Unveiling emissive H-aggregates of benzocoronenediimide, their photophysics and ultrafast exciton dynamics†
Received
24th October 2024
, Accepted 19th November 2024
First published on 20th November 2024
Abstract
H- and J-aggregates of many molecules can be considered ordered mesoscopic structures that behave like a single entity. This is due to coherent electronic coupling between electronic excitations of aggregated molecules, resulting in distinct electronic properties compared to the monomer. H-aggregates are generally non-emissive and, due to this property, they are considered unfit for optoelectronics applications, but they have found applications in organic light-emitting transistors. Herein, we designed t-butyl-substituted benzocoronenediimide (t-But-BCDI) forming rare emissive H-aggregates. The tertiary butyl groups are placed to inhibit the formation of strong aggregates. Photophysical studies showed that t-But-BCDI forms H-aggregates in a concentrated solution in a THF/CHCl3 mixture. A blue shift in absorption along with a decrease in the A0–0/A0–1 ratio and red-shifted weaker emission are observed for the aggregate compared to the monomer. Ultrafast transient absorption studies revealed biphasic relaxation with lifetimes of 150 (±10) fs and 13 (±2) ps, which are attributed to a higher-to-lower state transition and vibrational cooling, respectively. The transient spectral signature suggests the Frenkel-type (localized to a monomer) character of the exciton. Faster evolution at the tens of picosecond timescale suggests relaxation of the exciton state within the H-type exciton band. An extraordinarily long emission lifetime from the H-aggregated state is observed.
1. Introduction
Organic π-conjugated materials are of immense interest due to their applications in organic electronics, biological science, etc.1–4 Thin-film-forming properties, exciton diffusion, charge carrier properties, etc. of π-conjugated organic materials are essential for almost all organic electronic devices.5–7 Quite obviously, the electronic properties of organics in a solid thin film are often different from those in their solution phase. For organic π-conjugated materials, the electronic properties in the solid state are strongly influenced by their aggregation pattern and solid-state arrangement.8–11 Aggregates of many molecules can be considered as ordered mesoscopic structures that behave like a single entity. This is due to coherent electronic coupling between electronic excitations of aggregated molecules.12 Understanding electronic coupling in aggregates is important for the development of materials for organic-based devices, such as organic light emitting devices (OLEDs), solar cells, organic light emitting transistors (OLETs), light-harvesting in plants, molecular wires, and polymers.13–18 Based on molecular exciton coupling theory, aggregates exhibit distinct features in their absorption and emission bands compared to their monomers.19–21 According to Kasha's model, aggregates showing a bathochromic shift (red shift) in absorption features are called J-aggregates while those showing a hypsochromic shift (blue shift) are termed H-aggregates.19,20,22,23 The relaxation of the lowest excited state in J-aggregates is dipole allowed and results in a red shift in emission and a shorter excited state lifetime compared to the monomer.24,25 Many different classes of dyes, e.g., cyanine dyes,26 thiacarbocyanine,27 porphyrin,28 an perylenediimides,29 have been studied for J-aggregates. Oligothiophenes30 and poly-(3-hexylthiophene),31 which are capable of forming polymers by π-stacking, show the properties of H-aggregates.
Absorption and emission spectra of aggregates possess valuable information and a comparison of their vibronic features with the non-aggregated (monomer) spectrum provides insights into J- and H-aggregation. Kopainsky et al. first observed changes in vibronic features, which Scherer and Fischer later connected to J-aggregation.32,33 F. C. Spano and others presented the impact of vibronic coupling on the absorption and emission spectra of J- and H-aggregates.34,35 These studies revealed the distinguishing features of J- and H-aggregates. According to their work, J-aggregates show higher A0–0/A0–1 values than their monomers, which increase with the size of the aggregate. In contrast, H-aggregates show a decrease in A0–0/A0–1 compared to their monomers. The contrasting absorption features of J- and H-aggregates provide guidelines for studying the complex arrangement patterns in aggregates. J-aggregates of several linear perylene-3,4-9,10-bis(dicarboximide) derivatives showed an increase in A0–0/A0–1, as expected for J-aggregates.36,37 In the co-facial orientation of a covalently linked PDI derivative, a decrease in A0–0/A0–1 was observed and attributed to H-aggregation.38
Co-facial, parallel transition dipoles in H-aggregates usually exhibit fluorescence quenching, but these aggregates are advantageous in increasing the charge carrier mobility, which is greatly needed in organic electronic devices such as OLETs.15,20,21,34,35 Excimer state emission in aggregates has been investigated, but studies on the ultrafast dynamics of the initial exciton in H-aggregates are limited.38–44 Although H-aggregates are generally non-emissive, there are a few examples (such as p-distyrylbenzene cyclic triimidazole, and naphthalenediimides) of fluorescent H-aggregates.33–35 Considering the reports in the literature, we designed t-butyl-substituted benzocoronenediimide (t-But-BCDI, Fig. 1) and expected it to have face-to-face π–π interaction between two molecules due to its highly conjugated, planar, and symmetric core. The tertiary butyl groups are placed to inhibit the formation of strong aggregates. Our photophysical studies showed that t-But-BCDI forms H-aggregates in a concentrated solution in a THF/CHCl3 mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Compared to the monomer, a blue shift in absorption along with a decrease in the A0–0/A0–1 ratio and weaker, red-shifted emission are observed on increasing the concentration of aggregate. It is important to note that the transient spectral signature of the t-But-BCDI aggregate is very similar to that of the monomer, suggesting the Frenkel-type (localized to a monomer) character of the exciton. However, faster evolution at the tens of picosecond timescale suggests relaxation of the exciton state within the H-type exciton band. An extraordinarily long emission lifetime from the H-aggregated state is observed.
:1). Compared to the monomer, a blue shift in absorption along with a decrease in the A0–0/A0–1 ratio and weaker, red-shifted emission are observed on increasing the concentration of aggregate. It is important to note that the transient spectral signature of the t-But-BCDI aggregate is very similar to that of the monomer, suggesting the Frenkel-type (localized to a monomer) character of the exciton. However, faster evolution at the tens of picosecond timescale suggests relaxation of the exciton state within the H-type exciton band. An extraordinarily long emission lifetime from the H-aggregated state is observed.
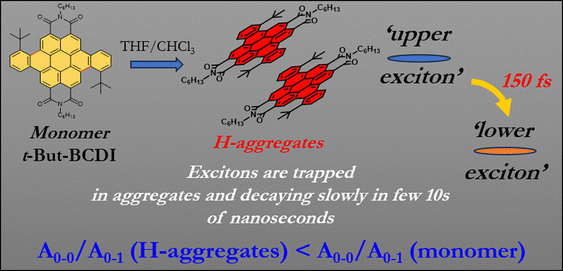 |
| | Fig. 1 Representation of H-aggregation in t-But-BCDI and its salient photophysical features obtained in the present study. | |
2. Results and discussion
2.1. Design and synthesis
J-aggregates have been investigated thoroughly; but such studies are rare for H-aggregates due to their non-emissive properties. To mitigate the non-radiative pathway, we synthesized symmetrical tertiary-butyl-substituted benzo-coronenediimide (t-But-BCDI) with a rigid structure with enhanced π-conjugation. The tertiary butyl groups were placed to inhibit stronger assembly formation and are expected to enhance its solubility in organic solvents. This design enabled us to facilitate the controlled aggregation of t-But-BCDI in selected solvents, and it shows remarkable photophysical properties, such as red-shifted emission from H-type aggregates with an extraordinarily long lifetime. The t-But-BCDI was synthesized in a four-step process involving bromination, imidization, Suzuki coupling, and cyclization (Scheme 1). The 1,7-dibromoperylene-3,4,9,10-tetracarboxydianhydride was converted to 3-t-but-aryl-substituted PDI (t-Bu-PDI).21,45 Under visible light irradiation using a halogen lamp, the aryl-substituted PDI core underwent C–C coupling without any metal catalyst in about 99% yield. Purified t-But-BCDI was characterized by 1H-NMR, FTIR, and MALDI-TOF. 13C-NMR could not be obtained due to lack of solubility. Tertiary butyl groups helped in marginally increasing the solubility compared to unsubstituted benzocoronenediimide, which is almost insoluble in common organic solvents. The solubility of t-But-BCDI is still poor (∼2 mg mL−1) in chloroform-d1. However, the solubility was slightly higher in tetrachloroethylene at 150 °C and thus deuterated tetrachloroethylene was used for 1H-NMR. Spectral characterizations are given in Fig. S1–S6 (ESI†).
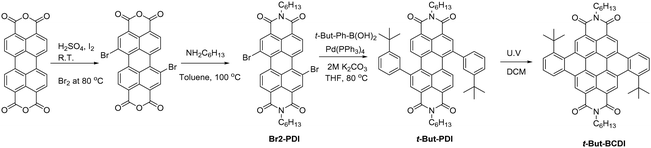 |
| | Scheme 1 Synthetic scheme of t-But-BCDI. | |
2.2. Absorption and emission studies
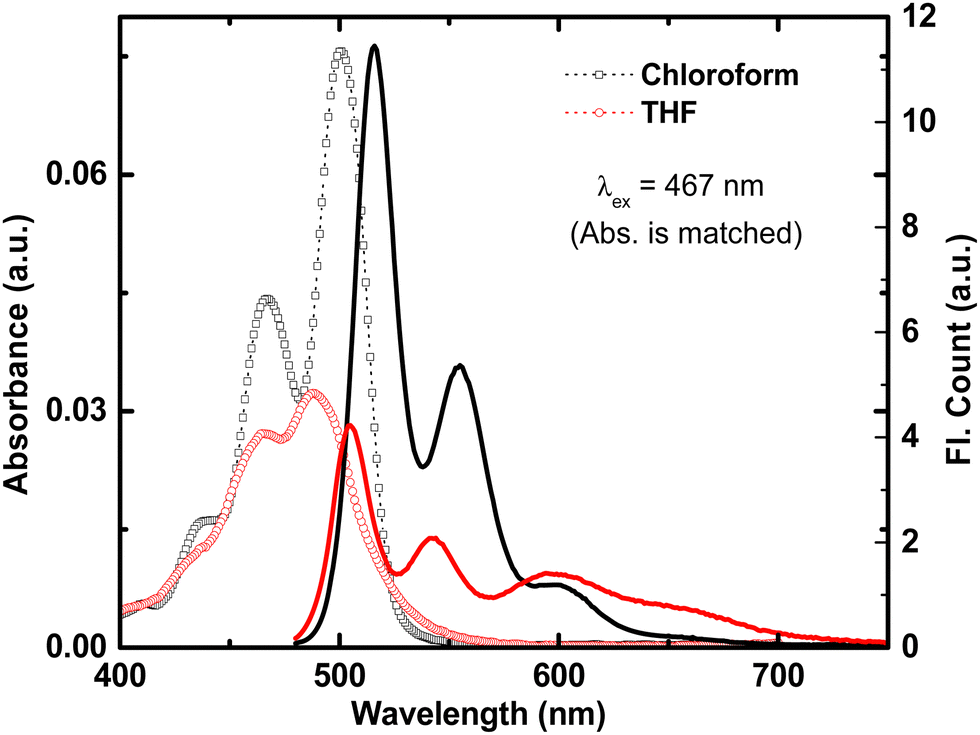
The photophysical properties of t-But-BCDI were elucidated by conducting absorption and emission studies in chloroform and THF (Fig. S7, ESI†). t-But-BCDI is slightly soluble in chloroform (2 mg mL−1) but has poor solubility in THF. Absorption and emission spectra are shown in Fig. 2 and the data are summarized in Table 1. The absorption spectra of t-But-BCDI in chloroform and THF show well-resolved vibronic bands, characteristic of benzocoronenediimide dyes. Absorption maxima for t-But-BCDI are observed at 500 and 487 nm in chloroform and THF, respectively. The vibronic band intensity ratio of the first two peaks (A0–0/A0–1 = 1.66) confirms the monomeric form of t-But-BCDI in chloroform. Accordingly, the emission spectrum of t-But-BCDI in chloroform also show well-resolved vibronic peaks at 515 (maximum) and 555 nm and a shoulder at 600 nm (Fig. 2). However, in THF, the absorption and emission features of t-But-BCDI are remarkably different. Vibronic peaks in absorption are less resolved in THF along with a reduction in absorption intensity (Fig. 2). On the other hand, the emission spectrum in THF shows a broad feature at 590–690 nm, along with vibronically resolved monomer emission with peaks at 505 and 542 nm (Fig. 2).
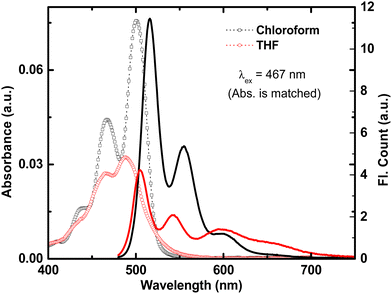 |
| | Fig. 2 Absorption (open symbols) and emission (solid line) in chloroform (black) and THF (red) for t-But-BCDI. Concentrations in both solvents are adjusted to show similar absorbance at λex, i.e., 467 nm. | |
Table 1 Photophysical data of t-But-BCDI
| Solvent |
λ
abs nm |
λ
em nm |
ϕ
(%) |
τ
1 (a1) ns (%) |
τ
2 (a2) ns (%) |
τ
3 (a3) ns (%) |
τ
avg (ns) |
Chi sq |
|
With respect to rhodamine 6B and measured at a low concentration (∼1 μM).
|
| CHCl3 |
436, 467, 500 |
512 |
98 |
5.94 (100.0) |
— |
— |
5.94 |
1.00 |
| 556 |
|
5.74 (91.05) |
9.65 (8.95) |
— |
5.96 |
1.00 |
| THF |
463, 487 |
502 |
75 |
3.48 (9.76) |
7.11 (90.24) |
— |
6.45 |
1.08 |
|
|
|
538 |
|
2.32 (4.6) |
6.81 (91.45) |
28.67 (3.94) |
6.43 |
1.06 |
|
|
|
600 |
|
5.37 (9.83) |
21.42 (12.21) |
55.52 (77.96) |
26.29 |
1.09 |
The considerable differences in absorption and emission features of t-But-BCDI in chloroform and THF prompted us to investigate them further. An excitation scan provides information about emissive species associated with multiple emission peaks in solution. Therefore, excitation spectra of t-But-BCDI solutions (∼2 μM) in chloroform and THF were recorded at different emission wavelengths. Fig. 3 shows a comparison of the excitation spectra of t-But-BCDI in chloroform and THF. In chloroform, the excitation spectrum (λem = 555 nm) is found to be identical to the absorption spectrum, indicating that the emissive species are the same (Fig. 3a). In the case of THF, the excitation spectrum at λem = 545 nm (excitation 1 in Fig. 3b) matches the monomer absorption spectrum, i.e., ∼488 nm. Surprisingly, the excitation spectrum monitored for λem = 650 nm (excitation 2 in Fig. 3b) is broad, has no vibrational features, and is blue-shifted by 11 nm (maximum at 477 nm) compared to the monomer absorption spectrum. It is important to note that the fluorescence quantum yield of the t-But-BCDI monomer in chloroform is very high (QY ∼ 0.95). In THF, the fluorescence yield is significantly less and it decreases with an increase in concentration (Table 1 and Fig. 4). The broad blue-shifted absorption and red-shifted weak fluorescence could be due to H-aggregation. In most cases, the fluorescent H-type aggregates are limited to smaller aggregates with a broad absorption band.19
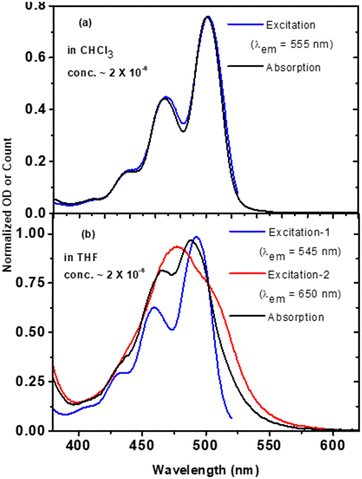 |
| | Fig. 3 Comparison of normalized absorption and excitation spectra of t-But-BCDI in (a) chloroform and (b) THF. In THF, excitation-1 and excitation-2 are the excitation spectra at different wavelengths. The concentration of the solution is 2 × 10−6 M. | |
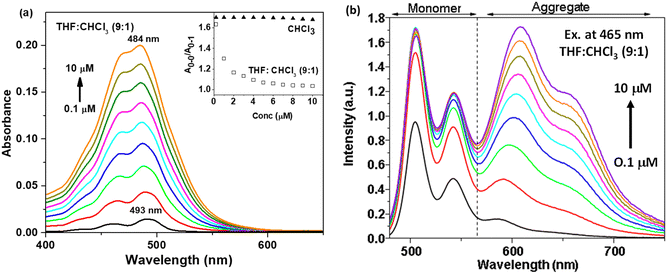 |
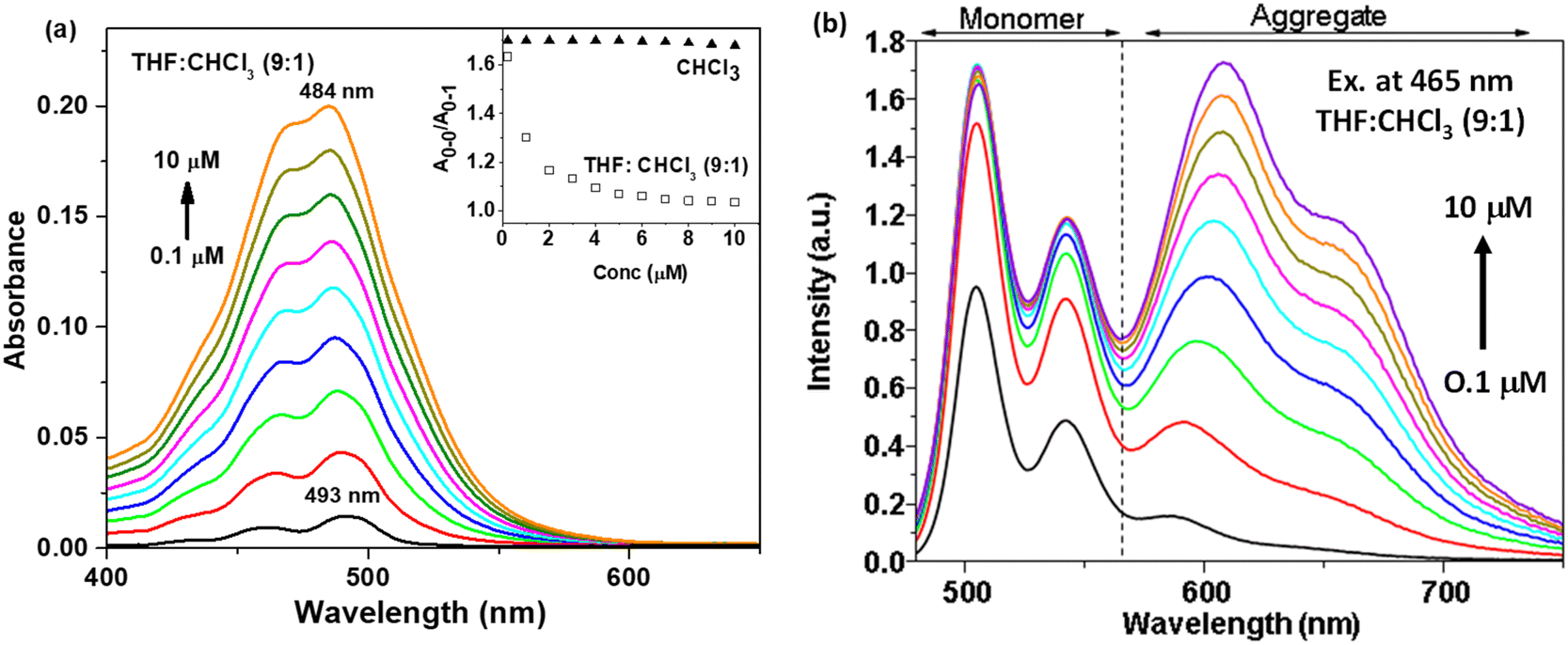
| | Fig. 4 (a) Absorption and (b) emission spectra at different concentrations (0.1 to 10 μM) of t-But-BCDI in THF:CHCl3 (9:1). The inset of (a) shows a plot between A0–0/A0–1 of t-But-BCDI in CHCl3 and THF:CHCl3 (9:1) at 0.1 to 10 μM concentrations. | |
Absorption and emission studies at different concentrations (0.1 to 10 μM) of t-But-BCDI were carried out in THF and chloroform. Fig. 4a shows the absorption spectra of t-But-BCDI (0.1 to 10 μM) in a 9:1 THF and chloroform mixture. It shows a gradual blue shift (∼9 nm) with the increase in concentration: from 493 nm at 0.1 μM to 484 nm at 10 μM. The inset of Fig. 4a shows the variation in A0–0/A0–1 for t-But-BCDI in CHCl3 and THF–CHCl3 (90:10 vol%) in 0.1 to 10 μM concentrations. In CHCl3, A0–0/A0–1 is ∼1.66 and it does not change with concentration. On the other hand, A0–0/A0–1 decreases rapidly with the increase in concentration in THF/CHCl3, suggesting an aggregation event in the solvent. Concentration-dependent emission studies in THF are shown in Fig. 4b. With the increase in concentration, the contribution of the 590–690 nm emission band increases, suggesting its origin to be different from that of the monomer of t-But-BCDI and it is expected to be due to aggregation. We carried out further emission studies at different ratios of good (chloroform) and bad (THF) solvents. The related figure is shown in Fig. S10 (ESI†). Quenching of emission from the monomer is observed on increasing the bad solvent (THF), and a clear new emission is found at 603 nm. Quenching of monomeric emission and a simultaneous increase in red-shifted emission with an increase in THF show the formation of emissive aggregates. Broad blue-shifted absorption and red-shifted weak fluorescence with an increase of concentration in THF are clear indications of H-aggregation.
2.3. Excited state decay studies
We further studied the lifetime decay of the excited state to gain insight into its nature. The fluorescence decays of t-But-BCDI in chloroform and THF at different emission wavelengths are shown in Fig. S8 and S9 (ESI†). Fluorescence lifetimes in chloroform were monitored at 520 and 550 nm and were found to be of similar magnitude (∼5.9 ns) confirming the common excited state of both these peaks. The decay times for 505 and 544 nm in THF are identical (∼6.45 ns) while a triexponential fit to decay at 600 nm revealed lifetimes of 5.3, 21.4, and 55.5 ns. The average decay time for emission in THF was found to be 26 ns, which is much longer than expected. Lifetime data suggests that broad emission at 590–600 nm arises from a different excited state. At this stage, we assume this could be due to excimer formation in THF, as t-But-BCDI has poor solubility in THF. With these observations, we can assign a blue shift of ∼11 nm in concentration-dependent absorption studies in THF to H-aggregation. Nevertheless, it is important to mention that red-shifted emission in H-aggregates is reported via excimer formation.16,46
2.4. TRES and TRANES studies of t-But-BCDI
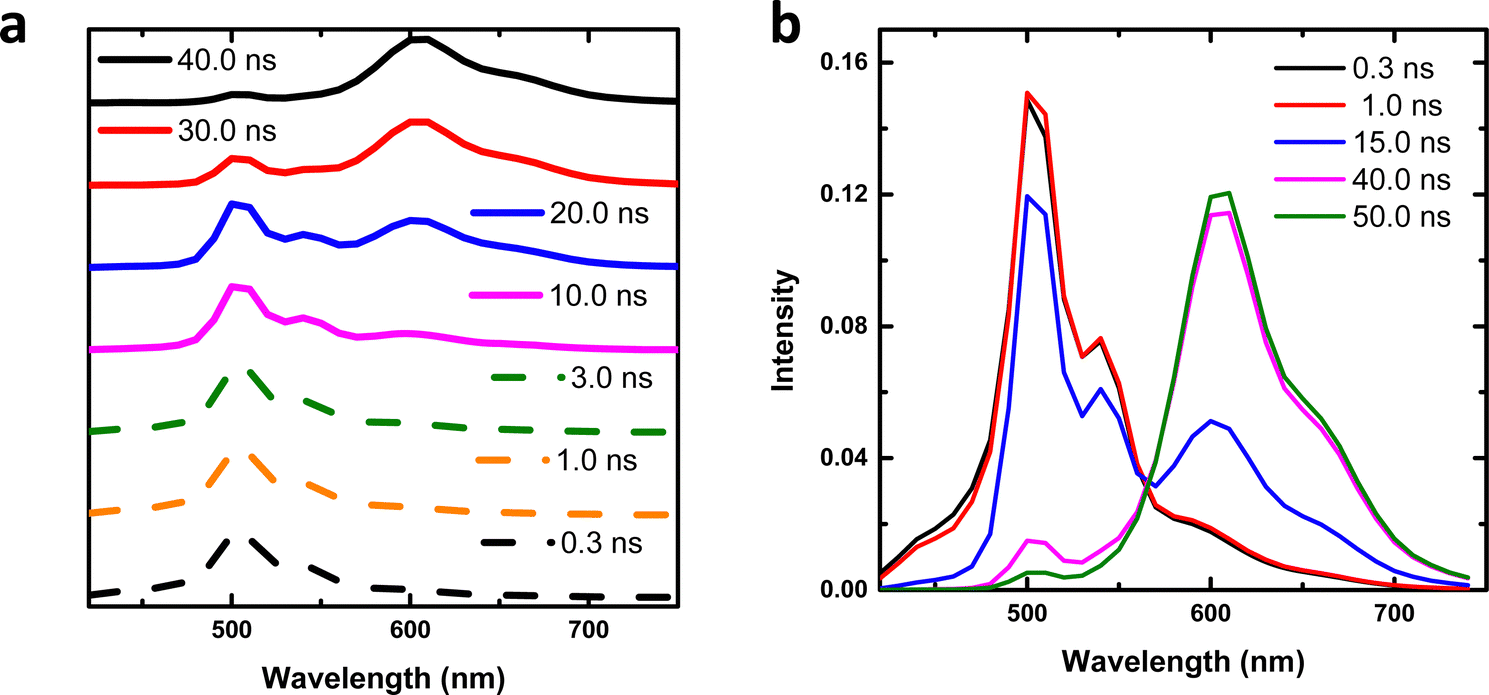
We further studied the excited state dynamics of t-But-BCDI by time-resolved emission spectra (TRES), as shown in Fig. 5a. At the initial time, an emission maximum was seen at 502 nm (up to ∼1 ns) with a long low-intensity tail. At a delay of 3 ns, a new emission at 600 ns was seen, which evolved completely in ∼40 ns. During this period, emission at 502 nm was almost completely quenched. The emission peak at 600 nm (longer lifetime of ∼26 ns) could be attributed to the excimer of electronically coupled t-But-BCDI molecules in the excited state. Stronger electronic coupling shows large exciton splitting, increasing the transition energy from the ground to the upper excited state.46 Absorption studies of t-But-BCDI showed that A0–0/A0–1 decreases with an increase in concentration, i.e., H-aggregation. To get an idea of the emissive species, time-resolved area normalized emission spectra (TRANES) were constructed from the TRES data (Fig. 5b).47–49 An iso-emissive point at 565 nm was observed. The iso-emissive point in TRANES confirms the involvement of two emissive species, i.e., monomeric and aggregated t-But-BCDI, which are in excited-state equilibrium. TRES and TRANES studies suggest that aggregate emission evolves from the monomeric exciton.
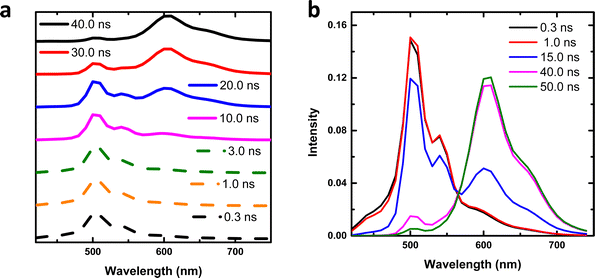 |
| | Fig. 5 (a) TRES and (b) TRANES traces for t-But-BCDI recorded in THF. | |
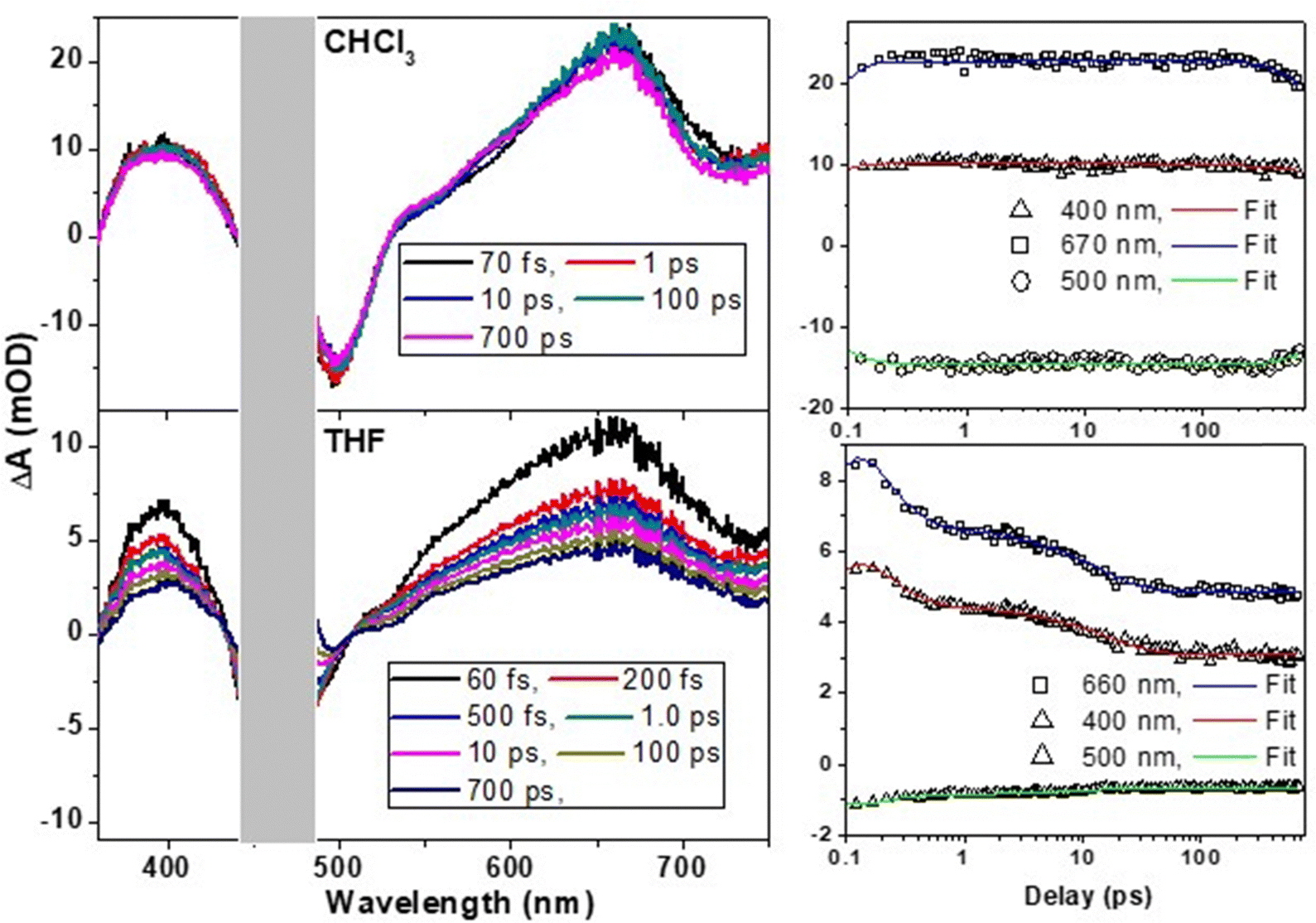
2.5. Ultrafast transient absorption studies
To understand the difference in excited-state dynamics of the t-But-BCDI monomer and aggregated states, ultrafast transient absorption experiments were carried out (Fig. 6). In chloroform, the t-But-BCDI monomer exhibits excited-state absorption peaks at 660 and 400 nm and a negative transient signal at 500 nm. A strong ESA signal in the 660 nm region is characteristic of the perylenediimide kind of chromophore. A negative signal in the 500 nm region is attributed to ground-state bleaching and the stimulated emission signal. The transient absorption signal of the t-But-BCDI monomer exhibits very weak evolution in the first few picoseconds (ps) and then very slowly decays at longer than a few nanoseconds, consistent with a long fluorescence lifetime. On the other hand, the t-Bu-BCDI aggregate in THF shows changes in signal in the early timescale (up to 30 ps) before stabilizing to a very long-lived state.
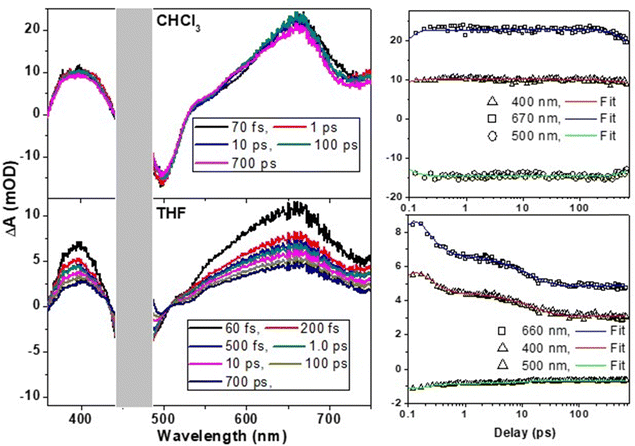 |
| | Fig. 6 Ultrafast transient spectral dynamics of the t-But-BCDI monomer in chloroform and aggregates in tetrahydrofuran. Samples were excited with an fs laser pulse at 470 nm. | |
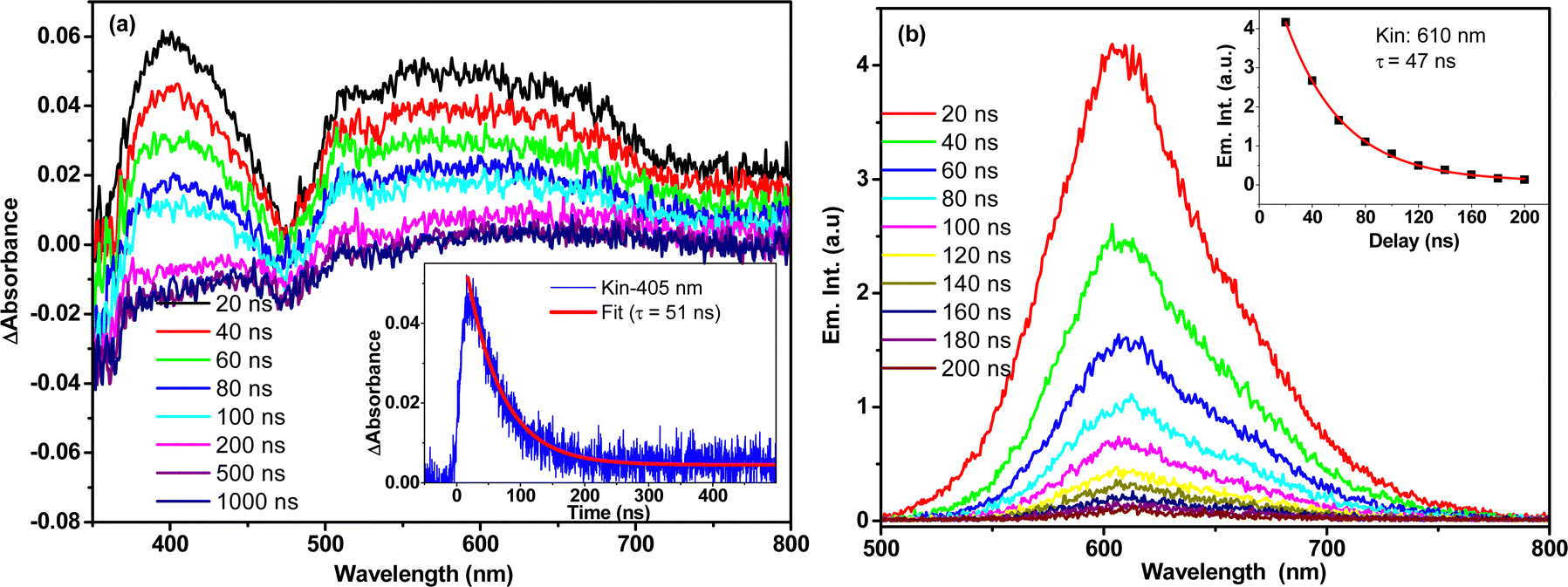
Kinetic fitting of the temporal kinetics recorded at selected wavelengths revealed biphasic relaxation with lifetimes of 150 (±10) fs and 13 (±2) ps, which are attributed to an upper-to-lower state transition and vibrational cooling, respectively. It is important to note that the transient spectral signature of the t-But-BCDI aggregate is very similar to that of the monomer, suggesting the Frenkel-type (localized to a monomer) character of the exciton. However, faster evolution at the tens of picosecond timescale suggests relaxation of the excited state within the H-type exciton band.39–41,50 Once the exciton reaches the exciton band minimum, it does not evolve and remains for a much longer time (beyond our instrumental delay of 2 ns), consistent with an extraordinarily long emission lifetime from the H-aggregated state. This was confirmed by nanosecond transient absorption measurement in a laser flash photolysis experiment (Fig. 7).
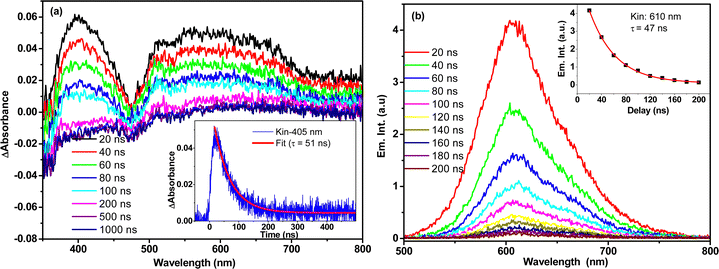 |
| | Fig. 7 (a) Nanosecond transient absorption (a) and emission (b) spectral measurement of t-Bu-BCDI in THF measured by the nanosecond laser flash photolysis technique. The sample was excited by a 7-ns 532-nm laser pulse. | |
3. Conclusions
Emissive H-aggregation with a long lifetime is demonstrated in a newly synthesized planar benzocoronenediimide derivative. Broad blue-shifted absorption is observed with an increase in concentration in THF, which is a clear indication of H-aggregation. Absorption spectra with vibronic features showed a blue shift along with a decrease in the A0–0/A0–1 ratio compared to the monomer. A decrease in the A0–0/A0–1 ratio and red-shifted weak fluorescence are observed in THF with an increase in concentration. The red-shifted emission from the aggregated state of t-But-BCDI has a very long excited-state lifetime (∼50 ns). Ultrafast transient absorption studies revealed biphasic relaxation with lifetimes of 150 (±10) fs and 13 (±2) ps, which are attributed to a bright-to-dark state transition and vibrational cooling, respectively. The transient spectral signature suggests the Frenkel-type (localized to a monomer) character of the exciton. Faster evolution at a tens of picosecond timescale clearly suggests relaxation of the exciton state within the H-type exciton band. From the bottom of the band, excitons decay, radiatively and non-radiatively, with a lifetime of ∼55 ns.
Author contributions
SJND and NA designed and conceptualized the work. SJND synthesized and characterized the materials. SJND, RG, and NA carried out photophysical studies and analysed the results. All transient absorption studies were carried out and analysed by RG. The manuscript was written by NA and RG in consultation with SJND.
Data availability
The additional characterization data, spectra, figures etc. supporting this article have been included as part of the ESI.† Raw data can be made available on request.
Conflicts of interest
There are no conflicts of interest.
References
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- B. S. Basel, C. Hetzer, J. Zirzlmeier, D. Thiel, R. Guldi, F. Hampel, A. Kahnt, T. Clark, D. M. Guldi and R. R. Tykwinski, Chem. Sci., 2019, 10, 3854–3863 RSC.
- D. Koylu, S. Sarrafpour, J. Zhang, S. Ramjattan, M. J. Panzer and S. W. Thomas, Chem. Commun., 2012, 48, 9489–9491 RSC.
- Q. T. Siddiqui, A. A. Awasthi, P. Bhui, M. Muneer, K. R. Chandrakumar, S. Bose and N. Agarwal, J. Phys. Chem. C, 2018, 123, 1003–1014 CrossRef.
- S. R. Forrest, M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley and M. E. Thompson, Nature, 1998, 395, 151–154 CrossRef.
- S. Ilic, E. S. Brown, Y. Xie, S. Maldonado and K. D. Glussac, J. Phys. Chem. C, 2016, 120, 3145–3155 CrossRef CAS.
- M. L. I. Ibrahim, Z. Ahmad, K. Sulaiman and S. V. Muniandy, AIP Adv., 2014, 4, 057133 CrossRef.
- K. V. Barhate, A. P. Wadawale, K. R. S. Chandrakumar and N. Agarwal, Chem. Commun., 2024, 60, 1408–1411 RSC.
- N. Ryu, Y. Okazaki, E. Pouget, M. Takafuji, S. Nagaoka, H. Ihara and R. Oda, Chem. Commun., 2017, 53, 8870–8873 RSC.
- R. M. Hochstrasser and M. Kasha, Photochem. Photobiol., 1964, 3, 317–331 CrossRef CAS.
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC.
- F. C. Spano, J. Chem. Phys., 2005, 122, 234701 CrossRef PubMed.
- T. Eder, T. Stangl, M. Gmelch, K. Remmerssen, D. Laux, S. Höger, J. M. Lupton and J. Vogelsang, Nat. Commun., 2017, 8, 1641–1651 CrossRef PubMed.
- G. D. Scholes, Faraday Discuss., 2020, 221, 265–280 RSC.
- J. Deng, Z. Zhang, P. Sang, S. Yin, S. Zhang, Y. Li, B. Yang, C. Gu and Y. Ma, Aggregate, 2023, 4, e313 CrossRef CAS.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef.
- L. J. Patalag, J. Hoche, M. Holzapfel, A. Schmiedel, R. Mitric, C. Lambert and D. B. Werz, J. Am. Chem. Soc., 2021, 143, 7414–7425 CrossRef CAS PubMed.
- V. A. Montes, C. P. Bolivar, N. Agarwal and P. Anzenbacher, Jr., J. Am. Chem. Soc., 2006, 128, 12436–12438 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. Bayoumi, Pure Appl. Chem., 1965, 11, 371 CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- S. J. N. Dixit, A. A. Awasthi, K. R. S. Chandrakumar, B. Manna and N. Agarwal, J. Phys. Chem. C, 2021, 125, 20405–20415 CrossRef CAS.
-
J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, New York, NY, 3rd edn, 2006, pp. 1–12 Search PubMed.
- Z. Xu, K. S. Park, J. J. Kwok, O. Lin, B. B. Patel, P. Kafle, D. W. Davies, Q. Chen and Y. Diao, Adv. Mater., 2022, 34, 2203055 CrossRef CAS.
- S. Ma, S. Du, G. Pan, S. Dai, B. Xu and W. Tian, Aggregate, 2021, 2, e96 CrossRef CAS.
- L. Wang, Y. Shen, M. Yang, X. Zhang, W. Xu, Q. Zhu, J. Wu, Y. Tiana and H. Zhou, Chem. Commun., 2014, 50, 8723 RSC.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- H. V. Berlepsch and C. Böttcher, Phys. Chem. Chem. Phys., 2018, 20, 18969–18977 RSC.
- O. Ohno, Y. Kaizu and H. Kobayashi, J. Chem. Phys., 1993, 99, 4128–4139 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- P. Leclère, M. Surin, P. Viville, R. Lazzaroni, A. F. M. Kilbinger, O. Henze, W. J. Feast, M. Cavallini, F. Biscarini, A. P. H. J. Schenning and E. W. Meijer, Chem. Mat., 2004, 16, 4452–4466 CrossRef.
- Edwards T. Niles, John D. Roehling, Hajime Yamagata, Adam J. Wise, Frank C. Spano, Adam J. Moulé and John K. Grey, J. Phys. Chem. Lett., 2012, 3, 259–263 CrossRef CAS.
- B. Kopainsky, J. K. Hallermeier and W. Kaiser, Chem. Phys. Lett., 1981, 83, 498–502 CrossRef CAS.
- P. O. J. Scherer and S. F. Fischer, Chem. Phys., 1984, 86, 269–283 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS.
- H. Langhals and W. Jona, Angew. Chem., Int. Ed., 1998, 37, 952–955 CrossRef CAS.
- H. Yoo, S. Furumaki, J. Yang, J.-E. Lee, H. Chung, T. Oba, H. Kobayashi, B. Rybtchinski, T. M. Wilson and M. R. Wasielewski, J. Phys. Chem. B, 2012, 116, 12878–12886 CrossRef CAS.
- E. A. Margulies, L. E. Shoer, S. W. Eaton and M. R. Wasielewski, Phys. Chem. Chem. Phys., 2014, 16, 23735–23742 RSC.
- M. Son, K. H. Park, C. Shao, F. Würthner and D. Kim, J. Phys. Chem. Lett., 2014, 5, 3601–3607 CrossRef CAS.
- J. Sung, P. Kim, B. Fimmel, F. Würthner and D. Kim, Nat. Commun., 2015, 6, 8646 CrossRef CAS PubMed.
- A. Austin, N. J. Hestand, I. G. McKendry, C. Zhong, X. Zhu, M. J. Zdilla, F. C. Spano and J. M. Szarko, J. Phys. Chem. Lett., 2017, 8, 1118–1123 CrossRef PubMed.
- S. Basak, N. Nandi, K. Bhattacharyya, A. Datta and A. Banerjee, Phys. Chem. Chem. Phys., 2015, 17, 30398 RSC.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed.
- J. Gierschner, B. Milian-Medina, D. Oelkrug and H.-J. Egelhaaf, J. Phys. Chem. Lett., 2013, 4, 2686–2697 CrossRef CAS.
- W. Jiang, Y. Li, W. Yue, Y. Zhen, J. Qu and Z. Wang, Org. Lett., 2012, 12, 131–228 Search PubMed.
- K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588–2593 CrossRef CAS.
- A. S. R. Koti, M. M. G. Krishna and N. Periasamy, J. Phys. Chem. A, 2001, 105, 1767–1771 CrossRef CAS.
- S. J. N. Dixit, A. P. Wadawale, R. Ghosh and N. Agarwal, J. Photochem. Photobiol., A, 2024, 447, 115179 CrossRef CAS.
- S. Mandal, J. R. Biswal, B. Kommula and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2024, 16, 36763–36773 CrossRef CAS.
- H. Zhou, T. Watanabe, A. Mito, I. Honma, K. Asai and K. Ishigure, Mater. Lett., 2002, 57, 589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectra related to characterization and photophysical studies is provided. See DOI: https://doi.org/10.1039/d4cp04084h |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a