
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA hairpin base-flipping dynamics drives APOBEC3A recognition and selectivity†
Mark A.
Hix
 a,
A. G. Pramoda
Sahankumari
a,
Ashok S.
Bhagwat
ab and
Alice R.
Walker
*a
a,
A. G. Pramoda
Sahankumari
a,
Ashok S.
Bhagwat
ab and
Alice R.
Walker
*a
aDepartment of Chemistry, Wayne State University, 5101 Cass Ave., Detroit, MI 48202, USA. E-mail: arwalker@wayne.edu
bDepartment of Biochemistry, Microbiology and Immunology, Wayne State University School of Medicine, Detroit, MI 48201, USA
First published on 9th May 2025
Abstract
APOBEC3A is a deoxycytidine deaminase which acts preferentially upon a 5′-TC-3′ motif, and was recently shown to prefer hairpin loops over unstructured single-stranded DNA. However, the underlying molecular details for its substrate specificity remain unclear. In this work we apply classical molecular dynamics to 212 unique hairpin loops in solvent, with lengths of 3 and 4 nucleotides in the loop, and a representative subset of 23 bound to APOBEC3A. This allows us to gain statistical insight into the types of motions and sequences potentially important for APOBEC3A activity. We demonstrate that base-flipping occurs in solvent before binding to APOBEC3A semi-dependent on the hairpin loop sequence, and that binding does not necessarily require the presence of a cytosine on the hairpin loop. Furthermore, we show measurable physical metrics, such as RMSD and sugar puckering, which may aid in identifying sequences which will exhibit higher rates of base-flipping commensurate with increased APOBEC3A activity.
1. Introduction
The APOBEC3 family of enzymes are deoxycytidine deaminases which catalyze the deamination reaction of deoxycytidine into uracil in single-stranded DNA (ssDNA) as part of a number of important biological processes including immune response, epigenetic regulation, and anti-viral cellular activity.1–4 Members of the APOBEC3 family have a conserved subunit structure consisting of five β-sheets surrounded by six α-helices.4APOBEC3s are known to exhibit preferential binding to 5′-T![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -3′ motifs.5–10 The physical driver behind the recognition mechanism for specific sequences by individual members of the APOBEC3 family is under ongoing study. The specific amino acids on loop 7 have been shown to interact favorably with specific cytosine-containing motifs.6,7,10–13 It has also been shown that APOBEC3s preferentially act upon hairpin loops (HPLs) of ssDNA, which allow the target cytosine to be more exposed and easier to bind than unstructured sequences.6,14–18 Additionally, the recognition mechanism of APOBEC proteins has been recently demonstrated to include sugar-specific interactions, effectively distinguishing against ssDNA and RNA.19 HPLs are single stranded nucleic acid sequences which are able to fold and self-pair, and can have a small section of unpaired bases linking them to larger duplex DNA (see Fig. 1). HPLs are of increasing interest in pharmaceutical design, sensing, and targeted mutagenesis by subsequent cellular processes.20–27 The highly tunable nature of hairpin loops arises from the complex interplay between stem binding affinity, loop size,23 loop sequence,24 and local environment.
-3′ motifs.5–10 The physical driver behind the recognition mechanism for specific sequences by individual members of the APOBEC3 family is under ongoing study. The specific amino acids on loop 7 have been shown to interact favorably with specific cytosine-containing motifs.6,7,10–13 It has also been shown that APOBEC3s preferentially act upon hairpin loops (HPLs) of ssDNA, which allow the target cytosine to be more exposed and easier to bind than unstructured sequences.6,14–18 Additionally, the recognition mechanism of APOBEC proteins has been recently demonstrated to include sugar-specific interactions, effectively distinguishing against ssDNA and RNA.19 HPLs are single stranded nucleic acid sequences which are able to fold and self-pair, and can have a small section of unpaired bases linking them to larger duplex DNA (see Fig. 1). HPLs are of increasing interest in pharmaceutical design, sensing, and targeted mutagenesis by subsequent cellular processes.20–27 The highly tunable nature of hairpin loops arises from the complex interplay between stem binding affinity, loop size,23 loop sequence,24 and local environment.
 | ||
| Fig. 1 An example of a hairpin loop (HPL) and nucleotide flipped in (left) and out (right) of an HPL. | ||
The preferential activity upon hairpin loops suggests the possibility of “hotspot” sequences in DNA that may be preferred APOBEC3 substrates.18 Bases in nucleic acid sequences are known to undergo flips (see Fig. 1) through rotation about the sugar-phosphate backbone, where they become oriented away from adjacent nucleobases.28 The flipped-out base therefore has increased solvent exposure and reduced interference from adjacent nucleobases in active site binding. Furthermore, this “flipped out” conformation is known to be required for deamination activity, as it is the only way to bring the target cytosine within range of a reactive water in the active site. We hypothesize that these base flips must occur before substrate binding in order to facilitate deamination, and that there are observable physical characteristics related to specific hairpin sequences that may explain why certain sequences exhibit higher rates of base flipping.
The number of unique cytosine-containing sequences that may be obtained in an n-nucleotide loop is given by the formula Nunique = 4n − 3n. Therefore, a 3-nucleotide (3NT) loop yields 37 unique sequences, a 4NT loop yields 175, a 5NT loop yields 781, a 6NT loop results in 3367, and so on. As the number of sequences increases combinatorially, it becomes intractable and costly to experimentally test and analyze each sequence. Computational methods allow for large-scale screening of these unique hairpin sequences. Molecular dynamics (MD) simulations can provide insight into specific atomic-level motions of the HPLs, including base-flipping events, that can drive protein–substrate binding.
In this work we apply MD simulations to identify physical characteristics of loops which drive base-flipping events in solution, assess binding energies of cytosine-containing hairpin loops with APOBEC3A, compare to non-cytosine-containing loops with APOBEC3A, and demonstrate the potential release pathway of the product from the enzyme. We use unbiased MD on a relatively short timescale (100 ns per trajectory), and show that base-flipping is accessible and consistent on this timescale with replicate simulations. This allows us to comprehensively investigate all 3NT and 4NT loop sequences containing one cytosine, along with some additional non-cytosine containing loops for comparison.
2. Methods
2.1. DNA hairpins in solvent
We generated starting structures using Chimera v1.1635, creating a double-helix “stem” with the sequence 5′-TTAGCATG-AAA-CATGCTAA-3′, where the triadenine group acted as the template for the new hairpin loop sequences, and each base in the stem was paired.29 All 3NT sequences containing at least one cytosine were generated from this initial structure by removing the nucleobase atoms from the template, renaming each nucleotide in the PDB file, and using the tleap program in AmberTools2230 to regenerate the nucleotide based on standard atomic positions. This resulted in 37 unique 3NT loops containing at least one cytosine. The same method was also applied using the same stem sequence with an AAAA loop as the initial template structure for 4NT loops, which resulted in 175 unique 4NT sequences containing at least one cytosine. We also prepared the hairpins AGG, TTU and AAAA as non-cytosine containing examples for 3/4NT loops by the same method. Each initial structure for 3NT and 4NT loops was prepared for molecular dynamics simulation using the OL15 forcefield for DNA, TIP3P forcefield for solvent water, and the Joung Cheathem counterion parameters.31,32 OL15 has been shown to have reasonable base-flipping dynamics for double helices.28,33–43 We also ran several comparative trajectories with parm99bsc1.44 Each system was neutralized with K+ ions to a neutral charge, then solvated with TIP3P waters to a minimum distance of 20 Å between the nucleotide and the edge of the solvent box. We first minimized each system to eliminate potential bad contacts and clashes, then allowed the solvent box to equilibrate around restrained nucleotides. We heated each system iteratively, allowing the pressure to equilibrate at each 15 °C increment between 0 °C and 300 °C. Once each system was equilibrated with constant pressure, we performed classical molecular dynamics (MD) simulations with a Langevin thermostat (friction constant = 5 ps−1) in an NVT ensemble at 300 °C and 1 atm using a 1 fs timestep for 100 ns of production using Amber22's pmemd.cuda module.30 The electrostatic interactions were computed with particle mesh Ewald and the nonbonded interaction cutoff was set to 10 Å. The combined simulations of 212 unique hairpin loops in solvent yielded 21.2 μs of sampling.2.2. Selected DNA hairpins bound to APOBEC3A
We extracted representative frames from the MD trajectories of selected sequences (ATC, CTC, TAC, TTC, ACGG, ATTC, GCAA, TTTC) which had the target cytosine base in both the “flipped-in” and “flipped-out” conformations. We also extracted frames from simulations of AGG and AAAA hairpins to act as non-cytosine-containing examples, allowing us to differentiate between interactions required for binding versus catalysis. These sequences were chosen to support future experimental work and provide a representative set. Additionally, we included the 3NT-HPL (TTU) system as a post-catalytic example. Each of these frames was overlaid on the crystal structure of APOBEC3A (PDB ID: 5KEG), which was selected since the crystal already included a bound DNA 4-mer which served as a guide for our own manual docking.45 The existing tetramer was removed from the crystal after alignment, resulting in a new PDB which contained the protein and the extracted DNA hairpin bound in the active site. Upon release of AlphaFold3 (AF3), which added the ability to use machine learning methods to identify protein/nucleotide interfaces,46 we also performed test calculations on ATC, CTC and ATTC sequences with AF3 to compare to the manual docking. The sequence of the crystallized protein includes two mutations (E72A, catalytically inactivating it, and C171A, to make the protein more soluble) which were not expected to significantly affect the ability of APOBEC3A to bind the target cytosine-containing motifs. Each protein/substrate complex was prepared for additional MD simulations in the same manner as above, and post-equilibration MD ran for 100 ns and five replicates, totaling 500 ns of sampling per system. The MD simulations of 12 unique sequences docked to APOBEC3A in both flipped-in and flipped-out orientations resulted in a total of 11.4 μs of sampling.To obtain qualitative binding energies, we computed enthalpies via MM/GBSA with the MMPBSA.py program available in Amber22.47 The absolute affinities cannot be compared directly with experimental values, as the affinity increases based on size of system and our obtained values are in the range of −10 to 100 kcal mol−1. However, as previously noted with MM/GBSA methods, we do find that they qualitatively reproduce relative binding affinity trends for our systems, given that they are sufficiently similar as with our sets of hairpin loops.48
3. Results and discussion
3.1. DNA hairpins in solution
-3′. This range overlaps the angular region defined as C2′-endo, which exhibits greater phosphate–phosphate distance across the nucleic acid backbone and allows for the base to easily flip from anti to syn conformations.50 While the key positioning of the deoxycytidine for the APOBEC3s is known, these results suggest that combining sugar puckering and RMSD values can be used to predict base-flipping behavior in DNA hairpins in general.
 | ||
| Fig. 2 (top) Trimer HPLs (a) average RMSD of each base at each position, (b) average sugar puckering of each base and position, (c) comparison of RMSD and sugar puckering grouped by position. (bottom) Tetramer HPLs (d) average RMSD of each base at each position in a tetramer HPL, (e) average sugar puckering of each base and position, and (f) comparison of RMSD and sugar puckering grouped by position. | ||
Recent work by Nakauma-González et al. identified several interesting trends related to “hot-spot” mutations on HPLs by APOBEC enzymes.51 In that work, the authors illustrate that cytosines in the third or further position from the 5′ start of the HPL are frequently mutated by APOBECs. We observed in our simulations that bases at these positions exhibit a combination of higher RMSD values and maintain a narrower range of sugar pucker values, suggesting that base flips are more favorable in these positions and a possible relationship to the sequence-specificity of APOBEC enzymatic activity. By comparison, bases in other positions with lower RMSD values exhibit a broader range of sugar pucker values. Together, this suggests that base flips may be less favorable at positions closer to the stem of the HPL as the phosphate bonds are less able to rotate, causing the deoxyribose to explore a larger conformational space instead.
3.2. DNA hairpins in complex with APOBEC3A
 | ||
| Fig. 3 HPL with GCAA loop sequence docked to APOBEC3A active site in (a) flipped-out and (b) flipped-in orientations. Grey structures represent target cytosine at all timesteps of simulation relative to active site position. | ||
Additional simulations were performed with 3NT sequences ATC, CTC, GTC, TTC, and TAC. Similar behaviors were observed in sequences in which the cytosine was initially flipped into the hairpin loop, most notably the higher level of mobility and the inaccessibility of the active site. By contrast to the larger 4NT sequences, the 3NT sequences which began flipped out of the loop and bound in the active site exhibited lower stability as well, with somewhat more flexibility and motion in the active site itself (see Fig. 4). However, as with the 4NT sequence, none of the 3NT sequences were able to bind or unbind from the active site in opposition to their starting orientation, again indicating that once bound, DNA sequences are not able to undergo base-flipping events which will allow cytosine to enter the active site. This further supports our hypothesis that hairpin loops must undergo base-flip events in solvent before protein binding in order to allow for the deamination reaction to proceed.
 | ||
| Fig. 4 Dynamic motions of selected 3NT sequences docked to APOBEC3A in flipped-in and flipped-out conformations. | ||
3.3. Cytosine is not necessary for A3A/HPL binding
To determine if the native cytosine substrate was necessary for binding, we performed additional simulations on hairpin sequences which did not contain cytosine (AGG and AAAA), then repeated binding simulations and analyses as above. For the 3NT loop AGG, we selected frames based on the base-flipped conformation of the 3′ guanine to be consistent with the cytosine-containing 3NT sequences. For the 4NT loop AAAA, we selected frames based on the second loop position, consistent with GCAA above. The AGG loop exhibits similar behavior to the GTC and TAC sequences, with the flipped-out initial configuration showing reduced binding affinity compared to the flipped-in. By contrast, the AAAA loop appears to have much stronger binding affinity when the nucleotide is in the active site. We attribute these disparities to the respective functional groups of the different nucleotides. The carbonyl group of the guanine is analogous to the uracil which would be present after successful deamination, while the amine group of the adenine mimics that of the cytosine. With these structural features in mind, it is reasonable to expect an enzyme would exhibit reduction in binding favorability with the the product of its catalyzed reaction, as this would encourage dissociation and subsequently free the binding site for further catalysis. The AAAA loop functions similarly to the TTU disfavored binding and eventual unbinding pathway, as discussed above.3.4. Base flips prior to binding determine binding affinity
In order to relate sequence to binding affinity more directly, we calculated binding affinities for several hairpin sequences in APOBEC3A using MM/GBSA. These binding energies include the entire hairpin sequence (19 nucleotides for 3NT loops) against the entire receptor complex (188 amino acids, 1 Zn2+ ion, and 1 coordinated water). Each trajectory was analyzed via a bootstrapping method and the resulting binding energies compared across all systems (see Fig. 5). For the ATC, CTC, and TAC sequences, the flipped-out conformations have more favorable binding energies than their flipped-in counterparts, while the opposite is true for the GTC and TTC sequences. However, the difference in favorability is considerably smaller for the GTC/TTC sequences as compared to ATC/CTC/TAC, supporting the idea that base-flipping generally improves binding. In examining the 4NT loops, we find that all tested sequences exhibit tighter binding after base-flipping than to their unflipped counterparts. Given that the phosphate backbone is able to interact with positively charged surface residues near the active site in both conformations, the difference is likely driven by favorable interactions with active site residues involved in the catalytic process. | ||
| Fig. 5 Binding energies (MMGBSA) for selected sequences docked to APOBEC3A in flipped-in and flipped-out conformations. | ||
3.5. Uracilated loops exhibit lower binding energies
To mimic the system after uracilation, we modified the final frame of the TTC-containing HPL docked to APOBEC3A by mutating the cytosine to uracil. We then performed MD to compare how going from reactant to product for catalysis affected substrate binding. We compared nearby residues in the first and last frame of the product (TTU) HPL and noted that many critical interactions which were present for the entire reactant (TTC) simulation were lost (see Fig. 6). The orientation of H61 changed during the unbinding of TTU, with K21 and K51 stabilizing the phosphate backbone as the HPL begins to move away from the active site. At this point R19 is able to engage with the HPL and continue guiding it away from the active site. At the start of the TTU simulation, the uracil is 4.15 Å from the α-carbon of H61, moving to 6.82 Å by the end. As shown in Fig. 5, the TTU sequence exhibits less favorable binding than the TTC motif. These motions are likely crucial for substrate release post-catalysis. | ||
| Fig. 6 (a) Initial binding orientation of TTC HPL in APOBEC3A, (b) critical protein–substrate interactions on binding, (c) first and last frame of TTU HPL in APOBEC3A, (d) zoomed in active site of first frame for TTU HPL, and (e) zoomed in active site of last frame of TTU HPL. | ||
3.6. Manually docked systems are closely matched by AlphaFold3 models
We compared the results of the manually docked ATC-containing hairpin loop to APOBEC3A with those provided by AlphaFold3 and found that there is good agreement between the systems (see Fig. 7). Specifically, the protein structure is highly conserved between the two systems, AlphaFold3 correctly identifies the nucleotide sequence as a hairpin, identifies the target cytosine in the hairpin loop, and properly flips the base into the active site. The major difference is visible only in the exact direction in which the hairpin stem extends into solvent. However, during MD simulations this direction is highly mobile, and the AF3 prediction is within its typical range of motion. Therefore, future works to dock hairpin loops to APOBEC3A may be made significantly simpler by using AlphaFold3 rather than performing classical dynamics on solvated loops and manually docking representative frames.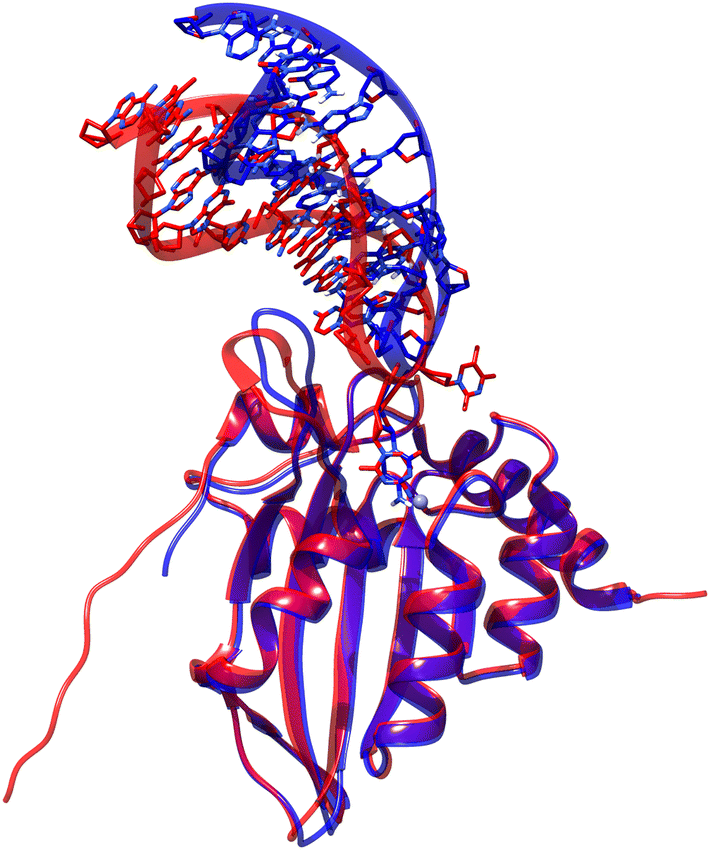 | ||
| Fig. 7 Comparison of manually docked ATC-containing sequence with APOBEC3A (blue) and results of AlphaFold3 provided with sequences of protein and nucleotide only (red). Notably, the protein structures are almost exactly conserved, the nucleotide sequence is correctly identified as a hairpin loop, and the target cytosine is correctly identified, flipped out of the loop, and docked into the active site of APOBEC3A. | ||
3.7. DNA forcefield selection may impact observed dynamics in free nucleotide simulations
We performed test simulations on the ATC hairpin loop using OL2152 and parmbsc144 forcefields to compare against OL15 used in this study. In comparing the resulting simulations, we find that OL21 produces dynamics similar to those from OL15, with comparable base-flipping behavior and nucleotide backbone motion. By contrast, the parmbsc1 simulation exhibits a highly constrained backbone with considerably less base-flipping behavior at the cytosine position. Interestingly, the 5′ thymine flips out of the hairpin loop early in the simulation and remains exposed for the duration. Meanwhile, the cytosine appears to pi-stack with the adenine in the hairpin and remain in that configuration throughout the simulation. Given this difference in base-flipping behavior and apparent rigidity of the phosphate backbone with parmbsc1, we are confident in our selection of the OL15 forcefield used for this study.4. Conclusions
Simulating hairpin loop motion in solvent can provide insights into the physical drivers of base-flipping events, providing a pathway to relate large-scale sequence data to APOBEC substrate preferences. In this work, we have generated a complete set of 3NT and 4NT hairpin loops, investigated their dynamics with relatively short timescale unbiased MD (100 ns per trajectory), and provided insights with both a manually and AF3-docked subset of these loops to demonstrate the physical connection between base-flipping and substrate binding. We find that 100 ns is sufficient to see multiple base-flipping events and to differentiate between hairpin loop sequences that are preferred by APOBEC3A. We note that choice of force field can affect these results, but that OL15 has been shown to successfully replicate base-flips in duplex DNA in previous work, even with nonstandard DNA bases.40,53 We also find that while MM/GBSA will not accurately compute the absolute free energies for the hairpin loops bound to APOBEC3A, their relative ordering by affinity is consistent with experimental biochemical data for hairpin loop preferences. The use of sugar puckering and RMSD provide guidance for the prediction of base-flipping events, which in turn correlate with binding energies of HPL to APOBEC3A. We find that while cytosine is required for deamination to uracil, it is not necessary for binding to APOBEC3A. We also find that base-flipping of a nucleotide in a hairpin loop must occur before binding in order to facilitate the subsequent enzyme catalysis.Author contributions
M. A. H. performed free-hairpin molecular dynamics simulations, complexed simulations, and analyses. P. G. performed additional complexed simulations and related analyses. A. S. B. initiated the project, contributed to the discussion of ideas and data, and provided biological expertise. A. R. W. supervised and directed the work and provided computational resources and funding.Data availability
CSV files containing RMSD and sugar puckering data for all free nucleotide simulations may be found at https://doi.org/10.5281/zenodo.13256543. Graphs of RMSD and sugar puckering data, including replicates, can be found in the ESI.†Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank the Wayne State University Grid for high performance computing resources. Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R35GM154949 to ARW, R21CA252858 and R21AI180183 to ASB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- L. Chelico, P. Pham, P. Calabrese and M. F. Goodman, Nat. Struct. Mol. Biol., 2006, 13, 392–399 CrossRef CAS PubMed
.
- L. Chelico, C. Prochnow, D. A. Erie, X. S. Chen and M. F. Goodman, J. Biol. Chem., 2010, 285, 16195–16205 CrossRef CAS PubMed
- Y. N. Lee, M. H. Malim and P. D. Bieniasz, J. Virol., 2008, 82, 8762–8770 Search PubMed
- S. G. Conticello, Genome Biol., 2008, 9, 229 Search PubMed
- M. A. Carpenter, E. Rajagurubandara, P. Wijesinghe and A. S. Bhagwat, DNA Repair, 2010, 9, 579–587 CrossRef CAS PubMed
- K. Shi, M. A. Carpenter, S. Banerjee, N. M. Shaban, K. Kurahashi, D. J. Salamango, J. L. McCann, G. J. Starrett, J. V. Duffy, O. Demir, R. E. Amaro, D. A. Harki, R. S. Harris and H. Aihara, Nat. Struct. Mol. Biol., 2017, 24, 131–139 CrossRef CAS PubMed
- M. Liu, A. Mallinger, M. Tortorici, Y. Newbatt, M. Richards, A. Mirza, R. L. Van Montfort, R. Burke, J. Blagg and T. Kaserer, ACS Chem. Biol., 2018, 13, 2427–2432 CrossRef CAS PubMed
- D. Ebrahimi, H. Alinejad-Rokny and M. P. Davenport, PLoS One, 2014, 9, 1–9 Search PubMed
- A. Rathore, M. A. Carpenter, O. Demir, T. Ikeda, M. Li, N. M. Shaban, E. K. Law, D. Anokhin, W. L. Brown, R. E. Amaro and R. S. Harris, J. Mol. Biol., 2013, 425, 4442–4454 CrossRef CAS PubMed
- E. C. Logue, N. Bloch, E. Dhuey, R. Zhang, P. Cao, C. Herate, L. Chauveau, S. R. Hubbard and N. R. Landau, PLoS One, 2014, 9, 1–10 CrossRef PubMed
- R. M. Kohli, R. W. Maul, A. F. Guminski, R. L. McClure, K. S. Gajula, H. Saribasak, M. A. McMahon, R. F. Siliciano, P. J. Gearhart and J. T. Stivers, J. Biol. Chem., 2010, 285, 40956–40964 CrossRef CAS PubMed
- J. A. Bohn, K. Thummar, A. York, A. Raymond, W. C. Brown, P. D. Bieniasz, T. Hatziioannou and J. L. Smith, Nat. Commun., 2017, 8, 1021 CrossRef PubMed
- Y. Feng, R. P. Love, A. Ara, T. T. Baig, M. B. Adolph and L. Chelico, J. Biol. Chem., 2015, 290, 27188–27203 CrossRef CAS PubMed
- A. Langenbucher, D. Bowen, R. Sakhtemani, E. Bournique, J. F. Wise, L. Zou, A. S. Bhagwat, R. Buisson and M. S. Lawrence, Nat. Commun., 2021, 12, 1602 CrossRef CAS PubMed
- L. Wong, A. Sami and L. Chelico, Nucl. Acids Res., 2022, 50, 12039–12057 CrossRef CAS PubMed
- R. Buisson, A. Langenbucher, D. Bowen, E. E. Kwan, C. H. Benes, L. Zou and M. S. Lawrence, Science, 2019, 364, eaaw2872 CrossRef CAS PubMed
- A. L. Brown, C. D. Collins, S. Thompson, M. Coxon, T. M. Mertz and S. A. Roberts, Sci. Rep., 2021, 11, 21008 CrossRef CAS PubMed
- R. Pecori, S. Di Giorgio, J. Paulo Lorenzo and F. Nina Papavasiliou, Nat. Rev. Genet., 2022, 23, 505–518 CrossRef CAS PubMed
- M. K. Wyllie, C. K. Morris, N. H. Moeller, H. A. M. Schares, R. Moorthy, C. A. Belica, M. J. Grillo, Ö. Demir, A. M. Ayoub, M. A. Carpenter, H. Aihara, R. S. Harris, R. E. Amaro and D. A. Harki, ACS Chem. Biol., 2025, 20, 117–127 Search PubMed
- H. Zahraee, Z. Khoshbin, F. Mohammadi, M. Mashreghi, K. Abnous and S. M. Taghdisi, Trends Anal. Chem., 2023, 165, 117160 CrossRef CAS
- K. Deiorio-Haggar, J. Anthony and M. M. Meyer, RNA Biol., 2013, 10, 1180–1184 CrossRef PubMed
- Y. Shen, S. V. Kuznetsov and A. Ansari, J. Phys. Chem. B, 2001, 105, 12202–12211 Search PubMed
- I. Prislan, H.-T. Lee, C. Lee and L. A. Marky, J. Phys. Chem. B, 2015, 119, 96–104 CrossRef CAS PubMed
- L. E. Xodo, G. Manzini, F. Quadrifoglio, G. Van Der Marel and J. Van Boom, Nucl. Acids Res., 1991, 19, 1505–1511 Search PubMed
- B. Nguyen and W. D. Wilson, J. Phys. Chem. B, 2009, 113, 14329–14335 Search PubMed
- N. E. Broude, K. Woodward, R. Cavallo, C. R. Cantor and D. Englert, Nucl. Acids Res., 2001, 29, e92 Search PubMed
- G. Wang and K. M. Vasquez, Nat. Rev. Genet., 2023, 24, 211–234 CrossRef CAS PubMed
- Nicy, D. Chakraborty and D. J. Wales, J. Phys. Chem. B, 2022, 126, 3012–3028 CrossRef CAS PubMed
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed
- M. Zgarbová, J. Šponer, M. Otyepka, T. E. I. Cheatham, R. Galindo-Murillo and P. Jurečka, J. Chem. Theory Comput., 2015, 11, 5723–5736 CrossRef PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- D. Sharma, K. Sharma, A. Mishra, P. Siwach, A. Mittal and B. Jayaram, Phys. Chem. Chem. Phys., 2023, 25, 7323–7337 RSC
- G. Kudo, T. Hirao, R. Harada, T. Hirokawa, Y. Shigeta and R. Yoshino, Sci. Rep., 2024, 14, 13508 CrossRef CAS PubMed
- C. Selvaraj, U. Panwar, D. C. Dinesh, E. Boura, P. Singh, V. K. Dubey and S. K. Singh, Front. Chem., 2021, 8, 595273 CrossRef PubMed
- J. C. Flores-Canales, N. A. Simakov and M. Kurnikova, Microsecond Molecular Dynamics Simulations of Diphtheria Toxin Translocation T-Domain pH-Dependent Unfolding in Solution, biorxiv, preprint, 2019 DOI:10.1101/572040.
- H. Kim and Y. Pak, J. Chem. Inf. Model., 2024, 64, 4511–4517 CrossRef CAS PubMed
- F. Pan, Y. Zhang, V. H. Man, C. Roland and C. Sagui, Nucl. Acids Res., 2018, 46, 942–955 CrossRef CAS PubMed
- Z. Sun and J. Z. Zhang, CCS Chem., 2021, 3, 1026–1039 Search PubMed
- Z. Sun, X. Wang, J. Z. H. Zhang and Q. He, Phys. Chem. Chem. Phys., 2019, 21, 14923–14940 Search PubMed
- X. Wang and Z. Sun, J. Chem. Inf. Model., 2019, 59, 2980–2994 CrossRef CAS PubMed
- Y. Z. Chen, V. Mohan and R. H. Griffey, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 1133–1137 Search PubMed
- R. Galindo-Murillo, J. C. Robertson, M. Zgarbová, J. Šponer, M. Otyepka, P. Jurečka and T. E. Cheatham, J. Chem. Theory Comput., 2016, 12, 4114–4127 CrossRef CAS PubMed
- I. Ivani, P. D. Dans, A. Noy, A. Pérez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goñi, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpí, C. González, M. Vendruscolo, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Meth., 2016, 13, 55–58 CAS
- T. Kouno, T. V. Silvas, B. J. Hilbert, S. M. D. Shandilya, M. F. Bohn, B. A. Kelch, W. E. Royer, M. Somasundaran, N. Kurt Yilmaz, H. Matsuo and C. A. Schiffer, Nat. Commun., 2017, 8, 15024 CrossRef PubMed
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. ONeill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed
- B. R. I. Miller, T. D. J. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333–2344 Search PubMed
-
P. S. Ho and M. Carter, in DNA Structure: Alphabet Soup for the Cellular Soul, ed. H. Seligmann, IntechOpen, Rijeka, 2011, ch. 1 Search PubMed
- J. A. Nakauma-González, M. Rijnders, M. T. Noordsij, J. W. Martens, A. A. Van Der Veldt, M. P. Lolkema, J. L. Boormans and H. J. Van De Werken, Cell Genom., 2024, 4, 100528 CrossRef PubMed
- M. Zgarbová, J. Šponer and P. Jurečka, J. Chem. Theory Comput., 2021, 17, 6292–6301 Search PubMed
- O. Love, R. Galindo-Murillo, M. Zgarbová, J. Šponer, P. Jurečka and T. E. I. Cheatham, J. Chem. Theory Comput., 2023, 19, 4299–4307 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00321k |
| This journal is © the Owner Societies 2025 |