
Unveiling ultraviolet photodissociation dynamics of SiO from a laser-ablated supersonic beam with time-sliced ion velocity imaging†
Yujie
Ma
,
Fangfang
Li
,
Dong
Yan
,
Ang
Xu
,
Ti
Zhou
,
Jiaxing
Liu
and
Fengyan
Wang
 *
*
Department of Chemistry and Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Collaborative Innovation Centre of Chemistry for Energy Materials (iChEM), Fudan University, Shanghai, 200438, China. E-mail: fengyanwang@fudan.edu.cn
First published on 25th November 2024
Abstract
SiO is a widespread molecule found in interstellar space, and its dissociation requires a substantial input of energy due to its high bond energy of 8.34 eV. The present study initially demonstrated across a broad range of ultraviolet (UV) wavelengths (243–288 nm) the one-photon and two-photon dissociation of SiO molecules, which were generated from the laser ablation of a Si rod colliding with an oxygen molecular beam. The images of Si products obtained through time-sliced ion velocity mapping have revealed the existence of distinct dissociation channels, encompassing Si(3P) + O(3P), Si(1D) + O(3P), Si(1D) + O(1D) and Si(1S) + O(1D) from the photodissociation of vibrationally excited SiO(X1Σ+, v) and low-lying electronically excited SiO(C1Σ−, D1Δ, a3Σ+, b3Π, d3Δ and e3Σ−) states. These findings contribute to a more comprehensive understanding of silicon chemistry during the combustion of silica-rich meteorites in the Earth's atmosphere, and have wider implications in the fields of atmospheric chemistry, astrochemistry, and combustion science.
Introduction
Silicon monoxide (SiO) is a prevalent silicon-containing molecule that has been identified in the interstellar medium and various astronomical environments through its rovibrational spectra.1–9 It is considered as a crucial precursor and basic building block for larger silicon oxides and interstellar silicate nanoparticles and is predicted to be the primary form of silicon produced during the combustion of silicate-rich meteors entering Earth's atmosphere.9 However, in a spaceborne ultraviolet 251–384 nm spectroscopy study of a meteor during the 1997 Leonid shower, SiO was not observed in the UV spectra resulting from the high-velocity burning of the meteorite in Earth's atmosphere.8,10 This highlights the necessity for further experimental measurements and accurate theoretical calculations to facilitate a comprehensive understanding of SiO spectra and its chemical evolution behavior.The electronic structure and spectroscopic properties of SiO have been extensively studied over the past century due to its astrophysical importance and similarity to CO.11–35 The UV spectra of SiO were first observed by Jevon in 1924,11 and since then, various absorption or emission spectra of SiO from different sources have been recorded and analyzed. A chemiluminescent flame of SiO has revealed band systems similar to CO, with both spin-allowed transitions, such as A1Π–X1Σ+, and spin-forbidden transitions, such as a3Σ+–X1Σ+ and b3Π–X1Σ+.28,32,33 Additionally, transitions between excited triplet states, such as 3Σ+–3Π and 3Π–3Π, have also been reported.20,23,30,33 In conjunction with spectroscopic studies, the potential energy curves (PECs) for various electronic states of SiO were calculated and different transition probabilities were given for dipole-allowed or spin-forbidden transitions.16,36,37 Among the transitions, the b3Π–X1Σ+, e3Σ−–X1Σ+ spin-forbidden transitions are weak in intensity, and the radiative partial lifetimes of the triplets states are in the order of milliseconds. In contrast, spin-allowed bands like A1Π–X1Σ+ have a large Franck–Condon factor with rotation-less radiative lifetimes of approximately 10 ns for vibrational levels of A1Π, E1Σ+ and 21Π states. This suggests that spontaneous vibronic emissions occur easily from these excited states to X1Σ+.16,36,37Table 1 summarizes the seven low-lying electronic states of SiO, specifically X1Σ+, a3Σ+, b3Π, d3Δ, e3Σ−, C1Σ−, and D1Δ, in which C1Σ− and D1Δ states have close electronic energy and vibrational constants.37
| Electronic state | T e (cm−1) | ω e (cm−1) | ω e x e (cm−1) |
|---|---|---|---|
| X1Σ+ | 0 | 1242 | 5.966 |
| a3Σ+ | 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 630 630 |
790 | 4.1 |
| b3Π0,1,2 | 33875, 33947, 34018 |
1014 | 7.5 |
| d3Δ | 36487 |
767 | 4.1 |
| e3Σ− | 38309 |
748 | 4.19 |
| C1Σ− | 38624 |
740 | 4.27 |
| D1Δ | 38823 |
730 | 3.9 |
The dissociation of SiO to its constituent silicon and oxygen atoms necessitates a considerably high energy input. In recent work, we used laser ablation of a silicon rod to observe the photodissociation of highly vibrationally excited SiO(X1Σ+, v > 13) at 193 nm and obtained the dissociation energy of SiO to be 67253 ± 110 cm−1 (8.34 ± 0.01 eV),38 in agreement with previous thermochemical data.37 In the present study, we utilized time-sliced ion velocity imaging to observe SiO photodissociation dynamics across a broad range of UV excitation wavelengths (243–288 nm), which revealed distinct dissociation channels from low-lying electronic states.
Experimental setup
We performed the photodissociation experiment of SiO in the home-built supersonic crossed molecular beam and time-sliced ion velocity map imaging setup.38–40 In the photodissociation experiments, only a laser ablation beam with a carrier gas was used. To quickly generate SiO in the molecular beam experiment, we utilized the barrierless reaction Si(3P) + O2(X3Σg−) → SiO(X1Σ+) + O(3P, 1D), previously studied by He et al.7,39,41 Electronically excited Si atoms in laser-ablated plasma can also participate in the reaction with O2 to produce the electronically excited SiO molecules. The reaction rate of the ground reaction Si + O2 → SiO + O was determined to be kSi+O2 = 1.7 × 10−10 cm3 molecule−1 s−1.41 We generated the Si plasma by ablation of a silicon rod (99.999%, PrMat) using a 532 nm laser (pulse energy ca. 5 mJ) from Continuum Minilite II, which then encountered the pure O2 supersonic molecular beam introduced through a pulsed Even–Lavie valve with a back pressure of 14 atm and flowed through the ablation region. The molecular beam expanded into the main vacuum chamber through a 3 mm skimmer (Beam dynamics model no. 50). The SiO molecular beam was then intersected with the dissociating/detecting laser focused by a circular convex lens (f = 35 cm) at a 45° angle. The dissociating/ionizing laser was at 243–288 nm (pulse energy ca. 1.2 mJ), generated from a Continuum Sunlite OPO/OPA laser pumped using a 355 nm Nd:YAG laser. Additionally, the formation of Si2 molecules via the possible process Si + Si + M → Si2 + M with a rate constant of kSi+Si+M = 7.4 × 10−34 cm6 molecule−2 s−1 (ref. 42) (M represents the third body for any species) was also present. Although the rate constant for Si2 formation is lower than that for SiO formation, the Si products from Si2 photodissociation has to be identified and excluded in the study of photodissociation processes of SiO. To achieve this, the use of Ar as a carrier gas instead of O2 is necessary in acquiring Si images for a comparison. The different speeds of the molecular beams indicate the existence of a Doppler effect at the intersection of the laser and the seeded SiO molecules, which is reduced with a slight tune of the photolysis laser wavelength in a small range of 2–6 cm−1, with the difference being negligible when compared to the obtained full width at half-maximum (FWHM) of the Si products in the 50–500 cm−1 range from the kinetic energy distributions. Images of Si+ ionized by the same laser were acquired using a position sensitive detector composed of two microchannel plates (MCP, 75 mm diameter, Photek), a phosphor screen and a CCD camera (La Vision E-lite 1.4M) under the focus of ion optics with a total voltage of 1800 V. The center slice image of the Si+ signal was accumulated with a gate of 30 ns and event counting in the software “Davis 8.2”.Results and discussion
In order to differentiate the signals of Si/Si+ from the UV photodissociation of SiO and Si2, raw slice images of mass-selected Si were obtained using two carrier gases: pure O2 and pure Ar. Fig. 1(a) displays the raw slice images of Si/Si+ ions with pure O2 and Ar carrier gases at wavelengths of 243.838 (243.800 for Ar) and 288.173 (288.158 for Ar) nm, respectively. The minor alterations in the excitation wavelength within the relatively narrow energy range of 10 cm−1 can be neglected in the photodissociation experiment, given the considerable kinetic energy release observed in the products. The normalized speed distributions of Si/Si+ products resulting from SiO and Si2 photolysis were obtained by integrating the signal in the images within the angular range of θ = 10–30°, as shown in Fig. 1(b). The angle θ refers to the angle between the recoil velocity of the fragment and the polarization direction of the dissociation laser. The use of pure O2 as a carrier gas for the ablation of silicon does not entirely eliminate the presence of the Si2 photolysis signal as Si2 can be formed by the Si + Si + M → Si2 + M collision processes prior to collision with O2. However, the rapid formation of SiO from Si and O2 results in a significantly stronger signal than the Si2 signal, as shown in the time-sliced ion velocity images of Si products in Fig. 1. The SiO photodissociation signal was obtained based on the expression I(SiO/Si2)–RI(Si2), where I(SiO/Si2) and I(Si2) are the normalized Si/Si+ signals from pure O2 and pure Ar, respectively, and R is the factor used to fully subtract the well-resolved * tagged peaks inclusive from the Si2 photodissociation.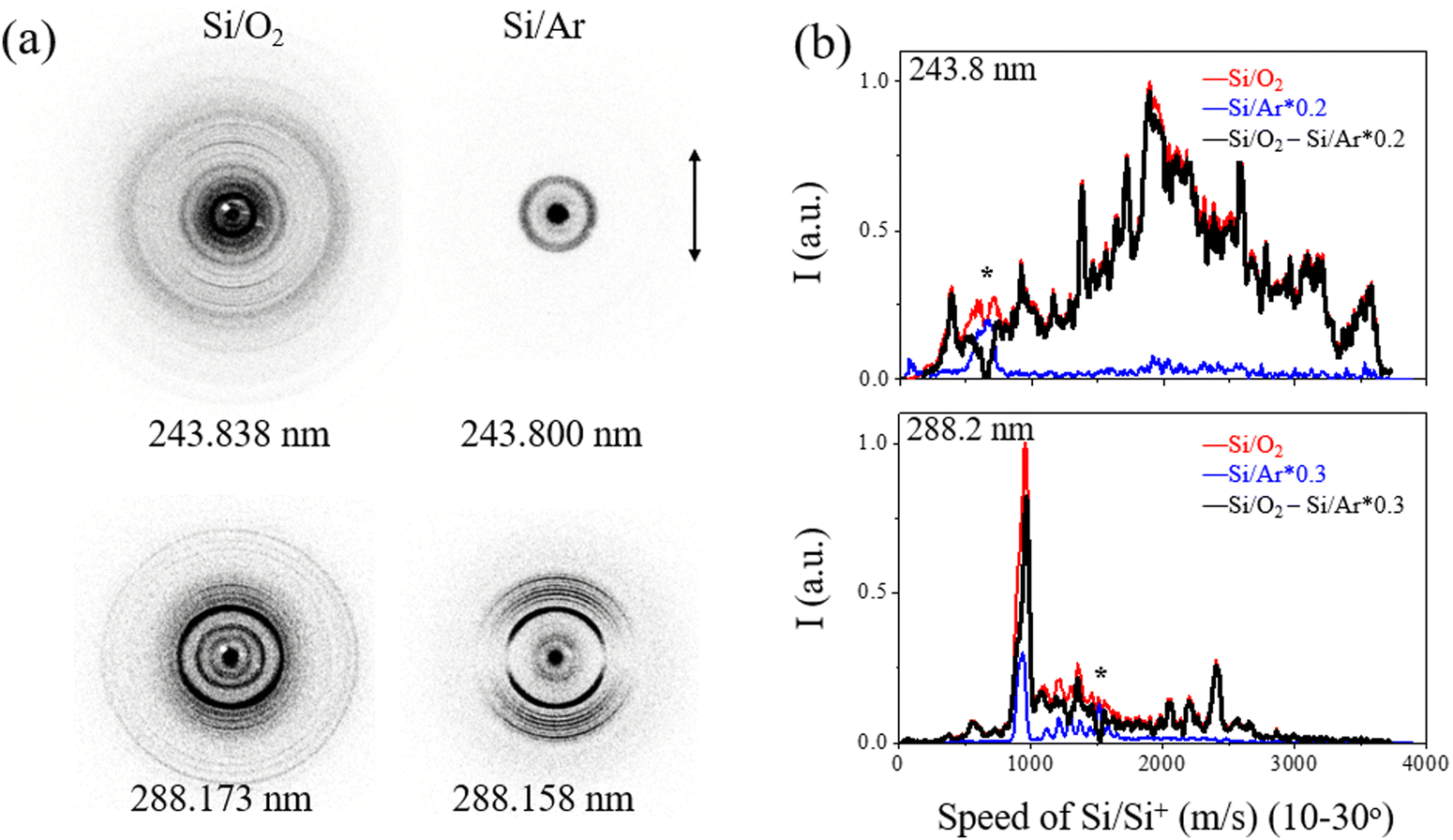 | ||
| Fig. 1 (a) The raw slice images of Si+ ions obtained at two wavelengths with O2 and Ar used as carrier gases of the ablated Si molecular beam, respectively. A small amount of signal from the photolysis of Si2 was recorded and subtracted with an appropriate ratio. The double-headed arrow presents the polarization direction of the photolysis laser. (b) The speed distributions of the Si/Si+ products are shown in red for O2 and in blue for Ar as carrier gases, and the Si signal from photolysis of SiO is shown in black. The signal of Si2 was subtracted according to the indicative peaks with * markers, which are inclusively from the photolysis of Si2. | ||
The total kinetic energy release (TKER) distribution P(E) of Si + O products was obtained from the corresponding speed distributions P(v) by following the conservation of a recoil momentum with E = 1/2mSi(1 + mSi/mo)v2 and P(E) ∝ P(v)/v, as shown in Fig. 2. The equation, Eavai = nhν + Te(SiO) + Evib(SiO) – De(SiO) = ET(Si + O) + Eint(Si + O), where n is the number of absorbed photons involved in the photolysis, hν is the photon energy, Te(SiO) and Evib(SiO) represents the electronic and vibrational energies of the SiO parent molecule, respectively, De(SiO) is the dissociation energy for Si(3P) + O(3P) with a value of 67253 ± 110 cm−1, and ET(Si + O) and Eint(Si + O) correspond to the total kinetic energy and internal energy of the photofragments, gives the available energy (Eavai) based on energy conservation. The equation allows for the assignment of specific channels from different electronic states of SiO.
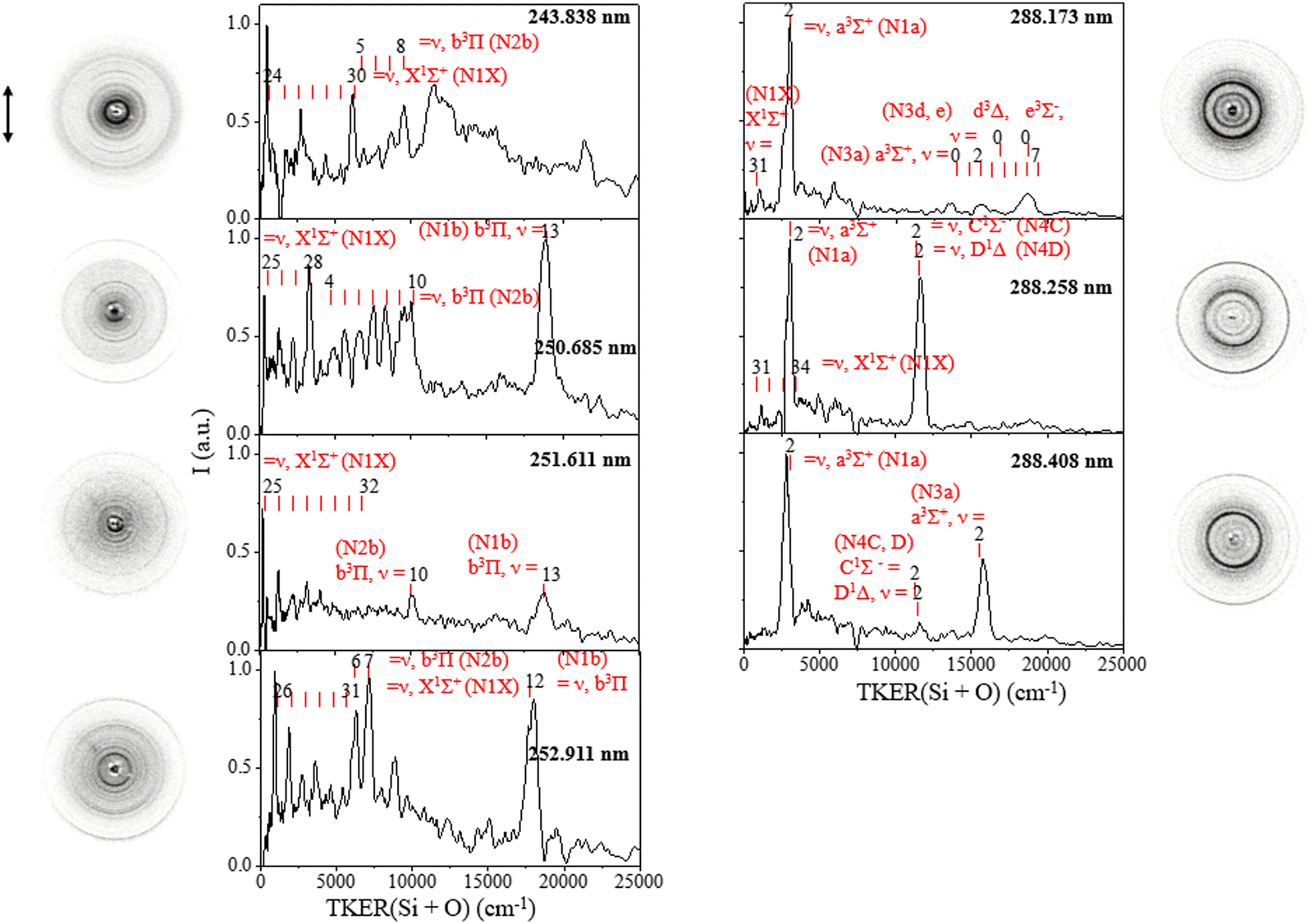 | ||
| Fig. 2 Total kinetic energy release (TKER) distributions of Si + O products in the UV region of 243–288 nm. The corresponding channels and vibrational states of the SiO reagent molecules were assigned. Raw slice images after background subtraction with double-headed arrows depicting the polarization direction of the photolysis laser are shown in the figure. | ||
From the TKER spectra, we observed the one-photon dissociation of vibrationally excited SiO(X1Σ+, high v) at the studied UV excitation wavelength in the range of 243–288 nm, leading to the Si(3P) + O(3P) ground channel in accordance with energy conservation. The TKER distributions at 243.838–252.911 nm in the left panel of Fig. 2 show products with peaks at <6160 cm−1 arising from the dissociation of SiO(X1Σ+, ν = 24–32) + hv → Si(3P) + O(3P) (N1X). For example, at 252.911 nm, TKER peaks at 988 and 1900 cm−1 correspond to SiO(X1Σ+, ν = 26 and 27), respectively, where their energy difference aligns with the difference between the two vibrational levels. At lower excitation energy, specifically, at 288.173 and 288.258 nm, we detected a weak signal for SiO(X1Σ+, ν = 31) + hv → Si(3P) + O(3P) (N1X) as shown in the right panel of Fig. 2.
Additionally, the results have yielded new insights into the photodissociation of electronically excited SiO in accordance with energy conservation. Specifically, the TKER spectra show the one-photon dissociation of SiO(a3Σ+, v = 2) at adjacent wavelengths of 288.173, 288.258, and 288.408 nm, producing Si(3P) + O(3P) (N1a), and two-photon excitation of SiO(a3Σ+) leading to Si(1D) + O(1D) (N3a). At 288.173 nm, two-photon excitation of SiO(a3Σ+, v = 0–7) also produced Si(1D) + O(1D) (N3a or N3d, e) with product kinetic energies in the range of 13100–19300 cm−1, and two-photon excitation of SiO(d3Δ, ν = 0; e3Σ−, ν = 0) gave the product in the same TKER range due to the close internal energy levels of SiO(a3Σ+, v = 0–7) and SiO(d3Δ, ν = 0; e3Σ−, ν = 0). Moreover, we observed the photodissociation of SiO(b3Π, v), resulting in Si(1D) + O(3P) (N2b) at wavelengths of 243.838 nm, 250.685, 251.611 and 252.911 nm and leading to Si(3P) + O(3P) (N1b) at 250.685, 251.611 and 252.911 nm. At 250.685 nm, the N2b channel reveals clear vibrational structures of SiO(b3Π, ωe = 1014 cm−1). We also observed the strong two-photon dissociation signal of singlet excited SiO(D1Δ & C1Σ−, v = 2) leading to Si(1S) + O(1D) (N4C, D) at a wavelength of 288.258 nm, which becomes weak at 288.408 nm.
It can be inferred that the lifetime of the excited electronic states observed in SiO photodissociation exceeds the flight time of the molecule, which is about 560 μs from the laser ablation zone to the center of the reaction chamber. In the molecular beam experiment, the seven low-lying electronic states in Table 1 with the corresponding dissociation channels in Table 2 were all observed. Table 2 shows the energy required for four dissociation channels from excitation of various electronic states: Si(3P) + O(3P) (N1), Si(1D) + O(3P) (N2), Si(1D) + O(1D)(N3), and Si(1S) + O(1D)(N4). After one-photon or two-photon dissociation of SiO, the Si products can be ionized via two-photon absorption at the same wavelength with an ionization energy (i.e. = 8.15 eV) for Si(3P). Broad peaks with high kinetic energy appear at a high excitation energy of 243.838 nm, which may originate from the multiphoton dissociation channels of SiO or SiO+. Assigning these signals is relatively difficult due to their complexity and the lack of high-potential energy surface theory in this region.
| Label | States of SiO reactants | Products channel of Si + O | ΔH (cm−1) | Photon(s) |
|---|---|---|---|---|
| N1X | X1Σ+ | 3P + 3P | 66634 |
1 |
| N1a | a3Σ+ | 3P + 3P | 33229 |
1 |
| N1b | b3Π | 3P + 3P | 32801 |
1 |
| N2b | b3Π | 1D + 3P | 39100 |
1 |
| N3a | a3Σ+ | 1D + 1D | 55396 |
2 |
| N3d | d3Δ | 1D + 1D | 52551 |
2 |
| N3e | e3Σ− | 1D + 1D | 50738 |
2 |
| N4C | C1Σ− | 1S + 1D | 59522 |
2 |
| N4D | D1Δ | 1S + 1D | 59328 |
2 |
The angular distribution of photofragments from time-sliced ion velocity images can be fitted with anisotropic parameters β using the expression 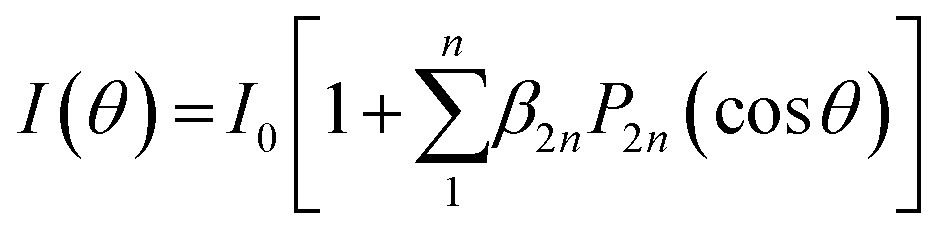 , where P2n(cosθ) is the 2n-th Legendre polynomial with n = 1 for one-photon dissociation and n = 2 for two-photon dissociation.43,44 For one-photon dissociation, the range of β value is from −1 to 2, with positive values indicating dominant parallel transition and negative values indicating dominant perpendicular transition. For two-photon dissociation of SiO, the near-zero value of β4 suggests that it is negligible in data analysis, simplifying β2 as β for comparison with one-photon dissociation dynamics, while the large value of β4 indicates the strong mixing transition characteristics with the involvement of a long-lived intermediate state. Fig. 3 shows the angular distributions of Si + O products recorded at 288.173 nm with a fit β. The ground product channel Si(3P) + O(3P) (N1) from the one-photon excitation of SiO(X1Σ+, ν = 31) shows a dominant perpendicular transition, while the channel from the one-photon excitation of excited SiO(a3Σ+, ν = 2) exhibits a dominant parallel transition. The excited product channel Si(1D) + O(1D) (N3) from the two-photon excitation of electronically excited states of a3Σ+, v = 0 shows a parallel angular distribution, while the vibrationally excited states show an almost isotropic angular distribution, indicating a slower dissociation process or a mixed parallel and perpendicular transition. The energetically overlapped vibrationally excited state of a3Σ+ with d3Δ or e3Σ− states may lead to the isotropic angular distributions. The angular distributions of Si products in different channels from other wavelengths are shown in Fig. S1–S4 (ESI†), with anisotropy parameter fits provided in the ESI.†
, where P2n(cosθ) is the 2n-th Legendre polynomial with n = 1 for one-photon dissociation and n = 2 for two-photon dissociation.43,44 For one-photon dissociation, the range of β value is from −1 to 2, with positive values indicating dominant parallel transition and negative values indicating dominant perpendicular transition. For two-photon dissociation of SiO, the near-zero value of β4 suggests that it is negligible in data analysis, simplifying β2 as β for comparison with one-photon dissociation dynamics, while the large value of β4 indicates the strong mixing transition characteristics with the involvement of a long-lived intermediate state. Fig. 3 shows the angular distributions of Si + O products recorded at 288.173 nm with a fit β. The ground product channel Si(3P) + O(3P) (N1) from the one-photon excitation of SiO(X1Σ+, ν = 31) shows a dominant perpendicular transition, while the channel from the one-photon excitation of excited SiO(a3Σ+, ν = 2) exhibits a dominant parallel transition. The excited product channel Si(1D) + O(1D) (N3) from the two-photon excitation of electronically excited states of a3Σ+, v = 0 shows a parallel angular distribution, while the vibrationally excited states show an almost isotropic angular distribution, indicating a slower dissociation process or a mixed parallel and perpendicular transition. The energetically overlapped vibrationally excited state of a3Σ+ with d3Δ or e3Σ− states may lead to the isotropic angular distributions. The angular distributions of Si products in different channels from other wavelengths are shown in Fig. S1–S4 (ESI†), with anisotropy parameter fits provided in the ESI.†
 | ||
| Fig. 3 Angular distributions of Si + O products at 288.173 nm with the fitted anisotropy parameters for the corresponding dissociation channels of SiO. | ||
In Table 3, we summarize the obtained β values for distinct dissociation channels at different excitation wavelengths, showing clear dependence on the initial states. The parallel angular distribution of the Si products, indicated by positive β values, suggests a fast dissociation process with an electronic transition of ΔΩ = 0, where Ω is the sum of the quantum numbers of the projections of the electronic orbital and electronic spin angular momentum along the diatomic internuclear axis. In contrast, the perpendicular angular distribution of the Si products, indicated by negative β values, suggests a fast dissociation process with an electronic transition of ΔΩ = ±1. Finally, the isotropic angular distribution suggests that the dissociation processes are slow or mixed ΔΩ = 0 and ±1 transitions.
| SiO + hv → Si(3P) + O(3P) (N1) | β 2 ± 0.10 |
|---|---|
| 243.838 nm (N1X) X1Σ+ | |
| ν = 24 | 0.06 |
| ν = 25 | −0.07 |
| ν = 26 | −0.14 |
| 250.685 nm (N1X) X1Σ+ | |
| ν = 25 | 0.39 |
| ν = 26 | 0.19 |
| ν = 27 | 0.08 |
| ν = 28 | 0.26 |
| (N1b) b3Π | |
| ν = 13 | 0.97 |
| 251.611 nm (N1X) X1Σ+ | |
| ν = 26 | −0.11 |
| ν = 27 | 0.05 |
| ν = 28 | −0.01 |
| ν = 29 | −0.05 |
| ν = 30 | −0.16 |
| (N1b) b3Π | |
| ν = 13 | 0.39 |
| 252.911 nm (N1X) X1Σ+ | |
| ν = 26 | 0.40 |
| ν = 27 | 0.14 |
| ν = 28 | −0.03 |
| ν = 29 | −0.06 |
| (N1b) b3Π | |
| ν = 13 | 0.60 |
| 288.173 nm (N1X) X1Σ+ | |
| ν = 31 | −0.12 |
| (N1a) a3Σ+ | |
| ν = 2 | 0.55 |
| 288.258 nm (N1X) X1Σ+ | |
| ν = 31 | 0.43 |
| (N1a) a3Σ+ | |
| ν = 2 | 0.59 |
| 288.408 nm (N1a) a3Σ+ | |
| ν = 2 | 0.65 |
| SiO + hv → Si(1D) + O(3P) (N2) | β 2 ± 0.10 |
|---|---|
| 243.838 nm (N2b) b3Π | |
| ν = 5–8 | 0.19 |
| 250.685 nm (N2b) b3Π | |
| ν = 4–10 | 0.16 |
| 251.611 nm (N2b) b3Π | |
| ν = 10 | −0.22 |
| 252.911 nm (N2b) b3Π | |
| ν = 6–7 | 0.17–0.25 |
| SiO + 2hv → Si(1D) + O(1D) (N3) | β 2 ± 0.10 | β 4 ± 0.10 |
|---|---|---|
| 288.173 nm (N3a) a3Σ+ | ||
| ν = 0 | 0.22 | −0.09 |
| (N3a) a3Σ+ | ||
| ν = 2 | −0.12 | −0.02 |
| (N3d) d3Δ | ||
| ν = 0 | ||
| (N3a) a3Σ+ | ||
| ν = 6 | 0.06 | −0.03 |
| (N3e) e3Σ− | ||
| ν = 0 | ||
| 288.408 nm (N3a) a3Σ+ | ||
| ν = 2 | 0.96 | 0.27 |
| SiO + 2hv → Si(1S) + O(1D) (N4) | β 2 ± 0.10 | β 4 ± 0.10 |
|---|---|---|
| 288.258 nm (N4C, D) C1Σ−, D1Δ | ||
| ν = 2 | 0.29 | 0.04 |
| 288.408 nm (N4C, D) C1Σ−, D1Δ | ||
| ν = 2 | −0.20 | −0.03 |
From theoretical calculations of the PECs for singlet and triplet states of SiO (as shown in Fig. 4),37,45 the vibrationally excited SiO(X1Σ) can be dissociated through one-photon excitation at 243–288 nm to the repulsive walls of the quasi-bound A1Π (Te = 42835 cm−1, ωe = 852.8 cm−1, and ωexe = 6.4 cm−1)29 or E1Σ+ (Te = 52860.9 cm−1, ωe = 675.5 cm−1, and ωexe = 4.2 cm−1) states, via a perpendicular transition or a parallel transition.46 The A1Π state has a lower energy than the E1Σ+ state. Both states display potential wells along the dissociation path, with the E1Σ+ state exhibiting a shallower well than the A1Π state. This results in a faster dissociation rate of SiO along the E1Σ+ state. The products with small TKER near zero from the dissociation of A1Π/E1Σ+ states indicate that no barrier exists above the dissociation limit along the dissociation path. Based on the analysis of the TKER and angular distributions, the excitation of SiO(X1Σ+) to the repulsive wall of the upper state of A1Π results in a perpendicular transition, while excitation to the higher upper state E1Σ+ results in a parallel transition.44 Consequently, excitation of different vibrationally excited SiO(X1Σ+) to the A1Π and E1Σ+ states can alter the product angular distributions.
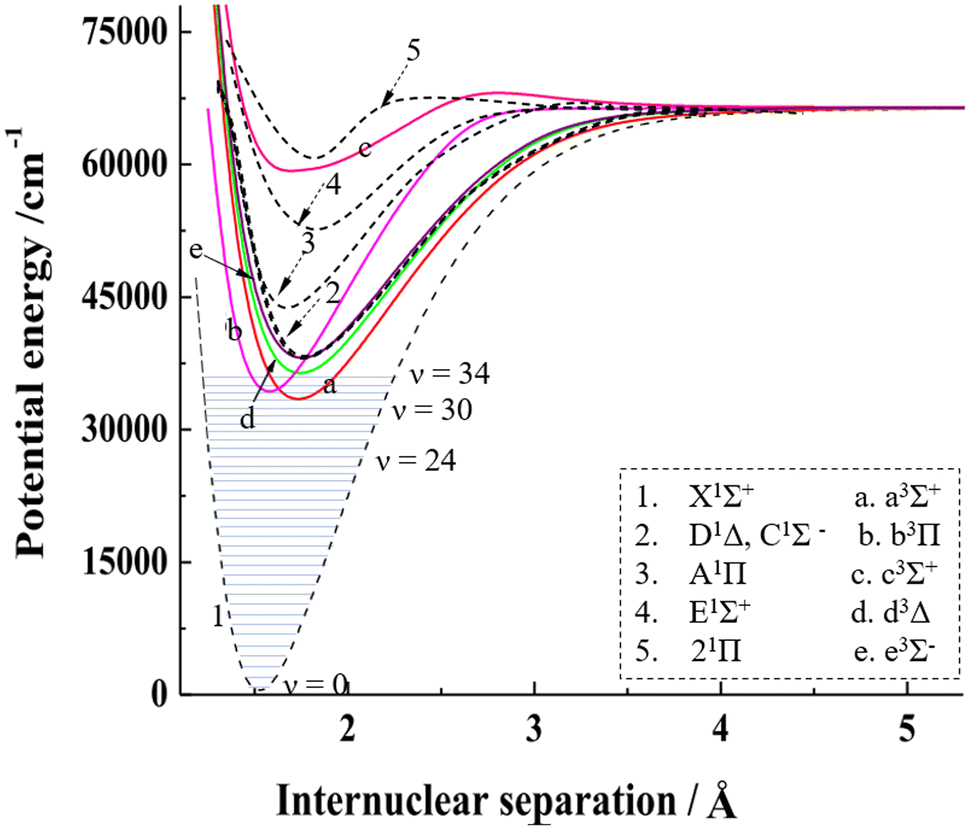 | ||
| Fig. 4 The PECs of SiO for singlet and triplet states in the 0–75000 cm−1 energy region with the vibrational levels for the ground electronic state. The combined PECs are from Feng and Zhu's theoretical results.37,45 The PECs in the figure are modified with permission from ref. 37 and 45 at Elsevier. | ||
The PECs indicate that the bound electronically excited states with long-lived lifetimes, such as C1Σ−, D1Δ, a3Σ+, b3Π, d3Δ and e3Σ−, can be involved in dissociation when they absorb one or more photons, explaining the dissociation behavior observed in a wide range of excitation wavelengths.47 For two-photon processes, such as channels (N3a, d, and e) at 288 nm, the angular distributions would reflect the symmetry of both the intermediate and final states. SiO(D1Δ) is excited to the highly excited SiO(1Σ, 1Π, 1Δ) and then dissociates to Si(1S) + O(1D) based on the building-up principles of the molecular state from atomic states.48 However, there is a likely competition between autoionization and neutral pre-dissociation along the highly excited SiO(1Δ), as the dissociation limit of Si(1S) + O(1D) with 97896 cm−1 is above the adiabatic ionization energy of SiO (i.e. = 92675 cm−1).49 According to the description of the multi-photon dissociation process from Dixon,43 the anisotropy parameters for the Si(1S) + O(1D) channel (N4C, D) indicate that the two-photon transitions from the initial C1Σ− and D1Δ are mainly through Σ and Δ intermediate states, respectively, at 288.258 nm according to the positive β2, while mainly through the Π or Φ intermediate state at 288.408 nm according to the negative β2. The intensity of photofragments depends on various factors, including the vibrational distribution of the parent molecules, the transition probability between the initial state and the upper state, and the dissociation rates along different excited states. Further theoretical research on highly excited electronic states of SiO is necessary to fully understand the detailed dissociation process.
Vibrationally excited SiO(X1Σ+) was observed in previous spectroscopic experiments. Mollaaghababa et al.50 utilized millimeter wave spectroscopy and observed rotational transitions in excited vibrational states up to v = 40 of the X1Σ+ electronic ground state in a glow discharge free-space cell. Sanz et al.51 measured highly excited vibrational states (v > 30) of SiO in a DC electric discharge molecular beam source using SiH4/O2 as the precursor gas and Ne as the carrier gas. The abundance of O2 in the discharge experiments is considerably higher than that of SiO, leading to O2 + SiO collisions being significant for vibrational ladder heating. However, in the present experiment, which involves single collisions for crossed-beams between the laser-ablated Si beam and an O2 supersonic beam, we suggest that electronically excited states of SiO may serve as an alternative source for the vibrational excitation of SiO(X1Σ+). According to theoretical calculations,37,45 some highly excited states of SiO with lifetimes in the teens or tens of nanoseconds, including A1Π, E1Σ+ and 21Π, can rapidly jump back to the X1Σ+ state, resulting in vibrational excitation of SiO. In the case of A1Π to X1Σ+ deexcitation, vibrationally excited SiO can have an internal energy range of 4000–37000 cm−1 and ν = 3–35 in the Franck–Condon region, thereby providing a rationale for the photolysis of SiO(X1Σ+, high v) via the excitation to the A1Π or E1Σ+ quasi-bound states.
A comprehensive understanding of the photodissociation dynamics of SiO molecules from various excited electronic states, including the vibrationally excited X1Σ+ state, is critical for elucidating the chemical behavior of silicon-containing meteoroids entering the Earth's atmosphere. These meteoroids interact with the uppermost atmospheric layers, resulting in rapid surface heating, surface ablation, high-velocity expansion of vapor, and reactions due to collisions with atmospheric molecules.52,53 Similarly, in the case of the laser-ablated supersonic beam, the formation of highly vibrationally and electronically excited SiO, resulting from the interaction of a laser-ablated silicon rod with O2, is followed by the multi-channel photodissociation and represents a significant challenge in the detection of SiO during the meteoroid entry into the atmosphere. The photodissociation of highly vibrationally and electronically excited SiO provides novel insights into the origin and composition of meteoroids and their relationship with other bodies in the solar system.
Conclusions
The use of laser ablation and time-sliced ion velocity imaging techniques enabled the observation of SiO molecules with high vibrational and electronic state excitation. The high excitation levels provided access to dissociation channels that are not observed at lower excitation levels. The observed dissociation channels included one-photon and two-photon dissociation, which led to the production of highly excited and reactive products. The one-photon dissociation produced Si(3P) + O(3P) and Si(1D) + O(3P) from SiO(X1Σ+, high ν ≥ 24) and SiO(a3Σ+, b3Π) at 243–288 nm. The two-photon dissociation produced Si(1D) + O(1D) and Si(1S) + O(1D) from SiO(a3Σ+, d3Δ, e3Σ−, C1Σ−/D1Δ), respectively. These dissociation channels were specific to the highly excited electronic states of SiO, and the coupling between these states was found to be weak. The observed dissociation channels provide insights into the PECs of SiO and the mechanisms that influence its formation and photodissociation behavior during meteor entry.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22073019 and 21327901), the Shanghai Key Laboratory Foundation of Molecular Catalysis and Innovative Materials, and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning.References
- E. J. Barton, S. N. Yurchenko and J. Tennyson, Mon. Not. R. Astron. Soc., 2013, 434, 1469–1475 CrossRef CAS.
- J. Nakashima, S. Deguchi, H. Imai, A. Kemball and B. M. Lewis, Astrophys. J., 2011, 728, 76 CrossRef.
- J. M. Campbell, D. Klapstein, M. Dulick and P. F. Bernath, Astrophys. J., Suppl. Ser., 1995, 101, 237–254 CrossRef CAS.
- J. Martinpintado, R. Bachiller and A. Fuente, Astron. Astrophys., 1992, 254, 315–326 CAS.
- C. W. Bauschlicher, Chem. Phys. Lett., 2016, 658, 76–79 CrossRef CAS.
- A. Mishra and A. G. Li, Astrophys. J., 2017, 850, 138 CrossRef.
- C. He, Y. H. Luo, S. Doddipatla, Z. H. Yang, T. J. Millar, R. Sun and R. I. Kaiser, Phys. Chem. Chem. Phys., 2022, 24, 19761–19772 RSC.
- A. A. Berezhnoy and J. Borovicka, Icarus, 2010, 210, 150–157 CrossRef CAS.
- J. M. C. Plane, J. C. Gómez-Martín, W. H. Feng and D. Janches, J. Geophys. Res. Atmos, 2016, 121, 3718–3728 CrossRef CAS PubMed.
- P. Jenniskens, E. Tedesco, J. Murthy, C. O. Laux and S. Price, Meteorit. Planet. Sci., 2002, 37, 1071–1078 CrossRef CAS.
- W. Jevons, Proc. R. Soc. London, Ser. A, 1924, 106, 174–194 CAS.
- W. H. B. Cameron, Philos. Mag., 1927, 3, 110–115 CAS.
- P. G. Saper, Phys. Rev., 1932, 42, 0498–0508 CrossRef CAS.
- H. C. Rowlinson and R. F. Barrow, J. Chem. Phys., 1953, 21, 378–379 CrossRef CAS.
- R. F. Barrow and H. C. Rowlinson, Proc. R. Soc. London, Ser. A, 1954, 224, 374–388 CAS.
- A. T. Mcgregor, W. R. Jarmain and R. W. Nicholls, Can. J. Phys., 1961, 39, 1215–1216 CrossRef CAS.
- R. D. Verma and R. S. Mulliken, Can. J. Phys., 1961, 39, 908–916 CrossRef CAS.
- S. Nagaraj and R. D. Verma, Can. J. Phys., 1970, 48, 1436–1440 CrossRef CAS.
- W. H. Smith and H. S. Liszt, J. Quant. Spectrosc. Radiat. Transfer, 1972, 12, 505–509 CrossRef CAS.
- R. Cornet and I. Dubois, Can. J. Phys., 1972, 50, 630–635 CrossRef CAS.
- H. Bredohl, R. Cornet, I. Dubois and F. Remy, Can. J. Phys., 1973, 51, 2332–2335 CrossRef CAS.
- A. Lagerqvist, I. Renhorn and N. Elander, J. Mol. Spectrosc., 1973, 46, 285–315 CrossRef CAS.
- M. Singh, H. Bredohl, F. Remy and I. Dubois, Can. J. Phys., 1974, 52, 569–574 CrossRef CAS.
- H. Bredohl, R. Cornet, I. Dubois and F. Remy, J. Phys. B: At., Mol. Opt. Phys., 1974, 7, L66–L68 CrossRef CAS.
- A. Lagerqvist and I. Renhorn, J. Mol. Spectrosc., 1974, 49, 157–166 CrossRef CAS.
- G. Hager, L. E. Wilson and S. G. Hadley, Chem. Phys. Lett., 1974, 27, 439–441 CrossRef CAS.
- R. F. Barrow and T. J. Stone, J. Phys. B: At., Mol. Opt. Phys., 1975, 8, L13–L15 CrossRef CAS.
- G. Hager, R. Harris and S. G. Hadley, J. Chem. Phys., 1975, 63, 2810–2820 CrossRef CAS.
- R. W. Field, A. Lagerqvist and I. Renhorn, Phys. Scr., 1976, 14, 298–319 CrossRef CAS.
- R. D. Verma and R. Shanker, J. Mol. Spectrosc., 1976, 63, 553–563 CrossRef CAS.
- J. Oddershede and N. Elander, J. Chem. Phys., 1976, 65, 3495–3505 CrossRef CAS.
- C. Linton and G. A. Capelle, J. Mol. Spectrosc., 1977, 66, 62–68 CrossRef CAS.
- R. Shanker, C. Linton and R. D. Verma, J. Mol. Spectrosc., 1976, 60, 197–209 CrossRef CAS.
- J. Hormes, M. Sauer and R. Scullman, J. Mol. Spectrosc., 1983, 98, 1–19 CrossRef CAS.
- C. S. Park, D. R. Crosley, D. J. Eckstrom and K. R. Heere, J. Quant. Spectrosc. Radiat. Transfer, 1993, 49, 349–360 CrossRef CAS.
- S. Chattopadhyaya, A. Chattopadhyay and K. K. Das, J. Phys. Chem. A, 2003, 107, 148–158 CrossRef CAS.
- Y. Feng and Z. Zhu, J. Quant. Spectrosc. Radiat. Transfer, 2019, 236, 106576 CrossRef CAS.
- M. Yujie, L. Fangfang, Y. Dong, X. Ang, Z. Ti, L. Jiaxing and W. Fengyan, Chin. J. Chem. Phys., 2024 DOI:10.1063/1674-0068/cjcp2407096.
- A. Xu, Y. Ma, D. Yan, F. Li, T. Zhou, J. Liu and F. Wang, J. Phys. Chem. A, 2024, 128, 3848–3854 CrossRef CAS PubMed.
- A. Xu, Y. Ma, D. Yan, F. Li, F. Song, T. Zhou, Z. Yuan, X. Liu, J. Liu and F. Wang, J. Phys. Chem. Lett., 2024, 15, 8721–8727 CrossRef CAS PubMed.
- S. D. Le Picard, A. Canosa, D. Reignier and T. Stoecklin, Phys. Chem. Chem. Phys., 2002, 4, 3659–3664 RSC.
- D. L. Martin, D. L. Thompson and L. M. Raff, J. Chem. Phys., 1986, 84, 4426–4428 CrossRef CAS.
- R. N. Dixon, J. Chem. Phys., 2005, 122, 194302 CrossRef.
- A. J. Alexander and R. N. Zare, Acc. Chem. Res., 2000, 33, 199–205 CrossRef CAS.
- Y. Feng and Z. Zhu, J. Quant. Spectrosc. Radiat. Transfer, 2019, 239, 106647 CrossRef CAS.
- S. N. Yurchenko, J. Tennyson, A.-M. Syme, A. Y. Adam, V. H. J. Clark, B. Cooper, C. P. Dobney, S. T. E. Donnelly, M. N. Gorman, A. E. Lynas-Gray, T. Meltzer, A. Owens, Q. Qu, M. Semenov, W. Somogyi, A. Upadhyay, S. Wright and J. C. Zapata Trujillo, Mon. Not. R. Astron. Soc., 2022, 510, 903–919 CrossRef CAS.
- T. G. Heil and H. F. Schaefer, J. Chem. Phys., 1972, 56, 958–968 CrossRef CAS.
- R. S. Mulliken, Rev. Mod. Phys., 1932, 4, 0001–0086 CrossRef CAS.
- Y. Hikosaka, P. Lablanquie, M. Ahmad, R. I. Hall, J. G. Lambourne, F. Penent and J. H. D. Eland, J. Phys. B: At., Mol. Opt. Phys., 2003, 36, 4311–4326 CrossRef CAS.
- R. Mollaaghababa, C. A. Gottlieb, J. M. Vrtilek and P. Thaddeus, Astrophys. J., 1991, 368, L19–L22 CrossRef CAS.
- M. E. Sanz, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2003, 119, 11715–11727 CrossRef CAS.
- L. P. Dyrud, K. Denney, S. Close, M. Oppenheim, J. Chau and L. Ray, Atmos. Chem. Phys., 2004, 4, 817–824 CrossRef CAS.
- A. Krivková, L. Petera, V. Laitl, P. Kubelík, E. Chatzitheodoridis, L. Lenza, J. Koukal, A. Knízek, R. Dudzák, D. Páclík, S. Civis, M. Krus and M. Ferus, Exp. Astron, 2021, 51, 425–451 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03843f |
| This journal is © the Owner Societies 2025 |