
Interplay between conformational flexibility, intermolecular H-bonding and 3d-metal cation extraction ability in a series of thiacalix[4]arene lower rim disubstituted Schiff base derivatives†
Iuliia V.
Strelnikova
 ab,
Alexander S.
Ovsyannikov
*a,
Aidar T.
Gubaidullin
a,
Artem S.
Agarkov
a,
Sophiya R.
Kleshnina
b,
A. A.
Iova
b,
Victor L.
Furer
c,
Alexander E.
Vandyukov
a,
Svetlana E.
Solovieva
a and
Igor S.
Antipin
b
ab,
Alexander S.
Ovsyannikov
*a,
Aidar T.
Gubaidullin
a,
Artem S.
Agarkov
a,
Sophiya R.
Kleshnina
b,
A. A.
Iova
b,
Victor L.
Furer
c,
Alexander E.
Vandyukov
a,
Svetlana E.
Solovieva
a and
Igor S.
Antipin
b
aArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, Arbuzova 8 str, Kazan 420088, Russian Federation. E-mail: osaalex2007@rambler.ru
bKazan Federal University, Kremlevskaya 18 str, Kazan 420008, Russian Federation
cKazan State Architect and Civil Engineering University, 1 Zelenaya Str., 420043 Kazan, Russian Federation
First published on 20th November 2024
Abstract
The rational design of organic ligands with the aim to control their binding abilities towards different metal ions can be considered as one of the key concepts in supramolecular coordination chemistry. Regarding the macrocyclic compounds of thiacalix[4]arene family, this can be achieved via the targeted modulation of macrocyclic platform rigidity as well as the proper choice of appended binding sites. Four macrocyclic salen-type ligands based on lower rim disubstituted thiacalix[4]arene derivatives, adopted in a cone conformation, bearing highly coordinating iminophenolic or catecholic groups and two –CH2– moieties as spacers but presenting different abilities to form H-bonds, were chosen to elucidate the interplay between the conformational flexibility of the macrocyclic ligands, propensity to participate in the intermolecular H-bonding and the extraction ability of 3d-metal cations. X-ray diffraction analysis, theoretical DFT calculations, IR and Raman spectroscopies, and dynamic light scattering (DLS) studies performed in combination with liquid–liquid metal extraction study revealed that compounds 4, and 6, based on a thiacalix[4]arene macrocyclic platform, display a higher extraction ability towards all studied 3d-metal ions, caused by enhanced conformational flexibility. This is in good accordance with the ability of 6 to form H-bonded supramolecular assemblies in solution and crystalline phases due to recognition between the catecholic moieties.
1. Introduction
Supramolecular chemistry, in which molecular recognition and self-assembly are the general principles, provides exceptionally powerful tools for constructing various building blocks, displaying the well-defined number and geometry of the anchored binding sites and enabling us to construct different supramolecular architectures in a solution or crystalline phase, due to relatively weak non-covalent interactions.1–8 In this respect, the macrocyclic organic compounds, presenting the preorganized and tunable molecular structures, represent attractive molecular building blocks.9–11 Among them, calix[4]arene12–14 (X = CH2; Fig. 1) and its structural analogue thiacalix[4]arene15 (X = S; Fig. 1), which is composed of four phenolate moieties connected by methylene bridging groups or S-atoms, respectively, are of particular interest. The binding ability of these fascinating organic receptors can be efficiently tuned up via a proper choice of the suitable conformation (cone, partial cone, 1,2-alternate or 1,3-alternate), both with functionalization of the upper or lower rims of the macrocyclic platform, by appending the binding sites of varying nature. In particular, this approach has been intensively applied to synthesize a series of thiacalix[4]arene-based macrocycles, which are capable of participating in molecular sensing, transport, or separation.16–29 Moreover, a considerably huge amount of 0D–3D metal–organic compounds, able to exhibit different attractive properties such as adsorption, single-molecule magnetic behavior, and catalysis or luminescence, were designed using the combination of these molecular building blocks with 3d–4f metal ions and different organic or inorganic auxiliary ligands.30–40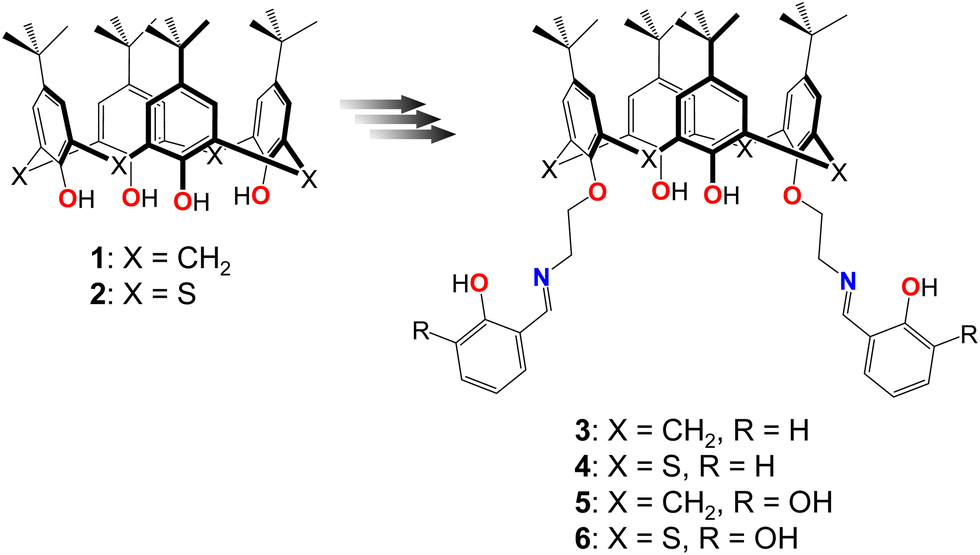 | ||
| Fig. 1 Representation of calix[4]arene (1), thiacalix[4]arene (2) and corresponding disubstituted Schiff base derivatives bearing phenolic (3, 5) or catecholic coordinating sites (4, 6). adopted in cone conformation. | ||
One of the rational approaches, allowing to control the selectivity of these macrocyclic compounds with respect to binding with metal ions, is the use of lower rim 1,3-disubstituted thiacalix[4]arenes, decorated with appended chelating coordinating sites. Coordinating groups, bearing O-, N- or S-donor atoms, such as salicylideneamine, ester, phosphonate, pyridyl, amid, crown and thia-crown ether, quinolyl, benzothiazolyl or benzimidazolyl were chosen to be incorporated into the lower rim of disubstituted thiacalix[4]arene derivatives in order to increase their selectivity towards Zn2+, Cu2+, Cd2+, Fe3+, Hg2+, Pb2+, Al3+, Ag+, U4+, alkali, alkaline earth, and lanthanide metal ions.41–51 Owing to a high coordinating ability, the 1,3-disubsustituted thiacalix[4]arene derivatives, bearing Schiff base moieties, are still gaining growing interest to design polydentate ligands, resulting in the formation of 3d–4f metal complexes displaying intriguing functions. In particular, the use of the calix[4]arenes with salicylideneamine-appended groups led to the formation of Fe(III)-based dinuclear diamond core clusters,52 exhibiting the tunable energy band gap, and mono- or dinuclear Zn(II) coordination compounds, demonstrating interesting solid-phase luminescence properties53 or the ability to promote dye photooxidation reactions via the 1O2 species generation,54 respectively, which highlights the extremely high importance of thiacalix[4]arene Schiff base coordination chemistry. It should be noted that the coordination ability of the calix[4]arene Schiff base was found to be strongly related to the flexibility of its molecular platform, causing its conformational adaptiveness of the ligand for binding with metal ions, which was confirmed by the X-ray diffraction analysis with both DFT calculations and liquid–liquid extraction techniques.55–57 Furthermore, it was established that not only the ligand flexibility but also its propensity to form the stable aggregates under rather weak intermolecular hydrophobic interactions can play a significant role in the metal ion binding process in solutions.58 However, no data devoted to study of interplay between the conformational behavior of the macrocyclic ligand, its ability to form H-bonded supramolecular architectures and its propensity to bind with 3d-metal ions was not documented up to date.
In this contribution, we report on the interplay between conformational flexibility, intermolecular H-bonding and 3d-metal cation extraction ability of a series of analogous calix[4]arene lower rim disubstituted Schiff base derivatives, differing by the nature of the macrocyclic platform (classical or thia-analogues) and the presence of α-hydroxy group in the salicylideneamine coordinating sites (salicylic and catecholic fragments), connected with the macrocyclic backbone via the spacer, composed of two methylene groups. The incorporation of additional OH-group allows the involvement of the macrocyclic ligand in extended H-bonded supramolecular architectures formation, exploiting the catechol–catechol recognition. As it was reported earlier, the use of a relatively long spacer group, composed of three methylene bridges, in the case of tetrasubstituted catecholic derivatives of thiacalix[4]arene ligands, allowed obtaining supramolecular H-bonded architectures in the crystalline phase.59 However, the presence of two –CH2– bridges in a spacer group was found to be quite enough to remarkably demonstrate the differences in the conformational behavior of the classical calix[4]arene, respective to thiacalix[4]arene analogues, when the huge tert-butyl groups were anchored at the lower rim appended salicylideneamine moieties.60
2. Experimental section
2.1. General
All chemicals were purchased from commercial suppliers and used without additional purification. Solvents were purified according to standard procedures.61p-tert-Butylcalix[4]arene (1) was synthesized following the reported procedure.12 Substance purity and reaction progress were monitored by TLC using Merck UV 254 plates and visualized by exposure to UV using a VL-6.LC lamp (Vilber, Marne-la-Vallée, France).For routine experiments, 1H and 13C NMR spectra were recorded using an AVANCE IITM 400/100 MHz BRUKER BioSpin (Germany) with signals from residual protons of DMSO-d6 or CDCl3 as the internal standard.
The mass spectra were recorded using an Ultraflex III TOF/TOF mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany). p-Nitroaniline (PNA) was used as a matrix.
Infrared (IR) and Raman spectra of milled crystalline samples in KBr pellets were recorded using a Vertex 70 FTIR spectrometer with a Bruker FT-Raman RAM II module. Raman spectra in the region of 3500–10 cm−1 were excited by the 1064 nm line of a Nd-YAG laser with a power of 300 mW on the sample.
Elemental analysis was performed using an EuroEA 3028-HT-OM Eurovector S.p.A. (Italy).
A melting point was measured by a simultaneous TGA/DSC method using a TGA/DSC NETZSCH (Selb, Germany) STA449 F3 analyser. First, approximately 15 mg sample was placed in an Al crucible with a pre-hole on the lid and heated from 25 to 500 °C. The same empty crucible was used as the reference sample. High-purity argon was used at a gas flow rate of 50 mL min−1. TGA measurement was performed at heating rates of 5 K min−1.
2.2. Synthesis of ligands
To a suspension of the corresponding precursor bis-(2-aminoethoxy)-p-tert-Butylcalix[4]arene62 (0.2 g, 0.3 mmol) in ethanol (20 mL), 2,3-dihydroxysalicylic aldehyde (0.08 g, 0.6 mmol) was added. The mixture was stirred at room temperature for 10 h. The reaction mixture was concentrated to half of its original volume under reduced pressure. A yellow precipitate of 5 was formed, filtered off, and washed with cold ethanol (10 mL), resulting in a pure product (0.19 g, 72%). Melting point 139.9 °C. 1H NMR (CDCl3, 400 MHz, 25 °C) δ, ppm: 0.93 (18H, s, -t-Bu). 1.29 (18H, s, -t-Bu), 3.31 (2H, d, 2J = 13,6 Hz, –CH2–), 4.05–4.08 (4H, m, –CH2–), 4.21–4.28 (4H, m, –CHbackbone– and –CH2–), 6.67 (2H, t, CHAr), 6.76 (4H, s, CHAr), 6.84 (2H, br d, CHAr), 6.93 (2H, brd, HAr), 7.04 (4H, s, HAr), 8.49 (2H, s, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 13C NMR (CDCl3, 100 MHz, 25 °C) δ, ppm: 167.28, 152.70, 150.54, 149.71, 147.41, 145.86, 141.83, 132.40, 127.88, 125.82, 125.25, 122.59, 117.74, 117.37, 116.69, 75.10, 56.92, 34.06, 33.98, 31.83, 31.10. MALDI TOF, m/z = 976.9 [M]+. EA calculated for C62H74O8N2: C, 76.36%, H, 7.65%, N, 2.87%, found: C, 76.15%; H, 7.81%; N, 2.80%.
N). 13C NMR (CDCl3, 100 MHz, 25 °C) δ, ppm: 167.28, 152.70, 150.54, 149.71, 147.41, 145.86, 141.83, 132.40, 127.88, 125.82, 125.25, 122.59, 117.74, 117.37, 116.69, 75.10, 56.92, 34.06, 33.98, 31.83, 31.10. MALDI TOF, m/z = 976.9 [M]+. EA calculated for C62H74O8N2: C, 76.36%, H, 7.65%, N, 2.87%, found: C, 76.15%; H, 7.81%; N, 2.80%.
2.3. Crystallization conditions
Compounds 4 [C58H66N2O6S4], 5a·[C62H74N2O8, CH2Cl2, 2C2H5OH] and 6 [C58H66N2O8S4,1.5C2H5OH]: In a crystallization vial, compound 4 (5 mg, 4.92 mmol), 5 (5 mg, 5.13 mmol) or 6 (5 mg, 4.77 mmol) was dissolved in a CH2Cl2–EtOH mixture (v/v = 1/1, 4 mL). The slow evaporation of the obtained solution at room temperature afforded yellow monocrystals, suitable for X-ray diffraction, after several days.Compound 5b·[C62H74N2O8, C2H5OH]: In a crystallization vial, compound 5 (5 mg, 5.13 mmol) was suspended in an EtOH solution (4 mL). Then, the solution was refluxed until the total solvation of compound was observed. The slow cooling till room temperature of the obtained solution during 12 h afforded yellow monocrystals, suitable for X-ray diffraction.
2.3. X-ray diffraction on single crystals
The X-ray diffraction data for the crystals of compounds 4, 5a, 5b and 6 were recorded using a Bruker D8 Quest single-crystal X-ray diffractometer equipped with an Incoatec IμS microfocus source (Mo Kα, λ = 0.71073 Å), a multilayer optics monochromator, and a PHOTON III area detector, in the ω and φ-scan modes at 296(2) for 4 and at 100(2) K for other samples. The frames were integrated using the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects by the Multi-Scan method using the SADABS program.63 The crystal data, data collection, and the refinement parameters are presented in Table 1. The structures were solved by a direct method using SHELXS and refined by the full-matrix least-squares using the SHELXTL programs.64 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were inserted at calculated positions and refined as riding atoms except the hydrogen atoms on hydroxyl groups, which were determined from the electronic density distribution. Data collection: images were indexed and integrated using the APEX3 data reduction package.65 All calculations were performed on PC using the WinGX suit of programs.66 The analysis of the intermolecular interactions was performed using the PLATON program.67 The Mercury program package68 was used for the description of the geometrical characteristics of studied compounds in the crystalline phase.| Compound name | 4 | 5a | 5b | 6 |
|---|---|---|---|---|
| Crystal formula | C58H66N2O6S4 | C62H74N2O8, CH2Cl2, 2C2H6O | C62H74N2O8, C2H5OH | C58H66N2O8S4,1.5C2H6O |
| M (g mol−1) | 1015.36 | 1152.29 | 1021.30 | 1122.45 |
| Temperature, K | 296(2) | 100(2) | 100(1) | 100(1) |
| Crystal class | Triclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | P212121 |
P |
| Crystal size, mm3 | 0.17 × 0.23 × 0.35 | 0.14 × 0.24 × 0.61 mm3 | 0.33 × 0.34 × 0.39 mm3 | 0.05 × 0.27 × 0.39 mm3 |
| Z, Z′ | 2, 1 | 4, 1 | 4, 1 | 2, 1 |
| a, Å | 12.2827(8) | 22.939(5) | 12.3553(14) | 10.3498(4) |
| b, Å | 13.3684(9) | 15.200(4) | 15.0554(19) | 16.2726(6) |
| c, Å | 16.9340(10) | 18.614(4) | 29.977(3) | 19.6183(7) |
| α,° | 99.331(4) | 90 | 90 | 67.9830(10) |
| β,° | 90.112(4) | 106.333(9) | 90 | 84.705(2) |
| γ,° | 94.896(4) | 90 | 90 | 72.065(2) |
| V, Å3 | 2733.4(3) | 6228(2) | 5576.1(11) | 2913.27(19) |
| F(000) | 1080 | 2472 | 2201 | 1194 |
| ρ calc, g cm−3 | 1.234 | 1.229 | 1.218 | 1.280 |
| μ, cm−1 | 2.25 | 1.63 | 0.80 | 2.22 |
| θ, ° | 2.371 ≤ θ ≤ 28.481 | 2.132 ≤ θ ≤ 27.342 | 1.514 ≤ θ ≤ 28.641 | 2.100 ≤ θ ≤ 33.226 |
| Refl. measured | 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 112 112 |
16540 |
160794 |
275438 |
| Independ. refl./Rint | 13467/0.1123 |
5507/0.0716 | 14103/0.1268 |
22174/0.1545 |
| Param./restraints | 660/0 | 385/0 | 720/95 | 772/4 |
| Reflections [I > 2σ(I)] | 6864 | 4187 | 10935 |
12371 |
| R 1/wR2 [I > 2σ(I)] | 0.0732/0.1372 | 0.0848/0.2238 | 0.1192/0.2869 | 0.0719/0.1712 |
| R 1/wR2 (all refl.) | 0.1686/0.1746 | 0.1063/0.2409 | 0.1443/0.3058 | 0.1470/0.2064 |
| Goodness-of-fit on F2 | 0.959 | 1.046 | 1.047 | 1.026 |
| ρ max/ρmin (e Å−3) | 0.692/−0.487 | 1.618/−0.690 | 0.647/−0.574 | 1.434/−1.205 |
Crystallographic data (excluding structure factors) for the studied structures have been deposited in the Cambridge Crystallographic Data Centre with CCDC Numbers 2345274 (4), 2345276 (5a), 2345275 (5b), and 2345277 (6),† respectively. The corresponding cif-files are available free of charge at https://www.ccdc.cam.ac.uk.
2.4. DFT calculations
The DFT optimization of geometry models and energy of conformations for 3–6 was carried out using the PRIRODA program,69 the PBE functional and the TZ2P basis. The bands in the vibrational spectra were assigned based on the potential energy distribution analysis.702.5. Dynamic light scattering
The DLS measurements of 4 × 10−3 M solutions of studied compounds in CH2Cl2 were carried out using a Zetasizer Nano ZS instrument (Malvern Panalytical, UK) with a 4 mW 633 nm He–Ne laser light source and a light scattering angle of 173° at RT. The solutions were filtered through an 800 nm filter before measurements to remove dust. Three scans were performed to calculate the average hydrodynamic diameter (d, nm) of forming aggregates in solutions. The DLS data analysis was performed using the Malvern DTS Software.2.6. Liquid–liquid metal extraction study
Transition metal picrates (C(MPicn) = 10−4 M) were obtained by mixing aqueous solutions of picric acid (C(PicH) = 10−3 M), a nitrate salt of the corresponding metals (Cu(NO3)2·3H2O, Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, Mn(NO3)2·4H2O, Co(NO3)2·6H2O, Zn(NO3)2·6H2O = 5 × 10−4 M), previously titrated with a 0.05 M solution of Trilon B, and Tris buffer (CTris = 0.05 M, pH = 5.8). Before starting the metal extraction experiments, the blank experiments were also performed, to demonstrate that neither the picrate ion nor the macrocyclic ligands individually pass into the organic phase. The aqueous solution of the prepared metal picrate (5 mL) and of a solution of ligand in methylene chloride (5 mL) was stirred in a closed flask for 30 minutes at 20 °C. The optical densities A0 and Ai of the aqueous phase before and after extraction, respectively, were measured at a wavelength in the range of 300–500 nm with a maximum at 354.91 nm. The extraction percentage (E%) was calculated according to the formula E, % = 100 × (A0 − Ai)/A0.3. Results and discussion
3.1. Synthesis of ligands
Compounds 3,714,72 and 673 were synthesized by following the earlier reported protocols via the preparation of the intermediate diamine derivatives: bis(2-aminoethyl)-p-tert-Butylcalix[4]arene62 and bis(2-aminoethyl)-p-tert-Butylthiacalix[4]arene.74 The condensation reaction of the obtained diamines with salicylic or 2,3-dihydroxysalicylic aldehydes afforded 3, 4, 5 and 6. The 1H NMR spectra obtained for 3, 4 and 6 were in good agreement with those reported in the literature, by proving the high purity of the prepared compounds (Fig. S1–S3, ESI†). A novel compound 5 was characterized by 1H/13C NMR, IR-spectroscopy, MALDI-TOF-mass spectrometry (Fig. S4–S6, ESI†), and elemental analysis (see Experimental part).3.2. Single-crystal X-ray diffraction study
The crystal structure of 3 was reported earlier.75 In order to study the structures of 4–6 in the crystalline phase, their monocrystals were obtained, using a slow evaporation technique (see Experimental part). It was revealed that thiacalix[4]arene-based compounds 4 and 6 crystallize in a triclinic P space group, whereas the calix[4]arene Schiff base derivative demonstrates more symmetrical monoclinic and orthorhombic crystal systems (C2/c or P212121, Table 1). For 4, no solvent molecules are found in the crystal lattice. In contrast, compounds 5 and 6 form solvates with EtOH and CH2Cl2 molecules. Moreover, depending on the crystallization conditions, compound 5 presents the formation of two distinct crystalline solvates, differing by the number and nature of solvate molecules. In all studied cases, the hydroxyl groups of the appended phenolic groups are still protonated and form an intermolecular H-bond with the adjacent N-imine atom (dO⋯N = 2.53(1)–2.65(2) Å). Particular attention has been paid to the solid-state conformation behaviour of the obtained compounds, which will be described below.
For 4, only one independent thiacalix[4]arene molecule, located in the general position, is found in the unit cell (Fig. 2a and Fig. S7a, ESI†). The macrocycle adopts a pinched cone conformation, displaying dihedral angles between the opposite aryl units, equal to −3.9(2)° and 110.3(2)°, and stabilized by three-centered intramolecular H-bonding, involving two OH groups of the distal phenolic moieties and one O-atom belonging to adjacent ether group (dO⋯O = 2.848(3) Å and 2.904(3) Å). The Δ parameter, denoted as the difference between the corresponding dihedral angles, is equal to 114.2°, presenting the highest value, compared to its analogue 3, based on the classical calix[4]arene platform (Table 2). Such kind of the H-bonding is typical for lower rim substituted thiacalix[4]arene compounds.76 The first alkyl C-atoms at O-ether junctions of appended lower rim substituents display endo and exo orientation with respect to the calix[4]arene cavity (Fig. 2b), which was earlier observed for the familiar thiacalix[4]arene compounds.60 Interestingly, one appended substituent is found to be directed inwards, whereas the second one outwards with respect to the macrocyclic cavity. For 4, the increase in the distance between the lower–rim-coordinating sites was established, compared to 3 (Table 3).
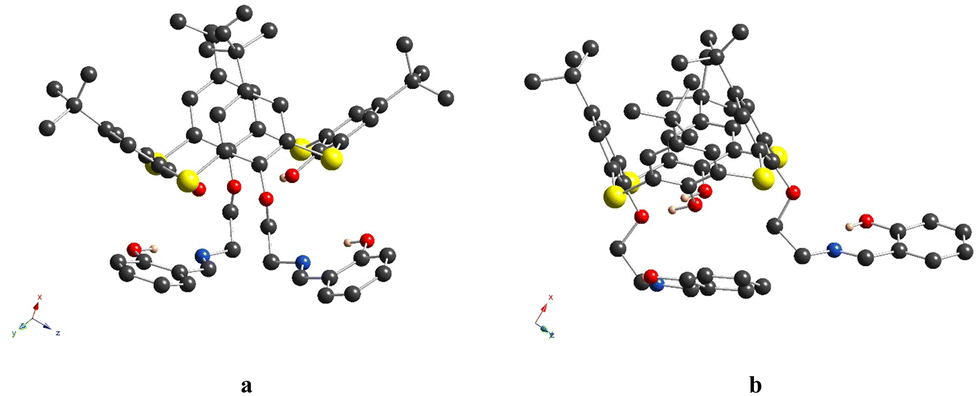 | ||
| Fig. 2 Crystal structure of 4 showing the pinched cone conformation of the macrocyclic platform (a) and exo- and endo-cavity orientation of first alkyl C-atoms grafted to O-ether junctions (b). C-, O-, N-, S- and H-atoms are represented by dark grey, red, blue, yellow and pale rose spheres. The H-atoms, which are not involved in H-bonding, are omitted for clarity. | ||
| Compound name | 3 | 4 | 5a | 5b | 6 |
|---|---|---|---|---|---|
| ∠A–C° | 52.1(2) | −3.9(2) | 39.8(1) | 40.5(1) | −2.1(1) |
| 53.0(1) | |||||
| ∠B–D° | 56.5(1) | 110.3(2) | 86.1(1) | 89.7(1) | 92.3(1) |
| 63.1(2) | |||||
| Δ° | 4.4(2) | 114.2(2) | 46.3(1) | 49.2(1) | 94.4(1) |
| 10.1(2) |
| Compound name | 3 | 4 | 5a | 5b | 6 |
|---|---|---|---|---|---|
| d ONcentr⋯ONcentr, Å | 5.412(2) | 8.453(3) | 3.768(3) | 5.33(2) | 7.210(3) |
| 5.643(2) | 5.42(2) |
In crystal, the calix[4]arene molecules are arranged into the layers along the x0y plane, in the alternate fashion, without any specific interactions (Fig. 3).
 | ||
| Fig. 3 For 4, the fragment of crystal packing shows the antiparallel arrangement of macrocyclic molecules along the 0y axis. The co-orientated molecules are presented in red and blue colors. The H-atoms are omitted for clarity. | ||
For 5a, the calix[4]arene molecule is located in a special position in the C2-axis, thus affording a very symmetrical crystal structure (Fig. 4a and Fig. S7b, ESI†). The macrocyclic platform adopts a distorted cone conformation with the dihedral angles between the opposite aryl units equal to 39.8(1)° and 86.1(1)° and accommodates one CH2Cl2 molecule, due to weak CH/π and Cl/π-interactions (dC6centr⋯C1S = 3.463(3) Å, dC6centr⋯Cl1 = 3.413(1) Å). Two H-bonds are observed between the adjacent O-hydroxyl and O-ether atoms (dO⋯O = 2.708(4) Å), leading to the strengthening of cone conformation distortion. The O-ether junctions display only an exo type of orientation of the first alkyl C-atom, leading to the symmetrical disposition of the appended lower rim substituents along the C2-axis, passing through the center of the calix[4]arene backbone (Fig. 4b). The symmetrical conformation of the molecule leads to a rather short distance between the ON centroids of coordinating sites (Table 3). Two ethanol molecules are involved in three-centered H-bonding formation with α-hydroxy groups of appended substituents (dO⋯O = 2.676(4) Å and 2.809(4) Å) (Fig. 4a). In the crystal, the calix[4]arene molecules are stacked in antiparallel rows running along the 0y axis (Fig. 5). No specific interactions are observed between the calix[4]arene molecules within the crystal.
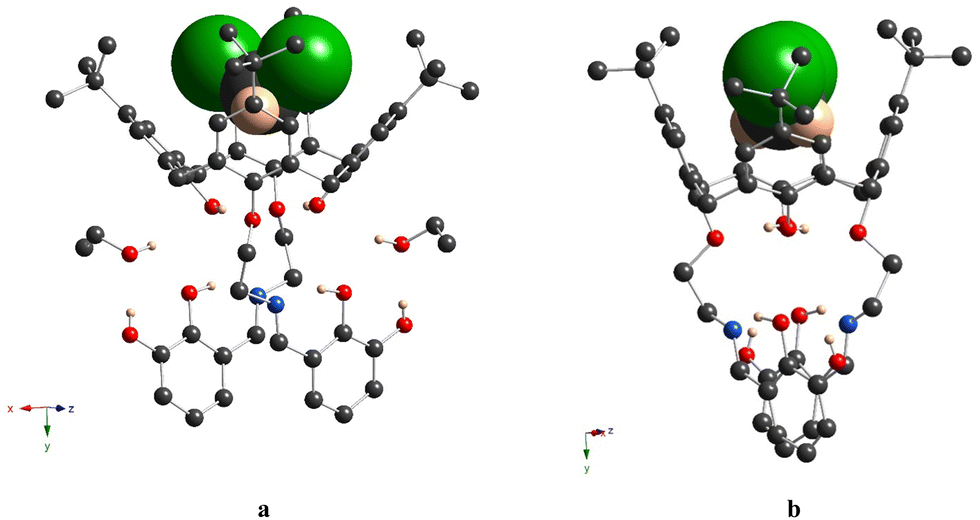 | ||
| Fig. 4 Crystal structure of 5a, showing the distorted cone conformation of the macrocyclic platform (a) and exo-cavity orientation of C-alkyl atoms belonging to O-ether junctions (b). C-, O-, N-, Cl- and H-atoms are represented by dark grey, red, blue, green and pale rose spheres. The H-atoms, which are not involved in H-bonding, are omitted for clarity. The CH2Cl2 molecule, accommodated within the cavity, is presented in a space-filling mode. | ||
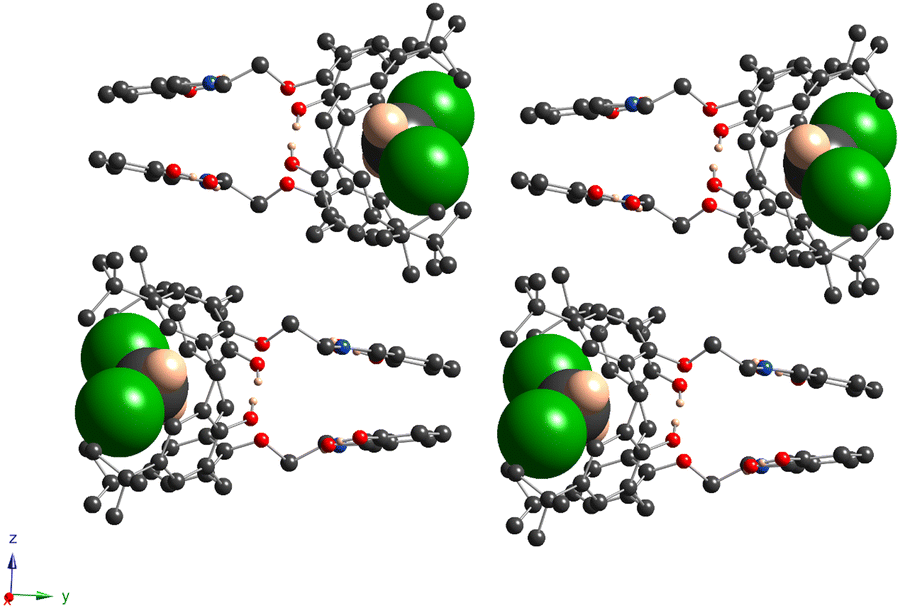 | ||
| Fig. 5 For 5a, the fragment of crystal packing shows the antiparallel arrangement of macrocyclic molecules along the 0y axis. The C-, O-, N-, Cl- and H-atoms are represented by dark grey, red, blue, green and pale rose spheres. The CH2Cl2 molecule, accommodated within the cavity, is presented in a space-filling mode. The H-atoms, which are not involved in H-bonding, as well as EtOH molecules are omitted for clarity. | ||
The crystal unit of 5b is composed of highly disordered molecules of the macrocycle, which are caused by the superposition of two left-handed and right-handed conformational enantiomers, derived from the rotation of the appendant substituent along the main calix[4]arene axis, passing through the centre of the macrocyclic cavity (Fig. S7c, ESI†). The macrocycle backbone of both isomers adopts a distorted cone conformation, which is very similar to that observed for 5a (Fig. 6 and Fig. S7c (ESI†) and Table 2). As for 5a, both C29 and C30-atoms, connected to O-ether junctions, unambiguously demonstrate exo orientation with respect to the calix[4]arene cavity. Within the cavity, the trapped solvent molecule (modeled as EtOH) is bound by weak CH/π-interactions with calix[4]arene aryl units (dC2V⋯C6centr = 3.50(1) Å, 3.53(1) Å). Similar to 5a, the crystal packing of 5b is formed by the staking of the macrocycle molecules into the 1D chains, running along the 0x axis (Fig. 7). Since the α-hydroxysalicylidenamine groups of the calixarene molecule and the solvate EtOH molecule are disordered over two positions; in some crystal units, the intermolecular H-bonding is observed between the neighboring inclusion complexes (dO⋯O = 2.64(4) Å, 2.85(4) Å).
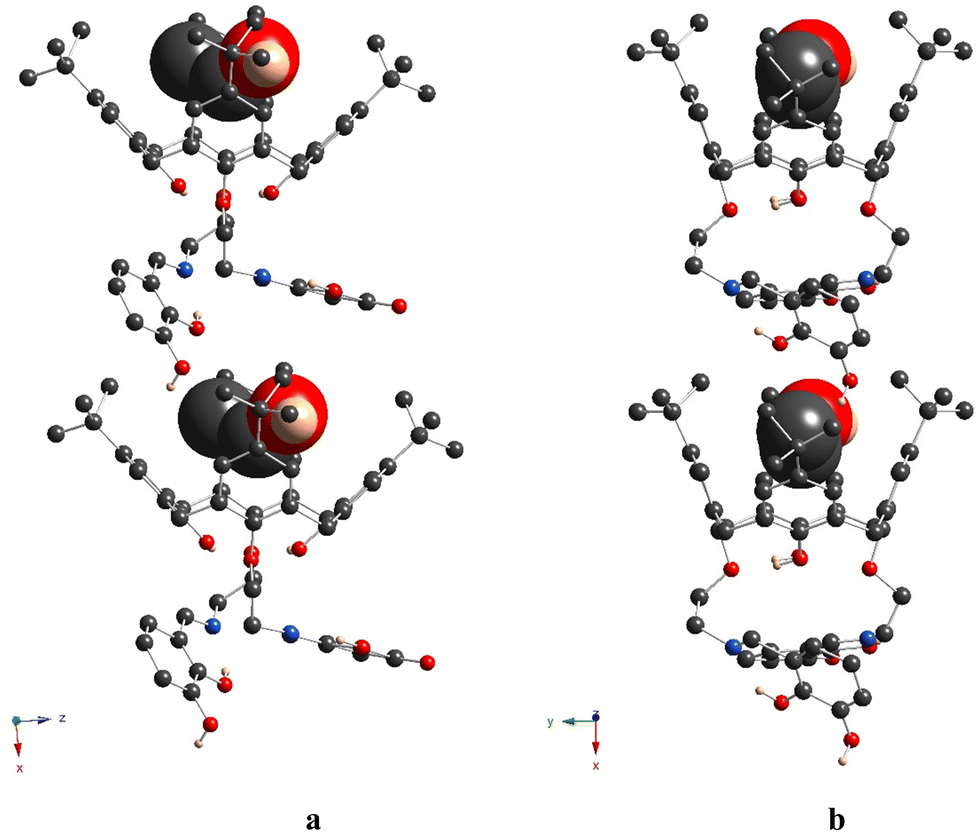 | ||
| Fig. 6 Crystal structure of 5b, showing the distorted cone conformation of the macrocyclic platform (a) and exo-cavity orientation of the C-alkyl atoms, attached to the O-ether junctions (b). The C-, O-, N- and H-atoms are represented by dark grey, red, blue and pale rose spheres. The H-atoms, which are not involved in H-bonding, as well as disordered fragments are omitted for clarity. The EtOH molecule, accommodated within the cavity, is presented in a space-filling mode. Only the atoms with the highest occupancies are presented. | ||
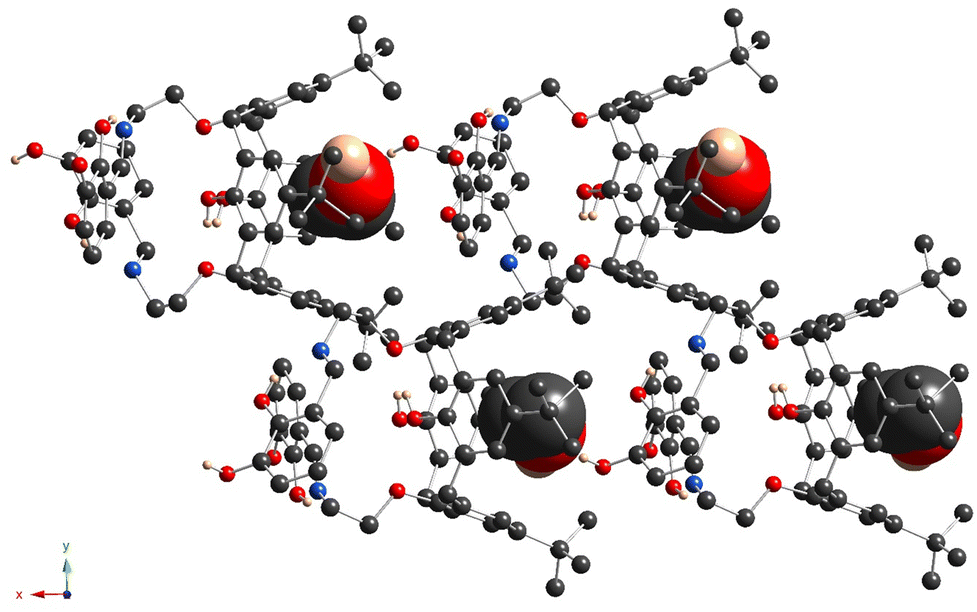 | ||
| Fig. 7 For 5b, the fragment of crystal packing, showing the stacking of the 1D chains, formed by macrocycle molecules, along the 0x axis. The C-, O-, N-, Cl- and H-atoms are represented by dark grey, red, blue, green and pale rose spheres. The accommodated EtOH molecules are presented in space-filling mode. The H-atoms, which are not involved in H-bonding, and the disordered fragments are omitted for clarity. The EtOH molecule, accommodated within the cavity, is presented in a space-filling mode. Only the atoms with the highest occupancies are presented. | ||
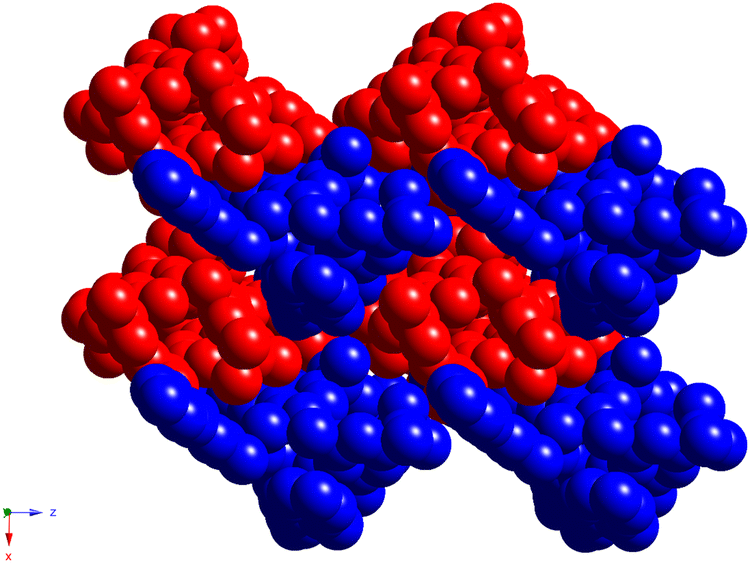
Finally, compound 6, representing the thiacalix[4]arene analogue of 5, displays a crystal structure, which is very similar to this one observed for 4. As for 4, the thiacalix[4]arene adopts a pinched cone conformation of the macrocyclic platform, which is confirmed by the analysis of the dihedral angles between the opposite aryl units (Fig. 8a and Fig. S7d (ESI†) and Table 2). Moreover, the same conformation of the O-ether junctions was evidenced, displaying the exo and endo orientations of the C-alkyl atoms, with respect to the inner macrocyclic cavity. As a result, it leads to similarity with 4 conformation behavior of the pendant units: the first one is directed outwardly and the second inwardly, with respect to the macrocycle platform, leading to a considerably large distance between the ON centroids (Table 3). As for 4, the three-centered H-bonding, stabilizing the pinched cone conformation, is observed in the case of 6 (dO⋯O = 2.892(2) Å and 2.915(2) Å).
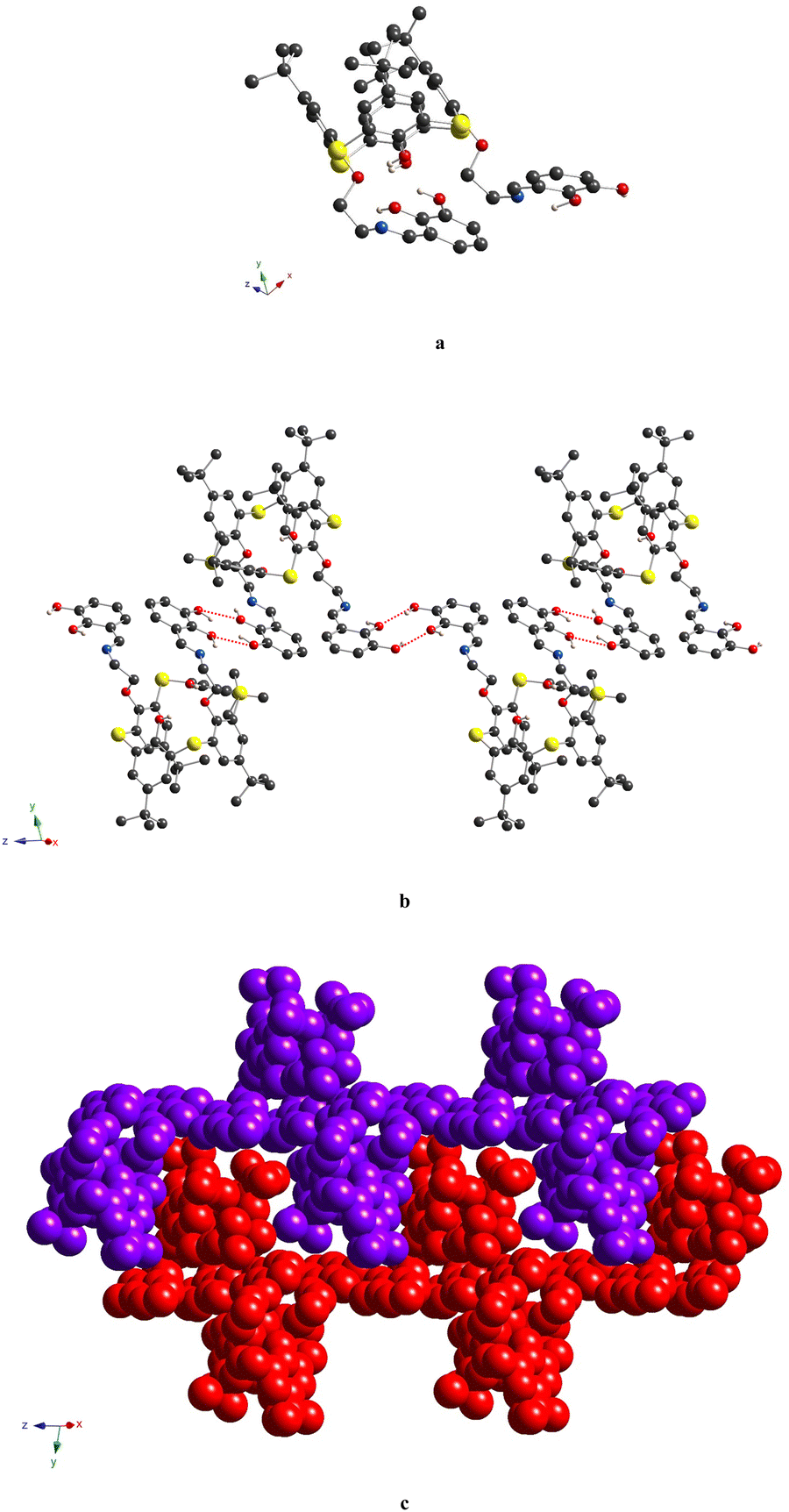 | ||
| Fig. 8 Crystal structure of 6, showing exo- and endo-cavity orientation of the C-alkyl atoms belonging to the O-ether junctions (a), formation of the 1D chain, resulting from the H-bonding between the OH-groups belonging to catecholic moieties of adjacent thiacalix[4]arene molecules (b), and crystal packing of adjacent H-bonded 1D chains (depicted in violet and red colors) (c). The C-, O-, N-, S- and H-atoms are represented by dark grey, red, blue, yellow and pale rose spheres. The H-atoms, which are not involved in H-bonding, as well as disordered tert-butyl fragments are omitted for clarity. | ||
A distinguished feature, which emphasizes the particularity of compound 6 in the row of studied compounds, is the formation of the 1D chains, resulting from the intermolecular H-bonding between the catecholic OH-groups, belonging to adjacent macrocyclic molecules, in the crystalline phase. One of two α-hydroxy groups of the catecholic moieties acts as the H-donor and another one as the H-acceptor, leading to the bis-monohapto binding mode of the H-bonding (Fig. 8b). Thus, the crystal packing of 6 is formed by the stacking of the obtained 1D chains in an antiparallel fashion (Fig. 8c). Additionally, the EtOH molecules are involved in the H-bonding with α-hydroxy groups of catecholic fragments with an O⋯O distance equal to 2.869(5) Å and 2.922(5) Å.
Thus, one may conclude that thiacalix[4]arene-based compounds 4 and 6 exhibit the enhanced conformational flexibility in terms of distortions of the macrocytic platform, compared to their calix[4]arene analogues 3 and 5, which is caused by the increase in the macrocyclic cavity size, due to the insertion of S-atoms (for 4), instead of –CH2– bridging groups. Additionally, it was observed that for 4 and 6, the exo and endo orientated the first alkyl C-atoms, belonging to appended substituents, whereas the calix[4]arenes 375 and 5 present only the exo orientation. Moreover, it should be noted that the hydrophobic macrocyclic cavity of calix[4]arenes 3 and 5 seems more suitable for the accommodation of the solvent molecules, due to noncovalent interactions, which can also contribute to a high conformational rigidity of the calix[4]arene compounds.
3.3. DFT study
In order to perform the conformational analysis and evaluate the molecular flexibility of compounds 3–6, the DFT calculations were carried out. As attested by X-ray diffraction analysis, two limited rotamers for studied lower rim disubstituted calix[4]arene and thiacalix[4]arene derivatives can be distinguished, depending on the orientation of the first C-alkyl atom at O-ether junctions, belonging to appendant substituents, with respect to the inner macrocyclic cavity, i.e. exo–exo and endo–exo rotamers (Fig. 9 and Fig. S8–S11, ESI†). The rotamer featured by endo–endo orientation of the appendant substituents was excluded from the calculations due to steric factor, resulting in enhanced repulsion of the alkyl substituents. The energy of transitions between the mentioned rotamers can help establish the adaptiveness of the ligands for binding with the appropriate metal cations.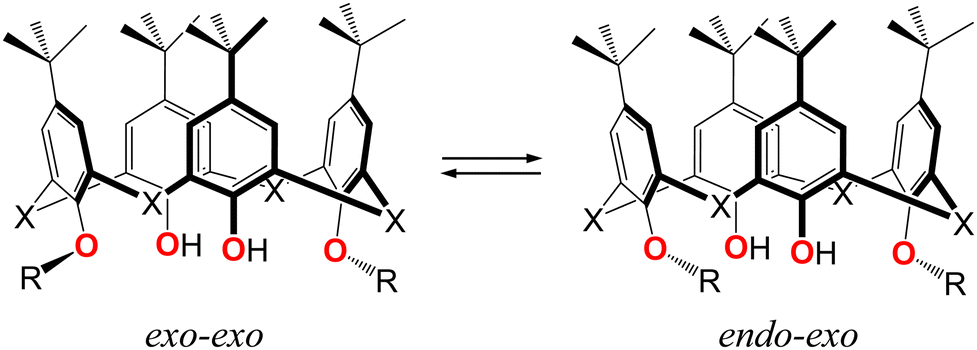 | ||
| Fig. 9 Two main possible conformers for 3 and 5 (X = CH2) and 4 and 6 (X = S), determined by the rotation of the appendant lower rim substituents around the C–O single bond and the tilting of the opposite aryl units of the macrocyclic backbone. | ||
As it was established, for calix[4]arenes 3 and 5, the exo–exo rotamer (Fig. S8a and S10a, ESI†) is considered as the most stable one. The energy barrier corresponding to transition from exo–exo to endo–exo rotamer (Fig. S8b and S10b, ESI†) was estimated as 5.37 kcal mol−1 and 5.96 kcal mol−1, respectively (Table 4). Moreover, such structural transformation undergoes significant distortion of the macrocyclic backbone, resulting in the dihedral angle increase of the unsubstituted aryl moieties (Tables S1 and S2, ESI†).
| Compound | Conformation | Total energy, kcal mol−1 | Energy difference, kcal mol−1 | E HOMO, eV | E LUMO, eV | Dipole moment, D |
|---|---|---|---|---|---|---|
| 3 | Exo–exo | −1860632.14 |
0.00 | −6.122 | −0.756 | 4.17 |
| Endo–exo | −1860626.77 |
5.37 | −6.276 | −0.817 | 3.57 | |
| 4 | Exo–exo | −2761142.79 |
2.06 | −6.260 | −0.822 | 9.05 |
| Endo–exo | −2761144.85 |
0.00 | −6.235 | −0.902 | 6.56 | |
| 5 | Exo–exo | −1954982.94 |
0.00 | −6.312 | −0.519 | 4.47 |
| Endo–exo | −1954976.98 |
5.96 | −6.186 | −0.795 | 4.44 | |
| 6 | Exo–exo | −2855493.37 |
1.96 | −6.151 | −0.854 | 9.05 |
| Endo–exo | −2855495.33 |
0.00 | −6.155 | −0.903 | 12.10 |
In contrast, for thiacalix[4]arenes 4 and 6, the endo–exo rotamer is the most stable one (Table 4). The energy of transition between the corresponding rotamers was estimated as 2.06 kcal mol−1 and 1.96 kcal mol−1 for 4 and 6, respectively.
It should be noted that the role of the H-bonding, which can occur between the OH-phenolic groups of the macrocycle platform and O-ether moieties of adjacent aryl units in the stabilization of the studied rotamers. For 3 and 5, the exo–exo rotamers display H-bonding between O-ether atoms, belonging to the lower rim substituents of the macrocycle backbone, with only one adjacent OH-phenolic center with O⋯O distances of 2.710 Å, 2.716 Å and 2.726 Å, 2.730 Å, respectively. In a meanwhile, for thiacalix[4]arenes 4 and 6, both OH-groups of the macrocycle backbone are involved in H-bonding with only one O-ether moiety of adjacent aryl units, exhibiting the exo orientation of the alkyl chain, with dO⋯O equal to 2.895 Å, 2.905 Å and 2.871 Å, 2.933 Å, respectively, which is in good agreement with the X-ray diffraction data (Fig. 2a and 8a).
Thus, one can conclude that the difference in conformational behavior between calix[4]arene and thiacalix[4]arene-based derivatives, revealed by X-ray diffraction, is additionally confirmed by DFT calculations, suggesting that involvement of the solvent molecules, in particular CH2Cl2 solvent, in interaction with the macrocyclic backbone does not significantly influence the transitions between exo–exo and endo–exo rotamers, when passing from the crystalline phase to the gas phase. Therefore, it can be assumed that the preferred most stable conformation, established for studied macrocycles, can also persist upon solubilization in a CH2Cl2 solvent that was used for DLS and metal extraction studies.
3.4. Molecular vibration analysis
The IR and Raman spectroscopies are excellent tools to perform the molecular vibrational analysis of compounds 3–6 in order to study their conformation behavior in the crystalline phase,77–79 which, being performed in combination with appropriate DFT calculations, allows not only assign the signals observed in IR-/Raman spectra with the molecular structure and specific conformations of studied compounds but also confirm the homogeneity of studied solid-state sample.For 3–6, the DFT-calculated data of vibrational characteristics are in good accordance with the experimentally obtained IR-/Raman spectra (Fig. S12–S21 and Tables S3–S6, ESI†), studied for crystalline samples, thus proving the homogeneity of the studied solid-state sample. In the region of the high wavenumbers of IR-/Raman spectra, the vibrational spectra of calix[4]arenes and thiacalix[4]arenes are quite similar (Fig. S12 and S13, ESI†). One can notice the asymmetric and symmetric stretching vibration bands corresponding to CH groups with frequencies in the range from 2962 to 2868 cm−1. The stretching vibration bands attributed to the OH groups are observed at 3433 cm−1 and 3389 cm−1, for calix[4]arenes 3 and 5, and 3420 cm−1 and 3386 cm−1 for thiacalix[4]arenes 4 and 6, indicating the shifting of the signals to lower wavenumbers, evidencing the decrease in the H-bonding strength for thiacalix[4]arenes 4 and 6, and the catecholic derivatives 5 and 6.
The intensive characteristic stretching vibration bands of the CN bonds are found to be quite similar for 3–6 and are located in the range of 1633 cm−1–1637 cm−1 and 1640 cm−1–1638 cm−1 in the IR and Raman spectra, respectively (Fig. S12 and S13, ESI†).
In the middle part of the IR spectra (Fig. S14, S16, S18, S20, ESI†), the vibration bands corresponding to symmetric and asymmetric stretching and bending vibrations of C–C, C–H, C–O, C–O–H and C–C–H bonds of aromatic units are found to be present at 1582–1195 cm−1, demonstrating a good match within the series of calix[4]arene and thiacalix[4]arene macrocycles. The Raman spectra demonstrate more specific characters depending on the compound structure at this wavenumber region (Fig. S15, S17, S19, S21, ESI†). The similar trend is observed for IR- and Raman spectra in the lower wavenumber region, displaying more specific vibrations, attributed to bending and twisting vibrations of the aromatic fragments, depending on the nature of the macrocyclic platform and the appended substituents.
Thus, the obtained results of vibrational analysis demonstrated a good agreement between theoretically calculated and experimentally obtained X-ray diffraction data evidencing a significant difference in conformation, observed for calix[4]arenes 3, 5 and thiacalix[4]arenes 4, 6 in the crystalline phase.
3.5. Metal extraction study
The ability of macrocyclic ligands to bind various cations of 3d-metals has been studied in solutions by a liquid extraction method (see Experimental part). Mn2+, Fe3+, Co2+, Ni2+, Cu2+ and Zn2+ were chosen to reveal the influence of the nature of the cations, their softness/hardness with respect to HSAB theory, charges and electronic properties on the coordination behavior of the ligand. In general, it was established, that thiacalix[4]arenes 4 and 6 demonstrated a higher extraction ability than that of their calix[4]arene-based analogues 3 and 5, respectively (Table 5, Fig. 10), whereas 4 extracts Mn2+, Fe3+, and Co2+ ions more efficiently with an extraction rate >91%, and 3 demonstrates the highest extraction towards Fe3+-ions with an extraction rate of 84%, followed by the decreasing extraction ability, when passing to Mn2+ and Co2+ ions (64% and 74%, respectively). Interestingly, it was found that in the case of the Cu2+ and Zn2+ ions, which are considered softer than Mn2+, Fe3+, and Co2+, the extraction rate displays the reverse dependence, when comparing 3 and 4 (Fig. 10b). Compound 3 showed superior extraction rates towards Cu2+ and Zn2+ ions, equal to 75% and 63%, respectively. The Ni2+ cations were bound by both macrocycles 3 and 4 with relatively lower extraction rates. Considering the results of liquid extraction, obtained for 3 and 4, one may conclude that compound 3 demonstrates a similar extraction ability, with respect to Mn2+, Co2+, Cu2+ and Zn2+, which may evidence the formation of the complexes of similar coordination motif and stability constants. For 4, the flexibility of the macrocyclic ligand starts to play a paramount role, displaying the differentiation in extraction ability between the Mn2+/Fe3+/Co2+ and Ni2+/Cu2+/Zn2+ groups of ions.| Metal ion | Mn2+ | Fe3+ | Co2+ | Ni2+ | Cu2+ | Zn2+ |
|---|---|---|---|---|---|---|
| 3 | 64 | 84 | 74 | 31 | 75 | 63 |
| 4 | 91 | 96 | 97 | 38 | 70 | 42 |
| 5 | 38 | 69 | 41 | 27 | 65 | 28 |
| 6 | 70 | 97 | 74 | 53 | 85 | 62 |
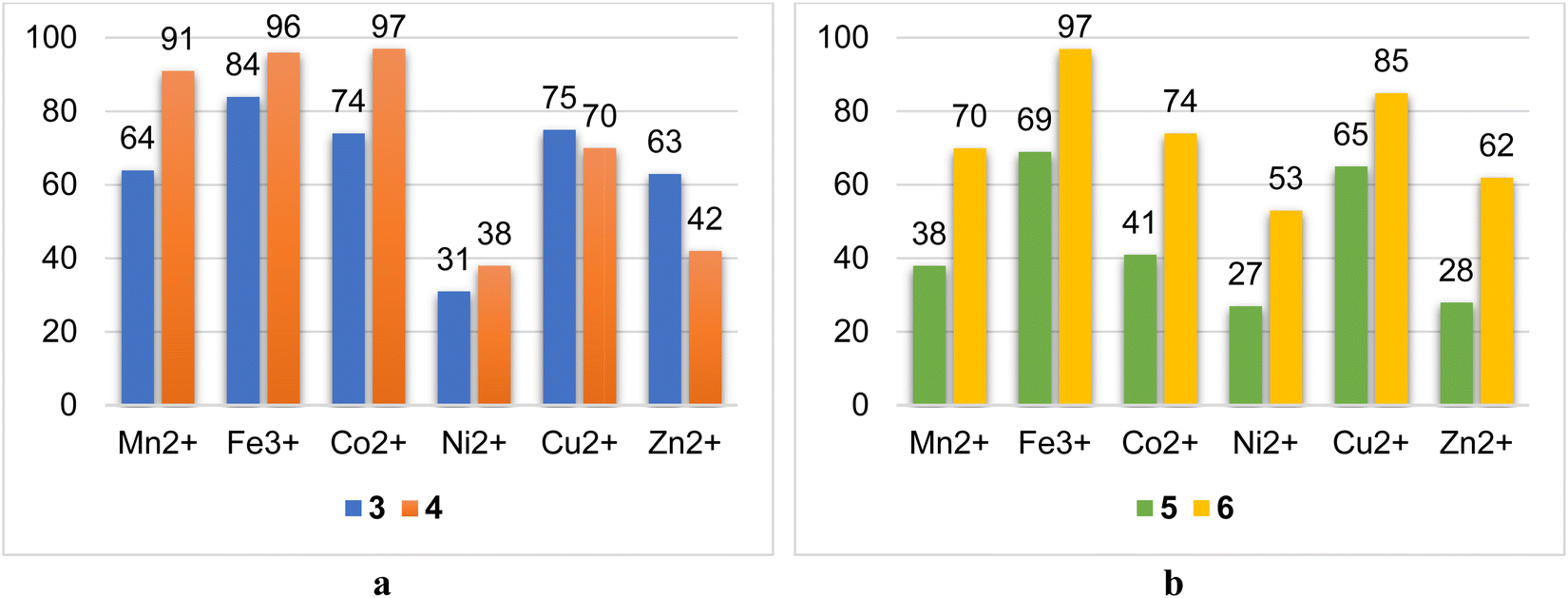 | ||
| Fig. 10 Diagrams showing the comparison of extraction rates for pairs 3, 4 (a) and 5, 6 (b) towards different 3d-metal cations. | ||
Meanwhile, it was revealed that the anchoring of an α-OH-group into the salicylideneamine coordinating sites (catecholic fragments) leads to reduced extraction ability of the calix[4]arene-based ligand, as shown by the example of 5 (Table 5 and Fig. 10b), which can be explained by the repulsion of OH-groups in the formed complexes, resulting in the diminishing of their stability constants. However, when switching from calix[4]arene 5 to thiacalix[4]arene 6, a drastic increase in the extraction rates was observed for Ni2+, Cu2+, and Zn2+ ions, which may indicate the involvement of α-OH-groups in coordination with metal ions.
All studied ligands have demonstrated the best extraction rates towards Fe3+ ions, which is evidently caused by the increase in the ion charge.
3.6. DLS study
As it was reported earlier,58 the metal cation extraction ability of calix[4]arene ligands can be related to their propensity to aggregate in solutions. In order to establish the influence of this factor on extraction behavior, the aggregation ability of 3–6 in the CH2Cl2 solution was studied by the standard method of dynamic light scattering (DLS) (see Experimental part). The DLS analysis of the freshly prepared solutions of the macrocycles with a concentration of 4 × 10−3 M was performed under room temperature conditions. It was found out that 3–5 do not form stable aggregates in the CH2Cl2 solution (Fig. S21, ESI†), whereas for 6, the associates exhibiting a hydrodynamic diameter of 28.29 ± 3.53 nm and relatively low polydispersity (PDI = 0.393) were observed (Fig. 11). The obtained results are in accordance with the X-ray diffraction data and highlight the propensity of ligand 6 to form 1D H-bonded supramolecular polymers, displaying an intermolecular catechol–catechol recognition pattern, which is derived from the enhanced flexibility of the thiacalix[4]arene macrocyclic backbone. In contrast, according to the X-ray diffraction data, compounds 3–5 are not prone to be involved in the intermolecular H-bonding, which may also reflect the decrease in their extraction ability towards the metal cations, compared to 6 (Fig. 8c).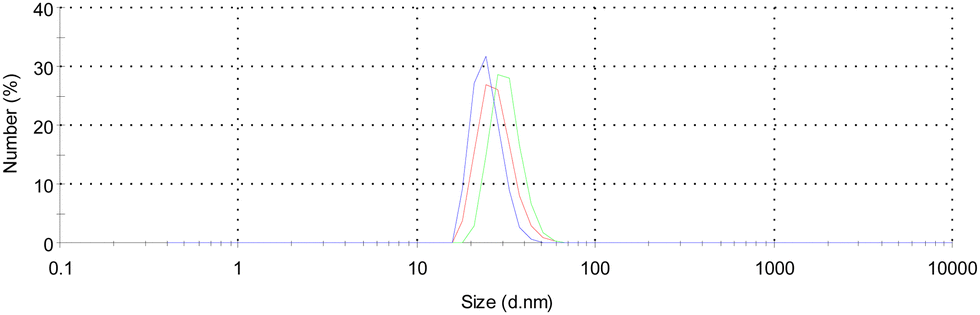 | ||
| Fig. 11 For 6, the DLS number distribution spectra for 6, showing the size of nanoaggregates, presenting in CH2Cl2 solution. The blue, red and green colored lines correspond to three consecutively recorded DLS spectra, indicating a relatively high convergence of results. | ||
4. Conclusion
For the first time, the correlation between the conformational flexibility, intermolecular H-bonding and 3d-metal cation extraction ability was established in a series of lower rim disubstituted thiacalix[4]arene ligands decorated with iminophenolic or catecholic coordinating binding sites. For classical calix[4]arenes 3 and 5, the X-ray diffraction, DFT calculations, and IR and Raman spectroscopy study revealed the most favorable conformation (PC) of the macrocycle ligand, when the first alkyl C-atoms of spacer groups adopt exo–exo orientation with respect to the calix[4]arene cavity. Contrary to that, for thiacalix[4]arenes 4 and 6, the endo–exo orientation of the first alkyl C-atoms of spacer groups is the most favorable, due to the high distortion of the macrocyclic platform, supported by the intramolecular H-bonding between two OH-phenolic H-donor centers of the macrocyclic platform and proximal O-ether junction, located in-between.In terms of supramolecular behavior in the crystalline phase, due to high rigidity of the macrocyclic platform, the classical calix[4]arene based compounds 3 and 5, regardless of the nature of the coordinating sites, are not involved in any intermolecular H-bonding. For thiacalix[4]arene 4, bearing iminophenolic groups, no extended molecular structures were observed in the crystalline phase. In contrast, in the case of compound 6, the catechol–catechol recognition between the appended moieties allowed obtaining the 1D H-bonded supramolecular architecture. Additionally, for 6, the DLS experiments confirmed the possibility to form the extended supramolecular aggregates in the solution.
The liquid–liquid extraction showed the highest extraction rate for compound 6 towards all studied metal ions. The obtained results allow us to better understand the tricky interplay between the conformational flexibility of the macrocyclic ligand of the thiacalix[4]arene family and their coordination and H-bonding behaviors, providing new pathways for the rational design of macrocyclic ligands with the desired coordination and aggregation properties.
Author's contributions
A. S. O., S. E. S., I. S. A. designed project, conception and methodology, A. T. G. performed X-ray diffraction analysis, I. V. S., A. S. A., A. A. I. carried out the synthesis of studied compounds and acquired data, V. L. F. performed quantum chemical calculations, A. E. V. performed IR-/Raman spectroscopies studies, S. R. K. performed the liquid–liquid extraction and DLS experiments, A. S. O., I. V. S., V. L. F. contributed in draft version of the article writing. All authors have read and agreed to the published version of the manuscript.Data availability
All ESI† is available and contains spectral characterization of the ligands (1H/13C NMR and MALDI TOF mass spectra), ORTEP views of asymmetric units, DFT-optimized geometry of the ligands, solid-state IR and Raman spectra and their characteristics, and tables with experimental and theoretically calculated structural characteristics for studied ligands. See DOI: https://doi.org/10.1039/D4CP03393K.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the State Assignment to Arbuzov Institute of Organic and Physical Chemistry of FRC Kazan Scientific Center of RAS (No. 122011800132-5). The authors are grateful to Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for their help and support in IR-, Raman, EA, TGA and XRD-studies. Dr Mariia V. Kniazeva is warmly acknowledged for her help in preparation of single crystals of studied compounds.References
- J.-M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379, 10.1039/C7CS00115K.
- E. Mattia and S. Otto, Nat. Nanotech., 2015, 10, 111–119, DOI:10.1038/nnano.2014.337.
- J. Vicens and Q. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 251–274, DOI:10.1007/s10847-011-0001-z.
- T. Haino, Polym. J., 2013, 45, 363–383, DOI:10.1038/pj.2012.144.
- I. S. Antipin, M. V. Alfimov, V. V. Arslanov, V. A. Burilov, S. Z. Vatsadze, Y. Z. Voloshin, K. P. Volcho, V. V. Gorbatchuk, Y. G. Gorbunova, S. P. Gromov, S. V. Dudkin, S. Y. Zaitsev, L. Ya. Zakharova, M. A. Ziganshin, A. V. Zolotukhina, M. A. Kalinina, E. A. Karakhanov, R. R. Kashapov, O. I. Koifman, A. I. Konovalov, V. S. Korenev, A. L. Maksimov, N. Z. Mamardashvili, G. M. Mamardashvili, A. G. Martynov, A. R. Mustafina, R. I. Nugmanov, A. S. Ovsyannikov, P. L. Padnya, A. S. Potapov, S. L. Selektor, M. N. Sokolov, S. E. Solovieva, I. I. Stoikov, P. A. Stuzhin, E. V. Suslov, E. N. Ushakov, V. P. Fedin, S. V. Fedorenko, O. A. Fedorova, Y. V. Fedorov, S. N. Chvalun, A. Yu. Tsivadze, S. N. Shtykov, D. N. Shurpik, M. A. Shcherbina and L. S. Yakimova, Russ. Chem. Rev., 2021, 90, 895, DOI:10.1070/RCR5011.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111(11), 6810–6918, DOI:10.1021/cr200077m.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111(9), 5784–5814, DOI:10.1021/cr100357h.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637, 10.1039/C7CS00063D.
- H. Zhu, L. Chen, B. Sun, M. Wang, H. Li, J. F. Stoddart and F. Huang, Nat. Rev. Chem., 2023, 7, 768–782, DOI:10.1038/s41570-023-00531-9.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121(4), 2445–2514, DOI:10.1021/acs.chemrev.0c00522.
- G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737–2763, 10.1039/D0CS00948B.
- C. D. Gutsche, M. Iqbal and D. Stewart, J. Org. Chem., 1986, 51(5), 742–745, DOI:10.1021/jo00355a033.
- J. Vicens, J. Harrowfield and L. Baklouti, Calixarenes in the nanoworld, Springer, Dordrecht, Netherlands, 2007 DOI:10.1007/978-1-4020-5022-4.
- P. Neri, J. L. Sessler and M.-X. Wang, Calixarenes and Beyond, Springer, Netherlands, 2016. DOI:10.1007/978-3-319-31867-7.
- N. Iki, C. Kabuto, T. Fukushima, H. Kumagai, H. Takeya, S. Miyanari, T. Miyashi and S. Miyano, Tetrahedron, 2000, 56, 1437–1443, DOI:10.1016/S0040-4020(00)00030-2.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119(16), 9657–9721, DOI:10.1021/acs.chemrev.8b00605.
- R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870, 10.1039/C4CS00068D.
- X. Fan and X. Guo, J. Mol. Liq., 2021, 325, 115246, DOI:10.1016/j.molliq.2020.115246.
- M. A. Hussain, M. U. Ashraf, G. Muhammad, M. N. Tahir and S. N. A. Bukhari, Curr. Pharm. Des., 2017, 23, 2377–2388, DOI:10.2174/1381612822666160928143328.
- C. Jiang, Z. Song, L. Yu, S. Ye and H. He, TrAC, Trends Anal. Chem., 2020, 133, 116086, DOI:10.1016/j.trac.2020.116086.
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107(9), 3780–3799, DOI:10.1021/cr068046j.
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386, 10.1039/C2CS35233H.
- B. Mokhtari and K. Pourabdollah, Asian J. Chem., 2013, 25, 1–12, DOI:10.14233/ajchem.2013.12058.
- D. M. Homden and Carl Redshaw, Chem. Rev., 2008, 108(12), 5086–5130, DOI:10.1021/cr8002196.
- W. Sliwa and T. Girek, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 15–41, DOI:10.1007/s10847-009-9678-7.
- O. A. Mostovaya, P. L. Padnya, A. A. Vavilova, D. N. Shurpik, B. I. Khairutdinov, T. A. Mukhametzyanov, A. A. Khannanov, M. P. Kutyreva and I. I. Stoikov, New J. Chem., 2018, 42, 177–183, 10.1039/C7NJ03953K.
- Y. Razuvayeva, R. Kashapov and L. Zakharova, Supramol. Chem., 2020, 178–206, DOI:10.1080/10610278.2020.1725515.
- D. Quaglio, F. Polli, C. Del Plato, G. Cianfoni, C. Tortora, F. Mazzei, B. Botta, A. Calcaterra and F. Ghirga, Supramol. Chem., 2021, 1–25, DOI:10.1080/10610278.2021.2011283.
- S. E. Solovieva, V. A. Burilov and I. S. Antipin, Macroheterocycles, 2017, 10, 134–146, DOI:10.6060/mhc170512a.
- T. Kajiwara, N. Iki and M. Yamashita, Coord. Chem. Rev., 2007, 251, 1734–1746, DOI:10.1016/j.ccr.2007.01.001.
- I. V. Khariushin, V. Bulach, S. E. Solovieva, I. S. Antipin, A. S. Ovsyannikov and S. Ferlay, Coord. Chem. Rev., 2024, 513, 215846, DOI:10.1016/j.ccr.2024.215846.
- Y. Bi, S. Du and W. Liao, Coord. Chem. Rev., 2014, 276, 61–72, DOI:10.1016/j.ccr.2014.06.011.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119(1), 322–477, DOI:10.1021/acs.chemrev.8b00260.
- A. Ovsyannikov, S. Solovieva, I. Antipin and S. Ferlay, Coord. Chem. Rev., 2017, 352, 151–186, DOI:10.1016/j.ccr.2017.09.004.
- X. Hang and Y. Bi, Dalton Trans., 2021, 50, 3749–3758, 10.1039/D0DT04233A.
- R. O. Fuller, G. A. Koutsantonis and M. I. Ogden, Coord. Chem. Rev., 2020, 402, 213066, DOI:10.1016/j.ccr.2019.213066.
- O. Santoro and C. Redshaw, Coord. Chem. Rev., 2021, 448, 214173, DOI:10.1016/j.ccr.2021.214173.
- L. R. B. Wilson, M. Coletta, M. Evangelisiti, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Dalton Trans., 2022, 51, 4213–4226, 10.1039/D2DT00152G.
- A. S. Ovsyannikov, I. V. Khariushin, S. E. Solovieva, I. S. Antipin, H. Komiya, N. Marets, H. Tanaka, H. Ohmagari, M. Hasegawa, J. J. Zakrzewski, S. Chorazy, N. Kyritsakas, M. W. Hosseini and S. Ferlay, RSC Adv., 2020, 10, 11755–11765, 10.1039/D0RA01263G.
- S. M. Aldoshin, I. S. Antipin, M. V. Kniazeva, D. V. Korchagin, R. B. Morgunov, A. S. Ovsyannikov, A. V. Palii, N. A. Sanina, G. V. Shilov and S. E. Solovieva, Isr. J. Chem., 2020, 60, 1–8, DOI:10.1002/ijch.201900155.
- R. Joseph and C. P. Rao, Chem. Rev., 2011, 111(8), 4658–4702, DOI:10.1021/cr1004524.
- A. Muravev, A. Yakupov, T. Gerasimova, D. Islamov, V. Lazarenko, A. Shokurov, A. Ovsyannikov, P. Dorovatovskii, Y. Zubavichus, A. Naumkin, S. Selektor, S. E. Solovieva and I. S. Antipin, Int. J. Mol. Sci., 2022, 23, 2341, DOI:10.3390/ijms23042341.
- W. Sliwa and T. Girek, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 15–41, DOI:10.1007/s10847-009-9678-7.
- R. Gramage-Doria, D. Armspach and D. Matt, Coord. Chem. Rev., 2013, 257, 776–816, DOI:10.1016/j.ccr.2012.10.006.
- A. Bauer, A. Jäschke, S. S. A. Azzam, F. Glasneck, S. Ullmann, B. Kersting, V. Brendler, K. Schmeide and T. Stumpf, Sep. Purif. Technol., 2019, 213, 246, DOI:10.1016/j.seppur.2018.12.041.
- A. Bauer, A. Jäschke, S. Schöne, R. Barthen, J. März, K. Schmeide, M. Patzschke, B. Kersting, K. Fahmy, J. Oertel, V. Brendler and T. Stumpf, ChemistryOpen, 2018, 7, 467, DOI:10.1002/open.201800085.
- V. V. S. Mummidivarapu, V. K. Hinge and C. P. Rao, Dalton Trans., 2015, 44, 1130, 10.1039/C4DT01726A.
- R. K. Pathak, V. K. Hinge, P. Mondal and C. P. Rao, Dalton Trans., 2012, 41, 10652, 10.1039/c2dt30432e.
- R. K. Pathak, J. Dessingou, V. K. Hinge, A. G. Thawari, S. K. Basu and C. P. Rao, Anal. Chem., 2013, 85(7), 3707–3714, DOI:10.1021/ac400059w.
- A. Jaeschke, M. Kischel, A. Mansel and B. Kersting, Eur. J. Inorg. Chem., 2017, 894, DOI:10.1002/ejic.201601326.
- A. Muravev, A. Yakupov, T. Gerasimova, R. Nugmanov, E. Trushina, O. Babaeva, G. Nizameeva, V. Syakaev, S. Katsyuba, S. Selektor, S. E. Solovieva and I. S. Antipin, Int. J. Mol. Sci., 2021, 22, 3535, DOI:10.3390/ijms22073535.
- I. V. Strelnikova, I. D. Shutilov, A. S. Ovsyannikov, D. R. Islamov, A. V. Pyataev, T. P. Gerasimova, A. R. Khamatgalimov, M. N. Khrizanforov, A. T. Gubaidullin, V. A. Burilov, S. E. Solovieva and I. S. Antipin, CrystEngComm, 2024, 26, 3973–3988, 10.1039/D4CE00347K.
- S. Ullmann, R. Schnorr, C. Laube, B. Abel and B. Kersting, Dalton Trans., 2018, 47, 5801, 10.1039/C8DT00757H.
- C. Laube, J. Taut, J. Kretzschmar, S. Zahn, W. Knolle, S. Ullmann, A. Kahnt, B. Kersting and B. Abel, Inorg. Chem. Front., 2020, 7, 4333–4346, 10.1039/D0QI00980F.
- J. P. Chinta, B. Ramanujam and C. P. Rao, Coord. Chem. Rev., 2012, 256, 2762–2794, DOI:10.1016/j.ccr.2012.09.001.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962, DOI:10.1016/j.ccr.2008.06.008.
- S. E. Solovieva, S. R. Kleshnina, M. N. Kozlova, L. F. Galiullina, A. T. Gubaidullin, Sh. K. Latypov, I. S. Antipin and A. I. Konovalov, Russ. Chem. Bull., 2008, 57, 1477–1485, DOI:10.1007/s11172-008-0191-8.
- A. I. Konovalov and I. S. Antipin, Mendeleev Commun., 2008, 18(5), 229–237, DOI:10.1016/j.mencom.2008.09.001.
- M. H. Noamane, S. Ferlay, R. Abidi, N. Kyritsakas and M. W. Hosseini, Inorg. Chim. Acta, 2017, 468, 260, DOI:10.1016/j.ica.2017.04.047.
- I. V. Strelnikova, I. D. Shutilov, A. S. Ovsyannikov, F. B. Gabdrakhmanova, A. S. Agarkov, A. T. Gubaidullin, A. R. Khamatgalimov, S. E. Solovieva and I. S. Antipin, Russ. Chem. Bull., 2024, 73, 653–668, DOI:10.1007/s11172-024-4175-0.
- H. G. Becker, W. Berger and G. Domschke, ORGANIKUM. Organisch-chemisches Grundpraktikum, Deutscher Verlag der Wissenschaften, Berlin, 1965 Search PubMed.
- A. P. Luk’yanenko, E. A. Alekseeva, S. S. Basok, A. V. Mazepa and A. I. Gren’, Russ. J. Org. Chem., 2011, 47, 527–529, DOI:10.1134/S1070428011040105.
- G. Sheldrick, SADABS, Program for empirical X-ray absorption correction, Bruker-Nonius, 2004.
- G. Sheldrick, SHELXTL v.6.12, Structure Determination Software Suite, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- APEX2 (Version 2.1), SAINTPlus, Data Reduction and Correction Program (Version 7.31A), BrukerAXS Inc., Madison, Wisconsin, USA, 2006 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838, DOI:10.1107/S0021889899006020.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13, DOI:10.1107/S0021889802022112.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., 2002, B58, 389–397, DOI:10.1107/S0108768102003324.
- D. N. Laikov and Yu. A. Ustynyuk, Russ. Chem. Bull., 2005, 54, 820–826, DOI:10.1007/s11172-005-0329-x.
- V. A. Sipachev, Calculation of shrinkage corrections in harmonic approximation, J. Mol. Struct. Theochem., 1985, 121, 143–151, DOI:10.1016/0166-1280(85)80054-3.
- M. Dey, J. P. Chinta, G. J. Long and C. P. Rao, Indian J. Chem., 2009, 48, 1484–1491 Search PubMed.
- E. N. Zapolotsky, S. P. Babailov, M. V. Kniazeva, Y. V. Strelnikova, A. S. Ovsyannikov, A. T. Gubaidullin, S. E. Solovieva, I. S. Antipin, E. S. Fomin and I. P. Chuikov, Inorg. Chim. Acta, 2022, 545, 121267, DOI:10.1016/j.ica.2022.121267.
- V. L. Furer, A. E. Vandyukov, A. S. Ovsyannikov, I. V. Strelnikova, A. S. Agarkov, S. E. Solovieva and I. S. Antipin, Struct. Chem., 2024, 1–13, DOI:10.1007/s11224-024-02298-1.
- V. Bhalla, M. Kumar, T. Hattori and S. Miyano, Tetrahedron, 2004, 60(28), 5881–5887, DOI:10.1016/j.tet.2004.05.035.
- L. Li, W.-W. Gu and C.-G. Yan, Chem. Res. Chin. Univ., 2010, 26, 38, DOI:10.5517/ccrzlpz.
- S. E. Solovieva, E. V. Popova, A. O. Omran, A. T. Gubaidullin, S. V. Kharlamov, Sh. K. Latypov, I. S. Antipin and A. I. Konovalov, Russ. Chem. Bull., 2011, 60, 486–498, DOI:10.1007/s11172-011-0076-0.
- I. Mohammed-Ziegler and F. Billes, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 19–42, DOI:10.1007/s10847-006-9132-z.
- V. L. Furer, E. I. Borisoglebskaya, V. V. Zverev and V. I. Kovalenko, Spectrochim. Acta, 2006, 63, 207–212, DOI:10.1016/j.saa.2005.05.006.
- V. I. Kovalenko, A. V. Chernova, E. I. Borisoglebskaya, S. A. Katsyuba, V. V. Zverev, R. R. Shagidullin, I. S. Antipin, S. E. Solovieva, I. I. Stoikov and A. I. Konovalov, Russ. Chem. Bull., 2002, 51, 825–827, DOI:10.1023/A:1016084817527.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2345274–2345277. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp03393k |
| This journal is © the Owner Societies 2025 |