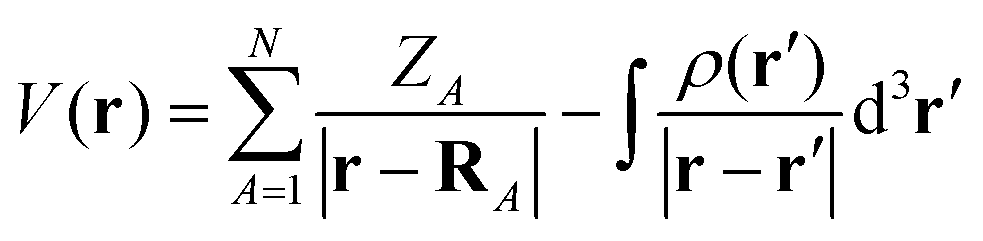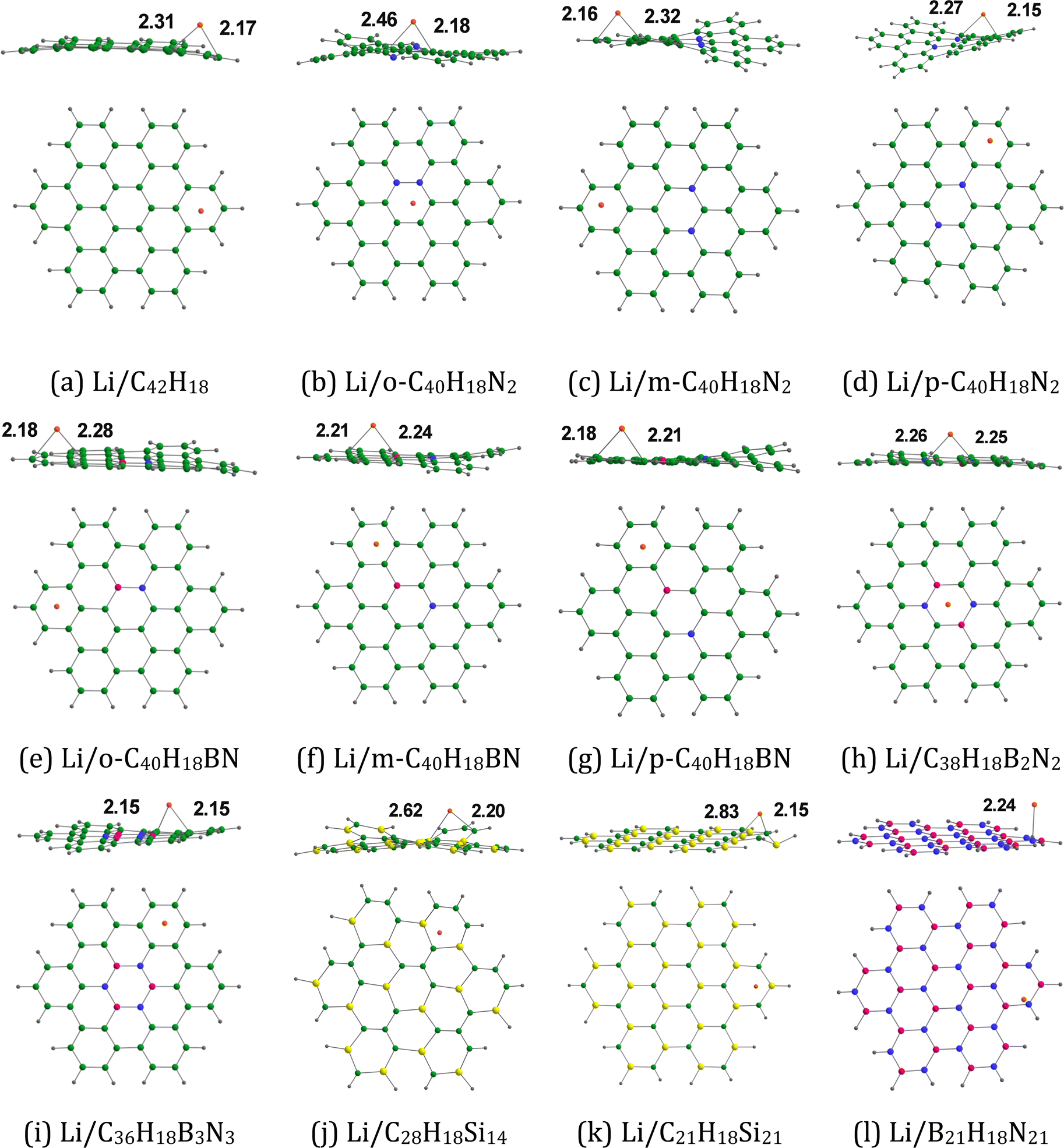Doped hexa-peri-hexabenzocoronene as anode materials for lithium- and magnesium-ion batteries†
Received
25th October 2024
, Accepted 25th November 2024
First published on 4th December 2024
Abstract
The adsorption processes of Li+, Li, Mg2+, and Mg on twelve adsorbents (pristine and N/BN/Si-doped hexa-peri-hexabenzocoronene (HBC) molecules) were studied using density functional theory. The molecular electrostatic potential (MESP) analyses show that the replacement of C atoms of HBC by N/BN/Si units can provide a more electron-rich system than the parent HBC molecule. Li+ and Mg2+ exhibit strong adsorption on pristine and doped HBC molecules. The adsorption energy of cations on these nanoflakes (Eads-1) was in the range of −247.44 (Mg2+/m-C40H18N2 system) to −47.65 kcal mol−1 (Li+/B21H18N21 system). Importantly, our results suggest the weaker interactions of Li+ and Mg2+ with the nanoflakes as the MESP minimum values of the nanoflakes became less negative. In all studied systems, we observed electron donation from the nanoflakes to Li+ and Mg2+. For the metal/nanoflake systems, the adsorption energy of metals on the nanoflakes (Eads-2) was in the range of −33.94 (Li/C38H18B2N2 system) to −2.14 kcal mol−1 (Mg/B21H18N21 system). Among the studied anode materials for lithium-ion batteries (LIBs), the highest cell voltage (Vcell) of 1.90 V was obtained for B21H18N21. Among the studied anode materials for magnesium-ion batteries (MIBs), the highest Vcell value of 5.29 V was obtained for m-C40H18N2. Eads-2 has a significant effect on the variation of Vcell of LIBs, while Eads-1 has a significant effect on the variation of Vcell of MIBs.
1. Introduction
Rechargeable energy storage devices like lithium-ion batteries (LIBs) are in high demand due to their potential applications in electronic devices and electric transportation.1–3 Since the commercialization by Sony Corporation in 1991,1–3 LIBs have dominated the markets of portable electronic devices and electric vehicles because of their high energy densities and long cycle lives. LIBs are mainly composed of an anode, a cathode, and an electrolyte. Lithium cobalt oxide, lithium iron phosphate, and lithium manganese oxide are typically used as cathode materials for LIBs. The electrolyte of LIBs is mostly based on a lithium salt dissolved in organic solvents. Carbon materials like graphene, carbon nanofibers, carbon nanotubes, polycyclic aromatic hydrocarbons (PAHs), etc. have been frequently employed for the construction of anode materials for LIBs.4–9 For instance, the high surface areas, electrical conductivities, and mechanical flexibilities of graphene make it a good candidate for anode materials in LIBs.5 Park et al. achieved good rate capabilities and cycling stability for LIBs by employing PAH in the design of anode materials.7 Arya et al. also obtained good cycling stability, high specific capacity, and excellent rate capability for LIBs by employing PAHs in the design of anode materials.9 Doped carbon materials have also been employed for the construction of anode materials for LIBs.10–13 Recently, divalent metal-ion batteries such as magnesium-ion batteries (MIBs) have attracted considerable interest.14,15 Mg could donate two valence electrons and therefore higher storage capacities may be realized in MIBs than in LIBs. Additional features of MIBs such as natural abundance of Mg, lower costs, and good safety make them an excellent alternative to LIBs.16
PAHs consist of multiple aromatic rings and are rich in delocalized π-electrons. Planar PAHs like coronene, hexa-peri-hexabenzocoronene (HBC), and circumcoronene are formed by fusion of 7, 13, and 19 benzene rings, respectively. These planar PAHs may be considered as finite-size models of graphene. Great progress has been made in the design and synthesis of different heteroatom-doped PAHs.17–20 For example, the synthesis of HBC with a central borazine core was reported by Krieg et al. in 2015.17 There have been several studies of molecular adsorption on pristine and doped PAHs using density functional theory (DFT).21,22 There have also been studies of the application of pristine and doped PAHs as anode materials in metal-ion batteries using DFT.23–29 Li+ and Li were observed to be bound to the peripheral benzene rings of coronene and circumcoronene.23 On the basis of cell voltage (Vcell), PAHs such as circumbiphenyl and coronene were recommended as good anode materials for LIBs.24 Li has a higher interaction with circumcoronene than with BN-circumcoronene.23 Li+ and Li were found to be bound to the peripheral rings of HBC.25 Li+ and Li were also bound to the peripheral rings of BN-doped HBC.26 Mg2+ was chemically adsorbed on HBC, while Mg was physically adsorbed on HBC.27 For the sodium-ion batteries, the Vcell value obtained using HBC with a central borazine core was lower than that obtained using the pristine HBC.28 A similar result was obtained for the calcium-ion batteries.29 However, the interactions of Mg2+ and Mg with adsorbents like Si-doped HBC have not yet been investigated.
In the present study, DFT calculations were carried out to understand the adsorption process of Li+, Li, Mg2+, and Mg on twelve nanoflakes (pristine and N/BN/Si-doped HBC molecules). The molecular electrostatic potential (MESP) minimum point (designated as Vmin) is often observed at the electron-rich sites like lone pairs and π-regions.30–32 Importantly, our results suggest the weaker interactions of Li+ and Mg2+ with the nanoflakes as the MESP Vmin values of the nanoflakes become less negative. For LIBs, the highest Vcell value of 1.90 V was obtained for B21H18N21 and for MIBs, the highest Vcell value of 5.29 V was obtained for m-C40H18N2. This study can provide some new insights into the design and exploration of anode materials suitable for metal ion batteries.
2. Computational details
All DFT computations were conducted through the Gaussian 16 program.33 In the present study, we use a total of twelve adsorbents (pristine and N/BN/Si-doped HBC molecules C42H18, o-C40H18N2, m-C40H18N2, p-C40H18N2, o-C40H18BN, m-C40H18BN, p-C40H18BN, C38H18B2N2, C36H18B3N3, C28H18Si14, C21H18Si21, and B21H18N21) and four adsorbates (Li+, Li, Mg2+, and Mg). The adsorbents were obtained by the replacement of C atoms of HBC by N/BN/Si units. The structural isomers of, for instance, C40H18N2 (two nitrogen atoms occupied adjacent positions in o-C40H18N2) were considered in this work. Note that, C28H18Si14, C21H18Si21, and B21H18N21 may be considered as finite-size models of graphene-like SiC2, SiC, and BN nanosheets, respectively. All structures are optimized at the M062X/6-31G(d,p) level34 and characterized as energy minima by frequency calculations. M06-2X is a hybrid meta functional with 54% of exact Hartree–Fock (HF) exchange. It is a high-nonlocality functional with double the amount of nonlocal exchange (2X) and it also considers the dispersion forces. The M06-2X functional is parameterized for nonmetals and recommended for the study of noncovalent interactions, kinetics, and main-group thermochemistry. The 6-31G(d,p) basis set was used for the geometry optimization calculations. In the geometry optimization process, the geometry is adjusted until a stationary point on the potential surface is found. Vibrational frequency analysis has been carried out to ensure that all the optimized structures correspond to the local minima containing only positive vibrational frequencies.
MESP V(r)30–32 is described by the following expression:
| | 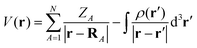 | (1) |
where
ZA denotes the charge on nucleus
A positioned at
RA and
ρ(
r) denotes the electron density. The first and second terms in
eqn (1) represent the nuclear and electronic contributions to the MESP, respectively.
V(
r) is positive when the first term in
eqn (1) dominates and negative when the second term dominates. The MESP analysis was performed at the M062X/6-31G(d,p) level of theory.
The adsorption energy (Eads) is described by the following expression:
| | | Eads = Eadsorbate/nanoflake − (Enanoflake + Eadsorbate) + EBSSE | (2) |
where
Eadsorbate/nanoflake,
Enanoflake, and
Eadsorbate denote the energies of the adsorbed system, adsorbent (
e.g., HBC), and adsorbate (Li
+/Li/Mg
2+/Mg), respectively.
EBSSE denotes the basis set superposition error energy calculated using the counterpoise approach.
35 The
Eads of Li
+/Mg
2+ and Li/Mg on the nanoflakes was denoted as
Eads-1 and
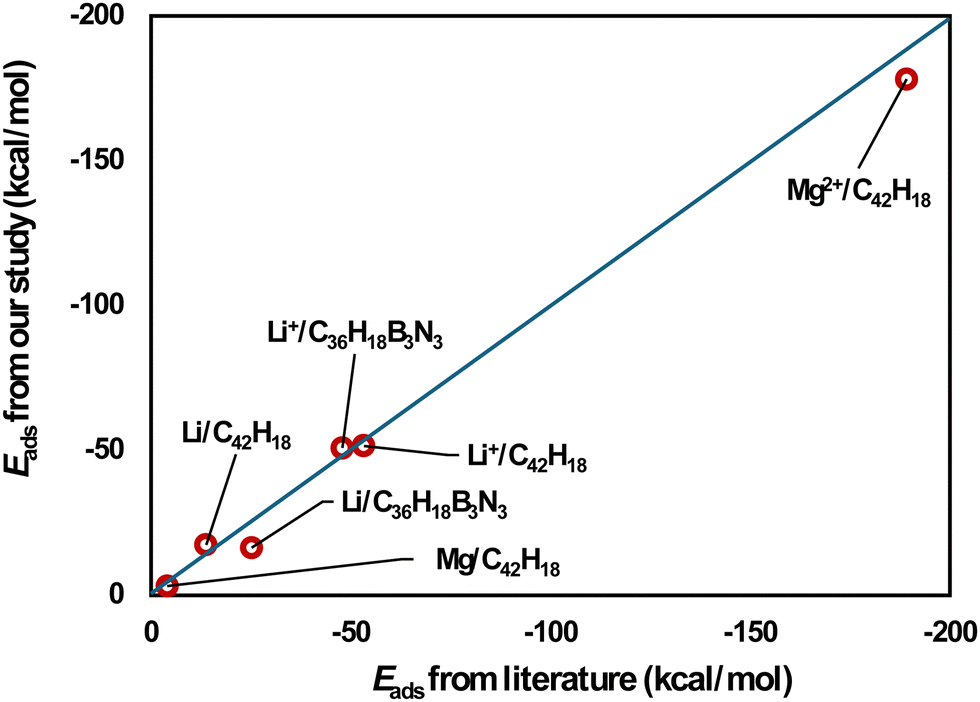
Eads-2, respectively. The adsorption energies calculated in our study are in line with previous DFT data
25–27 (
Fig. 1 and Table S1, ESI
†). The adsorption free energy (
Gads) is described by the following expression:
| | | Gads = Gadsorbate/nanoflake − (Gnanoflake + Gadsorbate) | (3) |
where
Gadsorbate/nanoflake,
Gadsorbate, and
Gnanoflake denote the free energies of the adsorbed system, adsorbent, and adsorbate, respectively.
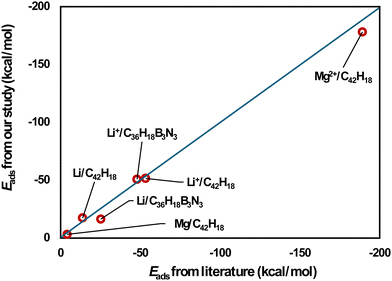 |
| | Fig. 1 Comparison of our results for adsorption energies with literature values.25–27 | |
The highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy gap (Eg) can be calculated using the below equation:
where
EHOMO and
ELUMO denote the HOMO and LUMO energies, respectively. The
Eg of the pristine nanoflake, the Li
+/Mg
2+-adsorbed nanoflake, and the Li/Mg-adsorbed nanoflake is denoted by
Eg-1,
Eg-2, and
Eg-3, respectively.
The percentage change in the HOMO–LUMO energy gap can be calculated using the below equations:
| | | ΔEg-1 = [(Eg-2 − Eg-1)/Eg-1] × 100 | (5) |
| | | ΔEg-2 = [(Eg-3 − Eg-1)/Eg-1] × 100 | (6) |
For using a nanoflake as an anode material in, for example, MIBs, the reactions in the anode and cathode can be written as:
| | | Anode: Mg/nanoflake ↔ Mg2+/nanoflake + 2e− | (7) |
| | | Cathode: Mg2+ + 2e− ↔ Mg | (8) |
Therefore, the total cell reaction is:
| | | Mg/nanoflake + Mg2+ ↔ Mg2+/nanoflake + Mg + ΔGcell | (9) |
The Gibbs free energy change for the total cell reaction (Δ
Gcell) is described by the following expression:
| | | ΔGcell = ΔEcell + PΔV − TΔS | (10) |
where
| | | ΔEcell = Eads-1 − Eads-2 | (11) |
The cell voltage can be calculated using the Nernst equation:
where
F is the Faraday constant (96485.3 C mol
−1) and
z is the valence of Li
+/Mg
2+. It might be assumed that Δ
Gcell ≈ Δ
Ecell, because the contributions of the entropy effect and the volume effect to
Vcell are expected to be negligible.
36
3. Results and discussion
3.1 Doped HBC molecules as anode materials in lithium-ion batteries
3.1.1. Structural properties of doped HBC molecules.
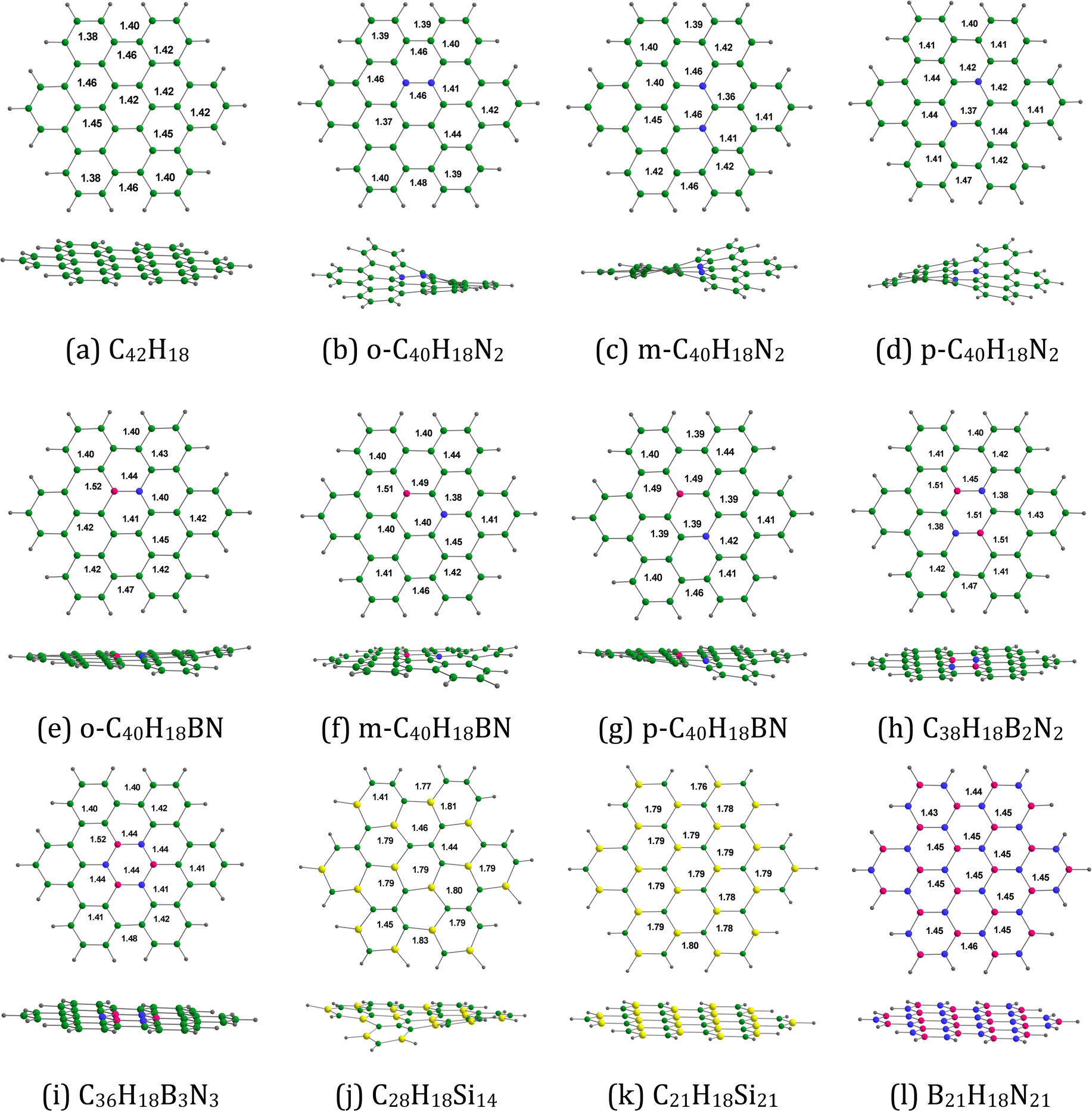
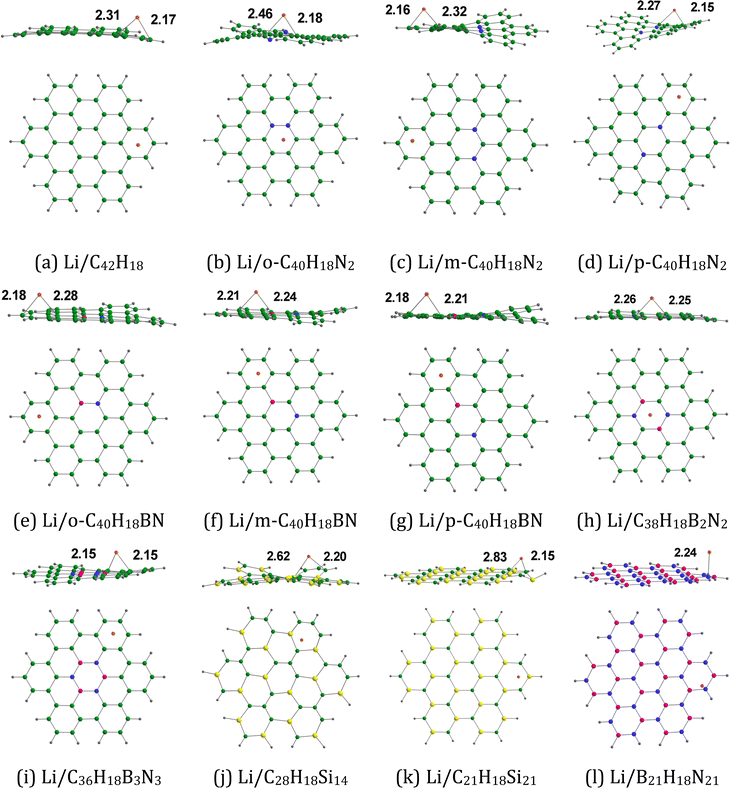
Fig. 2 provides the optimized configurations and the relevant bond lengths for the pristine and doped HBC molecules. The XY (X = C, B; Y = C, N, Si) bond lengths in the pristine and doped HBC molecules span from 1.36 to 1.83 Å. The carbon–carbon bond lengths in HBC span from 1.38 to 1.46 Å. This result is in good agreement with the experimental data.37 Clar's aromatic sextet rule can be applied for HBC38 and thus the carbon–carbon bond length in the central and the six peripheral benzene rings of HBC spans from 1.38 to 1.42 Å. For comparison, the carbon–carbon bond length in benzene is about 1.395 Å.39 In all cases, the carbon–silicon bond lengths (>1.76 Å) are significantly longer than the carbon–carbon bond lengths.
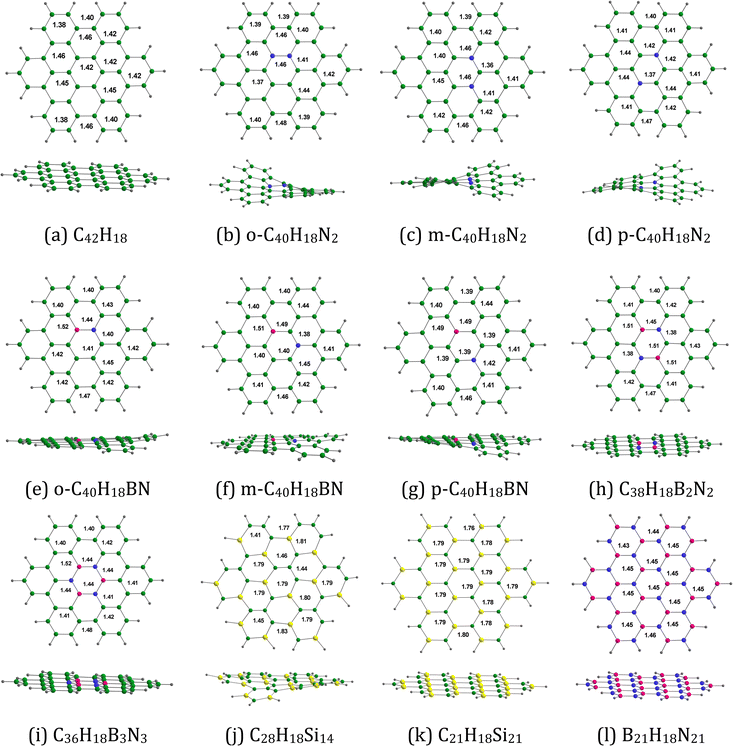 |
| | Fig. 2 Optimized structures of (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The bond distances are given in Å. Color code: green – C, purple – B, blue – N, gray – H, and yellow- – Si. | |
Table 1 lists the EHOMO, ELUMO, and Eg-1 values of the pristine and doped HBC molecules. Here the EHOMO, ELUMO, and Eg-1 values are in the ranges of −8.10 (B21H18N21) to −4.01 eV (m-C40H18N2), −1.74 (C38H18B2N2) to 0.89 eV (B21H18N21), and 2.46 (m-C40H18N2) to 9.00 eV (B21H18N21), respectively. The EHOMO, ELUMO, and Eg-1 values of HBC are −6.39, −1.00, and 5.40 eV, respectively. These results are in good agreement with previous theoretical calculations.40 Overall, the EHOMO (ELUMO) values of the doped HBC molecules are higher (lower) than those of the pristine HBC. Also, in general, the Eg-1 values of the doped HBC molecules are lower than those of the pristine HBC. A smaller Eg-1 value usually indicates a higher electronic conductivity. However, the Eg-1 values of C36H18B3N3 and B21H18N21 are higher than those of the pristine HBC.
Table 1
V
min, EHOMO, ELUMO and Eg-1 values of doped HBCsa
| Nanoflake |
V
min
|
E
HOMO
|
E
LUMO
|
E
g-1
|
|
The values are given in eV and Vmin in kcal mol−1.
|
| C42H18 |
−15.56 |
−6.39 |
−1.00 |
5.40 |
|
o-C40H18N2 |
−19.70 |
−4.89 |
−1.11 |
3.78 |
|
m-C40H18N2 |
−24.97 |
−4.01 |
−1.55 |
2.46 |
|
p-C40H18N2 |
−16.44 |
−4.74 |
−0.95 |
3.80 |
|
o-C40H18BN |
−18.64 |
−6.09 |
−1.22 |
4.87 |
|
m-C40H18BN |
−23.41 |
−5.90 |
−1.41 |
4.49 |
|
p-C40H18BN |
−22.53 |
−6.37 |
−0.99 |
5.38 |
| C38H18B2N2 |
−18.14 |
−5.45 |
−1.74 |
3.71 |
| C36H18B3N3 |
−16.32 |
−6.73 |
−0.69 |
6.05 |
| C28H18Si14 |
−31.38 |
−5.71 |
−1.71 |
4.00 |
| C21H18Si21 |
−28.43 |
−6.44 |
−1.14 |
5.30 |
| B21H18N21 |
−17.57 |
−8.10 |
0.89 |
9.00 |
3.1.2. MESP of doped HBC molecules.
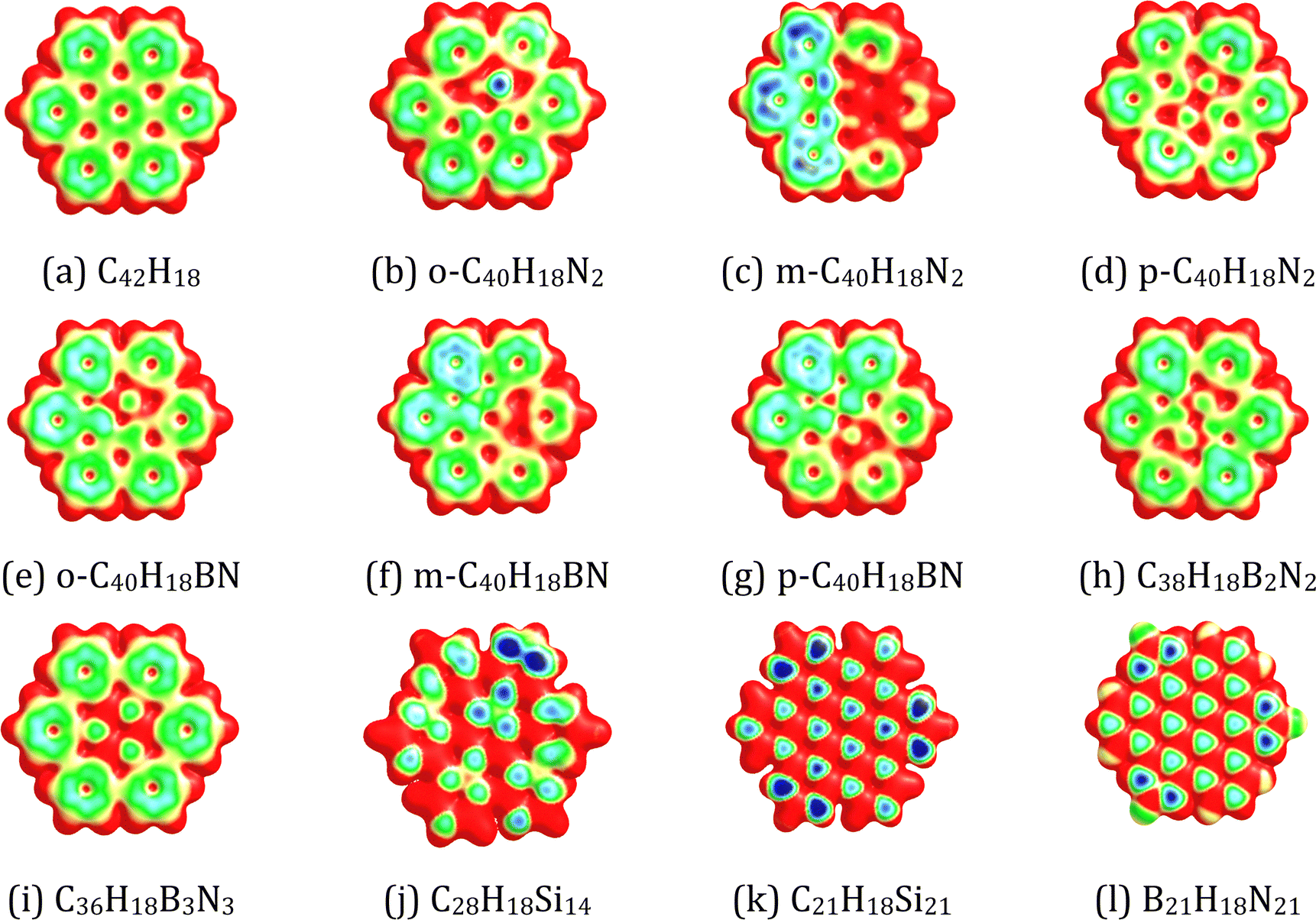
Fig. 3 displays the MESP surfaces of the pristine and doped HBC molecules. The MESP plots show the electron-rich region (e.g., green region) on the central and peripheral benzene rings of HBC arising from the cyclic delocalization of π-electrons. Our results show that the replacement of C atoms of HBC by N/BN/Si units can provide a more electron-rich environment (e.g., the blue region). Fig. S1, ESI,† provides the location of the MESP Vmin of the pristine and doped HBC molecules. The MESP Vmin points were situated near the six peripheral benzene rings of HBC. This indicates the higher electron-richness of the peripheral benzene rings of HBC as compared to the inner rings of HBC. This is due to the fact that the peripheral benzene rings of HBC also have carbon–hydrogen bonds and hydrogen is less electronegative than the sp2 carbon. Overall, the MESP Vmin points of all doped HBC molecules were also situated near the peripheral rings. However, the MESP Vmin points of o-C40H18N2 are situated near the inner N atoms. The MESP Vmin values of the pristine and doped HBC molecules are listed in Table 1. A higher negative value of MESP Vmin reflects a more electron rich nature of the nanoflake. These values were in the range of −31.38 (C28H18Si14) to −15.56 kcal mol−1 (C42H18). A higher negative value of MESP Vmin reflects a more electron rich nature of the nanoflake. The MESP Vmin value of the doped HBC molecules was more negative than that of the pristine HBC. In the case of N-doped HBC molecules, the absolute value of MESP Vmin followed the order p-C40H18N2 < o-C40H18N2 < m-C40H18N2. In the case of BN-doped HBC molecules, the absolute value of MESP Vmin followed the order C36H18B3N3 < B21H18N21 < C38H18B2N2 < o-C40H18BN < p-C40H18BN < m-C40H18BN. The MESP Vmin value of C21H18Si21 (−28.43 kcal mol−1) was less negative than that of C28H18Si14 (−31.38 kcal mol−1).
 |
| | Fig. 3 MESP mapped on the 0.01 a.u. electron density isosurface of (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The colour coding from blue to red indicates MESP values in the range −0.03 to 0.03 a.u. The colours at the blue end of the spectrum indicate the electron-rich regions, while those toward the red indicate the electron-deficient regions. | |
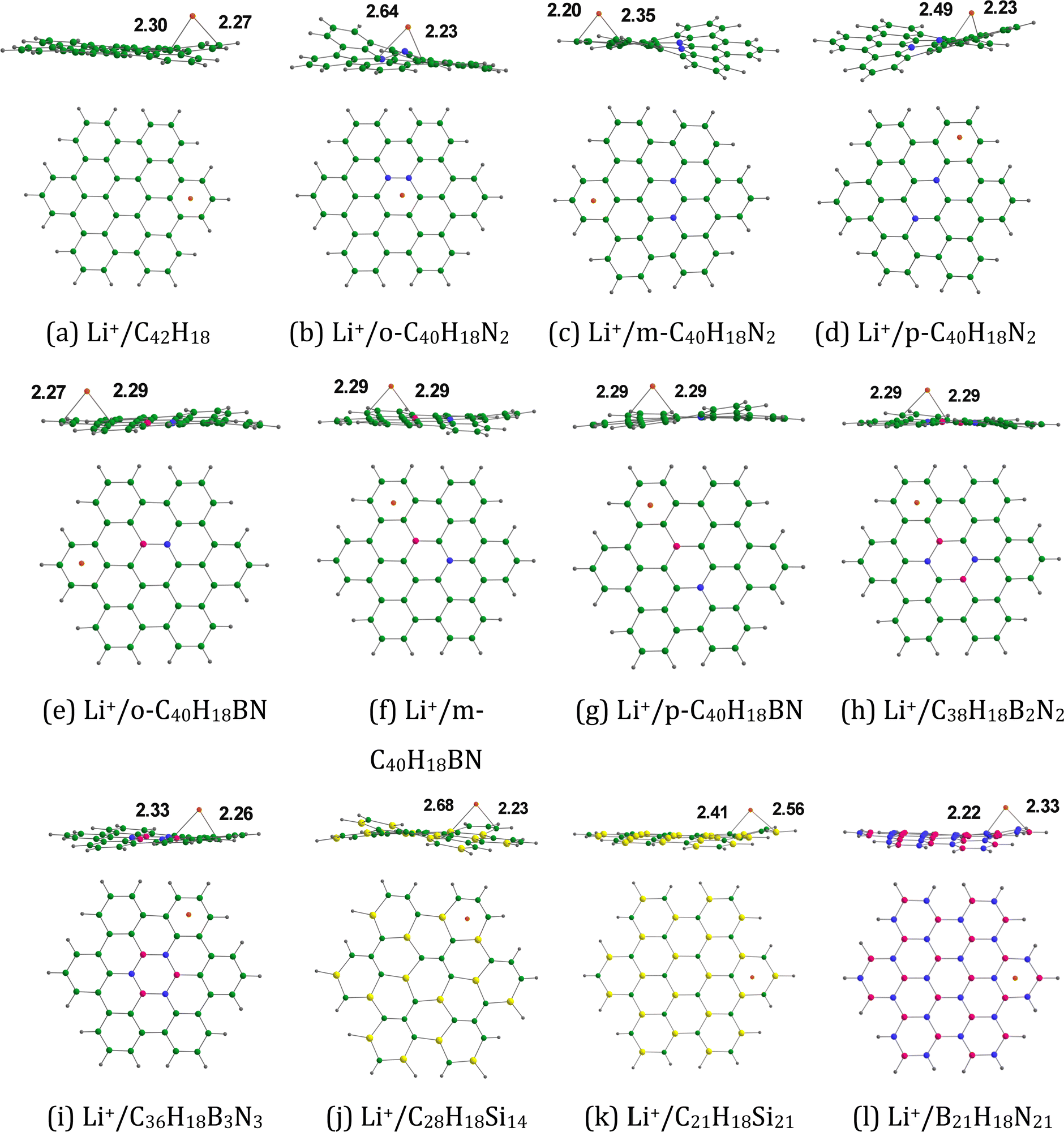
3.1.3. Li+ adsorption on doped HBC molecules.
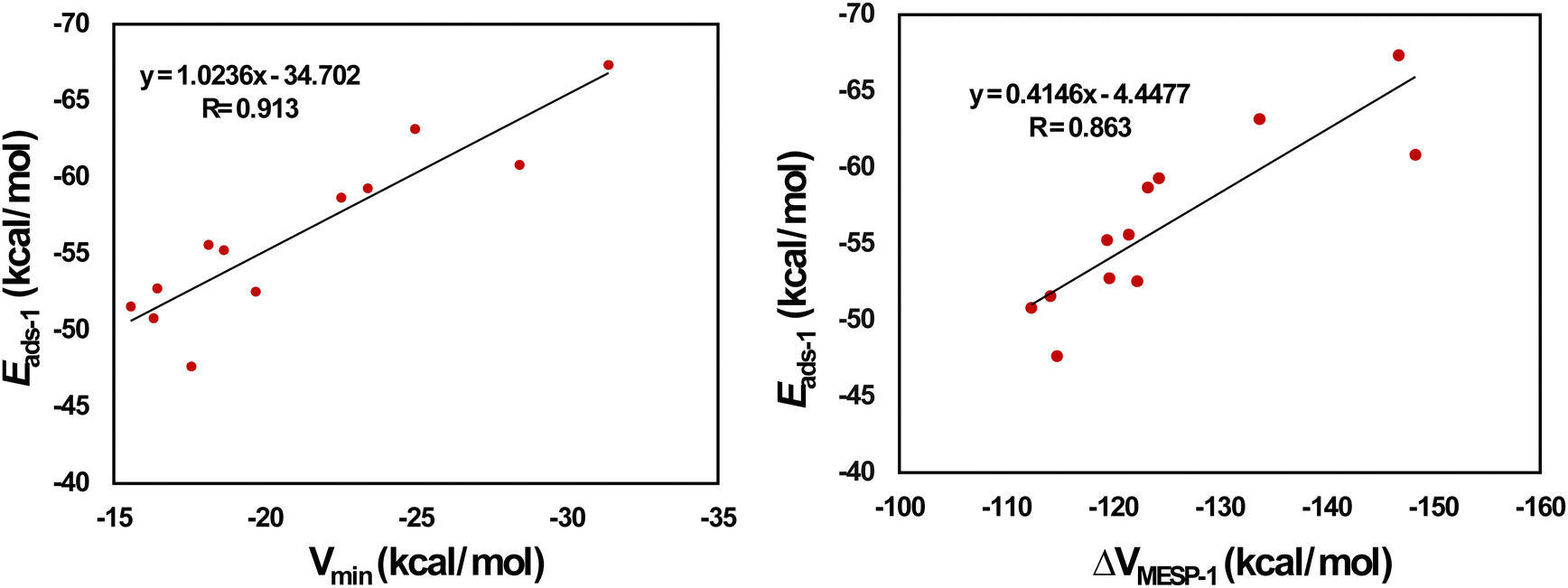
Fig. 4 provides the optimized configurations and the adsorption distances for the adsorption of Li+ on pristine and doped HBC molecules. We see that Li+ is bound to the peripheral benzene rings of HBC. This is possibly due to the higher electron-richness of the peripheral benzene rings of HBC as compared to the inner rings of HBC (see the above). These adsorption processes involve the cation–π interactions.24,41 These interactions are predominantly electrostatic in nature, involving the interaction of Li+ with the electron cloud of the π-system. Overall, Li+ is also bound to the peripheral rings of all doped HBC molecules. However, Li+ is bound to the central ring of o-C40H18N2. We observe that Li+ exhibits a relatively strong adsorption on pristine and doped HBC molecules. For instance, the adsorption distances of Li+ on these nanoflakes span from 2.20 (Li+/m-C40H18N2 system) to 2.41 Å (Li+/C21H18Si21 system). The adsorption energy data (Table 2) also support this possibility. Here the Eads-1 values were in the range of −67.34 (Li+/C28H18Si14 system) to −47.65 kcal mol−1 (Li+/B21H18N21 system). A higher negative value of Eads-1 typically implies a stronger interaction between Li+ and the nanoflake. The Eads-1 value of the Li+/HBC system was −51.57 kcal mol−1. This value is more negative than that of the Li+/C36H18B3N3 and Li+/B21H18N21 systems. In the case of Li+ adsorption on N-doped HBC molecules, the absolute values of Eads-1 followed the order o-C40H18N2 < p-C40H18N2 < m-C40H18N2. In the case of Li+ adsorption on BN-doped HBC molecules, the absolute values of Eads-1 followed the order B21H18N21 < C36H18B3N3 < o-C40H18BN < C38H18B2N2 < p-C40H18BN < m-C40H18BN. The Eads-1 value of the Li+/C21H18Si21 system (−60.82 kcal mol−1) was less negative than that of the Li+/C28H18Si14 system. Notably, a linear relationship between Eads-1 and MESP Vmin values of the nanoflakes exists with a correlation coefficient of 0.913 (Fig. 5a). This finding reflects the weaker interactions of Li+ with the nanoflakes as the MESP Vmin values of the nanoflakes become less negative. For all cases, the adsorption free energies were negative, indicating the spontaneous nature of the adsorption process (see Table 2). Here the Gads-1 values were in the range of −58.02 (Li+/C28H18Si14 system) to −37.15 kcal mol−1 (Li+/B21H18N21 system).
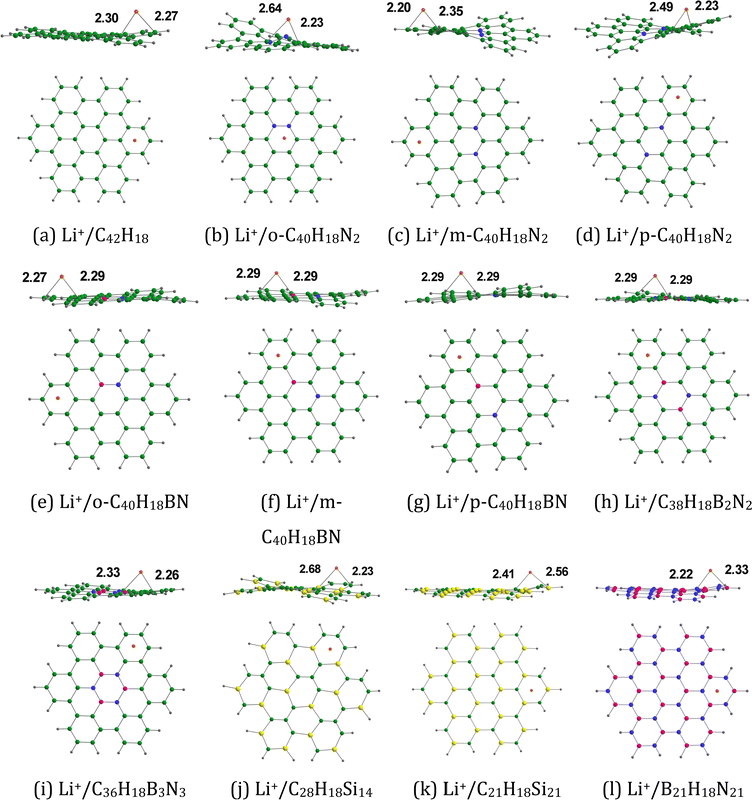 |
| | Fig. 4 Optimized structures of Li+ adsorbed on (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The bond distances are given in Å. The color code is the same as in Fig. 2. In addition, Li is denoted by the orange color. | |
Table 2
E
ads-1, ΔVMESP-1, EHOMOELUMO, Eg-2 and ΔEg-1 values of Li+ adsorbed HBCsa
| Nanoflake |
E
ads-1
|
G
ads-1
|
ΔVMESP-1 |
E
HOMO
|
E
LUMO
|
E
g-2
|
ΔEg-1 |
|
The values of Eads-1, Gads-1 and ΔVMESP-1 in kcal mol−1; EHOMO, ELUMO, and Eg-2 in eV; ΔEg-1 in %.
|
| C42H18 |
−51.57 |
−46.58 |
−114.15 |
−8.98 |
−4.54 |
4.44 |
−17.69 |
|
o-C40H18N2 |
−52.54 |
−43.76 |
−122.27 |
−8.67 |
−4.41 |
4.26 |
12.73 |
|
m-C40H18N2 |
−63.17 |
−52.97 |
−133.74 |
−7.06 |
−4.25 |
2.81 |
14.03 |
|
p-C40H18N2 |
−52.75 |
−43.65 |
−119.68 |
−7.65 |
−4.20 |
3.45 |
−9.10 |
|
o-C40H18BN |
−55.25 |
−45.65 |
−119.46 |
−8.80 |
−4.30 |
4.50 |
−7.65 |
|
m-C40H18BN |
−59.30 |
−49.77 |
−124.32 |
−8.67 |
−4.37 |
4.31 |
−4.12 |
|
p-C40H18BN |
−58.67 |
−49.26 |
−123.26 |
−9.05 |
−4.07 |
4.98 |
−7.53 |
| C38H18B2N2 |
−55.60 |
−47.70 |
−121.48 |
−8.24 |
−4.62 |
3.62 |
−2.49 |
| C36H18B3N3 |
−50.81 |
−45.48 |
−112.35 |
−9.05 |
−4.26 |
4.79 |
−20.83 |
| C28H18Si14 |
−67.34 |
−58.02 |
−146.76 |
−8.30 |
−4.15 |
4.15 |
3.81 |
| C21H18Si21 |
−60.82 |
−51.17 |
−148.34 |
−8.40 |
−3.76 |
4.64 |
−12.42 |
| B21H18N21 |
−47.65 |
−37.15 |
−114.78 |
−10.45 |
−4.11 |
6.35 |
−29.45 |
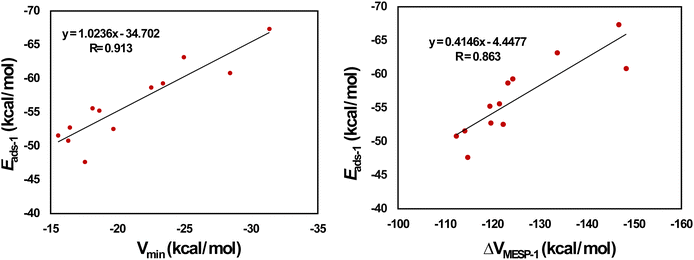 |
| | Fig. 5 Correlation between (a) Vmin and Eads-1 and (b) ΔVMESP-1 and Eads-1 for Li+-adsorbed HBCs. | |
The electron donation from the nanoflakes to Li+ can be assessed by determining the MESP at the nucleus of Li+ before and after adsorption. Therefore, ΔVMESP-1 is calculated by subtracting the MESP at the nucleus of free Li+ (−5.36 a.u.) from that of Li+ in the Li+/nanoflake system. For all cases, the ΔVMESP-1 values were negative, indicating the electron donation from the nanoflakes to Li+ (see Table 2). Here the ΔVMESP-1 values are in the range of −148.34 (Li+/C21H18Si21 system) to −112.35 kcal mol−1 (Li+/C36H18B3N3 system). The ΔVMESP-1 value of the Li+/HBC system is −114.15 kcal mol−1. This value is more negative than that of the Li+/C36H18B3N3 system. A linear relationship between Eads-1 and ΔVMESP-1 values exists with a correlation coefficient of 0.863 (Fig. 5b). This finding reflects that stronger interactions of Li+ with the nanoflakes push more electron density toward Li+.
Table 2 also lists the EHOMO, ELUMO, and Eg-2 values of the Li+-adsorbed nanoflakes. Here the EHOMO, ELUMO, and Eg-2 values are in the ranges of −10.45 (Li+/B21H18N21 system) to −8.24 eV (Li+/C38H18B2N2 system), −4.62 (Li+/C38H18B2N2 system) to −3.76 eV (Li+/C21H18Si21 system), and 2.81 (Li+/m-C40H18N2 system) to 6.35 eV (Li+/B21H18N21 system), respectively. The EHOMO, ELUMO, and Eg-2 values of the Li+/HBC system are −8.98, −4.54, and 4.44 eV, respectively. The EHOMO and ELUMO values of all the Li+-adsorbed nanoflakes are lower than those of the corresponding Li+-free nanoflakes. In general, a similar trend was obtained for Eg-2. The HOMO–LUMO gap decreased by 17.69% due to Li+ adsorption on HBC (see the ΔEg-1 values in Table 2). The maximum decrease in the HOMO–LUMO gap is obtained for Li+ adsorption on B21H18N21 (29.45%). The maximum increase in the HOMO–LUMO gap is obtained for Li+ adsorption on m-C40H18N2 (14.03%).
3.1.4. Li adsorption on doped HBC molecules.
Fig. 6 provides the optimized configurations and the adsorption distances for the adsorption of Li on pristine and doped HBC molecules. Li is mostly bound to the peripheral rings of these nanoflakes, as observed in the case of Li+. The adsorption distances of Li on these nanoflakes span from 2.15 (Li/p-C40H18N2 system) to 2.25 Å (Li/C38H18B2N2 system). For these nanoflakes, the adsorption distances of Li+ (see Fig. 4) are typically larger than those of Li. For instance, the adsorption distances of Li+ and Li on HBC are 2.27 and 2.17 Å, respectively. However, Li+ was strongly adsorbed on these nanoflakes in comparison with Li. The adsorption energy data (see Tables 2 and 3) support this possibility. Here, the Eads-2 values are in the range of −33.94 (Li/C38H18B2N2 system) to −3.93 kcal mol−1 (Li/B21H18N21 system). The Eads-2 value of the Li/HBC system is −17.39 kcal mol−1. This value is more negative than those of the Li/C36H18B3N3 and Li/B21H18N21 systems. In the case of Li adsorption on N-doped HBC molecules, the absolute values of Eads-2 followed the order p-C40H18N2 < o-C40H18N2 < m-C40H18N2. In the case of Li adsorption on BN-doped HBC molecules, the absolute values of Eads-2 followed the order B21H18N21 < C36H18B3N3 < p-C40H18BN < o-C40H18BN < m-C40H18BN < C38H18B2N2. The Eads-2 value of the Li/C21H18Si21 system (−30.84 kcal mol−1) was less negative than that of the Li/C28H18Si14 system (−32.42 kcal mol−1). Here, Eads-2 is not well correlated with the MESP Vmin values of the nanoflakes (Fig. S2, ESI†). Overall, the adsorption free energies were negative, indicating the spontaneous nature of the adsorption process (see Table 3). However, the Gads-2 value of the Li/B21H18N21 system is positive (3.87 kcal mol−1), suggesting that the adsorption of Li on B21H18N21 is not spontaneous. ΔVMESP-2 is calculated by subtracting the MESP at the nucleus of free Li (−5.72 a.u.) from that of Li in the Li/nanoflake system. Overall, the ΔVMESP-2 values were positive, indicating the electron density transfer from Li to the nanoflakes (see Table 3). However, the ΔVMESP-2 values were negative for Li adsorption on C21H18Si21 and B21H18N21. Unlike Eads-1, Eads-2 did not display a correlation with ΔVMESP-2 (see Fig. S2, ESI†).
 |
| | Fig. 6 Optimized structures of Li adsorbed on (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The bond distances are given in Å. The color code is the same as in Fig. 2. In addition, Li is denoted by the orange color. | |
Table 3
E
ads-2, ΔVMESP-2, EHOMOELUMO, Eg-3 and ΔEg-2 values of Li adsorbed HBCs, along with ΔEcell and Vcella
| Nanoflake |
E
ads-2
|
G
ads-2
|
ΔVMESP-2 |
E
HOMO
|
E
LUMO
|
E
g-3
|
ΔEg-2 |
ΔEcell |
V
cell
|
|
The values of Eads-2, Gads-2, ΔVMESP-2, and ΔEcell in kcal mol−1; EHOMO, ELUMO, and Eg-3 in eV; ΔEg-2 in %; and Vcell in V.
|
| C42H18 |
−17.39 |
−15.32 |
16.73 |
−3.59 |
−0.98 |
2.62 |
−51.51 |
−34.18 |
1.48 |
|
o-C40H18N2 |
−23.10 |
−16.24 |
14.88 |
−3.90 |
−0.88 |
3.02 |
−20.22 |
−29.44 |
1.28 |
|
m-C40H18N2 |
−30.68 |
−23.05 |
20.44 |
−3.82 |
−1.04 |
2.79 |
13.26 |
−32.49 |
1.41 |
|
p-C40H18N2 |
−18.02 |
−11.74 |
13.71 |
−3.64 |
−0.82 |
2.82 |
−25.72 |
−34.72 |
1.51 |
|
o-C40H18BN |
−20.81 |
−13.73 |
16.42 |
−3.58 |
−1.02 |
2.56 |
−47.43 |
−34.44 |
1.49 |
|
m-C40H18BN |
−26.64 |
−19.27 |
18.51 |
−3.64 |
−0.97 |
2.67 |
−40.52 |
−32.66 |
1.42 |
|
p-C40H18BN |
−18.95 |
−12.63 |
9.50 |
−3.35 |
−1.08 |
2.27 |
−57.81 |
−39.73 |
1.72 |
| C38H18B2N2 |
−33.94 |
−28.74 |
21.50 |
−4.39 |
−0.63 |
3.76 |
1.39 |
−21.66 |
0.94 |
| C36H18B3N3 |
−16.40 |
−13.51 |
13.21 |
−3.61 |
−0.84 |
2.77 |
−54.25 |
−34.40 |
1.49 |
| C28H18Si14 |
−32.42 |
−25.10 |
6.07 |
−3.73 |
−1.39 |
2.34 |
−41.36 |
−34.92 |
1.51 |
| C21H18Si21 |
−30.84 |
−22.75 |
−18.32 |
−4.71 |
−1.15 |
3.56 |
−32.85 |
−29.98 |
1.30 |
| B21H18N21 |
−3.93 |
3.87 |
−25.98 |
−3.10 |
0.32 |
3.42 |
−61.99 |
−43.72 |
1.90 |
Table 3 also lists the EHOMO, ELUMO, and Eg-3 values of the Li-adsorbed nanoflakes. Here the EHOMO, ELUMO, and Eg-3 values are in the ranges of −4.71 (Li/C21H18Si21 system) to −3.10 eV (Li/B21H18N21 system), −0.63 (Li/C38H18B2N2 system) to 0.32 eV (Li/B21H18N21 system), and 2.27 (Li/p-C40H18BN system) to 3.76 eV (Li/C38H18B2N2 system), respectively. The EHOMO, ELUMO, and Eg-3 values of the Li/HBC system are −3.59, −0.98, and 2.62 eV, respectively. The EHOMO values of all the Li-adsorbed nanoflakes are higher than those of the corresponding Li-free nanoflakes. In general, a similar trend was obtained for Eg-3. For all nanoflakes, the ELUMO was not much influenced by the presence of Li. The HOMO–LUMO gap decreased by 51.51% due to Li adsorption on HBC (see ΔEg-2 values in Table 3). The maximum decrease in the HOMO–LUMO gap is obtained for Li adsorption on B21H18N21 (61.99%). However, Eg-1 was lower than Eg-3 for Li adsorption on m-C40H18N2 (ΔEg-2 of 13.26%) and C38H18B2N2 (ΔEg-2 of 1.39%).
3.1.5. Cell voltage.
The ΔEcell and Vcell values of LIBs based on the pristine and doped HBC molecules are provided in Table 3. Here the ΔEcell and Vcell values were in the ranges of −43.72 (B21H18N21) to −21.66 kcal mol−1 (C38H18B2N2) and 0.94 (C38H18B2N2) to 1.90 V (B21H18N21), respectively. The less negative the ΔEcell, the lower the Vcell of the system. Thus, a nanoflake with a strong interaction with Li+ and a weak interaction with Li might be regarded as a good candidate for the anode material in metal ion batteries. For instance, in the case of B21H18N21, a high Vcell value of 1.90 V was obtained because of a high Eads-1 value of −47.65 kcal mol−1 (see Table 2) and a low Eads-2 value of −3.93 kcal mol−1 (see Table 3). In the case of C38H18B2N2, a low Vcell value of 0.94 V was obtained due to the higher contribution from Eads-2 (Eads-1 value of −55.60 kcal mol−1 and Eads-2 value of −33.94 kcal mol−1). The ΔEcell and Vcell values of HBC were −34.18 kcal mol−1 and 1.48 V, respectively. In the case of N-doped HBC molecules, the Vcell values followed the order o-C40H18N2 < m-C40H18N2 < p-C40H18N2. In the case of BN-doped HBC molecules, the Vcell values followed the order C38H18B2N2 < m-C40H18BN < C36H18B3N3 < o-C40H18BN < p-C40H18BN < B21H18N21. The Vcell value of C21H18Si21 (1.30 V) was lower than that of C28H18Si14 (1.51 V). When considering all the nanoflakes, unlike Eads-1, Eads-2 shows large variations (see Tables 2 and 3). Thus, Eads-2 can have a significant effect on the variation of Vcell. A linear relationship between Eads-2 and Vcell with a correlation coefficient of 0.765 further supports the significant effect of Eads-2 on the variation of Vcell (Fig. S3a, ESI†).
3.2 HBCs and their analogues as anode materials in magnesium-ion batteries
3.2.1. Mg2+ adsorption on doped HBC molecules.
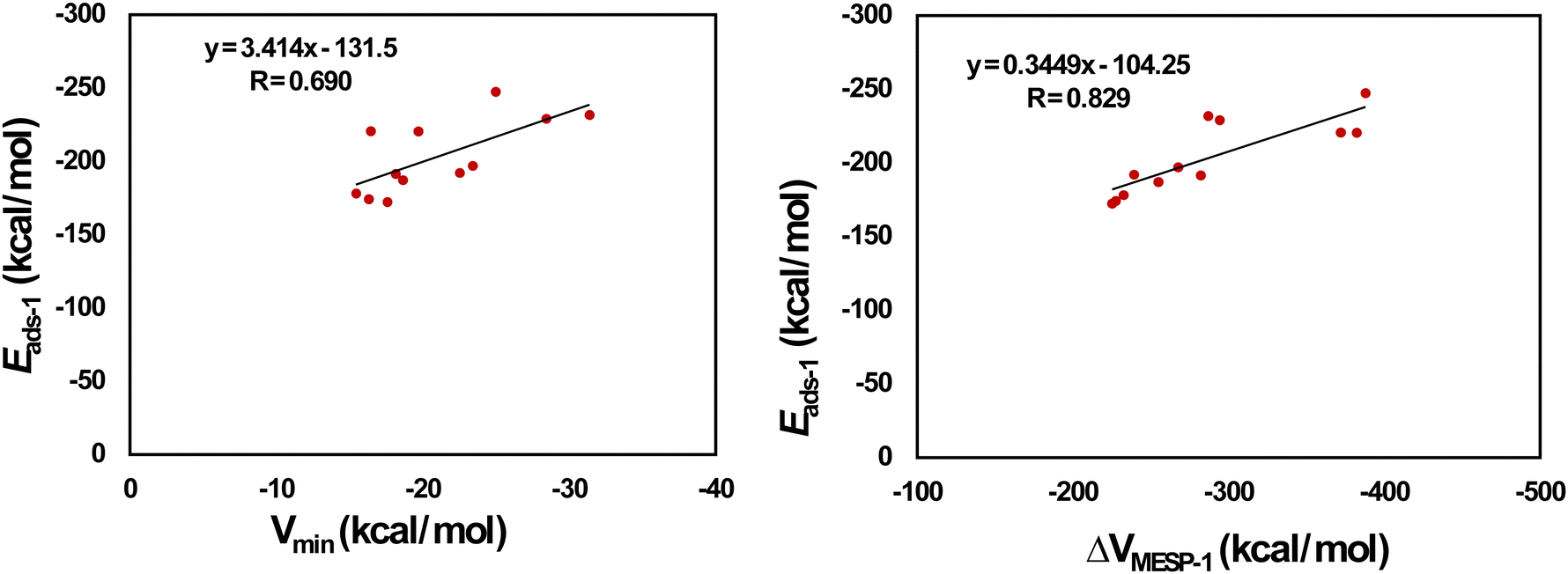
Fig. 7 provides the optimized configurations and the adsorption distances for the adsorption of Mg2+ on pristine and doped HBC molecules. We see that Mg2+ is bound to the peripheral benzene rings of HBC, similar to the case of Li+. Mg2+ is also bound to the peripheral rings of all doped HBC molecules except o-C40H18N2, o-C40H18N2, and p-C40H18N2. We find that Mg2+ is strongly adsorbed on pristine and doped HBC molecules. For instance, the adsorption distances of Mg2+ on these nanoflakes span from 3.69 (Mg2+/m-C40H18N2 system) to 2.14 Å (Mg2+/B21H18N21 system). The adsorption energy data (Table 4) also support this possibility. Here the Eads-1 values were in the range of −247.44 (Mg2+/m-C40H18N2 system) to −172.35 kcal mol−1 (Mg2+/B21H18N21 system). A higher negative value of Eads-1 usually implies a stronger interaction between Mg2+ and the nanoflake. The Eads-1 value of the Mg2+/HBC system is −178.09 kcal mol−1. This value is more negative than that of the Mg2+/C36H18B3N3 and Mg2+/B21H18N21 systems. In the case of Mg2+ adsorption on N-doped HBC molecules, the absolute values of Eads-1 followed the order o-C40H18N2 < p-C40H18N2 < m-C40H18N2. In the case of Mg2+ adsorption on BN-doped HBC molecules, the absolute values of Eads-1 followed the order B21H18N21 < C36H18B3N3 < o-C40H18BN < C38H18B2N2 < p-C40H18BN < m-C40H18BN. The Eads-1 value of the Mg2+/C21H18Si21 system (−229.05 kcal mol−1) was less negative than that of the Mg2+/C28H18Si14 system (−231.69 kcal mol−1). Here a linear relationship between Eads-1 and MESP Vmin values of the nanoflakes exists with a correlation coefficient of 0.690 (Fig. 8a). This finding reflects the weaker interactions of Mg2+ with the nanoflakes as the MESP Vmin values of the nanoflakes become less negative. Notably a better correlation was obtained for the adsorption of Li+ on pristine and doped HBC molecules (see Fig. 5a). For all cases, the adsorption free energies were negative, indicating the spontaneous nature of the adsorption process (see Table 4). Here the Gads-1 values were in the range of −238.37 (Mg2+/m-C40H18N2 system) to −161.41 kcal mol−1 (Mg2+/B21H18N21 system).
 |
| | Fig. 7 Optimized structures of Mg2+ adsorbed on (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The bond distances are given in Å. The color code is the same as in Fig. 2. In addition, Mg is denoted by the cyan color. | |
Table 4
E
ads-1, ΔVMESP-1, EHOMO, ELUMO, Eg-2 and ΔEg-1 values of Mg2+ adsorbed HBCsa
| Nanoflake |
E
ads-1
|
G
ads-1
|
ΔVMESP-1 |
E
HOMO
|
E
LUMO
|
E
g-2
|
ΔEg-1 |
|
The values of Eads-1, Gads-1 and ΔVMESP-1 in kcal mol−1; EHOMO, ELUMO, and Eg-2 in eV; ΔEg-1 in %.
|
| C42H18 |
−178.09 |
−173.45 |
−232.55 |
−11.39 |
−9.74 |
1.65 |
−69.35 |
|
o-C40H18N2 |
−220.45 |
−213.17 |
−382.29 |
−11.00 |
−8.62 |
2.38 |
−37.05 |
|
m-C40H18N2 |
−247.44 |
−238.37 |
−387.90 |
−10.86 |
−8.26 |
2.60 |
5.64 |
|
p-C40H18N2 |
−220.61 |
−213.27 |
−371.99 |
−11.30 |
−8.68 |
2.62 |
−31.09 |
|
o-C40H18BN |
−187.15 |
−177.46 |
−254.82 |
−11.48 |
−9.64 |
1.84 |
−62.28 |
|
m-C40H18BN |
−196.96 |
−187.18 |
−267.51 |
−11.79 |
−9.31 |
2.48 |
−44.70 |
|
p-C40H18BN |
−192.08 |
−182.61 |
−239.19 |
−11.60 |
−9.50 |
2.09 |
−61.14 |
| C38H18B2N2 |
−191.56 |
−183.82 |
−282.07 |
−11.12 |
−9.29 |
1.83 |
−50.62 |
| C36H18B3N3 |
−174.18 |
−168.92 |
−227.43 |
−11.32 |
−9.96 |
1.35 |
−77.60 |
| C28H18Si14 |
−231.69 |
−220.39 |
−286.89 |
−10.53 |
−8.20 |
2.33 |
−41.70 |
| C21H18Si21 |
−229.05 |
−218.53 |
−294.22 |
−10.31 |
−8.05 |
2.26 |
−57.34 |
| B21H18N21 |
−172.35 |
−161.41 |
−225.22 |
−12.62 |
−10.02 |
2.60 |
−71.06 |
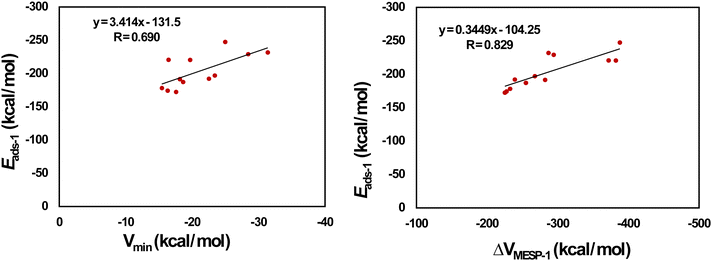 |
| | Fig. 8 Correlation between (a) Vmin and Eads-1 (b) ΔVMESP-1 and Eads-1 for Mg2+-adsorbed HBCs. | |
ΔVMESP-1 is calculated here by subtracting the MESP at the nucleus of free Mg2+ (−39.11 a.u.) from that of Mg2+ in the Mg2+/nanoflake system. For all cases, the ΔVMESP-1 values were negative, indicating the electron donation from the nanoflakes to Mg2+ (see Table 4). Here the ΔVMESP-1 values are in the range of −387.90 (Mg2+/m-C40H18N2 system) to −225.22 kcal mol−1 (Mg2+/C36H18B3N3 system). The ΔVMESP-1 value of the Mg2+/HBC system was −232.55 kcal mol−1. This value is more negative than those of the Mg2+/C36H18B3N3 and Mg2+/B21H18N21 systems. A linear relationship between Eads-1 and ΔVMESP-1 values exists with a correlation coefficient of 0.829 (Fig. 8b). This finding reflects that stronger interactions of Mg2+ with the nanoflakes push more electron density toward Mg2+.
Table 4 also lists the EHOMO, ELUMO, and Eg-2 values of the Mg2+-adsorbed nanoflakes. Here the EHOMO, ELUMO, and Eg-2 values are in the ranges of −12.62 (Mg2+/B21H18N21 system) to −10.31 eV (Mg2+/C21H18Si21 system), −10.02 (Mg2+/B21H18N21 system) to −8.05 eV (Mg2+/C21H18Si21 system), and 1.35 (Mg2+/C36H18B3N3 system) to 2.62 eV (Mg2+/p-C40H18N2 system), respectively. The EHOMO, ELUMO, and Eg-2 values of the Mg2+/HBC system are −11.39, −9.74, and 1.65 eV, respectively. The EHOMO and ELUMO values of all the Mg2+-adsorbed nanoflakes are lower than those of the corresponding Mg2+-free nanoflakes. In general, a similar trend was obtained for Eg-2. The HOMO–LUMO gap decreased by 69.35% due to Mg2+ adsorption on HBC (see ΔEg-1 values in Table 4). The maximum decrease in the HOMO–LUMO gap is obtained for Mg2+ adsorption on C36H18B3N3 (77.60%). However, Eg-1 was lower than Eg-2 for Mg2+ adsorption on m-C40H18N2 (ΔEg-1 of 5.64%).
3.2.2. Mg adsorption on doped HBC molecules.
Fig. 9 provides the optimized configurations and the adsorption distances for the adsorption of Mg on pristine and doped HBC molecules. Mg is mostly bound to the peripheral rings of these nanoflakes, as observed in the case of Mg2+. The adsorption distances of Mg on these nanoflakes span from 3.10 (Mg/C28H18Si14 system) to 3.88 Å (Mg/C21H18Si21 system). For these nanoflakes, the adsorption distances of Mg2+ (see Fig. 7) are typically smaller than those of Mg. For instance, the adsorption distances of Mg2+ and Mg on HBC are 2.30 and 3.61 Å, respectively. Mg2+ was strongly adsorbed on these nanoflakes in comparison with Mg. The adsorption energy data (see Tables 4 and 5) support this possibility. Here the Eads-2 values were in the range of −5.11 (Mg/C28H18Si14 system) to −2.14 kcal mol−1 (Mg/B21H18N21 system). The Eads-2 value of the Mg/HBC system was −3.17 kcal mol−1. This value is more negative than that of the Mg/C36H18B3N3 and Mg/B21H18N21 systems. In the case of Mg adsorption on N-doped HBC molecules, the absolute values of Eads-2 followed the order p-C40H18N2 < o-C40H18N2 < m-C40H18N2. In the case of Mg adsorption on BN-doped HBC molecules, the absolute values of Eads-2 followed the order B21H18N21 < C36H18B3N3 < o-C40H18BN < p-C40H18BN < m-C40H18BN < C38H18B2N2. The Eads-2 value of the Mg/C21H18Si21 system (−3.58 kcal mol−1) was less negative than that of the Mg/C28H18Si14 system. Here Eads-2 is not well correlated with the MESP Vmin values of the nanoflakes (Fig. S4, ESI†). Overall, the adsorption free energies were positive, suggesting that the adsorption of Mg on nanoflakes is not spontaneous (see Table 5). However, the Gads-2 values of the Mg/C42H18 and Mg/C36H18B3N3 systems are negative, indicating the spontaneous nature of the adsorption process. ΔVMESP-2 is calculated by subtracting the MESP at the nucleus of free Mg (−39.91 a.u.) from that of Mg in the Mg/nanoflake system. The ΔVMESP-2 values were negative, indicating the electron donation from the nanoflakes to Mg (see Table 5). Unlike Eads-1, Eads-2 did not display a correlation with ΔVMESP-2 (see Fig. S4, ESI†).
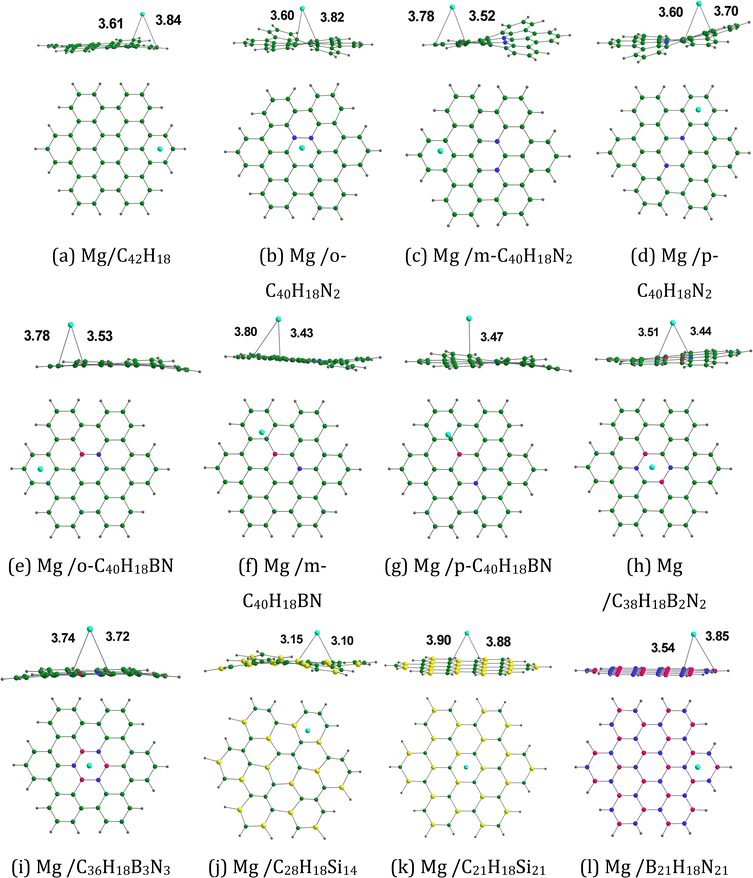 |
| | Fig. 9 Optimized structures of Mg adsorbed on (a) C42H18, (b) o-C40H18N2, (c) m-C40H18N2 (d) p-C40H18N2, (e) o-C40H18BN, (f) m-C40H18BN, (g) p-C40H18BN, (h) C38H18B2N2, (i) C36H18B3N3, (j) C28H18Si14, (k) C21H18Si21, and (l) B21H18N21. The bond distances are given in Å. The color code is the same as in Fig. 2. In addition, Mg is denoted by the cyan color. | |
Table 5
E
ads-2, ΔVMESP-2, EHOMOELUMO, Eg-3 and ΔEg-2 values of Mg adsorbed HBCs, along with ΔEcell and Vcella
| Nanoflake |
E
ads-2
|
G
ads-2
|
ΔVMESP-2 |
E
HOMO
|
E
LUMO
|
E
g-3
|
ΔEg-2 |
ΔEcell |
V
cell
|
|
The values of Eads-2, Gads-2, ΔVMESP-2, and ΔEcell in kcal mol−1; EHOMO, ELUMO, and Eg-3 in eV; ΔEg-2 in %; and Vcell in V.
|
| C42H18 |
−3.17 |
−0.65 |
−12.38 |
−5.57 |
−1.08 |
4.49 |
−16.73 |
−174.91 |
3.79 |
|
o-C40H18N2 |
−3.37 |
2.21 |
−12.13 |
−4.95 |
−1.18 |
3.77 |
−0.29 |
−217.08 |
4.71 |
|
m-C40H18N2 |
−3.50 |
2.56 |
−18.73 |
−4.10 |
−1.62 |
2.48 |
0.66 |
−243.95 |
5.29 |
|
p-C40H18N2 |
−3.34 |
2.94 |
−11.96 |
−4.83 |
−1.03 |
3.80 |
0.07 |
−217.27 |
4.71 |
|
o-C40H18BN |
−3.20 |
3.03 |
−14.90 |
−5.45 |
−1.31 |
4.14 |
−15.01 |
−183.95 |
3.99 |
|
m-C40H18BN |
−3.61 |
2.58 |
−17.56 |
−5.30 |
−1.50 |
3.80 |
−15.38 |
−193.36 |
4.19 |
|
p-C40H18BN |
−3.48 |
2.93 |
−17.12 |
−5.33 |
−1.09 |
4.24 |
−21.27 |
−188.60 |
4.09 |
| C38H18B2N2 |
−4.73 |
0.07 |
−9.53 |
−5.54 |
−1.80 |
3.74 |
0.81 |
−186.83 |
4.05 |
| C36H18B3N3 |
−2.57 |
−2.06 |
−7.58 |
−5.74 |
−0.71 |
5.03 |
−16.82 |
−171.62 |
3.72 |
| C28H18Si14 |
−5.11 |
1.81 |
−10.95 |
−5.00 |
−1.76 |
3.24 |
−18.98 |
−226.58 |
4.91 |
| C21H18Si21 |
−3.58 |
1.95 |
−3.77 |
−5.80 |
−1.15 |
4.65 |
−12.27 |
−225.46 |
4.89 |
| B21H18N21 |
−2.14 |
3.89 |
−7.07 |
−5.76 |
0.12 |
5.88 |
−34.65 |
−170.21 |
3.69 |
Table 5 also lists the EHOMO, ELUMO, and Eg-3 values of the Mg-adsorbed nanoflakes. Here the EHOMO, ELUMO, and Eg-3 values are in the ranges of −5.80 (Mg/C21H18Si21 system) to −4.10 eV (Mg/m-C40H18N2 system), −1.80 (Mg/C38H18B2N2 system) to 0.12 eV (Mg/B21H18N21 system), and 2.48 (Mg/m-C40H18N2 system) to 5.88 eV (Mg/B21H18N21 system), respectively. The EHOMO, ELUMO, and Eg-3 values of the Mg/HBC system are −5.57, −1.08, and 4.49 eV, respectively. Overall, the EHUMO, ELUMO and Eg values of all the nanoflakes are not much influenced by the presence of Mg. The HOMO–LUMO gap decreased by 16.73% due to Mg adsorption on HBC (see ΔEg-2 values in Table 5). The maximum decrease in the HOMO–LUMO gap is obtained for Mg adsorption on B21H18N21 (34.65%).
3.2.3. Cell voltage.
The ΔEcell and Vcell values of MIBs based on the pristine and doped HBC molecules are provided in Table 5. Here the ΔEcell and Vcell values were in the ranges of −243.95 (m-C40H18N2) to −170.21 kcal mol−1 (B21H18N21) and 3.69 (B21H18N21) to 5.29 V (m-C40H18N2), respectively. As previously noted, a nanoflake with a strong interaction with Mg2+ and a weak interaction with Mg might be regarded as a good candidate for the anode material in metal ion batteries. For instance, in the case of m-C40H18N2, a high Vcell value of 5.29 V was obtained because of a high Eads-1 value of −247.44 kcal mol−1 (see Table 4) and a low Eads-2 value of −3.50 kcal mol−1 (see Table 5). The ΔEcell and Vcell values of HBC were −174.91 kcal mol−1 and 3.79 V, respectively. In the case of N-doped HBC molecules, the Vcell values followed the order o-C40H18N2 < p-C40H18N2 < m-C40H18N2. In the case of BN-doped HBC molecules, the Vcell values followed the order B21H18N21 < C36H18B3N3 < o-C40H18BN < C38H18B2N2 < p-C40H18BN < m-C40H18BN. The Vcell value of C21H18Si21 (4.89 V) was slightly lower than that of C28H18Si14 (4.91 V). When considering all the nanoflakes, unlike Eads-2, Eads-1 shows large variations (see Tables 4 and 5). Thus, here, Eads-1 can have a significant effect on the variation of Vcell. A strong linear relationship between Eads-1 and Vcell further supports the significant effect of Eads-1 on the variation of Vcell (see Fig. S3b, ESI†).
The changes in Eg could influence the electrical conductivity (σ) of the material. The relationship between σ and Eg is given as:42
where
k is the Boltzmann constant and
T is the temperature. Our results show that the
Eg-1 values of the doped HBC molecules are lower than those of the pristine HBC. A smaller
Eg-1 value indicates a higher electronic conductivity. Furthermore, the
Eg values of the cation/metal-adsorbed nanoflakes were usually lower than those of the cation/metal-free nanoflakes.
The MESP is a real physical property which can be determined using X-ray diffraction techniques or computational methods. The MESP analyses showed that the replacement of the C atoms of HBC by N/BN/Si units provides a more electron-rich system than the parent HBC molecule. Importantly, we observed stronger interactions of Li+ and Mg2+ with the nanoflakes as the MESP minimum values of the nanoflakes became more negative. We observed weaker interactions of Li+ and Mg2+ with the nanoflakes as the MESP minimum values of the nanoflakes become less negative. The adsorption energy of Li has a significant effect on the variation of Vcell of LIBs, while the adsorption energy of Mg2+ has a significant effect on the variation of Vcell of MIBs. Furthermore, the storage capacity (C = (n × z × F × 103)/M, where n is the maximum number of adsorbed metal atoms and M is the molecular mass of the electrode) is an important factor for the performance of metal-ion batteries.23 Such nanoflakes are promising anode materials for high-capacity metal-ion batteries. For example, the lithium storage capacity of coronene was 536.2 mA h g−1 and the magnesium storage capacity of HBC was 923.1 mA h g−1.23,27 For a comparison, the lithium storage capacity of graphite is 372 mA h g−1, that of Sc2C MXene is 462 mA h g−1, that of graphene-like C2N is 671.7 mA h g−1, and that of pentagraphyne is 687 mA h g−1.23 This work could provide insights into the design and exploration of anode materials suitable for metal ion batteries.
The doping process and/or the adsorption process could affect the planar structure of the HBC molecule. The formation of non-planar structures is more evident in o-C40H18N2, m-C40H18N2, and p-C40H18N2 (see Fig. 2) and Mg2+-adsorbed nanoflakes (see Fig. 7). The diffusion of Li+/Li/Mg2+/Mg on the nanoflake surface could be an important factor for battery applications.43 For example, the lower diffusion barrier of Li+ accelerates the charging and discharging process of the battery.43 We plan to study the diffusion processes in a future publication.
4. Conclusions
DFT studies were carried out to understand the adsorption process of Li+, Li, Mg2+, and Mg on twelve adsorbents (pristine and N/BN/Si-doped HBC molecules C42H18, o-C40H18N2, m-C40H18N2, p-C40H18N2, o-C40H18BN, m-C40H18BN, p-C40H18BN, C38H18B2N2, C36H18B3N3, C28H18Si14, C21H18Si21, and B21H18N21). The XY (X = C, B; Y = C, N, Si) bond lengths in the pristine and doped HBC molecules span from 1.36 to 1.83 Å. In all cases, the carbon–silicon bond lengths (>1.76 Å) are significantly longer than the carbon–carbon bond lengths. The MESP analyses show that the replacement of C atoms of HBC by N/BN/Si units can provide a more electron-rich system than the parent HBC molecule.
Li+ exhibits a relatively strong adsorption on pristine and doped HBC molecules. The adsorption distances of Li+ on these nanoflakes span from 2.20 (m-C40H18N2) to 2.41 Å (C21H18Si21). Here the Eads-1 values were in the range of −67.34 (Li+/C28H18Si14 system) to −47.65 kcal mol−1 (Li+/B21H18N21 system). For the Li/nanoflake system, the Eads-2 values were in the range of −33.94 (Li/C38H18B2N2 system) to −3.93 kcal mol−1 (Li/B21H18N21 system). Mg2+ also exhibits a relatively strong adsorption on the pristine and doped HBC molecules. Here the Eads-1 values were in the range of −247.44 (Mg2+/m-C40H18N2 system) to −172.35 kcal mol−1 (Mg2+/B21H18N21 system). Our results suggest the weaker interactions of Li+ and Mg2+ with the nanoflakes as the MESP Vmin values of the nanoflakes become less negative. In all studied systems, we found electron donation from the nanoflakes to Li+ and Mg2+. For the Mg/nanoflake system, the Eads-2 values were in the range of −5.11 (Mg/C28H18Si14 system) to −2.14 kcal mol−1 (Mg/B21H18N21 system). The HOMO–LUMO gap of the cation/metal-adsorbed nanoflakes was usually lower than that of the cation/metal-free nanoflakes.
A nanoflake with a strong interaction with Li+/Mg2+ and a weak interaction with Li/Mg might be regarded as a good candidate for the anode material in metal ion batteries. Among the studied anode materials for LIBs, the highest Vcell value of 1.90 V was obtained for B21H18N21 because of the high Eads-1 value of −47.65 kcal mol−1 and the low Eads-2 value of −3.93 kcal mol−1. Among the studied anode materials for MIBs, the highest Vcell value of 5.29 V was obtained for m-C40H18N2 because of the high Eads-1 value of −247.44 kcal mol−1 and the low Eads-2 value of −3.50 kcal mol−1. For LIBs, Eads-2 has a significant effect on the variation of Vcell, while for MIBs, Eads-1 has a significant effect on the variation of Vcell.
Data availability
The data that support the findings of this study are available upon request from the corresponding author.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. ORFS-2022-CRG11-5028. R. G. S. N. and A. K. N. N. would like to thank KAUST for providing computational resources of the Shaheen II supercomputer.
References
- G. E. Blomgren, J. Electrochem. Soc., 2017, 164, A5019 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- V. Ruiz, A. Pfrang, A. Kriston, N. Omar, P. Van den Bossche and L. Boon-Brett, Renewable Sustainable Energy Rev., 2018, 81, 1427–1452 CrossRef CAS.
- W. Li, M. Li, K. R. Adair, X. Sun and Y. Yu, J. Mater. Chem. A, 2017, 5, 13882–13906 RSC.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- S. Chang, X. Jin, Q. He, T. Liu, J. Fang, Z. Shen, Z. Li, S. Zhang, M. Dahbi, J. Alami, K. Amine, A.-D. Li, H. Zhang and J. Lu, Nano Lett., 2022, 22, 263–270 CrossRef CAS.
- J. Park, C. W. Lee, S. H. Joo, J. H. Park, C. Hwang, H.-K. Song, Y. S. Park, S. K. Kwak, S. Ahn and S. J. Kang, J. Mater. Chem. A, 2018, 6, 12589–12597 RSC.
- W. Yuan, Y. Zhang, L. Cheng, H. Wu, L. Zheng and D. Zhao, J. Mater. Chem. A, 2016, 4, 8932–8951 RSC.
- A. Arya, S.-L. Hsu, C.-Y. Liu, M.-Y. Chang, J.-K. Chang, E. Y.-T. Li and Y.-S. Su, Small Struct., 2024, 2400273 CrossRef.
- L. G. Bulusheva, A. V. Okotrub, A. G. Kurenya, H. Zhang, H. Zhang, X. Chen and H. Song, Carbon, 2011, 49, 4013–4023 CrossRef CAS.
- L. Zhang, G. Xia, Z. Guo, X. Li, D. Sun and X. Yu, Int. J. Hydrogen Energy, 2016, 41, 14252–14260 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- H. Yue, F. Li, Z. Yang, J. Tang, X. Li and D. He, Mater. Lett., 2014, 120, 39–42 CrossRef CAS.
- G. Li, B. Huang, Z. Pan, X. Su, Z. Shao and L. An, Energy Environ. Sci., 2019, 12, 2030–2053 RSC.
- J. A. Blázquez, R. R. Maça, O. Leonet, E. Azaceta, A. Mukherjee, Z. Zhao-Karger, Z. Li, A. Kovalevsky, A. Fernández-Barquín and A. R. Mainar, Energy Environ. Sci., 2023, 16, 1964–1981 RSC.
- V. S. Thakur and S. Banerjee, J. Phys. Chem. C, 2024, 128, 7865–7875 CrossRef CAS.
- M. Krieg, F. Reicherter, P. Haiss, M. Ströbele, K. Eichele, M.-J. Treanor, R. Schaub and H. F. Bettinger, Angew. Chem., Int. Ed., 2015, 54, 8284–8286 CrossRef CAS.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- A. Hosseinian, A. Bekhradnia, E. Vessally, L. Edjlali and M. D. Esrafili, J. Mol. Graphics Modell., 2017, 73, 101–107 CrossRef CAS PubMed.
- W. Zhang, G. Liu, J. Cao, Y. Chen, L. Gao, G. Liu, G. Dai and Q. Wang, Chem. – Asian J., 2022, 17, e202200340 CrossRef CAS.
- D. Wang and J. Zhao, Comput. Theor. Chem., 2020, 1185, 112880 CrossRef CAS.
- A. Hosseinian, E. Vessally, M. Babazadeh, L. Edjlali and M. Es’haghi, J. Phys. Chem. Solids, 2018, 115, 277–282 CrossRef CAS.
- R. Geetha Sadasivan Nair, A. K. Narayanan Nair and S. Sun, Sci. Rep., 2024, 14, 15220 CrossRef CAS PubMed.
- P. K. Ramya and C. H. Suresh, J. Phys. Chem. A, 2023, 127, 2511–2522 CrossRef CAS PubMed.
- X. Wu, Z. Zhang and H. Soleymanabadi, Solid State Commun., 2020, 306, 113770 CrossRef CAS.
- R. Rahimi and M. Solimannejad, J. Mol. Model., 2020, 26, 157 CrossRef CAS.
- A. Hassanpour, N. Farhami, M. Derakhshande, P. Delir Kheirollahi Nezhad, A. Ebadi and S. Ebrahimiasl, Inorg. Chem. Commun., 2021, 129, 108656 CrossRef CAS.
- A. A. Peyghan and J. Beheshtian, Thin Solid Films, 2020, 704, 137979 CrossRef CAS.
- L. Buenaño, Y. Ajaj, C. Padilla, B. Vaca Barahona, N. Mejía, R. B. D. Real, B. S. R. Oviedo, J. J. F. Fiallos and S. K. Saraswat, Chem. Phys., 2024, 579, 112194 CrossRef.
- C. H. Suresh, G. S. Remya and P. K. Anjalikrishna, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2022, 12, e1601 CAS.
- G. S. Remya and C. H. Suresh, New J. Chem., 2018, 42, 3602–3608 RSC.
- G. S. Remya and C. H. Suresh, New J. Chem., 2019, 43, 14634–14642 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone and G. A. Petersson, et al., Gaussian 16, Rev.B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- Y.-X. Yu, J. Phys. Chem. C, 2016, 120, 5288–5296 CrossRef CAS.
- R. Goddard, M. W. Haenel, W. C. Herndon, C. Krueger and M. Zander, J. Am. Chem. Soc., 1995, 117, 30–41 CrossRef CAS.
- E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, Appl. Surf. Sci., 2017, 396, 740–745 CrossRef CAS.
- F. Mittendorfer and J. Hafner, Surf. Sci., 2001, 472, 133–153 CrossRef CAS.
- A. E. Kuznetsov, Comput. Theor. Chem., 2020, 1188, 112973 CrossRef CAS.
- F. B. Sayyed and C. H. Suresh, J. Phys. Chem. A, 2011, 115, 9300–9307 CrossRef CAS PubMed.
- Z. A. Alothman, M. M. Alam, M. Naushad and R. Bushra, Int. J. Electrochem. Sci., 2015, 10, 2663–2684 CrossRef.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Arun Kumar
Narayanan Nair
*,
Arun Kumar
Narayanan Nair