First principles density functional theory study of tritium species adsorption on Ni(111) surface and diffusion in nickel-sublayer for tritium storage
Received
18th November 2024
, Accepted 3rd December 2024
First published on 4th December 2024
Abstract
The nickel-plated zircaloy-4 is used as a tritium (3H) getter in the tritium-producing burnable absorber rods (TPBARs) to capture 3H produced in the 6Li-riched annular γ-LiAlO2 pellet under neutron irradiation. The experimental data and our previous theoretical results showed that the 3H species produced from the γ-LiAlO2 pellet were mainly 3H2 and 3H2O. These 3H species diffuse from the surface of the LiAlO2 pellet across vacuum to the nickel-plated zircaloy-4 getter and then further diffuse into the getter to chemically form metal hydrides. While a number of studies show that oxygen binds strongly as compared to 3H on the nickel (Ni) layer, the detailed mechanism of 3H species absorption and diffusion across the Ni plate and Ni/Zr interface are still unclear. By employing density functional theory calculations, here we explored the 3H2 and 3H2O species adsorption and dissociation on the Ni(111) surface and diffusion into the Ni sublayer. Our results indicated that the 3H2 and 3H2O dissociate on the Ni(111) surface. The NiOx and Ni(O3H)x could be formed in the Ni layer due to the higher oxygen (O) diffusion energy barrier and formation of Ni vacancy defects. The oxygen was found to be retained in the Ni layer from diffusing across the Ni–Zr interface. This was revealed by comparing the diffusion barriers for 3H with O. 3H was found to have nearly three times smaller diffusion barrier than for O, making 3H comparatively easier to diffuse through the Ni layer. The obtained results provide guidelines for experimental measurements on 3H retention behavior in TPBARs and may open further avenues to explore the impurity effects on 3H diffusion and storage at the Ni/zircaloy interfaces.
1. Introduction
Zirconium (Zr) and its alloys (zircaloy-4) are widely used in nuclear reactors due to their low neutron absorption cross-section and excellent corrosion resistance.1–3 In tritium-producing burnable absorber rods (TPBARs), the metal getter tube located between the cladding and the γ-LiAlO2 pellets is composed of nickel (Ni)-plated zircaloy-4, which is used to capture tritium (3H) species (mainly 3H2 and 3H2O) generated from γ-LiAlO2 pellets during irradiation.4 The 3H-related products transfer to the surface of metal Ni upon adsorption and dissociation to form new 3H species and diffuse into the zircaloy-4 getters to form metal hydrides (Zr3Hx). Therefore, exploring 3H species (3H2, 3H2O) dissociation on the surface of Ni and diffusion across the interface of Ni-plated zircaloy-4 getters can provide a better understanding of 3H species formation and transport from pellets into the getters.
In recent years, we conducted a series of studies on γ-LiAlO2 pellets to understand the tritium products and their transport through the pellet materials.2,5–7 Aside from studying the surface properties of γ-LiAlO2, we also explored the diffusion of 3H and O3H species on its defective (100) and (101) surfaces, and the 3H2O formation and desorption from the γ-LiAlO2(101) and LiAl5O8(111) surfaces.8–13 In our earlier studies, we found that the 3H2 molecule is the main product initially released from the pellets. With increasing the number of Li vacancies and 3H atoms under irradiation, 3H2O can be formed and released from the pellets as well. Although we explored 3H diffusion in different zirconium hydrides,3,14,15 there remained open questions that needed answers: (i) how and which 3H species (3H, O3H, O, or 3H2, etc.) do diffuse into zircaloy-4 from the surface of γ-LiAlO2 pellets? (ii) In the case of 3H2O, can the Ni coating layer retain O by forming NiOx or Ni(O3H)x to prevent O diffusion into zircalory-4 getter? Detail answers to these questions provide understanding of NiOx or Ni(O3H)x phase formation at various concentration of O and its impact on the 3H and O adsorption and diffusion in Ni layer.
In TPBARs, the 3H2 and 3H2O molecules generated from the γ-LiAlO2 pellets8–13 are transported to the Ni-plated zircaloy-4 getters. The adsorption and diffusion processes in Ni layer directly impacts the 3H retention rate at the getter. To capture the chemical adsorption kinetics in Ni layer and diffusion process across the Ni and Zrcaloy-4 interface, it is essential to include accurate electronic and magnetic properties of Ni. While the classical diffusion model where diffusion coefficient is extracted from the Brownian motion combined with adsorption/desorption and particle displacement distributions at single molecular levels could provide insight migration and displacement mechanisms for large interfaces, and fluid systems,16 better understanding of diffusion mechanisms in solid systems with accurate electronic structures could be achieved using density functional theory (DFT) level of calculations. The 3H2O molecules dissociate on Ni(111) surface according to the reaction mechanism: 3H2O → O3H + 3H and O3H + 3H → O + 3H + 3H. Previous studies showed that the first dissociation requires an activation energy of 0.68 eV, where 3H2O is dissociated into an O3H and 3H species.17 The second process involves the dissociation of O3H into O and 3H and requires a higher activation barrier of 1.25 eV.17 The 3H2O species are adsorbed on Ni(111) by occupying a top of a Ni atom, whereas the other three species, O3H, 3H, and O occupy a three-fold hollow site face centred cubic (fcc). As 3H2 reaches the Ni(111) surface it dissociates into two atomic 3H (3H2 → 3H + 3H) with a low activation energy. Shirazi et al. obtained activation energies of 0.03 and 0.09 eV for H-coverage of 0.125 and 0.25 monolayer (ML), respectively.18
To fully address aforementioned open questions, in this study, we explored the 3H2 and 3H2O molecules’ dissociation on the surfaces of Ni-plated zircaloy-4 getters to clarify the 3H species that dissolve into the metal getters and calculated the diffusion pathways of these 3H species in the bulk Ni phase to identify species that diffuse across the interface of Ni-plated zircalory-4. While there are comprehensive studies of H2 and H2O adsorption on the surfaces of Ni available in the literature,19,20 here we targeted on the possibility of the nickel oxide/hydroxide (NiOx, Ni(O3H)x) phase formation in the Ni-coated layer to prevent O diffusion into the zircaloy-4 getter. We calculated the diffusion pathways of 3H across such oxide/hydroxide layer into the zircaloy-4 getter. By taking Ni(111) surface, we provided detail mechanisms for adsorption and diffusion pathways for 3H. Our hypothesis here is that 3H has a lower diffusion barrier in Ni than O, leading to higher chance of 3H diffusion into the zircalloy getter. Particularly, we determined if O from 3H2O could be retained in the Ni layer to form the NiOx/Ni(O3H)x phase and let only 3H diffuse to the zircaloy-4 getter to form metal hydrides.
This paper is organized as follows. We first present theoretical and computational methodologies implemented in this work. Then we present and discuss the results of our study on NiO species formation and the diffusion mechanisms for 3H in Ni surfaces and subsurface. Finally, we summarize our work and provide further outlook.
2. Theoretical and computational methods
All the calculations were performed based on the DFT with the plane-wave basis set and the pseudopotential as implemented in the Vienna ab initio simulation package (VASP).21–23 The electron–ion interaction is described with the projector augmented wave method (PAW).24,25 The exchange and correction energies are described within generalized gradient approximation using the spin-polarized Perdew–Burke–Ernzerhof formulation (GGA-PBE).26 A cutoff energy of 400 eV was used for the plane wave expansion. The convergence criteria for geometry optimizations of total energy and force were 10−5 eV and 0.01 eV Å−1, respectively. The pseudo-potential of 3H was obtained by modifying the mass in the standard hydrogen (1H) pseudo-potential. The surface slab was generated from the fully relaxed bulk structure. For the Ni(111) surfaces this study used a five-layered 4 × 4 supercell slab with a vacuum region of 12 Å along the z-axis to avoid interactions between periodically repeated slabs. The bottom two layers were kept fixed in their bulk positions as the rest of the slab including adsorbates were relaxed. The k-point sampling in a reciprocal space was generated using Monkhorst–Pack method and a 3 × 3 × 1 grid size.
Diffusion barriers for reactions were calculated using the climbing nudged elastic band (c-NEB) method.27 For each simulation a minimum of five images were used. The energy difference between the two optimized minima gave the reaction energy. The diffusion barrier was defined as the difference between the transition state and the initial state energies.
3. Results and discussion
3.1. Stability and formation of NiOx and/or Ni(O3H)x phase
Based on literature report,17,18 we determined the mechanisms of dissociation of 3H2 and 3H2O molecules on the Ni surfaces to clarify the 3H species that dissolved into the metal getters. With such information, then we focused on the diffusion of these 3H species in the Ni bulk and explored a possibility of forming NiOx and/or Ni(O3H)x phases. The stability of Ni metal and its derived compounds, including oxides, hydroxides, and oxyhydroxides can be predicted from DFT combined with thermodynamic analysis. To predict the stability and formation of the NiOx and/or Ni(O3H)x phase, we explored the most stable compounds of Ni oxides, hydroxides and oxyhydroxides observed experimentally (Table 1). We computed the equilibrium crystal structures, electronic structures, and thermodynamic energies using DFT with different options of exchange correlation potentials. In addition to the PBE functional, we used the hybrid functional HSE0628 and a PBE+U functional. The HSE06 tends to perform better especially when dealing with oxides. Electronic structures and thermodynamic analysis are combined together to determine the chemical potential domains of Ni(O3H)x stability and phase formation.
Table 1 Space groups, calculated total and formation energies (eV per f.u.) of Ni–O–H based compounds. Isotopes for H are specifically written as 1H and 3H
| Compound |
Space group |
Formula unit |
Total energy HSE06 |
Total energy PBE |
Total energy PBE+U |
ΔH HSE06 |
ΔH PBE |
ΔH PBE corrected |
ΔH PBE+U corrected |
| Ni |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
1 |
−6.448 |
−5.459 |
−1.842 |
|
|
|
|
|
1H2 |
|
1 |
−7.713 |
−6.772 |
−6.772 |
|
|
|
|
| O2 |
|
1 |
−17.036 |
−9.862 |
−9.862 |
|
|
|
|
| NiO |
Fmm |
2 |
−17.970 |
−11.655 |
−10.278 |
−3.004 |
−1.265 |
−1.945 |
−4.184 |
| NiO2 |
Pm1 |
1 |
−25.491 |
−17.350 |
−14.277 |
−2.007 |
−2.029 |
−3.389 |
−3.933 |
| Ni2O3 |
Rc |
2 |
−42.851 |
−29.648 |
−24.132 |
−4.402 |
−3.937 |
−5.977 |
−7.694 |
| Ni3O4 |
Fdm |
8 |
−61.366 |
−41.783 |
−34.696 |
−7.950 |
−5.681 |
−8.401 |
−12.165 |
| Ni(O3H)2 |
Pm1 |
1 |
−37.451 |
−26.220 |
−25.071 |
−6.255 |
−4.127 |
−5.487 |
−7.955 |
| NiOO3H |
Rm |
2 |
−31.462 |
−22.124 |
−19.789 |
−4.122 |
−3.417 |
−4.777 |
−6.059 |
3.1.1. Calculations of total and formation energies.
The formation energy of a NixOyHz compound can be defined as follows:| | | ΔH(NixOyHz) = EDFT(NixOyHz) − xμsolidNi − yμgasO − zμgasH | (1) |
where EDFT (NixOyHz) is the DFT calculated total energy of the compound; μsolidNi, μgasO, and μgasH, are the chemical potentials of Ni, O, and H in their stable elemental solid/gas state; x, y, and z are coefficients of proportionality. Table 1 lists the DFT calculated total and formation energies of the most stable compounds of nickel oxides and hydroxides using different exchange correlation functionals. The formation energy ΔH for the PBE corrected and PBE+U corrected used a corrected value for the O2 molecule. Our results for the formation energies are in good agreement with literature data.29,30 The computed total energies and formation energies of these compounds are used to determine the chemical potential domains for the formation of Ni(O3H)x.
3.1.2. Chemical potential domains for the stability and formation of Ni(O3H)x.
The formation and stability of a system depend on the chemical potential of its constituent elements, which varies with specific equilibrium conditions. In the thermodynamic limits, the chemical potential domains for Ni(O3H)2 can be determined. The stability of Ni(O3H)2 against decomposition into its elemental constituents requires a smaller atomic chemical potential than the corresponding elemental solid, i.e.,| | | μi ≤ μsolid/gasi, i = Ni, H, O. | (2) |
Setting Δμi = μi − μsolid/gasi, the above condition becomes
| | | Δμi ≤ 0, i = Ni, H, O. | (3) |
For Δ
μi = 0 we obtain a maximum
i-rich condition. Thermodynamic equilibrium of a system requires that a sum of the chemical potentials of its constituent atoms be equal to its formation energy. Therefore, we can write:
| | | ΔμNi + 2ΔμO + 2ΔμH = ΔH(Ni(OH)2) | (4) |
where Δ
H(Ni(OH)
2) is the formation energy of bulk Ni(O
3H)
2.
To avoid the formation of other oxide compounds (such as NiO, NiO2, Ni2O3, Ni3O4, NiOOH), the ranges of chemical potentials for the stability of Ni(O3H)x are subject to the following additional criteria:
| | | ΔμNi + 2ΔμO ≤ ΔH(NiO2) | (6) |
| | | 2ΔμNi + 3ΔμO ≤ ΔH(Ni2O3) | (7) |
| | | 3ΔμNi + 4ΔμO ≤ ΔH(Ni3O4) | (8) |
| | | ΔμNi + 2ΔμO ≤ ΔμH ≤ ΔH(NiOOH) | (9) |
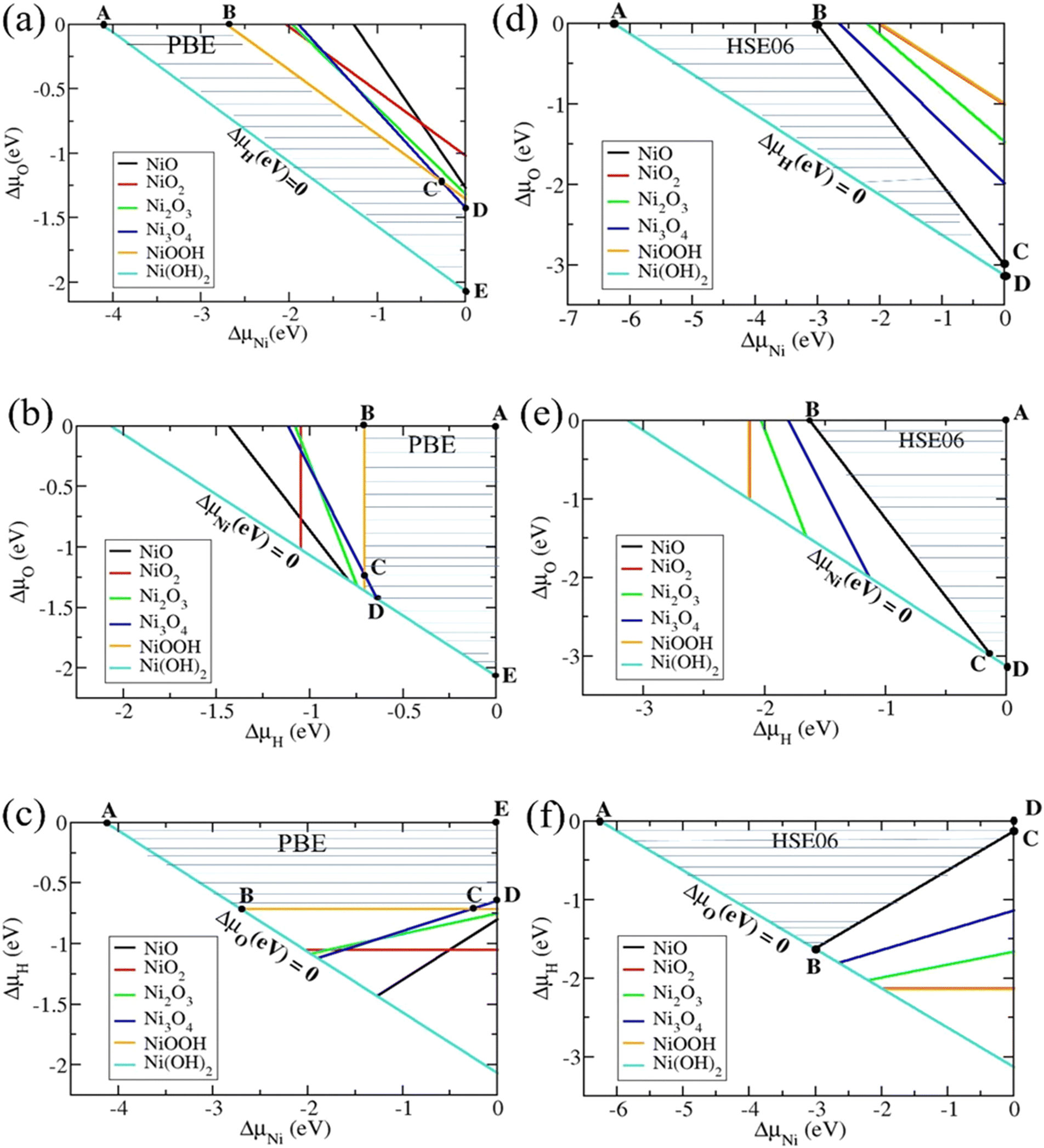
Combining eqn (4) and (5)–(9), we computed the chemical potential domains for the formation and stability of Ni(O3H)2 using the total energies and formation energies calculated above. Fig. 1 shows the stable chemical potential region of the bulk Ni(O3H)2 in terms of the chemical potentials ΔμO, ΔμNi, and ΔμH using PBE and HSE06 functionals. The shaded area is the allowed chemical potential range for Ni(O3H)2 to be a stable phase. In the PBE functional, the limiting stable conditions are defined by five points with coordinates (ΔμNi, ΔμO, and ΔμH) namely A(−4.13, 0, 0), B(−2.71, 0, −0.71), C(−0.27, −1.22, −0.71), D(0, −1.42, −0.64), and E(0, −2.06, 0) with boundary lines formed by Ni(O3H)2, NiOO3H, and Ni3O4. In contrast, the limiting stable conditions using the HSE06 functional is defined by the following four points with coordinates (ΔμNi, ΔμO, and ΔμH): A(−6.26, 0, 0), B(−3.00, 0, −1.63), C(0, −3.00, −0.12), and D(0, −3.13, 0) with boundary lines corresponding to Ni(O3H)2 and NiO.
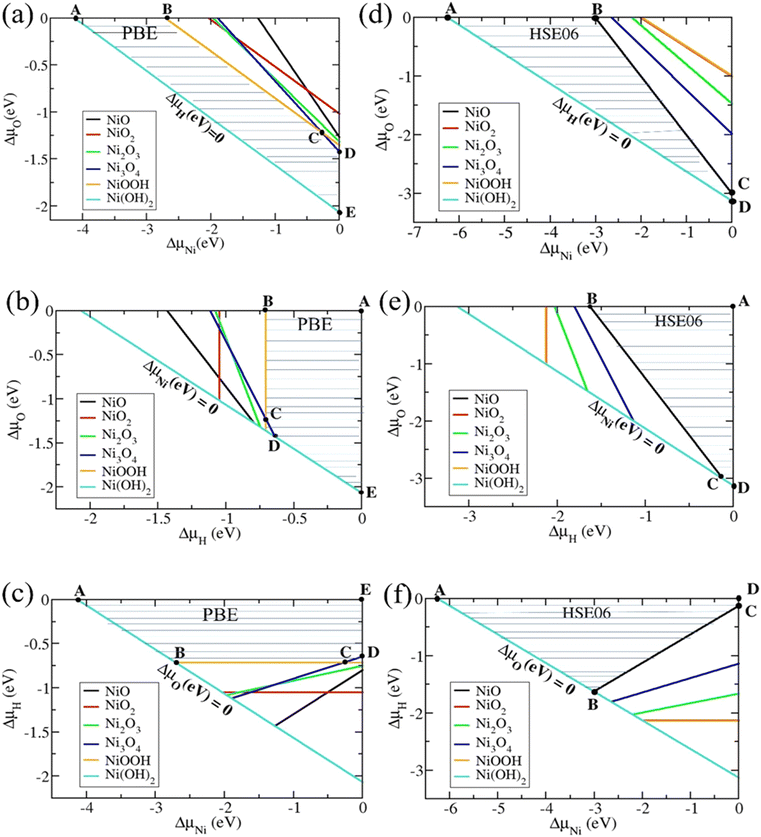 |
| | Fig. 1 The allowed chemical potential domains (shaded regions determined by A, B, C, D, and E) for Ni(O3H)2 to be stable. (a)–(c) Are results obtained using the PBE functional. (d)–(f) Are results obtained using the HSE06 functional. The secondary phases which are indicated by color-coded lines restrict the stable phase. | |
To understand the discrepancy between the results from the PBE and HSE06 functionals, we look at the formation energy per atom for nickel oxide compounds. In Fig. 2, we compare the formation energy per atom of the oxide compounds obtained using PBE, HSE06, and PBE+U functionals. The results show that HSE06 correctly predicts NiO as the most stable among all Ni oxides in agreement with the experiments, whereas PBE incorrectly assigns the lowest formation energy to Ni3O4.31 PBE+U follows the same trend as HSE06. Our results also show that the formation energy per atom of Ni(O3H)2 is lower than that of NiOO3H in HSE06 (−1.25 eV per at vs. −1.03 eV per at), but it is the reverse in PBE (−0.82 eV per at vs. −0.85 eV per at). Our results agree with that of Huang et al. who studied the role of the exchange interactions on the accuracy of the electrochemical phase diagrams of Ni using first-principles methods.29 They observed that the simulation accuracy tends to increase going from semi-local functionals to non-local hybrid functionals due to the increased electronic exchange attraction. Overall, we found that the HSE06 and PBE+U functionals correctly predict NiO as the most stable oxide. The chemical potential domains are better described using the HSE06 functional.
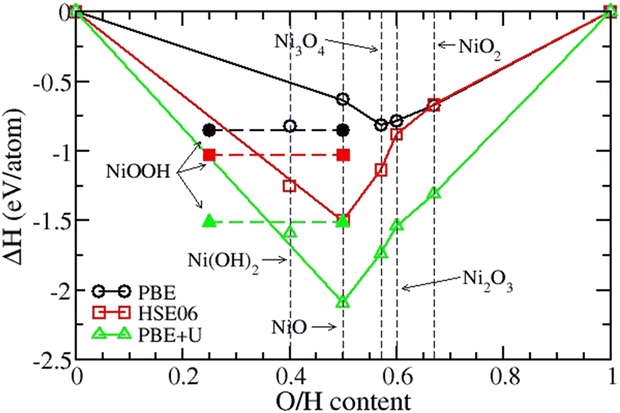 |
| | Fig. 2 Formation energies per atom from PBE, HSE06, and PBE+U functionals for nickel oxide compounds. The blue dashed line connects the formation energies of Ni(O3H)2 with equal O and 3H content, 0.4. Horizontal dashed lines with solid symbols connect the formation energies of NiOO3H with O content 0.5 and 3H content 0.25. | |
3.2. Determining the 3H species dissolve from surface into Ni bulk
3.2.1. Tritium (3H) diffusion from Ni(111) surface to bulk.
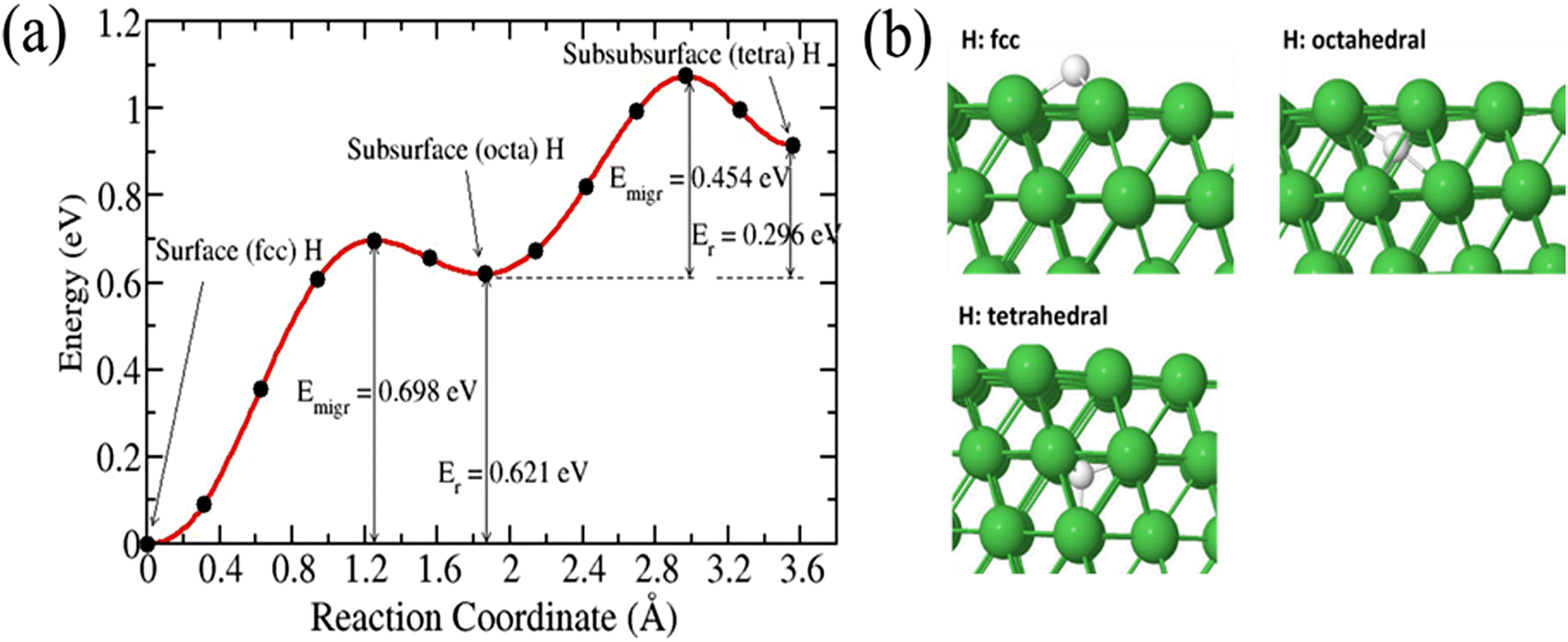
We first calculated the diffusion of a single 3H atom into a pure Ni(111) surface. 3H species diffused on the surface and occupied an interstitial site in the subsurface. There are two possible interstitial sites in the bulk Ni: a tetrahedral (denoted tetra) site and an octahedral (denoted octah) site. Fig. 3 shows the migration of 3H species from the surface into bulk. The calculated migration barrier of 3H from a fcc surface to a subsurface octahedral site is 0.698 eV. The reaction is endothermic by 0.621 eV with activation energy for resurfacing 0.077 eV. This result is in good agreement with previous calculations.18,32–34 Shirazi et al. found an activation energy of 0.64 eV for a H-coverage of 0.50 monolayer and a resurfacing energy barrier of 0.05 eV.18 Henkelman et al. calculated energy barriers of 0.6 eV and 0.1 eV for the diffusion of H to the subsurface and the resurfacing of H, respectively.32 An octahedral subsurface H can diffuse into the bulk via a tetrahedral site with an activation energy of 0.454 eV. Greeley and Mavrikakis found an energy barrier of 0.52 eV.35
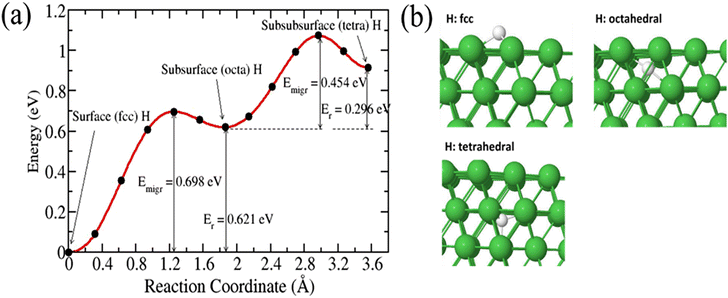 |
| | Fig. 3 (a) Energy barrier and transition pathway for 3H diffusing into a pure Ni(111) surface, (b) initial and final structural configurations. | |
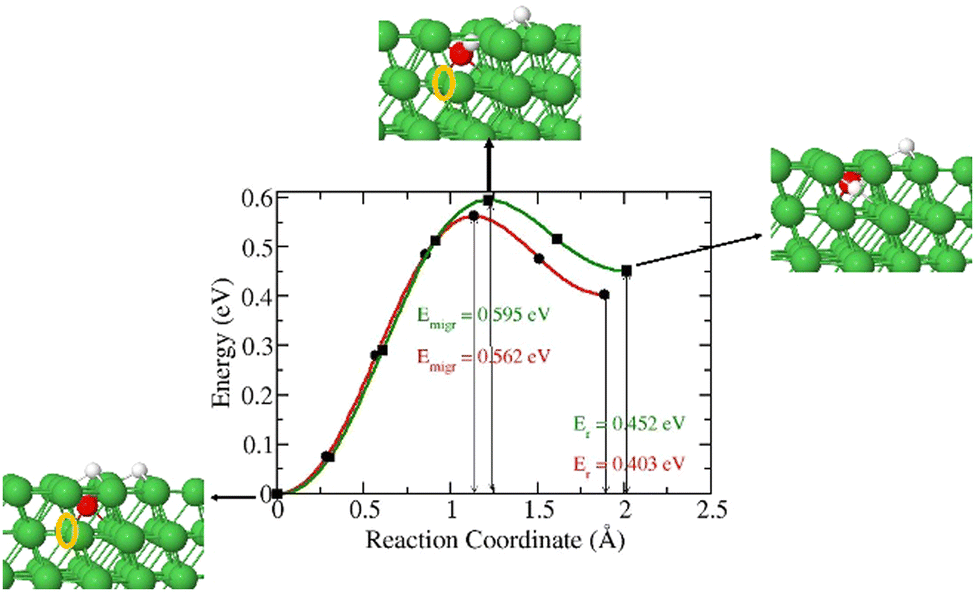
3.2.2. Migration of 3H species from surface to subsurface after 3H2O dissociation.
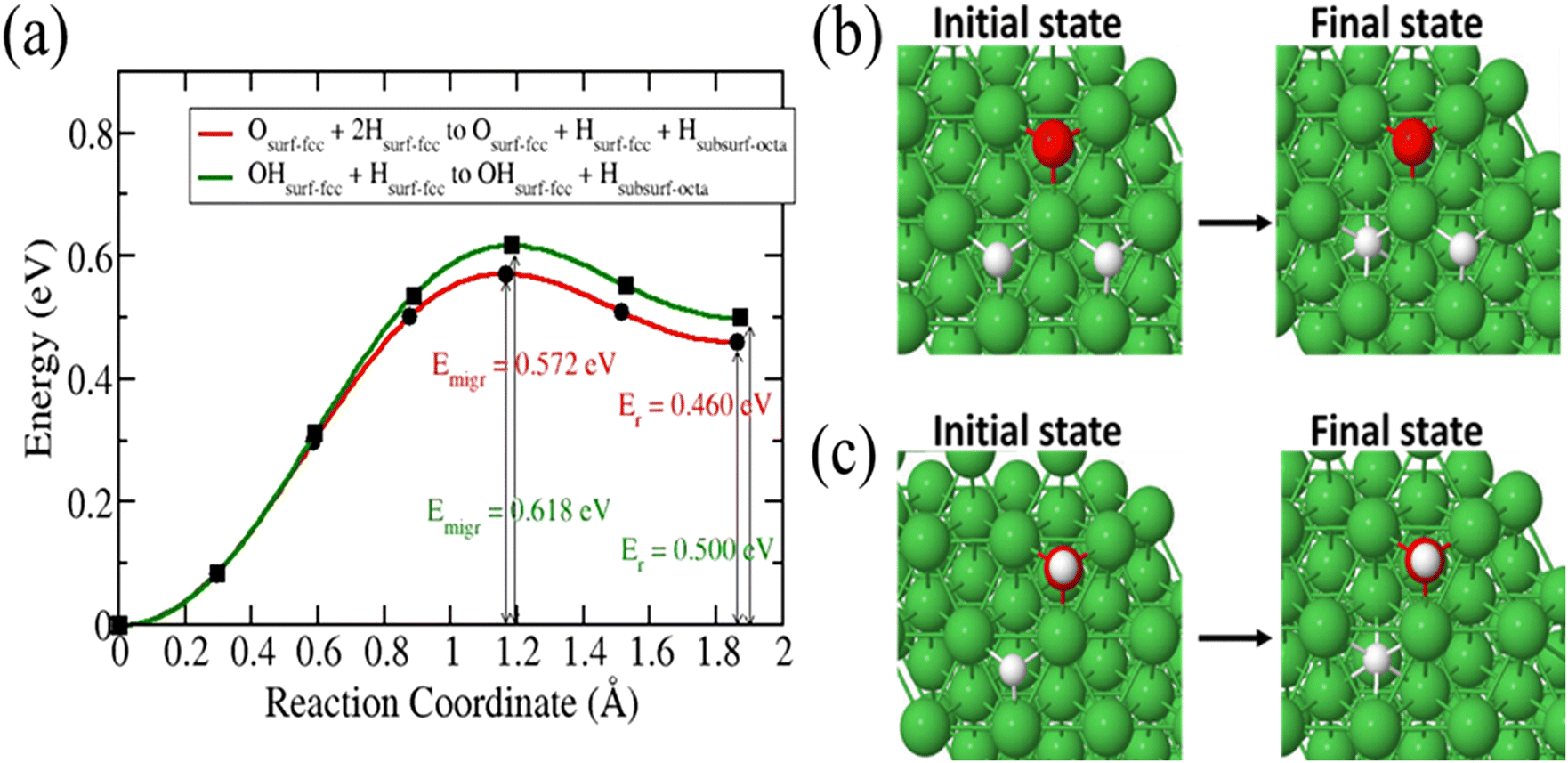
Next, we investigated the diffusion of 3H into the surface after a 3H2O dissociation at partial and complete dissociation. In the partial dissociation, we have an 3H and O3H species lying on the surface. In complete dissociation, we have two 3H species and an O species lying on the surface. Our results in Fig. 4 show a slightly lower diffusion barrier for 3H in the case of a complete dissociation (0.572 eV) compared to that in the partial dissociation (0.618 eV). These two barriers are lower than that of a single 3H atom on the surface diffusing into the subsurface. As 3H2O dissociates, surface sites are occupied which increases surface coverage leading to a redistribution of electron density. The increase of surface coverage is accompanied by a reduction in activation energy of the diffusion of a surface-bound 3H-atom to the subsurface.
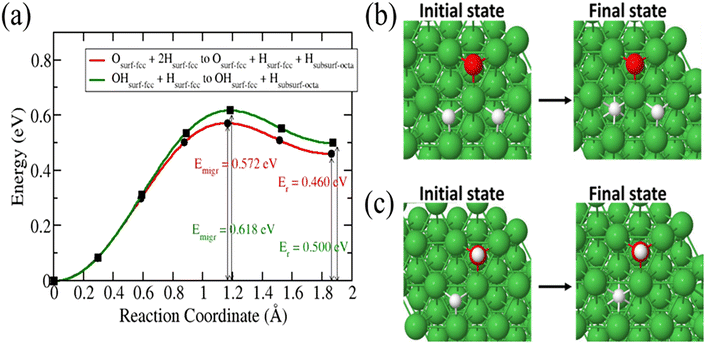 |
| | Fig. 4 Energy barrier of 3H species migration from surface to subsurface after 3H2O dissociation (a). Structures of the initial and final states (b and c). | |
3.2.3. Migration of O species from surface to subsurface.
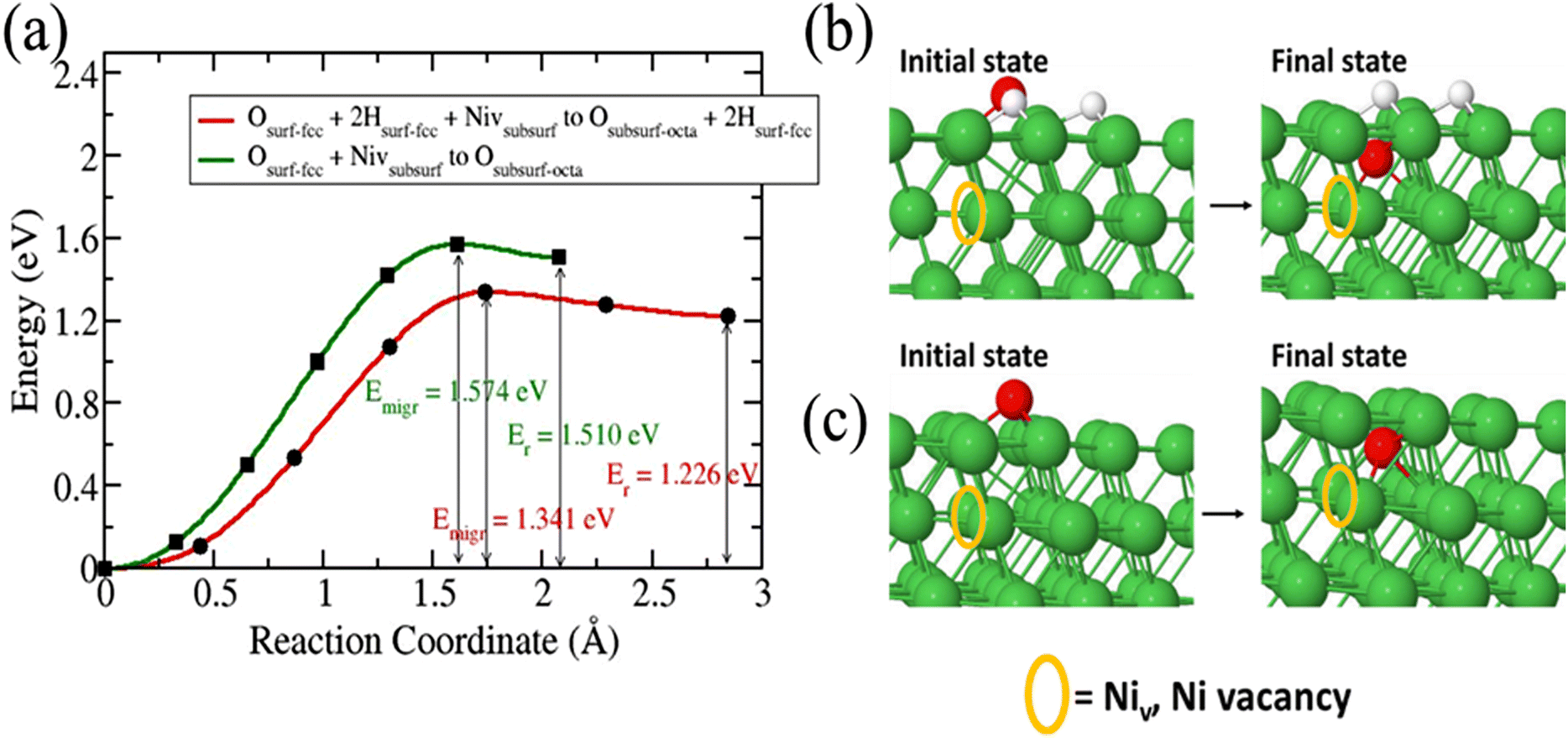
The migration of O species from a fcc surface site to a subsurface octahedral requires the presence of a defect such as Ni vacancy.36 Our study reveals that in the absence of Ni vacancy O diffusion into the surface is unfavourable. We studied the diffusion of O species in a pure Ni surface with a subsurface VNi and the diffusion of O after an 3H2O dissociation (Fig. 5). Our calculations reveal that presence of 3H helps to reduce the diffusion barrier for O in the Ni(111) surface than in presence of a subsurface VNi and without 3H. In the presence of 3H the activation energy and reaction energy are 1.341 eV and 1.226 eV, respectively, as compared to 1.574 eV and 1.510 eV for pure Ni surface with a subsurface VNi.
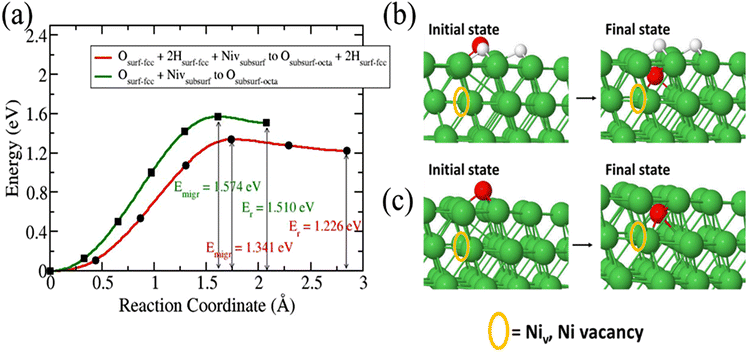 |
| | Fig. 5 Migration barrier of O species diffusion into Ni(111) surface. Structures of the initial and final states (b) after 3H2O dissociation and (c) in bare surface. Orange circle denotes the location of Ni vacancy. | |
The diffusion barrier for 3H is lower than that for O on the Ni surface and bulk. Therefore, it is favourable for 3H to pass through the Ni coating and reach the Ni–zircalory-4 interface. The calculated diffusion barrier for O is nearly three times that for the diffusion of 3H. Experimentally, Ebisuzki et al.37 and Lu et al.38 obtained the diffusion barriers for H in Ni to be 0.42 and 0.19 eV, respectively. Lu et al.38 also predicted the trapping of H in Ni using electrochemical permeation test and thermal desorption spectroscopy (TDS) and found the reversible trapping energy to be 0.26 eV. Similarly, Zholobov et al.39 and Park et al.40 experimentally predicted the diffusion barrier for O in Ni to be 1.89 and 1.70 eV, respectively. These results together with our calculated values indicate that 3H has much higher probability to diffuse through the Ni surface as compared to O.
3.2.4. Diffusion of two 3H species into nickel surface in the presence of O surface species.
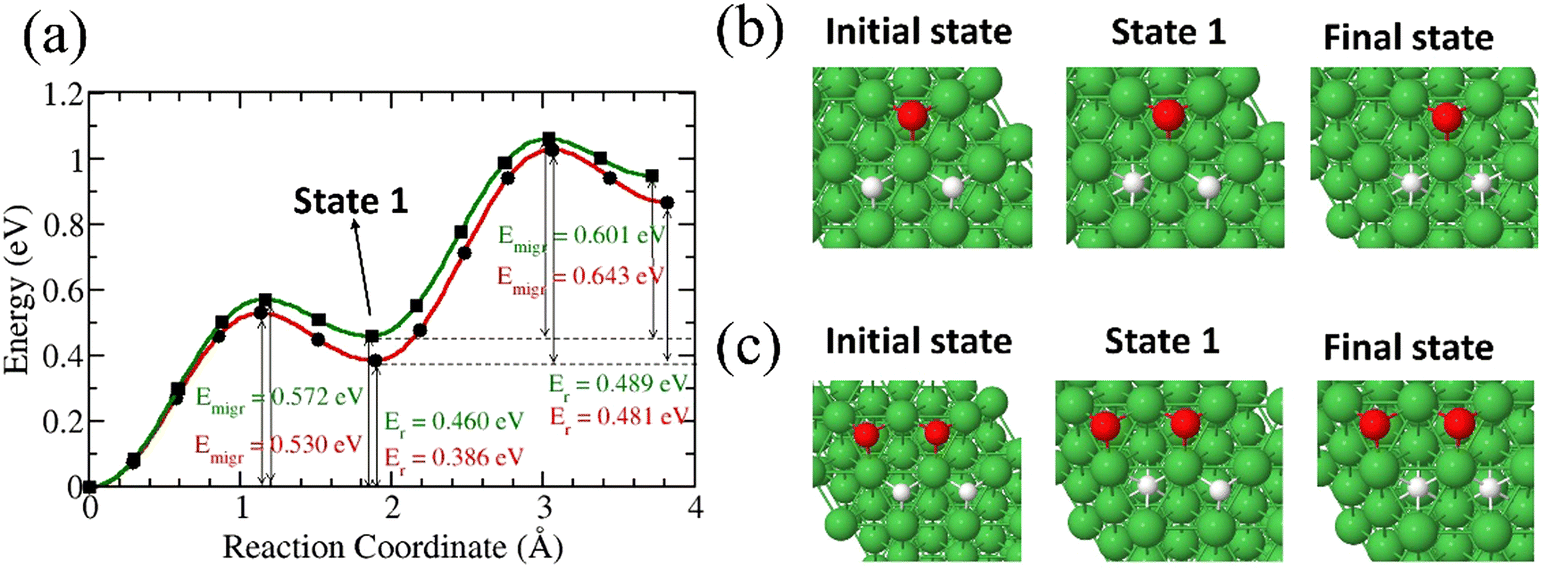
In this section, we studied the diffusion of two 3H species into Ni(111) surface in the presence of surface O and O3H species. Three different cases were investigated.
3.2.4.1. One O surface species.
We studied the diffusion of two 3H atoms into Ni(111) surface after a 3H2O dissociation. The first 3H is found to migrate into the subsurface with an energy barrier of 0.572 eV while the second 3H is found to have an energy barrier of 0.601 eV (Fig. 6). The presence of a second 3H on the surface contributes to the lowering of the migration barrier for the first 3H.
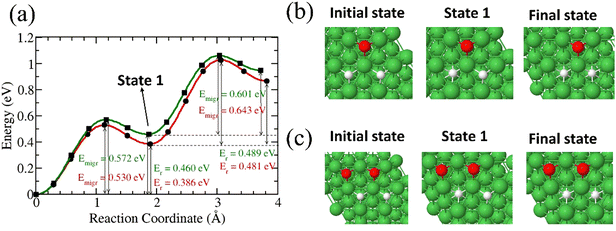 |
| | Fig. 6 (a) Migration barrier and reaction energies of two 3H species on Ni surface after 3H2O dissociation,41 and in the presence of two surface O species (green) Structures of the initial and final states (b) for the two 3H species on Ni surface after 3H2O dissociation and (c) in the presence of two surface O species. | |
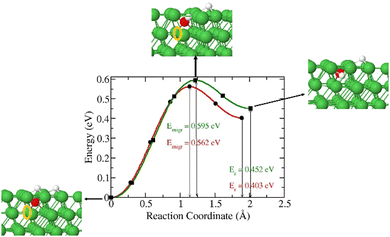 |
| | Fig. 7 Migration barrier and reaction energy of the diffusion of 3H on the surface in presence of O and O3H species41 and in the presence of an O in a subsurface octahedral (green) The insets are the initial, transition and final states for the O in a subsurface octahedral. | |
In all three cases, the diffusion of 3H on the surface is favourable compared to that of O. We also investigated the diffusion of 3H from the surface to the subsurface in the presence of an O at the octahedral site (Fig. 7). Our results reveal that 3H diffuses into a subsurface octahedral site with an energy barrier of 0.595 eV and endothermic reaction energy of 0.452 eV.
3.3. Identifying the energy barrier of 3H species diffusion in the Ni bulk
In this section, we investigate the diffusion of 3H in Ni bulk and the formation of Ni(O3H)x. We started with the assumption that an O migrates into the subsurface and we studied the diffusion of a 3H species toward O to form Ni(O3H)x. We studied three different cases with the third case consisting of diffusion of O3H from the surface into the subsurface. However, O3H diffusion into the surface is unfavourable.
3.3.1. Case 1: formation of Ni(O3H)x through diffusion of a subsurface H species.
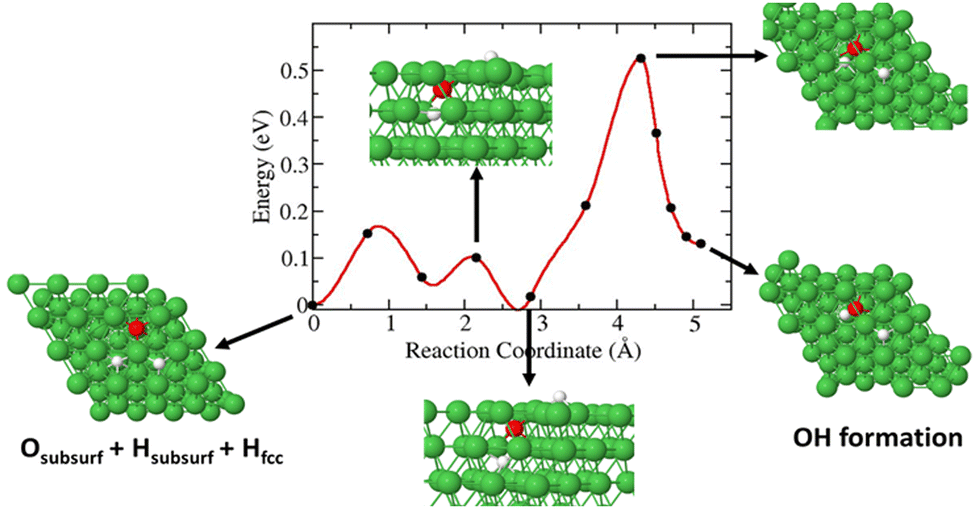
In this case, a 3H species in a subsurface octahedral site is allowed to diffuse toward O to form a Ni(O3H)x unit. We obtained an activation barrier of ∼0.55 eV for the diffusion of 3H and formation of Ni(O3H)x. The reaction is endothermic by 0.125 eV as shown in Fig. 8.
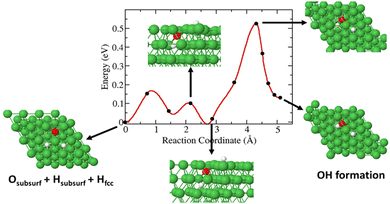 |
| | Fig. 8 Diffusion barrier and diffusion pathways for subsurface 3H diffusion and Ni(O3H)x formation. | |
3.3.2. Case 2: formation of Ni(O3H)x through diffusion of a surface 3H species.
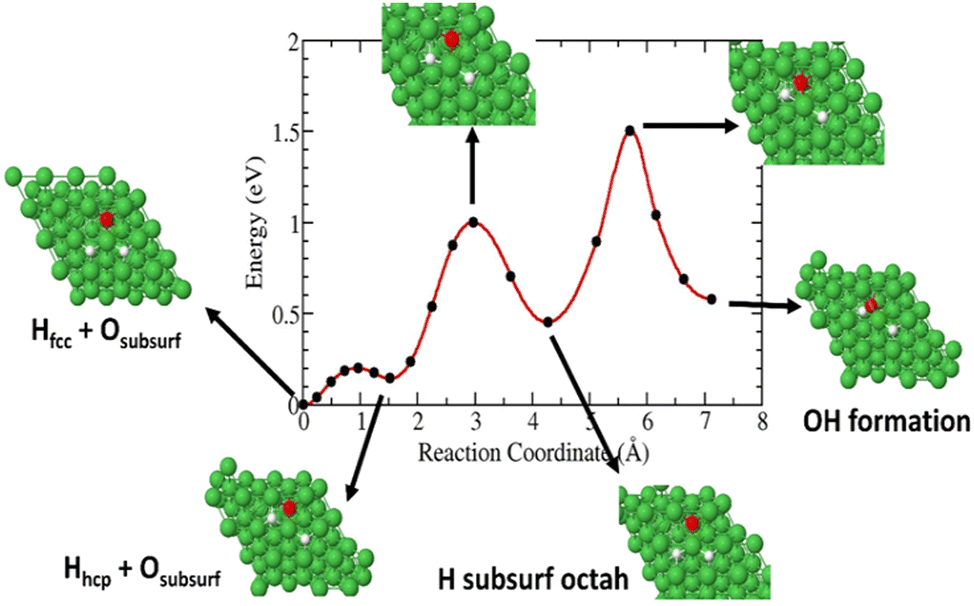
In this case, a surface 3H species is allowed to diffuse in the surface through a hexagonal closest packed (hcp) site close to the subsurface O species. As shown in Fig. 9, our calculated pathways show that 3H on a fcc site migrate to a hcp surface site with an energy barrier of ∼0.2 eV. Then it diffuses into a subsurface octahedral site away from the subsurface O with an activation energy of 0.87 eV. This barrier is 0.27 eV higher than that of a 3H, migrating from a surface fcc site (Fig. 7). Finally, the diffusing 3H must overcome a barrier of ∼1 eV to form a Ni(O3H)x complex.
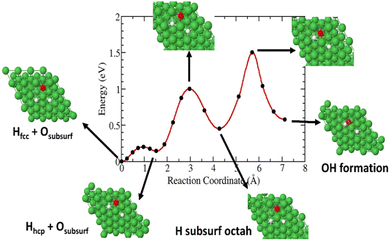 |
| | Fig. 9 Diffusion barrier and transition pathways for 3H diffusion into surface and subsurface formation of Ni(O3H)x. | |
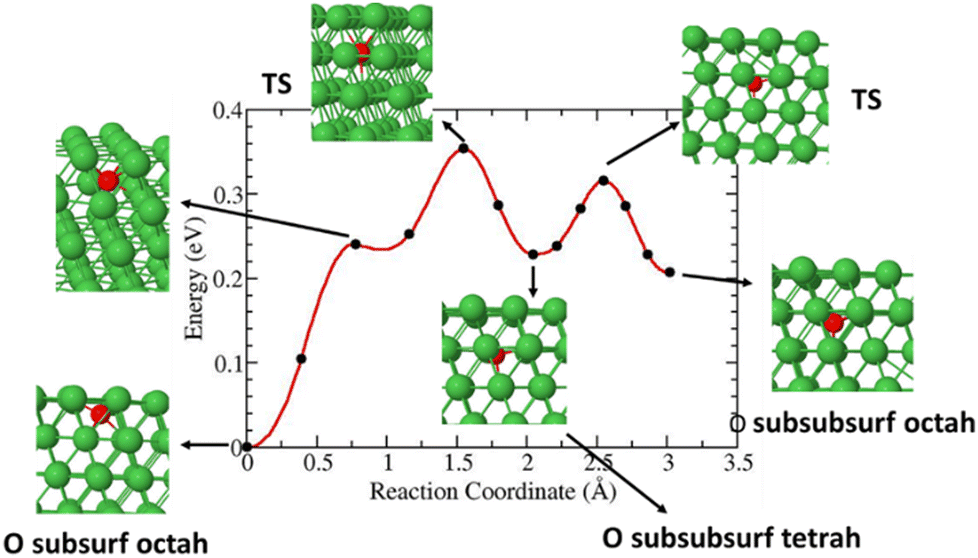
This study also investigated the diffusion of O species from a subsurface octahedral to a sub-subsurface octahedral. The results show a diffusion barrier of less than 0.4 eV as shown in Fig. 10.
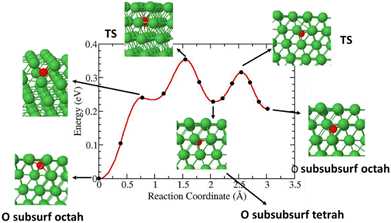 |
| | Fig. 10 Diffusion barrier and reaction pathways of O species from a subsurface octahedral to a sub-subsurface octahedral. | |
David et al. calculated the migration energies of the diffusion along octah–tetrah for O atoms in Ni bulk and found 0.75 eV.42 Previous work from Fang et al. revealed that for O to diffuse into nickel bulk requires an activation energy of at least 1.68 eV.43
4. Summary and conclusions
Formation of chemical compounds O3H, 3H2O, and metal oxide on the Ni surface and subsurface regions can significantly affect adsorption, desorption, diffusion, and hinder 3H and 3H2 reaching out to the getter material. Experimental evidence shows the presence of above compounds including hydrocarbons forming in Ni and getter materials. In this study, we explored 3H2 and 3H2O binding sites and their dissociation on the pure and defective (111) surface of Ni. As it is evident from the earlier studies, the possible dissociation steps of 3H2O are 3H2O → O3H + 3H → O + 3H + 3H, and 3H2 → 3H + 3H. Our calculations also showed that there is a stable chemical potential region where NiOx or Ni(O3H)x phases could form. We found that the HSE06 and PBE+U functionals more accurately predict NiO as the most stable oxide as compared other methods employed in this study for the formation energy calculations. The 3H2 and 3H2O dissociated on the surface of Ni could diffuse into the subsurface region. We found that the presence of 3H on the surface reduces diffusion barriers for O by more than 15%. The results for the calculation on diffusion barriers showed that O prefers to chemically bsind in the Ni layer to form NiOx or Ni(O3H)x. This was evident from the fact that it the calculated diffusion barrier for 3H is 0.48 eV, which is nearly three times smaller than the corresponding value for O in the Ni(111) surface. In addition, we also found that formation of NiOx or Ni(O3H)x phase in Ni subsurface layer is limited by diffusion of O and formation of Ni vacancies. We conclude that 3H has higher probability of diffusion through the Ni layer and reaching out to the getter materials as compared to O.
Author contributions
Conceptualization, Y. D. and H. P.; methodology, D. T.; software, Y. D.; validation, D. T.; formal analysis, D. T.; investigation, D. T.; resources, Y. D., D. S., and A. C.; data curation, D. T.; writing – original draft preparation, D. T.; writing – review and editing, H.P., D. S., A. C. and Y. D.; visualization, D. T.; supervision, Y. D.; project administration, Y. D and A. C.; funding acquisition, D. S. and A. C. All authors have read and agreed to the published version of the manuscript.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations but may be obtained by contacting corresponding author. The VASP software can be found at https://www.vasp.at. The version of VASP employed for this study is 5.4.4.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the National Nuclear Security Administration (NNSA) of the U. S. Department of Energy (DOE) through the Tritium Science Research Supporting the Tritium Modernization Program managed by Pacific Northwest National Laboratory (PNNL). This project was also funded by the United States Department of Energy, National Energy Technology Laboratory, in part, through a site support contract. Neither the United States Government nor any agency thereof, nor any of their employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favouring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Notes and references
- J. J. Kearns, J. Nucl. Mater., 1972, 43, 330–338 CrossRef CAS.
- H. Paudel, Y. L. Lee, D. Senor and Y. Duan, J. Phys. Chem. C, 2018, 122, 9755–9765 CrossRef CAS.
- C. M. Andolina, W. A. Saidi, H. P. Paudel, D. J. Senor and Y. Duan, Comput. Mater. Sci., 2022, 209, 111384 CrossRef CAS.
-
D. J. Senor, Recommendations for Science and Technology in Support of the Tritium Sustainment Program, PNNL-27216, Rev.1, PNNL, 2018 Search PubMed.
- H. P. Paudel and Y. Duan, J. Phys. Chem. C, 2018, 122, 28447–28459 CrossRef CAS.
- Y.-L. Lee, J. Holber, H. P. Paudel, D. C. Sorescu, D. J. Senor and Y. Duan, J. Nucl. Mater., 2018, 511, 375–389 CrossRef CAS.
- Y. Duan, D. C. Sorescu, W. L. Jiang and D. J. Senor, J. Nucl. Mater., 2020, 530, 152963 CrossRef.
- T. Jia, D. J. Senor and Y. Duan, Comput. Mater. Sci., 2020, 181, 109748 CrossRef CAS.
- T. Jia, D. J. Senor and Y. Duan, J. Nucl. Mater., 2020, 540, 152394 CrossRef CAS.
- T. Jia, D. J. Senor and Y. Duan, Appl. Surf. Sci. Adv., 2021, 5, 100114 CrossRef.
- T. Jia, D. J. Senor and Y. Duan, J. Nucl. Mater., 2021, 555, 153111 CrossRef CAS.
- T. Jia, Z. Zeng, H. Paudel, D. J. Senor and Y. Duan, J. Nucl. Mater., 2019, 522, 1–10 CrossRef CAS.
- H. P. Paudel, D. J. Senor and Y. Duan, Comput. Mater. Sci., 2021, 193, 110419 CrossRef CAS.
- T. Jia, H. P. Paudel, D. J. Senor and Y. Duan, Comput. Mater. Sci., 2022, 203, 111158 CrossRef CAS.
- H. P. Paudel, T. Jia, W. A. Saidi, D. J. Senor, A. M. Casella and Y. Duan, J. Phys. Chem. C, 2023, 127, 12435–12443 CrossRef CAS.
- A. Díez Fernández, P. Charchar, A. G. Cherstvy, R. Metzler and M. W. Finnis, Phys. Chem. Chem. Phys., 2020, 22, 27955–27965 RSC.
- L. Zhu, C. Liu, X. Wen, Y.-W. Li and H. Jiao, Catal. Sci. Technol., 2019, 9, 199–212 RSC.
- M. Shirazi, A. Bogaerts and E. C. Neyts, Phys. Chem. Chem. Phys., 2017, 19, 19150–19158 RSC.
- J. Carrasco, A. Hodgson and A. Michaelides, Nat. Mater., 2012, 11, 667–674 CrossRef CAS.
- K. Christmann, R. J. Behm, G. Ertl, M. A. Van Hove and W. H. Weinberg, J. Chem. Phys., 2008, 70, 4168–4184 CrossRef.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- L.-F. Huang and J. M. Rondinelli, J. Phys.: Condens. Matter, 2017, 29, 475501 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39, 969–980 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, J. Chem. Phys., 2006, 124, 2161193 CrossRef.
- A. Michaelides, P. Hu and A. Alavi, J. Chem. Phys., 1999, 111, 1343–1345 CrossRef CAS.
- V. Ledentu, W. Dong and P. Sautet, J. Am. Chem. Soc., 2000, 122, 1796–1801 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Surf. Sci., 2003, 540, 215–229 CrossRef CAS.
- V. R. Galakhov, E. Z. Kurmaev, K. Kuepper, M. Neumann, J. A. McLeod, A. Moewes, I. A. Leonidov and V. L. Kozhevnikov, J. Phys. Chem. C, 2010, 114, 5154–5159 CrossRef CAS.
- Y. Ebisuzaki, W. J. Kass and M. O'Keeffe, J. Chem. Phys., 1967, 46, 1378–1381 CrossRef CAS.
- X. Lu, T. Depover and R. Johnsen, Int. J. Hydrogen Energy, 2022, 47, 31673–31683 CrossRef CAS.
- S. P. Zholobov and M. D. Malev, Soviet Phys. Tech. Phys., 1971, 16, 488 Search PubMed.
- J.-W. Park and C. J. Altstetter, Metall. Trans. A, 1987, 18, 43–50 CrossRef.
- C. Mitra, T. Meyer, H. N. Lee and F. A. Reboredo, J. Chem. Phys., 2014, 141, 084710 CrossRef PubMed.
- M. David, A. Prillieux, D. Monceau and D. Connétable, J. Alloys Compd., 2020, 822, 153555 CrossRef CAS.
- H. Z. Fang, S. L. Shang, Y. Wang, Z. K. Liu, D. Alfonso, D. E. Alman, Y. K. Shin, C. Y. Zou, A. C. T. van Duin, Y. K. Lei and G. F. Wang, J. Appl. Phys., 2014, 115, 043501 CrossRef.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Hari P.
Paudel
cd,
David J.
Senor
e,
Andrew M.
Casella
e and
Yuhua
Duan
ab,
Hari P.
Paudel
cd,
David J.
Senor
e,
Andrew M.
Casella
e and
Yuhua
Duan