
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revising exciton diffusion lengths in polymer dot photocatalysts†
Andjela
Brnovic
 ,
Leigh Anna
Hunt
,
Haining
Tian
and
Leif
Hammarström
*
,
Leigh Anna
Hunt
,
Haining
Tian
and
Leif
Hammarström
*
Department of Chemistry, Ångström Laboratory, Uppsala University, SE 751 20 Uppsala, Sweden. E-mail: leif.hammarstrom@kemi.uu.se
First published on 2nd December 2024
Abstract
Exciton migration in organic polymer dots (Pdots) is crucial for optimizing photocatalytic reactions at the particle surface, such as hydrogen evolution and carbon dioxide reduction. Despite the use of Pdots in photocatalysis, there is still a need for better understanding of exciton diffusion within these systems. This study investigates the exciton diffusion in PFBT Pdots stabilized with different weight percentages of the co-polymer surfactant PS–PEG–COOH and doped with perylene red as an internal quencher. Time-resolved fluorescence quenching data yields a quenching volume that the excitons explore during their lifetime (Vq), which is comparable to the volume of the hydrophobic core of PFBT Pdots. This indicates that excitons can migrate to the particle surface with high probability and suggests that the intrinsic exciton diffusion length (LD ≈ 19 nm) for PFBT is significantly larger than previously reported in Pdot studies from the literature (5.3 and 8.6 nm). Additionally, a larger quenching rate constant (kq) and smaller volume (Vq) is observed for the higher PS–PEG–COOH weight ratio, which are attributed to their smaller core. The study provides insights into the exciton migration within Pdots, with important implications for photocatalysis.
Introduction
Organic polymer dots (Pdots) are π-conjugated, semiconducting polymers prepared as nanoparticles with particle size less than 100 nm. They have attracted attention in recent years for the photocatalytic synthesis of solar fuels from water and carbon dioxide.1–3 Photoexcitation of Pdots results in the formation of tightly bound and localized Frenkel excitons within one of the polymer segments, followed by non-radiative energy transfer between the segments in a series of incoherent hopping steps, a process known as exciton diffusion. Of particular importance in this study is the diffusion of singlet excitons, which is facilitated by Förster resonance energy transfer (FRET).4–11 In the context of solar photochemistry, the efficiency of light-harvesting relies, among several other factors, on the diffusion of excitons to the Pdots–water interface where photocatalytic reactions take place. The self-assembly of conjugated polymers into Pdots allows them to be effectively dispersed in water where its high dielectric constant promotes dissociation of excitons at the interface.9,12 Thus, a long exciton diffusion length (LD), i.e., long-range excitation energy transport, with LD being at least similar to the Pdot radius is necessary for their optimal photocatalytic performance.12,13Accurately measuring exciton diffusion in Pdots, however, has been challenging due to inconsistencies in the existing literature regarding modeling approaches.14–22 To address this, we used semiconducting polymer PFBT, as it has been intensively investigated for different photocatalytic proton and carbon dioxide reduction reactions in single-component and heterojunction Pdots.23–26 Additionally, PFBT Pdots have been used previously as a model system to gain better understanding of exciton diffusion in Pdots.19,21,26 In a study by Groff et al., LD of PFBT Pdots was investigated through doping with the internal quencher perylene red.19 The Stern–Volmer analysis of steady-state fluorescence data yielded LD = 5.3 nm, while simulations incorporating energy transfer quenching by both perylene red and defects in PFBT resulted in LD = 12 nm for hypothetically defect-free PFBT Pdots. The authors concluded that existing methods for determining LD experimentally might distort their accurate values, attributing this to quenching by volumetrically distributed defects in aggregated systems such as Pdots. Significant quenching by defects was supported by the much shorter reported average excited state lifetime of PFBT Pdots (0.8 ns) compared to PFBT in a good solvent such as THF (ca. 3.0 ns). Building upon this study, Ponzio et al. determined LD of PFBT Pdots through titration with the external quencher rhodamine B.21 A more detailed theoretical modeling was performed, yielding a LD value of 8.6 nm, and the quenching by defect sites distributed on the Pdot surface rather than volume was proposed. However, the excited state lifetime of Pdots was reported as ca. 2.8 ns, suggesting that the defect density was much lower than in Groff et al. Agreement between the simulations and experiments was fair, but time-resolved fluorescence data with rhodamine B were lacking, and defect quenching kinetics were poorly fit by the model used.
Here, we use analytical model to fit time-resolved fluorescence data of Pdots to determine the quenching volume (Vq) that the excitons explore during their lifetime and estimate LD. The model was originally developed independently by Infelta and Tachiya for fluorescence quenching in micelles.27,28 To our knowledge, this is the first study to utilize the Infelta–Tachiya model for investigating exciton diffusion in Pdots. We use PFBT (average Mn ≤ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) Pdots stabilized with different weight ratios of the amphiphilic co-polymer surfactant PS–PEG–COOH (Mw 36500) (1:1 or 1:3 PFBT:PS–PEG–COOH (wt/wt)) and doped with perylene red as internal energy transfer quencher (Förster radius R0 = 3 nm (ref. 19)) distributed within the hydrophobic core of the particle. PS–PEG–COOH provides electrostatic and steric stabilization to the particles surface, ensures colloidal stability, and is not photo- or redox active.24,29 From time-resolved fluorescence decay traces at different quencher concentrations, we estimated Vq and the rate constant for quenching in the exciton volume (kq).
000) Pdots stabilized with different weight ratios of the amphiphilic co-polymer surfactant PS–PEG–COOH (Mw 36500) (1:1 or 1:3 PFBT:PS–PEG–COOH (wt/wt)) and doped with perylene red as internal energy transfer quencher (Förster radius R0 = 3 nm (ref. 19)) distributed within the hydrophobic core of the particle. PS–PEG–COOH provides electrostatic and steric stabilization to the particles surface, ensures colloidal stability, and is not photo- or redox active.24,29 From time-resolved fluorescence decay traces at different quencher concentrations, we estimated Vq and the rate constant for quenching in the exciton volume (kq).
Results and discussion
Pdots were prepared by a nanoprecipitation method, with different concentrations of the quencher perylene red (a detailed procedure is provided in the ESI†).2,24 Chemical structures of PFBT, perylene red, and PS–PEG–COOH are presented in Fig. 1. The quencher was assumed to be randomly distributed within the hydrophobic particle core, as both the excited probe (PFBT) and the quencher have hydrophobic properties. The hydrodynamic radii of PFBT Pdots were determined using dynamic light scattering (DLS) (Fig. S1a and b, ESI†). Both single-component PFBT and doped PFBT Pdots had an average radius of 15 nm for both 1:1 or 1:3 (wt/wt) ratios, which suggests that the number of Pdots per volume in the latter solution was larger for the same concentration of PFBT. PS–PEG–COOH has stabilizing properties in colloids where its sterically demanding PEG groups prevent aggregation. We suggest that the higher PS–PEG–COOH content likely favoured the formation of smaller Pdots with increased surface curvature, as it provided larger volume per PEG chain. Thus, instead of forming larger Pdots at the 1:3 (wt/wt) ratio, the core volume decreased, maintaining a hydrodynamic radius comparable to that of the 1:1 (wt/wt) ratio. The UV-Vis absorption spectra of Pdots (Fig. 2a and b) showed broad absorption extending into the visible region up to 540 nm, with two distinct absorption bands at ∼310 nm and ∼470 nm. Upon doping with perylene red, a weak, red-shifted absorption band appeared between 550 and 620 nm, corresponding to perylene red absorption (Fig. S2, ESI†) and showed a steady increase with increasing perylene red concentration. The doping concentrations of perylene red used were 7, 22, 36, 71, 102, and 127 nM for the 1:1 (wt/wt) ratio, and 7.8, 22, 37, 66, 103, and 129 nM for the 1:3 (wt/wt) ratio, determined from the amount of perylene red added during Pdots preparation.
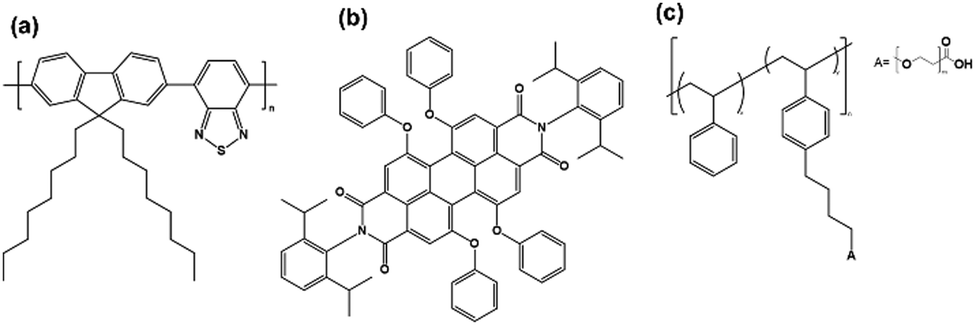 | ||
| Fig. 1 Chemical structures of (a) PFBT, (b) perylene red and (c) PS–PEG–COOH. | ||
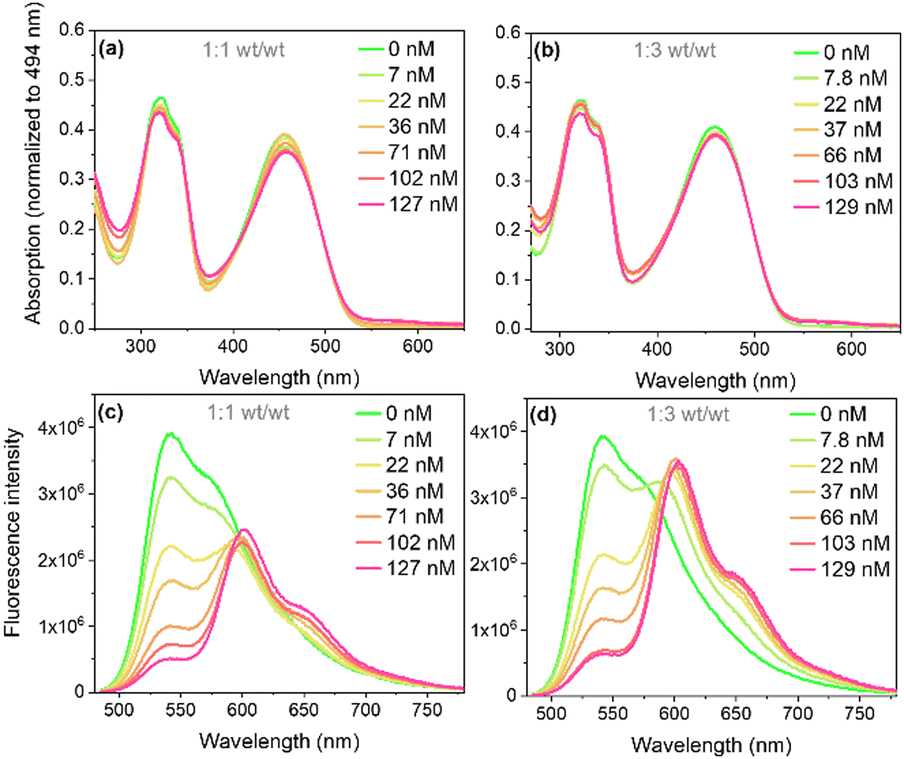 | ||
| Fig. 2 Absorption (a) and (b) and fluorescence (c) and (d) spectra with excitation at 473 nm (Abs = 0.08) of single-component and doped PFBT Pdots at 1:1 and 1:3 (wt/wt) ratios. | ||
The steady state fluorescence spectra of PFBT and doped PFBT Pdots under selective excitation of PFBT at 473 nm is shown in Fig. 2c and d. The PFBT fluorescence peak at 540 nm decreased with increasing perylene red concentration, while an additional fluorescence band from perylene red appeared with a peak at 600 nm and a shoulder around 660 nm (cf. Fig. S2, ESI†), which suggests FRET between PFBT and perylene red within Pdots. The occurrence of energy transfer was supported by overlaying the corrected excitation spectra for perylene red emission onto the absorption spectra of doped PFBT Pdots (Fig. S3, ESI†).
Time-resolved fluorescence measurements revealed that the decrease in fluorescence emission of PFBT in the presence of perylene red was followed by a near equivalent reduction of fluorescence lifetime, which is consistent with dynamic (diffusional) quenching (Fig. 3a and b). Fluorescence decay traces of PFBT Pdots with no quencher present were fit with a double-exponential decay function with time constants of τ1 = 1.4 ns (A1 = 0.76) and τ2 = 3.4 ns (A2 = 0.24) for 1:1 (wt/wt) ratio and of τ1 = 1.3 ns (A1 = 0.76) and τ2 = 3.6 ns (A2 = 0.24) for 1:3 (wt/wt) ratio. The double-exponential behavior suggests inhomogeneous probe environments. In contrast to PFBT dissolved in THF, where polymeric chains are isolated by solvent molecules and exhibit a single exponential fluorescence decay with an excited state lifetime of 2.8 ns (Fig. S4, ESI†), the polymeric chains in Pdots are densely packed.
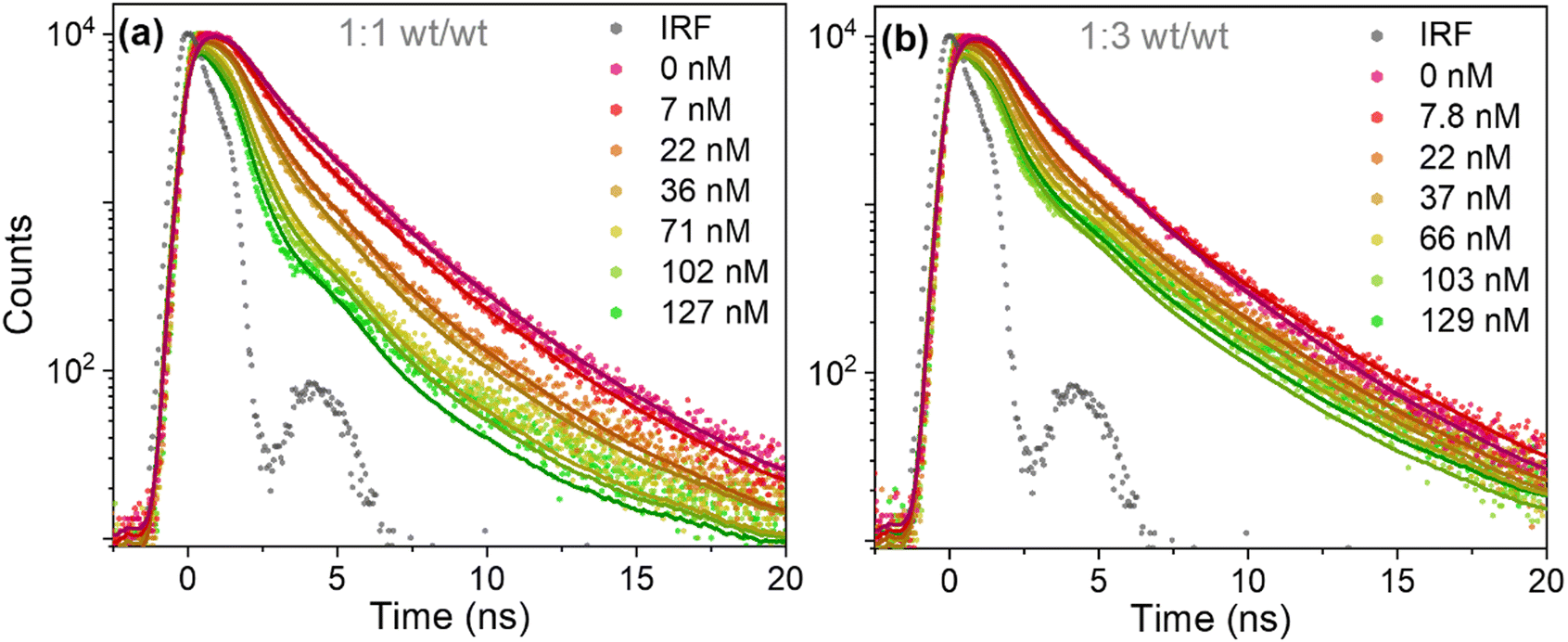 | ||
| Fig. 3 Fluorescence decay traces of PFBT Pdots at different quencher concentrations, at 1:1 (a) and 1:3 (b) (wt/wt) ratios. The fits according to eqn (1) are convoluted with the IRF, with freely adjustable kq and 〈n〉 parameters. | ||
The fluorescence lifetimes were reduced as the perylene red concentration was increased (Fig. 3a and b). Fluorescence decay traces of doped PFBT Pdots were well described by the Infelta–Tachiya model (eqn (1)),27,28 that in its original form has been employed to describe fluorescence quenching kinetics in micellar solutions. The model assumes a random (Poisson) distribution of quenchers over the Pdots and in its simplest form, which is appropriate here, assumes no migration of excitons or quenchers between the particles. An important difference to micelles is that the Pdot volume can be larger than the exciton quenching volume, i.e. the volume the exciton can explore during its lifetime. Only excitons that contain a quencher within its Vq will be quenched, and the quenching is faster the more quenchers that are in the volume. In eqn (1), we use a sum of two Infelta–Tachiya terms to account for the biexponential decay behavior of the Pdots fluorescence in the absence of quenchers:
| I(t,n,kq) = A1·exp(−k1·t + 〈n〉·(exp(−kq·t) − 1)) + A2·exp(−k2·t + 〈n〉·(exp(−kq·t) − 1)) | (1) |
Here, 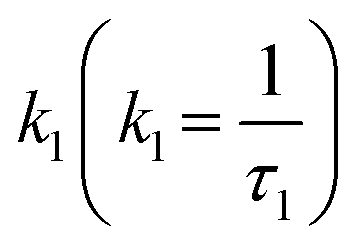 and
and  are rate constants for the sample without added quencher and their relative amplitudes A1 and A2, n is the average number of quenchers per Vq, and kq is the first-order quenching rate constant for quenching in volumes with at least one quencher. To obtain robust fits, the value of kq was assumed to be the same in both terms.
are rate constants for the sample without added quencher and their relative amplitudes A1 and A2, n is the average number of quenchers per Vq, and kq is the first-order quenching rate constant for quenching in volumes with at least one quencher. To obtain robust fits, the value of kq was assumed to be the same in both terms.
Firstly, single trace fits were performed according to eqn (1) with all parameters freely adjustable, as shown in Fig. 3a and b (solid line). Here, the values of k1, k2, A1 and A2 were fixed to their values in the absence of quenchers, while kq and n were floating parameters. The values of kq and n obtained for different perylene red doping concentrations are summarized in Table 1. With increasing doping concentrations above 40 nM, n increased much less than proportionally with concentration and kq increased. This deviation from the model could be attributed to the perylene red aggregated species formation when there is more than one quencher per Pdot present.19,30 Aggregation can also explain why the perylene red fluorescence intensity in Fig. 2c and d did not increase after 40 nM was reached, although the PFBT fluorescence was further quenched.
| Perylene red/nM | Single trace fitting | Global fitting | |||
|---|---|---|---|---|---|
| k q/ns−1 | 〈n〉 | k q/ns−1 | 〈n〉 | ||
| 1:1 (wt/wt) Pdots |
7.2 | 0.37 | 0.4 | 0.47 | 0.3 |
| 22 | 0.42 | 1.1 | 0.47 | 1 | |
| 36 | 0.47 | 1.4 | 0.47 | 1.3 | |
| 71 | 0.57 | 2.2 | — | — | |
| 102 | 0.67 | 2.4 | — | — | |
| 127 | 0.77 | 2.7 | — | — | |
| 1:3 (wt/wt) Pdots |
7.8 | 0.75 | 0.15 | 0.89 | 0.084 |
| 22 | 0.73 | 0.77 | 0.89 | 0.71 | |
| 37 | 0.86 | 0.97 | 0.89 | 0.91 | |
| 66 | 1.2 | 1.2 | — | — | |
| 103 | 1.3 | 1.6 | — | — | |
| 129 | 1.4 | 1.5 | — | — | |
For the global fit analysis, we used only the data with <40 nM quencher, where aggregation effects should be small and kq invariant with quencher concentration as expected in the Infelta–Tachiya model (Fig. 4b and c). The fluorescence decay parameters k1, k2, A1, and A2 were fixed to values obtained from the double-exponential fitting of PFBT Pdots without a quencher. The assessment of the goodness of the fit to the model was based on the weighted residuals. The residuals showed some deviation (only <5 cps compared to 104 in the maximum channel) at <5 ns and were randomly distributed around zero for both 1:1 and 1:3 (wt/wt) ratios (Fig. 4d and e). From the values of n obtained and the estimated number of moles of quencher added, we calculated the number of quenching volumes in the sample. With the known mass of PFBT in the sample and its corresponding density, we could then calculate the value of Vq (see ESI†). From the global fit results in Table 1, we calculated Vq values as 2300 nm3 and 1400 nm3, corresponding to spheres with radii of 8.2 nm and 6.9 nm for the 1:1 and 1:3 (wt/wt) ratios, respectively. The hydrodynamic radii from DLS (15 nm for both (wt/wt) ratios, Fig. S1a and b, ESI†) were larger because DLS captures not only the hydrophobic core but also included the PS–PEG–COOH shell and a solvent layer of certain thickness (Fig. 4a). A precise estimate of the shell thickness is not available, but the molecular weights of the PFBT and the PEG-part of PS–PEG–COOH are very similar (ca. 24000 g mol−1; see ESI†) while the PS-part is smaller (8500 g mol−1). As the PEG-layer is much less dense than the core, it will constitute a major part of the particle volume even for the 1:1 (wt/wt) ratio Pdots. Thus, a conservative estimate of the core radius in the 1:1(wt/wt) Pdots is <75% of the 15 nm value (<42% of the volume), i.e. <10.5 nm. Thus, it is likely that the observed Vq is actually limited by the physical size of the hydrophobic core.
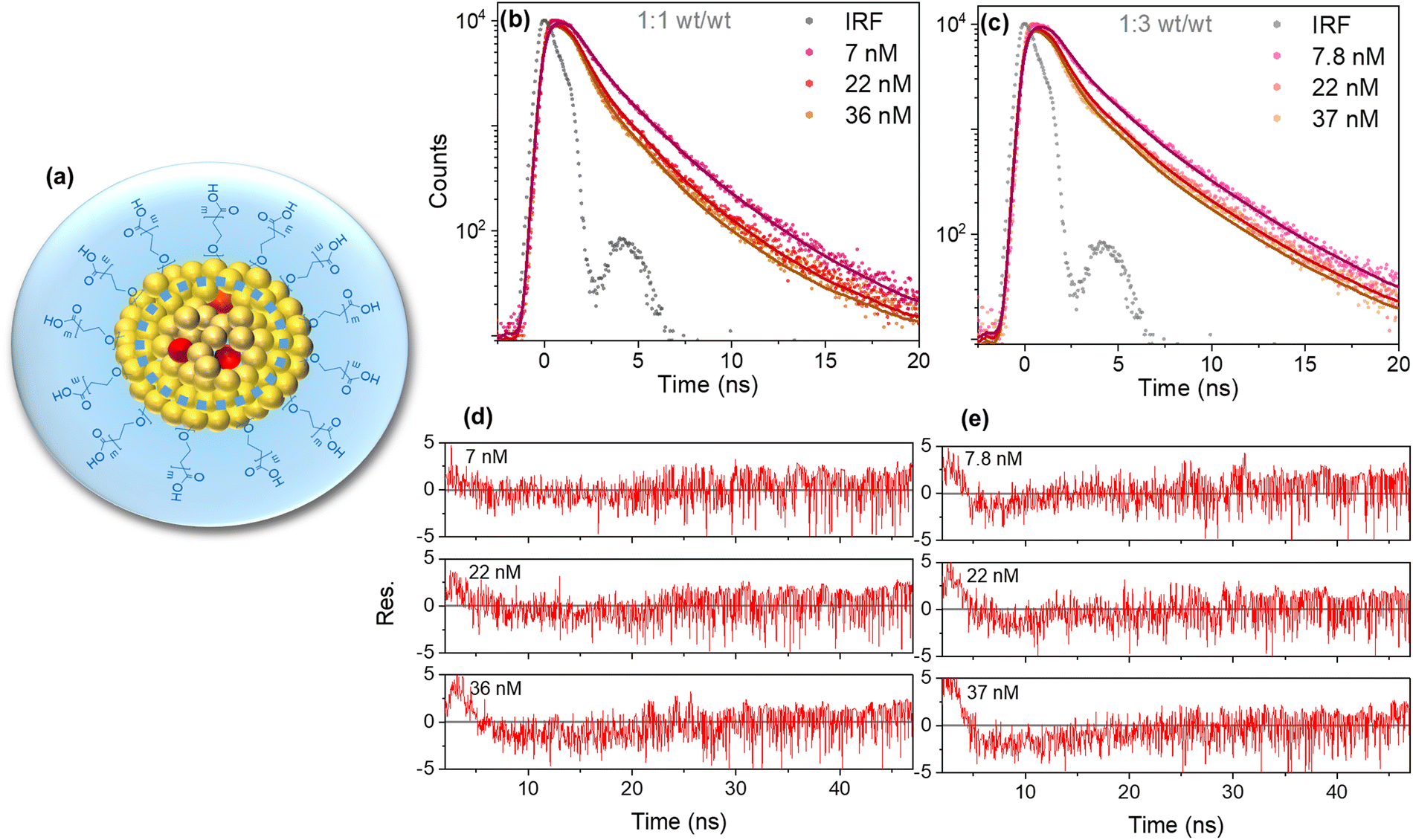 | ||
| Fig. 4 Illustration depicting the proposed morphology of Pdots for 1:1 and 1:3 (wt/wt) ratio. The dashed blue circle indicates the reduction in Vq for PFBT Pdots at 1:3 (wt/wt) ratio (a). Fluorescence decay traces for three different doping concentrations of PFBT Pdots at 1:1 (b) and 1:3 (c) (wt/wt) ratios convoluted with IRF and globally fit with eqn (1) and their corresponding residuals (d and e). | ||
The larger kq values obtained as PFBT is diluted with more PS–PEG–COOH was initially surprising, as it could instead be expected to lead to longer exciton hoping distances and slower quenching. It is not likely to be due to less energetic heterogeneity, as the absorption and fluorescence spectra are essentially identical for the two samples. Instead, the difference in kq can be attributed to a smaller core volume with the 1:3 (wt/wt) ratio, since the particle size did not change when the concentration of PS–PEG–COOH used for preparation was increased and thus the Pdots concentration must have increased for the same amount of PFBT (Fig. S1a and b, ESI†). Fig. 4a illustrates the proposed reduction in Vq and Pdots morphology. The larger kq with more PS–PEG–COOH can also explain the similar extent of quenching at similar perylene red concentrations in Fig. 2c and d, although the number of Pdots is larger and thus n is smaller. The smaller Vq obtained with the 1:3 ratio, in spite of a larger kq, is also consistent with Vq being limited by the physical size of the hydrophobic cores.
Fluorescence quenching efficiencies from steady-state data were compared with those from the integrated surface areas under the decay curves from time-resolved fluorescence measurements at both (wt/wt) ratios (Fig. 5a and b). The integrated surface areas under the normalized decay curves were estimated from the average lifetimes obtained from eqn (S1) (ESI†) for undoped and eqn (1) for doped Pdots. Steady-state and time-resolved experiments yielded very similar extent of quenching showing that static quenching (not resolved by TCSPC) is less important. Nevertheless, the somewhat larger steady-state intensity quenching suggests that n, and thus Vq, are somewhat underestimated. It is important to highlight that both Pdots without and with quenchers (>36 nM) contribute significantly to the observed fluorescence as Pdots with quenchers only exhibit moderate quenching. This is different from the model of complete quenching within a sphere-of-action, where the fluorescence intensity ratio versus concentration plot shows characteristic upward exponential increase.31
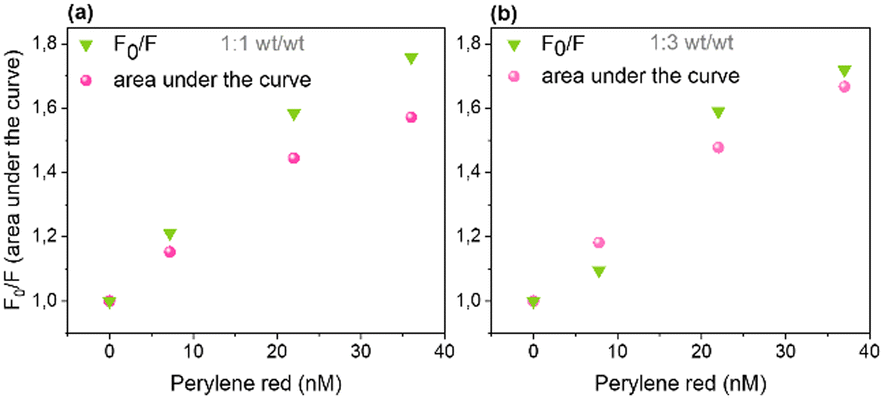 | ||
| Fig. 5 Comparing fluorescence quenching efficiencies from steady-state data with that from the integrated surface area under the decay curves from time-resolved fluorescence measurements at 1:1 (a) and 1:3 (b) (wt/wt) ratios. | ||
Conclusions
In summary, our study uses the Infelta–Tachiya model for fluorescence quenching in particles to provide a new analysis and understanding of exciton migration within PFBT Pdots stabilized with different (wt/wt) ratios of PS–PEG–COOH. In contrast to previous work on PBFT Pdots that have used numerical simulations of steady-state fluorescence intensity data, we use an analytical expression to fit time-resolved fluorescence data. The model's applicability was further confirmed by agreement from both steady-state and time-resolved fluorescence experiments. The estimated Vq value of 2300 nm3 for 1:1 (wt/wt) PBFT:PS–PEG–COOH Pdots, to a spherical radius of 8.2 nm, is similar to the estimated radius of the hydrophobic core, and agrees well with LD = 8.6 nm reported by Ponzio et al. using the external quencher rhodamine B.21 However, our results suggest a significantly longer LD than that, for the following reasons: (1) the value of Vq obtained in the present study seems to be limited by the radius of the hydrophobic core, as discussed above; and (2) the random walk of excitons with 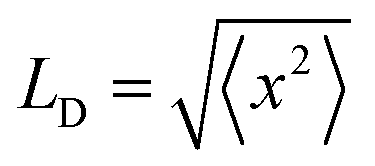 (r.m.s. displacement in three dimensions) explores a volume much smaller than that of a sphere with radius = LD. The volume explored by random walk, using the Förster radius (3.0 nm) as the reaction radius, corresponds to the so-called Wiener sausage volume.32 This suggests that the value of LD is at least twice the radius of our particle core, LD ≈ 19 nm (see ESI† for details), similar to the value estimated from Monte Carlo simulations of neat PFBT thin films (16 nm).33
(r.m.s. displacement in three dimensions) explores a volume much smaller than that of a sphere with radius = LD. The volume explored by random walk, using the Förster radius (3.0 nm) as the reaction radius, corresponds to the so-called Wiener sausage volume.32 This suggests that the value of LD is at least twice the radius of our particle core, LD ≈ 19 nm (see ESI† for details), similar to the value estimated from Monte Carlo simulations of neat PFBT thin films (16 nm).33
Our results suggest that excitons diffuse to the Pdot surface with high probability, which in addition to their porous properties,34 are important for photocatalytic applications. This contrasts with light-emitting diode applications, where the exciton diffusion to the particle surface is not required. Moreover, quenching by surface defects, as suggested by Ponzio et al.,21 could be outcompeted by a rapid catalytic reaction. Finally, as excitons diffuse to the Pdot surface to a high extent, we hypothesize that using PS–PEG–COOH could potentially reduce overall defect density, which is supported by the comparable lifetimes between PFBT Pdots in water and PFBT in THF. The study contributes valuable insights into the dynamics of exciton migration and quenching behavior within Pdots, enhancing our understanding for potential applications in energy conversion.
Author contributions
AB and LAH: investigation, formal analysis, validation, writing review and editing. AB: writing original draft. LH and HT: conceptualization, project administration, supervision, writing review and editing.Data availability
Data available on request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the K&A Wallenberg foundation (Wallenberg Academy Fellow grant 2019.0156 to H. T.; Project grant 2019.0071 to L. H.). We are thankful to Prof. Christer Elvingson for the discussion.Notes and references
- M. V. Pavliuk, S. Wrede, A. Liu, A. Brnovic, S. Wang, M. Axelsson and H. Tian, Preparation, Characterization, Evaluation and Mechanistic Study of Organic Polymer Nano-Photocatalysts for Solar Fuel Production, Chem. Soc. Rev., 2022, 51, 6909–6935 RSC.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Organic Polymer Dots as Photocatalysts for Visible Light-Driven Hydrogen Generation, Angew. Chem., 2016, 128, 12494–12498 CrossRef.
- P. J. Tseng, C. L. Chang, Y. H. Chan, L. Y. Ting, P. Y. Chen, C. H. Liao, M. L. Tsai and H. H. Chou, Design and Synthesis of Cycloplatinated Polymer Dots as Photocatalysts for Visible-Light-Driven Hydrogen Evolution, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- S. Bhattacharyya, B. Paramanik and A. Patra, Energy Transfer and Confined Motion of Dyes Trapped in Semiconducting Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2011, 115, 20832–20839 CrossRef CAS.
- S. Grigalevicius, M. Forster, S. Ellinger, K. Landfester and U. Scherf, Excitation Energy Transfer from Semi-Conducting Polymer Nanoparticles to Surface-Bound Fluorescent Dyes, Macromol. Rapid Commun., 2006, 27, 200–202 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Lessons from Nature about Solar Light Harvesting, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- G. D. Scholes, Long-Range Resonance Energy Transfer in Molecular Systems, Annu. Rev. Phys. Chem., 2003, 54, 57–87 CrossRef CAS PubMed.
- O. V. Mikhnenko, P. W. M. Blom and T. Q. Nguyen, Exciton Diffusion in Organic Semiconductors, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- G. D. Scholes and G. Rumbles, Excitons in Nanoscale Systems, Nat. Mater., 2006, 5, 683–696 CrossRef CAS PubMed.
- B. Jana, A. Ghosh and A. Patra, Photon Harvesting in Conjugated Polymer-Based Functional Nanoparticles, J. Phys. Chem. Lett., 2017, 8, 4608–4620 CrossRef CAS PubMed.
- T. Förster, Zwischenmolekulare Energiewanderung Und Fluoreszenz, Ann. Phys., 1948, 437, 55–75 CrossRef.
- M. Rahman, H. Tian and T. Edvinsson, Revisiting the Limiting Factors for Overall Water-Splitting on Organic Photocatalysts, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- S. Kundu and A. Patra, Nanoscale Strategies for Light Harvesting, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- X. Wang, L. C. Groff and J. D. McNeill, Multiple Energy Transfer Dynamics in Blended Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2014, 118, 25731–25739 CrossRef CAS.
- K. Lix, K. D. Krause, H. Kim and W. R. Algar, Investigation of the Energy Transfer Mechanism between Semiconducting Polymer Dots and Organic Dyes, J. Phys. Chem. C, 2020, 124, 17387–17400 CrossRef CAS.
- S. Wang, A. Thorn and G. Redmond, Photophysical Probing of Dye Microenvironment, Diffusion Dynamics, and Energy Transfer, J. Phys. Chem. C, 2018, 122, 6900–6911 CrossRef CAS.
- J. A. Bjorgaard and M. E. Köse, Amplified Quenching of Conjugated Polymer Nanoparticle Photoluminescence for Robust Measurement of Exciton Diffusion Length, J. Appl. Phys., 2013, 113, 203707 CrossRef.
- K. Trofymchuk, L. Prodi, A. Reisch, Y. Mély, K. Altenhöner, J. Mattay and A. S. Klymchenko, Exploiting Fast Exciton Diffusion in Dye-Doped Polymer Nanoparticles to Engineer Efficient Photoswitching, J. Phys. Chem. Lett., 2015, 6, 2259–2264 CrossRef CAS PubMed.
- L. C. Groff, X. Wang and J. D. McNeill, Measurement of Exciton Transport in Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2013, 117, 25748–25755 CrossRef CAS.
- A. Ghosh, B. Jana, A. Kumar, S. Ghosh and A. Patra, Manipulation of the Exciton Diffusion Length of Conjugated Polymer Nanoparticles: Role of Electron and Hole Scavenger Molecules, Bull. Mater. Sci., 2020, 43, 174 CrossRef CAS.
- R. A. Ponzio, R. M. Spada, A. B. Wendel, M. V. Forcone, F. D. Stefani, C. A. Chesta and R. E. Palacios, Exciton Diffusion, Antenna Effect, and Quenching Defects in Superficially Dye-Doped Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2021, 125, 23299–23312 CrossRef CAS.
- C. Wu, Y. Zheng, C. Szymanski and J. D. McNeill, Energy Transfer in a Nanoscale Multichromophoric System: Fluorescent Dye-Doped Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2008, 112, 1772–1781 CrossRef CAS PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Organic Polymer Dots as Photocatalysts for Visible Light-Driven Hydrogen Generation, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- A. Liu, L. Gedda, M. Axelsson, M. V. Pavliuk, K. Edwards, L. Hammarström and H. Tian, Panchromatic Ternary Polymer Dots Involving Sub-Picosecond Energy and Charge Transfer for Efficient and Stable Photocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef CAS PubMed.
- B. Cai, M. Axelsson, S. Zhan, M. V. Pavliuk, S. Wang, J. Li and H. Tian, Organic Polymer Dots Photocatalyze CO2 Reduction in Aqueous Solution, Angew. Chem., Int. Ed., 2023, 135, e202312276 CrossRef.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, Tracking Charge Transfer to Residual Metal Clusters in Conjugated Polymers for Photocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- M. Tachiya, Application of a Generating Function to Reaction Kinetics of Quenching of Luminescent Probes in Micelles, Chem. Phys. Lett., 1975, 33, 289 CrossRef CAS.
- P. P. Infelta, M. Gratzel and J. K. Thomas, Luminescence Decay of Hydrophobic Molecules Solubilized in Aqueous Micellar Systems A Kinetic Model, J. Phys. Chem., 1973, 78, 190–195 CrossRef.
- A. Brnovic, L. Hammarström and H. Tian, Mechanistic Insights into the Photocatalytic Hydrogen Production of Y5 and Y6 Nanoparticles, J. Phys. Chem. C, 2023, 127, 12631–12639 CrossRef CAS.
- F. Würthner, Perylene Bisimide Dyes as Versatile Building Blocks for Functional Supramolecular Architectures, Chem. Commun., 2004, 1564–1579 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, US, Boston, MA, 2006 Search PubMed.
- A. M. Berezhkovskii, Yu. A. Makhnovskii and R. A. Suris, Wiener Sausage Volume Moments, J. Stat. Phys., 1989, 57, 333–346 CrossRef.
- S. Westenhoff, I. A. Howard and R. H. Friend, Probing the Morphology and Energy Landscape of Blends of Conjugated Polymers with Sub-10 nm, Resolution, Phys. Rev. Lett., 2008, 101, 016102 CrossRef PubMed.
- A. Liu, C. W. Tai, K. Holá and H. Tian, Hollow Polymer Dots: Nature-Mimicking Architecture for Efficient Photocatalytic Hydrogen Evolution Reaction, J. Mater. Chem. A, 2019, 7, 4797–4803 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04108a |
| This journal is © the Owner Societies 2025 |