
Light driven photoswitches: three classes of molecular systems that result in a single photoproduct via a conical intersection and an exothermic reverse reaction
Shmuel
Zilberg

Department of Chemical Sciences, Materials Research Center, Ariel University, 4076414 Ariel, Israel. E-mail: shmuelz@ariel.ac.il
First published on 20th November 2024
Abstract
A common feature of molecular photoswitches is the selectivity of their photo-processes. The photoswitching model must combine a selective photochemical direct route and a thermal reverse reaction from the product back to the parent reactant. The conical intersection model is an appropriate approach to this problem. Valence bond analysis of the ground state reactions between the photoswitching isomers provides a chemically oriented approach to locate the conical intersection and to define its two coordinates. Three different classes of molecular photoswitches have been identified:(1) A reactant and product are connected by two distinct reaction routes with two different transition states. The conical intersection is situated inside this phase-inverting loop. An example of this class of photoswitches is an isomerization around a polar double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN or NN bond. The capacity to store energy is indicated by the energy gap between the reactant and product. However, this can also result in the destabilisation of the product. For instance, the addition of bulky substituents can disrupt the planar fragment around the double bond, leading to the loss of π-conjugation. Two non-equivalent isomers with different contributions of polar and biradical forms can exhibit a highly distorted conical intersection topology. (2) The photoreaction leading to two photoproducts is a regular case. However, this case could be a “quasi single product”, if two products or the parent reactant and one of the products are equivalent isomers. This is the second type of photoswitch. (3) If one of the two products is at a higher energy level than both the reactant and the main product, then this is also a possible molecular photoswitching mechanism. The second high energy product is likely to be unstable and it is close to conical intersection. The norbornadiene–quadricyclane pair is an example of this type of photoswitch.
C, CN or NN bond. The capacity to store energy is indicated by the energy gap between the reactant and product. However, this can also result in the destabilisation of the product. For instance, the addition of bulky substituents can disrupt the planar fragment around the double bond, leading to the loss of π-conjugation. Two non-equivalent isomers with different contributions of polar and biradical forms can exhibit a highly distorted conical intersection topology. (2) The photoreaction leading to two photoproducts is a regular case. However, this case could be a “quasi single product”, if two products or the parent reactant and one of the products are equivalent isomers. This is the second type of photoswitch. (3) If one of the two products is at a higher energy level than both the reactant and the main product, then this is also a possible molecular photoswitching mechanism. The second high energy product is likely to be unstable and it is close to conical intersection. The norbornadiene–quadricyclane pair is an example of this type of photoswitch.
Introduction
Photoswitches are a class of compounds that undergo reversible photo-transformation, rendering them a potential material for information storage. From another side, photoinduced isomerization has been proposed as a possible way to store solar energy.The amazing natural photoswitch–rhodopsin is based on an elementary chemical step, the photochemical cis–trans isomerization around a carbon–carbon double bond in the retinal chromophore as was specially pointed by B. Feringa in his Nobel lecture.1 Among the most studied photoswitches are compounds with polar double bonds that are capable of efficient interconversion between cis- and trans-isomers.2 The efficient cis–trans interconversion was detected for compounds with CC (stilbenes3 and merocyanine (MRCN)4) and NN double bonds (azobenzenes5,6). Photoisomerization cyclisation is a common phenomenon observed in a variety of systems, including those documented for diarylethenes,7,8 fulgimides,9–11 hydrazones12,13 arylazoimidazole,14,15 arylazopyrazole16,17 and spiropyrans (SPP).18
The intramolecular interconversion between norbornadiene (NBD) and its higher energy isomer quadricyclane (QC) is an important model system for molecular solar thermal energy storage. This photoswitchable pair of NBD-QC isomers looks very attractive as a molecular battery, because a significant energy difference ΔE(NBD-QC) could provide effective conversion of solar energy into thermal energy.19–24
Molecular photoswitches, conical intersection and valence bond description
Molecular photoswitches have two routes between the same reactant–product pair: (1) direct photochemical transition and (2) reverse thermal route:
Various photoswitchable systems have been reported in the literature, but usually the unique electronic mechanism has been proposed for each case. This work aims to present a common approach to the analysis of the electronic mechanism of molecular photoswitches. Experimental studies and quantum chemical calculations of the different photochromic processes lead to the conclusion that this photoprocess passes through the ConInt. A common feature of the previously listed photoswitches is the selectivity of their photo-processes, only a single product is observed. Consequently, the photoswitching model must combine the coexistence of both selective processes: a selective photoreaction, as well as a thermal back route from the product to the parent reactant. This key feature of photoswitches is contradicted the rules based on the correlation between reactant and product, like Woodward–Hoffmann rules.25,26 It should be noted that the assumption of a unique transition state (TS) is common to all correlation models. The correlation between reactant and product along the reaction coordinate is an indication of the preferable TS for the ground state reaction.25,26 This leads to a dichotomy in the analysis of chemical reactions: what is allowed thermally is forbidden photochemically, and vice versa. However, a photochemical reaction is going through the electronic degeneracy that occurs along two specific coordinates as was proposed by Teller and named the conical intersection (ConInt).27
The topology of the multidimensional surface in the vicinity of the electronic degeneracy (ConInt) has a significant effect on the probability of transitions between electronic states,28,29 and how the systems evolve towards the final stable structures on the electronic ground state.30
Computational evidence for the existence of ConInts in many polyatomic systems including photoswitches is known. Nevertheless, the interpretation of photoreaction mechanism is often the result of serendipity as was pointed by González.31 Qualitative models present the photochemical reaction vs. the thermal ground state reaction as a dichotomy.25,26,32,33
Longuet-Higgins formulated the phase change theorem,34,35 which describes the special property of electronic degeneracy: “a conical intersection necessarily arises within a region enclosed by a loop, provided the total electronic wave function changes phase when transported along a complete circle around the loop”.34 If the electronic wave function (EWF) is real, the phase change is expressed as a sign change.
A chemically oriented approach for the location of ConInt was proposed at the end of the 1990s.36 This method is based on valence bond (VB) analysis, using the dominant Lewis structures of reactants and products, for the description of the ground state and the lowest excited state.37–40 Elementary 2D domains surrounded by reaction coordinates define the two- or three-legged loops that was first introduced by Longuet-Higgins for the analysis of electronic degeneracy of the H3 system. It has been shown34 that the total electronic wave function changes continuously during the reaction from that of the reactant (φR) to that of the product (φP). The electronic wave function of the TS can be represented by a linear combination of the electronic wave functions of the reactant and product.
| ΦPP = φR + φP (phase preserving (PP) TS) | (1) |
| ΦPI = φR − φP (phase inverting (PI) TS) | (2) |
We assume that all TSs between anchor structures of this loop and electronic degeneracy inside the loop can be described as a linear combination of the dominant VB functions of the anchors of an elementary loop.41,42
Consider the reactant R and a desired product P1 as two ‘points’ on the potential energy surface (PES). The reaction coordinate leading from R to P1 is the first coordinate. To complete a loop, it is necessary to add a third ‘point’ P2 on the PES which is chemically distinct from both R and P1. The reaction coordinate leading from P1 to P2 (or from R to P2) may be chosen as the second coordinate (Fig. 1a). This analysis may be conducted in a straightforward manner, using the VB anchor structures for the description of the selected elementary domain on the ground state PES.38,39
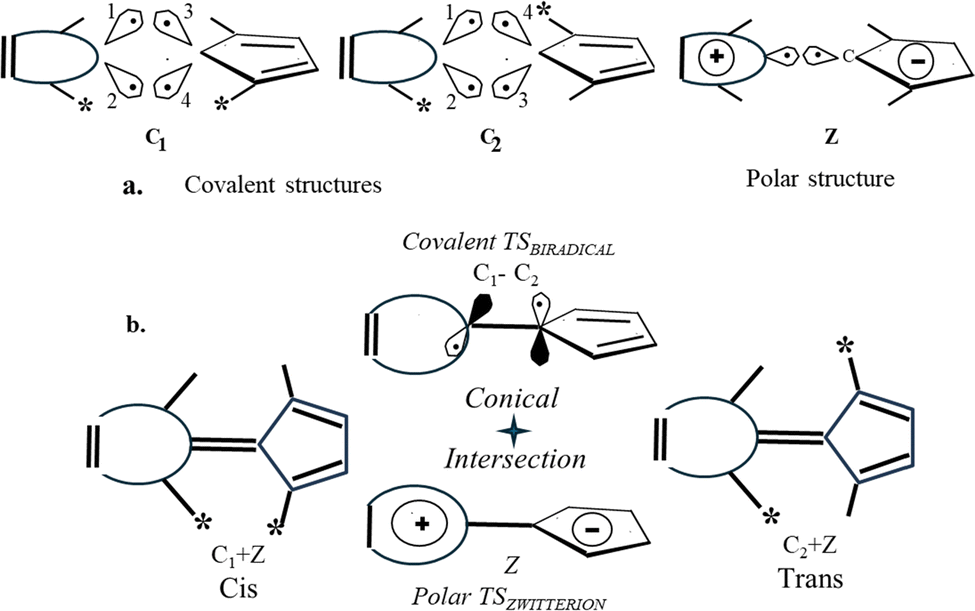 | ||
| Fig. 1 (a) Three dominant VB forms describing the cis- and trans-isomers of the molecule with a polar double bond: two covalent contributions C1 (cis), C2 (trans), and a polar form Z (common for both isomers). (b) ConInt inside the phase-inverting loop formed by two different cis–trans isomerization coordinates. Both TSs, the phase-inverting non-polar TSBIRADICAL and the phase-preserving polar TSZW have a dominant contribution of the covalent (C1 − C2) and polar Z forms respectively. | ||
A Longuet-Higgins loop is formed by the transport of the electronic wave function Φ along the trajectory R → P1 → P2 → R. The overall phase change is given by the combination of the phase changes incurred in the individual reactions.
A three-legged phase-inverting loop (Scheme 1a and c) is a key point for the understanding of the numerous organic photoreactions leading to two photoproducts.37,38,43–45
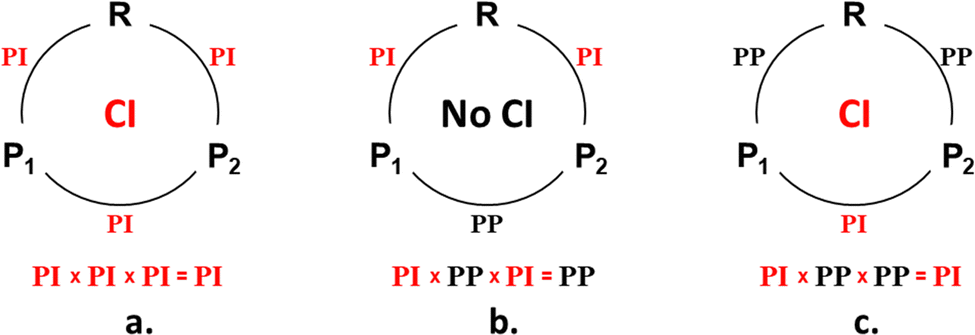 | ||
| Scheme 1 Three possible three-legged loops, where R is the reactant and P1 the desired product and P2 is a second possible product. Loops in which a ConInt may be found are designated as phase inverting (PI) (a) and (c). The loop that does not include a ConInt is designated as phase preserving (PP) (b). | ||
ConInt of single product's photoreaction has a unique topography which arises from the fact that the reactant R is connected to the product P via two different reaction coordinates (Scheme 2).46–48 The VB approach provides a lucid rationalization of the chemical reaction with two TSs.49 If the reactant and product pair has been described by three dominant VB structures, they can be connected by two different TSs. The different resonance possibilities – in-phase and out-of-phase combinations, allow for the distinction between two different TSs. To illustrate, if one minimum is described by a single anchor A and the second one by a combination of B and C (Scheme 2a). To illustrate, the two-legged loop will be phase-inverting and ConInt inside if one minimum is described by a single structure A and the second one by a combination of B and C of two dominant forms (Scheme 2a). The PES topography can be inverted, if the AC (A ↔ C) and AB (A ↔ B) combinations are two equivalent minima, whereas the two different TSs are A≠ and BC≠ structures (Scheme 2b).
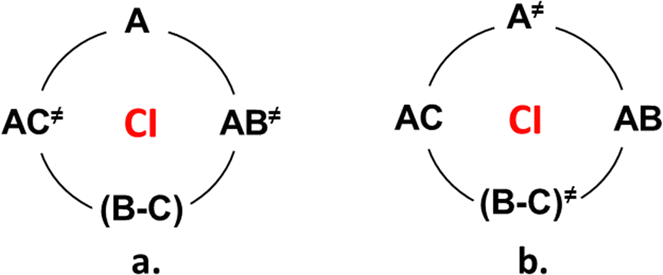 | ||
| Scheme 2 Two types of two-legged domains describing by three VB anchors system: (a) two different isomers (A and BC) connected by two TSs AC≠ and AB≠; and (b) two isomers AC and AB connected by two different TSs – A≠ and BC≠. | ||
Both loops (Scheme 2a and b) are phase inverting if B ↔ C resonance is described via anti-combination (B–C) in the ground state. These are two models of single product photosystems with two TSs and ConInt inside the loop.
Results and discussion
The location of the ConInt of the photoswitch is a key point in the study of the photochemical mechanism. VB analysis of the ground state reactions between the photoswitching isomers provides a chemically oriented approach to describing the ConInt. The coordinates of ConInt are the corresponding elementary reaction coordinates connecting reactant and product.37–40,49Cis–trans isomerization around double polar bond
The cis–trans interconversion is a most studied single product photoreaction. The different cis–trans photo-isomerization reactions are known as ultrafast selective photo-processes, which occur via ConInt.2,29,36,40,47The VB model of cis–trans isomerization around a polar CC double bond predicts that two different TSs are possible if three dominant VB structures – the covalent component CC (cis) versus CT (trans), whereas a third term, polar form (Z), is common to both:47,49
| |Reactant〉 = |CC〉 + |Z〉 | (1a) |
| |Product〉 = |CT〉 + |Z〉 | (1b) |
If covalent and polar forms make similar contributions to the cis–trans isomers with a polar double bond, they can produce two TS structures between reactant and product where one can be described as a phase-inverting TS(PI) (2a) and second as a phase-preserving TS(PP) combination (2b):
| |TS(PI)〉 = |Reactant〉 − |Product〉 = |C1〉 − |C2〉 | (2a) |
| |TS(PP)〉 = |Reactant〉 + |Product〉 = 2|Z〉 + |C1〉 + |C2〉 | (2b) |
Thus, the reactant can be photochemically transformed into the product via the ConInt, because the loop is phase inverting:47,49
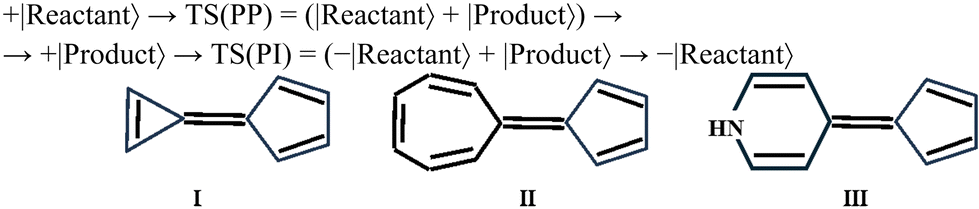 | (3) |
This condition is fulfilled for the molecules with two conjugated π-rings: one of them is the π-acceptor cyclopentadiene (right fragment for I–III) and the left one is the π-donor: cyclopropene for (I), cycloheptatriene for (II) and dihydropyridine for (III). The π-donor–acceptor conjugation can lead to a very effective π-charge transfer and produce the low lying zwitterionic form (Z).
Two covalent structures have a different pairing scheme for the cis- (C1) and for the trans- (C2) isomer (Fig. 1a), whereas the polar structure Z is common for both isomers (Fig. 1a).
The loop presented in Fig. 1b is a particular case of a cis–trans isomerization phase-inverting loop (3):
(C 1 + Z) → TSZW (Z) → (C2 + Z) → TSBIRADICAL (C2 – C1) → −(C1+ Z) is phase-inverting49 and the ConInt exists inside this loop.
In all cases (I–III) ConInt between biradical (11A2) and zwitterionic (11A1) states was detected at the CASSCF level of theory. However, the position of the ConInt is different, and it depends on the relative energy of polar vs. covalent forms in the reactant/product. The studied case shows the separation between covalent and zwitterionic forms in two different TSs as was predicted early.49 Our CASSCF calculations show that the lowest TS of (I) is zwitterionic (E = 18 kcal mol−1, μ = 10.2D), of (II) is biradical (E = 10 kcal mol−1, μ = 0.6D), while in (III) the biradical structure (μ = 2.2D) is only 2.4 kcal mol−1 more stable than polar TS (μ = 16.3D). Calculations predict that molecules I, II and III have a different topology of ConInt.
Molecule III has a peaked ConInt with two equidistant TSs whereas molecules I and II show a strong distortion from the peaked topology, because in both cases one of electronic states are significantly higher than other. Thus, the lowest point of biradical state is close to the ConInt in molecule I. It means that de facto only polar TS will be operative in the case of I. The picture is reversed in molecule II. It has a non-polar biradical TS but the lowest point of polar state in the vicinity of the ConInt.
Consequently, molecular systems I–III predict the different effects of electric field or polar solvent on the photo-processes as has been theoretically predicted50a,b and experimentally confirmed for the isomerization around the polar double bond.50c Thus, the stabilization of the polar state, including the polar TS, by electric field or solvent, leads to an acceleration of the thermal reaction, but it can annihilate the crossing, destroy the ConInt funnel, and strongly delay the photo-transition. This is the electric field/solvent effect in the case of molecule I (Fig. 2). Increasing of the electric field/solvent effect shows the opposite picture in case II. There is no change in the photo-process, but the barrier in the ground state will be higher due to the stabilisation of the reactant relative to the non-polar biradical TS. The weak solvent effect can’t change the ultrafast photoreaction and only slightly accelerates the thermal reaction in case III.
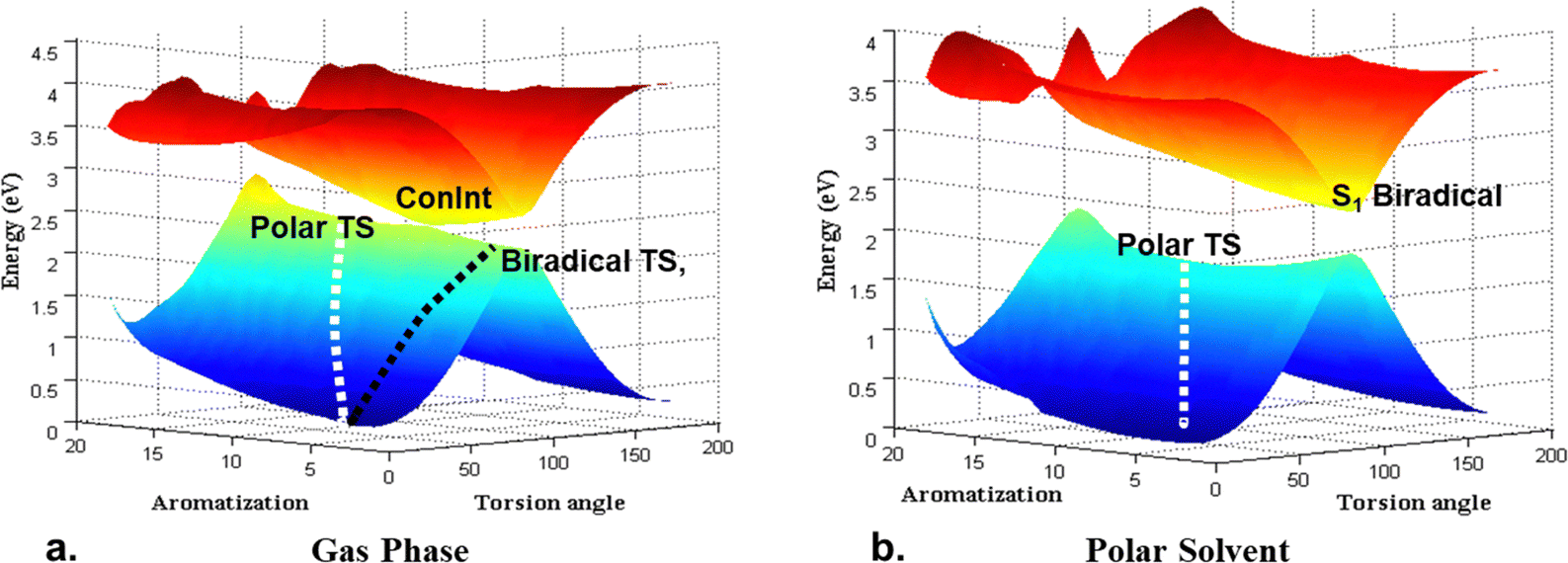 | ||
| Fig. 2 Schematic presentation of the ground S0 and the lowest excited state S1 PESs for cis–trans isomerization around polar double bond in a model structure I: (a) gas phase; and (b) upon a polar solvent. | ||
Molecules with a polar double bond serve as a prototype for the various photoswitches.1
Merocyanine: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C photoisomerization and regioselectivity
C photoisomerization and regioselectivity
A push–pull MRCN51 is a molecule substituted at opposite ends of a butadiene fragment by a donor and an acceptor. Such a molecule is expected to have a significant zwitterion contribution and therefore to be strongly influenced by the polar environment. Two isomerization processes are possible around the two double bonds of the butadiene backbone via the different S0/S1 ConInts.4
The transition through the different ConInt to the different products constitutes a demonstration of the possibility to tune the regioselectivity: thus, the photoproduct Pr-A is favoured in the gas phase via the CI-A (Fig. 3a); photoproduct Pr-B via the CI-B is favoured in a low polar solvent (Fig. 3b) and the photoreaction disappears in a high polar environment (Fig. 3c).
 | ||
| Fig. 3 Schematic representation of the solvent effect on the photoisomerization mechanism of model MRCN. The photoproduct Pr-A is favoured in the gas phase (a), whereas in the low polar solvent the photoproduct Pr-B is favoured (b). In the polar solvent, the ConInt disappears (c), leading to the enhanced fluorescence yield. | ||
CASSCF calculations50b show that the contribution of the zwitterionic structure in the ground state increases with increasing the polarity of the solvent or with increasing strength of an external electric field. At high electric fields, the ground state structure can be transformed to a pure zwitterionic form (Fig. 3c).
In MRCNs solvent dependencies of the quantum yields have been observed and possible relaxation mechanisms have been discussed.52–55 An ultrafast study of a MRCN molecule has shown that the fluorescence lifetime can be tuned by changing the solvent polarity.50c,56 This fully confirms a theoretical prediction50a,b that the fluorescence lifetime is significantly shortened when the polarity of the solvent is reduced due to tuning of the ConInt properties. The solvent dependence of the regioselectivity of the MRCN affects the photoswitching character, up to its disappearance.
The spiropyran–merocyanine conversion
Spiropyrans (SP) are one of several classes of photochromic compounds that are effective photoswitches (Scheme 3).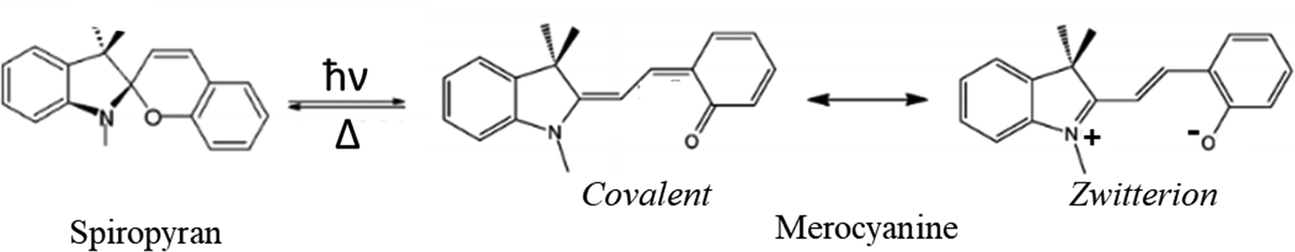 | ||
| Scheme 3 Spiropyran–merocyanine interconversion. Two dominant VB forms in MRCN molecules. | ||
Ring-opened zwitterionic MRCNs and ring-closed SPs are interconvertible isomers. Ultrafast studies have provided a picture of the dynamics of the photochromic process in indolinobenzospiropyrans.56,57 The photoinduced ring-opening SP-MRCN process through the ConInt has been studied by Robb and coworkers58 at the CASSCF level of theory. This selective ultrafast photoreaction can be explained by the strong mixing of the zwitterionic form with the covalent Lewis structure of MRCN, which together with VB form of the SP fulfils the conditions for the localization of ConInt inside the SP-MRCN two-legged loop. This case is similar to the case of cis–trans isomerization around a polar double bond, which also leads to a single photoproduct (Scheme 2a).
Isomerization around a CN and a NN double bond
Two different TSs are possible for syn–anti isomerization around NN or CN double bonds: a perpendicular biradical TS and an in-plane inversion TS (Fig. 4). In-plane H-atom motion provides syn–anti isomerization by N-atom inversion without changing chemical bonding including π-bonds. Torsion leads to the biradical transition structure TStorsion. The in-plane TSinversion has a low barrier whereas the barrier of the biradical TStorsion is considerably higher. These two reaction pathways constitute the phase-inverting loop with a ConInt inside (Fig. 4).47,49
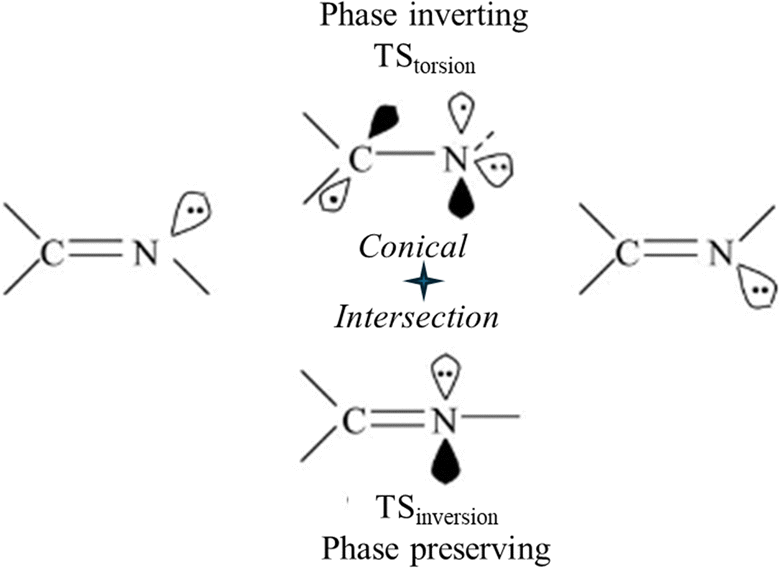 | ||
| Fig. 4 A two-legged phase inverting loop for syn–anti isomerization around the CN double bond, which connects two isomers through two TSs. | ||
This model can serve as a common scheme for different photoswitches with NN or CN fragment.
Norbornadiene–quadricyclane photoswitches
The intramolecular NBD—QC and their derivatives interconversions (Scheme 4) were studied for a long time.19,20 The NBD molecule, which photoconverts to a more strained QC, is of great interest due to its high energy storage density. On the other hand, QC is a stable compound and has the potential to store energy for a very long time. The parent NBD has some practical limitations, but various NBD derivatives show considerably enhanced characteristics.21,22 The light-induced switching between NBD and QC isomers is an ultrafast photoprocess.59,60 The model [2+2]-ethylene dimerization is photochemically allowed according to Woodward–Hoffmann rules25 and the HOMO–LUMO crossing has been predicted at the rectangular structure. In contrast to this assumption the ConInt geometry according to MCSCF calculations in ethylene dimerization is not rectangular but is distorted to a rhombus.61 The ultrafast pathway is going the crossing between two valence states S0–S1 ConInt, which leads to both the reactant (NBD) and the product (QC). Computational studies on the CASSCF level of theory show that the structure of S0–S1 ConInt has a distinct rhombic distortion.62,63 Recent ultrafast dynamics studies have supported this interpretation.60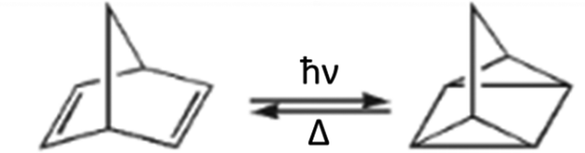 | ||
| Scheme 4 Norbornadiene–quadricyclane rearrangement. | ||
The photo-transformations of butadiene – a parent molecule with two π-bonds leads to two photo-products – cyclobutene and bicyclobutane. Every transformation: butadiene–cyclobutene; cyclobutene–bicyclobutane and bicyclobutane–butadiene describe by two electron pairs involving in resonance, which means the TS structure is a phase-inverting for all three elementary reactions according to the VB analysis.41,42 These three anchor structures define the elementary phase inverting loop that includes ConInt according to the Longuet-Higgins theorem.34,35 This ConInt was detected on the CASSCF level of theory.64
The NBD and QC pair is analogous to the butadiene–cyclobutene pair, but the rigid polycyclic frame provides a strong additional strain in the QC isomer. The strain effect produces a substantial difference between these two systems, because it doesn't allow construction of the bicyclobutane fragment in the rigid NBD polycycle. Such a type of bonding could be reached in the rhombic biradical structure of NBD but calculations detected electronic degeneracy in this geometry. This is a reason for the selective (single product) photoreaction of NBD.
Benzene–benzvalene potential molecular photoswitches
The benzene–benzvalene pair is a potential photoswitch with high solar energy storage capacity. Benzvalene is a product of the photolysis of benzene (Fig. 5), but it is thermally converted to benzene with a large release of heat. Benzvalene has a high steric deformation energy, which is 71 kcal mol−1 higher than that of benzene.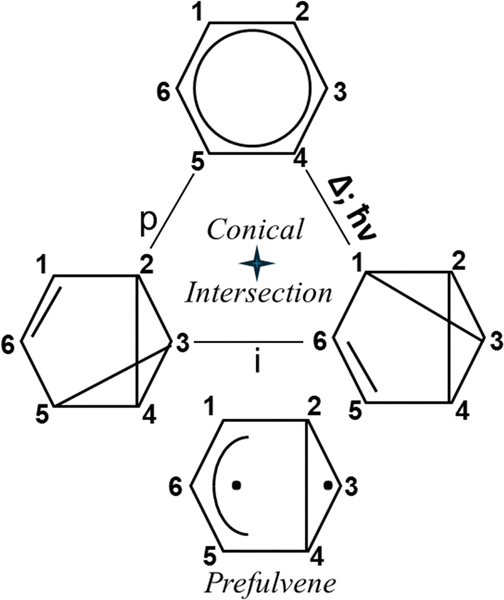 | ||
| Fig. 5 The photoisomerization of benzene to benzvalene via prefulvene ConInt. | ||
VB and MO approaches showed that the benzvalene–benzene transformation has an aromatic TS, and this is an allowed reaction in the ground state. Consequently, the photochemical valence isomerization of benzene to benzvalene is forbidden according to the MO/VB correlation approach.65
The photochemical transformation of benzene, which leads to benzvalene, was explained by the prefulvene S0/S1 ConInt66,67 (Fig. 5). The prefulvene structure plays a central role in photochemical valence isomerization of benzene to form benzvalene (Fig. 5). Prefulvenic ConInt structures have been identified computationally in toluene,68 phenol,69 isomeric o-, m- and p-chloroanilines,70 and hexafluorobenzene.71 Substituents variation affects the prefulvenic funnel and this indicates the approach to the possible design of effective molecular benzene type photoswitches.
The photochemical transformation of benzene to benzvalene is different from the previous photoswitching cases because two isomeric products are formed. Nevertheless, it is also a selective photoreaction because both products are equivalent.
Consequently, the benzene–benzvalene pair is a potential photoswitch, particularly for solar energy storage, if chemical modification will provide its thermal stability.
Irradiation of cyclooctatetraene (COT) yields semibullvalene (SB), in spite of the fact that this photochemical reaction is forbidden by orbital conservation rules.36,72,73 The cyclooctatetraene (COT)–semibullvalene (SB) photo-rearrangement (Fig. 6) includes two isomeric Kekule type structures of COT, and one SB structure. COT is an antiaromatic molecule and the transition between the two COT isomers going through the planar symmetric configuration describing as an out-of-phase combination of two Kekule forms.40,41 The transition from either one to semibullvalene is phase-preserving, since only six electrons are involved in the transition state because one pair is “frozen” in this reaction. The Longuet-Higgins loop for this system is phase inverting (Fig. 6) and the photoreaction is made possible by the fact that COT valence isomerization, a phase-inverting reaction, takes place simultaneously. This COT → SB system satisfies the single product photoswitching condition because the reactant exists in two equivalent isomeric forms – COT(L) and COT(R) (Fig. 6).
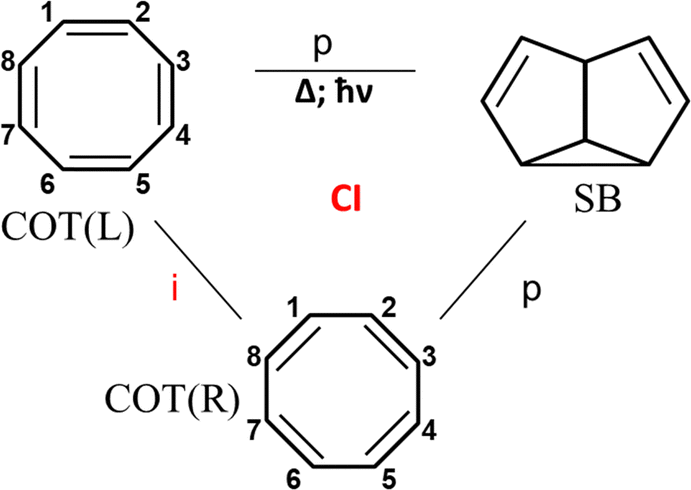 | ||
| Fig. 6 The cyclooctatetraene to semibullvalene reaction. | ||
Three classes of molecular photoswitches: topology of potential energy surface in the vicinity of ConInt for and the problem of solar energy storage
Three different classes of photoswitches have been identified in the cases discussed above:(1) A reactant–product pair connected by two different reaction routes with two different TSs on the ground state PES and ConInt inside the loop (Scheme 2 and Fig. 7a)).49 A cis–trans isomerization around the polar double bond is an example of this class of photoswitches. If two isomers exhibit comparable contributions of polar and covalent forms, this can result in polar and non-polar biradical TS (Fig. 7a). The ability to store energy is indicated by the energy gap between reactant and product. However, it can also lead to the destabilization of the product. For example, the addition of the bulky substituents can disrupt the planar fragment around the double bond, leading to the loss of π-conjugation. Two non-equivalent isomers with different contributions of polar and biradical forms, can have a highly distorted ConInt topology.50a,b
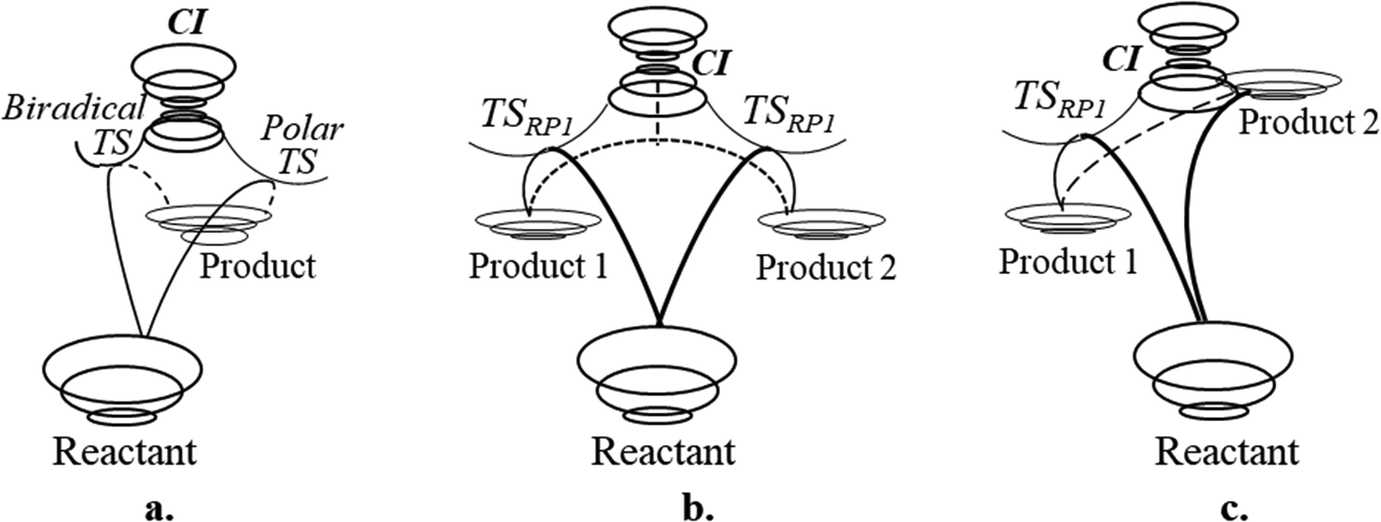 | ||
| Fig. 7 Selective photoprocess passing through ConInt: (a) Reactant and product connected by two reaction paths through biradical and polar TSs and ConInt between them. (b) Reactant and two equivalent isomeric products constructing a three-legged loop and ConInt inside. (c) ConInt leading to stable Product 1. Second Product 2 in the vicinity of ConInt is unstable and cannot be observed. | ||
(2) The three-legged loop molecular systems – reactant and two products – could be “quasi single product” if two products (Fig. 7b, see also Fig. 5 and 6) or the parent reactant and one of the products are equivalent isomers.
(3) A reactant and two different products, but one of the products is at a higher energy than the other two (Fig. 7c). This is a regular case in organic photochemistry. It is obvious that the highly strained product can be located close to the ConInt. In this case, the second product is probably unstable.
Conclusions
The key point of the activity of the photoswitches is the selectivity of the photoconversion and the formation of the single product. On the other hand, there is a thermal back reaction from the product to the reactant, which allows the release of part of the solar energy storage. Two different reaction paths connecting the reactant to a single product indicate the existence of a ConInt inside the loop according to Longuet-Higgins phase change rule.34,35 This ConInt, which connects a lowest excited state to a ground state, combines both the photo and thermal paths of a photoswitch. The chemically oriented VB model makes it possible to check the existence of the ConInt for the chosen photoprocess by analysing the ground state reactions that can create a phase-inverting loop.36 A cis–trans isomerization around polar double bond is an example of a reactant–product pair connected by two different reaction routes with two different TSs on the ground state PES and ConInt inside the loop.49 This is the first class of photoswitches.Two products, or reactant and one product, could be equivalent isomers. This is a second class of possible photoswitches that can be defined as a “quasi single product” case.
The case of a reactant and two different products is a regular case in organic photochemistry. However, one of the products can possess a higher energy than the other two, and this highly strained product can be situated in close proximity to the ConInt. In this case, the second product is a high energy unstable intermediate. This is a third class of molecular photoswitches.
The delicate balance between covalent and zwitterionic contributions is of paramount importance for the photoswitching structure. This is the reason that the photoswitches, in principle, will exhibit a strong solvent/electric field dependence on the regioselectivity, up to its disappearance.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The author declares no competing financial interest.References
- B. L. Feringa, The Art of Building Small: From Molecular Switches to Motors (Nobel Lecture), Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS.
- (a) A. Lennartson, A. Roffey and K. Moth-Poulsen, Designing photoswitches for molecular solar thermal energy storage, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS; (b) C. Dugave and L. Demange, Cis−Trans Isomerization of Organic Molecules and Biomolecules: Implications and Applications, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- (a) D. H. Waldeck, Photoisomerization dynamics of stilbenes, Chem. Rev., 1991, 91, 415–436 CrossRef CAS; (b) H. Görner and H. J. Kuhn, Cis–Trans Photoisomerization of Stilbenes and Stilbene-Like Molecules, Adv. Photochem., 1995, 19, 1–117 Search PubMed.
- N. Blaise, J. A. Green, C. Benitez-Martin, C. Kaiser, M. Braun, J. M. Schaible, J. Andréasson, I. Burghardt and J. Wachtveitl, Isomerization dynamics of a novel cis/trans-only Merocyanine, ChemPhotoChem, 2024, 8, e202300327 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- K. Matsuda and M. Irie, Diarylethene as a photoswitching unit, J. Photochem. Photobiol., C, 2004, 5, 169–182 CrossRef CAS.
- M. Irie, Diarylethene Molecular Photoswitches: Concepts and Functionalities, Wiley-VCH, 2021 Search PubMed.
- Y. Yokoyama, Fulgides for Memories and Switches, Chem. Rev., 2000, 100(5), 1717–1740 CrossRef CAS PubMed.
- S. Draxler, T. Brust, S. Malkmus, J. A. DiGirolamo, W. J. Lees, W. Zinth and M. Braun, Ring-opening reaction of a trifluorinated indolylfulgide: mode-specific photochemistry after pre-excitation, Phys. Chem. Chem. Phys., 2009, 11, 5019–5027 RSC.
- C. Slavov, N. Bellakbil, J. Wahl, K. Mayer, K. Rück-Braun, I. Burghardt, J. Wachtveitl and M. Braun, Ultrafast coherent oscillations reveal a reactive mode in the ring-opening reaction of fulgides, Phys. Chem. Chem. Phys., 2015, 17, 14045–14053 RSC.
- H. Qian, S. Pramanik and I. Aprahamian, Photochromic Hydrazone Switches with Extremely Long Thermal Half-Lives, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, Photon Energy Storage in Strained Cyclic Hydrazones: Emerging Molecular Solar Thermal Energy Storage Compounds, J. Am. Chem. Soc., 2022, 144(28), 12627–12631 CrossRef CAS PubMed.
- J. Otsuki, K. Suwa, K. Narutaki, C. Sinha, I. Yoshikawa and K. Araki, Photochromism of 2-(phenylazo)imidazoles, J. Phys. Chem. A, 2005, 109, 8064–8069 CrossRef CAS PubMed.
- J. Otsuki, K. Suwa, K. K. Sarker and C. Sinha, Photoisomerization and thermal isomerization of arylazoimidazoles, J. Phys. Chem. A, 2007, 111, 1403–1409 CrossRef CAS.
- C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, Arylazopyrazoles: azoheteroarene photoswitches offering quantitative isomerization and long thermal half-lives, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, Arylazopyrazoles as Light-Responsive Molecular Switches in Cyclodextrin-Based Supramolecular Systems, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- V. A. Bren′, A. D. Dubonosov, V. I. Minkin and V. A. Chernoivanov, Norbornadiene–quadricyclane—an effective molecular system for the storage of solar energy, Russ. Chem. Rev., 1991, 60, 451–469 CrossRef.
- K. Maruyama and Y. Kubo, in Organic Photochemistry, ed. W. M. Horspool and P.-S. Song, CRC Press, Boca Raton, 1995, p. 222 Search PubMed.
- K. Jorner, A. Dreos, R. Emanuelsson, O. El Bakouri, I. F. Galvan, K. Börjesson, F. Feixas, R. Lindh, B. Zietz, K. Moth-Poulsen and H. Ottosson, Unravelling factors leading to efficient norbornadiene–quadricyclane molecular solar-thermal energy storage systems, J. Mater. Chem. A, 2017, 5, 12369–12378 RSC.
- A. Dreos, Z. Wang, J. Udmark, A. Ström, P. Erhart, K. Börjesson, M. B. Nielsen and K. Moth-Poulsenet, Liquid norbornadiene photoswitches for solar energy storage, Adv. Energy Mater., 2018, 8, 1703401 CrossRef.
- J. Orrego-Hernández, J. Dreos and K. Moth-Poulsen, Engineering of norbornadiene/quadricyclane photoswitches for molecular solar thermal energy storage applications, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef.
- F. Hemauer, H.-P. Steinrück and C. Papp, The Norbornadiene/Quadricyclane Pair as Molecular Solar Thermal Energy Storage System: Surface Science Investigations, ChemPhysChem, 2024, 25, e202300806 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag chemie, Weiheim, 1970 Search PubMed.
- D. M. Silver, Hierarchy of symmetry conservation rules governing chemical reaction systems, J. Am. Chem. Soc., 1974, 96, 5959–5967 CrossRef CAS.
- (a) E. Teller, The crossing of potential surfaces, J. Phys. Chem., 1937, 41, 109–116 CrossRef CAS; (b) E. Teller, Internal conversion in polyatomic molecules, Isr. J. Chem., 1969, 7, 227–235 CrossRef CAS.
- G. J. Atchity, S. S. Xantheas and K. Ruedenberg, Potential energy surfaces near intersections, J. Chem. Phys., 1991, 95, 1862–1877 CrossRef.
- M. Ben-Nun, F. Molnar, K. Schulten and T. J. Martínez, The role of intersection topography in bond selectivity of cis–trans photoisomerization, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1769–1773 CrossRef CAS.
- C. A. Farfan and D. B. Turner, A systematic model study quantifying how conical intersection topography modulates photochemical reactions, Phys. Chem. Chem. Phys., 2020, 22, 20265–20283 RSC.
- S. Mai and L. González, Molecular Photochemistry: Recent Developments in Theory, Angew. Chem., Int. Ed., 2020, 59, 2–17 CrossRef PubMed.
- L. Salem, Electrons in Chemical Reactions: First Principles, John Wiley & Sons, Inc., New York, 1982 Search PubMed.
- W. A. Goddard, Selection rules for chemical reactions using the orbital phase continuity principle, J. Am. Chem. Soc., 1972, 94, 793–807 CrossRef CAS.
- G. Herzberg and H. C. Longuet-Higgins, Intersection of potential energy surfaces in polyatomic molecules, Dis. Faraday Soc., 1963, 35, 77–82 RSC.
- H. C. Longuet-Higgins, The intersection of potential energy surfaces in polyatomic molecules, Proc. R. Soc. London, Ser. A, 1975, 344, 147–156 CAS.
- S. Zilberg and Y. Haas, Molecular Photochemistry: A General Method for Localizing Conical Intersections Using the Phase-Change Rule, Chem. – Eur. J., 1999, 5, 1755–1765 CrossRef CAS.
- Y. Haas and S. Zilberg, Photochemistry by conical intersections: a practical guide for experimentalists, J. Photochem. Photobiol., A, 2001, 144, 221–228 CrossRef CAS.
- Y. Haas, S. Cogan and S. Zilberg, The use of elementary reaction coordinates in the search for conical intersections, Int. J. Quantum Chem., 2005, 102, 961–970 CrossRef CAS.
- S. Zilberg and Y. Haas, Locating electronic degeneracies of polyatomic molecules: a general method for non-symmetric molecules, J. Phys. Chem. A, 2003, 107, 1222–1227 CrossRef CAS.
- S. Zilberg and B. Dick, in Mechanistic Photochemistry and Conical Intersections, Excited States and Photodynamic Simulations from Photobiology to Photomaterials, ed. M. Yáñez and R. J. Boyd, Comprehensive Computational Chemistry, Elsevier, 1st edn, 2024, vol.4, pp. 25–54 Search PubMed.
- S. Zilberg and Y. Haas, A two state model of antiaromaticity: The low lying singlet states, J. Phys. Chem., 1998, 102, 10843–10850 CrossRef CAS.
- S. Zilberg and Y. Haas, The electron pair origin of antiaromaticity: spectroscopic manifestations, Int. J. Quantum Chem., 1999, 71, 133–145 CrossRef CAS.
- F. Bernardi, M. Olivucci and M. A. Robb, Potential energy surface crossings in organic photochemistry, Chem. Soc. Rev., 1996, 25, 321–328 RSC.
- M. A. Robb and M. Olivucci, Photochemical processes: Potential energy surface topology and rationalization using VB arguments, J. Photochem. Photobiol., A, 2001, 144, 237–243 CrossRef CAS.
- M. A. Robb, This molecule there must be a conical intersection, Adv. Phys. Org. Chem., 2014, 48, 189–228 CrossRef.
- (a) L. De Viko, C. S. Page and M. Garavelli, et al., Reaction path analysis of the “Tunable” photoisomerization selectivity of free and locked retinal chromophores, J. Am. Chem. Soc., 2002, 124, 4124–4134 CrossRef; (b) L. De Viko, M. Garavelli, F. Bernardi and M. Olivucci, Photoisomerization mechanism of 11-cis-locked artificial retinal chromophores: Acceleration and primary photoproduct assignment, J. Am. Chem. Soc., 2005, 127, 2433–2442 CrossRef PubMed.
- S. Zilberg and Y. Haas, Isomerization around a C = N double bond and a C = C double bond with a nitrogen atom attached: thermal and photochemical routes, Photochem. Photobiol. Sci., 2003, 2, 1256–1263 CrossRef CAS.
- O. Tishchenko, D. G. Thrular, A. Ceulemans and M. T. Nguen, A unified perspective on the hydrogen atom transfer and proton-coupled electron transfer mechanisms in terms of topographic features of the ground and excited potential energy surfaces as exemplified by the reaction between phenol and radicals, J. Am. Chem. Soc., 2008, 130, 7000–7010 CrossRef CAS PubMed.
- S. Zilberg, Chemical Reaction with Two Different Elementary Transition States, Int. J. Quantum Chem., 2014, 114, 1162–1168 CrossRef CAS.
- (a) S. Alfalah, O. Deeb, S. Zilberg and Y. Haas, Solvent effect on the conical intersection of 4-cyclopentadienylidene-1,4-dihydropyridine, Chem. Phys. Lett., 2008, 459, 100–104 CrossRef CAS; (b) X. Xu, A. Kahan, S. Zilberg and Y. Haas, Photo-reactivity of a push-pull merocyanine in static electric fields: A three state model of isomerization reactions involving conical intersections, J. Phys. Chem. A, 2009, 113, 9779–9791 CrossRef CAS PubMed; (c) A. Kahan, A. Wand, S. Ruhman, S. Zilberg and Y. Haas, Solvent tuning of a conical intersection: Direct experimental verification of a theoretical prediction, J. Phys. Chem. A, 2011, 115, 10854–10861 CrossRef CAS.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Cyanines during the 1990s:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A Review, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS PubMed.
A Review, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS PubMed. - S. Upadhyayula, V. Nuñez, E. M. Espinoza, J. M. Larsen, D. Bao, D. Shi, J. T. Mac, B. Anvari and V. I. Vullev, Photoinduced dynamics of a cyanine dye: parallel pathways of non-radiative deactivation involving multiple excited-state twisted transients, Chem. Sci., 2015, 6, 2237–2251 RSC.
- S. Wiedbrauk, B. Maerz, E. Samoylova, A. Reiner, F. Trommer, P. Mayer, W. Zinth and H. Dube, Twisted Hemithioindigo Photoswitches: Solvent Polarity Determines the Type of Light-Induced Rotations, J. Am. Chem. Soc., 2016, 138, 12219–12227 CrossRef CAS.
- C. Petermayer and H. Dube, Indigoid Photoswitches: Visible Light Responsive Molecular Tools, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- J. Hoche, A. Schulz, L. M. Dietrich, A. Humeniuk, M. Stolte, D. Schmidt, T. Brixner, F. Würthner and R. Mitric, The origin of the solvent dependence of fluorescence quantum yields in dipolar merocyanine dyes, Chem. Sci., 2019, 10, 11013–11022 RSC.
- J. Z. Zhang, B. J. Schwartz, J. C. King and C. B. Harris, Ultrafast Studies of Photochromic Spiropyrans in Solution, J. Am. Chem. Soc., 1992, 114, 10921–10927 CrossRef CAS.
- N. P. Ernsting and T. Arthen-Engeland, Photochemical Ring-Opening Reaction of Indollnosplropyrans Studied by Subpicosecond Transient Absorption, J. Phys. Chem., 1991, 95, 5502–5509 CrossRef CAS.
- (a) P. Celani, F. Bernardi, M. Olivucci and M. A. Robb, Conical Intersection Mechanism for Photochemical Ring Opening in Benzospiropyran Compounds, J. Am. Chem. Soc., 1997, 119, 10815–10820 CrossRef CAS; (b) I. Gómez, M. Reguero and M. A. Robb, Efficient Photochemical Merocyanine-to-Spiropyran Ring Closure Mechanism through an Extended Conical Intersection Seam. A Model CASSCF/CASPT2 Study, J. Phys. Chem. A, 2006, 110, 3986–3991 CrossRef.
- W. Fuß, K. K. Pushpa, W. E. Schmid and S. A. Trushin, Ultrafast [2 + 2]-cycloaddition in norbornadiene, Photochem. Photobiol. Sci., 2002, 1, 60–66 CrossRef PubMed.
- K. D. Borne, J. C. Cooper, M. N. R. Ashfold, J. Bachmann, S. Bhattacharyya, R. Boll, M. Bonanomi, M. Bosch and C. Callegari, et al., Ultrafast electronic relaxation pathways of the molecular photoswitch quadricyclane, Nat. Chem., 2024, 16, 499–505 CrossRef CAS PubMed.
- (a) F. Bernardi, S. De, M. Olivucci and M. A. Robb, The mechanism of ground-state-forbidden photochemical pericyclic reactions: evidence for real conical intersections, J. Am. Chem. Soc., 1990, 112, 1737–1744 CrossRef CAS; (b) F. Bernardi, M. Olivucci and M. A. Robb, Predicting forbidden and allowed cycloaddition reactions: potential surface topology and its rationalization, Acc. Chem. Res., 1990, 23, 405–412 CrossRef CAS.
- I. Antol, Photodeactivation Paths in Norbornadiene, J. Comp. Chem., 2013, 34, 1439–1445 CrossRef CAS.
- F. J. Hernández, J. M. Cox, J. Li, R. Crespo-Otero and S. A. Lopez, Multiconfigurational calculations and photodynamics describe norbornadiene photochemistry, J. Org. Chem., 2023, 88, 5311–5320 CrossRef.
- M. Olivucci, I. N. Ragazos, F. Bernardi and M. A. Robb, A conical intersection mechanism for the photochemistry of butadiene. A MC-SCF Study, J. Am. Chem. Soc., 1993, 115, 3710–3721 CrossRef CAS.
- E. A. Halevi, Orbital Symmetry and Reaction Mechanism, Ch. 5.3.2. The OCAMS View, Springer-Verlag, (GmbH), Berlin Heidelberg, 1992 Search PubMed.
- I. Ohmine, Mechanisms of nonadiabatic transitions in photoisomerization processes of conjugated molecules: Role of hydrogen migrations, J. Chem. Phys., 1985, 83, 2348–2362 CrossRef CAS.
- L. Blancafort and M. A. Robb, A valence bond description of the prefulvene extended conical intersection seam of benzene, J. Chem. Theor. Comput., 2012, 8, 4922–4930 CrossRef CAS PubMed.
- S. Cogan, Y. Haas and S. Zilberg, Intersystem crossing at singlet conical intersections, J. Photochem. Photobiol., A, 2007, 190, 200–206 CrossRef CAS.
- O. P. J. Vieuxmaire, Z. Lan, A. L. Sobolewski and W. Domcke, Ab initio characterization of the conical intersections involved in the photochemistry of phenol, J. Chem. Phys., 2008, 129, 224307 CrossRef PubMed.
- C. Nitu, J. J. van der Waals, N. Kaul, J. D. Steen, L. Hammarström, M. Fagnoni and S. Crespi, Meta-Ortho Effect on the Excited State Pathways of Chloroanilines, Eur. J. Org. Chem., 2024, e202300461 CrossRef CAS.
- J. M. Cox, M. Bain, M. Kellogg, S. E. Bradforth and S. A. Lopez, Role of the Perfluoro Effect in the Selective Photochemical Isomerization of Hexafluorobenzene, J. Am. Chem. Soc., 2021, 143, 7002–7012 CrossRef CAS PubMed.
- H. E. Zimmerman and H. Iwamura, Thermal and photochemical interconversions of cyclooctatetraenes and semibullvalenes. Exploratory organic photochemistry. LII., J. Am. Chem. Soc., 1970, 92, 2015–2022 CrossRef CAS.
- (a) M. Garavelli, F. Bernardi and A. Cembran, et al., Cyclooctatetraene computational photo- and thermal chemistry: A reactivity model for conjugated hydrocarbons, J. Am. Chem. Soc., 2002, 124, 13770–13789 CrossRef CAS; (b) D. Andrae, I. Barth and T. Bredtmann, et al., Electronic quantum fluxes during pericyclic reactions exemplified for the cope rearrangement of semibullvalene, J. Phys. Chem. B, 2011, 115, 5476–5483 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2025 |