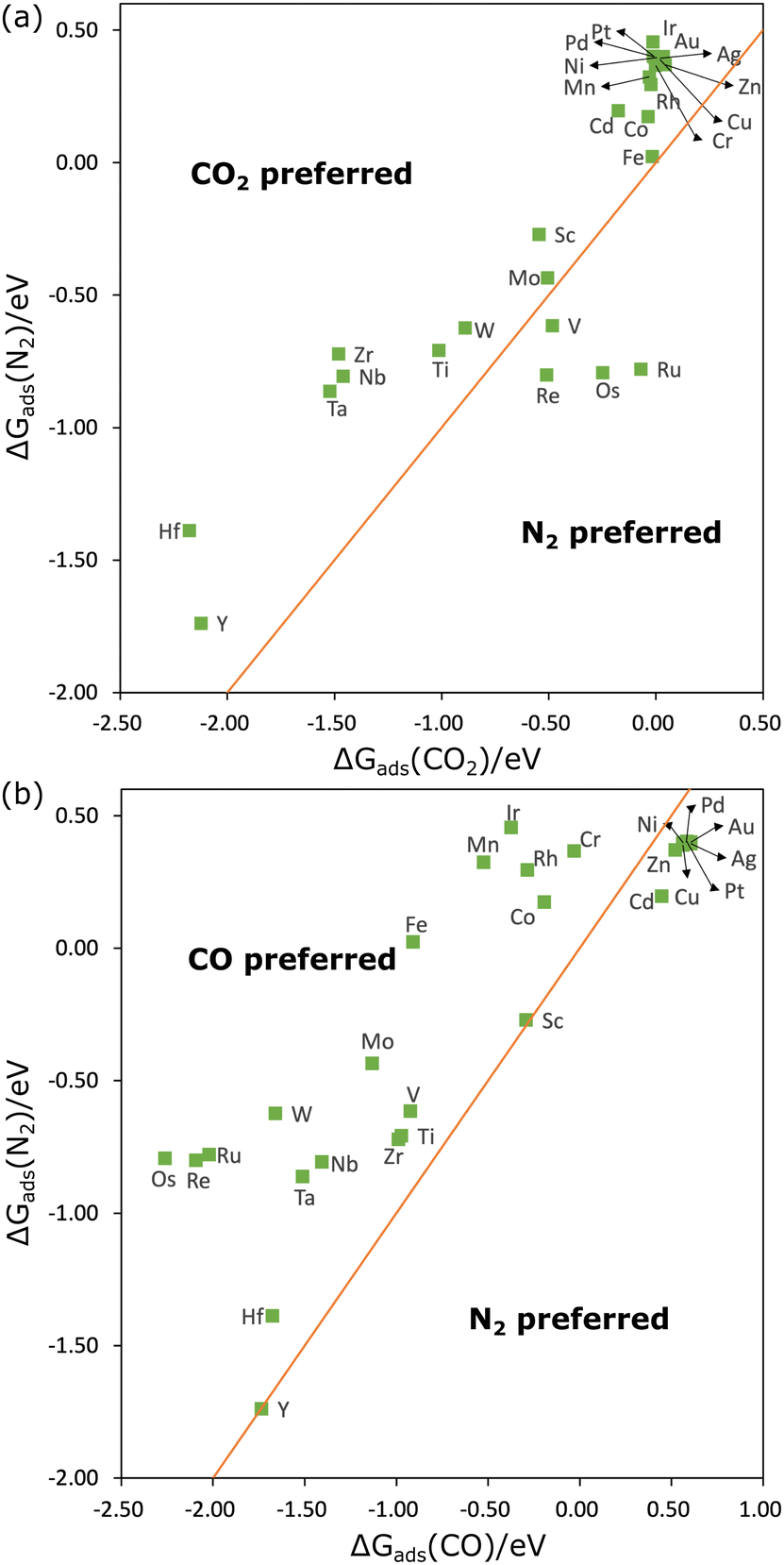 | ||
| Fig. 2 The adsorption free energy of N2 against that of CO2 (a) and CO (b). The orange line is the parity line (y = x). | ||
The parity plot comparing ΔGads of CO and N2 is shown in Fig. 2(b). Interestingly, ΔGads(N2) is lower than ΔGads(CO) when both molecules are physically adsorbed (on group 10–12 elements). When chemically adsorbed (on group 3–9 elements), CO adsorption is preferred in almost all cases. This is probably because of the better match of the electronic structure of CO with those substrates. For example, we have included the projected density of states of CO and N2 adsorbed on FePc in Fig. S1 (ESI†). It was found that the PDOS of N2 is sharper and more discrete than that of CO, suggesting that the interaction of N2 with FePc is weaker than CO. On the other hand, the net Bader40 charge of adsorbed CO is 0.18e−, while that on adsorbed N2 is 0.11e−, which is also consistent with a stronger interaction with CO. For ScPc and YPc, ΔGads(N2) is nearly equivalent to ΔGads(CO), and on Ti, V, Cr, Co, Hf, and Zr sites, ΔGads(N2) is also very close to ΔGads(CO), suggesting N2 can compete with CO at these catalysts. This competition is beneficial for the formation of C–N bonds, as it suggests that N2 may be efficiently activated for C–N coupling. This could be one of the reasons why CoPc on MoS2 can effectively reduce CO and N2 to urea.26
3.2 Competition between CO/N2 reduction and C–N coupling
In the reaction, CO2 is proposed to be reduced to CO, which is expected to couple with N2 to form C–N bonds.5–15 However, the reduction of CO may compete with the C–N coupling step. Fig. 3 compares the free energy changes associated with CO reduction and C–N coupling. We define ΔGi as the reaction free energy for reaction (i), in which ΔG1 and ΔG2 are the reaction free energies for CO* reduction to CHO*, and CO* coupling with N2(g) respectively.| CO* + H+ + e− → CHO* | (1) |
| CO* + N2(g) → CON2* | (2) |
| N2* + H+ + e− → HNN* | (3) |
| N2* + CO(g) → CON2* | (4) |
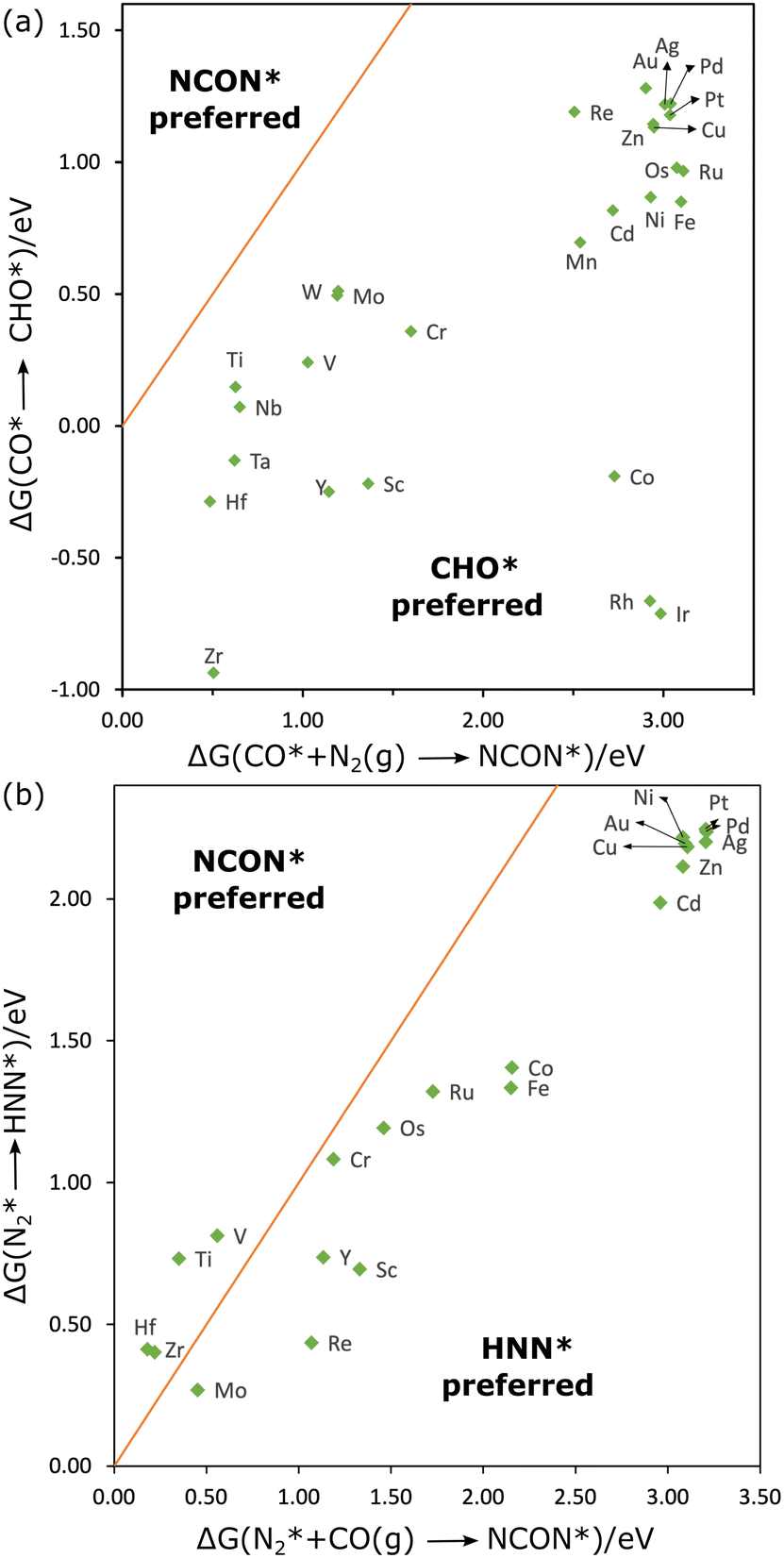 | ||
| Fig. 3 Competition of CO–N2 coupling with CO reduction (a) and N2 reduction (b), NCON includes CON2. | ||
As illustrated in Fig. 3(a), ΔG1 is significantly lower than ΔG2 for all the MPc catalysts, suggesting that the C–N coupling is not favored over CO reduction when CO is preferentially adsorbed. Furthermore, the CO*–N2(g) coupling at the group 7–12 metal sites is endothermic by more than 2.00 eV, suggesting that the sites are not active for urea synthesis. The ΔG2 value is lower than 1.00 eV only on TiPc, HfPc, NbPc, TaPc, and ZrPc. Notably, ΔG2 on CuPc is about 3.05 eV which agrees with the results in the previous study by Ghorai et al.27 This discrepancy may imply that urea production in their study may occur via an alternative mechanism, such as CO–NH2 coupling, or that additional factors, like the field effect from ions23,41, could be influential.
The reaction can also proceed in a tandem style in which one catalyst site efficiently reduces CO2 to CO, and the other converts nitrogen-containing molecules to intermediates that can couple with CO.42,43 Subsequently, CO produced by other catalysts could desorb and react with adsorbed N2 at the MPc. If N2 is initially adsorbed at the metal site, its reduction will compete with C–N coupling. We defined ΔG3 and ΔG4 as the reaction free energies for N2* reduction to HNN* and that for N2* coupling with CO(g), respectively. Fig. 3(b) presents the parity plot between ΔG3 and ΔG4 for group 10–12 elements which have comparable ΔGads for CO and N2 (Fig. 2(b)), and prefer N2 over CO2 (Fig. 2(a)). For Ti, V, Hf, and ZrPc, ΔG3 values are significantly higher than ΔG4, and ΔG4 is relatively lower on these sites, suggesting their potential as efficient catalysts for C–N coupling. An energy-decomposition analysis (Fig. S2, ESI†) reveals that C–N coupling is only preferred on these four MPcs owing to the formation of NCON, requiring too much energy input which can only be compensated when the adsorption of NCON is strong enough. However, since CO adsorption is still favored over N2 on these four metal sites, these catalysts may still exhibit low selectivity for the desired C–N coupling reaction.
3.3 Free energy surfaces for urea synthesis
The free energy surfaces (FES) for CO–N2 coupling to urea are shown in Fig. 4, and the optimized structures of the intermediates are summarized in Fig. S3 (ESI†). We start with N2 adsorption because the reaction is likely to proceed selectively towards CO reduction instead of C–N coupling if CO is adsorbed before N2 (the FES for CO2 reduction to CO can be found in Fig. S4 (ESI†) which shows that TiPc, VPc and HfPc can efficiently reduce CO2 to CO). On TiPc, the reduction of NCON* to NCONH* is uphill by 0.39 eV. Then NCONH* can be reduced via the distal path to form NCONH2*, or via the alternating path to form NHCONH*. The reaction is highly exothermic to either of the two intermediates, but NHCONH* is more favored by 0.35 eV. Subsequent reduction of NHCONH* to NHCONH2*, and NHCONH2* to NH2CONH2* is endothermic by 0.06 and 0.53 eV, respectively. Therefore, the potential-limiting step is the reduction of NHCONH2* to NH2CONH2* with a limiting potential of −0.53 V. This step was also identified as potential-limiting on MoP5 and single-atom catalysts.16 The profile of the free energy surface on VPc is quite similar to that on TiPc, but with weaker adsorption of the intermediates and a preference for the distal path over the alternating path. The potential-limiting step remains the reduction of NHCONH* to NHCONH2*, but with a smaller limiting potential of −0.25 V. Although this is smaller than that on TiPc, the C–N coupling is more difficult than TiPc, suggesting that TiPc may be more active than VPc if the rate-limiting step is the C–N coupling.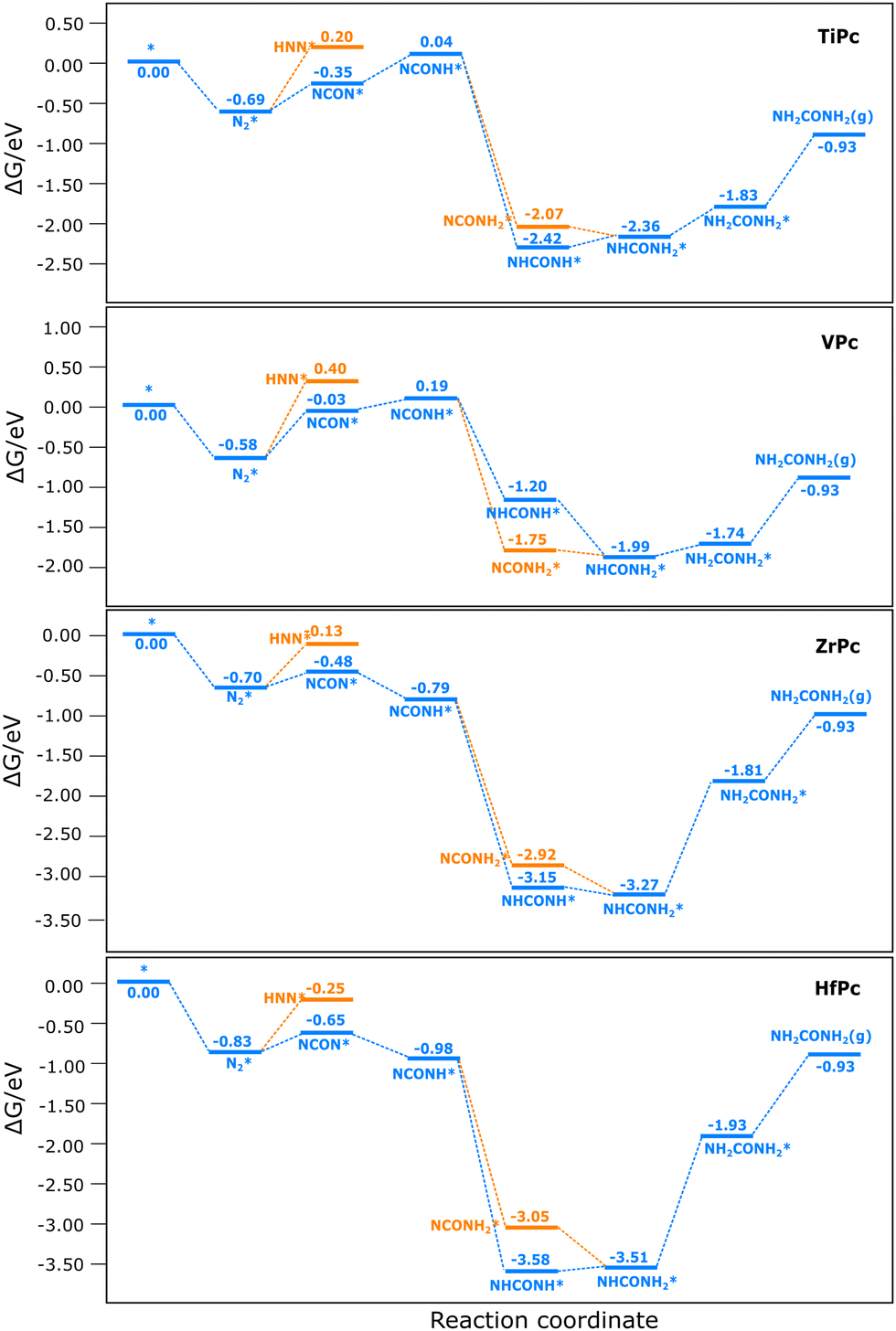 | ||
| Fig. 4 The free energy surfaces for CO and N2 coupling to urea on Ti, V, Zr and HfPc. | ||
The free energy surface on ZrPc is similar to that on HfPc but significantly different from that on TiPc and VPc. Firstly, the adsorption of the intermediates is generally much stronger. Secondly, the reduction of NCON* to NCONH* is no longer uphill. Thirdly, though the C–N coupling becomes more favored than TiPc and VPc, the reduction of NHCONH2* to NH2CONH2* becomes much more difficult with a limiting potential of −1.46 eV for TiPc and −1.58 eV for HfPc, indicating that these catalysts are not very active for urea synthesis. Therefore, the potential-limiting step on these four catalysts is the reduction of NHCONH2* to NH2CONH2* because they bind the intermediates strongly.44 Additionally, the desorption of urea is endergonic by around 0.80 to 1.00 eV, which may limit the reaction.
3.4 C–N coupling
Although the CO(g)–N2* coupling is thermodynamically preferred over N2 reduction, we need to further estimate its activity by calculating the kinetic barrier for the coupling. Fig. 5(a) shows the free energy change and the structures for CO–N2 coupling on the four MPcs. We have only shown the structures on ZrPc because they are quite similar on the four catalysts. It is found that the product of the coupling reaction is in a linear form (CON2), as has been observed in other studies,45,46 rather than a triangular shape (NCON, like CO32−). We found that for M in group 4–6 elements (except for Cr), the MPc prefers the linear CON2, while other MPcs prefer NCON (in an η1 structure on group 7–12 MPc, and η2 structure on Sc and YPc, Fig. S5, ESI†). Both CON2 and NCON are adsorbed via the N atom, suggesting that CO must desorb for N2 to be adsorbed if the reaction starts with CO*–N2(g).17,27 Despite the C–N coupling reaction being endothermic by only 0.18–0.56 eV, the kinetic barrier is calculated to be 1.42–2.09 eV, indicating that the reaction is unfavorable at room temperature. The magnitude of the barrier agrees well with the results on other atomic catalysts.16,17,44,47,48 Overall, the barrier increases with the reaction energy in Fig. 5(a), and fits the Brønsted–Evans–Polanyi (BEP) relation well in Fig. 5(b). Since Hf and Zr have the lowest coupling reaction energy, the barrier will not be lower than 1.40 eV on other MPc catalysts. Note that if the reaction starts with CO*–N2(g), the reaction should have the same transition state, but the barrier becomes higher because CO*–N2(g) is lower in energy than CO(g)–N2* for almost all MPcs. With such a high kinetic barrier, the MPcs are not active for urea synthesis via electrocatalytic coupling of CO with N2 (even if the barrier is lower than that for CO* reduction to CHO*). However, Fig. 5(b) shows that the barrier also scales well with the adsorption energy of N2, indicating that enhancing the adsorption of N2 may further promote the activity of the catalysts for urea synthesis. This could be achieved by modifying MPc molecules with function groups21 or via the field effect.23,41 For example, the substitution of H with the OMe functional group21 or the field effect from a nearby potassium ion was shown to significantly stabilize the COOH* intermediate which greatly promotes the activity of NiPc for CO2 reduction.23 Similar strategies may be applied to MPc molecules.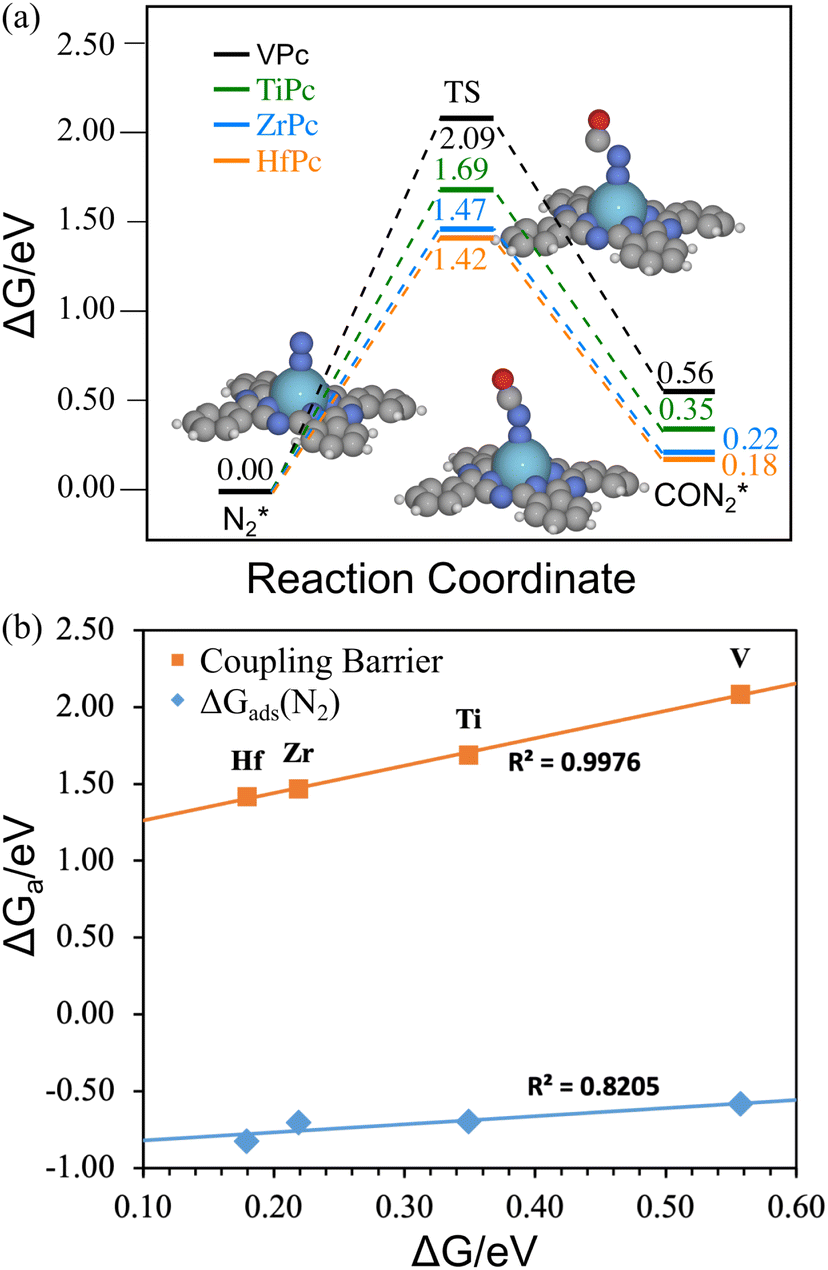 | ||
| Fig. 5 (a) The free energy surface for CO–N2* coupling, only the structures on ZrPc are shown, TS stands for transition state; (b) the BEP relation between the reaction energy and barrier for CO–N2* coupling, and the scaling relation between the barrier and N2 adsorption free energy. | ||
On the other hand, the work on FeN4/graphene49 and Ru(0001) surfaces50 suggests that the kinetic barrier for the hydrogenation of the nitrogen reduction intermediates via proton-coupled electron transfer (PCET) is normally small if the reaction is exothermic, or close to the reaction energy if the reaction is endothermic. Subsequently, the kinetic barrier for the reduction of N2* to HNN* may be lower than that for CO(g)–N2* coupling, especially under certain potentials, indicating that N2 reduction will become more favorable than the C–N coupling which further lowers the urea selectivity.
Another nitrogen intermediate that has been proposed frequently for C–N coupling, especially for electrocatalytic co-reduction of CO2 with NOx, is NH2*.2,43 To form urea, one CO couples with NH2 to form the intermediate CONH2, requiring either CO or NH2 to have a low desorption barrier for diffusion and coupling. And to form NH2CONH2, either CONH2 or NH2 should have a low desorption barrier. Therefore, if the adsorption of NH2 is weak, it can easily desorb to couple with CO and CONH2; if its adsorption is strong, it would require the adsorption of CO and CONH2 to be weak enough. We have calculated the adsorption free energy of NH2 and CONH2 for all the 3d–5d MPcs. As shown in Fig. 6(a) (blue region), the adsorption of NH2 is rather weak at the group 10–12 elements, this is probably one of the reasons that they showed significantly higher efficiency for the co-reduction of CO2 and nitrite to urea.25 However, these metal phthalocyanines have a high limiting potential (Fig. 6(b)) for N2* reduction to HNN* (around −2.20 eV)51 which makes them unfavorable for the co-reduction of CO2 and N2. For early transition meal phthalocyanines, the strong adsorption of CO and CONH2 hinders the desorption and coupling of the intermediates. This is in agreement with the experimental findings that Ti, V, Mo, and Ru phthalocyanines cannot co-reduce CO2 and nitrite to urea.25 To couple efficiently, ΔGads of NH2 and CONH2 should be probably no lower than −1.50 eV (because FePc was found active but CrPc was not25 (Fig. 6(a) orange region). The reduction of N2* to HNN* by Mn, Fe, and CoPc has a limiting potential of −1.16 V, −1.33 V and −1.40 V (Fig. 6(b)), which agrees well with the finding that CoPc is also active for CO2–N2 coupling under certain potentials.26 However, the reduction of NH2* to NH3* on these three phthalocyanines is quite exothermic (Fig. 6(b)), suggesting that urea won’t be more selective than ammonia.26 In a word, the co-reduction of CO2 and N2 to urea via the CO–NH2 coupling is not efficient as well.
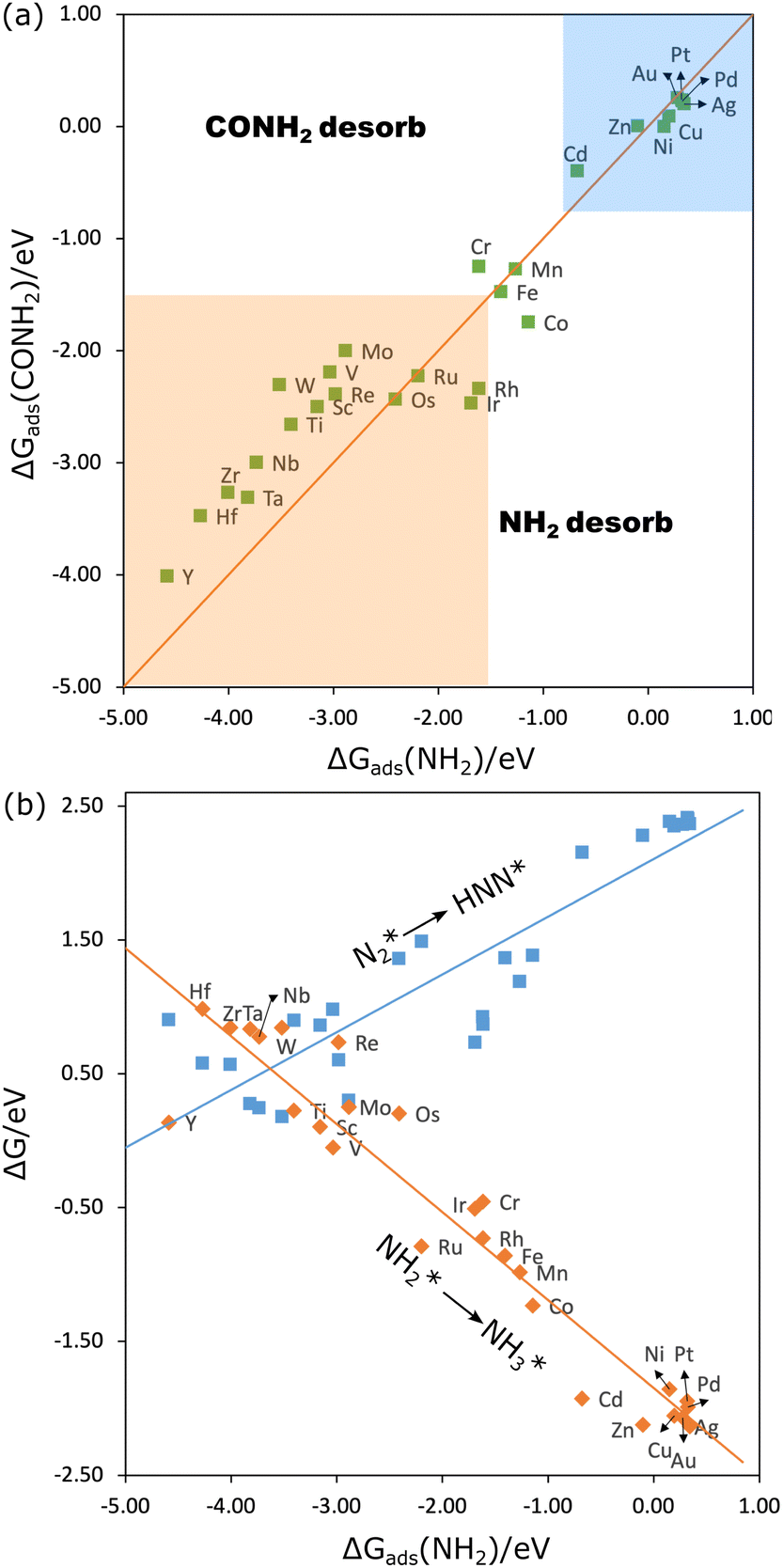 | ||
| Fig. 6 (a) The adsorption free energies of NH2 and CONH2, the blue and orange regions are where the binding of the intermediates is too weak and too strong. (b) The reaction free energies for N2* to HNN* and NH2* to NH3*. | ||
Although we cannot completely exclude all other alternative C–N coupling pathways,47 a comprehensive investigation of these possibilities would require the calculation of all the kinetic barriers for the C–N coupling step and the competing PCET steps,52 which requires extensive computational resources. Moreover, such alternative pathways are rarely reported in existing experimental studies for electrocatalytic CO2 and N2 coupling, and thus are not included in this work.
4 Conclusions
In summary, our study has thoroughly examined the catalytic efficacy of 3d–5d transition metal phthalocyanine catalysts for the electrocatalytic synthesis of urea from CO2 and N2. The findings indicate several challenges that limit the efficiency of MPc catalysts in this reaction: firstly, in most instances, N2 fails to compete with CO2 and CO for adsorption on the transition metal sites, hindering the activation of N2, even in the context of tandem catalysts; secondly, CO–N2 coupling cannot compete with CO reduction and/or N2 reduction in most cases; thirdly, the kinetic barrier for C–N coupling is no lower than that on ZrPc and HfPc (about 1.40 eV) which is not surmountable at room temperature, and the potential may further lower the urea selectivity by promoting the PCET steps. Therefore, it is very challenging to design efficient single-atom catalysts for urea synthesis. In addition, the results suggest that co-reduction of CO2 and N2 to urea via the CO–NH2 coupling is not efficient as well because of the high over-potential for N2 reduction or because of the low selectivity compared to ammonia formation. However, the results of this work suggest that future work could focus on the development of catalysts with tailored electronic properties (by using functional groups) or the use of external fields to influence the adsorption and desorption of key intermediates. Additionally, experimental validation of the theoretical predictions presented here could provide further insights into the design of efficient catalysts for electrocatalytic urea synthesis from CO2 and N2.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province (2023A1515012238 and 2023A1515010051), the National Natural Science Foundation of China (22273024), and the Guangzhou Municipal Science and Technology Bureau (202201010374, 202201010612, and 202201020145).References
- C. Yang, Z. Yang, W. Zhang, A. Chen and Y. Li, Chem. Commun., 2024, 60, 5666–5682 RSC.
- C. Yang, Z. Li, J. Xu, Y. Jiang and W. Zhu, Green Chem., 2024, 26, 4908–4933 RSC.
- W. Meng, D. Wang, H. Zhou, Y. Yang, H. Li, Z. Liao, S. Yang, X. Hong and G. Li, Energy, 2023, 278, 127537 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS.
- D. Jiao, Y. Dong, X. Cui, Q. Cai, C. R. Cabrera, J. Zhao and Z. Chen, J. Mater. Chem. A, 2023, 11, 232–240 RSC.
- X. Chen, S. Lv, J. Kang, Z. Wang, T. Guo, Y. Wang, G. Teobaldi, L.-M. Liu and L. Guo, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2306841120 CrossRef CAS PubMed.
- Z. Lv, S. Zhou, L. Zhao, Z. Liu, J. Liu, W. Xu, L. Wang and J. Lai, Adv. Energy Mater., 2023, 13, 2300946 CrossRef CAS.
- W. Wu, Y. Yang, Y. Wang, T. Lu, Q. Dong, J. Zhao, J. Niu, Q. Liu, Z. Hao and S. Song, Chem Catal., 2022, 2, 3225–3238 CrossRef CAS.
- P. Xing, S. Wei, Y. Zhang, X. Chen, L. Dai and Y. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 22101–22111 CrossRef CAS.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- S. Su, X. Li, W. Ding, Y. Cao, S. Yuan, Z. Liu, Y. Yang, Y. Ding and M. Luo, J. Mater. Chem. A, 2024, 278, 15300–15310 RSC.
- L. Pan, J. Wang, F. Lu, Q. Liu, Y. Gao, Y. Wang, J. Jiang, S. Chao, J. Wang and X. Wang, Angew. Chem., Int. Ed., 2022, 62, e202216835 CrossRef PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, K. Cheng, J. Zheng, S. Li, X. Tu, W. Chen, C. Xie, X. Wei, D. Wang, Y. Liu, P. Chen, S. P. Jiang, Y. Li, Q. Liu, C. Li and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305447 CrossRef CAS PubMed.
- Z. Xiong, Y. Xiao and C. Shen, Chem. Mater., 2022, 34, 9402–9413 CrossRef CAS.
- C. Zhu, M. Wang, C. Wen, M. Zhang, Y. Geng, G. Zhu and Z. Su, Adv. Sci., 2022, 9, 2105697 CrossRef CAS.
- J. Liu, X. Guo, T. Frauenheim, Y. Gu and L. Kou, Adv. Funct. Mater., 2024, 34, 2313420 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- A. Adalder, S. Paul, B. Ghorai, S. Kapse, R. Thapa, A. Nagendra and U. K. Ghorai, ACS Appl. Mater. Interfaces, 2023, 15, 34642–34650 CrossRef CAS.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- C. Chen, X. Wang, C. Wang, H. Zhang, X. Yang, Y. Sun, X. She, X. Zhao and D. Yang, J. Environ. Chem. Eng., 2023, 11, 109084 CrossRef CAS.
- S. Wei, H. Zou, W. Rong, F. Zhang, Y. Ji and L. Duan, Appl. Catal., B, 2021, 284, 119739 CrossRef CAS.
- D. Zhang, Z. Wang, F. Liu, P. Yi, L. Peng, Y. Chen, L. Wei and H. Li, J. Am. Chem. Soc., 2024, 146, 3210–3219 CrossRef CAS.
- M. Shibata and N. Furuya, J. Electroanal. Chem., 2001, 507, 177–184 CrossRef CAS.
- S. Paul, S. Sarkar, A. Adalder, A. Banerjee and U. K. Ghorai, J. Mater. Chem. A, 2023, 11, 13249–13254 RSC.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- P. E. Blöchl, C. J. Först and J. Schimpl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- X. Tu, X. Zhu, S. Bo, X. Zhang, R. Miao, G. Wen, C. Chen, J. Li, Y. Zhou, Q. Liu, D. Chen, H. Shao, D. Yan, Y. Li, J. Jia and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317087 CrossRef CAS.
- J. Fu, Y. Yang and J.-S. Hu, ACS Mater. Lett., 2021, 3, 1468–1476 CrossRef CAS.
- Y. Gao, J. Wang, M. Sun, Y. Jing, L. Chen, Z. Liang, Y. Yang, C. Zhang, J. Yao and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202402215 CrossRef CAS PubMed.
- L. Kong, D. Jiao, Z. Wang, Y. Liu, Y. Shang, L. Yin, Q. Cai and J. Zhao, Chem. Eng. J., 2023, 451, 138885 CrossRef CAS.
- Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Natl. Sci. Rev., 2020, 7, 1564–1583 CrossRef CAS PubMed.
- M. Wang, L.-Y. Chu, Z.-Y. Li, A. M. Messinis, Y.-Q. Ding, L. Hu and J.-B. Ma, J. Phys. Chem. Lett., 2021, 12, 3490–3496 CrossRef CAS PubMed.
- C. Zhu, C. Wen, M. Wang, M. Zhang, Y. Geng and Z. Su, Chem. Sci., 2022, 13, 1342–1354 RSC.
- Z. Chen, Y. Liu and T. Wang, Chem. Sci., 2023, 14, 12707–12714 RSC.
- C. Choi, G. H. Gu, J. Noh, H. S. Park and Y. Jung, Nat. Commun., 2021, 12, 4353 CrossRef CAS PubMed.
- E. Tayyebi, Y. Abghoui and E. Skúlason, ACS Catal., 2019, 9, 11137–11145 CrossRef CAS.
- C.-X. Huang, G. Li, L.-M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2021, 13, 608–621 CrossRef CAS.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2022, 13, 5471 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04047c |
| This journal is © the Owner Societies 2025 |