
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the properties, types, and performance of atomic site catalysts in electrochemical hydrogen evolution reactions
M. Nur
Hossain
 a,
Lei
Zhang
*a,
Roberto
Neagu
a and
Shuhui
Sun
*b
a,
Lei
Zhang
*a,
Roberto
Neagu
a and
Shuhui
Sun
*b
aEnergy, Mining and Environment, National Research Council of Canada, Vancouver, BC V6T 1W5, Canada. E-mail: lei.zhang@nrc-cnrc.gc.ca
bInstitut National de la Recherche Scientifque (INRS), Center Énergie Matériaux Télécommunications, Varennes, QC J3X 1P7, Canada. E-mail: shuhui.sun@inrs.ca
First published on 21st February 2025
Abstract
Atomic site catalysts (ASCs) have recently gained prominence for their potential in the electrochemical hydrogen evolution reaction (HER) due to their exceptional activity, selectivity, and stability. ASCs with individual atoms dispersed on a support material, offer expanded surface areas and increased mass efficiency. This is because each atom in these catalysts serves as an active site, which enhances their catalytic activity. This review is focused on providing a detailed analysis of ASCs in the context of the HER. It will delve into their properties, types, and performance to provide a comprehensive understanding of their role in electrochemical HER processes. The introduction part underscores HER's significance in transitioning to sustainable energy sources and emphasizes the need for innovative catalysts like ASCs. The fundamentals of the HER section emphasizes the importance of understanding the HER and highlights the key role that catalysts play in HER. The review also explores the properties of ASCs with a specific emphasis on their atomic structure and categorizes the types based on their composition and structure. Within each category of ASCs, the review discusses their potential as catalysts for the HER. The performance section focuses on a thorough evaluation of ASCs in terms of their activity, selectivity, and stability in HER. The performance section assesses ASCs in terms of activity, selectivity, and stability, delving into reaction mechanisms via experimental and theoretical approaches, including density functional theory (DFT) studies. The review concludes by addressing ASC-related challenges in HER and proposing future research directions, aiming to inspire further innovation in sustainable catalysts for electrochemical HER.
 M. Nur Hossain | M. Nur Hossain is a Research Officer at the National Research Council of Canada. He earned his PhD in Chemistry and Materials Science from Lakehead University. After completing his PhD, he pursued a postdoctoral fellowship in the Department of Physical and Environmental Sciences at the University of Toronto Scarborough. Nur's research interests encompass Electrochemistry, Catalysis, Materials Science, and Nanotechnology. |
 Lei Zhang | Lei Zhang is a Senior Research Officer at the National Research Council Canada (NRC), Clean Energy Innovation Research Center, Canada. Lei's main research interests include Electrocatalysis and Advanced Materials for Energy Conversion and Storage, including PEM fuel cells, Supercapacitors, Li-ion batteries, Metal–air batteries, CO2 Capture and Electrolysis, and Water Electrolysis. She is a Fellow of the Royal Society of Chemistry (FRSC) and a Fellow of the Industry Academy of the International Artificial Intelligence Industry Alliance. |
 Roberto Neagu | Roberto Neagu is a senior researcher with the National Research Council of Canada's Clean Energy, Innovation Research Centre in Vancouver, British Columbia. He has a MASc in chemical processes engineering from the Polytechnic University of Bucharest, and a PhD in Electrochemistry from the National Polytechnic Institute in Grenoble. He works on additive manufacturing of electrochemical devices and leads the redox flow battery development at NRC. |
 Shuhui Sun | Shuhui Sun is a Full Professor at the Institut National de la Recherche Scientifique (INRS), center for Energy, Materials, and Telecommunications, Canada. He is a Fellow of both the Royal Society of Canada (RSC) and the Canadian Academy of Engineering. His current research interests focus on multifunctional nanomaterials for energy conversion and storage applications, including H2 fuel cells, lithium-metal batteries, metal-ion (Li+, Na+, Zn2+) batteries, metal–air batteries, solid-state batteries, etc. He is also interested in nanostructured photo- and electro-catalysts for H2 production, CO2 reduction, and water treatment, as well as in situ characterization. |
1. Introduction
1.1. Background
Given concerns over climate change and environmental impact from non-renewable fossil fuel consumption, exploring clean, sustainable alternatives is crucial. Hydrogen, with its high energy density, zero greenhouse gas emissions, and renewability, stands out as a promising substitute.1–8 It can be either directly burned as a fuel or utilized in fuel cells to generate clean electrical energy.9–13 Most hydrogen production relies on fossil fuels, causing issues like impurity, high costs, instability, and low efficiency, undermining sustainability and depleting fossil fuel reserves, while generating waste.14–23 Producing green hydrogen through non-polluting and eco-friendly methods is crucial. Electro-catalytic water splitting, utilizing electricity from renewable sources, offers several advantages, including high hydrogen purity, device simplicity, and renewability. Geothermal, sea-wave, waterfall, and wind power are vital renewable energy that can support electro-catalytic water splitting, thereby boosting the production of green hydrogen.24–29 However, water electrolysis is both energy-intensive and slow, requiring efficient electrocatalysts to lower activation energy and enhance conversion efficiency in the cathodic reaction. Therefore, the use of efficient electrocatalysts is crucial to reduce the overpotential and overcome activation energy, enabling a cost-effective and energy-efficient HER in water splitting processes. Fig. 1 illustrates the production and utilization of hydrogen. Hydrogen can be synthesized through water electrolysis powered by renewable energy sources and facilitated by efficient electrocatalysts. The generated hydrogen serves as a versatile green fuel, suitable for various applications such as fuel cells, aircraft propulsion, industrial processes, household energy needs and energy storage solutions.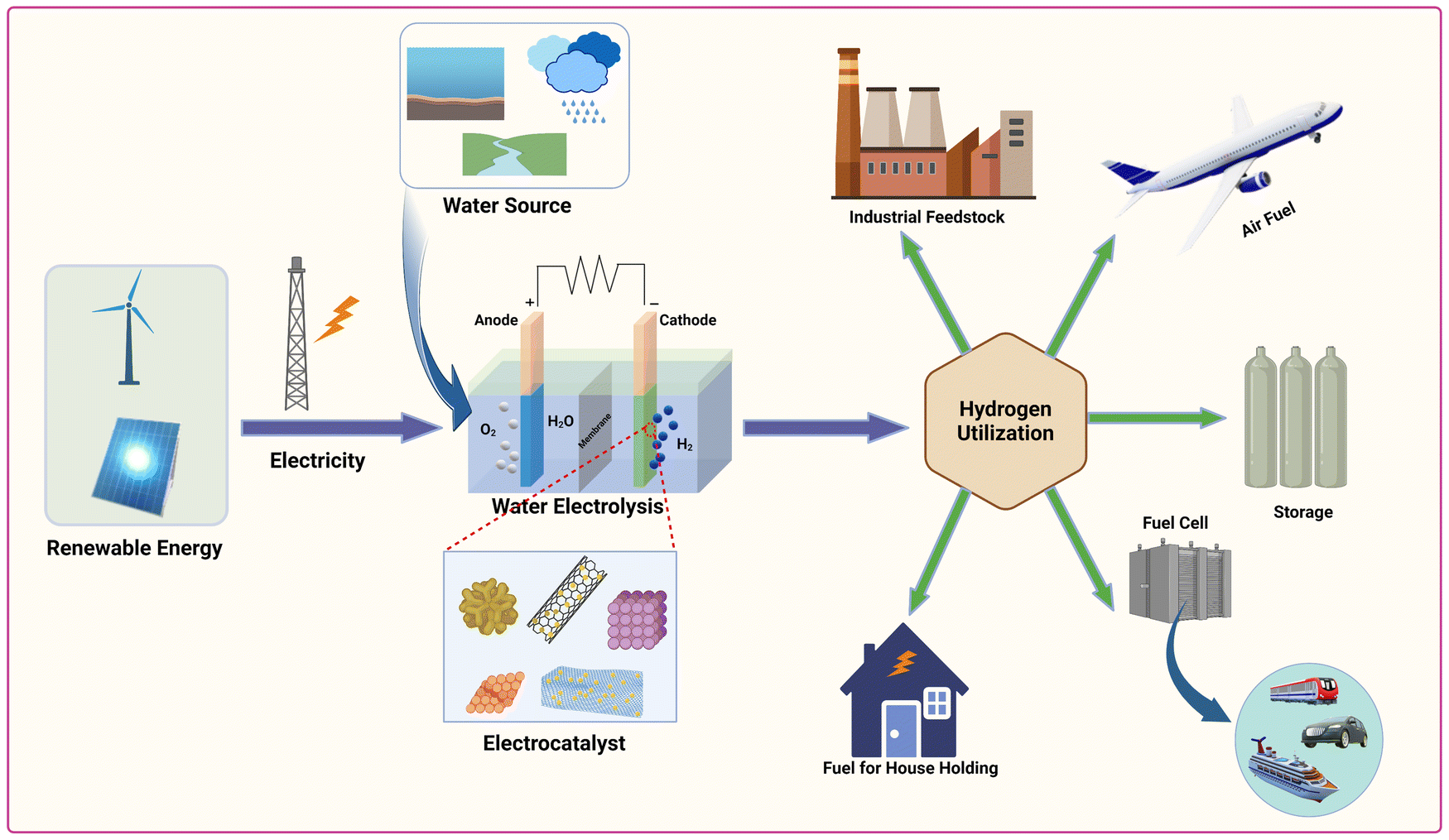 | ||
| Fig. 1 Illustration of sustainable green hydrogen (H2) production and utilization processes. | ||
Ideally, an HER electrocatalyst should possess desirable traits like low cost, ease of recycling, facile synthesis, high durability, and robust activity across a broad pH range. While Pt group metals (PGMs) are considered as benchmark catalysts for HER due to their remarkable catalytic performance, their large-scale application is limited by drawbacks such as high cost, natural scarcity, limited durability, and susceptibility to poisoning. In contrast, non-PGM catalysts, such as Ir, Co, Fr, Cu nanodendrites, Co–C3N4, Mo2C, Ni2P, MoS2/rGO show significant potential for advancing sustainable energy development.30–37 In this regard, various strategies have been proven effective in enhancing the activity and stability of non-PGM catalysts.38–40 These include introducing crystal defects to enhance conductivity, increasing porosity for efficient charge and mass transfer, promoting electrical contact between the electrode and electrolyte, exposing abundant catalytic active sites, and optimizing specific surface area. Furthermore, the size of the metal plays a crucial role in determining its physicochemical properties and subsequent electrocatalytic performance.
1.2. The current status of traditional catalysts
In recent years, extensive research has focused on various earth-abundant and non-PGM catalysts for HER, such as alloys, chalcogenides, borides, carbides, phosphates, borophosphates, phosphites, phosphides, phosphonates, and intermetallics, and metal-free electrocatalysts.38–49 Despite significant progress, the catalytic performance of these materials still requires improvement in both activity and stability. Transition metal compounds, such as nitrides, sulfides, selenides, and phosphides, have emerged as promising alternatives to precious metal catalysts for HER. Among these, transition metal phosphides have attracted significant research attention due to their unique metalloid properties and superior electrocatalytic performance. Researchers are actively exploring their potential as efficient catalysts for HER in water splitting processes.50 Moreover, recent research has highlighted the critical status of certain elements, considering their availability and usage in the electronic industry (Fig. 2).51 Elements such as Zn, Ga, Ge, As, In, Te, and Ag are at risk of being depleted entirely within the next 100 years. Additionally, Co and Cr are facing an increasing threat due to their growing usage. Elements such as Li, Mg, P, Mn, Ni, and Cu are at risk of limited availability, raising concerns about future supply.52,53 These ‘endangered elements’ not only show excellent electrocatalytic activity but also pose sustainability challenges. | ||
| Fig. 2 Aster plot depicting endangered element species and their potential applications as electrocatalysts for water splitting. Reproduced with permission from ref. 51 Copyright 2022, Elsevier. | ||
In response to these challenges, substantial research efforts have been devoted to developing electrocatalysts using advanced nanomaterials. The focus has been put on low-cost, non-PGM alternatives, including transition metals, metal alloys, core–shell structures, transition metal oxides, carbides, nitrides, phosphides, dichalcogenides, borides, and metal-free composite catalysts.54–64 Research efforts have explored various synthesis protocols and approaches to enhance the intrinsic activity of each active site and increase the number of active sites for advanced HER catalysts. Studies have also investigated the relationship between catalytic activity, morphology, structure, composition, and synthesis methods, aiming to optimize the design and fabrication of nanostructured catalysts. Practical design strategies and advancements in nanostructured catalysts offer significant promise for the development of efficient and cost-effective electrocatalysts, propelling progress toward a sustainable energy future. Specifically, the size of the metal used as a catalyst significantly influences its physicochemical properties and electrocatalytic performance.65–67 Reducing the metal size from the nanometer scale to the single atom level not only enhances the geometric effect, maximizing atom utilization, but also increases the surface-to-volume ratio and active site concentration, both of which benefit HER activity.68 Consequently, in recent advancements in electrocatalysis, ASCs have gained increasing significance, particularly for HER applications. The chronological progression of ASCs for HER showcases their journey from foundational concepts to state-of-the-art technologies, propelled by advancements in materials science, innovative synthesis techniques, and computational methodologies (Fig. 3A).69–83 Each atom in an ASC serves as an active site, offering exceptional catalytic efficiency and maximizing atom utilization, which in turn enhances both catalytic activity and efficiency (Fig. 3B and C). Furthermore, the precise atomic dispersion inherent to ASCs enables controlled reaction environments, reducing energy requirements (overpotentials) and improving selectivity towards specific reaction pathways. Additionally, ASCs demonstrate remarkable durability and stability during electrocatalysis. This robustness is primarily due to the strong anchoring of individual atoms to the support material, which effectively mitigates the risk of atom aggregation or detachment under operational conditions, thereby preserving the catalyst's integrity and effectiveness. However, it is important to note that reducing the size to the atomic level does not always result in favorable changes to the electronic structure for catalytic applications. While discrete energy levels can replace the continuous energy spectrum, this modification does not always lead to an enhancement in catalytic performance. Computational quantum chemistry has been integral in providing insights into the catalytic mechanisms involved in ASCs and their interaction with HER processes. By investigating the relationship between catalytic activity and physical properties such as morphology, structure, and composition, researchers have been able to tailor the design of ASCs for optimized HER performance. Despite these advancements, transitioning to atomic levels introduces complexities in the electronic structure that may not always be beneficial for catalysis, highlighting the need for meticulous evaluation of each ASC type. This refined understanding underscores the growing interest in metal-based ASCs as potential candidates for electrocatalytic HER processes. The discussions and findings presented in this review offer valuable insights and open up possibilities for future breakthroughs in ASCs development for HER.
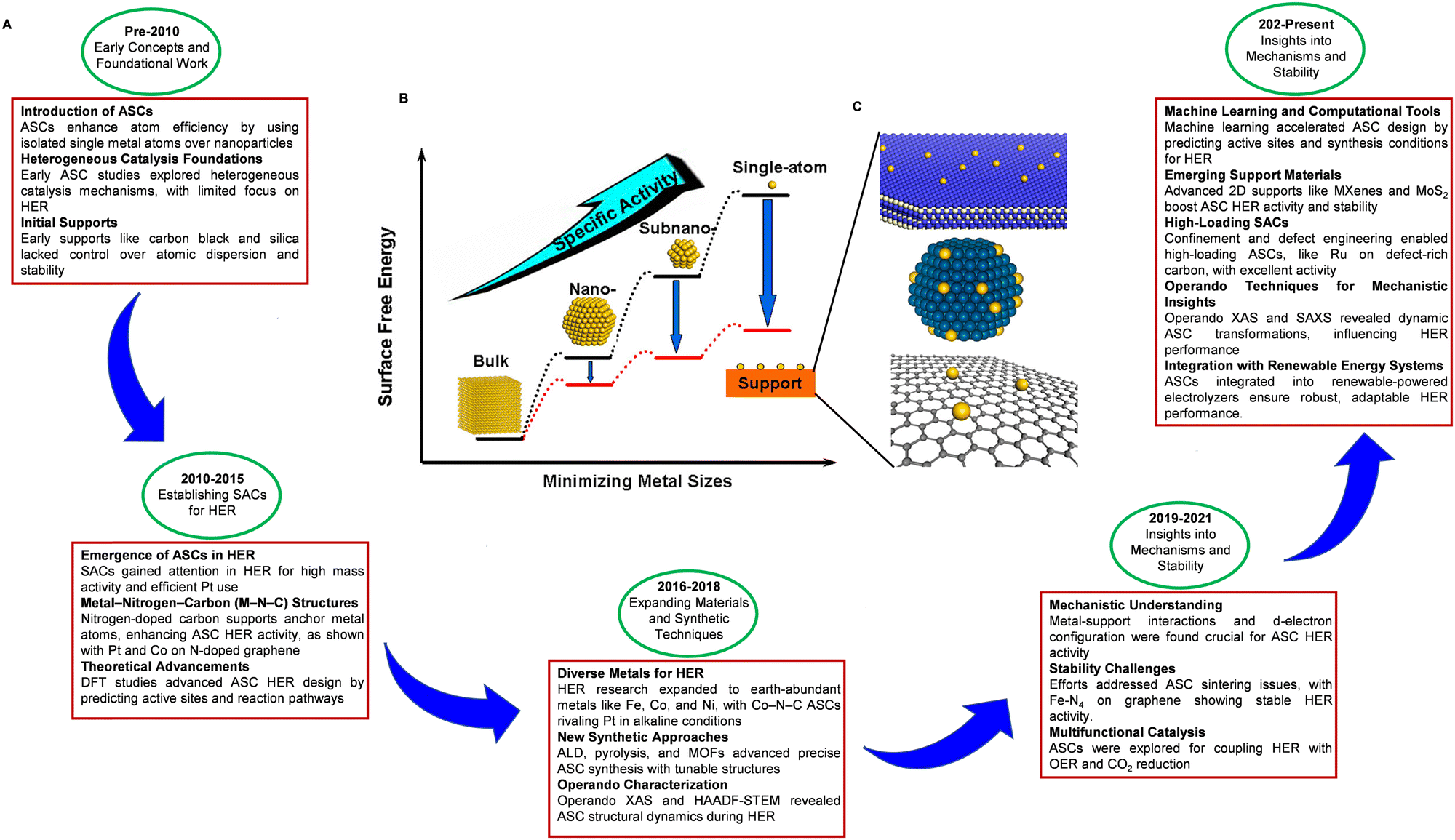 | ||
| Fig. 3 (A) Chronological development of ASCs for HER highlights the evolution from early concepts to cutting-edge technologies, driven by advances in materials science, synthetic methods, and computational tools.69–83 (B) Schematic representation illustrating how surface free energy and specific activity per metal atom vary with metal particle size. (C) Schematic diagrams illustrating the support effects that stabilize the ASCs: metal single atoms anchored to metal oxide (top), metal surfaces (middle), and graphene (bottom). Reproduced with permission from ref. 84 Copyright 2013, American Chemical Society. | ||
2. Fundamentals of HER
2.1. Mechanism of HER
The electrochemical HER is a fundamental step in water electrolysis, occurring at the electrode surface, where hydrogen ions are reduced to produce hydrogen gas. Influenced by factors like electrode material properties, electrolyte pH, and applied potential, this complex reaction follows a two-step process.28,85,86 It involves the adsorption of hydrogen from a hydronium ion (in acidic medium) or a dissociated water molecule (in alkaline medium), followed by H2 desorption.65,87 The adsorption step forms crucial intermediate species on the electrode surface, impacting the overall reaction. The reduction step, influenced by the electrode material and environment, is a key determinant of HER rate. It is a two-electron transfer reaction that necessitates active catalysts to lower the energy barriers in each step. The rate of HER is primarily determined by the adsorption step, which is dependent on the surface area, composition, and morphology of the electrode material. The commonly accepted pathways for the HER involve the adsorption/desorption of a hydrogen intermediate (H*) through either the Volmer–Heyrovsky or the Volmer–Tafel mechanism.88–91 The pathways for the HER under acidic and alkaline conditions are shown in Fig. 4.92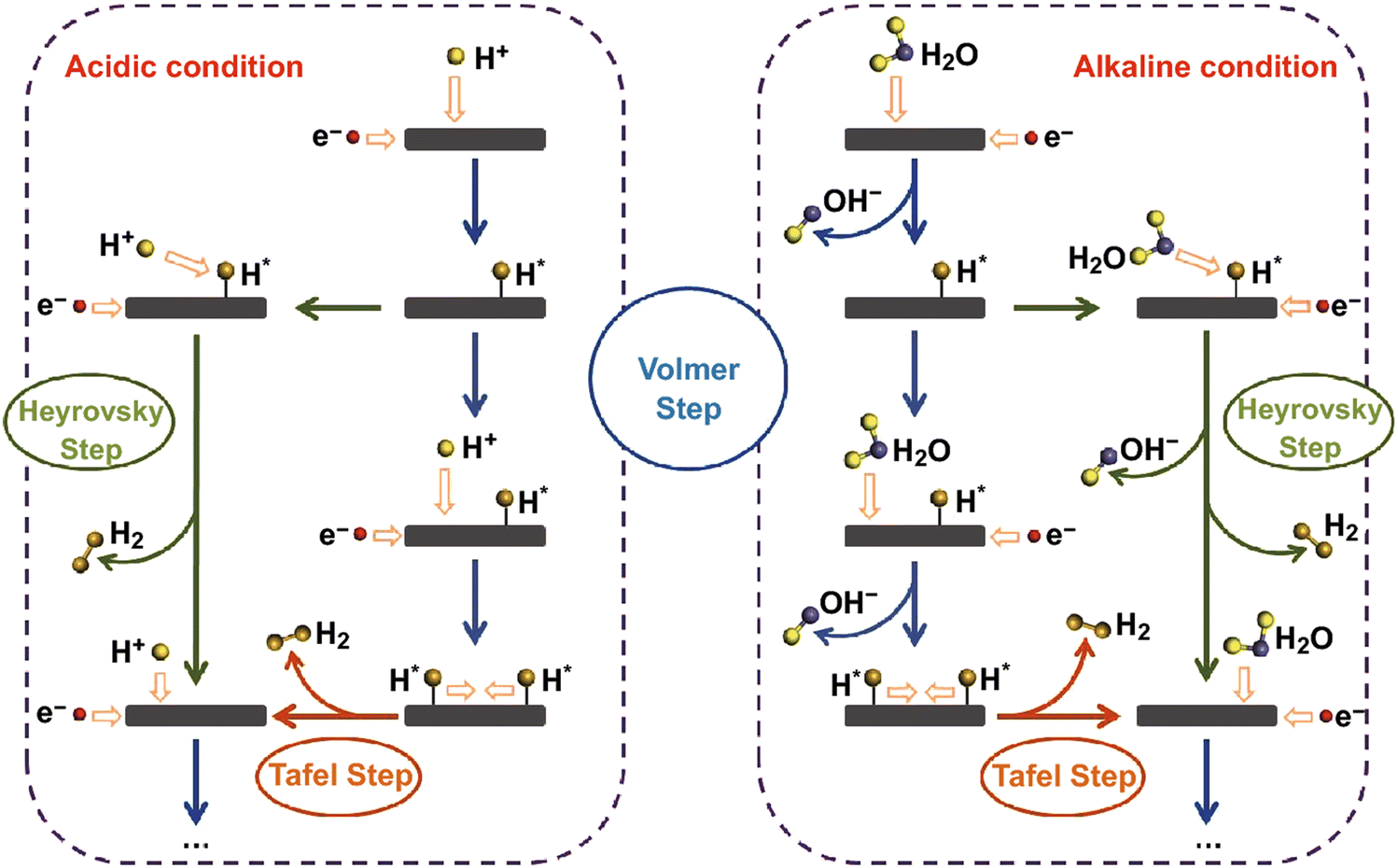 | ||
| Fig. 4 Schematic representations of pathways for the hydrogen evolution reaction under both acidic and alkaline conditions. Reproduced with permission from ref. 92 Copyright 2018, Springer. | ||
The reaction pathway in the HER is determined by the Tafel slope, obtained from a Tafel plot derived from the HER polarization curve.86,93,94 A Tafel slope around 29 mV dec−1 indicates a rapid and facile Tafel pathway, while a high density of active sites favors the Volmer–Tafel mechanism.94 Conversely, a low coverage of adsorbed hydrogen atoms leads to the Volmer–Heyrovsky mechanism. However, the Tafel slope does not solely represent the catalyst's intrinsic activity and can change with catalyst loading or hydrogen coverage.95,96 The pH of the electrolyte plays a significant role, affecting the solubility of hydrogen ions and the required reduction potential. The use of acidic or alkaline electrolytes can enhance HER performance by creating a favorable environment for proton adsorption and reduction. The reaction pathways for the HER in the acidic solution are represented as:92
| Volmer step: H+ + e− → H* | (1) |
| Heyrovsky step: H* + H+ + e− → H2 | (2) |
| Tafel step: H* + H* → H2 | (3) |
The HER in acidic media involves two distinct steps at the active site of the electrocatalyst surface. In the first step, a proton combines with an electron to form an adsorbed hydrogen intermediate, referred to as the Volmer step. Subsequently, the second step occurs through either electrochemical desorption, where the adsorbed hydrogen reacts with an electron and a proton to liberate H2, known as the Heyrovsky step, or through chemical desorption, where two adjacent adsorbed hydrogen intermediates undergo a chemical reaction to generate H2, known as the Tafel step. The asterisk (*) denotes the active site on the electrocatalyst surface, where these reactions take place. The reaction pathways for the HER in the basic/neutral solution are represented as:92
| Volmer step: H2O + e− → H* + OH− | (4) |
| Heyrovsky step: H2O + e− + H* → H2 + OH− | (5) |
| Tafel step: H* + H* → H2 | (6) |
The proposed reaction mechanism for the HER in alkaline electrolyte can be described as follows. In the first step, known as the Volmer step, a water molecule interacts with an electron instead of a proton on the active surface of the catalyst, resulting in the formation of an adsorbed hydrogen atom. This introduces an additional step of water dissociation to generate protons, which subsequently adsorb onto the active site. In the second step, there are two possible pathways. The first is the Heyrovsky reaction, where the adsorbed hydrogen atom reacts simultaneously with a water molecule and an electron, leading to the production of a hydrogen molecule. The second pathway is the Tafel reaction, which involves the coupling of adjacent adsorbed hydrogen intermediates, ultimately releasing H2. These steps collectively contribute to the overall HER process in an alkaline electrolyte. Understanding these mechanisms is crucial for designing efficient electrocatalysts for HER applications.
2.2. Role of catalysts
The efficiency of the HER is significantly impacted by catalyst properties, including surface morphology, composition, electrochemically active surface area, and the presence of active sites. The surface area and composition of the catalyst material directly influence proton adsorption rates and overall reaction efficiency. Utilizing catalysts with larger surface areas and specific compositions can enhance HER performance.20,97–101 The Sabatier principle suggests that an optimal catalytic surface should bind reaction intermediates neither too weak nor too strong.102 This principle can be quantified using calculated or measured adsorption energies for relevant intermediates at specific active sites on the surface. Fig. 5A illustrates the relationship between theoretical adsorption energies and experimental HER activity data for pure metal surfaces.103 The trends in measured HER activity can be explained by the hydrogen binding energy (ΔEH) as a descriptor, determined through density functional theory (DFT) calculations. According to this approach, the ideal electrocatalytic sites for HER should bind *H slightly weaker (around 0.09 eV) than those of Pd, Rh, or Pt. The electronic properties of metal surfaces can be modulated through methods such as preparing bulk alloys or selectively positioning atomic layers of solute metals at the surface to form overlayers, surface alloys, or subsurface alloys.104–106 For instance, the pseudocapacitive behavior of electrocatalysts plays a crucial role in the performance of the HER because hydrogen typically needs to be adsorbed on the electrocatalyst surface before the HER occurs.97,107 In this sense, most potential HER electrocatalysts exhibit good pseudocapacitive behavior in the hydrogen adsorption region. An efficient pseudocapacitor should desorb all the adsorbed hydrogen in the reverse scan, maximizing its efficiency. While in an energy storage system, adsorbed hydrogen is returned back to the electrolyte, the HER is an irreversible process where the overaccumulation of adsorbed hydrogen leads to hydrogen evolution. This similarity between the HER and a hydrogen-based pseudocapacitor is noteworthy. The adsorption of hydrogen, also known as underpotential deposition of hydrogen, is a prerequisite for the HER, making studies on the pseudocapacitive behavior prior to the HER potential valuable for understanding electrocatalytic activity. Comparing the pseudocapacitive behavior of Pt-based electrocatalysts at positive potentials before the HER potential reveals that ideal behavior, represented by a rectangular shape in cyclic voltammograms (CVs), correlates directly with better HER electrocatalytic activity and lower overpotential (Fig. 5B).107 The pseudocapacitive behavior in the H adsorption potential region provides crucial information about the electrocatalysts' ability to efficiently adsorb and desorb hydrogen with high coulombic efficiency, indicating an appropriate H adsorption energy. The voltammogram for near surface alloy (NSA) with 1 mL Cu initially deposited shows significantly higher hydrogen evolution activity than Pt(111) in Ar-saturated electrolytes, indicating clearer activity trends with minimal experimental interference (Fig. 5C). Additionally, examining the rate capability through CVs at different potential scan rates offers insights into the HER rate capability, characterized by the Tafel slope. Pseudocapacitance investigation provides insights into the adsorption capabilities of electrocatalysts without considering factors like gas evolution, mechanical perturbation, and the mechanical stability of the electroactive film, which are involved in the harsh conditions of the HER.91,107,108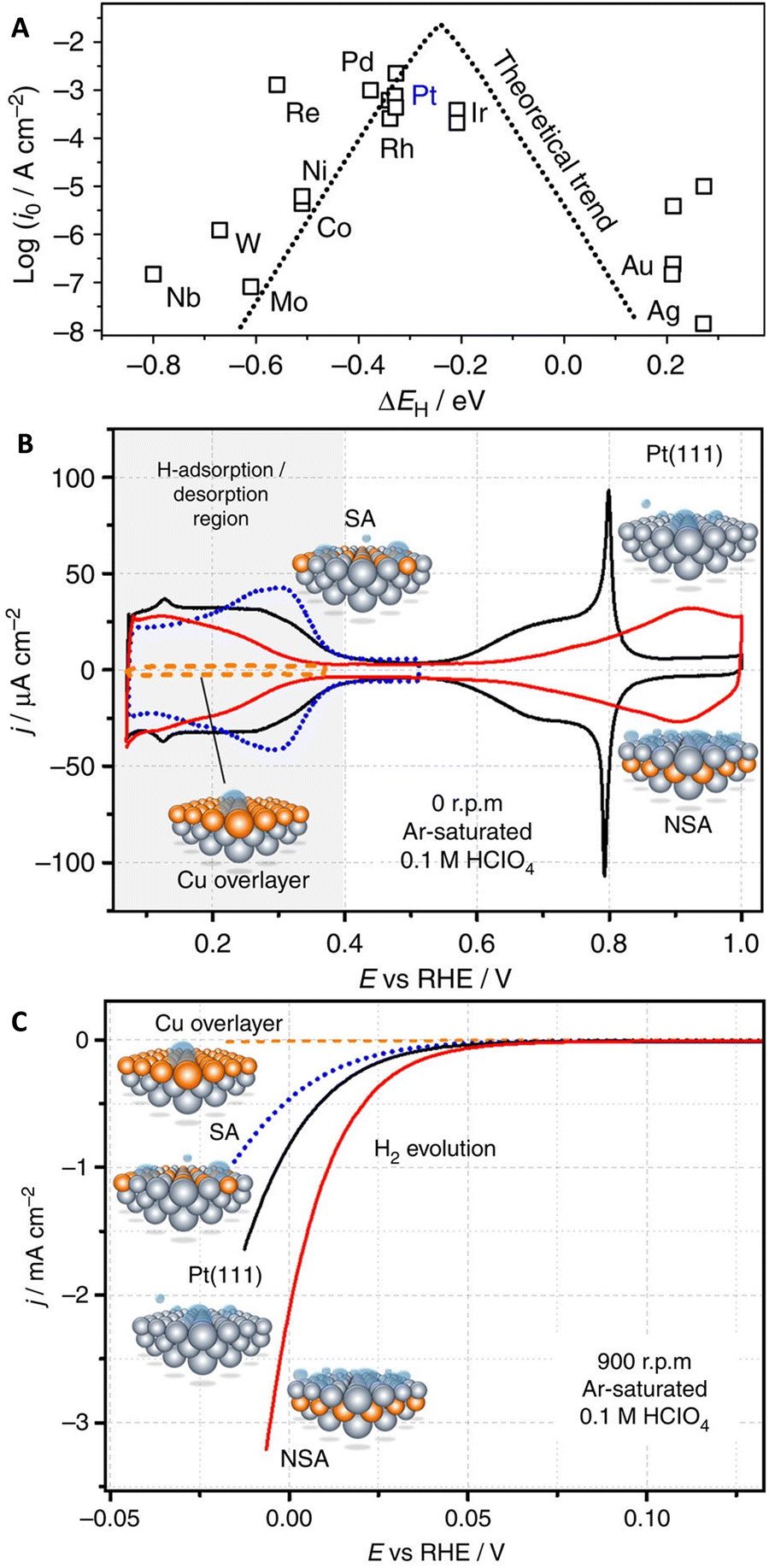 | ||
| Fig. 5 (A) Experimental HER activity in terms of exchange current density (log(i0)) for various metal surfaces against the calculated *H chemisorption energy (ΔEH). The results of a simple theoretical kinetic model are also depicted as a dotted line. (B) CVs and (C) RDE voltammetry for Cu overlayer, Pt(111), surface alloy (SA), and NSA in Ar-saturated 0.1 M HClO4, illustrating the correlation between HER activity and the position of Cu atomic layers relative to the topmost Pt layer. Reproduced with permission from ref. 107 Copyright 2016, Nature Publishing Group. | ||
The performance of HER is influenced by various surface processes, which highlights the significance of electrocatalyst morphology, even in well-defined catalysts like Pt.109 Research indicates that particle morphology, particularly at the nanoscale, significantly impacts HER performance due to its effect on surface properties and reaction dynamics.110–112 This impact is observed not only in traditional catalysts but also in ASCs, where the local environment of the active sites can profoundly affect activity. Consequently, defect engineering has emerged as a highly promising approach for improving HER performance, potentially offering greater benefits than simply enhancing morphology to increase the availability of edge sites. Precisely manipulating of these defects can optimize the density and functionality of active sites, directly boosting electrocatalytic activity. This approach aligns with findings that electrocatalytic performance is closely related to the number of accessible and efficient active sites, which can be strategically increased through targeted defect engineering within the catalyst structure.113,114 Furthermore, the high surface area and excellent electrical conductivity of the catalysts contribute to enhanced HER performance.115,116 Increasing the electrochemically active surface area (ECSA) of catalysts can lead to an increase in the current density of the HER. Studies on metal-based HER catalysts such as Cu, Ag, and Au nanodendrites (NDs) have shown that higher ECSA correlates with higher HER activity.32,117,118 For instance, Cu NDs demonstrated a significantly higher double-layer capacitance (12.6 mF cm−2) compared to Cu wire (0.043 mF cm−2) (Fig. 6A and B). Similarly, Ag NDs exhibited a much higher current density and approximately 10 times higher ECSA compared to Ag wire (Fig. 6C). The synthesized Au NDs also showed a high ECSA (826.17 mC) compared to polycrystalline Au wire (5.06 mC).118 The superior catalytic activity of Au NDs was confirmed by their higher current density (10 mA cm−2) at a lower overpotential (67 mV) compared to Ag and Cu NDs (Fig. 6E and F). These findings suggest that the increased ECSA contributes to the enhanced HER performance of the synthesized Au NDs. The presence of abundant O vacancies and Mn3+ active sites in ultrathin MnO2 nanosheets has been found to significantly enhance the HER activity by promoting conductivity and favorable H+ adsorption.119,120 Additionally, the introduction of boron into the N-doped graphene matrix, forming borocarbonitrides (BxCyNz), has been shown to further enhance electron transfer by modulating the band structure and increasing the number of active sites BC2N7 and B2C5N3.120,121 The core–shell structure with multiple active sites has also been identified as a key factor contributing to the high catalytic performance of HER in alkaline media.122 Therefore, key descriptors, including Gibbs free-energy changes (ΔGH) value, exchange current density, overpotential, faradaic efficiency, ECSA, mass activity, turnover frequency, and stability, are commonly employed to evaluate the overall performance of HER.
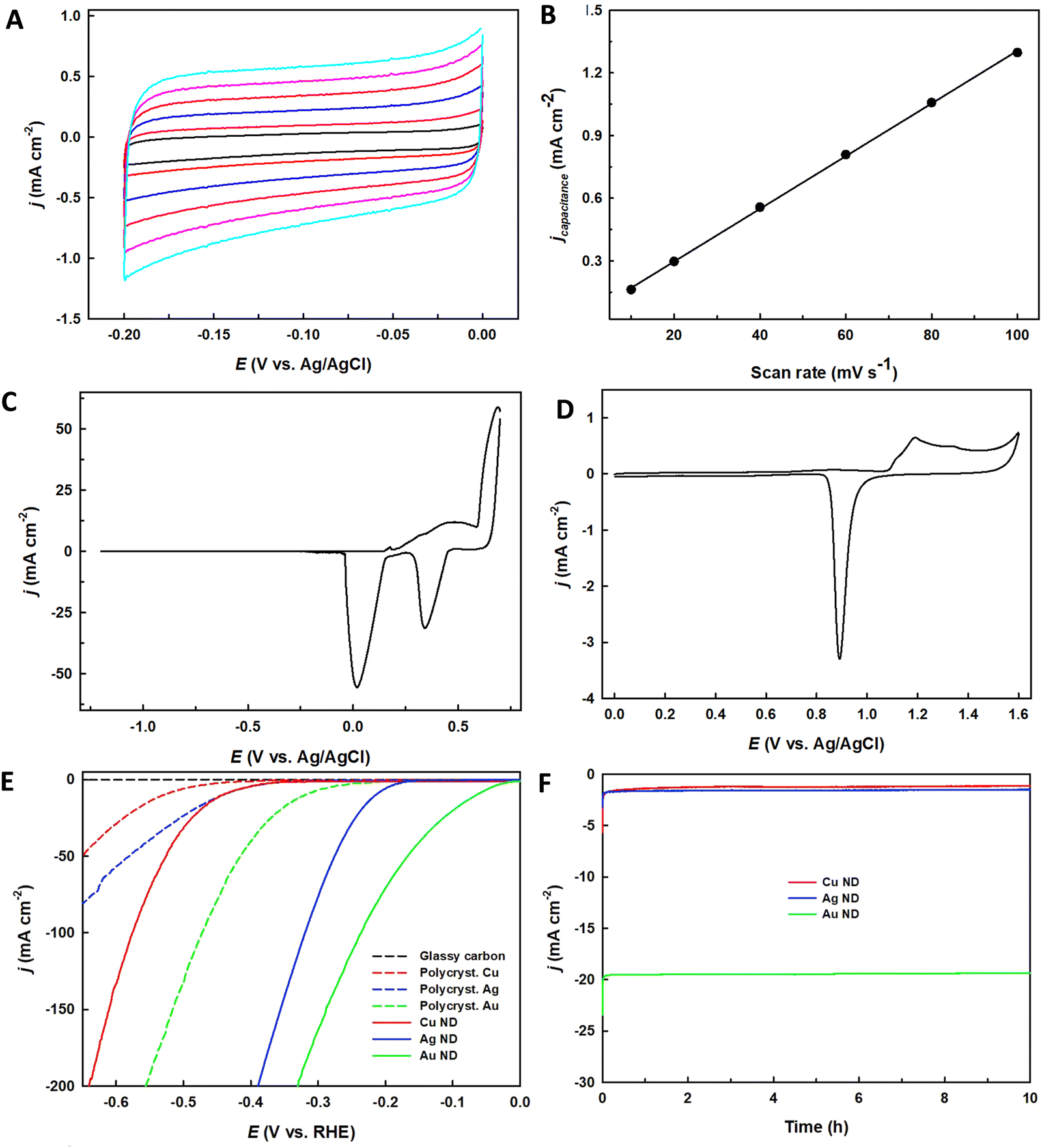 | ||
| Fig. 6 ECSA measurements of metal nanodendrites (NDs). (A) CVs at the different scan rates and (B) the current density vs. the applied scan rate of Cu NDs recorded in 0.1 M HClO4 solution. (C) CVs recorded in 1.0 M KOH for Ag NDs and (D) CVs of Au NDs recorded in 0.1 M H2SO4. (E) The LSVs recorded at 20 mV s−1 on glassy carbon plate, various polycrystalline metal wires and synthesized ND electrodes, (F) Chronoamperometry (CA) recorded at an overpotential of 0.1 V for the formed metal NDs in 0.5 M H2SO4 solution. Reproduced with permission from ref. 118 Copyright 2021, Elsevier. | ||
3. Properties of ASCs
3.1. Atomic structure of ASCs
ASCs offer great potential for the efficient use of metal resources and achieving atomic economy. With identical active sites, ASCs enable selective catalysis of specific reactions. Furthermore, they exhibit remarkable atom utilization efficiency, approaching a 100% utilization rate. This enhances catalytic activity per atom while reducing the consumption of metal resources.123,124 However, while ASCs present a seemingly simpler structural model, characterizing their active sites introduces significant challenges. These sites are often stabilized by surface defects on supports, such as carbon, metal oxides, or other inorganic materials, complicating direct observations of their atomistic structures. Unlike traditional metal-based catalysts, ASCs require sophisticated characterization techniques. Methods such as high-angle annular dark-field scanning transmission electron microscopy, X-ray absorption spectroscopy, and advanced computational modeling are crucial for uncovering the atomistic details of ASCs. These techniques help elucidate the structures of ASCs, enhancing our understanding of their relationship with catalytic performance. The isolated nature of the metal atoms in ASCs allows for a high surface area to volume ratio, leading to their high activity in catalyzing reactions. Moreover, ASCs exhibit unique adsorption properties due to their discrete electronic structure, offering highly tunable and selective adsorption of reactive molecules like H+. By customizing the coordination environment and substrates, the adsorption behavior on ASCs can be precisely controlled, which will positively influence the catalytic selectivity and activity. The electronic structure of ASCs, influenced by factors such as electronegativity and ionic radius, directly impacts adsorption behavior and catalytic performance. DFT calculations play a crucial role in predicting atomic-level properties and catalytic mechanisms of ASCs, allowing for a deeper understanding of their catalytic behavior. By leveraging DFT calculations, the entire reaction cycle, energy barriers for each elementary step, and the electronic structures of active sites can be analyzed, unveiling the structure–performance relationship. Computational modeling using DFT calculations has extensively contributed to the study of ASCs, revealing explicit insights into the reaction processes.125–129 The adsorption patterns of Pt single atoms on the C12A7 support are investigated through initial DFT calculations, focusing on the energetics of various adsorption models. Four distinct adsorption sites near a cavity structure on the C12A7 (001) surface are analyzed, along with the corresponding adsorption energies (Eads) of these anchored Pt single atoms (Fig. 7A–C).130 Position 3 emerges as the most stable, boasting an Eads of −9.53 eV, significantly surpassing the cohesive energy of bulk Pt (−5.85 eV). This underscores the thermodynamic stability of the anchored Pt single atom against sintering, preventing aggregation and the formation of Pt nanoparticles.131,132 The formation of Pt dimers and trimers is ruled out due to elevated dimerization and trimerization energies. In configuration 3, the Pt single atom is ensnared in the middle of a cavity by two exposed oxygen ions, representing a unique arrangement compared to other positions. The surface cavity with unsaturated oxygen ions of C12A7 is identified as providing a distinctive structure for securely confining the Pt single atoms. Consequently, the theoretical modeling suggests the potential fabrication of thermally stable, atomically dispersed Pt on the surface of C12A7.133–135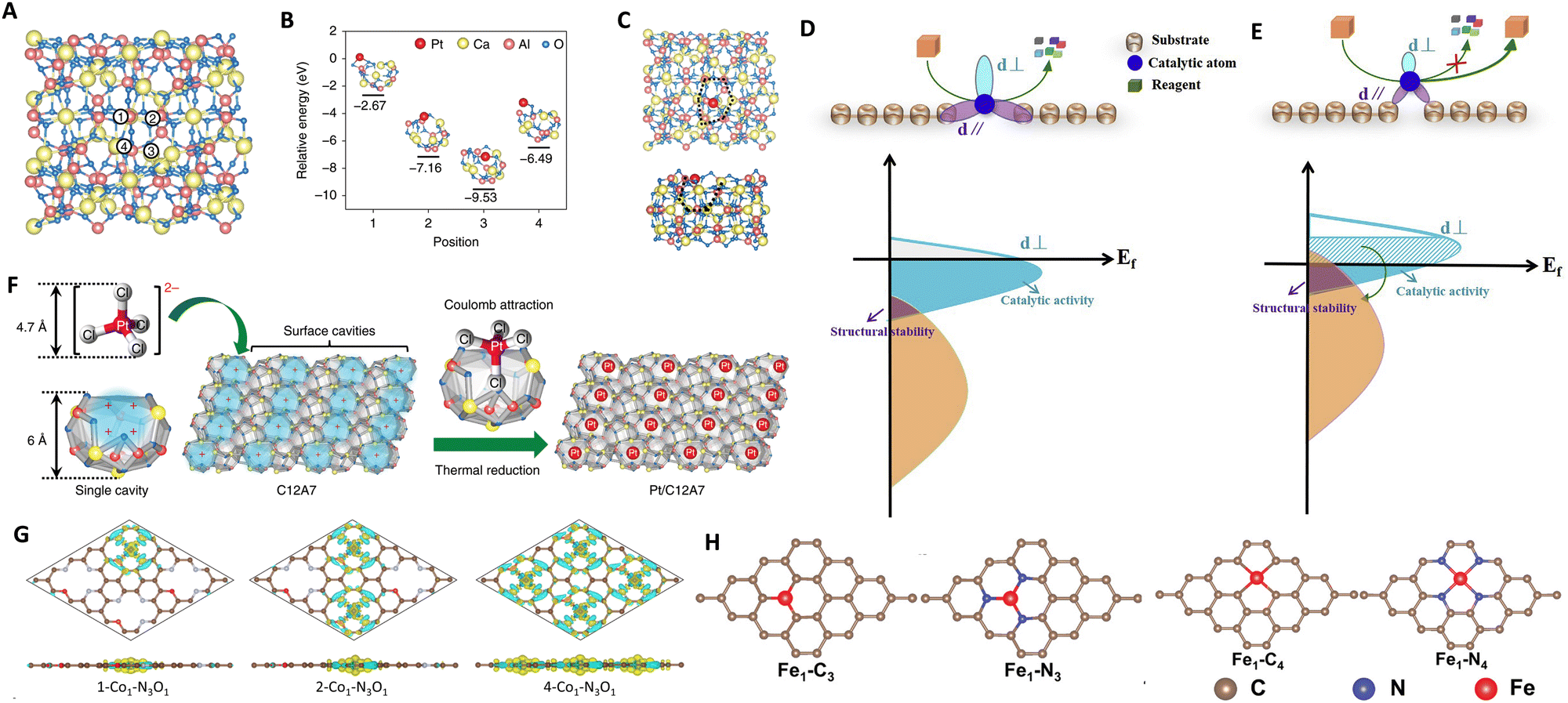 | ||
| Fig. 7 Computed adsorption models for Pt/C12A7 synthesis. (A) Illustration of adsorption sites and (B) energetic characteristics of Pt single atoms adsorbed on the (001) surface of C12A7 as determined by DFT calculations. (C) Top and side views illustrating the configurations of the most stable Pt single atom anchored on the surface. Black dotted lines highlight the open mouths of surface cavities. Reproduced with permission from ref. 130 Copyright 2020, Nature Publishing Group. Depiction of the competitive relationship between (D) catalytic activity and (E) structural stability of ASCs. Reproduced with permission from ref. 135 Copyright 2019, Elsevier. (F) Effective stabilization of single Pt atoms through the interaction between negatively charged [PtCl4]2− ions and positively charged surface cavities of C12A7. Reproduced with permission from ref. 130 Copyright 2020, Nature Publishing Group. (G) Variations in the electronic structure of Co ASC at different densities. Reproduced with permission from ref. 136 Copyright 2023, Nature Publishing Group. (H) Geometric configurations of Fe ASC across four distinct coordination environments. Reproduced with permission from ref. 137 Copyright 2022, Wiley-VCH. | ||
In a study conducted by Wang et al., a rational description was developed to understand the structural stability and catalytic activity of ASCs.135 The researchers demonstrated that the partial electrons, represented in purple, with the same orbital symmetry as the substrate atoms could chemically bind to them, resulting in a loss of catalytic ability. By adjusting the relative positions between the active atom and the substrate, the distribution of free electrons near the Fermi level could be tuned to simultaneously optimize stability and catalysis, as depicted in Fig. 7D and E. The competitive distribution of free electrons near the Fermi level was identified as a crucial factor that should be considered when optimizing both the structural stability and catalytic activity of ASCs. Experimental synthesis of ASCs for various reactions has demonstrated their tremendous potential in catalysis.138–142 The process for preparing the single-atomic Pt-loaded C12A7 catalyst is depicted in Fig. 7F.130 The Pt/C12A7 catalyst was synthesized using a simple wet impregnation method with K2PtCl4 as the precursor. C12A7, possessing a lattice framework with a positive charge [Ca24Al28O64]4+, has its positive charge balanced by anions within subnanometer cages. Upon reduction, the atomic Pt species are expected to be directly anchored to the surface cavities of C12A7, as predicted by the DFT results (Fig. 7A–C). Despite their exceptional catalytic performance, the intrinsic factors governing ASCs' activity remain unclear. Optimizing the atomic structure of ASCs, including the arrangement of atoms within the catalyst material, is crucial for enhancing their catalytic performance. This can be achieved through careful selection of metal atoms, support materials, and synthesis methods, allowing for precise control over properties such as electronic structure and reactivity. A comprehensive understanding of the atomic structure of ASCs is essential for maximizing their catalytic performance in specific applications. The study of Co ASCs reveals how charge density and changes in electronic structure, due to interactions between active sites, significantly influence their properties and optimize structural performance.136Fig. 7G illustrates the variation in charge distribution across different densities of Co1–N3O1 sites on the catalyst's support. With an increase from one to four Co1–N3O1 sites, there is a noticeable enhancement in charge connectivity across the support, facilitating a redistribution of charge. This effect is quantitatively supported by Bader charge analysis, which indicates a decrease in the average charge transfer from Co atoms as the density of Co1–N3O1 sites increases. This observation aligns with the results from XAFS and XPS, confirming that the electronic structure of Co atoms is adjustable through charge redistribution in densely populated arrays. Further, the analysis extends to the impact of local coordination environments on the catalytic performance of metal ASCs.137,143,144 As demonstrated in Fig. 7H, four distinct Fe ASC configurations Fe1–C3, Fe1–C4, Fe1–N3, and Fe1–N4 were examined for their adsorption characteristics with various reactants.137 The adsorption energies varied significantly across these sites, with strong chemisorption observed for O2, NO, SO2, and SO3 on Fe1–C3, Fe1–C4, and Fe1–N3 sites. This indicates potential sulfur poisoning at these sites due to the stable chemisorption of SO2 and SO3. In contrast, the Fe1–N4 sites exhibit weak physical adsorption for SO2 and SO3 (Eads < −0.5 eV), while maintaining robust chemisorption for O2 and NO. This selective adsorption behavior suggests that Fe1–N4 sites could be particularly resistant to sulfur poisoning, favoring the adsorption of NO and O2. Such findings underscore the importance of tailored site configurations in optimizing the catalytic efficacy and selectivity of ASCs, guided by both experimental findings and theoretical predictions.
3.2. Unique properties of ASCs for HER
The performance of HER catalysts is influenced by the chemical nature and density of active centers. To address challenges in reaction kinetics and stability, it is crucial to develop HER electrocatalysts that can tune surface chemistry through alterations in electronic structure, morphology, composition, and porosity/active surface area.1,145–147 ASCs exhibit distinct characteristics compared to cluster catalysts and nanoparticle catalysts.148,149 The surface of ASCs, including atoms at defect sites, edges, and vertices, plays a crucial role in influencing catalytic reactions. Downsizing nanoparticles and increasing the number of undercoordinated surface atoms have been employed to enhance catalyst efficiency. The smaller size, structural effects, and coordination environment of ASCs contribute to their promising physicochemical properties. Since their introduction in 2011, ASCs have gained momentum, particularly in the electrocatalytic sector.150 ASCs offer excellent atom utilization and unique size quantum effects, generating interest in catalysis and chemical transformations. The development of a flexible and simple synthesis approach that modifies the interaction between metal centers and supports can be used to tune the inherent features of ASCs.151,152 However, key challenges in HER electrocatalysis for ASCs include precise control over the local structure of single-atomic sites and increasing the density of active sites. Rational design of atomic structures greatly influences their intrinsic activity, affecting the activation and adsorption of reactants. Increasing the metal loading of ASCs can significantly boost the density of active sites and related mass activity.147,153The chemical properties of ASCs are determined by the specific element used. Tailoring the electronic properties and structures of ASCs enables effective control over adsorption behavior during the HER process and, consequently, catalytic activities.139,154 DFT calculations have been employed to systematically investigate the HER performance of various single transition-metal atoms implanted in phosphorus carbide monolayers, revealing a volcano-shaped relationship between Gibbs free energy for hydrogen adsorption and HER activity (Fig. 8A).155 The coordination environment also profoundly influences the catalytic performance of ASCs.84,156,157 Determining the optimum coordination scenario can be challenging due to the diverse range of ASCs and supports.158 The achievement of advanced catalytic activity and stability in ASCs relies on secure and close interactions between the SAs and coordinating atoms on the support. The adjustment of coordinating atoms is as critical as the variation of metal atoms, as it can influence the electronic structures of HER catalysts. Coordinating atoms such as C, N, O, S, P, and B are commonly used in conjunction with carbonaceous materials and defective metal oxides/sulfides to modulate the electronic properties.138,159,160 ASCs dispersed on carbon-based materials, such as graphene, graphdiyne, or graphitized carbon, have shown promise as hybrid nanocatalysts for various applications.161 Metal–carbon coordination, formed between transition metal like Ni or Fe and carbon materials, contributes to improved electrochemical HER activities.162–164 Defective sites on carbonaceous materials act as anchoring sites for ASCs, providing unique electronic structures that enhance catalytic activity. For example, Ni ASCs on nanoporous graphene demonstrated excellent HER performance due to stable coordination between Ni ASCs and carbon atoms on the support.165 N-coordinating ASCs, prepared through the straightforward process of N-doped carbon materials, have also been extensively studied.166–169 The electronegative N atoms possess lone-pair electrons that can bond with the empty d-orbitals of transition metals, offering stable and abundant anchoring sites on carbon materials without SA aggregation.170,171 Theoretical studies utilizing DFT calculations have explored the relationship between TM–N coordination and N-doped graphene (NG) supports in terms of HER activity. Screening the performance of transition metal (TM)-based ASCs (TM = Co, Cr, Fe, Rh, and V) supported on NG, Hossain et al. calculated the respective Gibbs free energy change (ΔGH) values.170,172 Among the tested samples, Co-based ASCs showed high promise for HER, with a calculated ΔGH of +0.13 eV in a four N atoms-coordinating model. Sredojević et al. conducted a computational search for efficient ASCs by embedding TM atoms into monovacancies of graphene and hexagonal boron–nitride (h-BN) structures.173 Point defects were employed to stabilize the metal adatoms and prevent their aggregation into larger clusters. The efficiency of these catalysts was evaluated based on their ability to adsorb hydrogen atoms and facilitate their recombination into H2 in a thermodynamically neutral manner. By modelling the critical steps of the HER using nine different metal adatoms embedded in three-point defects of graphene and h-BN, they identified several ASCs with nearly flat potential energy landscapes for HER, comparable to or even more favourable than those found on Pt surfaces. In the study of ASCs for HER, the recombination of H atoms into a molecule plays a crucial role. ASCs capable of catalyzing HER should accommodate one or two H atoms, depending on the reaction mechanism. The initial step involves the chemisorption of the first H atom, and adsorption geometries are categorized as A1 and B1, depending on the H coordination number (Fig. 8B–H). The A1 geometry corresponds to H binding solely to the metal atom, while the B1 configuration represents a bridge site between the metal adatom and the neighbouring surface atom. The binding of an H atom to the metals attached to certain defects is found to be weak and unfavourable, except for Os at VC and Ru, Os, and Pt at VB. The majority of the studied metals exhibit higher reactivity when attached to VN than VC or VB, indicating stronger metal binding at the former two defects (Fig. 8D). The ability of ASCs to simultaneously adsorb two H atoms and their binding energies are also analyzed. Among the metals attached to VC, only Os can accommodate two H atoms, but the binding is weaker compared to Os attached to VN. Pd and Pt at VB strongly bind two H atoms, but one of the hydrogen atoms adsorbs to the neighbouring B atom, leading to a large separation that necessitates high energy barriers for recombination. The simultaneous binding of two H atoms to other metal atoms embedded into VC and VB is weak. Thus, the study primarily focuses on ASCs where the metals are attached to VN when investigating the Volmer–Tafel (VT) mechanism of HER. The H adsorption energies of the studied ASCs reveal that most of them exhibit stronger binding for the first hydrogen atom compared to the second one. Fe, Ru, Os, Co, Ni, Pd, and Pt adatoms attached to VN exhibit band gaps ranging from 2.1 to 3.3 eV and favourably bind the first H atom (Fig. 8E). Metal atoms with preadsorbed H demonstrate larger band gaps, resulting in weaker binding of additional H atoms. Fe, Ru, Os, and Co show modest increases in band gaps but can still accommodate two H atoms and facilitate the VT type of H2 recombination. However, preadsorbed H significantly increases the band gaps of ASCs based on Ni, Pd, and Pt, making the binding of the second H unfavourable for Pd and Pt and less disadvantageous for Ni. ASCs based on Rh and Ir have their unique characteristics, with large band gap values rendering the binding of the first H atom unfavourable. However, the presence of the H adsorbate decreases the band gaps and enables stronger bonding of the second atom. Nevertheless, the overall H binding in these ASCs remains weaker than that in the free H2 molecule.
 | ||
| Fig. 8 (A) Relationship between theoretical overpotential (ηHER) and the hydrogen adsorption free energy of transition metals. Reproduced with permission from ref. 155 Copyright 2021, Elsevier. (B) Adsorption configurations for one H atom and (C) two H atoms at ASCs. H and metal atoms are shown as small yellow and large blue spheres, respectively, with gray spheres representing surface atoms and distances indicated by dH–M and dH–S. (D) Atomic structures of graphene and h-BN with missing atoms, denoted by light blue, reddish-brown, and blue spheres for C, B, and N, respectively. (E) Correlation between electronic band gap and adsorption energy of an H atom on pristine (orange circles) or preadsorbed (blue squares) metal adatoms at VN; (F)–(H) similar correlation for three ASC groups, highlighting changes in H binding from the first to the second chemisorbed H at metal adatoms with arrows. Reproduced with permission from ref. 173 Copyright 2020, American Chemical Society. | ||
4. Advancement of ASCs
4.1. Recent emergence of ASCs
The size of metal particles plays a crucial role in determining catalyst performance. Specifically, the specific activity per metal atom increases as the particle size decreases, mainly because smaller particles expose more metal atoms with unoccupied coordination sites on their surface. These under-coordinated or low-coordinated metal atoms are often highly reactive and serve as active sites for catalytic reactions.174–178 However, as metal particles decrease in size, their surface free energy increases, which promotes the aggregation of small clusters or nanoparticles. To prevent aggregation, researchers employ various strategies to mitigate this issue. By carefully controlling synthesis conditions and employing support materials, it's possible to harness the enhanced reactivity of small metal nanoparticles while minimizing aggregation, thereby optimizing their catalytic performance. Practical supported metal catalysts, however, are typically heterogeneous in the size and distribution of metal particles on the support material, consisting of a mixture of metal particle sizes ranging from nanoparticles to subnanometer clusters. This heterogeneity not only reduces the efficiency of metal atom utilization but also often results in undesired side reactions, posing challenging to identify and control the active sites of interest. ASCs represent the ultimate size limit for metal particles, consisting of individually dispersed metal atoms anchored on support materials. ASCs maximize the efficiency of metal atom utilization, particularly important for supported noble metal catalysts.84 Furthermore, ASCs offer great potential for achieving high activity and selectivity due to their well-defined and uniform single-atom dispersion. One of the main advantages of ASCs is their high mass efficiency, with nearly 100% catalyst utilization, as every atom serves as an active site. This characteristic can lead to significant cost savings compared to traditional catalysts. Additionally, ASCs feature a well-defined and specific atomic structure, which enables high selectivity toward the adsorption and desorption of certain intermediates during catalysis, thereby improving both reaction efficiency and selectivity. Moreover, ASCs offer higher uniformity of active sites compared to nanoparticles, leading to improved performance.179–181 Consequently, ASCs have emerged as highly efficient and tunable catalysts for energy conversion and storage applications. Despite these advantages, ASCs face several challenges, including the complexity of their synthesis. The process involved are intricate, requiring precise procedures to ensure the stability and dispersion of single atoms on supports. These steps are technically demanding and crucial for achieving consistent catalytic performance. Additionally, the aggregation of single atoms into nanoparticles, particularly when using transition metal oxide supports, can hinder effective electron transfer and lead to instability under harsh environments.182,183 Although theoretical understanding of ASCs has advanced, it still lacks complete clarity regarding the dynamics and interactions at the atomic level during catalysis. This knowledge gap hinders the ability to reliably predict and enhance catalyst performance. Furthermore, reproducing these catalysts with consistent quality and activity across different research settings and scaling them for commercial use, remains a significant challenge.Recent advancements have significantly enhanced the design and application of ASCs for electrochemical HER.184–187 However, due to high surface free energy, specially under harsh synthesis and reaction conditions of HER, the rapid diffusion and agglomeration of isolated single atoms on support materials are the most critical challenge that ASCs are currently facing. This agglomeration process diminishes the catalytic efficiency and stability of ASCs in HER. In recent years, substantial progress has been made to address this issue. For example, nitrogenized carbon materials with high conductivity and surface area have been explored as effective supports. These materials provide strong anchoring sites for single atoms and facilitate electronic structure modulation, thereby enhancing the stability and performance of ASCs.182 Another effective strategy involves exploring innovative support materials, including metal–organic frameworks (MOFs), MXenes, metal dichalcogenides, advanced carbon-based supports, and metal oxides. These supports stabilize isolated single atoms, preventing aggregation and enhancing electrocatalytic performance.68,188–190 They offer numerous anchoring sites for single atoms and can be engineered to increase metal loading, thereby influencing the catalytic activity and selectivity of the active metal centers in ASCs. Modifying these supports can adjust the electronic and geometric structure of the catalysts through metal–support interactions, significantly enhancing their efficiency and selectivity.1,20,145,153,191,192 ASCs are primarily distinguished by the type of central metal atom, such as iron, copper, nickel, or platinum, plays a crucial role in determining their catalytic behavior and potential applications.193,194 The active site of a ASC is typically characterized by its coordination environment, which includes the number and type of neighboring atoms bonded to the central metal. This environment significantly influences the electronic structure of the metal atom and, in turn, its reactivity.195,196 For instance, iron atoms coordinated with nitrogen in a graphene matrix can exhibit distinct catalytic properties compared to those coordinated with carbon in a MOF structure.197,198 Moreover, the specific arrangement of these neighboring atoms – whether forming a square planar, tetrahedral, or octahedral geometry – can significantly impact the catalyst's effectiveness and selectivity.199 These advancements have not only deepened the fundamental understanding of catalytic mechanisms at the atomic level but have also enabled the design of ASCs with tailored electronic structures for enhanced HER activity.
4.2. ASC synthesis strategies
Various synthesis approaches, such as wet-chemistry, atomic layer deposition (ALD) and electrodeposition, have been utilized to design ASCs with different metal atoms, aiming to enhance catalytic activities and stabilities for the HER.200–205 The wet impregnation method, for example, is effective and does not require specialized equipment, making it suitable for large-scale production.206 However, it has limitations in achieving high metal loading. High-temperature pyrolysis methods, conducted under a protective atmosphere, have been widely employed for synthesizing atomically dispersed materials by decomposing selected precursors. These methods have been utilized to obtain ASCs from various precursors, including carbon sources, doped/coordination atoms, and metals.159,207–210 For example, MOFs and MOF-derived materials have been explored as precursors in pyrolysis processes. Beyond serving as precursors, MOFs also act as support materials in the MOF-derived method, enabling the synthesis of efficient ASCs. While MOFs typically suffer from low electrical conductivity, recent advancements have demonstrated their potential as electrocatalyst supports.211,212 The thermal transformation of MOF templates through pyrolysis leads to the stabilization of target single atoms on a heteroatom-doped carbon support. The presence of numerous organic linkers in MOFs provides potential anchoring sites for metal atoms, preventing their aggregation. MOFs offer unique characteristics such as tunable porosity, high surface area, structural diversity, and adjustable functionality, making them suitable for producing various functional ASCs.213–219 The use of conductive MOFs or hybrid structures combining MOFs with conductive materials, has shown improved electrocatalytic performance. For instance, the pyrolysis of Fe-containing MOFs resulted in the synthesis of Ni ASCs within MXene sheets. Additionally, the intercalation of Fe-MOF through a simple sonochemical method successfully produced a hybrid architectural framework of MXene and MOF, with Ni atoms coordinated on the MXene support (Fig. 9A and Fig. 10A), demonstrating excellent HER activity (Fig. 10B).220 Pyrolysis, particularly on nitrogen-doped carbon support, is a versatile and widely adopted technique for ASC preparation. Although the high temperature in pyrolysis can lead to metal aggregation, additional acid leaching can be applied to selectively remove redundant metal nanoparticles without affecting the anchored single metal atoms. These synthesis strategies have successfully yielded a range of ASCs, including both non-noble metal-based and noble metal-based ASCs.221–224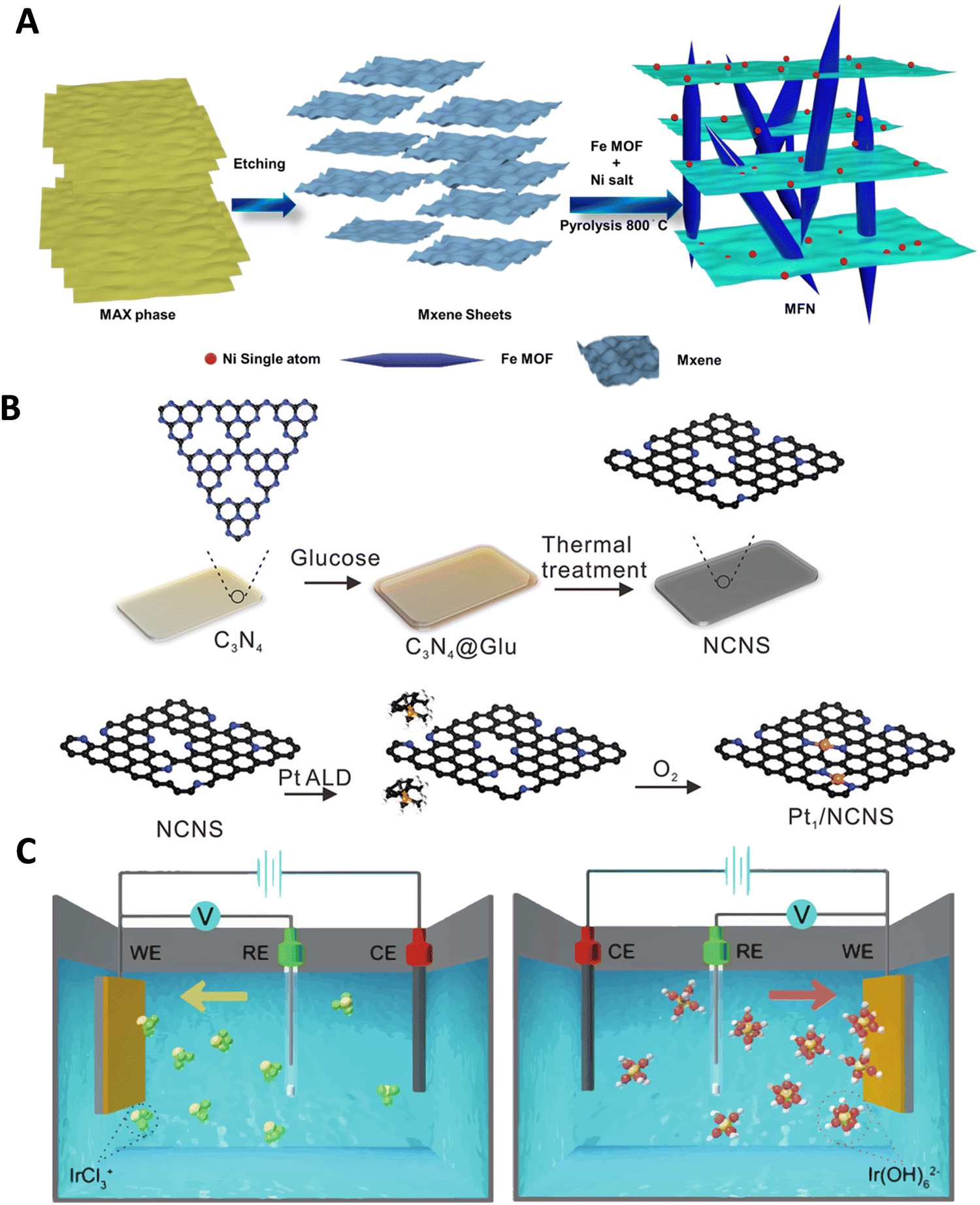 | ||
| Fig. 9 (A) Illustration of the formation process of MXene-Fe-MOF/Ni ASCs. Reproduced with permission from ref. 220 Copyright 2024, The Royal Society of Chemistry. (B) Schematic representation depicting the Pt ALD mechanism on nitrogen-doped carbon nanosheets (Pt1/NCNS). Reproduced with permission from ref. 225 Copyright 2021, Wiley-VCH. (C) Schematic representation of cathodic (left) and anodic (right) deposition of Ir species. Reproduced with permission from ref. 226 Copyright 2016, Nature Publishing Group. | ||
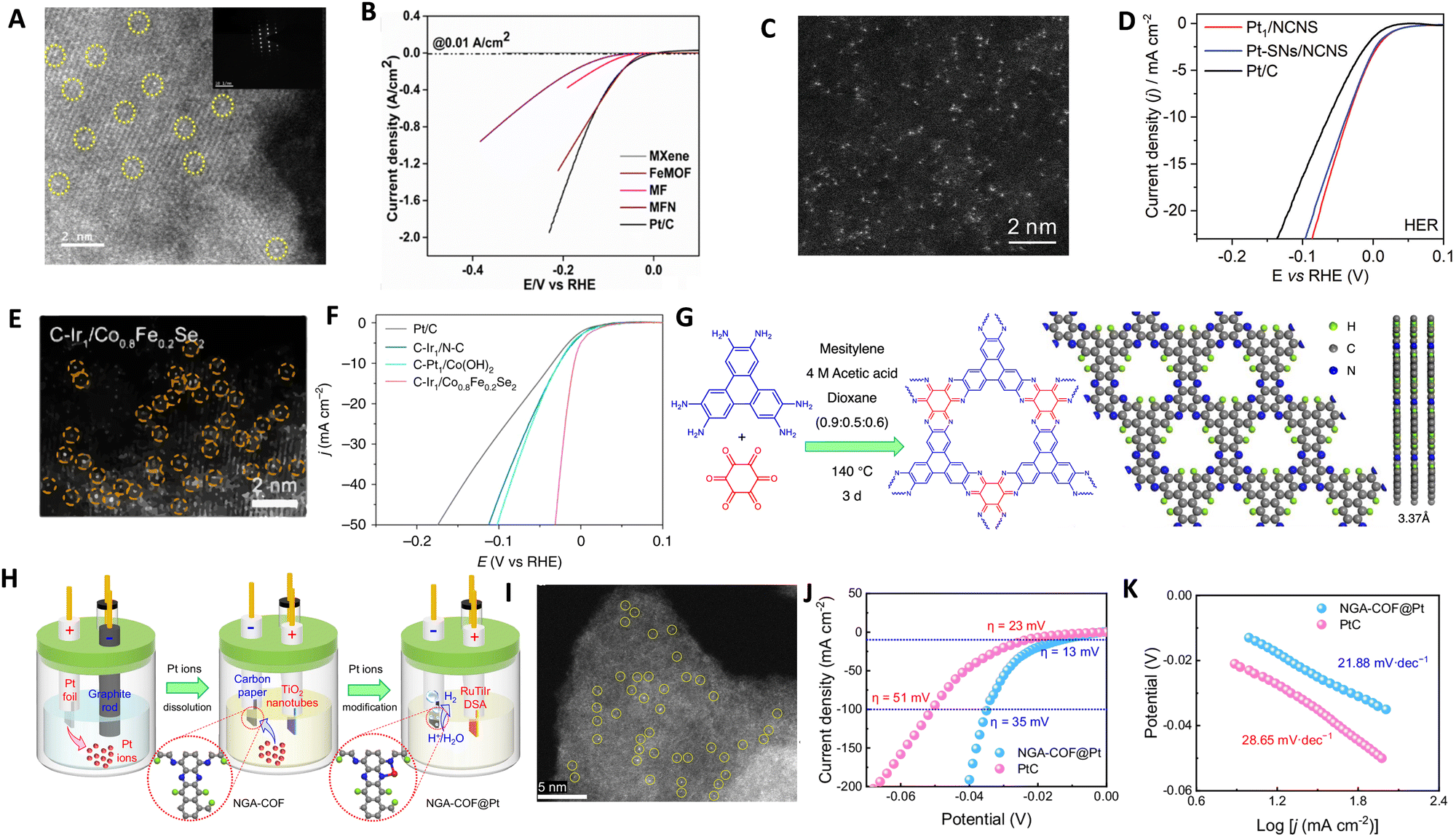 | ||
| Fig. 10 (A) HAADF-STEM images at high magnification of MXene-Fe-MOF/Ni ASCs, revealing the exclusive presence of individual Ni atoms in MXene-Fe-MOF/Ni ASCs. (B) LSVE polarization curves comparing the electrocatalytic performance of MXene-Fe-MOF/Ni ASCs with different materails, Co NPs-N/C, and Pt/C in 0.5 M H2SO4. Reproduced with permission from ref. 220 Copyright 2024, The Royal Society of Chemistry. (C) HAADF-STEM images of Pt deposited through the ALD process on NCNS. (D) The HER polarization curves for Pt1/NCNS, Pt with both single-atoms and nanoparticles and Pt/C catalysts obtained via LSV in 0.5 M H2SO4. Reproduced with permission from ref. 225 Copyright 2021, Wiley-VCH. (E) HAADF-STEM image of cathodically deposited Ir single atoms on Co0.8Fe0.2Se2 and (F) Polarization curves of cathodically deposited ASCs for HER on different supports. Reproduced with permission from ref. 227 Copyright 2016, Nature Publishing Group. Schematic illustration of the synthesis of (G) NGA-COF and (H) NGA-COF@Pt catalyst by electrochemical treatment, (I) HAADF-STEM image of NGA-COF@Pt, (J) LSV curves and (K) corresponding Tafel plots in 0.5 M H2SO4. Reproduced with permission from ref. 228 Copyright 2024, Nature Publishing Group. | ||
ALD technique has been employed to synthesize catalyst materials with precise control over size, composition, morphology, and structure, including nanoparticles, nanoclusters and ASCs.229–231 In one method, Pt ASCs were obtained by reacting oxygen atoms on graphene nanosheets with a platinum precursor in a two-step ALD cycle.232 The size and loading weight of Pt ASCs could be adjusted by changing the number of ALD cycles. Similar approaches were used to prepare Pt ASCs on nitrogen-doped carbon nanosheets (Fig. 9B and Fig. 10C) and exhibited superb HER activity (Fig. 10D).225 Another strategy involved utilizing ALD to produce Pt ASCs and Pt nanoparticles supported by nitrogen-doped graphene nanosheets, as well as Pd catalysts and Pt2 dimers on graphene.230,233 Although ALD technique enables the growth of noble metal ASCs, such as Ru, Pt, and Pd, on various substrates, it suffers from low yields, high equipment and precursor costs, which limit its widespread application.230,234,235 Electrochemical deposition is a widely used method to modify material surfaces. It offers advantages such as adjustable particle size, deposition on the outer surface of the substrate for better exposure of active sites, and fast, scalable, controllable, and efficient fabrication. However, non-uniform plating can be a drawback in some cases. Studies have successfully fabricated atomically dispersed Pt atoms on single-walled carbon nanotubes (SWCNT) and CoP nanotubes supported on Ni foam using electroplating and potential-cycling methods, respectively.201,202 Vacancies in the material can also stabilize single metal atoms during the electrochemical process.236,237 The cathodic reduction method allows for the reduction of various metal ions. For example, Pt atoms can be dissolved from Pt anodes and redeposited onto cathodes. Pt ASCs supported on CoP nanotube arrays have demonstrated high electrocatalytic activity for the HER process. Similarly, Pt ASCs on SWCNT and ASCs of Ni and Fe have been obtained through simple electroplating and electrodeposition methods, respectively.163,202 Ir ASCs were deposited on Co0.8Fe0.2Se2@Ni foam support using electrochemical deposition, while Pt ASCs were prepared on graphite carbon paper (CP) through a controllable electrodeposition approach (Fig. 9C and E) and exhibited higher HER activity compared to commercial Pt/C catalyst (Fig. 10F).227 Various alternative methods have been explored for synthesizing ASCs, including hydrothermal and photochemical reduction. Bao's group achieved Pt ASCs on MoS2 through hydrothermal reduction, improving their performance.74 Iced photochemical was used for reduction to avoid metal atom aggregation and successfully deposit Pt, Au and Ag ASCs on various substrates, respectively.203 Pd ASCs were synthesized on ultrathin titanium oxide nanosheets using room-temperature photochemical reduction by Zheng's group.238
Covalent organic frameworks (COFs) are increasingly utilized to synthesize ASCs for the HER due to their distinctive properties. These frameworks offer nanoscale pore structures, stable periodic arrangements, abundant coordination sites, and a high surface area, making them ideal for hosting catalytic active sites. Their robustness and environmentally friendly nature, along with resistance to a broad pH range, further enhance their suitability as electrocatalysts. Additionally, the ability to customize COFs enables precise control over the coordination environment, which significantly influences the catalytic behavior of the hosted metal atoms. This adaptability allows for the optimization of metal loading and the enhances the catalytic activity and selectivity of the active metal centers in ASCs.239–247 For instance, Zhang et al. developed an innovative Pt-based ASC using a nitrogen-rich graphene analogue COF (NGA-COF@Pt), synthesized through a conductive agent-free and pyrolysis-free process at room temperature.228 This process utilizes the COF itself as a homogeneous current collector, which not only facilitates charge transfer but also enhances the intrinsic activity of Pt single atoms. NGA-COF was meticulously synthesized through a reaction between 2,3,6,7,10,11-triphenylenehexamine and hexaketocyclohexane (Fig. 5G), followed by a controlled electrochemical modification process in 0.5 M H2SO4 (Fig. 10H). HAADF-STEM confirmed the presence of densely distributed Pt single atoms within the NGA-COF matrix, indicating a uniform atomic dispersion, as shown in Fig. 10I. When tested for HER in 0.5 M H2SO4, NGA-COF@Pt achieved a significantly lower overpotential of 13 mV at 10 mA cm−2 compared to the 20 wt% Pt/C benchmark (Fig. 10J), demonstrating enhanced catalytic efficiency. Additionally, Tafel analysis revealed a slope of 21.88 mV dec−1 for NGA-COF@Pt, underscoring its superior kinetic performance in HER compared to the 28.65 mV dec−1 observed for Pt/C (Fig. 10K). Ranjeesh et al. synthesized a porphyrinic squaraine-linked COF 2D nanosheet with bimetallic Co/Ni catalysts that produced 670 μmol H2 over 45 minutes with a faradaic efficiency of 97% and an overpotential of 105 mV.248 Yi et al. developed a porous porphyrinic triazine-based COF incorporating Co–N4 active sites, which exhibited a Tafel slope of 50 mV dec−1 and a low overpotential of 94 mV.243 Qu et al.'s work with N-rich COFs absorbing Ru3+ ions led to ultrafine Ru species embedded in nitrogen-doped carbons, showing remarkable HER performance in both alkaline and acidic conditions with mass activities significantly surpassing commercial Pt/C.239 Qiao and his team crafted a Zn-Salen COF via Schiff base condensation, subsequently enhancing its conductivity with poly(3,4-ethylenedioxythiophene) (PEDOT), which led to an overpotential of 150 mV and a Tafel slope of 43 mV dec−1 for the PEDOT@Mn-Salen COFEDA heterostructure.242 These studies underscore the powerful synergies between COF architectures and metal functionalities, paving the way for highly efficient and stable ASCs tailored for electrocatalytic applications. The impact of these innovations is profound, setting new benchmarks for ASCs performance in HER and encouraging further exploration across different electrochemical applications. Looking forward, the field is poised to explore more complex and hybrid ASC structures, leveraging the unique properties of newly developed support materials and synthesis techniques to further enhance the efficiency and sustainability of hydrogen production.
4.3. Design principles for enhanced catalytic activity in ASCs
The design of ASCs for HER relies on a comprehensive understanding of atomic-level dynamics and the precise tuning of electronic states at active sites. ASCs provide a versatile framework for investigating the relationship between structure and reactivity, as well as the complexities of catalytic mechanisms at the atomic scale. The sucessful deployment of ASCs in industrial settings depends on addressing challenges such as selecting appropriate supports and developing advanced design strategies. To engineer ASCs with tailored active centers, high selectivity, and optimized hydrogen output, several key design principles are essential.168,249–253 These principles include establishing homogeneous active sites to deepen our understanding of catalytic processes and boost activity and selectivity, fine-tuning the coordination configurations and oxidation states of the catalytic centers, managing the mass loading of ASCs to ensure high density and uniform dispersion of active sites, modifying local coordination environments through heteroatom doping, and meticulously engineering metal–support interactions via both strong and electronic modalities. In electrochemical HER, understanding catalyst behavior under varying operational conditions, such as temperature, pH, and electrical potential, is crucial due to the potential for catalyst reconstruction.254,255 These conditions complicate the delineation of catalytic mechanisms and require precise identification of active species. The performance of ASCs is significantly influenced by the synergistic effects between the metal atoms and their surrounding coordination atoms. Variations in the inherent properties of support materials and the non-uniformity of fabrication methods often result in the formation of different active metal species. This is evident in studies by Zhang and colleagues, where various forms of FeNx species were identified in Fe–N–C ASCs, each displaying unique catalytic properties.256 Modern characterization techniques coupled with theoretical modeling are vital for a comprehensive understanding of these systems. Techniques such as XRD, TEM, XPS, FTIR, and Raman spectroscopy play a crucial role in analyzing ASC structures.257 Specifically, AC-HAADF-STEM and X-ray absorption spectroscopy, including XANES and EXAFS, provide valuable insights into the atomic configuration and electronic states of ASCs.258,259 These methodologies help in identifying the coordination number and bond lengths of atoms, which are crucial for validating structural models derived from DFT calculations. This integrated approach enables a comprehensive understanding of the interactions between isolated atoms and supports, revealing how these interactions influence catalytic activity and stability.One of the main challenges faced by ASCs is aggregation at high temperatures, driven by their high surface energy and thermodynamic instability. In this regard, strategies to optimize ASC design include controlling metal loading and employing robust support materials to prevent aggregation. For instance, studies on the relationship between Pt loading and hydrogen evolution activity in ASCs show that optimal loading prevents cluster formation, which could otherwise reduce catalytic activity.260 Advanced synthesis methods, such as tunning annealing temperatures, can regulate defect distribution and improve the dispersion of metal atoms, thereby enhancing catalytic performance.261,262 The selection of metal atoms and their interaction with support materials are critical for designing ASCs with enhanced catalytic performance. Transition metals such as Fe, Co and Ni, as well as noble metals like Pt, are favored for their strong catalytic properties, which are significantly influenced by the choice of support material.263–265 Supports such as carbon-based materials, metal oxides, or COFs stabilize metal atoms while modifying their electronic properties through metal–support interactions (MSI).223,241,242,266,267 This interaction is crucial for optimizing the activity and stability of ASCs by influencing charge transfer and enhancing catalytic performance. Challenges in managing MSI include establishing effective interactions between metal sites and the support to maximize the utilization of exposed active sites and understanding the differing MSI characteristics between single atoms and nanoparticles. For example, studies on Pt/TiO2 have shown that while nanoparticles may lose adsorption capacity due to MSI effects, single atoms retain distinct catalytic functionalities after specific treatments. This underscores the complex interplay within ASCs that influences reactivity and selectivity.266
Doping strategies in materials chemistry, particularly for electrocatalysis, are pivotal for enhancing HER performance. Doping introduces foreign atoms into a material's structure, effectively altering its electronic and geometric properties.268–271 Non-metal atom doping, such as nitrogen (N), sulfur (S), or phosphorus (P), modifies the electronic structure of catalysts, facilitating charge redistribution and optimizing hydrogen adsorption energies.272–274 This process can significantly improve catalytic activity and selectivity for HER. On the other hand, metal atom doping focuses on incorporating metal atoms such as Co, Ni, or Fe into the catalysts.271,275–277 This type of doping typically increases the density of active sites and influences the catalytic process by altering electronic states and interactions with hydrogen atoms, which are crucial for achieving low overpotentials and high HER activity. Building on these principles, heteroatom doping is recognized as a powerful strategy to enhance the catalytic performance and selectivity of ASCs for HER. This approach modifies the coordination chemistry around active metal sites, optimizing hydrogen adsorption and desorption by fine-tuning the electronic configuration of the catalyst. These modifications affect not only the metal single atoms (M-SAs) directly involved in the reaction but also the neighboring atoms, which play a significant role in influencing catalytic outcomes.278 Changes in the electronic structure of metal sites, similar to the role of ligands in homogeneous catalysts, and the involvement of metal cations from the support in the reaction process are crucial for enhancing both catalytic performance and stability.200,279,280 Transition metals use their d-orbitals to form low-energy transition states, while non-metals such as carbon, oxygen, nitrogen, sulfur, or phosphorus contribute to stability and tunability.281,282 The variety in doping types, such as pyridinic-N, pyrrolic-N, or graphitic-N, allows for the tailored optimization of ASCs for specific tasks like HER, thereby enhancing their effectiveness and efficiency.283–285
4.4. Advancements in characterizing active sites of ASCs
Advancements in the characterization of active sites in ASCs have revolutionized our understanding of catalysis at the atomic level, largely due to the development and refinement of various analytical techniques. These techniques allow for precise determination of the electronic structure, atomic coordination, and real-time behavior of ASCs during chemical reactions, which is crucial for enhancing their design and functionality in applications like HER. In this context, we explore some of the key techniques employed in the detailed characterization of ASCs.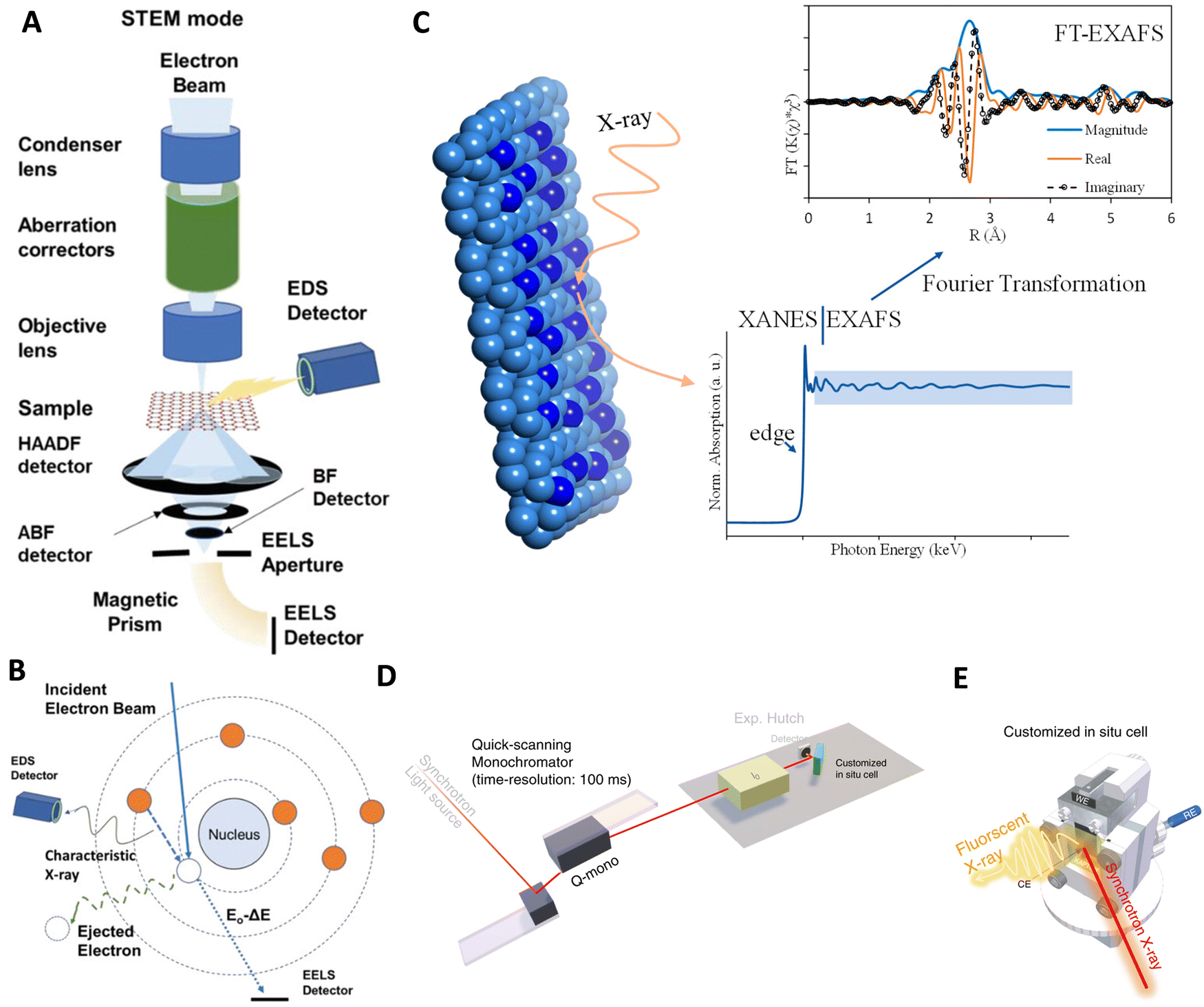 | ||
| Fig. 11 Technologies used for characterizing ASC active sites: (A) overview of an electron microscope in STEM configurations, equipped with components like condenser and objective lenses, aberration correctors, projector systems, retractable detectors, and devices for EDS and EELS acquisition. (B) Diagram showing inner shell ionization processes that generate characteristic X-rays for EDS and inelastic scattering for core-loss EELS signals. Reproduced with permission from ref. 290 Copyright 2014, National Academy of Sciences. (C) Depiction of typical XAS spectra at the bottom and FT-EXAFS spectra at the top. Reproduced with permission from ref. 294 Copyright 2024, American Chemical Society. (D) Diagrammatic representation of the setup for operando time-resolved XAS experiments. (E) Conceptual illustration of a customized operando XAS cell. Reproduced with permission from ref. 295 Copyright 2020, Nature Publishing Group. | ||
5. Types of ASCs for HER
Various ASCs have found extensive applications in electrocatalytic HER, including Pt, Co, Ni, Cu, Mo, Fe, W, Pd, and Mn-based ASCs.200–202,207,312–323 These catalysts can be categorized into three groups: metal-based ASCs, bimetal-based ASCs, and metal alloy ASCs. The development of ASCs in these categories offers opportunities to enhance the efficiency and selectivity of HER, opening up possibilities for improved catalytic performance in hydrogen evolution.5.1. Single metal-based ASCs
 | ||
| Fig. 12 (A) Schematic representation of the synthesis process for Pt SASs/AG. (B) Bright-field HR-TEM image and (C) AC HAADF-STEM images of Pt SASs/AG. (D)–(E) XANES and FT-EXAFS spectra of the Pt L3-edge for Pt SASs/AG and Pt/C. Reproduced with permission from ref. 332,335 Copyright 2019, The Royal Society of Chemistry. (F) Schematic representation of the synthesis process for NPCN–Pt. (G) TEM image and (H) HAADF-STEM images of NPCN–Pt. Reproduced with permission from ref. 335 Copyright 2021, Elsevier. (I) Proposed reaction mechanism for the synthesis of Pt SAs/DG. (J) Magnified HAADF STEM image of Pt SAs/DG. (K) and (L) R-space spectra and Pt L-edge XANES spectra. (M) Corresponding EXAFS fitting curve for Pt SAs/DG; inset shows the proposed Pt–C coordination environment. Reproduced with permission from ref. 336 Copyright 2019, American Chemical Society. | ||
A novel approach for generating atomically dispersed platinum on defective graphene (Pt SAs/DG) is presented, employing a thermal emitting method with bulk Pt metal as the precursor.336 The process involves the coordination of ammonia, produced by the pyrolysis of dicyandiamide, with platinum atoms, followed by anchoring the volatile Pt(NH3)x species onto the surface of defective graphene (Fig. 12I). The resulting Pt SAs/DG catalysts exhibit excellent activity for both the electrochemical hydrogen evolution reaction and the selective oxidation of various organosilanes. The synthesis procedure and proposed formation mechanism involve heating dicyandiamide (DCD), Pt net, and graphene oxidation (GO) in an Ar flow at 1100 °C. The pyrolysis of DCD generates ammonia gas, which coordinates strongly with Pt atoms to form volatile Pt(NH3)x species. Simultaneously, Pt0 species may be oxidized by oxygen-containing functional groups on the GO surface, resulting in Ptδ+ (0 < δ < 4) species. Subsequent thermal treatment removes most oxygen-containing functional groups, yielding defective graphene (DG). The Ptδ+ (0 < δ < 4) species are then trapped by the DG, leading to the formation of isolated Pt SAs/DG catalysts. The argon is unable to extract Pt atoms from bulk Pt, which is confirmed by scanning transmission electron microscopy (STEM) and aberration-corrected HAADF STEM analysis (Fig. 12J). The introduction of DCD enables the clear identification of isolated Pt atoms on DG through aberration-corrected HAADF STEM images. X-ray absorption fine structure (XAFS) analysis reveals the bond length of Pt–C in the as-prepared Pt SAs/DG samples, while Pt foil and PtO2 serve as reference materials (Fig. 12K). The Pt SAs/DG exhibit a main peak in the R-space spectrum of the EXAFS, matching the Pt–C bond length. In contrast, Pt foil shows a prominent peak corresponding to Pt–Pt coordination, while Pt NPs/DG display a shorter Pt–Pt bond length. XANES spectra indicate that the white-line intensity of Pt SAs/DG falls between Pt foil and PtO2, confirming the oxidation state of Pt (Ptδ+, 0 < δ < 4) in Pt SAs/DG (Fig. 12L). The fitting results of the R-space spectrum provide insights into the proposed coordination structure of Pt–C4 in Pt SAs/DG (Fig. 12M). The activity and performance of metallic particles in catalytic reactions are strongly influenced by their nature, size, placement, and stability.324,337,338 It is well-established that the corner and edge sites of catalysts exhibit superior activity.338 Motivated by this understanding, Liu et al. aimed to utilize highly curved supports to anchor single metal sites, mimicking the effect of metal sites at the corners and edges of particles.339 In this context, they employed onion-like nanospheres of carbon (OLC) as a support to anchor stable atomically dispersed Pt, creating a catalyst denoted as Pt1/OLC, for the HER. Their research demonstrates that the distribution of Pt single atoms on nanosized onion-like carbon supports (Pt1/OLC) results in highly efficient electrocatalysts for hydrogen evolution, surpassing the performance of a two-dimensional (2D) graphene-supported catalyst with a similar Pt loading. Simulations indicate that this architecture, characterized by high curvature, leads to the accumulation of electrons around the Pt regions, inducing a local electric field that enhances the catalytic kinetics. A method for anchoring atomically dispersed Pt atoms on carbon nanotubes (Pt-CNTs) involves an ultralow-temperature solution reduction approach.340 This approach aims to decelerate the diffusion rate of PtCl62− ions onto the carbon nanotubes and reduce the free energy of Pt atoms in the solution, thereby minimizing the probability of Pt aggregation. In this method, a specific volume of H2PtCl6 solution was injected into an alkaline microthermal reduction solution (N2H5OH, 5.0 M, and KOH, 0.05 M) at ultralow temperatures. The resulting mixture is allowed to react for 2 hours to generate atomically dispersed Pt-based catalysts. After dispersing CNTs in ethanol and further stirring for 3 hours, atomic Pt species are sufficiently anchored on the CNTs. The resulting sample was collected through vacuum filtration and naturally dried at room temperature. HAADF-STEM imaging reveals bright spots corresponding to individual heavy Pt atoms randomly dispersed on the CNT supports. The morphology of the CNTs is also clearly observed, with Pt atoms measuring approximately 0.2 nm dispersed between or on the lattice planes of the CNTs. This ultralow-temperature solution reduction approach effectively anchors atomically dispersed Pt atoms on CNTs, leading to improved catalytic activity for the HER. By decelerating the diffusion rate of PtCl62− ions onto the carbon nanotubes and lowering the free energy of Pt atoms in the solution, the approach helps prevent Pt atom aggregation in the solution, contributing to enhanced HER performance.
The utilization of hydroxide-supported atomic structures, especially single atoms, presents a promising avenue for enhancing catalytic performance through active microenvironmental tuning.341,342 However, there has been limited exploration of the electronic synergy between mono- and dual-hydroxides. To address this, Pei et al. developed a novel strategy to construct a Pt1/(Co,Ni)(OH)2/C ASC by stabilizing and anchoring Pt single-atoms, Pt1, on the surface of defective (Co,Ni)(OH)2, which is further supported on carbon black.343 Remarkably, this catalyst exhibits significantly superior HER activity compared to Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, Pt1/C, and even the commercial 20 wt% Pt/C catalyst. It demonstrates an almost negligible onset overpotential and remarkable electrocatalytic mass activity for the HER, showcasing its exceptional performance in this reaction. Recent research by Xu et al. introduced a novel approach combining atomically dispersed Pt single atoms with amorphous MoOx (denoted as Pt-SA/α-MoOx) for highly efficient HER.344 The Pt-SA/α-MoOx catalysts were synthesized through low-temperature pyrolysis of one-dimensional organic–inorganic hybrid nanorods, where (C2H10N2)[Mo3O10] served as the precursor. Surface functional groups on (C2H10N2)[Mo3O10] facilitated the adsorption of Pt precursors, followed by reduction and anchoring of single Pt atoms. The controlled pyrolysis temperature prevented the formation of metal nanoclusters or nanoparticles, making this method practical and versatile. Pt-SA/α-MoOx exhibited ultrahigh mass activity, Pt-like kinetics, a small Tafel slope, and remarkable long-term electrochemical stability. The synergistic effect between unsaturated Pt atoms and defective MoOx contributed to lowered reaction barriers, enhancing the overall HER kinetics. In a recent study, researchers introduced a unique ganoderma-like MoS2/NiS2 hetero-nanostructure that incorporates isolated Pt atoms.345 This novel heterostructure exhibits several advantageous characteristics. Firstly, it efficiently disperses and confines the few-layer MoS2 nanosheets, ensuring maximum exposure of the edge sites of MoS2. This arrangement creates more opportunities for the capture of Pt atoms. Additionally, the electronic structure of the catalyst can be modified, further enhancing its catalytic activity. The ganoderma-like structure also provides exceptional dispersibility and a large specific surface area, enabling the easy anchoring of single Pt atoms on the MoS2 nanosheets at ultrahigh loading. As a result of this unique architecture and the presence of platinum atoms, the catalyst demonstrates Pt-like catalytic activity in the hydrogen evolution reaction, exhibiting an ultralow overpotential and excellent durability, particularly when operating under a constant current density of 10 mA cm−2. A novel 3D hierarchical nanostructure, named Pt single atoms immobilized Ni2P nanosheet arrays on nickel foam (PtSA-NiP@NF), has been designed as an alkaline HER catalyst.346 This structure combines the advantages of a continuous nickel foam (NF) and a conductive Ni2P array support, creating an efficient pathway for electron transport in Pt single atoms. Consequently, the charge transfer impedance is significantly reduced, and the number of electrochemical active sites is increased, accelerating the HER process in alkaline electrolytes. Experimental results demonstrate the exceptional HER activity of PtSA-Ni2P@NF, with a low overpotential and high mass activity. An ultrahigh mass activity can be achieved through the strategic merging of isolated Pt active sites in ASCs. A catalyst was synthesized by thermodynamically driven diffusion and merging of phosphorus-doped carbon (PC) supported Pt single atoms (Pt1@PC) into Pt nanoclusters (PtM@PC).347 XAS analysis revealed that the merged nanoclusters exhibited significantly stronger interactions with the support compared to traditional methods, facilitating more efficient electron transfer. The optimized PtM@PC catalyst demonstrated an order of magnitude higher mass activity than Pt1@PC at the low overpotential in acidic media. A novel photochemical solid-phase reduction method was developed to synthesize well-defined isolated Pt atoms on a nitrogen-doped porous carbon (Pt1/NPC) catalyst.167 This approach offers a simple and efficient strategy for achieving Pt atom dispersion on the carbon support, eliminating the formation of clusters or nanoparticles. In the synthesis process, nitrogen-doped porous carbon (NPC) was mixed with an aqueous solution of H2PtCl6 and sonicated to disperse the components. After filtration and washing steps, the resulting precursor powder was spread as a thin film on a glass plate to allow light penetration. The thin film was then exposed to ultraviolet light from a mercury lamp for 1 hour at room temperature. Under the irradiation, the adsorbed PtCl62− ions on the NPC surface were directly reduced to isolated Pt atoms. The resulting Pt/NPC catalyst exhibited a high Pt loading of up to 3.8 wt% relative to the carbon. Remarkably, the Pt/NPC catalyst demonstrated ultrahigh electrocatalytic activity for the HER.
Various synthetic methods, including chemical vapor deposition, atomic layer deposition, pyrolysis, and wet impregnation method, have been employed to fabricate platinum-based ASCs like PtSA-graphene, and PtSA on titanium tungsten carbide nanoparticles.200,317 These ASCs have demonstrated remarkable performance in the electrocatalysis of the HER in acidic or alkaline media when combined with binders on cathodes.348,349 However, very few ASCs have been reported for HER electrocatalysis in neutral media. Commercial Pt/C catalysts composed of Pt nanoparticles on carbon have been considered the most effective benchmark for HER electrocatalysis in neutral media. Due to their greater mass utilization and the positive modulation of their electronic structures by substrates, the implementation of Pt ASCs in this domain has the potential to attain even higher levels of activity.350 It has been observed that Pt atoms can dissolve into acid or alkaline media from Pt anodes at high voltages and subsequently transfer onto cathodes.202,351,352 Inspired by this observation, a potential-cycling method utilizing a three-electrode cell with phosphate buffer solution (PBS) has been developed to synthesize a novel catalyst called PtSANT–NF, which consists of large-area Pt ASCs supported by CoP-based NT arrays on NF.201Fig. 13A illustrates the synthesis process of PtSA–NT–NF. In the three-electrode cell containing PBS at pH 7.2, a Pt foil serves as the counter electrode (CE), a saturated calomel electrode (SCE) acts as the reference electrode (RE), and a precursor sample, denoted as S1, comprises a NF with CoP-based nanorod/nanosheet arrays on its surface, serving as the working electrode (WE). Potential cycling between −1.5 and −0.668 V vs. SCE at 25 °C was performed at a scan rate of 150 mV s−1. After 5000 cycles, the changing trend in the potential faded, indicating the stability of the S1 structure. Thus, the 5000 potential cycles were employed to transform each precursor sample into the desired PtSA–NT–NF structure. Fig. 13 B and C displays the SEM, and TEM images of a sample obtained after the 5000 potential cycles. This sample exhibits centimeter-scale dimensions and features an array of NTs standing on a NF. The large-scale dimensions are determined by the size of the three-electrode cell and the autoclave used during precursor production, making the synthesis process scalable. The NTs have an average diameter of approximately 140 nm and a wall thickness of around 20 nm. AC-STEM with AR HAADF imaging reveals a high dispersion of single Pt atoms on the NTs, as shown in Fig. 13D. This dispersion of Pt atoms on the NTs is similar to previous reports and indicates the absence of Pt crystal grains or particles in the catalyst structure.90,353,354 The X-ray diffraction (XRD) pattern of a centimeter-scale piece of PtSA–NT–NF depicted in Fig. 13E, which exhibits signals from Ni(OH)2, CoP, and Co2P crystal grains but not from Pt. Additionally, Fig. 13F illustrates the X-ray photoelectron spectrum (XPS) of a commercial Pt/C catalyst containing Pt nanoparticles, displaying two Pt 4f peaks at 71.6 and 74.9 eV, characteristic of metallic Pt (Pt0).317,355,356 In contrast, the Pt 4f peaks of the PtSA–NT–NF sample are located at 72.3 and 75.6 eV, indicative of Pt2+ species.317,355,356 The absence of a detectable Pt0 signal suggests the exclusive presence of single Pt atoms rather than Pt crystal grains in PtSA–NT–NF, corroborating the absence of Pt crystal grain signals in the XRD and electron diffraction patterns. The Pt2+ valence arises from the interaction between the single Pt atoms and the substrate.317 The synthesized PtSA–NT–NF, which is directly used as the HER cathodes, exhibits comparable performance at low current densities and superior performance at high current densities in phosphate buffer solutions with a pH of 7.2 when compared to commercial Pt/C catalysts. The Pt mass activity of PtSA–NT–NF is four times higher than that of Pt/C, highlighting its enhanced catalytic efficiency. Additionally, PtSA–NT–NF demonstrates improved electrocatalytic stability compared to Pt/C, indicating its potential for prolonged and sustainable catalytic performance. PtSA–NiCo LDHs/NF catalyst was synthesized through the electrochemical deposition process, where Pt single atoms were captured by oxygen vacancy-enriched NiCo layered double hydroxides (LDHs) on Ni foam (Fig. 13G).357 The electrodeposition was carried out using a mixed precursor solution containing Ni and Co precursors along with K2PtCl4, and the deposition was performed at a constant potential of −1.0 V for 25 min. This process led to the generation of a large number of oxygen vacancies in the NiCo LDHs/NF, which interacted with the Pt precursor to form a stable Pt–O structure with oxygen. Aberration-corrected HAADF-STEM images revealed numerous bright dots corresponding to Pt single atoms on PtSA–NiCo LDH/NF (Fig. 13H). XANES spectra indicated the valence state of Pt in PtSA–NiCo LDH/NF to be approximately +4 (Fig. 13I), while the Fourier transform of the EXAFS spectra demonstrated Pt–O coordination at 1.66 Å (Fig. 13J). No Pt–Pt coordination was observed. The coordination number of Pt–O bonding in PtSA–NiCo LDH/NF was determined to be 5.3 (Fig. 13K). The presence of unsaturated coordination between Pt and O not only confirms the existence of atomic Pt in the structure but also enhances the catalyst's HER activity. In the process of HER, LDHs play a crucial role as active sites for cleaving H–OH bonds and dissociating water molecules. On the other hand, Pt single atoms function as binding sites for intermediate hydrogen species, and the Pt–O bonds play a vital role in facilitating the coupling of protons and electrons, ultimately promoting the release of H2 molecules. In the context of the HER in alkaline solution, the water dissociation step is often hindered by slow kinetics.358–360 Nevertheless, the obstacle was tackled in a recent investigation where single-atomic Pt was successfully synthesized and immobilized within the lattice of CoP mesoporous nanosheets (MNSs) that were grown on carbon fiber cloth (CFC) through the electrodeposition process.361 This catalyst, known as Ptat–CoP MNSs/CFC, demonstrated remarkable electrocatalytic performance for HER in alkaline solution, despite its ultralow Pt loading of 0.7 wt% relative to CoP (5.89 mg cmgeo−2). The strong interaction between Ptat and CoP facilitated spontaneous water dissociation, resulting in a significantly reduced HER kinetic barrier. Ptat–CoP MNSs/CFC exhibited outstanding HER performance, characterized by negligible onset potential, exceptionally high catalytic activity, and excellent durability surpassing that of the commercial 20 wt% Pt/C catalyst. Additionally, Ptat–CoP MNSs/CFC showcased excellent catalytic activity and stability when employed in seawater electrolysis.
 | ||
| Fig. 13 (A) Schematic representation of the synthesis process for PtSA–NT–NF. (B) Optical and SEM images of PtSA–NT–NF. (C) TEM image of a NT of PtSA–NT–NF. (D) Atomic-resolution HAADF image of the NT highlighting Pt atoms. (E) XRD graph of PtSA–NT–NF and Pt. (F) HR-XPS spectra of PtSA–NT–NF and commercial 20 wt% Pt/C. Reproduced with permission from ref. 201 Copyright 2017, Wiley-VCH. (G) Schematic for PtSA–NiCo LDH/NF fabrication. (H) AC-HAADF-STEM images of PtSA–NiCo LDH/NF. (I) XANES spectra, (J) EXAFS spectra in R-space, and (K) EXAFS spectra in k-space for PtSA–NiCo LDH/NF, PtO2, and Pt foil. Reproduced with permission from ref. 357 Copyright 2023, Elsevier. | ||
A novel approach was developed to embed Ru nanoparticles (RuNP) and single atoms (RuSA) into a three-dimensional crystalline fullerene network (CFN), resulting in the formation of RuNP–RuSA@CFN-800.382 By taking advantage of the synergistic effect between RuNP and RuSA, the synthesized RuNP–RuSA@CFN-800 demonstrated remarkably superior HER activity. The construction process involved confining Ru species within the crystal lattice of the fullerene network (Fig. 14A). Initially, a solvothermal process was used to assemble Ru3+ ions with imidazole and C60 (Ru–Im–C60), forming a 3D architecture called Ru–Im–CFN. The C60 crystallized into a face-centered cubic (fcc) network. In the subsequent pyrolysis step, the Ru species underwent in situ transformation, resulting in the formation of hexagonal RuNP and RuSA confined within the crystal lattice of the C60 network. Characterized by the presence of bright dots representing single-atomic Ru and hexagonal RuNP with distinct crystal planes (002) and (101), the RuNP–RuSA@CFN-800 exhibited the coexistence of Ru NP and SA (Fig. 14B). The XANES spectra displayed in Fig. 14C indicate that the RuNP–RuSA@CFN-800 exhibits a similar absorption edge to Ru foil, suggesting a close chemical valence of Ru in the material, which is primarily attributed to the presence of RuNP. However, in the range of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 131–22150 eV, the spectrum of RuNP–RuSA@CFN-800 shows a flattened profile between Ru foil and RuO2, indicating the coexistence of Ru NP and SA.364 Fourier-transformed Ru K-edge EXAFS analysis provides further insights into the chemical coordination state of RuNP–RuSA@CFN-800 (Fig. 14D). The dominant peak at around 2.34 Å corresponds to the Ru–Ru bond in Ru NP, while the peak at 1.53 Å corresponds to atomic Ru coordinated with N derived from imidazole.383 The coordination numbers (CN) for Ru–Ru and Ru–N were estimated to be 5.0 and 1.5, respectively, indicating the presence of a Ru–N4 moiety in RuNP–RuSA@CFN-800 (Fig. 14E). Additionally, the Ru atomic ratio between single atoms and nanoparticles was determined to be approximately 0.86, confirming the coexistence of Ru NP and single atomic RuN4 species.364,384 Analysis of wavelet transform for the Ru K-edge EXAFS spectra reveals a dominant peak at 8.13 Å−1 corresponding to the Ru–Ru bond and a minor peak at 4.02 Å−1 assigned to single atomic Ru, providing further confirmation of the presence of both Ru NP and single atomic RuN4 species in RuNP–RuSA@CFN-800 (Fig. 14F).385
131–22150 eV, the spectrum of RuNP–RuSA@CFN-800 shows a flattened profile between Ru foil and RuO2, indicating the coexistence of Ru NP and SA.364 Fourier-transformed Ru K-edge EXAFS analysis provides further insights into the chemical coordination state of RuNP–RuSA@CFN-800 (Fig. 14D). The dominant peak at around 2.34 Å corresponds to the Ru–Ru bond in Ru NP, while the peak at 1.53 Å corresponds to atomic Ru coordinated with N derived from imidazole.383 The coordination numbers (CN) for Ru–Ru and Ru–N were estimated to be 5.0 and 1.5, respectively, indicating the presence of a Ru–N4 moiety in RuNP–RuSA@CFN-800 (Fig. 14E). Additionally, the Ru atomic ratio between single atoms and nanoparticles was determined to be approximately 0.86, confirming the coexistence of Ru NP and single atomic RuN4 species.364,384 Analysis of wavelet transform for the Ru K-edge EXAFS spectra reveals a dominant peak at 8.13 Å−1 corresponding to the Ru–Ru bond and a minor peak at 4.02 Å−1 assigned to single atomic Ru, providing further confirmation of the presence of both Ru NP and single atomic RuN4 species in RuNP–RuSA@CFN-800 (Fig. 14F).385
 | ||
| Fig. 14 (A) Schematic synthesis of RuNP–RuSA@CFN-800. (B) AC-HAADF-STEM images showing single-atomic Ru (yellow circles (C)and (D) XANES and Fourier-transformed EXAFS spectra of RuNP–RuSA@CFN-800. (E) EXAFS fitting in R space, highlighting Ru–Ru (black) and Ru–N (blue) paths. (F) Wavelet transform of Ru K-edge EXAFS for RuNP–RuSA@CFN-800. Reproduced with permission from ref. 382 Copyright 2013, Wiley-VCH. (G) Preparation of Rud=9.3Å, Rud=7.0Å, and Rud=2.4Å. (H) AC-HAADF-STEM image of Rud=7.0Å. (I) EXAFS spectra for Ru K-edge of Ru foil, RuO2, Rud=9.3Å, Rud=7.0Å, and Rud=2.4Å. Reproduced with permission from ref. 386 Copyright 2024, Wiley-VCH. (J) Synthesis illustration (K) TEM image (L) AC-TEM images of Ru1/D-NiFe LDH. (M) and (N) XANES spectra at Fe and Ni K-edges for Ru1/D-NiFe LDH, Fe/Ni foil, and Fe2O3/NiO. (O) and (P) Fourier-transform EXAFS spectra from (M) and (N). Reproduced with permission from ref. 387 Copyright 2021, Nature Publishing Group. | ||
Recent advancements in correlated single-atomic site catalysts (c-ASCs) with tailored intersite metal–metal interactions show significant potential for optimizing catalytic performance. Unlike traditional ASCs, c-ASCs feature metal atoms in paired, site-synergistic configurations at higher ASC loadings, enhancing catalytic properties.388 A few studies demonstrated that quasipaired single sites significantly improve the activity and selectivity of c-ASCs in electrocatalytic reactions.389–391 Long-range interaction (LRI) between adjacent metal atoms in c-ASCs, influenced by electrostatic forces and single-atomic interdistance (SAD), plays a crucial role in shaping the local geometries and electronic structures of catalytic metal atoms, thereby impacting performance.392,393 Research has shown that modulating SADs near peaks or valleys of a wave intensifies LRI, enhancing catalytic reaction kinetics. A recent study regulated SADs to assess LRI's impact on c-ASCs' electrocatalytic reactivity, designing and synthesizing three Ru-based catalysts with SADs of 2.4 Å, 7.0 Å, and 9.3 Å using planar organometallic molecular design and π–π molecule–carbon nanotube confinement.386Fig. 14G outlines the synthetic approach for preparing c-ASCs with varying SADs between two neighboring Ru atoms. Continuing efforts to develop versatile N-heterocyclic carbene (NHC) ligands for catalysis led to the design of two NHC ligands: one with a phenyl ring and the other with a naphthyl group. Ru was selected for its coordination ability with various NHC ligands, generating well-defined NHC–Ru complexes as ideal Ru precursors. The dinuclear NHC–Ru complexes ([Ru]–benzene and [Ru]–naphthalene) with atomic distances of 9.3 and 7.0 Å, respectively, were successfully obtained. Additionally, the commercial [Ru(p-cymene)I2]2 with a shorter atomic distance (2.4 Å) was used. AC HAADF-STEM study confirmed the presence of paired, uniform bright dots corresponding to the Ru precursors (Fig. 14H). Ru K-edge XANES spectra indicated an average valence state between Ru(0) and Ru(4). EXAFS spectra provided a comprehensive understanding of the local structure of the samples, showing good fittings for the coordination environment of each Ru atom (Fig. 14I). The findings highlight that manipulating the coordination environment and SAD in c-ASCs can significantly enhance their catalytic efficiency and stability, advancing electrocatalysis for sustainable energy applications. The study found that decreasing SADs reduced the overpotential of the alkaline HER. Specifically, the Ru catalyst with an SAD of 7.0 Å showed the highest mass activity over 100 A mg−1 (at η = 100 mV) and intrinsic activity, with HER performance reaching 17.92 s−1 (at η = 50 mV). This performance was 143.4 times and 228.0 times higher than that of 20 wt% Pt/C and 5 wt% Ru/C, respectively, showcasing exceptionally high electrocatalytic activity among the Ru-based catalysts.
Layered double hydroxides (LDHs) based on 3d transition metals have shown promise as electrocatalysts due to their unique structure and abundant active sites.394–396 Various strategies have been developed to enhance the catalytic activity of LDHs, including defect engineering, morphology control, and charge transfer regulation.397–399 Defect engineering is particularly effective in stabilizing ASCs that are prone to aggregation. 2D LDHs offer an ideal platform for ASC stabilization due to their flat facet, thin structure, and large surface area.400,401 However, LDH catalysts for the HER in alkaline media often face challenges such as high energy barriers and slow water dissociation kinetics. Stabilizing single atoms on LDHs for both water oxidation and reduction in the same alkaline electrolyte remains a challenge. While various synthesis techniques have been explored, there is still a need for a simple large-scale protocol to stabilize single atoms on supports.150,339 One promising approach is introducing defects into the support, facilitating the interaction and stabilization of single atoms.402,403 By combining defect engineering with 2D LDHs supported single atoms, it becomes possible to design highly active single atoms stabilized on defective LDH supports, enabling exceptional performance in overall HER in alkaline electrolytes. For example, a ruthenium catalyst supported on defective nickel–iron LDH nanosheets (Ru1/D-NiFe LDH) has demonstrated remarkable HER performance, outperforming commercial Pt/C catalysts.387 Ru1/D-NiFe LDH was synthesized through a straightforward electrodeposition and etching procedure (Fig. 14J). Detailed characterization using HAADF-STEM and XXAFS spectroscopy confirmed the presence of single Ru atoms and provided insights into the local atomic structures of Ni, Fe, and Ru sites (Fig. 14K–P). Carbon aerogels, which contain atomically dispersed Ru sites and Ru nanoclusters, were fabricated pyrolytically using biomass hydrogels. These aerogels demonstrated remarkable performance in the HER across a wide pH range (0 to 14), surpassing commercial Pt/C catalysts in terms of ultrahigh mass activity and stability.404 Interestingly, the HER activity exhibited a significant increase with a higher fraction of Ru single atom sites. Additionally, a single atomic Ru doping technique was employed to introduce Ru into the WO3 lattice, resulting in an excellent HER activity spanning the entire pH range and remarkable stability even at high current densities. In acidic, alkaline, and neutral media, the single atomic Ru-doped WO3 catalyst exhibited very low overpotentials. This catalyst maintained its stability for 100 hours at ampere-level current density.405 Furthermore, leveraging theoretical predictions, a novel single-atomic Ru-based catalyst was developed, utilizing individual Ru atoms supported on a MoS2 nanosheets array anchored to a carbon cloth substrate. This scalable array-catalyst demonstrated outstanding HER performance, making it suitable for pH-universal hydrogen evolution as binder-free cathodes.406
:1 showing the best performance.
 | ||
| Fig. 15 (A) Schematic synthesis of Ni/Ni-SA-NC. (B) HAADF-STEM images of Ni/Ni-SA-NC. (C) XANES spectra at the Ni K-edge. Reproduced with permission from ref. 416 Copyright 2023, The Royal Society of Chemistry. (D) Schematic illustration for the preparation of Ni–N2+2–S and Ni–N4. (E) AC-HAADF-STEM of Ni–N2+2–S (red circles are Ni atoms). (F) k2-weighted FT spectra in the R space of Ni–N2+2–S. (G) WT contour plot of Ni–N2+2–S. Reproduced with permission from ref. 417 Copyright 2022, American Chemical Society. (H) Schematic illustration of Ni SA@N-Ti3C2Tx preparation. (I) HAADF-STEM image of Ni-SAM. (J) EDS elemental mapping of Ni SA@N-Ti3C2Tx. (K) Ni K-edge EXAFS spectra of NiO, Ni foil, and Ni SA@N-Ti3C2Tx. Reproduced with permission from ref. 418 Copyright 2024, The Royal Society of Chemistry. (L) Schematic synthesis MCM@MoS2–Ni. (I) MoS2 nanosheets growth, (II) Surface decoration of isolated Ni atoms. (M) Experimental XANES spectra and (N) Ni K-edge EXAFS spectra of MCM@MoS2–Ni and Ni foil. Reproduced with permission from ref. 419 Copyright 2018, WILEY-VCH. | ||
Further characterization using HAADF-STEM confirmed the uniform dispersion of single Ni atoms on the carbon substrate (Fig. 15B), while XANES analysis revealed changes in the valence state of Ni, indicative of Ni–N bond formation and the impact of AlN etching (Fig. 15C).77,420 While atomic Ni–N–C electrocatalysts have shown promise for alkaline hydrogen evolution reactions, their intrinsic activity and the specific characteristics contributing to their performance remain to be fully understood. Comparative analyses reveal that Ni–N–C ASCs typically exhibit lower HER activity than other transition metal-doped carbon catalysts, primarily due to their moderate reaction kinetics. This limitation arises from the inherently sluggish water adsorption and dissociation processes associated with Ni–N–C ASCs.421 To address this issue, hierarchical porous carbon microstructures were utilized as scaffolds to create unique Ni–N2+2–S active sites, which significantly enhance the Volmer reaction kinetics.417 DFT calculations revealed an upshift in the d-band center (εd) of the edge-hosted Ni–N2+2–S sites compared to pristine Ni–N4, resulting in more stabilized OH adsorption. Furthermore, the synergetic interplay between Ni and S atoms led to a decoupled regulation of intermediate species adsorption strength and facilitated energetic water dissociation kinetics. To achieve the construction of Ni–N2+2–S active sites, sodium thiosulfate was deliberately employed as an additive in the molten salt system during the synthesis process (Fig. 15D). Through high-temperature pyrolysis within a molten-salt reaction environment, deliberate formation of Ni–N2+2–S active sites was achieved, as evidenced by the confirmation from HAADF-STEM images and XANES analysis (Fig. 15E–G). The Ni–N2+2–S catalyst exhibited a low working overpotential and a small Tafel slope, comparable to transition metal-based electrodes, indicating its superior activity in alkaline media. These results establish a clear structure–activity correlation for Ni–N2+2–S and provide valuable insights for further investigations on edge-hosted atomic M–N2+2 active sites. A recent development involves the use of defective graphene (DG) with a high density of defects as a substrate for efficient anchor sites, facilitating strong charge transfer between metal atoms and carbon atoms.422 This concept led to the formation of an integrity known as aNi@defect, where atomic Ni species (aNi) are trapped within the graphene defects. This unique configuration results in aNi@Divacancy and aNi@5775 coordination, which have a significant impact on local electronic structures and enhance the electrocatalytic performance for hydrogen and oxygen evolution. A facile and cost-effective approach was used to fabricate a highly stable and atomically dispersed Ni catalyst on DG (A-Ni@DG) using an incipient wetness impregnation method followed by acid leaching.423 The Ni loading achieved was up to 1.24 wt%, as confirmed by HAADF-STEM analysis and linear combination fitting analysis of XANES. The HAADF-STEM analysis clearly demonstrated the trapping of aNi within the graphene defects, forming the integrity of aNi@defect.
Choosing an appropriate support is essential for optimizing the coordination configuration to boost catalytic performance. MXenes, a category of two-dimensional materials, are composed of transition-metal carbides, nitrides, or carbon nitrides.424,425 Exfoliated MXene sheets possess reactive vacancy defects that enhance their reductive properties, offer large specific surface areas, and feature unique surface characteristics. Their conductivity and hydrophilicity are improved through surface functionalization and tunable structures. Additionally, Ti-deficient defects in MXenes provide sites to host and stabilize single metal atoms, making MXene-based Ni ASCs highly desirable for enhancing HER performance.426 Yang et al. developed isolated Ni atoms supported on N-doped Ti3C2Tx (Ni SA@N-Ti3C2Tx) using a vacancy-anchoring and N-coordination strategy.418 This involved synthesizing Ti3C2Tx nanosheets by immersing the parent Ti3AlC2 MAX phase in a mixture of LiF and HCl, followed by ultrasonic exfoliation (Fig. 15H). Mixing the NiCl2·6H2O precursor with the Ti3C2Tx suspension facilitated the self-reduction of Ni2+. Subsequent annealing adjusted the coordination configuration of central Ni atoms. Ni SA@N-Ti3C2Tx, along with a comparison sample without N coordination (Ni SA@Ti3C2Tx), was prepared. HAADF-STEM imaging showed no Ni nanoparticles, and mapping images confirmed the homogeneous dispersion of Ti, C, N, and Ni across the Ti3C2Tx nanosheets. AC HAADF-STEM imaging indicated the presence of isolated Ni atoms firmly anchored within Ti vacancies (Fig. 15I and J). XAFS analysis revealed that Ni species could form a Ni–N1C1 configuration in Ni SA@N-Ti3C2Tx by adjusting the annealing temperature, achieving precise control over the coordination environment. The adsorption edge position indicated a unique electronic structure of Niδ+ due to metal–support interactions. FT-EXAFS displayed a clear peak at 1.45 Å due to Ni–C/N bonds and no detectable Ni–Ni coordination peaks, confirming the presence of isolated Ni atoms (Fig. 15K).427 Ni SA@N-Ti3C2Tx showed excellent HER activity with an overpotential of 63 mV at 10 mA cm−2, highlighting the potential of MXene-based ASCs in HER applications.
Metal oxides, metal phosphides and metal sulfides, apart from carbon-based substrates, have attracted attention due to their layered structure and unique electronic configuration. One such example is molybdenum disulfide (MoS2).428,429 However, the inactive basal plane and limited electron transfer capacity of MoS2 hinder its electrocatalytic performance.430 To overcome these limitations, authors present a novel approach involving the incorporation of isolated Ni atoms onto hierarchical MoS2 nanosheets supported by multichannel carbon matrix (MCM) nanofibers, denoted as MCM@MoS2–Ni.419 This integration of single-atom Ni modification effectively activates the S atoms in the basal plane, resulting in modified electronic structures that facilitate water dissociation and subsequent catalytic reactions. The synthesis process entails annealing electrospun polystyrene–polyacrylonitrile nanofibers to obtain MCM nanofibers, which serve as the carbonaceous matrix. The hybrid MCM@MoS2–Ni nanofibers optimize both the electronic structure and architecture, facilitating efficient electron transfer and enhancing hydrogen evolution activity (Fig. 15L). XANES analysis confirms electron transfer from Ni to S, leading to the formation of positively charged Ni centers (Fig. 15M). Notably, EXAFS spectra reveal the absence of Ni–Ni bonding and instead demonstrate the presence of Ni–S bonding, indicating the successful decoration of isolated Ni atoms (Fig. 15N).431 Leveraging the unique tubular structure and basal plane modulation, the newly developed MoS2 catalyst exhibits exceptional hydrogen evolution activity and remarkable stability. In a recent study, researchers have implemented a strategy known as self-templating transformation to synthesize Ni-doped CoP hollow polyhedron frames (HPFs) as catalysts for the HER in both acidic and alkaline media.432 Through precise control of the electronic structure and d-band center, the HER process was significantly accelerated. Notably, the Ni–CoP/HPFs catalyst exhibited remarkable electrocatalytic activity.
 | ||
| Fig. 16 (A) Schematic synthesis of CoNC-SA/N*−C using the doping–adsorption–pyrolysis strategy. (B) AC-HAADF-STEM, (C) Co K-edge spectra, (D) k3-weighted FT-EXAFS spectra and (E) WT-EXAFS of CoNC-SA/N*−C. Reproduced with permission from ref. 439 Copyright 2022, American Chemical Society. (F) Schematic preparation of Co-SAS/HOPNC. (G) AC-HAADF-STEM images of the Co-SAS/HOPNC (light-yellow circles are single Co atoms). (H) XANES spectra and (I) FTEXAFS curves and (J) WT-EXAFS of Co-SAS/HOPNC, Co foil, CoO, and Co3O4. Reproduced with permission from ref. 434 Copyright 2018, National Academy of Sciences. | ||
Aberration-corrected HAADF-STEM images confirmed the presence of isolated Co single atoms, providing clear evidence of their atomic dispersion (Fig. 16B). XANES analysis suggests that the chemical state of Co in the CoNC-SA/N*–C catalyst is a mix of Co0 and Co2+ (Fig. 16C). EXAFS analysis provided additional confirmation, revealing a Co–N coordination peak at ∼1.5 Å, characteristic of Co–N3 single-atomic sites (Fig. 16D). Furthermore, the EXAFS-derived coordination number of CoNC-SA/N–C (3) compared to CoSA/N–C (4) reinforced the hypothesis of fewer coordinated nitrogen atoms in the single-atomic sites. Notably, the EXAFS spectra also detected a Co–Co coordination peak at ∼2.2 Å, indicative of the encapsulated Co NCs present in the catalyst, demonstrating the synergistic coexistence of these two features. This observation was further supported by WT analysis, which effectively distinguished Co–Co contributions from Co–N interactions, highlighting the coexistence of single-atomic C–N3 sites and Co NCs (Fig. 16E).
An effective strategy for stabilizing single-metal atoms on a supporting substrate is through strong metal–substrate interactions, which is well-known in the field of electrocatalysis. The specific surface area and porous structure of the supporting material play a crucial role in the design of electrocatalysts, influencing reactant and product transport as well as the accessibility of active sites.139,440–442 While various porous structures have been explored to enhance catalytic properties, achieving precise control over the ordering and homogeneity of pores at multiple scales remains a challenge. To address this, a dual-template cooperative pyrolysis approach was employed to synthesize single-atomic Co sites embedded in hierarchically ordered porous N-doped carbon (Co-SAS/HOPNC).434 This catalyst possesses isolated atomic Co–N4 active sites, a large surface area, high porosity, and good conductivity, resulting in excellent catalytic performance. The fabrication process involved using a silica colloidal crystal and a triblock copolymer as templates, followed by pyrolysis and removal of the silica template (Fig. 16F). The AC HAADF-STEM image confirms the presence of isolated single Co atoms on the N-doped porous carbon matrix (Fig. 16G). The XANES spectra indicate that the valence of Co atoms in Co-SAS/HOPNC is between Co0 and Co2+ (Fig. 16H). The FT EXAFS profile shows a strong peak at approximately 1.32 Å, corresponding to Co–N/C coordination, and no Co–Co interaction peaks are observed (Fig. 16I). Wavelet transform analysis confirms the atomic dispersion of Co in Co-SAS/HOPNC, showing a single intensity maximum at 4 Å−1 for Co–N(C) coordination (Fig. 16J). The results demonstrated the atomically dispersed nature of Co atoms in the catalyst, confirming the effectiveness of the synthesis approach. To fully exploit the dispersing capability of graphene, achieving ASC is crucial. In this regard, a recent study introduced a cost-effective and scalable approach for dispersing Co onto nitrogen-doped graphene.159 The method involved heat-treating graphene oxide and small quantities of cobalt salts in a gaseous NH3 environment. As a result, the cobalt atoms became coordinated with nitrogen atoms on the graphene, effectively functioning as exceptional catalysts for the HER in both acidic and alkaline water environments. In the realm of synthesizing monodispersed atomic Co embedded in nitrogen-doped graphene, a rapid and convenient method has been developed.443 By employing a two-second microwave (MW) heating process, a mixture of amine-functionalized graphene oxide and metal salts undergoes reduction, nitrogen doping, and incorporation of metal atoms into the graphene lattice. This MW approach prevents metal diffusion and aggregation, resulting in the exclusive dispersion of single metal atoms within the graphene lattice. Electrochemical studies confirm the highly active electrocatalytic behavior of Co atoms supported on graphene towards the HER. Furthermore, the catalytic activities of N-doped Co@GY catalysts have been explored by precisely manipulating N doping modes in graphyne.444 The introduction of N induces charge redistribution on the surface of Co@N1-GY catalysts, leading to an increased number of active sites and enhanced HER activity. Additionally, single-atomic site catalysts consisting of Co-g-C3N4/rGO (Co-CNG) have been fabricated, exhibiting comparable HER performance to commercially available Pt/C catalysts.445 Notably, Co-CNG surpasses non-noble transition-metal single-atomic catalysts in alkaline conditions, displaying a mass activity four times higher than that of Pt/C and demonstrating excellent long-term durability. In the field of molybdenum disulfide (MoS2) electrocatalysis, innovative approaches have been employed to enhance catalytic performance.446–448 One method involves synthesizing GQD/Co-MoS2 through a hydrothermal reaction, resulting in a catalyst that exhibits comparable performance to commercial Pt/C catalysts in alkaline medium.447 Meanwhile, another study focuses on Co1/MoS2 catalysts, where Co single atoms are anchored on MoS2 nanosheets.448 This approach activates the MoS2 basal plane for hydrogen generation, leading to superior HER activity and long-term stability.
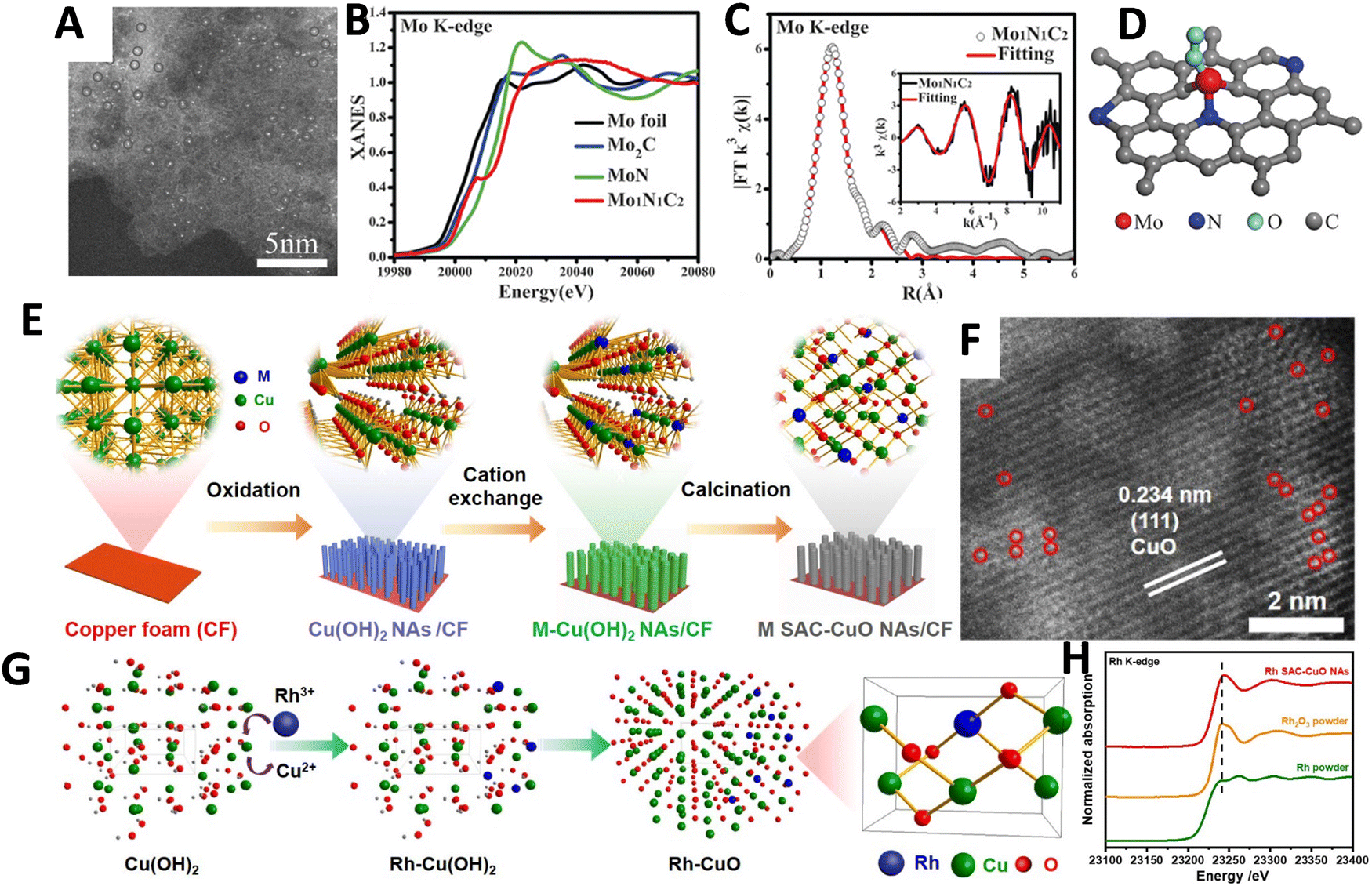 | ||
| Fig. 17 (A) AC-STEM image of Mo1N1C2 (B) XANES curves, (C) FTEXAFS fitting curves at Mo K-edge, inset: k space fitting and (D) Atomic structure model of the Mo1N1C2. Reproduced with permission from ref. 207 Copyright 2017, Wiley-VCH. (E) Schematic synthesis of Cu(OH)2 NAs/CF, Rh–Cu(OH)2 NAs/CF, and Rh ASC–CuO NAs/CF. (F) AC-HAADF-STEM image of Rh ASC–CuO NAs. (G) Scheme shows the cation exchange mechanism during the transformation from Cu(OH)2 to Rh–CuO. (H) K-edge XANES spectra of Rh ASC–CuO NAs. Reproduced with permission from ref. 453 Copyright 2020, American Chemical Society. | ||
In recent studies, researchers have explored the exceptional catalytic performance of rhodium (Rh) metal nanoparticles and phosphides in the HER reaction.454,455 Notably, Rh-doped SrTiO3 has demonstrated superior photocatalytic hydrogen evolution performance.453 In one study, Rh–N–C catalysts were successfully synthesized by annealing a mixture of graphene oxide (GO) and rhodium salt under an ammonia atmosphere. The presence of oxygen-rich functional groups on the GO surface facilitated high dispersion and coordination with Rh3+ ions, while a high-temperature nitriding strategy led to the formation of Rh–N bonds.456 The resulting atomic rhodium catalyst displayed remarkable stability and performance in both acidic and alkaline media for both the ORR and HER, exhibiting significantly low overpotentials. Another study utilized a cation exchange approach to fabricate single-atomic Rh-doped CuO nanowire arrays on a copper foam (CF) electrode for alkaline water splitting.456 The incorporation of Rh greatly enhanced the performance of CuO nanowires. Through a three-step procedure, three-dimensional Rh single-atomic site catalysts supported on CuO nanowires were achieved (Fig. 17E), with STEM images confirming the presence of monodispersed bright spots representing individual Rh atoms (Fig. 17F). XANES and EXAFS spectra provided insights into the atomic-level electronic and structural information of Rh species. The obtained results indicated the successful atomic dispersion of Rh within the CuO nanowires, while the chemical state of Rh in the hybrid material resembled Rh2O3 with a trivalent valence state (Fig. 17G). The Rh single-atomic site catalyst exhibited exceptional catalytic activity for HER, approaching the performance of Pt/C/CF, and demonstrated remarkable stability. In a different investigation, it was observed that the presence of rhodium single atoms confined within MoS2 (Rh–MoS2) can trigger a highly efficient HER activity specifically on the sulfur sites within the MoS2 lattice.457 The HER performance is enhanced through a synergistic effect that occurs at a controlled distance between the confined rhodium atoms. By optimizing the concentration of rhodium, an impressively low overpotential was achieved at a high current density. This exceptional performance not only approaches that of Pt but also surpasses the activity of most MoS2-based catalysts reported thus far in acidic solutions. These findings highlight the potential of Rh-based ASCs for efficient hydrogen evolution and water splitting applications.
Palladium (Pd0) catalysts play a crucial role in various catalytic reactions, including Heck and cross-coupling reactions.458,459 A recent study focused on exploring the atomic-level structure of these Pd0 catalysts and their correlation with HER activity.460 The study introduced a zero-valent electrocatalyst system by anchoring Pd0 onto GDY, leading to a remarkable enhancement in HER performance. The successful anchoring of individual Pd0 atoms onto the GDY substrate was confirmed through both experimental and theoretical investigations. To prepare the Pd0/GDY catalyst, the GDY substrate was securely attached to a working electrode clip, forming a three-electrode system with a graphite counter electrode and a saturated calomel reference electrode. Electrochemical deposition was performed by immersing the GDY electrode in a PdCl2 solution with H2SO4 electrolyte under galvanostatic conditions for 10 seconds at a current density of 2 mA cm−2. Subsequently, the Pd0/GDY catalyst underwent thorough washing with H2SO4, deionized water, and H2SO4 to remove any impurities before conducting electrochemical measurements. The HAADF-STEM images of Pd0/GDY nanosheets, obtained from different sample batches, clearly showed the uniform distribution of individual Pd atoms (depicted as white dots) on the GDY substrate, indicating the homogeneous dispersion of Pd atoms without any signs of aggregation (Fig. 18A and B). The EXAFS spectrum of Pd–GDY exhibited a prominent peak at approximately 1.5 Å, which is smaller than the Pd–Pd contribution observed in Pd foil, indicating the presence of exclusively dispersed Pd atoms in Pd–GDY (Fig. 18C). The XANES spectra of Pd/GDY and Pd foil displayed identical main peaks, providing strong evidence for the anchoring of zero-valent Pd atoms on the GDY substrate (Fig. 18D). The successful anchoring process of single Pd atoms on GDY was found to be consistent and reproducible across different regions. The analysis of HAADF images revealed that the individual Pd atoms anchored on GDY had an average diameter of 3.6 ± 0.1 Å, which aligns with the typical size of a Pd atom. Remarkably, the Pd0/GDY samples exhibited outstanding electrocatalytic activity, including a low overpotential, high mass activity, and a large turnover frequency, surpassing the performance of 20% Pt/C. This exceptional HER performance can be attributed to the unique and well-defined chemical and electronic structures of Pd0/GDY. In a recent study, a highly active and durable catalyst for the HER based on MoS2 was reported.461 The catalyst's surface basal plane was chemically activated through a thermodynamically spontaneous interfacial redox reaction between MoS2 and Pd(II). This reaction resulted in the atomically doping of Pd into the original Mo sites, leading to the generation of sulfur vacancies, conversion to the stabilized 1T phase, stabilization of defective sites, and activation of the 1T basal plane. The resulting Pd–MoS2 catalyst exhibited exceptional HER performance in acidic conditions, surpassing the performance of heteroatom-doped MoS2-based materials. It also demonstrated excellent cycling stability and remarkable resistance to leaching, making it a promising candidate for sustainable catalytic applications.
 | ||
| Fig. 18 (A) HAADF images and (B) STEM-HAADF image of the Pd0/GDY nanosheet. (C) and (A) EXAFS spectra and (D) XANES spectra at the Pd K-edge of Pd0/GDY and Pd foil. Reproduced with permission from ref. 460 Copyright 2019, Cell Press. (E) Synthesis representation of the synthesis process for Cu SA-modified 1T-MoS2. (F) HAADF-STEM analysis of Cuads@MoS2. (G) XAS analysis at the Cu K-edge and (H) Mo K-edge for Cuads@MoS2 and Cusub@MoS2. (I) FT-EXAFS spectra of Cuads@MoS2 in R-space (open circles) along with the corresponding best-fitting lines (solid lines). Reproduced with permission from ref. 462 Copyright 2022, American Chemical Society. | ||
Cu single atoms have also been investigated for their potential in the HER. In a recent study, the coordination structures of Cu SAs on 1T-MoS2 were examined, specifically the substitution model (Cusub@MoS2) and the adsorption model (Cuads@MoS2).462 These models involve Cu SAs replacing Mo atoms or adsorbing onto S atoms on the basal plane of MoS2, respectively. Distinguishing between these models and understanding their coordination geometries, electronic states, and catalytic activities has been a challenge in previous research. Cuads@MoS2 was synthesized using a one-pot hydrothermal method with an Anderson-type polyoxometalate precursor, while Cusub@MoS2 was synthesized through an implantation strategy involving the reaction of pre-synthesized 1T-MoS2 with CuSO4·5H2O (Fig. 18E). In HAADF-STEM image, elements with higher atomic numbers exhibit brighter intensities. The presence of a brighter dot (red arrow in L2) confirms the formation of Cuads@MoS2, where a Cu single atom is positioned above a Mo atom (Fig. 18F). The XANES spectra obtained at the Cu K-edge reveal that both Cuads@MoS2 and Cusub@MoS2 exhibit unique valence states between +1 and +2, as indicated by the absorption edges observed between Cu(I)Cl and Cu(II)Cl2 (Fig. 18G). The XANES spectra at the Mo K-edge (Fig. 18H) display discernible differences in the absorption edge positions between Cuads@MoS2 and Cusub@MoS2, indicating distinct local coordination environments surrounding the Mo center due to different Cu SA anchoring sites. The Fourier transforms EXAFS spectra of the Mo K-edge in k-space and R-space further reveal slight variations in the Mo–S bond length, with Cu@MoS2 exhibiting a longer bond length compared to Cusub@MoS2 (Fig. 18I). Additionally, the presence of Mo–O bonds is confirmed in the EXAFS spectra. The XAS results provided evidence of different coordination environments surrounding the Cu and Mo centers in Cuads@MoS2 and Cusub@MoS2. Remarkably, both Cuads@MoS2 and Cusub@MoS2 exhibited significantly reduced overpotentials for HER compared to 1T-MoS2 in acidic conditions, showcasing their enhanced HER performance.
5.2. Bimetal-based ASCs
While ASCs excel in maximizing metal atom utilization and achieving high reaction rates, their performance can be limited in reactions that require neighboring activated sites for synergistic effects. To overcome this limitation, bimetallic strategies have been explored to enhance catalytic properties. Bimetallic ASCs combine the complementary characteristics of two metal elements, resulting in improved activity and selectivity.463,464 Despite their advantages, agglomeration during catalytic reactions remains a significant challenge for ASCs, often compromising stability. A promising yet underexplored alternative involves immiscible bimetal systems, which inherently resist aggregation and provide enhanced durability. Additionally, dual-atom site catalysts (DASCs), composed of isolated atoms of two different metals with richer atomic coordination environments, have demonstrated superior catalytic activities compared to ASCs. DASCs enable the simultaneous regulation of complex reactions, such as overall water splitting, highlighting their potential as next-generation catalysts.465,466 Designing suitable supports with large surface areas and strong coordination environments is critical for stabilizing ASCs and DASCs. Carbon-based materials have been commonly used, but their electrochemical inertness and hydrophobicity hinder practical applications.467 Layered hydroxides, with their low cost and good hydrophilicity, present a promising alternative for ASC support.468 However, the coordination interaction between DASCs and layered hydroxides requires further investigation. Understanding this interaction and the synergistic effects between different single atoms is of paramount importance for advancing ASCs and achieving efficient HER performance. To tackle these challenges, researchers led by Kim developed high-performance electrocatalysts by harnessing the immiscibility of Cu/Ru bimetal atoms.469 Their synthesized electrocatalyst, known as Cu/Ru@GN, features dispersed Ru-SAs on an N-doped graphitic matrix (GN). Additionally, certain Cu-SAs form bridges to the surface of Ru NPs through nitrogen atoms. This unique configuration results in remarkable mass activity and excellent catalytic performance for the HER, surpassing the performance of commercial Pt20wt%/C catalysts. The Cu-SAs and Ru-SAs synergistically enhance electric conduction and charge-transfer rates within the GN template, facilitating fast kinetics. The immiscibility of Cu and Ru prevents aggregation of the Ru-NPs, while the coordination of Cu-SAs by N atoms on the Ru-NPs surface introduces new active sites, ensuring long-term stability even after 600 hours in acidic media. In another study, the researchers synthesized single-atomic Ir and Ru catalysts anchored on mesoporous graphitic carbon nitride (Ir-g-CN and Ru-g-CN) for the HER.470 Remarkably, Ru-g-CN exhibited a high turnover frequency, surpassing Ir-g-CN, commercial Pt/C benchmark, and other advanced HER catalysts. Furthermore, Ru-g-CN demonstrated exceptional mass activity in both acidic and alkaline solutions, along with a high apparent current density, making it highly suitable for practical applications. Both Ru-g-CN and Ir-g-CN exhibited outstanding catalytic stability, maintaining continuous HER activity for 120 hours in acidic and alkaline conditions with minimal degradation. Kumar et al. developed a facile strategy to obtain NiCo-SAD on N-doped carbon (NiCo-SAD-NC) by in situ trapping of metal ions followed by controlled annealing with precisely controlled N-moieties.471 The synthesis involved trapping Ni2+ and Co2+ ions into a polydopamine sphere, followed by annealing at 800 °C to transform the precursor into NiCo-NP encapsulated in an N-carbon matrix (NiCo-NP-NC) (Fig. 19A). Aberration-corrected HAADF-STEM imaging and EEL spectra confirmed the presence of isolated Ni–Co dimer sites along with some isolated Ni or Co atoms (Fig. 19B–E). XAS analysis revealed the emergence of Ni–Co coordination at the atomic level in NiCo-SAD-NC. The Ni K-edge XANES spectra demonstrated a shakedown transition of square-planar coordination with D4h local symmetry, indicating the coordination of Ni with four nearest guest atoms. Similarly, the Co K-edge XANES spectra suggested the presence of X-ray absorbing Co centers with four coordination (Fig. 19F–I). The obtained NiCo-SAD-NC catalyst exhibited exceptional pH-universal HER activity, demonstrating low overpotentials in both acidic and alkaline media. | ||
| Fig. 19 (A) Schematic synthesis strategy for the NiCo-SAD-NC and NiCo-NP-NC. (B) AC-HAADF-STEM image of the NiCo-SAD-NC, highlighting dimer and single Ni/Co atom sites (yellow squares and orange circles, respectively). (C) and (D) Intensity profile and EEL spectra at atomic-scale Ni and Co coordination sites. (E) Statistical Ni–Co distances in observed dimers. Experimental Ni K-edge (F) and Co K-edge (G) XANES spectra of NiCo-SAD-NC along with reference samples, inset zooming in on near-edge and white line features. Experimental Ni K-edge (H) and Co K-edge (I) FT-EXAFS spectra of NiCo-SAD-NC with reference samples. Reproduced with permission from ref. 471 Copyright 2021, Nature Publishing Group. | ||
A novel approach utilizing the proximity electronic effect (PEE) of Ni/Co dual-atom sites (DASs) anchored in an N-doped carbon (N–C) substrate, referred to as NiCo DASs/N–C, has been proposed to enhance the electrocatalytic HER.472 The PEE is achieved through the adjacent positioning of Ni atoms anchored by the Ni–N4 site, resulting in remarkable catalytic activity for HER. The synthesis of NiCo DASs/N–C was accomplished by Li et al. using a MOF-assisted host–guest strategy (Fig. 20A). Initially, Ni and Co atoms were encapsulated within the ZIF-8 cage as guests, while ZIF-8 served as the host material. Subsequent pyrolysis and carbonization steps yielded an N–C substrate with uniformly embedded Ni–N4 and Co–N4 sites in a three-dimensional porous carbon matrix. Comparative studies were conducted with single-atomic Ni–N4 site catalysts (Ni SAs/N–C) and single-atomic Co–N4 site catalysts (Co SAs/N–C) to elucidate the modulation effect of adjacent Ni–N4 moieties on the proximal active Co–N4 sites in NiCo DASs/N–C. The HAADF-STEM images confirmed the homogeneous distribution of Ni, Co, N, and C elements within the NiCo DASs/N–C catalyst, with abundant paired bright spots indicating the presence of diatomic sites at approximately 6.4 Å distances (Fig. 20B and C). These findings highlight the dispersal of active sites and the promising potential of NiCo DASs/N–C for efficient water electrocatalysis. Fang et al. developed a rod-like Ni3S2 material encapsulated with single-atomic Fe–Mo co-modified CoNi-based nanosheets, forming FeMo@CoNi–OH/Ni3S2.473 This dual-atom site catalyst exhibited improved performance for the HER due to the modification of its electronic structure and enhanced conductivity. XANES analysis confirmed the positive valence states of Fe and Mo single atoms in the catalyst (Fig. 20D and E). The successful synthesis of the FeMo@CoNi–OH/Ni3S2 DAC was confirmed by the uniform dispersion of Fe and Mo atoms within the CoNi–OH structure, achieved by lattice matching of Ni(OH)2 and Co(OH)2, resulting in the formation of FeMo@CoNi–OH/Ni3S2 (Fig. 20F). The FeMo@CoNi–OH/Ni3S2 catalyst showed low overpotentials in alkaline, acidic, and neutral solutions, demonstrating its effectiveness.
 | ||
| Fig. 20 (A) Schematic synthesis process of NiCo DASs/N–C. (B) AC-HAADF-STEM, (C) corresponding intensity distributions in areas A1, A2, and A3 shown in Fig. 19B. Reproduced with permission from ref. 472 Copyright 2022, Wiley-VCH. (D) The Fe XANES curves for Fe@CoNi–OH/Ni3S2, FeMo@CoNi–OH/Ni3S2 and reference samples. (E) The Mo XANES curves of Mo@CoNi–OH/Ni3S2, FeMo@CoNi–OH/Ni3S2 and reference samples. (F) Schematic representation of the formation process of FeMo@CoNi–OH/Ni3S2. Reproduced with permission from ref. 473 Copyright 2023, Elsevier. (G) HAADF-STEM images of Pt–Ni(OH)2. FT-HAADF-STEM images viewed from (H) and (I). Intensity profiles correspond to labeled lines in (H), (I) (J) and (K). (L) Fourier transform EXAFS spectra of NiPt, commercial Pt/C, and PtO2. Reproduced with permission from ref. 474 Copyright 2021, American Chemical Society. (M) HAADF-STEM images of (Ru–N)@Pt, (N) Normalized Ru K-edge XANES spectra and (O) normalized Pt L3-edge XANES spectra. Reproduced with permission from ref. 475 Copyright 2021, American Chemical Society. | ||
5.3. Metal alloy SACs
Single-atom alloys (SAAs) are promising materials for catalytic applications due to their unique properties. They consist of an atomically active metal dispersed within a less-active metal matrix, allowing for tailored electronic structure and enhanced catalytic performance. Modifying the coordination site or introducing secondary metal atoms further enhances the intrinsic activity of SAAs.476–481 These materials have shown excellent performance in various catalytic reactions, including oxygen reduction, hydrogen evolution, and CO2 reduction.482–484 SAAs exhibit well-defined coordination environments and can break the limits of scaling relationships. The spillover effect in SAAs enables reactant activation at single-atomic sites, followed by transfer to the matrix surface, preventing over-binding of adsorbates.485–487 Recent studies have demonstrated efficient bifunctional electrocatalysts, such as Pt–Ru dimers, Cu–Pt dual sites and RuAu SAAs, for various reactions.229,488,489 SAAs provide opportunities for further modification and expansion of their applications. Synthesis methods such as pyrolysis, atomic layer deposition and laser ablation in liquid have been employed to create stable and active SAA catalysts.229,489 Huo et al. employed Ni(OH)2 nanosheets as templates to produce ultrathin porous Ni nanosheets with a remarkably low Pt loading, referred to as NiPt, for catalyzing the alkaline HER.474 The unique structure of Ni(OH)2 served as an attractive layered template, preserving the structural integrity and preventing lattice expansion during the alloying process. This resulted in the formation of porous and ultrathin nanosheets with excellent mass transfer, electrical conductivity, and a high density of catalytic active sites. The Ni(OH)2 nanoarrays were uniformly prepared on a nickel foam substrate through hydrothermal treatment, and subsequent treatment with chloroplatinic acid pentahydrate solution yielded Pt–Ni(OH)2, which retained the original flake array shape. Further reduction in an Ar/H2 atmosphere transformed the Pt–Ni(OH)2 into porous NiPt alloy nanosheets. High-resolution STEM imaging demonstrated the even distribution of Pt single atoms occupying the lattice sites of Ni(OH)2, confirming the formation of a single-atom NiPt alloy catalyst (Fig. 20G–K). EXAFS spectroscopy confirmed the local structure of Pt single atoms in NiPt (Fig. 20L). A major peak at 2.27 Å indicated a shorter Pt bond length compared to Pt foil, attributed to electron loss in Pt. The NiPt single-atom alloy exhibited outstanding alkaline HER performance, surpassing commercial Pt/C and other reference catalysts, with low overpotential, high mass activity and exceptional stability. N-doped Pt–Ru single atom alloy electrocatalysts were synthesized by dispersing Pt single atoms onto Ru nanoparticles through thermal annealing in an ammonium atmosphere, resulting in the formation of (Ru–N)@Pt.475 HAADF-STEM imaging confirmed the presence of Pt single atoms. The heavier Pt atoms appeared as brighter spots compared to Ru and the carbon support, with isolated Pt atoms uniformly dispersed on the Ru/CNT surface, as shown in Fig. 20M. Synchrotron-based XANES and XAFS spectroscopy confirmed the incorporation of nitrogen and Pt into the Ru lattice, with Pt existing as individual atoms and nitrogen coordinating with both Ru and Pt metal sites (Fig. 20N and O). The (Ru–N)@Pt catalyst exhibited exceptional catalytic performance for the alkaline HER, demonstrating a remarkably low overpotential and a low Tafel slope in alkaline media. These results outperformed commercial Pt/C and Ru/C catalysts, underscoring the superior performance of the (Ru–N)@Pt single-atom alloy catalyst. Another study prepared atomically dispersed Cu–Pt dual sites alloyed with Pd nanorods involved a two-step approach.323 Initially, atomically dispersed Cu was generated on ultrathin Pd nanorods by introducing a Cu precursor into the in situ prepared Pd nanosheets, utilizing CO as a surface-confining and reducing agent. The resulting Pd/Cu aqueous dispersion was then mixed with Pt2+ precursor in an acid solution and subjected to ultrasonic treatment to synthesize the Cu–Pt dual sites alloyed with Pd nanorods. The XAFS measurements confirmed the ultrafine structure of the atomically dispersed Cu–Pt dual sites. The Pd/Cu–Pt nanorods exhibited remarkable HER properties in acidic solutions, demonstrating a low overpotential and a high activity. The catalyst displayed superior HER activity, surpassing that of commercial Pt/C catalysts.6. Performance of ASCs
6.1. Activity, selectivity, stability
ASCs have found widespread application in electrocatalytic HER, where their activity relies on the abundance of exposed active sites.315,316Table 1 presents a summary of ASCs recently reported in the literature for HER, detailing their overpotential and stability across different conditions. Among the metals, Pt stands out as the most efficient and active HER electrocatalyst, due to its remarkable properties such as low overpotential, small Tafel slope, and a strong hydrogen adsorption affinity.324 Carbon-based substrates, particularly graphene, offer a wealth of anchoring sites that effectively stabilize and disperse single atoms. Moreover, the carbon matrix enhances electrical conductivity and influences the electronic structure and charge density of adjacent metal atoms.490,491 Researchers have developed strategies to synthesize Pt single-site catalysts on graphene, such as Pt SAs/DG, which exhibit superior performance for the HER.336 Pt SAs/DG shows low overpotential (23 mV at 10 mA cm−2) and fast electron transfer. Another approach involves anchoring Pt atoms on carbon nanotubes using an ultralow-temperature solution reduction method, resulting in a low HER overpotential (41 mV at 10 mA cm−2).340 Onion-like nanospheres of carbon have also been used to anchor stable atomically dispersed Pt, forming Pt1/OLC catalyst with excellent HER performance (low overpotential of 38 mV at 10 mA cm−2 and high turnover frequencies).339 These catalysts outperform or rival commercial Pt/C catalysts. Heteroatoms like N exhibit strong coordination affinity, further facilitating the anchoring of non-noble metal single atoms.316 In a recent study, a facile microwave reduction method was employed to synthesize single Pt atoms anchored on aniline-stacked graphene with exceptional HER performance.332 This method involved aniline anchoring and microwave reduction, resulting in exclusive dispersion of Pt atomic sites on graphene. Notably, the presence of aniline molecules modified the d-electron structure of Pt atoms, crucially enhancing HER activity. The as-prepared catalyst, with a Pt loading of 0.44 wt%, exhibited unexpectedly high catalytic activity, showcasing negligible onset potential and an overpotential of only 12 mV at 10 mA cm−2 in a 0.5 M H2SO4 solution. Furthermore, it demonstrated excellent stability and durability, exhibiting no significant current deterioration or structural changes during long-term catalysis. A nitrogen-doped porous carbon nanocages catalyst was prepared by Wang et al. through etching metal–organic framework ZIF-8 with tannic acid, followed by high-temperature pyrolysis.335 Platinum single atoms and clusters were formed on the carbon nanocages after adsorption of chloroplatinic acid and calcination. The NPCN–Pt catalyst exhibited excellent alkaline HER catalytic activity, surpassing Pt/C, Pt clusters, and Pt particles supported on porous carbons. It demonstrated a low overpotential (39 mV at 10 mA cm−2) and significantly higher turnover frequency and mass activity compared to Pt/C. Jong-Boem Baek group demonstrated a method to achieve ultrahigh mass activity by merging Pt single atoms into Pt nanoclusters supported on phosphorus-doped carbon (PC).347 The catalytic activities of the Pt1@PC and PtM@PC catalysts were evaluated for the HER in an acidic solution. The PtM@PC800 catalyst with the highest merging level exhibited significantly enhanced catalytic activity (Fig. 21A), as indicated by the lower charge transfer resistance and smaller Tafel slope compared to Pt/C and Pt1@PC (Fig. 21B). It achieved low overpotentials for achieving current densities of 10 and 50 mA cm−2 (Fig. 21C). The mass activities of the catalysts were also evaluated, with PtM@PC800 showing a mass activity over 141 times higher than Pt1@PC and Pt/C (Fig. 21D). Furthermore, the mass activity of PtM@PC800 surpasses that of other exceptional catalysts reported in the literature (Fig. 21E).74,200,201,325,339,492–495 The merging of Pt single atoms into nanoclusters resulted in superior intrinsic activity, leading to ultrahigh mass activity. The PtM@PC800 catalyst also demonstrated good electrochemical stability, retaining its microstructure and elemental composition after long-term stability tests. Li et al. explored the impact of active site interaction on the intrinsic activity of ASCs for acidic hydrogen evolution Catalysis.496 By fine-tuning the spatial distance between Pt atoms on graphitic carbon supports (PtSA/GCs), they achieved enhanced active site interaction, resulting in significantly improved intrinsic activity. The PtSA/GCs with optimized active site interaction exhibited remarkable performance, including a high turnover frequency, surpassing commercial Pt/C and other reported catalysts. In another study, Pt atoms and clusters supported on NGNs were synthesized using atomic ALD method.200 The precise control of Pt size and density on NGNs led to superior hydrogen evolution activity compared to conventional Pt nanoparticle catalysts. Xiong et al. developed a novel strategy to stabilize isolated Pt atoms within a nitrogen-containing porous carbon matrix, resulting in a significantly enhanced mass activity for hydrogen evolution compared to commercial Pt/C catalysts.495 Zhang et al. demonstrated the construction of platinum single-site catalysts on hierarchical nitrogen-doped carbon nanocages using an impregnation-adsorption method.330 The optimized catalyst exhibited record-high electrocatalytic performance for hydrogen evolution, surpassing previous platinum-based catalysts. Li et al. utilized a photochemical solid-phase reduction method to fabricate well-defined isolated Pt atoms on a nitrogen-doped porous carbon.167 This catalyst displayed ultrahigh electrocatalytic activity for hydrogen evolution, with exceptional overpotential and mass activity, outperforming commercial Pt/C catalysts by a significant margin.| Catalyst | Substrate/current collector | Electrolyte | Cell for electrolysis | η at 10 mA cm−2 (mV vs. RHE) | Tafel slope (mV dec−1) | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| SAC, single-atom catalyst; NP, nanoparticles; GNF, graphite nanofiber; MEA, membrane electrode assembly; MS, melamine sponge; NW, nanowire; NS, nanosheet; NF, Ni foam; CP, carbon paper; CNT, carbon nanotubes; NC; nitrogen-doped carbon; MMO, mixed metal oxide; CC, carbon cloth; SAD, single-atom dimer; LDH, layered double hydroxide; VG, vertical graphene; NTA, Nanotube; PNPM, 3D porous N, P codoped Ti3C2TX MXene; ML, monolayer; MWCNT, multi-walled carbon nanotubes; SAA, single-atom alloy; m, metallene; CNF, porous carbon nanofibers; NW, nanowires | |||||||
| ASCs | |||||||
| Noble metal-based ASCs | |||||||
| Pt–Ni(OH)2-E | Ni(OH)2 | 1 M KOH | Three-electrode system | 21 | 51 | 80 h at 100 mA cm−2 | 177 |
| Pt SA-PNPM | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 35 | 31 | 5000 cycles | 497 |
| Ru@Co-SAs/N–C | Glassy carbon electrode | 1 M KOH | Three-electrode system | 7 | 30 | 3000 cycles | 498 |
| Pt/MoS2-NTA/ Ti3C2 | Anode aluminum oxide | 0.5 M H2SO4 | Three-electrode system | 32 | 35 | 3000 cycles | 499 |
| Pt@Co SAs-ZIF-NC | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 27 | 21 | 5000 cycles | 500 |
| Pt–Ru/RuO2 | Carbon paper | 1 M KOH | Three-electrode system and MEA | 18 | 18.5 | 100 h at 250 mA cm−2 | 501 |
| Pt–Ni(OH)x | Ni-BDC | 1 M KOH | Three-electrode system | 58 | 84 | 5000 cycles | 178 |
| PtSA@Mo2C@NC | N-doped carbon support | 1 M KOH | Three-electrode system and MEA | 66 (mV per 100 mA cm−2) | 27 | 150 h at 150 mA cm−2 | 502 |
| SA In-Pt NWs/C | Glassy carbon electrode | 1 M KOH | Three-electrode system | 46 | 32.4 | 5 h at 10 mA cm−2 | 503 |
| S-Ru@NiHO/Ni5 P4 | Ni foam | 1 M KOH | Three-electrode system and MEA | 16 | 35.2 | 150 h at 500 mA cm−2 | 504 |
| Pt SASs/AG | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 12 | 29.33 | 2000 cycles | 332 |
| Ru-LC-Ni(OH)2 | Ni foam | 1 M KOH (three-electrode system); 1 M KOH, 1 M KOH seawater (MEA) | Three-electrode system and MEA | 9 | 39.02 | 500 h at 500 mA cm−2 | 505 |
| Pt /NC-AF | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 139 | — | 5000 cycles | 506 |
| Ru–GaSA/N–C | Carbon paper | 1 M KOH | Three-electrode system and MEA | 4 | 21.1 | 100 h at 10 mA cm−2; 170 h at 1 A cm−2 | 507 |
| PtSA–NiSe–V | Carbon cloth (three-electrode system), GDL (MEA) | 1 M KOH | Three-electrode system and MEA | 45 | 52 | 80 h at 390 mA cm−2 | 508 |
| aPt/pNi-NOMC | N-doped ordered mesoporous carbon substrate | 1 M KOH (three-electrode system), 0.1 M KOH (MEA) | Three-electrode system and MEA | 142.2 (mV per 50 mA cm−2) | 40 | 210 h at 500 mA cm−2 | 185 |
| Pt/NGN | Rotating glassy carbon disk electrode | 0.5 M H2SO4 | Three-electrode system | 50 | 29 | 1000 cycles | 200 |
| Pt-SAs/MoSe2 | Glassy carbon electrode | 1 M KOH | Three-electrode system | 29 | 41 | — | 509 |
| Pt-CNT | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 41 | 48.57 | — | 340 |
| Au/NiFe LDH | Ti mesh | 1 M KOH | Three-electrode system | 10 | 25 | 2000 cycles | 401 |
| Cl-Pt/LDH | Ni foam | 1 M KOH | Three-electrode system | 25.2 | 24.33 | 100 h at −50 mA cm−2 | 510 |
| Pt/VG Ar-5 | Carbon fiber | 1 M KOH | Three-electrode system | 124 | 36 | 24 h at −100 mA cm−2 | 511 |
| Ru@MWCNT | Rotating ring-disk electrode | 0.5 M H2SO4 | Three-electrode system | 13 | 27 | 10000 cycles |
512 |
| Mo2TiC2Tx–PtSA | Carbon paper | 0.5 M H2SO4 | Three-electrode system | 30 | 30 | 10000 cycles |
513 |
| Pt-SA/ML-WO3 | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 22 | 27 | 10000 cycles |
514 |
| Non-noble metal-based ASCs | |||||||
| Mo1N1C2 | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 154 | 86 | 1000 cycles | 207 |
| Co–N/C | Glassy carbon electrode | 1 M KOH | Three-electrode system | 178 | 137 | 10 h at −0.3 V (vs. RHE) | 515 |
| Co4N@CoSA/N-CNT/CC | Carbon cloth | 0.5 M H2SO4 | Three-electrode system | 78 | 76.2 | 50 h at −10 mA cm−2 | 516 |
| Ni–MoS2 | Carbon cloth | 0.5 M H2SO4 | Three-electrode system | 110 | 74 | 2000 cycles | 517 |
| Fe1/NC | NC substrate | 1 M KOH | Three-electrode system | 111 | 86.1 | 20 h at −10 mA cm−2 | 518 |
| Ni-N,P/CNFs | Carbon cloth | 0.5 M H2SO4 | Three-electrode system | 38 | 24 | 519 | |
| MoS2/CoSAseNS-CNTs@CoS2/CC | Carbon cloth | 0.5 M H2SO4 | Three-electrode system | 72 | 60 | 5000 cycles | 520 |
| Mn/BN-CNT | Glassy carbon electrode | 1 M KOH | Three-electrode system | 51 | 51 | 36 h at −10 mA cm−2 | 316 |
| CoNx/C | 0.5 M H2SO4 | 133 | 57 | 5000 cycles | 73 | ||
| Ni–NP/PCN | Carbon-based support | 1 M KOH | Three-electrode system and two-electrode cell | 25 | 30 | 100 h at 500 mA cm−2 | 521 |
| Mo@NMCNFs | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 66 | 49 | 3000 cycles | 522 |
| Ni/SGNCs | Rotating glassy carbon disk electrode | 1 M KOH | Three-electrode system | 27 | 38 | 5000 cycles | 523 |
| NiO/Ni@NCNTs | Rotating glassy carbon disk electrode | 1 M KOH | Three-electrode system | 87.5 | 80 | 24 h at −100 mA cm−2 | 312 |
| CoSA/N,S-HCS | Rotating glassy carbon disk electrode | 1 M KOH | Three-electrode system | 165 | 96 | 48 h at −100 mA cm−2 | 524 |
| W-SAC | Glassy carbon electrode | 1 M KOH | Three-electrode system | 85 | 53 | 10000 cycles |
313 |
| Co-NG | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 147 | 82 | 1000 cycles | 159 |
| Bimetal-based ASCs | |||||||
| CuRu@GN | Rotating glassy carbon disk electrode | 0.5 M H2SO4 | Three-electrode system | 10 | 25 | 600 h | 469 |
| NiSAFeSAeNi50Fe/CNT | Carbon cloth | 1 M KOH | Three-electrode system | 64 | 48 | 65 h at −10 mA cm−2 | 525 |
| FeMo@CoNi–OH/Ni3S2 | Ni foam | 1 M KOH | Three-electrode system and two-electrode cell | 89 | 92.2 | 30 h at −15 mA cm−2, 75 h at 10 mA cm−2 | 473 |
| NiCo-SAD-NC | Ni foam/ carbon fiber paper | 1 M KOH | Three-electrode system | 61 | 55 | 15 h at −100 mA cm−2, 50 h at −50 mA cm−2 | 471 |
| Cu/Rh(SAs)+Cu2Rh-(NPs)/GN | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 8 | 27 | 500 h | 526 |
| W1–Mo1–NG | N-doped graphene | 1 M KOH | Three-electrode system | 67 | 45 | 28 h at −100 mA cm−2 (HER) | 527 |
| Ru–NiMoO4-Vo/NF | Glassy carbon electrode | 1 M KOH | Three-electrode system | 49 | 49.6 | 24 h at 10 mA cm−2 | 373 |
| NMCP@NF | Ni foam | 1 M KOH | Three-electrode system | 88 | 70 | 1000 cycles | 528 |
| FePt/NC | Rotating glassy carbon disk electrode | 0.5 M H2SO4 | Three-electrode system | 27 | 28 | 1 h at 10 mA cm−2 | 529 |
| RuAu-0.2 | Glassy carbon electrode | 1 M KOH | Three-electrode system | 202 | 37 | 1000 cycles | 530 |
| NiCo DASs/N–C | Rotating glassy carbon disk electrode | 1 M KOH | Three-electrode system | 189 | 72.5 | 24 h at 10 mA cm−2 | 531 |
| Pt0.47–Ru/Acet | Ni foam | 1 M KOH | Three-electrode system | 17 | 67 | 8000 cycles | 532 |
| Metal alloy SACs | |||||||
| PtSAA-NiFe | Ni foam | 1 M KOH | Three-electrode system | 18 | 29.14 | 48 h at 10 mA cm−2 | 533 |
| Co/Pdm-4 | Carbon Paper | 0.5 M KOH | Three-electrode system | 24.7 | 8.2 | 50 h at 50 mA cm−2 | 34 |
| NiPt | Glassy carbon electrode | 1 M KOH | Three-electrode system | 18 | 45 | 5000 cycles | 474 |
| (Ru–N)@Pt | — | 1 M KOH | Three-electrode system | 15 | 25 | 58 h at −0.1 V vs. RHE | 475 |
| Pd/Cu–Pt | Glassy carbon electrode | 0.5 M H2SO4 | Three-electrode system | 22.8 | 25 | 15 h at −0.24 mV vs. RHE | 323 |
 | ||
| Fig. 21 (A) Polarization curves were obtained for various catalysts in a 0.5 M aqueous H2SO4 solution. (B) Tafel plots corresponding to the polarization curves in (A). (C) Overpotentials measured at 10 and 50 mA cm−2 for different catalysts. (D) Mass activities at overpotentials of 5 and 10 mV, respectively. (E) Mass activities of Pt1@PC and PtM@PC800, compared with other reported noble metal-based catalysts in acidic media. Reproduced with permission from ref. 347 Copyright 2023, American Chemical Society. HER performance in 1 M KOH of various catalysts: (F and G) LSV polarization curves, (H) overpotentials at 10 mA cm−2 and 100 mA cm−2, (I) Tafel slopes, (J) EIS spectra, (K) electrochemical double-layer capacitances, Reproduced with permission from ref. 532 Copyright 2022, Elsevier. | ||
Various support materials, other than carbon-based, have been explored for using ASCs in HER applications. One such design involves a Pt single atom strongly coupled with amorphous MoOx, forming Pt-SA/α-MoOx.344 The synergistic effect between the unsaturated Pt atoms and defective MoOx significantly reduces the reaction barrier, resulting in rapid hydrogen evolution reaction kinetics. This catalyst exhibits an exceptionally high mass activity of 52.0 A mgPt−1 at an overpotential of 50 mV, outperforming previously reported Pt SA-based catalysts for acidic HER. Moreover, Pt-SA/α-MoOx shows Pt-like kinetics with a small Tafel slope of 123 mV dec−1 and maintains excellent long-term electrochemical stability for 20 hours. Chen et al. developed a highly efficient catalyst for HER by atomically dispersing Pt on ultrafine Ru nanoclusters using a one-step cold-plasma technique.532 The resulting Pt0.47–Ru/Acet catalyst demonstrated exceptional alkaline HER activity, outperforming commercial Ru/C and 20 wt% Pt/C. The cooperative effects between Ru nanoclusters and Pt single atoms contributed to the catalyst's enhanced performance (Fig. 21F–H). The Pt0.47–Ru/Acet catalyst displayed a small Tafel slope, indicating faster reaction kinetics with a Volmer–Heyrovsky mechanism (Fig. 21I). Electrochemical impedance spectroscopy and charge transfer resistance measurements confirmed efficient charge transfer at the catalyst–electrolyte interface (Fig. 21J). The catalyst also maintained a high electrochemical active surface area and demonstrated superior mass activity, surpassing other reported Ru-based catalysts (Fig. 21K). The Pt0.47–Ru/Acet catalyst's high TOF and cost-effectiveness make it a promising candidate for HER applications, outperforming previously reported electrocatalysts (Fig. 22A). Jiang et al. introduced a highly efficient electrocatalyst, Pt/np-Co0.85Se, for hydrogen evolution.534 The catalyst, composed of single-atomic platinum decorated nanoporous Co0.85Se, showed superior performance compared to commercial Pt/C at high overpotentials, thanks to its unique bicontinuous nanoporous structure, facilitating improved mass-transfer processes. Pt/np-Co0.85Se achieved an HER current density of −10 mA cm−2 with an overpotential of only −55 mV, whereas np-Co0.85Se required an overpotential of −264 mV for the same current density (Fig. 22B). The catalyst's TOF at −100 mV vs. RHE was 3.93 s−1, surpassing most reported catalysts (Fig. 22C). Gas chromatography analysis confirmed the Pt/np-Co0.85Se's close to 100% Faraday efficiency for H2 production under different applied potentials (Fig. 22D and E). In the research conducted by Liu group, they introduced single-atomic Pt immobilized within the lattice of CoP mesoporous nanosheets grown on carbon fiber cloth with an ultralow Pt loading.361 This catalyst displayed remarkable HER performance and demonstrated excellent durability. In another study, single-atomic Pt was anchored onto 2D Co(OH)2 nanosheets grown on Ag nanowires, leading to an excellent HER activity attributed to the presence of a continuous electron transport pathway and enhanced hydrogen adsorption.535 Additionally, a unique ganoderma-like MoS2/NiS2 hetero-nanostructure featuring isolated Pt atoms showed Pt-like catalytic activity for HER, exhibiting an ultralow overpotential.345 Deng et al. demonstrated the enhanced HER activity of Pt-doped few-layer MoS2 nanosheets (Pt–MoS2) due to the tuned adsorption behavior of H atoms on in-plane S sites.74 Moreover, Yi et al. utilized N-, B-, and F-atom-doped ReS2 supported on MoC2Tx MXene with single-atomic Pt loading, leading to extraordinary catalytic activity and stability for HER and oxygen-related reactions.536 Zhang et al. synthesized a binder-free catalyst comprising single Pt atoms on CoP-based nanotube arrays supported by Ni foam, showing excellent HER performance comparable to commercial Pt/C.201 Ren et al. designed a 3D hierarchical nanostructure with Pt single atoms immobilized on Ni2P nanosheet arrays on nickel foam, achieving outstanding HER activity with a low overpotential and significantly enhanced mass activity compared to commercial Pt/C.346
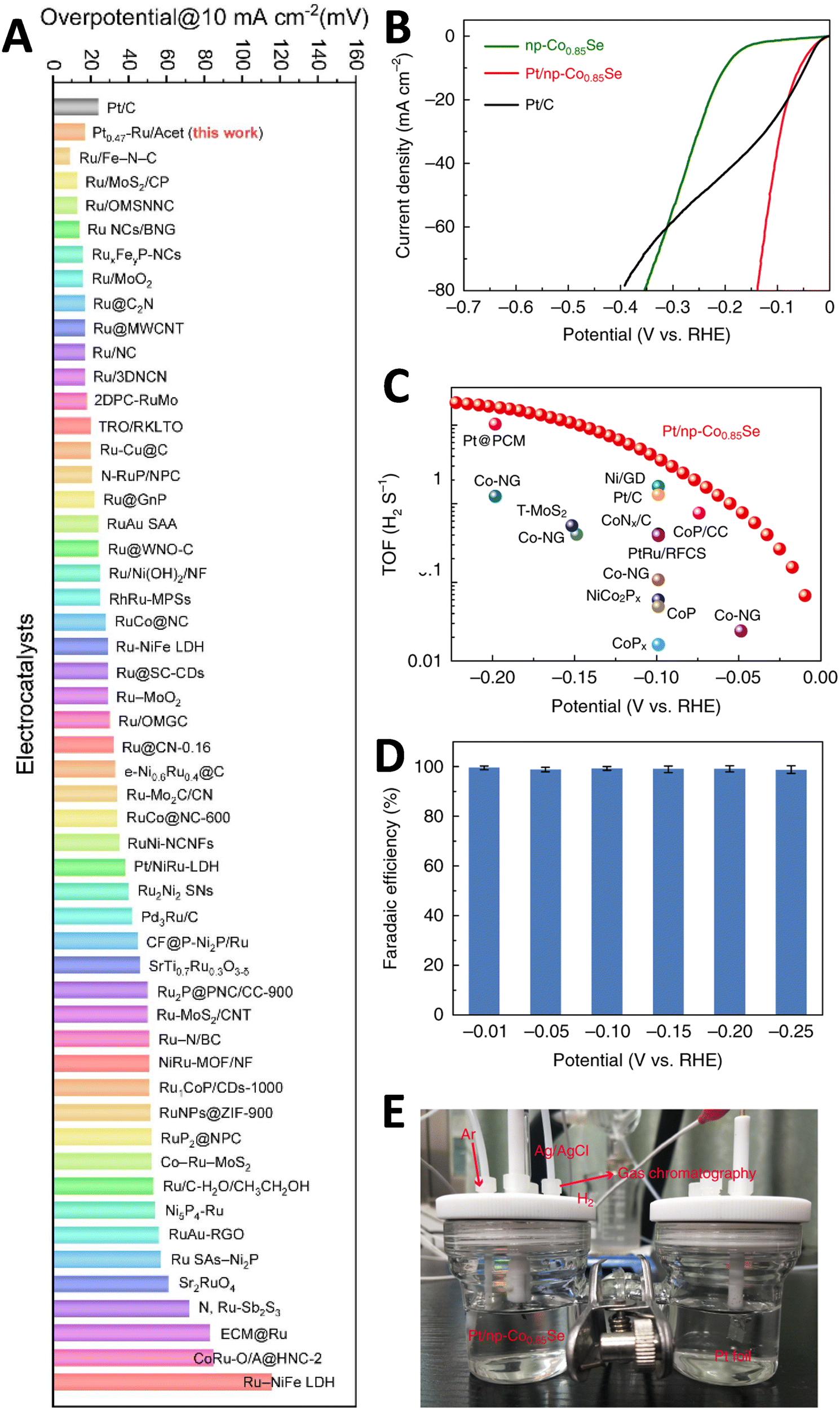 | ||
| Fig. 22 (A) Comparative HER activity for Ru-based electrocatalysts against reported in literature. Reproduced with permission from ref. 532 Copyright 2022, Elsevier. (B) HER polarization curves for np-Co0.85Se, Pt/np-Co0.85Se, and Pt/C. (C) TOF values of Pt/np-Co0.85Se, compared with reported electrocatalysts at −100 mV vs. RHE. (D) Faradaic efficiency of Pt/np-Co0.85Se at varied potentials. (E) Detailed measurement of hydrogen faradaic efficiency, including accelerated HER polarization curves for Pt/np-Co0.85Se. Reproduced with permission from ref. 534 Copyright 2019, Nature Publishing Group. | ||
Assessing the HER capabilities of the Co-CNG and NiCo DASs/N–C catalysts, Liu et al. conducted chronoamperometric tests under alkaline conditions (1 M KOH) with an Hg/HgO reference electrode.445 Notably, Co-CNG exhibited remarkable HER efficiency, achieving a current density of 10 mA cm−2 at an impressively low overpotential of around 47 mV, nearly identical to Pt/C (10 mA cm−2 at approximately 48 mV), showcasing its top-tier performance (Fig. 23A). Even under a high standard current density of 500 mA cm−2, Co-CNG displayed a lower overpotential (∼155 mV) compared to Pt/C (∼162 mV), underscoring the pivotal role of Co single atoms in the HER process (Fig. 23B). Tafel slope analysis further confirmed Co-CNG's excellent performance with a slope of 44 mV dec−1, akin to Pt/C (46 mV dec−1) (Fig. 23C). In comparison to other previously reported single-atomic site catalysts, Co-CNG exhibited superior HER performance with an exceptionally low overpotential and rapid kinetics. They bolstered the validation of Co-CNG's HER activity using large-area Pt mesh electrodes and subsequent XPS measurements. Co-CNG outperformed other catalysts, demonstrating a mass activity approximately four times greater than Pt/C and nine times greater than Co-CN at various overpotentials (Fig. 23D). Meanwhile, NiCo DASs/N–C displayed outstanding HER activity under both alkaline and acidic conditions, demanding significantly lower overpotentials than Ni SAs/N–C and Co SAs/N–C synthesized by Li et al. (Fig. 23E).472 This was corroborated by the remarkably low Tafel slopes of 72.5 and 82.4 mV dec−1 for NiCo DASs/N–C, signifying swift HER kinetics and efficient hydrogen production (Fig. 23F). Calculations revealed the exchange current density (j0) for NiCo DASs/N–C as 0.97 mA cm−2 in 1 M KOH and 0.99 mA cm−2 in 0.5 M H2SO4. Moreover, NiCo DASs/N–C, synthesized by Hu group, showcased a higher mass activity of 99.5 and 22.6 A g−1 at 250 mV (Fig. 23G).474 In evaluations of HER performance among the synthesized catalysts, it was NiPt alloy, containing just 0.3 wt% Pt single atoms, that demonstrated superior HER activity compared to other catalysts, attributable to the intriguing electronic interactions between atomic Pt and Ni nanosheets (Fig. 23H).
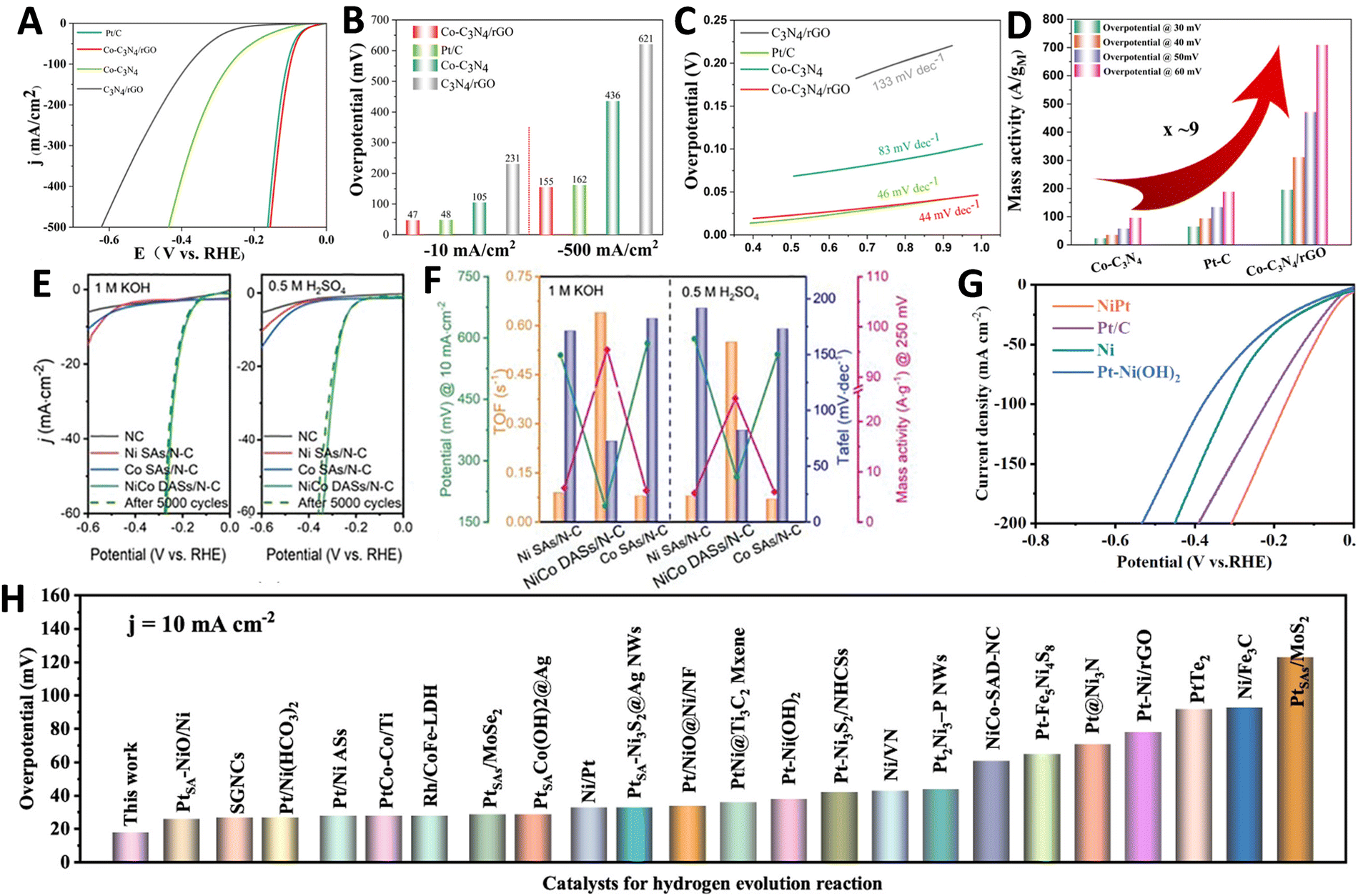 | ||
| Fig. 23 (A) LSV curves in 1.0 M KOH at 5 mV s−1 for Co–C3N4/rGO, Co–C3N4, C3N4/rGO, and Pt/C. (B) Overpotential comparison to achieve 10 and 500 mA cm−2. (C) Tafel plots derived from polarization curves in (A). (D) Mass activity at different overpotentials for Co–C3N4/rGO, Co–C3N4, C3N4/rGO, and Pt/C. Reproduced with permission from ref. 445 Copyright 2022, American Chemical Society. (E) LSV curves for HER and (F) Comparison of potential at 10 mA cm−2, TOF, Tafel slopes and mass activity of NiCo DASs/N–C. Reproduced with permission from ref. 472 Copyright 2022, Wiley-VCH. (G) HER polarization curves of NiPt, Pt–Ni(OH)2, Ni, and Pt/C. (H) HER activity comparison for NiPt against the reported catalysts. Reproduced with permission from ref. 474 Copyright 2022, American Chemical Society. | ||
Stability is a crucial parameter to assess the suitability of a catalyst for industrial applications. An ideal electrocatalyst should exhibit high chemical and electrochemical stability, low production cost, and support large-scale electrode fabrication. Pt-based metals are currently the state-of-the-art electrocatalysts for HER due to their optimal hydrogen adsorption properties.360,537,538 However, these precious-metal-based catalysts suffer from high cost and low catalytic stability in highly acidic and alkaline electrolytes. Various factors can limit the catalytic durability, such as leaching, aggregation, and poor mechanical stability.539,540 To evaluate intrinsic stability, researchers use cyclic voltammetry (CV) and chronopotentiometry (CP) or chronoamperometry (CA) methods. Liu et al. utilized onion-like nanospheres of carbon (OLC) to anchor atomically dispersed Pt, forming Pt1/OLC as a stable catalyst for HER.339 The durability of Pt1/OLC was assessed through 6000 CV sweeps, which revealed only a small shift in potential at the same current density as the initial curve, indicating excellent stability (inset, Fig. 24A). Additionally, the amperometric current density–time response showed that the Pt1/OLC electrode maintained a current density of 10 mA cm−2 for over 100 hours without any detectable loss, further demonstrating its robust stability in an acidic medium (Fig. 24A). This stability is attributed to the strong interaction between the Pt single atom and the OLC supports through the interfacial oxygen species. In a separate study, Yu et al. synthesized PtSA-NiCo LDHs/NF by anchoring oxygen vacancies in NiCo LDHs with Pt to form Pt–O5 coordination.357 The catalytic stability and durability of PtSA-NiCo LDH/NF were evaluated through 10000-cycle accelerated aging CV tests and a V–t test. The LSV curve after 10000 cycles nearly overlapped with the initial one, showcasing excellent stability (Fig. 24B). Furthermore, the durability test of PtSA–NiCo LDH/NF at 10 mA cm−2 for 50 hours exhibited excellent electrocatalyst durability (Fig. 24C). Feng et al. conducted operando synchrotron XAS to understand the evolution of single Pt atoms during the HER.541 They found that single Pt atoms dynamically release from the nitrogen-carbon substrate, affecting the catalyst's geometric structure and electronic properties. Pt1/N-C demonstrated superior HER activity with excellent long-time durability, making it a promising candidate for practical applications (Fig. 24D and E).
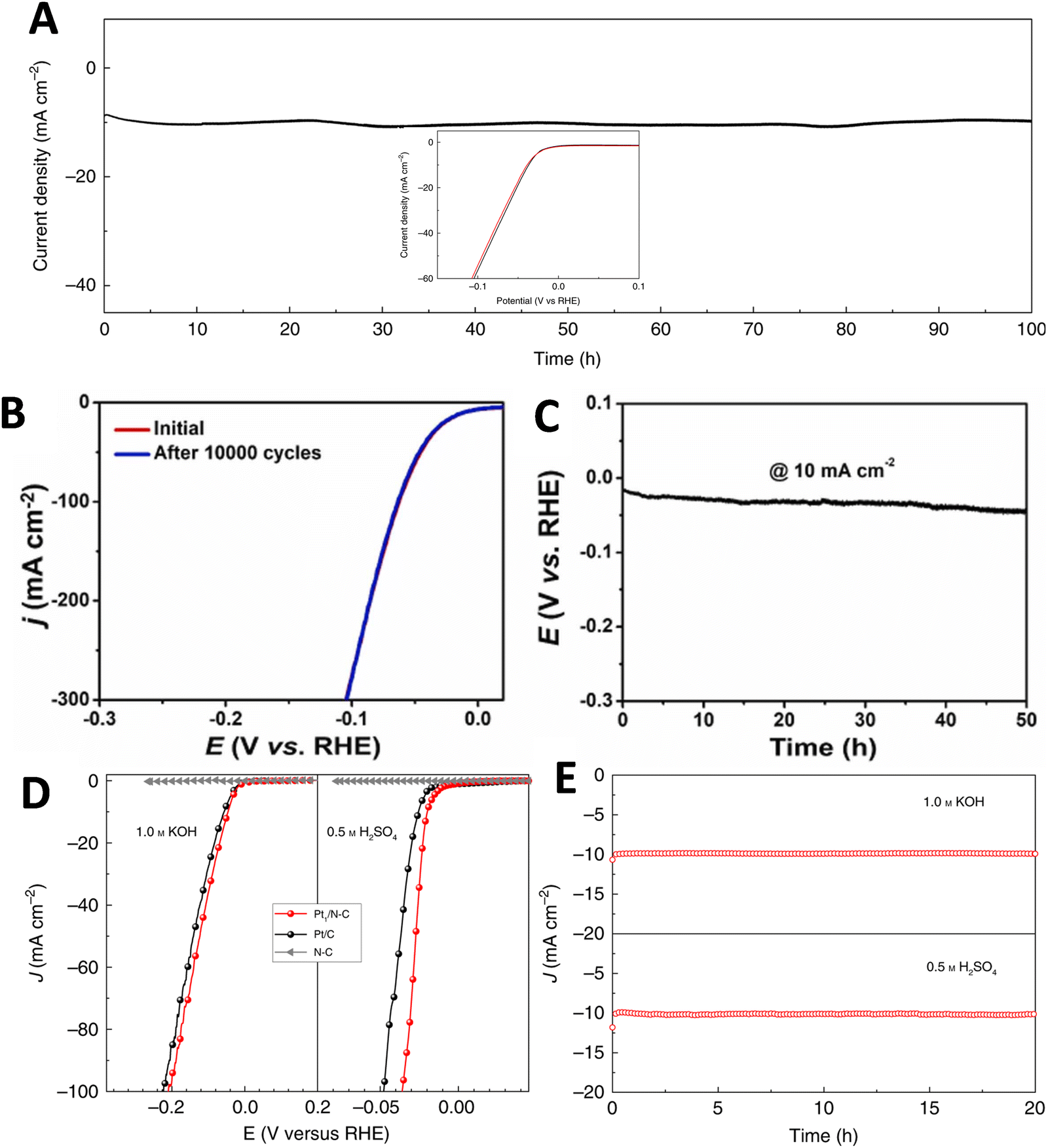 | ||
| Fig. 24 (A) Current density evolution for Pt1/OLC over 100 h under η = 40 mV in acidic conditions; (Polarization curves before (black) and after (red) 6000 CV sweeps in 0.5 M H2SO4 at 500 mV s−1). Reproduced with permission from ref. 339 Copyright 2019, Nature Publishing Group. (B) HER polarization curves of PtSA–NiCo LDH/NF pre and post durability test. (C) Chronopotentiometry for PtSA–NiCo LDH/NF at 10 mA cm−2 over 50 h. Reproduced with permission from ref. 357 Copyright 2023, Elsevier. LSV curves of the Pt1/N–C, Pt/C and N–C (D) and stability test of Pt1/N–C (E). Reproduced with permission from ref. 541 Copyright 2023, Nature Publishing Group. | ||
6.2. Reaction mechanism and structure–activity relationships
In recent years, the rapid advancement of in situ/operando characterization techniques has opened up new avenues for exploring the structural changes that occur in ASCs during catalytic reactions and for developing insights into reaction mechanisms. Additionally, theoretical investigations utilizing DFT calculations have been instrumental in deepening the understanding of these reaction mechanisms. In the context of HER, where molecular hydrogen is generated, researchers have made significant progress. For instance, Cao and colleagues have proposed a catalytic cycle specifically for alkaline HER over atomically dispersed Co into the phosphorized carbon nitride (HO–Co1/PCN) (as depicted in Fig. 25A).168 In this alkaline HER process, the catalytic cycle commences with the adsorption of H2O onto Co (step I), followed by its dissociation into adsorbed OH* and H* on Co and nearby N sites, respectively (step II, where the asterisk denotes the adsorption site). Subsequently, another proton originating from an adjacent H2O molecule interacts with the initially formed H* to produce H2 (steps III–V). It's worth noting that in alkaline HER, the initial rate-determining step is typically the catalytic dissociation of a water molecule, leading to the formation of H* (H2O + e− = H* + OH−).542,543 Therefore, a critical aspect of understanding alkaline HER on HO–Co1/PCN involves evaluating the energies associated with H2O and H adsorption on its surface. In an acidic solution, the catalyst facilitates the HER through a process illustrated in Fig. 25B. Initially, two H+ ions in the solution form Pt–H bonds during two Volmer steps (H+ + e− → H*).544 Subsequently, during the Tafel step (2H* → H2), the bonded protons combine, releasing H2 into the solution. Pt1/NPC, synthesized by Li et al., exhibits enhanced HER activity due to electron transfer from Pt atoms to coordinated N atoms, promoting H atom coverage on Pt surfaces.167 This increased H coverage improves HER kinetics by reducing H2 adsorption energy, thanks to Pt's unoccupied 5d orbitals facilitating electron pairing with H 1s orbitals.200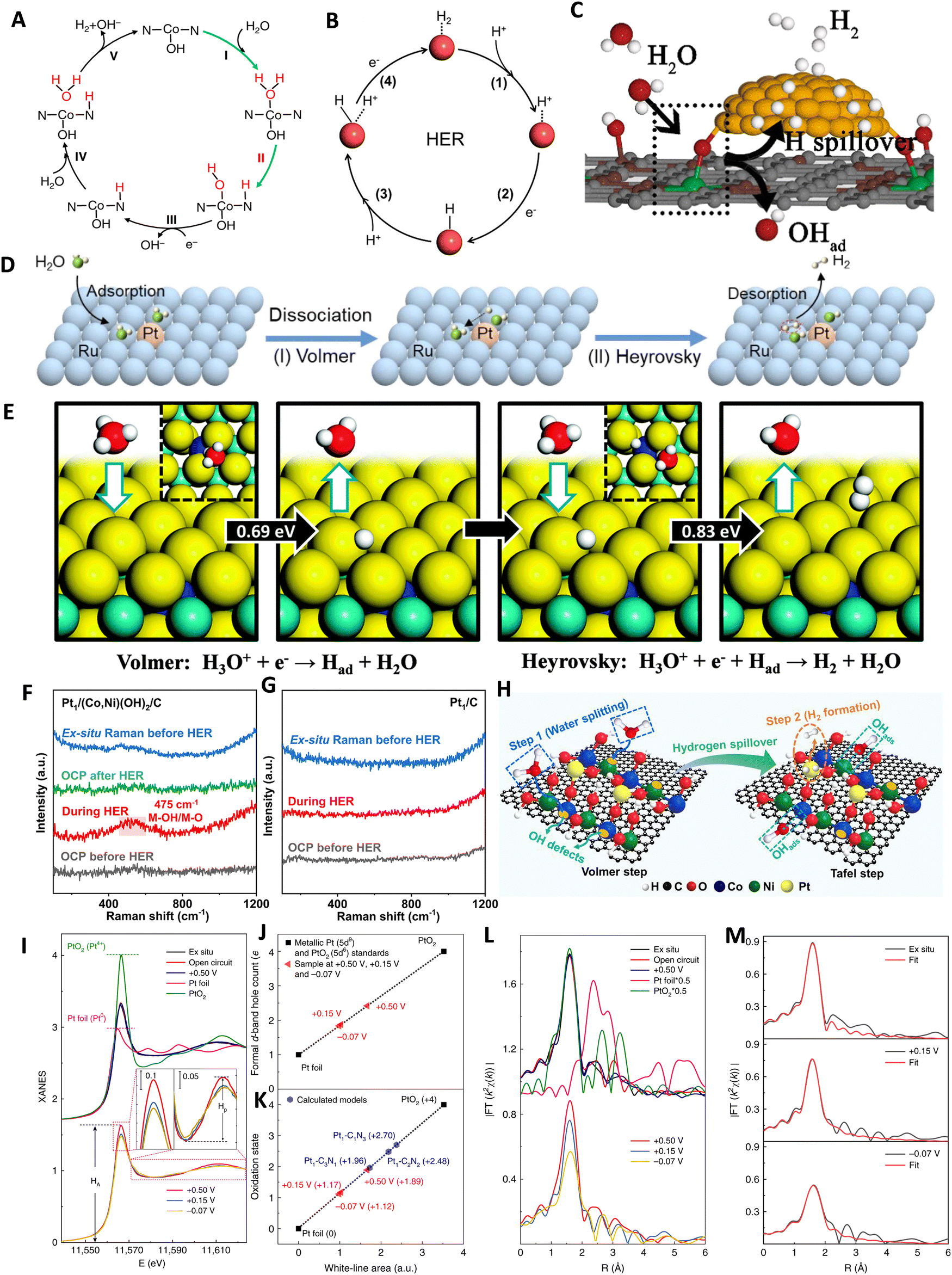 | ||
| Fig. 25 (A) Proposed alkaline HER mechanism on HO–Co1/PCN. Reproduced with permission from ref. 168 Copyright 2019, Nature Publishing Group. (B) Proposed acidic HER mechanism on Pt1/NPC. Reproduced with permission from ref. 167 Copyright 2018, American Chemical Society. (C) Enhanced HER activity via promoted water dissociation and Had recombination processes. Reproduced with permission from ref. 545 Copyright 2021, Elsevier. (D) Schematic of the alkaline HER mechanism on Pt–Ru/Acet. Reproduced with permission from ref. 532 Copyright 2022, Elsevier. (E) Pt–MoS2 catalyst's HER process, with reaction barriers shown. Reproduced with permission from ref. 74 Copyright 2015, The Royal Society of Chemistry. (F) Ex situ and Operando Raman spectra for Pt1/(Co,Ni)(OH)2/C during HER, (G) Ex situ and Operando Raman spectra for Pt1/C during HER. (H) HER mechanism on Pt1/(Co,Ni)(OH)2/C, highlighting synergies between (Co,Ni)(OH)2 surface and Pt single atoms. Reproduced with permission from ref. 343 Copyright 2023, The Royal Society of Chemistry. (I) XANES spectra recorded at different applied voltages, ranging from open-circuit conditions to −0.07 V during the HER, along with reference spectra of Pt foil and PtO2. Inset: Enlarged view of the white-line peak and post-edge XANES regions. (J) Fitted average formal d-band hole counts of Pt. (K) Oxidation states of Pt derived from ΔXANES spectra, compared with calculated oxidation states for Pt1–C3N1, Pt1–C2N2, and Pt1–C1N3 models, showing that Pt1–C3N1 closely matches the ex situ sample. (L) k2-weighted Fourier transform (FT) spectra corresponding to the XANES spectra in (I). (M) First-shell EXAFS fitting under ex situ, +0.15 V, and −0.07 V conditions. Reproduced with permission from ref. 541 Copyright 2020, Nature Publishing Group. | ||
Exploring the distinctive functions of Ni single atoms and Ni nanoparticles in the alkaline HER process is valuable, especially considering the significantly superior mass and specific activities of Ni–N–C/Pt relative to Ni NPs/Pt (Fig. 25C).545 Ni–N4 moieties, known for weak hydrogen adsorption, primarily facilitate water adsorption dissociation in the Ni–N–C/Pt electrocatalyst system.103,170 Subsequent hydrogen adsorption (Had) recombination occurs on metallic Pt sites. Conversely, in the Ni NPs/Pt system, strong Had adsorption hampers Had transfer and decelerates the Tafel or Heyrovsky step. Furthermore, active Ni species exposure is limited in Ni NPs/Pt, contributing to its lower activity. Ni–N4 acts as a co-catalyst in the alkaline HER process by facilitating H–OH bond cleavage, releasing active sites on metallic Pt. Oxidized Pt species also adsorb water and dissociate O–H bonds, supporting the release of H2.546,547 This process involves Had spillover from Ni–N4 or Pt–O moieties to metallic Pt sites, generating H2.
Pt single atoms play a crucial role in fine-tuning the electronic configuration of Ru clusters, resulting in the reduction of energy barriers during key alkaline HER stages, such as water dissociation (Volmer step) and subsequent hydrogen evolution (Heyrovsky step). This enhanced performance is illustrated in the alkaline HER mechanism on Pt–Ru/Acet (Fig. 25D).532 Initially, H2O molecules are adsorbed onto the Ru sites within the basic electrolyte, followed by their dissociation into intermediate OH* and H* through the Volmer step. Subsequently, the generated H* can react with another H2O molecule, forming H2via the Heyrovsky step. Computational simulations via DFT reveal that the introduction of atomically dispersed Pt onto the Ru nanocluster surface significantly influences the electronic structure of Ru, leading to reduced energy barriers for water dissociation and hydrogen evolution, ultimately enhancing the alkaline HER performance. Exploring the reaction pathway of the HER on Pt–MoS2 has provided valuable insights into the underlying mechanism (Fig. 25E).74 In this process, the unsaturated S atom exhibits a preference for absorbing a single H atom, with a barrier of approximately 0.69 eV. Subsequently, the absorbed H atom engages in a reaction with a hydrated proton, simultaneously receiving an electron, resulting in the formation of one H2 molecule with a moderate energy barrier of 0.83 eV. This sequence of events follows the Volmer–Heyrovsky mechanism, shedding light on the intricate steps involved in the hydrogen evolution reaction on Pt–MoS2.
The exploration of hydroxide-supported atomic structures, particularly single atoms, has opened up new avenues for enhancing catalytic performance through microenvironmental adjustments. A Pt1/(Co,Ni)(OH)2/C ASC was innovatively developed by Pei et al., featuring Pt single atoms (Pt1) securely stabilized and anchored onto the surface of defective (Co,Ni)(OH)2, with further support from carbon black.343 To delve into the HER mechanism of Pt1/(Co,Ni)(OH)2/C and Pt1/C, operando Raman spectrometry was employed. The operando Raman spectra of Pt1/(Co,Ni)(OH)2/C were meticulously recorded at various applied potentials in a 1.0 M KOH solution. A comprehensive analysis, involving ex situ Raman spectra collected before the HER and operando Raman spectra during and after the HER, provided compelling evidence that the initial step of water decomposition (breaking the H–OH bond) predominantly occurred on the surface of (Co,Ni)(OH)2 rather than due to the effect of lattice oxygen (Fig. 25F).548,549 The water molecules adsorbed on the hydrophilic (Co,Ni)(OH)2 surface dissociated to generate highly active M–*OH intermediates. Subsequent steps included the formation of Ni–*OH and Co–*OH intermediates, which facilitated water splitting and induced *H transfer to the single-atomic Pt sites, resulting in Pt–*H formation. The second step, the formation of H2, was characterized by the rapid interosculation of adjacent Pt–*H species (Tafel step). In contrast, Pt1/C exhibited no significant peaks during the HER, indicating inefficient dissociation of the H–OH on Pt species (Volmer step) (Fig. 25G). The Pt1/(Co,Ni)(OH)2/C showcased a robust synergistic effect between (Co,Ni)–(OH)2, rich in hydroxyl defect sitesand the Pt single atom: the former expedited the water decomposition step (Volmer step), while the latter, with its optimized hydrogen adsorption free energy, facilitated the hydrogen formation step (Tafel step) (Fig. 25H). This dynamic interaction underscores the catalyst's remarkable performance in the HER.
Recent advancements in characterization techniques and theoretical modeling have significantly enhanced our understanding of the activity–structure relationship in ASCs, sparking considerable interest in the field. These advancements have provided deeper insights into how the local coordination environment and electronic structure of active sites influence catalytic behavior, particularly in HER. This enhanced understanding has unveiled critical details about the fundamental mechanisms driving the performance of ASCs.263,550 The local coordination environment plays a critical role in determining the electronic structure and catalytic activity, with changes in coordination directly altering catalytic properties.78,494 Additionally, studies have highlighted the crucial role of support materials, which influence both the electronic and geometric properties of the catalyst.513,551,552 The application of d-orbital theory has further elucidated how electron arrangements around central metal atoms affect catalytic activity.330,553 Moreover, observations of dynamic structural changes during catalysis have provided valuable insights into the stability and durability of active sites. Interactions between ASCs and different substrates have provided valuable insights into selectivity and efficiency, particularly in the activation of various chemical bonds. Computational modeling and simulations have become essential tools in predicting ASC behavior, significantly aiding catalyst design.263,550 The size of nanoparticles in ASCs is closely linked to their catalytic activity, with smaller nanoparticles exhibiting higher activity due to a larger surface area and greater availability of active sites.225,554 Moreover, the use of advanced characterization techniques, such as in situ and operando XAS, has enabled real-time monitoring of structural and electronic changes, enhancing our understanding of catalyst dynamics. Operando XAFS measurements, as shown in Fig. 25I–M, provide valuable insights into the chemical state and structural dynamics of single-atomic Pt sites during HER.541 The XANES spectra (Fig. 25I and J) demonstrate a reduction in the white-line intensity as the applied potential shifts from +0.50 V to −0.07 V, indicating an increase in the d-electron occupancy of Pt and a corresponding reduction in its valence state. The valence state transitions from +1.89 at +0.50 V to +1.17 at +0.15 V, and further to +1.12 at −0.07 V, as shown in Fig. 25K. This shift reflects the dynamic weakening of Pt–support interactions during HER, with Pt moving towards a more metallic state under cathodic conditions.437,555 The Fourier transform spectra of k2-weighted EXAFS (24L) further reveal structural changes. The intensity of the Pt–N coordination peak decreases with increasing cathodic potential, indicating a reduction in coordination as the HER progresses. First-shell fitting of the EXAFS spectra (24M) confirms the presence of Pt–N bonds under ex situ conditions and at +0.50 V, but these bonds diminish during HER, as additional Pt–O bonds appear at +0.15 V, likely due to water adsorption. At −0.07 V, the coordination number reduces further, indicating hydrogen adsorption and water dissociation.556 These findings validate the reaction mechanism of single-atomic Pt sites during HER, highlighting the dynamic evolution of valence states and coordination environments. The results underscore the critical role of in situ techniques in probing the chemical states and structural changes of single-atomic site catalysts during catalytic reactions.
Metal doping has been explored to understand changes in electronic states and their effects on catalytic performance.74,432 In this regard, Jiang et al. introduced a ground-breaking electrocatalyst, Pt/np-Co0.85Se, comprising single-atomic platinum on nanoporous Co0.85Se, which exhibited exceptional efficiency in hydrogen evolution.534 In a neutral medium, Pt/np-Co0.85Se demonstrated remarkable catalytic performance, featuring an almost negligible onset overpotential, a low Tafel slope of 35 mV dec−1, and an impressive turnover frequency of 3.93 s−1 at −100 mV. This performance exceeded that of commercial Pt/C catalysts and other transition-metal-based compounds. To unravel the reasons behind its outstanding activity in a neutral electrolyte, the team conducted in situ and operando Co K-edge XANES and FT-EXAFS spectroscopy during the HER under working conditions. This involved cycling the working electrode potential from the open circuit voltage (OCV, ∼0.75 V vs. RHE) to −0.2 V vs. RHE and back to OCV. The Co K-edge XANES spectra of np-Co0.85Se and Pt/np-Co0.85Se, illustrated in Fig. 26A and B, respectively, unveiled distinct variations in the absorption onset with changing bias voltages. Particularly, Pt/np-Co0.85Se displayed a noticeable shift towards higher energy, as discerned through the first-order derivatives of the XANES spectra presented in Fig. 26C and D. The significant alteration observed in the radial distance of the Co–Se shell in the FT-EXAFS spectra, as seen in Fig. 26E and F, underscores the pivotal role of Pt and Co0.85Se electronic interactions in enhancing electron transfer from Co to Se atoms during the HER. This phenomenon was notably absent in np-Co0.85Se (Fig. 26E), signifying an increase in electron intensities on Pt/Co0.85Se.557 These findings suggest that the introduction of Pt optimizes the electronic structure of neighboring Co atoms, thereby expediting the adsorption and dissociation processes of water molecules during the HER. In Pt/np-Co0.85Se, the interaction of water molecules, composed of positively charged H atoms and a negatively charged oxygen atom, with positively charged Co atoms facilitates the adsorption and activation of water molecules. Moreover, the heightened intensity of the Co–OH shell during the HER in Pt/np-Co0.85Se implies a greater concentration of adsorbed OHads on Co atoms, particularly during the Volmer step of the HER, compared to Co0.85Se.558 These collective findings suggest that Pt dopants play a crucial role in optimizing the electronic structure of adjacent Co atoms, thereby expediting the adsorption and dissociation of H2O. According to the in situ and operando XAS results for Pt/np-Co0.85Se during the HER, H2O molecules selectively adsorb or bind to Co sites in the early HER potential stages (Step I in Fig. 26G). Subsequently, in a neutral medium, H2O molecules capture electrons and undergo dissociation into intermediate Hads and OHads through the Volmer step, facilitated by Co sites. Simultaneously, the generated Hads can be adsorbed on nearby vacant Co or Pt sites and efficiently converted into H2 through the Tafel step (Step II in Fig. 26G). These developments have significantly enhanced our understanding of ASCs, guiding the design of more efficient and selective catalysts for HER application.
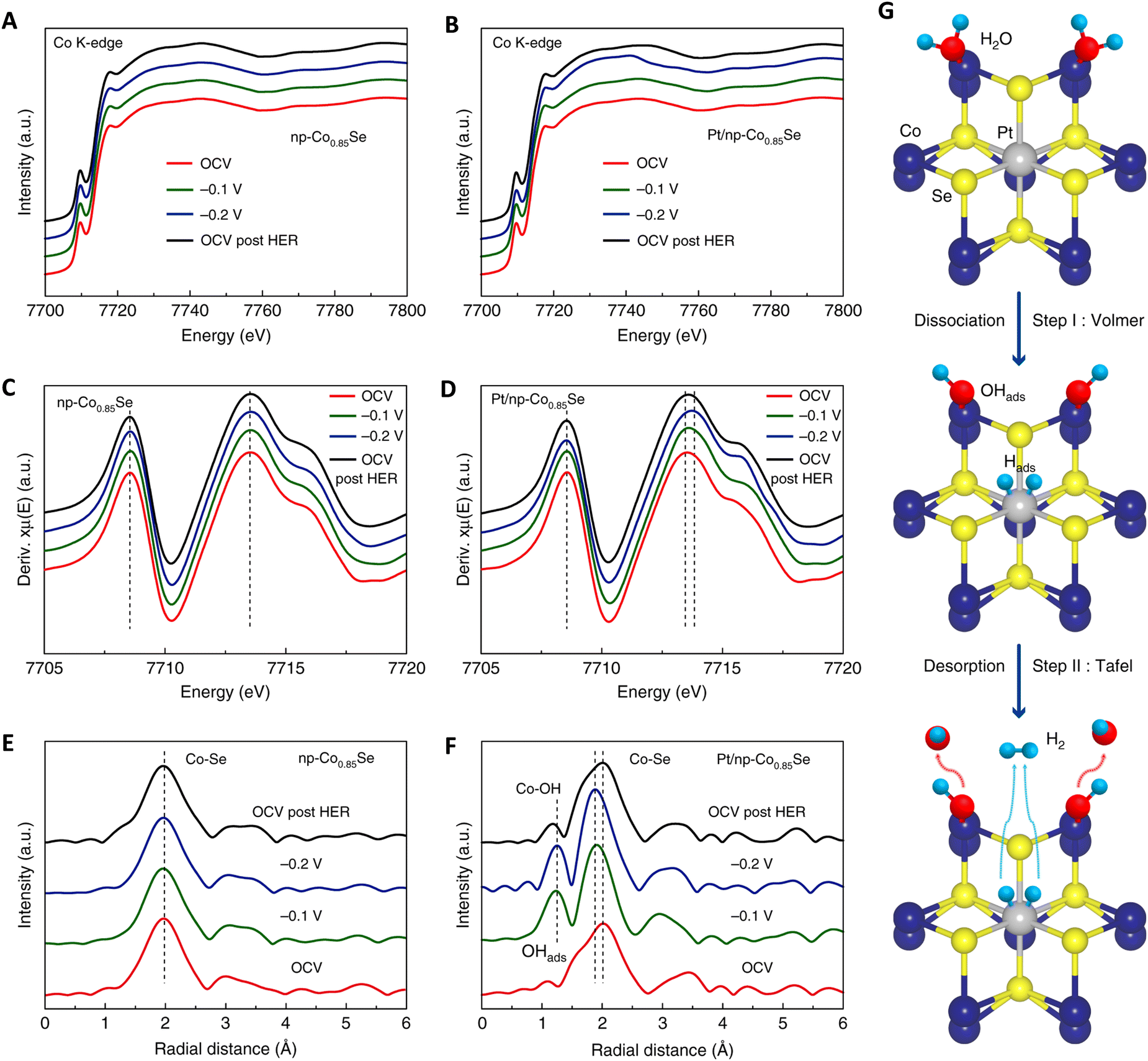 | ||
| Fig. 26 Co K-edge XANES spectra for np-Co0.85Se (A) and Pt/np-Co0.85Se (B), spanning from OCV to −0.2 V (vs. RHE) in 1.0 M PBS. First-order derivatives of XANES spectra for np-Co0.85Se (C) and Pt/np-Co0.85Se (D). FT-EXAFS spectra for np-Co0.85Se (E) and Pt/np-Co0.85Se (F). Schematic representation of HER mechanism as elucidated by in situ and operando XAS analysis of Pt/np-Co0.85Se in neutral media (G). Reproduced with permission from ref. 534 Copyright 2019, Nature Publishing Group. | ||
6.4. DFT studies
DFT calculations play a pivotal role in understanding the mechanisms behind high catalytic performance, particularly in the realm of ASCs. DFT enables the identification of true active sites and their catalytic activity through the computation of Gibbs free energy for hydrogen adsorption (ΔGH*). It also aids in predicting metal–support interactions by examining the charge density distribution of the metal atom within the support. Additionally, DFT assists in evaluating the stability of supported single atoms by calculating their formation or adsorption energies. These theoretical insights are invaluable for designing and synthesizing efficient catalysts, offering significant time and cost savings.559–562 Pei et al. devised a Pt1/(Co,Ni)(OH)2/C ASC featuring Pt single atoms hosted on defective (Co,Ni)(OH)2 and carbon black support.343 This ASC stood out in the HER due to a robust synergy between Pt single atoms and bimetallic (Co,Ni)(OH)2. DFT calculations validated the electronic synergy between Pt single atoms and bimetallic (Co,Ni)(OH)2, ensuring stable Pt anchoring and an ideal *H adsorption energy for efficient H2 generation. Various models were assessed, including (Co,Ni)(OH)2, (Co,Ni)(OH)2 with a Pt single atom (Pt1/(Co,Ni)(OH)2), and Pt(111) slabs. In contrast to pure Pt(111), where slow water dissociation posed a bottleneck for the HER, combining Pt with hydroxides facilitated the HER process by furnishing defect sites such as *OH for water adsorption and dissociation (Fig. 27D). Both defective (Co,Ni)(OH)2 with Pt single atom adsorption and pristine (Co,Ni)(OH)2 were scrutinized (Fig. 27A and B). Empirical observations indicated that Pt1/(Ni,Co)(OH)2 proficiently cleaved H2O into *OH and *H, forging a covalent connection with Pt1. This led to less adhesive *H binding on Pt1/(Ni,Co)(OH)2 compared to pure Pt, thereby invigorating the HER. Charge density difference analysis revealed a significant electron transfer from Pt to O atoms of (Co,Ni)(OH)2, fortifying the *H interaction (Fig. 27C). Consequently, *H adsorption on Pt1/(Ni,Co)(OH)2 was milder than on pure Pt, heightening the HER efficiency. Free energy diagrams and ΔGH* data lent support to Pt1/(Co,Ni)(OH)2/C as the superior performer in the HER. Lastly, the analysis of density of states showed that the d-band center shifted away from the Fermi level as ΔGH* decreased, in alignment with the ΔGH* trend. This underscored the role of hydroxides in boosting both H2O dissociation and H2 formation. | ||
| Fig. 27 Atomic structures of (A) (Co,Ni)(OH)2 and (B) Pt1/(Co,Ni)(OH)2, with (C) charge density difference for Pt adsorption. (D) ΔG profiles for HER on Pt(111), (Co,Ni)(OH)2, Pt1/Ni(OH)2, Pt1/Co(OH)2, and Pt1/(Co,Ni)(OH)2. Reproduced with permission from ref. 343 Copyright 2023, The Royal Society of Chemistry. (E) PDOS of Pt–N4 and Pt55 for d orbitals, with the inset showing their configurations. (F) The free energy path of HER on Pt–N4 and Pt55. Reproduced with permission from ref. 335 Copyright 2022, Elsevier. (G) Calculated free-energy diagram for HER at pH = 0, with H adsorption on Pt site. (H) Electric field map for H2Pt1/OLC at Pt site. (I) Proton concentration distribution based on the Gouy–Chapman–Stern model around Pt site in H2Pt1/OLC. Reproduced with permission from ref. 339 Copyright 2019, Nature Publishing Group. | ||
Wang et al. developed a unique material, NPCN–Pt, by co-loading Pt single atoms and Pt clusters onto double-layered N-doped porous carbon nanocages, aiming to enhance the HER.335 Pt clusters (Pt55) were found to have a d-band center closer to the Fermi level than Pt–N4, making them more favorable for water molecule adsorption and hydrogen–oxygen bond cleavage in the alkaline HER (Fig. 27E). Pt55 also showed better *OH desorption, creating space for subsequent catalytic reactions. However, Pt55 had a higher *H adsorption energy compared to Pt–N4 (Fig. 27F). Their theoretical findings showed that Pt clusters had a lower energy barrier for engaging water molecules, particularly favoring the HER's Volmer step.563,564 Pt single atoms exhibited a smaller ΔGH*, aiding H2 release. DFT calculations confirmed that Pt likely existed as Pt–N4 within NPCN–Pt. Additionally, Pt clusters facilitated water molecule decomposition, generating hydrogen radicals, while Pt single atoms promoted H2 formation through these radicals, contributing to NPCN–Pt's high catalytic activity in alkaline conditions. Lui et al. showcased the remarkable potential of Pt single atoms distributed on nanosized OLC supports (Pt1/OLC) as highly efficient electrocatalysts for hydrogen evolution, surpassing 2D graphene-supported catalysts with similar Pt loadings.339 Computer simulations revealed that this architecture's high curvature results in electron accumulation around the Pt regions, creating a localized electric field that boosts catalytic kinetics. To understand the Pt1/OLC catalyst's working mechanism, they conducted DFT-level simulations using the PtO2C295 model derived from XAFS results, considering the computational hydrogen evolution model at pH = 0.103 When applying a constant potential (U = 0) and utilizing a state-of-the-art constant potential algorithm with grand canonical DFT calculations, it was found that the first and second adsorbed hydrogen atoms had calculated ΔG of −0.92 and −0.50 eV, respectively (Fig. 27G).565,566 This indicates strong hydrogen adsorption on unsaturated Pt coordinates. In contrast, the third hydrogen adsorption had a calculated ΔG of −0.01 eV, suggesting the formation of H2Pt1/OLC. Effective mass transfer of protons to the active Pt site was another critical factor determining hydrogen evolution activity. Their calculations demonstrated the formation of a strong local electric field around the Pt site at equilibrium potential, protruding like a tip from the curved OLC surface (Fig. 27H). This tip effect enhanced proton mass transfer by creating a high local proton concentration around the active sites. This enriched proton concentration was not observed in the OLC configuration without Pt. Using the Gouy–Chapman–Stern model, they identified a significant proton concentration (>1.99 mol L−1) around Pt in PtO2C295, which can enhance proton transfer during the HER (Fig. 27I).567 In contrast, the Pt-absent OLC (C300) configuration lacked these effects.
Qian et al. have developed an innovative heteroatom-transfer strategy to precisely control phosphorus coordination in carbon-supported single-atomic site catalysts.249 By synthesizing self-assembled helical fibers with encapsulated cavities that chelate metal ions, this method ensures structural integrity during pyrolysis-based phosphidation, leading to the strategic transfer of heteroatoms into the catalyst structure. The resulting Co–P2N2–C catalysts demonstrate remarkable electrocatalytic performance for alkaline hydrogen evolution, comparable to noble metal catalysts, through the systematic tailoring of the coordination environment enabled by this method. To explore the enhancement of HER performance by P atoms in Co-based ASCs, Qian et al. utilized DFT to analyze Co–PxN4−x–C catalysts, where x ranges from 0 to 3. The computational models, represented in Fig. 28A, showcased a graphene layer with cobalt coordinated by both nitrogen and phosphorus atoms. DFT calculations demonstrated a preferential formation of para-positioned Co–P2N2, attributed to its lower formation energy, highlighting a structurally favored configuration for effective catalysis. Fig. 28B illustrates differential charge density maps, indicating enhanced electron-rich environments around cobalt in Co–PxN4−x–C, compared to Co–N4–C. This suggests lower oxidation states for cobalt, enhancing its ability to bond with water molecules. Notably, Fig. 28C and D reveal that phosphorus coordination results in both charge accumulation and depletion around cobalt atoms, altering the electron cloud density and symmetry of charge distribution. This adjustment in electronic structure is confirmed by Bader charge analysis,568 showing decreased cobalt valence states with increased phosphorus coordination. The study further quantified the impact of phosphorus on HER kinetics, with Fig. 28E demonstrating a volcanic relationship between the Bader charge of cobalt and overpotential of HER, where Co–P2N2–C exhibited optimal performance. The calculated Gibbs free energies for water dissociation (ΔGw) and adsorbed hydrogen (ΔGH*) across different configurations highlighted Co–P2N2–C as having the most optimized binding energies conducive for efficient HER. These results suggest that a moderate coordination of phosphorus in Co–P2N2–C not only optimizes the charge distribution around the cobalt center but also balances the energies required for effective water dissociation and hydrogen adsorption/desorption, key steps in the HER process. The synergy between defective graphene (DG) and anchored nickel atoms (aNi) significantly enhances the A-Ni@DG catalyst's performance in both the HER.423 DFT calculations reveal electron transfer from Ni to the surrounding carbon in NiFe-LDH@DG. Electronic structure analysis of aNi on DG shows distinct configurations, impacting its electronic properties (Fig. 28F–K). The d-band center theory demonstrates strong interactions between carbon and aNi, favoring adsorbate (e.g., H+ for HER) binding.569 aNi in specific sites exhibits lower desorption energy for H2, akin to platinum, indicating superior HER activity compared to other configurations (Fig. 28L and M). First-principles DFT calculations were employed to unravel the mechanism behind the enhanced HER performance facilitated by copper single atoms (Cu SAs) in 1T-MoS2.462 H* adsorption on 1T-MoS2, Cuads@MoS2, and Cusub@MoS2 revealed critical insights (Fig. 28N). The electronic structure analysis highlighted that Cu SAs induce electron transfer from Cu to neighboring S atoms, effectively altering the electronic structure of the original S active sites in 1T-MoS2. Subsequently, the optimal active states for H* adsorption and their ΔGH* were calculated (Fig. 28O). Cu SAs substantially increased ΔGH*, shifting from −1.9 eV (1T-MoS2) to 0.24 eV (Cuads@MoS2) and −0.07 eV (Cusub@MoS2), facilitating H* desorption. Further examination of electronic structure changes upon H* adsorption (Fig. 28P) demonstrated that Cu SAs transformed H* from an electron donor to an electron acceptor in Cuads@MoS2. This key insight clarifies the role of Cu SAs as main active sites in Cuads@MoS2 and the adjacent S atoms as the primary active sites in Cusub@MoS2, shedding light on the design of advanced catalysts.
 | ||
| Fig. 28 DFT Analysis of Cobalt–Phosphorus–Nitrogen-Doped Carbon Catalysts for HER. (A) Top view electron density difference and Bader charge analysis for Co–N4–C, Co–P1N3–C, Co–P2N2–C, and Co–P3N1–C configurations; yellow indicates electron accumulation while blue shows electron depletion. (B) Free energy profiles for the water dissociation process across different catalyst models. (C) Calculated hydrogen adsorption energies on P or N sites in various Co–PxN4−x–C (x = 0, 1, 2, 3) configurations. (D) Diagram showing ΔGH* across the catalysts studied. (E) Plots depicting the variation in ΔGH, ΔGOH*, and ΔGw for different Co–PxN4−x–C models, demonstrating the impact of phosphorus incorporation on catalytic activity. Reproduced under permission from ref. 249, Copyright 2024, Nature Publishing Group. (F)–(H) Catalytic active sites: single Ni atom on (F) perfect hexagons, (G) D5775, and (H) Di-vacancy. (I)–(K) PDOS for the three configurations. (L) and (M) Energy profiles for HER (L) and OER (M). Reproduced with permission from ref. 423 Copyright 2017, Elsevier. (N) Hydrogen adsorption modes on 1T-MoS2, Cuads@MoS2, and Cusub@MoS2. (O) Their ΔGH* for the HER at 0 V vs. RHE at pH = 0. (P) Electron density distinctions in hydrogen adsorption on 1T-MoS2, Cuads@MoS2, and Cusub@MoS2. Reproduced with permission from ref. 462 Copyright 2022, American Chemical Society. | ||
7. Challenges and future research directions
This review provides a comprehensive examination of ASCs in the context of the HER, focusing on their properties, types, and performance. It discusses the importance of HER in sustainable energy transition, compares ASCs to traditional catalysts, evaluates ASC performance, and explores reaction mechanisms. The review concludes by addressing challenges and suggesting future research directions for efficient and sustainable HER catalysts. Despite significant recent progress in the development of ASCs for the electrocatalytic HER, several substantial challenges remain that need to be effectively addressed for the design of ASCs with exceptional activity and durability. While substantial achievements have been made, there is still untapped potential for further enhancements in future research. The following sections outlines these challenges and suggests potential solutions.7.1. Challenges of ASCs for HER
One of the primary challenges is the stability of ASCs under the harsh conditions of electrochemical reactions. Although ASCs often exhibit excellent catalytic activity initially, they can degrade over time due to the agglomeration of single atoms into clusters, particularly under harsh reaction conditions, such as elevated temperatures or corrosive environments commonly found in industrial electrolyzers. These conditions can lead to rapid degradation, significantly reducing their effective lifespan and efficiency. While ASCs offer high mass efficiency, their synthesis is often complex and costly, which can offset the cost benefits derived from their efficient use of materials. These catalysts typically consist of isolated metal atoms anchored on supports, making it challenging to maintain their dispersion and structural integrity during prolonged electrocatalysis. This issue becomes particularly pronounced under the harsh operational conditions commonly found in industrial settings, where ASCs are exposed to extreme environments that can accelerate degradation. Furthermore, the controlled synthesis of ASCs with precise atomic dispersion remains a complex and resource-intensive process. Achieving uniform dispersion of single atoms on a support material while preventing aggregation is particularly challenging. Scaling up the production of ASCs for practical applications presents another obstacle, as methods that work effectively at small laboratory scales may not easily translate to large-scale industrial processes, including ensuring the reproducibility and stability of ASCs. For example, methods that achieve atomic dispersion effectively at the gram scale often fail when scaled to kilograms, limiting their industrial scalability. Despite their promising initial catalytic activity, ASCs can degrade over time due to exposure to reactive intermediates, resulting in a loss of activity and stability. The interactions between metal atoms and support materials significantly influence catalytic properties, making the enhancement of performance through the choice of metal atoms, supports, and synthesis methods a complex challenge. Optimizing these interactions for optimal performance is challenging and requires a deep understanding of the underlying mechanisms. For instance, achieving stable metal–support interactions for sustained HER applications remain a focus of active research, as reactive species generated during electrochemical reactions can disrupt these interactions. Efficient mass transport of reactants and products to and from the active sites on ASCs can be limited, particularly when single atoms are dispersed within a dense support matrix. Furthermore, ASCs can be highly sensitive to impurities or contaminants in the reactants, which may poison the active sites and reduce catalytic activity. For example, sulfur or chloride impurities in electrolytes can bind strongly to single metal atoms, leading to the deactivation of active sites. Characterizing ASCs at the atomic scale – identifying the nature of active sites and unraveling reaction mechanisms – is a complex task that requires advanced analytical techniques. Methods like HAADF-STEM, XAS, and EELS often face sensitivity limitations, particularly when detecting single atoms within complex or heterogeneous matrices. For example, EXAFS often struggles to identify minority species, such as isolated metal clusters, due to its averaging nature, which necessitates rhigh-quality data and extensive data ranges for accurate detection. Operando XAS is a valuable tool for real-time monitoring of dynamic changes, such as modifications in electronic states and coordination environments. However, further refinements are needed to accurately capture fast transient phenomena during catalytic processes. Additionally, characterizing interfacial chemistry at gas–solid or liquid–solid interfaces, which is critical for understanding catalytic mechanisms, remains an unresolved challenge. The synthesis and integration of ASCs into practical devices are further hindered by high costs and scalability challenges, limiting their widespread adoption. Moreover, sample preparation for advanced characterization techniques can introduce artifacts that alter the native state of ASCs, potentially leading to misinterpretation of their catalytic properties. While in situ XAS provides valuable insights into dynamic structural changes, it often generates large datasets of low quality, making data analysis and interpretation challenging. Moreover, aligning experimental data with simulated XANES spectra to validate structural models of ASCs demands advanced simulation techniques and accurate theoretical models, further increasing the complexity of characterization. Advancing materials design, refining synthesis techniques, and deepening the fundamental understanding of catalytic mechanisms will be crucial in overcoming these obstacles and unlocking the full potential of ASCs for HER and other sustainable energy applications.7.2. Future research directions
In the realm of ASCs for HER, future research should prioritize key avenues to tackle current challenges and drive the advancement of these promising catalysts. One of the primary challenges is the stability of ASCs under harsh operational conditions. Sintering effects, where single atoms aggregate into clusters, can significantly diminish catalytic efficiency. While ASCs may maintain stability for hours in laboratory settings, ensuring their dispersion over weeks under industrial conditions remains difficult. Addressing these stability issues will require the development of robust support materials, such as graphene, hybrid MOFs, or advanced carbides, that can effectively anchor single atoms and prevent agglomeration. Another critical challenge is the scalability of ASC synthesis, as current production methods are limited to gram-scale laboratory processes. To enable large-scale manufacturing while preserving atomic dispersion, novel, cost-effective synthetic strategies, such as atomic layer deposition or continuous-flow chemical methods, are essential. To deepen our understanding of the atomic-scale mechanisms governing ASC catalysis, advanced in situ and operando characterization techniques, such as operando XAS and STM, must be further refined to unravel the dynamic structural transformations of ASCs during electrocatalysis. Additionally, integrating machine learning algorithms can accelerate the discovery of optimal ASC configurations by analyzing both experimental and computational datasets. Machine learning and computational modeling can predict ASC behavior under various operational conditions, uncover subtle reaction trends, and aid in the design of new catalysts with enhanced properties, significantly advancing catalyst development. Practical applications of ASCs in devices like electrolysers and fuel cells for green hydrogen production offer immense potential. Catalytic design innovations, such as tailoring electronic properties through heteroatom doping or modifying coordination environments, could enhance selectivity and activity. This is particularly critical for HER applications in renewable energy systems, where ASCs must operate efficiently under the intermittent inputs of solar or wind energy. Furthermore, exploring cost-effective and environmentally friendly synthesis approaches is crucial for producing ASCs in large quantities without compromising atomic dispersion. Customizing ASCs with tailored active sites optimized for specific reaction pathways remains an essential area of research focus. This process involves precisely tuning the coordination environment and electronic structure of individual metal atoms to enhance their catalytic activity for HER. Understanding the synergistic interactions between ASCs and support materials is pivotal for maximizing catalytic performance. Additionally, strategies must be developed to mitigate catalyst degradation over time, potentially through the design of self-healing ASCs or protective coatings that maintain catalytic activity under challenging conditions. Integration with renewable energy sources, such as solar and wind, represents another critical avenue for ASC development. ASCs must operate efficiently under intermittent energy input conditions, necessitating the design of catalysts that exhibit high durability and responsiveness in variable operating environments. Moreover, the potential of ASCs for multifunctional catalysis – such as coupling HER with other electrochemical reactions like OER or CO2 reduction – could pave the way for integrated and efficient energy conversion systems. Advancing theoretical modeling, computational simulations, and accelerated materials discovery will remain invaluable in guiding the design of ASCs with enhanced catalytic properties. For instance, replacing noble metals like platinum with earth-abundant materials, such as iron or nickel, could reduce costs while maintaining catalytic activity. Finally, gaining deeper insights into interfacial chemistry at gas–solid and liquid–solid interfaces is crucial for understanding how reactants and intermediates interact with active sites. Advanced characterization techniques are essential to overcome challenges such as monitoring transient intermediates and dynamic structural changes during catalytic reactions. It is equally important to assess the environmental sustainability of ASC synthesis processes and their impact on green hydrogen production. Collaborative research across multiple disciplines, including materials science, catalysis, electrochemistry, and engineering, can advance these goals, bridging the gap between theory and practical applications for a greener energy future.Data Availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- Z. Pu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, S. Mu, W. Zhao, F. Su, G. Zhang, S. Liao and S. Sun, Nano-Micro Lett., 2020, 12, 21 CrossRef CAS PubMed
.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC
- J. Cai, R. Javed, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2020, 8, 22467–22487 RSC
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 6217 CrossRef PubMed
- M. Khalid, P. A. Bhardwaj, A. M. B. Honorato and H. Varela, Catal. Sci. Technol., 2020, 10, 6420–6448 RSC
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS
- M. Younas, M. Yar, H. AlMohamadi, T. Mahmood, K. Ayub, A. L. Khan, M. Yasin and M. A. Gilani, Int. J. Hydrogen Energy, 2024, 51, 758–773 CrossRef CAS
- E. Yu and Y. Pan, Int. J. Hydrogen Energy, 2024, 50, 920–931 CrossRef CAS
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- S. S. Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed
- K. Song, H. Zhang, Z. Lin, Z. Wang, L. Zhang, X. Shi, S. Shen, S. Chen and W. Zhong, Adv. Funct. Mater., 2024, 34, 2312672 CrossRef CAS
- Z. Wu, Q. Li, G. Xu, W. Jin, W. Xiao, Z. Li, T. Ma, S. Feng and L. Wang, Adv. Mater., 2024, 36, 2311018 CrossRef CAS PubMed
- J.-H. Wang, S.-W. Yang, F.-B. Ma, Y.-K. Zhao, S.-N. Zhao, Z.-Y. Xiong, D. Cai, H.-D. Shen, K. Zhu and Q.-Y. Zhang, Tungsten, 2024, 6, 114–123 CrossRef
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526–1536 CrossRef CAS
- Z. Li and K. Sibudjing, ChemCatChem, 2018, 10, 2994–3001 CrossRef CAS
- Z. Bian, I. Y. Suryawinata and S. Kawi, Appl. Catal., B, 2016, 195, 1–8 CrossRef CAS
- J. Ni, L. Chen, J. Lin and S. Kawi, Nano Energy, 2012, 1, 674–686 CrossRef CAS
- S. Das, A. Jangam, S. Xi, A. Borgna, K. Hidajat and S. Kawi, ACS Appl. Energy Mater., 2020, 3, 7719–7735 CrossRef CAS
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS
- S. Dwyer and S. Teske, Renewables 2018 Global Status Report.
- C. Energy, National Hydrogen Vision Meeting, Washington DC, 2002, 15–16.
- A. Indra, P. W. Menezes, I. Zaharieva, H. Dau and M. Driess, J. Mater. Chem. A, 2020, 8, 2637–2643 RSC
- A. K. Singh and D. Sarkar, Nanoscale, 2018, 10, 13130–13139 RSC
- P. W. Menezes, C. Walter, B. Chakraborty, J. N. Hausmann, I. Zaharieva, A. Frick, E. von Hauff, H. Dau and M. Driess, Adv. Mater., 2021, 33, 2004098 CrossRef CAS PubMed
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118(13), 6337–6408 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, A. Vasileff and S. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed
- S. Yang, Z. Si, G. Li, P. Zhan, C. Liu, L. Lu, B. Han, H. Xie and P. Qin, Small, 2023, 19(15), 2207651 CrossRef CAS PubMed
- S. Chen, J. Xu, J. Chen, Y. Yao and F. Wang, Small, 2024, 20, 2304681 CrossRef CAS PubMed
- P. Liu, X. Zhang, J. Fei, Y. Shi, J. Zhu, D. Zhang, L. Zhao, L. Wang and J. Lai, Adv. Mater., 2024, 36, 2310591 CrossRef CAS PubMed
- N. Kousar and L. K. Sannegowda, Int. J. Hydrogen Energy, 2024, 50, 37–47 CrossRef CAS
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. - Eur. J., 2017, 23, 10947–10961 CrossRef CAS PubMed
- K. Liu, H. Zhong, F. Meng, X. Zhang, J. Yan and Q. Jiang, Mater. Chem. Front., 2017, 1, 2155–2173 RSC
- X. Cheng, Y. Lu, L. Zheng, Y. Cui, M. Niibe, T. Tokushima, H. Li, Y. Zhang, G. Chen and S. Sun, Nano Energy, 2020, 73, 104739 CrossRef CAS
- Y. Zhou, Y. Leng, W. Zhou, J. Huang, M. Zhao, J. Zhan, C. Feng, Z. Tang, S. Chen and H. Liu, Nano Energy, 2015, 16, 357–366 CrossRef CAS
- S. Peng, L. Li, X. Han, W. Sun, M. Srinivasan, S. G. Mhaisalkar, F. Cheng, Q. Yan, J. Chen and S. Ramakrishna, Angew. Chem., 2014, 126, 12802–12807 CrossRef
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Sci., 2018, 5, 1700772 CrossRef PubMed
- Y. Li, Y. Bu, X. Chen, T. Zhu, J. Wang, S. Kawi and Q. Zhong, ChemCatChem, 2019, 11, 1320–1327 CrossRef CAS
- Y. Li, X. Bao, D. Chen, Z. Wang, N. Dewangan, M. Li, Z. Xu, J. Wang, S. Kawi and Q. Zhong, ChemCatChem, 2019, 11, 5913–5928 CrossRef CAS
- Y. Zhao, J. Zhang, W. Zhang and A. Wu, Int. J. Hydrogen Energy, 2021, 46, 35550–35558 CrossRef CAS
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed
- H. Vrubel, T. Moehl, M. Grätzel and X. Hu, Chem. Commun., 2013, 49, 8985–8987 RSC
- Y. Yang, X. Li, G. Liu, H. Liu, Y. Shi, C. Ye, Z. Fang, M. Ye and J. Shen, Adv. Mater., 2024, 36, 2307979 CrossRef CAS PubMed
- S. Liu, Z. Li, Y. Chang, M. Gyu Kim, H. Jang, J. Cho, L. Hou and X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202400069 CrossRef CAS PubMed
- P. E. Karthik, H. Rajan, V. R. Jothi, B. I. Sang and S. C. Yi, J. Hazard. Mater., 2022, 421, 126687 CrossRef CAS PubMed
- D. J. Cole-Hamilton, Chem. Int., 2019, 41, 23–28 CAS
- A. A. Yaroshevsky, Geochem. Int., 2006, 44, 48–55 CrossRef
- C. Panda, P. W. Menezes, S. Yao, J. Schmidt, C. Walter, J. N. Hausmann and M. Driess, J. Am. Chem. Soc., 2019, 141, 13306–13310 CrossRef CAS PubMed
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed
- R. Beltrán-Suito, V. Forstner, J. N. Hausmann, S. Mebs, J. Schmidt, I. Zaharieva, K. Laun, I. Zebger, H. Dau and P. W. Menezes, Chem. Sci., 2020, 11, 11834–11842 RSC
- R. Beltrán-Suito, P. W. Menezes and M. Driess, J. Mater. Chem. A, 2019, 7, 15749–15756 RSC
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS
- P. W. Menezes, A. Indra, I. Zaharieva, C. Walter, S. Loos, S. Hoffmann, R. Schlögl, H. Dau and M. Driess, Energy Environ. Sci., 2019, 12, 988–999 RSC
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287–1298 RSC
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., 2021, 133, 4690–4697 CrossRef
- J. N. Hausmann, R. A. Khalaniya, C. Das, I. Remy-Speckmann, S. Berendts, A. V. Shevelkov, M. Driess and P. W. Menezes, Chem. Commun., 2021, 57, 2184–2187 RSC
- Z. Xia, Nat Energy, 2016, 1, 1–2 Search PubMed
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 CrossRef CAS PubMed
- S. Yao, V. Forstner, P. W. Menezes, C. Panda, S. Mebs, E. M. Zolnhofer, M. E. Miehlich, T. Szilvási, N. A. Kumar and M. Haumann, Chem. Sci., 2018, 9, 8590–8597 RSC
- C. Panda, P. W. Menezes, M. Zheng, S. Orthmann and M. Driess, ACS Energy Lett., 2019, 4, 747–754 CrossRef CAS
- W. Zhang and W. Zheng, Adv. Funct. Mater., 2016, 26, 2988–2993 CrossRef CAS
- D. K. Böhme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44, 2336–2354 CrossRef PubMed
- M. Lübbe and W. Moritz, J. Phys.:Condens. Matter, 2009, 21, 134010 CrossRef PubMed
- V. Ortalan, A. Uzun, B. C. Gates and N. D. Browning, Nat. Nanotechnol., 2010, 5, 506–510 CrossRef CAS PubMed
- A. Morozan, V. Goellner, Y. Nedellec, J. Hannauer and F. Jaouen, J. Electrochem. Soc., 2015, 162, H719 CrossRef CAS
- H.-W. Liang, S. Brüller, R. Dong, J. Zhang, X. Feng and K. Müllen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC
- A. Shahraei, A. Moradabadi, I. Martinaiou, S. Lauterbach, S. Klemenz, S. Dolique, H.-J. Kleebe, P. Kaghazchi and U. I. Kramm, ACS Appl. Mater. Interfaces, 2017, 9, 25184–25193 CrossRef CAS PubMed
- N. Cheng and X. Sun, Chin. J. Catal., 2017, 38, 1508–1514 CrossRef CAS
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS PubMed
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef CAS
- H. Sun, C.-W. Tung, Y. Qiu, W. Zhang, Q. Wang, Z. Li, J. Tang, H.-C. Chen, C. Wang and H. M. Chen, J. Am. Chem. Soc., 2021, 144, 1174–1186 CrossRef PubMed
- G. M. Tomboc, T. Kim, S. Jung, H. J. Yoon and K. Lee, Small, 2022, 18(17), 2105680 CrossRef CAS PubMed
- Z. Fu, M. Wu, Q. Li, C. Ling and J. Wang, Mater. Horiz., 2023, 10, 852–858 RSC
- Z. Luo, X. Han, Z. Ma, B. Zhang, X. Zheng, Y. Liu, M. Gao, G. Zhao, Y. Lin and H. Pan, Angew. Chem., Int. Ed., 2024, e202406728 CAS
- Y. Liu, X. Su, J. Ding, J. Zhou, Z. Liu, X. Wei, H. Bin Yang and B. Liu, Chem. Soc. Rev., 2024, 53, 11850–11887 RSC
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60, 18981–19006 CrossRef CAS PubMed
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC
- S. Sultan, J. N. Tiwari, A. N. Singh, S. Zhumagali, M. Ha, C. W. Myung, P. Thangavel and K. S. Kim, Adv. Energy Mater., 2019, 9, 1900624 CrossRef
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef CAS
- M. Gong, D.-Y. Wang, C.-C. Chen, B.-J. Hwang and H. Dai, Nano Res, 2016, 9, 28–46 CrossRef CAS
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu and Y. W. Li, Nature, 2017, 544, 80–83 CrossRef CAS PubMed
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS
- J. Wei, M. Zhou, A. Long, Y. Xue, H. Liao, C. Wei and Z. J. Xu, Nanomicro Lett., 2018, 10, 1–15 Search PubMed
- S. Anantharaj, S. R. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, ACS Energy Lett., 2018, 3(7), 1713–1721 CrossRef CAS
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS
- H. Jin, X. Liu, S. Chen, A. Vasileff, L. Li, Y. Jiao, L. Song, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 805–810 CrossRef CAS
- J. Mohammed-Ibrahim and X. Sun, J. Energy Chem., 2019, 34, 111–160 CrossRef
- P. R. Jothi, Y. Zhang, K. Yubuta, D. B. Culver, M. Conley and B. P. T. Fokwa, ACS Appl. Energy Mater., 2018, 2, 176–181 CrossRef
- X. Chen, Z. Yu, L. Wei, Z. Zhou, S. Zhai, J. Chen, Y. Wang, Q. Huang, H. E. Karahan and X. Liao, J. Mater. Chem. A, 2019, 7, 764–774 RSC
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819 CrossRef
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed
- J. Greeley, J. K. Nørskov, L. A. Kibler, A. M. El-Aziz and D. M. Kolb, ChemPhysChem, 2006, 7, 1032–1035 CrossRef CAS PubMed
- J. Tymoczko, F. Calle-Vallejo, W. Schuhmann and A. S. Bandarenka, Nat. Commun., 2016, 7, 10990 CrossRef CAS PubMed
- V. Čolić, J. Tymoczko, A. Maljusch, A. Ganassin, W. Schuhmann and A. S. Bandarenka, ChemElectroChem, 2015, 2, 143–149 CrossRef
- Y. Li, H. Zhang, T. Xu, Z. Lu, X. Wu, P. Wan, X. Sun and L. Jiang, Adv. Funct. Mater., 2015, 25, 1737–1744 CrossRef CAS
- F. Meng, E. Hu, L. Zhang, K. Sasaki, J. T. Muckerman and E. Fujita, J. Mater. Chem. A, 2015, 3, 18572–18577 RSC
- Y. Liu, S. Liu, Y. Wang, Q. Zhang, L. Gu, S. Zhao, D. Xu, Y. Li, J. Bao and Z. Dai, J. Am. Chem. Soc., 2018, 140, 2731–2734 CrossRef CAS PubMed
- T. G. Kelly, S. T. Hunt, D. V. Esposito and J. G. Chen, Int. J. Hydrogen Energy, 2013, 38, 5638–5644 CrossRef CAS
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi and X. Meng, ACS Nano, 2016, 10, 684–694 CrossRef CAS PubMed
- X. Zhong, Y. Sun, X. Chen, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2016, 26, 5778–5786 CrossRef CAS
- S. Liu, X. Mu, H. Duan, C. Chen and H. Zhang, Eur. J. Inorg. Chem., 2017, 535–539 CrossRef CAS
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Nørskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC
- M. N. Hossain, S. Ahmad and H.-B. Kraatz, Int. J. Hydrogen Energy, 2021, 46, 2007–2017 CrossRef CAS
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef
- M. Chhetri, S. Maitra, H. Chakraborty, U. V. Waghmare and C. N. R. Rao, Energy Environ. Sci., 2016, 9, 95–101 RSC
- C. N. R. Rao and M. Chhetri, Adv. Mater., 2019, 31, 1803668 CrossRef PubMed
- Y. Luo, X. Luo, G. Wu, Z. Li, G. Wang, B. Jiang, Y. Hu, T. Chao, H. Ju, J. Zhu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, ACS Appl. Mater. Interfaces, 2018, 10, 34147–34152 CrossRef CAS PubMed
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- Y. Li, Green Energy Environ., 2020, 5, 4–5 CrossRef
- H. Gao, Appl. Surf. Sci., 2016, 379, 347–357 CrossRef CAS
- Z. Hao, S. Guo and L. Guo, Appl. Organomet. Chem., 2019, 33, e4960 CrossRef
- L. Wu, T. Guo and T. Li, J. Mater. Chem. A, 2020, 8, 19290–19299 RSC
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2019, 3, 1800376 CrossRef
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Perez-Ramirez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed
- T. N. Ye, Z. Xiao, J. Li, Y. Gong, H. Abe, Y. Niwa, M. Sasase, M. Kitano and H. Hosono, Nat. Commun., 2020, 11, 1020 CrossRef CAS PubMed
- B. Zhang, G. Sun, S. Ding, H. Asakura, J. Zhang, P. Sautet and N. Yan, J. Am. Chem. Soc., 2019, 141, 8185–8197 CrossRef CAS PubMed
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková and J. Mysliveček, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS PubMed
- M. S. Frei, C. Mondelli, R. García-Muelas, K. S. Kley, B. Puértolas, N. López, O. V. Safonova, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, Nat. Commun., 2019, 10, 3377 CrossRef PubMed
- E. Vorobyeva, E. Fako, Z. Chen, S. M. Collins, D. Johnstone, P. A. Midgley, R. Hauert, O. V. Safonova, G. Vilé and N. López, Angew. Chem., 2019, 131, 8816–8821 CrossRef
- Y. Wang, E. Song, W. Qiu, X. Zhao, Y. Zhou, J. Liu and W. Zhang, Prog. Nat. Sci.:Mater. Int., 2019, 29, 256–264 CrossRef CAS
- H. Jin, K. Zhou, R. Zhang, H. Cui, Y. Yu, P. Cui, W. Song and C. Cao, Nat. Commun., 2023, 14, 2494 CrossRef CAS PubMed
- W. Yang, X. Liu, X. Chen, Y. Cao, S. Cui, L. Jiao, C. Wu, C. Chen, D. Fu and I. D. Gates, Adv. Mater., 2022, 34, 2110123 CrossRef CAS PubMed
- Y. Li, Z. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang and H. Sun, Nat. Catal., 2018, 1, 63–72 CrossRef CAS
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard and G. Vilé, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard and L. Kovarik, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed
- J. Shin, Y. J. Lee, A. Jan, S. M. Choi, M. Y. Park, S. Choi, J. Y. Hwang, S. Hong, S. G. Park and H. J. Chang, Energy Environ. Sci., 2020, 13, 4903–4920 RSC
- W. Yang, S. Xu, K. Ma, C. Wu, I. D. Gates, X. Ding, W. Meng and Z. Gao, Nano Mater. Sci., 2020, 2, 120–131 CrossRef
- C. Chen, W. Ou, K. Yam, S. Xi, X. Zhao, S. Chen, J. Li, P. Lyu, L. Ma and Y. Du, Adv. Mater., 2021, 33, 2008471 CrossRef CAS PubMed
- Z. Ma, L. Niu, W. Jiang, C. Dong, G. Liu, D. Qu, L. An and Z. Sun, JPhys Mater., 2021, 4, 042002 CrossRef CAS
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., 2012, 124, 12663–12666 CrossRef
- Y. Zhou, G. Gao, J. Kang, W. Chu and L.-W. Wang, Nanoscale, 2019, 11, 18169–18175 RSC
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef
- X. Zhang, X. Xu, S. Yao, C. Hao, C. Pan, X. Xiang, Z. Q. Tian, P. K. Shen, Z. Shao and S. P. Jiang, Small, 2022, 18, 2105329 CrossRef CAS PubMed
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef PubMed
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef
- Y. Lei, Y. Wang, Y. Liu, C. Song, Q. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 20794–20812 CrossRef CAS PubMed
- Z. Chen and P. Zhang, ACS Omega, 2022, 7, 1585–1594 CrossRef CAS PubMed
- D. Chen, Z. Chen, X. Zhang, Z. Lu, S. Xiao, B. Xiao and C. V. Singh, J. Energy Chem., 2021, 52, 155–162 CrossRef CAS
- W. Liu, H. Zhang, C. Li, X. Wang, J. Liu and X. Zhang, J. Energy Chem., 2020, 47, 333–345 CrossRef
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga and X. I. Pereira Hernández, Science, 2016, 353, 150–154 CrossRef CAS PubMed
- J. Wu, N. Han, S. Ning, T. Chen, C. Zhu, C. Pan, H. Wu, S. J. Pennycook and C. Guan, ACS Sustainable Chem. Eng., 2020, 8, 14825–14832 CrossRef CAS
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed
- T. S. Zeleke, M.-C. Tsai, M. A. Weret, C.-J. Huang, M. K. Birhanu, T.-C. Liu, C.-P. Huang, Y.-L. Soo, Y.-W. Yang and W.-N. Su, ACS Nano, 2019, 13, 6720–6729 CrossRef CAS PubMed
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS
- R. Leng, Q. Yuan, Y. Wang, Q. Kuang and P. Ren, Sustainability, 2020, 12, 533 CrossRef CAS
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef PubMed
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed
- H.-J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., 2015, 127, 14237–14241 CrossRef
- Y. Peng, B. Lu, L. Chen, N. Wang, J. E. Lu, Y. Ping and S. Chen, J. Mater. Chem. A, 2017, 5, 18261–18269 RSC
- T. Li, J. Liu, Y. Song and F. Wang, ACS Catal., 2018, 8, 8450–8458 CrossRef CAS
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Nat. Catal., 2019, 2, 134–141 CrossRef CAS
- C. Lei, Y. Wang, Y. Hou, P. Liu, J. Yang, T. Zhang, X. Zhuang, M. Chen, B. Yang and L. Lei, Energy Environ. Sci., 2019, 12, 149–156 RSC
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. Sun and H. Wong, Adv. Energy Mater., 2019, 9, 1803689 CrossRef
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed
- V. Fung, G. Hu, Z. Wu and D. Jiang, J. Phys. Chem. C, 2020, 124, 19571–19578 CrossRef CAS
- D. N. Sredojević, M. R. Belić and Ž. Šljivančanin, J. Phys. Chem. C, 2020, 124, 16860–16867 CrossRef
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS
- G. A. Somorjai and J. Y. Park, Chem. Soc. Rev., 2008, 37, 2155–2162 RSC
- M. Crespo-Quesada, A. Yarulin, M. Jin, Y. Xia and L. Kiwi-Minsker, J. Am. Chem. Soc., 2011, 133, 12787–12794 CrossRef CAS PubMed
- C. Jin, L. Huo, J. Tang, S. Li, K. Jiang, Q. He, H. Dong, Y. Gong and Z. Hu, Small, 2024, 20, 2309509 CrossRef CAS PubMed
- Q. Li, Q. Zhang, W. Xu, R. Zhao, M. Jiang, Y. Gao, W. Zhong, K. Chen, Y. Chen and X. Li, Adv. Energy Mater., 2023, 13, 2203955 CrossRef CAS
- M. Ranocchiari, C. Lothschuetz, D. Grolimund and J. A. van Bokhoven, Proc. R. Soc. A, 2012, 468, 1985–1999 CrossRef CAS
- R.-Q. Zhang, T.-H. Lee, B.-D. Yu, C. Stampfl and A. Soon, Phys. Chem. Chem. Phys., 2012, 14, 16552–16557 RSC
- Z. Novotný, G. Argentero, Z. Wang, M. Schmid, U. Diebold and G. S. Parkinson, Phys. Rev. Lett., 2012, 108, 216103 CrossRef PubMed
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem Rev, 2018, 119, 1806–1854 CrossRef PubMed
- L. Zhang, X. Guo, S. Zhang, T. Frauenheim and S. Huang, Adv. Energy Mater., 2024, 14, 2302754 CrossRef CAS
- Y. Gao, Y. Xue, H. Wu, S. Chen, X. Zheng, C. Xing and Y. Li, J. Am. Chem. Soc., 2024, 146, 10573–10580 CrossRef CAS PubMed
- W.-G. Lim, H. N. Truong, J.-Y. Jeong, D. Kim, L. S. Oh, C. Jo, C. Kim, H. J. Kim, S. M. Choi and H. Shin, Appl. Catal., B, 2024, 343, 123568 CrossRef CAS
- W. Li, D. Yin, P. Li and X. Zhao, Phys. Chem. Chem. Phys., 2024, 26, 19822–19830 RSC
- W. Zhao, S. Shen, Y. Zhao, T. Wu, S. Ding and Y. Su, J. Mater. Chem. A, 2024, 12, 16476–16481 RSC
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC
- S. Hussain, S. H. Talib, S. Mohamed, R. Zhao, A. Qurashi, J. Li and Z. Lu, Int. J. Hydrogen Energy, 2024, 53, 969–978 CrossRef CAS
- C. Yao, Q. Wang, C. Peng, R. Wang, J. Liu, N. Tsidaeva and W. Wang, Chem. Eng. J., 2024, 479, 147924 CrossRef CAS
- A. Radwan, H. Jin, D. He and S. Mu, Nanomicro Lett., 2021, 13, 1–32 Search PubMed
- H. Liu, X. Peng and X. Liu, ChemElectroChem, 2018, 5, 2963–2974 CrossRef CAS
- S. Li, Z. Xin, Y. Luo, J. Pan, G. Liao, Q. Li, Y. Sun, Z. Feng and R. Tan, Int. J. Hydrogen Energy, 2024, 82, 1081–1100 CrossRef CAS
- P. Yin and B. You, Mater. Today Energy, 2021, 19, 100586 CrossRef CAS
- A. R. Woldu, A. G. Yohannes, Z. Huang, P. Kennepohl, D. Astruc, L. Hu and X. Huang, Adv. Mater., 2024, 2414169 CrossRef CAS PubMed
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed
- Y. Jia, C. Shi, W. Zhang, W. Xia, M. Hu, R. Huang and R. Qi, Nanomaterials, 2022, 12, 1593 CrossRef CAS PubMed
- X. Xiang, X. Zhang, B. Yan, K. Wang, Y. Wang, D. Lyu, S. Xi, Z. Q. Tian and P. K. Shen, Chem. Eng. J., 2022, 440, 135721 CrossRef CAS
- W. Zhang, Q. Fu, Q. Luo, L. Sheng and J. Yang, JACS Au, 2021, 1, 2130–2145 CrossRef CAS PubMed
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed
- L. Zhang, L. Han, H. Liu, X. Liu and J. Luo, Angew. Chem., 2017, 129, 13882–13886 CrossRef
- M. Tavakkoli, N. Holmberg, R. Kronberg, H. Jiang, J. Sainio, E. I. Kauppinen, T. Kallio and K. Laasonen, ACS Catal., 2017, 7, 3121–3130 CrossRef CAS
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li and M. Lei, Nat. Commun., 2017, 8, 1490 CrossRef PubMed
- Y. Qu, L. Wang, Z. Li, P. Li, Q. Zhang, Y. Lin, F. Zhou, H. Wang, Z. Yang, Y. Hu, M. Zhu, X. Zhao, X. Han, C. Wang, Q. Xu, L. Gu, J. Luo, L. Zheng and Y. Wu, Adv. Mater., 2019, 31, 1904496 CrossRef CAS PubMed
- L. Liu and A. Corma, Chem Rev, 2018, 118, 4981–5079 CrossRef CAS PubMed
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed
- W. Chen, J. Pei, C. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., 2017, 129, 16302–16306 CrossRef
- L. Zou, C. Hou, Q. Wang, Y. Wei, Z. Liu, J. Qin, H. Pang and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 19627–19632 CrossRef CAS PubMed
- X. X. Wang, D. A. Cullen, Y. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang and Y. He, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC
- X. Zhao, Y. Wang, D. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed
- X. Zhang, S. Zhang, Y. Tang, X. Huang and H. Pang, Composites, Part B, 2022, 230, 109532 CrossRef CAS
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed
- Y. V. Kaneti, J. Tang, R. R. Salunkhe, X. Jiang, A. Yu, K. C. Wu and Y. Yamauchi, Adv. Mater., 2017, 29, 1604898 CrossRef PubMed
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS
- L. Zou, Y. Wei, C. Hou, C. Li and Q. Xu, Small, 2021, 17, 2004809 CrossRef CAS PubMed
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC
- L. Zou, C.-C. Hou, Z. Liu, H. Pang and Q. Xu, J. Am. Chem. Soc., 2018, 140, 15393–15401 CrossRef CAS PubMed
- G. Chellasamy, S. K. Arumugasamy, S. Kuppusamy, V. Ekambaram, K. Rajagopalan, S. Venkateswarlu, P. Deivasigamani, M. J. Choi, S. Govindaraju and K. Yun, J. Mater. Chem. A, 2024, 12, 1115–1127 RSC
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long and M. Chen, Chem, 2019, 5, 2865–2878 CAS
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng and W. Yan, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed
- S. Ji, Y. Chen, S. Zhao, W. Chen, L. Shi, Y. Wang, J. Dong, Z. Li, F. Li and C. Chen, Angew. Chem., 2019, 131, 4315–4319 CrossRef
- J. Li, M. N. Banis, Z. Ren, K. R. Adair, K. Doyle-Davis, D. M. Meira, Y. Z. Finfrock, L. Zhang, F. Kong and T. Sham, Small, 2021, 17, 2007245 CrossRef CAS PubMed
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si and S. Zhou, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed
- Z. Zhang, Z. Zhang, C. Chen, R. Wang, M. Xie, S. Wan, R. Zhang, L. Cong, H. Lu and Y. Han, Nat. Commun., 2024, 15, 2556 CrossRef CAS PubMed
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song and J. Li, Nat. Commun., 2019, 10, 4936 CrossRef PubMed
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed
- B. J. O’Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng and M. N. Banis, Sci. Rep., 2013, 3, 1775 CrossRef
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li and X. Huang, Nat. Commun., 2017, 8, 1070 CrossRef PubMed
- M. Piernavieja-Hermida, Z. Lu, A. White, K.-B. Low, T. Wu, J. W. Elam, Z. Wu and Y. Lei, Nanoscale, 2016, 8, 15348–15356 RSC
- L. Zhang, Z.-J. Zhao, M. N. Banis, L. Li, Y. Zhao, Z. Song, Z. Wang, T.-K. Sham, R. Li and M. Zheng, J. Mater. Chem. A, 2018, 6, 24397–24406 RSC
- L. Zhang, Y. Jia, H. Liu, L. Zhuang, X. Yan, C. Lang, X. Wang, D. Yang, K. Huang and S. Feng, Angew. Chem., 2019, 131, 9504–9508 CrossRef
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu and L. Zhuang, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo and D. Zang, Science, 2016, 352, 797–800 CrossRef CAS PubMed
- K.-G. Qu, Z.-F. Chen, L.-H. Wang, H.-B. Li, S.-Y. Zeng, R. Li, L.-J. Meng, H.-Y. Chen and Q.-X. Yao, Rare Metals, 2024, 1–9 CAS
- I. Barlocco, G. Di Liberto and G. Pacchioni, Energy Adv., 2023, 2, 1022–1029 RSC
- Y. Wu, R. Wang and Y. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 66874–66899 CrossRef CAS PubMed
- B. Zhang, L. Chen, Z. Zhang, Q. Li, P. Khangale, D. Hildebrandt, X. Liu, Q. Feng and S. Qiao, Adv. Sci., 2022, 9, 2105912 CrossRef CAS PubMed
- J. D. Yi, R. Xu, G. L. Chai, T. Zhang, K. Zang, B. Nan, H. Lin, Y. L. Liang, J. Lv, J. Luo, R. Si, Y. B. Huang and R. Cao, J. Mater. Chem. A, 2019, 7, 1252–1259 RSC
- B. Ball, C. Chakravarty and P. Sarkar, J. Phys. Chem. Lett., 2020, 11, 1542–1549 CrossRef CAS PubMed
- K. Iwase, S. Nakanishi, M. Miyayama and K. Kamiya, ACS Appl. Energy Mater., 2020, 3, 1644–1652 CrossRef CAS
- C. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed
- K. Chandran Ranjeesh, R. Illathvalappil, V. Chandrakant Wakchaure, Goudappagouda, S. Kurungot and S. S. Babu, ACS Appl. Energy Mater., 2018, 1, 6442–6450 CrossRef CAS
- S. Qian, F. Xu, Y. Fan, N. Cheng, H. Xue, Y. Yuan, R. Gautier, T. Jiang and J. Tian, Nat. Commun., 2024, 15, 2774 CrossRef CAS PubMed
- F. Yang, R. Yao, Z. Lang, F. Yu, H. Dong, Y. Wang, Y. Li and H. Tan, ACS Energy Lett., 2023, 8, 5175–5183 CrossRef CAS
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek and J. F. Sanz, Science, 2014, 345, 546–550 CrossRef CAS PubMed
- B. Wang, X. Zhu, X. Pei, W. Liu, Y. Leng, X. Yu, C. Wang, L. Hu, Q. Su and C. Wu, J. Am. Chem. Soc., 2023, 145, 13788–13795 CrossRef CAS PubMed
- Y. Wu, T. Bo, H. Tu, L. Wang and W. Shi, Small, 2024, 2402847 CrossRef CAS PubMed
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig and V. S. Batista, Nat. Commun., 2018, 9, 415 CrossRef PubMed
- C. Lang, Y. Xu and X. Yao, Chin. J. Catal., 2024, 64, 4–31 CrossRef CAS
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC
- F. S. Hage, G. Radtke, D. M. Kepaptsoglou, M. Lazzeri and Q. M. Ramasse, Science, 2020, 367, 1124–1127 CrossRef CAS PubMed
- T. Zhang, Z. Chen, A. G. Walsh, Y. Li and P. Zhang, Adv. Mater., 2020, 32, 2002910 CrossRef CAS PubMed
- S. Qiu, Y. Shen, G. Wei, S. Yao, W. Xi, M. Shu, R. Si, M. Zhang, J. Zhu and C. An, Appl. Catal., B, 2019, 259, 118036 CrossRef CAS
- S. Hejazi, S. Mohajernia, B. Osuagwu, G. Zoppellaro, P. Andryskova, O. Tomanec, S. Kment, R. Zbořil and P. Schmuki, Adv. Mater., 2020, 32, 1908505 CrossRef CAS PubMed
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, S. Kment, R. Zboril and P. Schmuki, ACS Catal., 2018, 9, 345–364 CrossRef PubMed
- Z. Wang, X. Yuan, H. Guo, X. Zhang, J. Peng and Y. Pan, Energy Environ. Sci., 2024, 17, 8019–8056 RSC
- M. I. Hussain, M. U. Farooq, M. Xia, X. Ren, R. R. Zairov and M. Harb, Appl. Mater. Today, 2024, 36, 102037 CrossRef
- L. Zhang, J. Yang, X. Yang, A. Wang and T. Zhang, Chem. Catal., 2023, 3(3), 100560 CrossRef CAS
- B. Han, Y. Guo, Y. Huang, W. Xi, J. Xu, J. Luo, H. Qi, Y. Ren, X. Liu and B. Qiao, Angew. Chem., Int. Ed., 2020, 59, 11824–11829 CrossRef CAS PubMed
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan and J. Zhu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed
- Z. Liu, T. He, Q. Jiang, W. Wang and J. Tang, Int. J. Hydrogen Energy, 2022, 47, 29698–29729 CrossRef CAS
- T. H. M. Lau, J. S. Foord and S. C. E. Tsang, Nanoscale, 2020, 12, 10447–10455 RSC
- H. Jin, X. Liu, S. Chen, A. Vasileff, L. Li, Y. Jiao, L. Song, Y. Zheng and S. Z. Qiao, ACS Energy Lett., 2019, 4, 805–810 CrossRef CAS
- J. Yin, J. Jin, H. Zhang, M. Lu, Y. Peng, B. Huang, P. Xi and C. Yan, Angew. Chem., 2019, 131, 18849–18855 CrossRef
- K. Qu, Y. Zheng, X. Zhang, K. Davey, S. Dai and S. Z. Qiao, ACS Nano, 2017, 11, 7293–7300 CrossRef CAS PubMed
- G. Li, J. Yu, J. Jia, L. Yang, L. Zhao, W. Zhou and H. Liu, Adv. Funct. Mater., 2018, 28, 1801332 CrossRef
- G. Li, J. Wang, J. Yu, H. Liu, Q. Cao, J. Du, L. Zhao, J. Jia, H. Liu and W. Zhou, Appl. Catal., B, 2020, 261, 118147 CrossRef CAS
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed
- T. H. M. Lau, X. Lu, J. Kulhavý, S. Wu, L. Lu, T.-S. Wu, R. Kato, J. S. Foord, Y.-L. Soo and K. Suenaga, Chem. Sci., 2018, 9, 4769–4776 RSC
- P. Liu and N. Zheng, Nat. Sci. Rev., 2018, 5, 636–638 CrossRef CAS
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Small, 2020, 16, 1906782 CrossRef CAS PubMed
- V. Ramalingam, P. Varadhan, H. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma, Y. Wang and H. N. Alshareef, Adv. Mater., 2019, 31, 1903841 CrossRef CAS PubMed
- Y. Cao, S. Chen, Q. Luo, H. Yan, Y. Lin, W. Liu, L. Cao, J. Lu, J. Yang and T. Yao, Angew. Chem., Int. Ed., 2017, 56, 12191–12196 CrossRef CAS PubMed
- X. Huang, H. Yan, L. Huang, X. Zhang, Y. Lin, J. Li, Y. Xia, Y. Ma, Z. Sun and S. Wei, J. Phys. Chem. C, 2018, 123, 7922–7930 CrossRef
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song and J. Zhu, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed
- J. Zhu, W. Li, S. Li, J. Zhang, H. Zhou, C. Zhang, J. Zhang and S. Mu, Small, 2018, 14, 1800563 CrossRef PubMed
- M. Fan, Z.-Q. Feng, C. Zhu, X. Chen, C. Chen, J. Yang and D. Sun, J. Mater. Sci., 2016, 51, 10323–10349 CrossRef CAS
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng and A.-B. Xu, Nat. Energy, 2021, 6, 807–814 CrossRef CAS
- F. Kraushofer, N. Resch, M. Eder, A. Rafsanjani-Abbasi, S. Tobisch, Z. Jakub, G. Franceschi, M. Riva, M. Meier and M. Schmid, Adv. Mater. Interfaces, 2021, 8, 2001908 CrossRef CAS
- Q. Liu and Z. Zhang, Catal. Sci. Technol., 2019, 9, 4821–4834 RSC
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J.-S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS
- J. Zhao, Q. Deng, S. M. Avdoshenko, L. Fu, J. Eckert and M. H. Rümmeli, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15641–15646 CrossRef CAS PubMed
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang and L. Zheng, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng and M. Wei, Science, 2014, 344, 616–619 CrossRef CAS PubMed
- P. Qi, J. Wang, X. Djitcheu, D. He, H. Liu and Q. Zhang, RSC Adv., 2022, 12, 1216–1227 RSC
- Z. Chen, A. G. Walsh and P. Zhang, Acc. Chem. Res., 2024, 57, 521–532 CAS
- S.-C. Lin, C.-C. Chang, S.-Y. Chiu, H.-T. Pai, T.-Y. Liao, C.-S. Hsu, W.-H. Chiang, M.-K. Tsai and H. M. Chen, Nat. Commun., 2020, 11, 3525 CrossRef CAS PubMed
- R. F. Egerton and M. Watanabe, Ultramicroscopy, 2018, 193, 111–117 CrossRef CAS PubMed
- R. Senga and K. Suenaga, Ultramicroscopy, 2017, 180, 150–155 CrossRef CAS PubMed
- L. Cheng, H. Yin, C. Cai, J. Fan and Q. Xiang, Small, 2020, 16, 2002411 CrossRef CAS PubMed
- J. Xu, Y. Wang, D. Wang and J. Liu, Microsc. Microanal., 2020, 26, 1762–1763 CrossRef
- X. Liang, Z. Li, H. Xiao, T. Zhang, P. Xu, H. Zhang, Q. Gao and L. Zheng, Chem. Mater., 2021, 33, 5542–5554 CrossRef CAS
- J. Amsler, B. B. Sarma, G. Agostini, G. Prieto, P. N. Plessow and F. Studt, J. Am. Chem. Soc., 2020, 142, 5087–5096 CrossRef CAS PubMed
- T. K. Sham, Int. J. Nanotechnol., 2008, 5, 1194–1246 CrossRef CAS
- D. E. Sayers, E. A. Stern and F. W. Lytle, Phys. Rev. Lett., 1971, 27, 1204 CrossRef CAS
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou and J. Zhang, Angew. Chem., 2020, 132, 19618–19627 CrossRef
- Q. Sun, N. Wang, R. Bai, Y. Hui, T. Zhang, D. A. Do, P. Zhang, L. Song, S. Miao and J. Yu, Adv. Sci., 2019, 6, 1802350 CrossRef PubMed
- A. A. Guda, S. A. Guda, K. A. Lomachenko, M. A. Soldatov, I. A. Pankin, A. V. Soldatov, L. Braglia, A. L. Bugaev, A. Martini and M. Signorile, Catal. Today, 2019, 336, 3–21 CrossRef CAS
- J. Wei, S. Qin, J. Yang, H. Ya, W. Huang, H. Zhang, B. J. Hwang, Z. Tian and J. Li, Angew. Chem., Int. Ed., 2021, 60, 9306–9310 CrossRef CAS PubMed
- N. H. M. Dostagir, R. Rattanawan, M. Gao, J. Ota, J. Hasegawa, K. Asakura, A. Fukouka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef CAS
- B. B. Sarma, P. N. Plessow, G. Agostini, P. Concepción, N. Pfänder, L. Kang, F. R. Wang, F. Studt and G. Prieto, J. Am. Chem. Soc., 2020, 142, 14890–14902 CrossRef CAS PubMed
- G. Spezzati, Y. Su, J. P. Hofmann, A. D. Benavidez, A. T. DeLaRiva, J. McCabe, A. K. Datye and E. J. M. Hensen, ACS Catal., 2017, 7, 6887–6891 CrossRef CAS PubMed
- R. Lang, W. Xi, J.-C. Liu, Y.-T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li and L. Li, Nat. Commun., 2019, 10, 234 CrossRef PubMed
- J. Wang, X. Ge, L. Shao, J. Zhang, D. Peng, G. Zou, H. Hou, W. Deng, S. Xu and X. Ji, Mater. Today Energy, 2020, 17, 100436 CrossRef
- W. Chen, J. Pei, C. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W. Cheong and J. Mao, Adv. Mater., 2018, 30, 1800396 CrossRef PubMed
- L. Cao, X. Wang, C. Yang, J. Lu, X. Shi, H. Zhu and H.-P. Liang, ACS Sustainable Chem. Eng., 2020, 9, 189–196 CrossRef
- J. Lim, S. Back, C. Choi and Y. Jung, ChemCatChem, 2018, 10, 4450–4455 CrossRef CAS
- X. Peng, J. Hou, Y. Mi, J. Sun, G. Qi, Y. Qin, S. Zhang, Y. Qiu, J. Luo and X. Liu, Nanoscale, 2021, 13, 4767–4773 RSC
- S. T. Hunt, M. Milina, Z. Wang and Y. Román-Leshkov, Energy Environ. Sci., 2016, 9, 3290–3301 RSC
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef
- W. Liu, Q. Xu, P. Yan, J. Chen, Y. Du, S. Chu and J. Wang, ChemCatChem, 2018, 10, 946–950 CrossRef CAS
- Y. Zhang, W. Li, L. Lu, W. Song, C. Wang, L. Zhou, J. Liu, Y. Chen, H. Jin and Y. Zhang, Electrochim Acta, 2018, 265, 497–506 CrossRef CAS
- Y. Zhou, J. Li, X. Gao, W. Chu, G. Gao and L.-W. Wang, J. Mater. Chem. A, 2021, 9, 9979–9999 RSC
- T. He, C. Zhang and A. Du, Chem. Eng. Sci., 2019, 194, 58–63 CrossRef CAS
- T. Chao, X. Luo, W. Chen, B. Jiang, J. Ge, Y. Lin, G. Wu, X. Wang, Y. Hu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, Angew. Chem., 2017, 129, 16263–16267 CrossRef
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- X. Huang, Z. Zeng, S. Bao, M. Wang, X. Qi, Z. Fan and H. Zhang, Nat. Commun., 2013, 4, 1444 CrossRef PubMed
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed
- H.-Y. Cheng, P.-O. Åstrand, D. Chen, Y.-A. Zhu, X.-G. Zhou and P. Li, Chem. Phys. Lett., 2013, 575, 76–80 CrossRef CAS
- Y.-C. Zhang, M. Zhao, J. Wu, Y. Wang, L. Zheng, F. Gu, J.-J. Zou, J. Gao and X.-D. Zhu, ACS Catal., 2024, 14, 7867–7876 CrossRef CAS
- X. Liu, X. Song, G. Jiang, L. Tao, Z. Jin, F. Li, Y. He and F. Dong, Chem. Eng. J., 2024, 481, 148430 CrossRef CAS
- Z. Zhang, Y. Chen, L. Zhou, C. Chen, Z. Han, B. Zhang, Q. Wu, L. Yang, L. Du, Y. Bu, P. Wang, X. Wang, H. Yang and Z. Hu, Nat. Commun., 2019, 10, 1657 CrossRef PubMed
- Z. Zhang, Q. Wu, K. Mao, Y. Chen, L. Du, Y. Bu, O. Zhuo, L. Yang, X. Wang and Z. Hu, ACS Catal., 2018, 8, 8477–8483 CrossRef CAS
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC
- A. N. Mansour, J. W. Cook Jr and D. E. Sayers, J. Phys. Chem., 1984, 88, 2330–2334 CrossRef CAS
- T. K. Sham, S. J. Naftel and I. Coulthard, J. Appl. Phys., 1996, 79, 7134–7138 CrossRef CAS
- C. Wang, F. Chen, Q. Wang, X. Yang, H. Zang, N. Yu and B. Geng, Carbon, 2023, 201, 278–284 CrossRef CAS
- Y. Qu, B. Chen, Z. Li, X. Duan, L. Wang, Y. Lin, T. Yuan, F. Zhou, Y. Hu, Z. Yang, C. Zhao, J. Wang, C. Zhao, Y. Hu, G. Wu, Q. Zhang, Q. Xu, B. Liu, P. Gao, R. You, W. Huang, L. Zheng, L. Gu, Y. Wu and Y. Li, J. Am. Chem. Soc., 2019, 141, 4505–4509 CrossRef CAS PubMed
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS
- W. Zhong, W. Tu, Z. Wang, Z. Lin, A. Xu, X. Ye, D. Chen and B. Xiao, J. Energy Chem., 2020, 51, 280–284 CrossRef
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du and M. Song, Energy Environ. Sci., 2021, 14, 6428–6440 RSC
- D. Li, X. Chen, Y. Lv, G. Zhang, Y. Huang, W. Liu, Y. Li, R. Chen, C. Nuckolls and H. Ni, Appl. Catal., B, 2020, 269, 118824 CrossRef CAS
- A. Pei, R. Xie, Y. Zhang, Y. Feng, W. Wang, S. Zhang, Z. Huang, L. Zhu, G. Chai, Z. Yang, Q. Gao, H. Ye, C. Shang, B. H. Chen and Z. Guo, Energy Environ. Sci., 2023, 16, 1035–1048 RSC
- J. Xu, C. Zhang, H. Liu, J. Sun, R. Xie, Y. Qiu, F. Lü, Y. Liu, L. Zhuo, X. Liu and J. Luo, Nano Energy, 2020, 70, 104529 CrossRef CAS
- Y. Guan, Y. Feng, J. Wan, X. Yang, L. Fang, X. Gu, R. Liu, Z. Huang, J. Li, J. Luo, C. Li and Y. Wang, Small, 2018, 14, 1800697 CrossRef PubMed
- D. Ren, G. Wang, L. Li, Y. Jin, K. Zhou, C. Zeng, Q. Zhang, J. Liu, R. Wang, X. Ke, M. Sui and H. Wang, Chem. Eng. J., 2023, 454, 140557 CrossRef CAS
- F. Li, D. H. Kweon, G. F. Han, H. J. Noh, W. Che, I. Ahmad, H. Y. Jeong, Z. Fu, Y. Lu and J. B. Baek, ACS Nano, 2023, 17, 2923–2931 CrossRef CAS PubMed
- A. Wang and T. Zhang, Nat Energy, 2016, 1, 1–2 Search PubMed
- Y. Li, S. Chen, R. Long, H. Ju, Z. Wang, X. Yu, F. Gao, Z. Cai, C. Wang and Q. Xu, Nano Energy, 2017, 34, 306–312 CrossRef CAS
- B. Bayatsarmadi, Y. Zheng, A. Vasileff and S. Qiao, Small, 2017, 13, 1700191 CrossRef PubMed
- R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef CAS
- J. Li, H.-M. Yin, X.-B. Li, E. Okunishi, Y.-L. Shen, J. He, Z.-K. Tang, W.-X. Wang, E. Yücelen and C. Li, Nat Energy, 2017, 2, 1–9 CAS
- G. Xu, H. Wei, Y. Ren, J. Yin, A. Wang and T. Zhang, Green Chem., 2016, 18, 1332–1338 RSC
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček and V. Matolín, Nat. Commun., 2016, 7, 10801 CrossRef PubMed
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed
- H. Yu, W. Wang, Q. Mao, K. Deng, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Appl. Catal., B, 2023, 330, 122617 CrossRef CAS
- H. Jin, X. Liu, Y. Jiao, A. Vasileff, Y. Zheng and S.-Z. Qiao, Nano Energy, 2018, 53, 690–697 CrossRef CAS
- S. Ye, F. Luo, T. Xu, P. Zhang, H. Shi, S. Qin, J. Wu, C. He, X. Ouyang and Q. Zhang, Nano Energy, 2020, 68, 104301 CrossRef CAS
- X. Wang, Y. Zheng, W. Sheng, Z. J. Xu, M. Jaroniec and S.-Z. Qiao, Mater. Today, 2020, 36, 125–138 CrossRef CAS
- S. Ye, W. Xiong, P. Liao, L. Zheng, X. Ren, C. He, Q. Zhang and J. Liu, J. Mater. Chem. A, 2020, 8, 11246–11254 RSC
- T. Li, X. Zhang, Y. Chen, L. Zhong, S. Li, P. Zhang and C. Zhao, J. Mater. Chem. A, 2021, 9, 16967–16973 RSC
- J. Li, C. Zhang, C. Zhang, H. Ma, Y. Yang, Z. Guo, Y. Wang and H. Ma, Chem. Eng. J., 2022, 430, 132953 CrossRef CAS
- B. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. Chen, Nat. Commun., 2019, 10, 631 CrossRef CAS PubMed
- S.-W. Sun, G.-F. Wang, Y. Zhou, F.-B. Wang and X.-H. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 19176–19182 CrossRef CAS PubMed
- Z. Che, X. Lu, B. Cai, X. Xu, J. Bao and Y. Liu, Nano Res., 2022, 15, 1269–1275 CrossRef CAS
- S. Ajmal, H. T. D. Bui, V. Q. Bui, T. Yang, X. Shao, A. Kumar, S.-G. Kim and H. Lee, Chem. Eng. J., 2022, 429, 132282 CrossRef CAS
- C. Cai, K. Liu, Y. Zhu, P. Li, Q. Wang, B. Liu, S. Chen, H. Li, L. Zhu and H. Li, Angew. Chem., 2022, 134, e202113664 CrossRef
- Y. Liu, X. Li, Q. Zhang, W. Li, Y. Xie, H. Liu, L. Shang, Z. Liu, Z. Chen and L. Gu, Angew. Chem., Int. Ed., 2020, 59, 1718–1726 CrossRef CAS PubMed
- P. Li, W. Li, S. Zhao, Y. Huang, S. Tian and X. Huang, Chem. Eng. J., 2022, 429, 132557 CrossRef CAS
- Y. Li, F. Chu, Y. Bu, Y. Kong, Y. Tao, X. Zhou, H. Yu, J. Yu, L. Tang and Y. Qin, Chem. Commun., 2019, 55, 7828–7831 RSC
- X. Chen, J. Wan, J. Wang, Q. Zhang, L. Gu, L. Zheng, N. Wang and R. Yu, Adv. Mater., 2021, 33, 2104764 CrossRef CAS PubMed
- D. Sui, R. Luo, S. Xie, H. Zhang, T. Ma, H. Sun, T.-T. Jia, J. Sun and X. Li, Chem. Eng. J., 2024, 480, 148007 CrossRef CAS
- L. Zhang, H. Jang, Y. Wang, Z. Li, W. Zhang, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Adv. Sci., 2021, 8, 2004516 CrossRef CAS PubMed
- Q. He, Y. Zhou, H. Shou, X. Wang, P. Zhang, W. Xu, S. Qiao, C. Wu, H. Liu and D. Liu, Adv. Mater., 2022, 34, 2110604 CrossRef CAS PubMed
- V. Ramalingam, P. Varadhan, H. Fu, H. Kim, D. Zhang, S. Chen, L. Song, D. Ma, Y. Wang and H. N. Alshareef, Adv. Mater., 2019, 31, 1903841 CrossRef CAS PubMed
- B. Liu, R. Feng, M. Busch, S. Wang, H. Wu, P. Liu, J. Gu, A. Bahadoran, D. Matsumura and T. Tsuji, ACS Nano, 2022, 16, 14121–14133 CrossRef CAS PubMed
- S. Yin, J. Yang, Y. Han, G. Li, L. Wan, Y. Chen, C. Chen, X. Qu, Y. Jiang and S. Sun, Angew. Chem., 2020, 132, 22160–22163 CrossRef
- F. Li, Q. Wu, W. Yuan and Z. Chen, Dalton Trans., 2024, 53, 12022–12033 RSC
- Q. Wu, H. Li, Y. Zhou, S. Lv, T. Chen, S. Liu, W. Li and Z. Chen, Inorg. Chem., 2022, 61, 11011–11021 CrossRef CAS PubMed
- Y. Liu, J. Wu, Y. Zhang, X. Jin, J. Li, X. Xi, Y. Deng, S. Jiao, Z. Lei, X. Li and R. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 14240–14249 CAS
- T. Luo, J. Huang, Y. Hu, C. Yuan, J. Chen, L. Cao, K. Kajiyoshi, Y. Liu, Y. Zhao, Z. Li and Y. Feng, Adv. Funct. Mater., 2023, 33, 2213058 CrossRef CAS
- C. Hu, E. Song, M. Wang, W. Chen, F. Huang, Z. Feng, J. Liu and J. Wang, Adv. Sci., 2021, 8, 2001881 CrossRef CAS PubMed
- L. S. R. Kumara, O. Sakata, S. Kohara, A. Yang, C. Song, K. Kusada, H. Kobayashi and H. Kitagawa, Phys. Chem. Chem. Phys., 2016, 18, 30622–30629 RSC
- J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2010, 12, 5502 RSC
- B. Jiang, J. Zhu, Z. Xia, J. Lyu, X. Li, L. Zheng, C. Chen, S. Chaemchuen, T. Bu and F. Verpoort, Adv. Mater., 2024, 36, 2310699 CrossRef CAS PubMed
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed
- J. Shan, C. Ye, Y. Jiang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2022, 8, eabo0762 CrossRef CAS PubMed
- L. Zhang, T. Yang, W. Zang, Z. Kou, Y. Ma, M. Waqar, X. Liu, L. Zheng, S. J. Pennycook and Z. Liu, Adv. Sci., 2021, 8, 2101344 CrossRef CAS PubMed
- Y. Pan, C. Zhang, Z. Liu, C. Chen and Y. Li, Matter, 2020, 2, 78–110 CrossRef
- M. A. Hunter, J. M. T. A. Fischer, Q. Yuan, M. Hankel and D. J. Searles, ACS Catal., 2019, 9, 7660–7667 CrossRef CAS
- W. Li, J. Yang and D. Wang, Angew. Chem., 2022, 134, e202213318 CrossRef
- G. Li, C. Fu, W. Shi, L. Jiao, J. Wu, Q. Yang, R. Saha, M. E. Kamminga, A. K. Srivastava and E. Liu, Angew. Chem., 2019, 131, 13241–13246 CrossRef
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef PubMed
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 2373 CrossRef PubMed
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 1–7 Search PubMed
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 29, 1808367 CrossRef
- Y. Tang, Q. Liu, L. Dong, H. Bin Wu and X.-Y. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef CAS
- Z. Gao, J. Liu, X. Chen, X. Zheng, J. Mao, H. Liu, T. Ma, L. Li, W. Wang and X. Du, Adv. Mater., 2019, 31, 1804769 CrossRef PubMed
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma and Z. Feng, Nat. Commun., 2019, 10, 1711 CrossRef PubMed
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed
- J. Zhang, X. Wu, W.-C. Cheong, W. Chen, R. Lin, J. Li, L. Zheng, W. Yan, L. Gu and C. Chen, Nat. Commun., 2018, 9, 1002 CrossRef PubMed
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen and Q. Peng, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed
- T. He, Y. Song, Y. Chen, X. Song, B. Lu, Q. Liu, H. Liu, Y. Zhang, X. Ouyang and S. Chen, Chem. Eng. J., 2022, 442, 136337 CrossRef CAS
- H. Liu, G. Tan, M. Li, Z. Zhang, M. Getaye Sendeku, Y. Li, Y. Kuang and X. Sun, Chem. Eng. J., 2023, 458, 141414 CrossRef CAS
- D. Wang, Q. Li, C. Han, Z. Xing and X. Yang, Appl. Catal., B, 2019, 249, 91–97 CrossRef CAS
- S. Anantharaj, P. N. Reddy and S. Kundu, Inorg. Chem., 2017, 56, 1742–1756 CrossRef CAS PubMed
- Y. Pan, Y. Liu, J. Zhao, K. Yang, J. Liang, D. Liu, W. Hu, D. Liu, Y. Liu and C. Liu, J. Mater. Chem. A, 2015, 3, 1656–1665 RSC
- Z. Huang, Z. Chen, Z. Chen, C. Lv, H. Meng and C. Zhang, ACS Nano, 2014, 8, 8121–8129 CrossRef CAS PubMed
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed
- P. C. K. Vesborg, B. Seger and I. B. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed
- W. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed
- Y. Zhai, Z. Zhu and S. Dong, ChemCatChem, 2015, 7, 2806–2815 CrossRef CAS
- J.-M. Ge, B. Zhang, L.-B. Lv, H.-H. Wang, T.-N. Ye, X. Wei, J. Su, K.-X. Wang, X.-H. Li and J.-S. Chen, Nano Energy, 2015, 15, 567–575 CrossRef CAS
- L. Feng, Y. Li, C. Fu, D. Li, J. Huang, H. Yin, L. Cao and D. He, New J. Chem., 2023, 47, 17657–17665 RSC
- J. Guo, W. Shang, J. Hu, C. Xin, X. Cheng, J. Wei, C. Zhu, W. Liu and Y. Shi, ACS Appl. Mater. Interfaces, 2022, 14, 29822–29831 CrossRef CAS PubMed
- H. Yang, P. Wu, J. Pei, B. Peng and Q. Liu, Chem. Commun., 2024, 60, 718–721 RSC
- H. Zhang, L. Yu, T. Chen, W. Zhou and X. W. (David) Lou, Adv. Funct. Mater., 2018, 28, 1807086 CrossRef
- H. Bin Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai and R. Chen, Nat Energy, 2018, 3, 140–147 CrossRef
- J. W. Park, G. Park, M. Kim, M. Han, J. Jang, Y. Yamauchi, B. Yuliarto, P. Krüger, J. Kim, N. Park and H. Lim, Chem. Eng. J., 2023, 468, 143733 CrossRef CAS
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed
- H. Gu, X. Li, J. Zhang and W. Chen, Small, 2022, 18, 2105883 CrossRef CAS PubMed
- J. Liu, G. Zhang, K. Ye, K. Xu, Y. Sheng, C. Yu, H. Zhang, Q. Li, Z. Liang and K. Jiang, Chem. Commun., 2023, 59, 611–614 RSC
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed
- X. Zhang, W. X. Liu, Y. W. Zhou, Z. Da Meng, L. Luo and S. Q. Liu, J. Electroanal. Chem., 2021, 894, 115359 CrossRef CAS
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan and F. Abild-Pedersen, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis and J. H. Warner, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed
- Y. Pan, K. Sun, Y. Lin, X. Cao, Y. Cheng, S. Liu, L. Zeng, W.-C. Cheong, D. Zhao and K. Wu, Nano Energy, 2019, 56, 411–419 CrossRef CAS
- H. Feng, L. Tang, G. Zeng, J. Yu, Y. Deng, Y. Zhou, J. Wang, C. Feng, T. Luo and B. Shao, Nano Energy, 2020, 67, 104174 CrossRef CAS
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS PubMed
- W. Zang, T. Sun, T. Yang, S. Xi, M. Waqar, Z. Kou, Z. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33, 2003846 CrossRef CAS PubMed
- A. Montenegro, C. Dutta, M. Mammetkuliev, H. Shi, B. Hou, D. Bhattacharyya, B. Zhao, S. B. Cronin and A. V. Benderskii, Nature, 2021, 594, 62–65 CrossRef CAS PubMed
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. Koper, Nat Energy, 2017, 2, 1–7 Search PubMed
- X. Yang, J. Nash, N. Oliveira, Y. Yan and B. Xu, Angew. Chem., 2019, 131, 17882–17887 CrossRef
- M. Wang, K. Sun, W. Mi, C. Feng, Z. Guan, Y. Liu and Y. Pan, ACS Catal., 2022, 10771–10780 CrossRef CAS
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil and S. Mukerjee, Nat. Commun., 2017, 8, 957 CrossRef PubMed
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed
- T. Sun, N. Shan, L. Xu, J. Wang, J. Chen, A. A. Zakhidov and R. H. Baughman, Chem. Mater., 2018, 30, 1617–1624 CrossRef CAS
- H. Fei, J. Dong, C. Wan, Z. Zhao, X. Xu, Z. Lin, Y. Wang, H. Liu, K. Zang, J. Luo, S. Zhao, W. Hu, W. Yan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2018, 30, 1802146 CrossRef PubMed
- X. Gao, Y. Zhou, S. Liu, Z. Cheng, Y. Tan and Z. Shen, Appl. Surf. Sci., 2020, 502, 144155 CrossRef CAS
- X. Liu, Y. Deng, L. Zheng, M. R. Kesama, C. Tang and Y. Zhu, ACS Catal., 2022, 12, 5517–5526 CrossRef CAS
- K. Qi, X. Cui, L. Gu, S. Yu, X. Fan, M. Luo, S. Xu, N. Li, L. Zheng, Q. Zhang, J. Ma, Y. Gong, F. Lv, K. Wang, H. Huang, W. Zhang, S. Guo, W. Zheng and P. Liu, Nat. Commun., 2019, 5231 CrossRef PubMed
- J. Gong, Z. Zhang, S. Xi, W. Wang, J. Lu and P. Chen, Chem. Eng. J., 2023, 451, 138951 CrossRef CAS
- H. Duan, W. Liu, P. Guo, F. Tang, W. Yan and T. Yao, Radiat. Phys. Chem., 2020, 175, 108151 CrossRef CAS
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. - Eur. J., 2017, 23, 10947–10961 CrossRef CAS PubMed
- Y. Ouyang, C. Ling, Q. Chen, Z. Wang, L. Shi and J. Wang, Chem. Mater., 2016, 28, 4390–4396 CrossRef CAS
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131–2136 CrossRef CAS PubMed
- X. Gao, Y. Zhou, Y. Tan, B. Yang, Z. Cheng and Z. Shen, Int. J. Hydrogen Energy, 2019, 44, 14861–14868 CrossRef CAS
- Y. Zhang, Y. Li, D. Ni, Z. Chen, X. Wang, Y. Bu and J. Ao, Adv. Funct. Mater., 2019, 29, 1902101 CrossRef
- F. Yang, Y. Zhao, Y. Du, Y. Chen, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2018, 8, 1703489 CrossRef
- Q. Qin, H. Jang, L. Chen, G. Nam, X. Liu and J. Cho, Adv. Energy Mater., 2018, 8, 1801478 CrossRef
- H. Xu, T. Liu, S. Bai, L. Li, Y. Zhu, J. Wang, S. Yang, Y. Li, Q. Shao and X. Huang, Nano Lett., 2020, 20, 5482–5489 CrossRef CAS PubMed
- X. Meng, C. Ma, L. Jiang, R. Si, X. Meng, Y. Tu, L. Yu, X. Bao and D. Deng, Angew. Chem., 2020, 132, 10588–10593 CrossRef
- M. Kolter, K. Böck, K. Karaghiosoff and K. Koszinowski, Angew. Chem., Int. Ed., 2017, 56, 13244–13248 CrossRef CAS PubMed
- H. Zheng, Y. Zhu and Y. Shi, Angew. Chem., 2014, 126, 11462–11466 CrossRef
- H. Yu, Y. Xue, B. Huang, L. Hui, C. Zhang, Y. Fang, Y. Liu, Y. Zhao, Y. Li, H. Liu and Y. Li, iScience, 2019, 11, 31–41 CrossRef CAS PubMed
- Z. Luo, Y. Ouyang, H. Zhang, M. Xiao, J. Ge, Z. Jiang, J. Wang, D. Tang, X. Cao and C. Liu, Nat. Commun., 2018, 9, 2120 CrossRef PubMed
- Z. Li, X. Yan, D. He, W. Hu, S. Younan, Z. Ke, M. Patrick, X. Xiao, J. Huang, H. Wu, X. Pan and J. Gu, ACS Catal., 2022, 12, 7687–7695 CrossRef CAS
- S. Gong, C. Wang, P. Jiang, L. Hu, H. Lei and Q. Chen, J. Mater. Chem. A, 2018, 6, 13254–13262 RSC
- B. Wang, J. Zou, X. Shen, Y. Yang, G. Hu, W. Li, Z. Peng, D. Banham, A. Dong and D. Zhao, Nano Energy, 2019, 63, 103851 CrossRef CAS
- S. Wang, Z. Hu, Q. Wei, P. Cui, H. Zhang, W. Tang, Y. Sun, H. Duan, Z. Dai and Q. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 20669–20681 CrossRef CAS PubMed
- X. Peng, H.-X. Liu, Y. Zhang, Z.-Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang and C. Au, Chem. Sci., 2021, 12, 7125–7137 RSC
- M. Dhiman and V. Polshettiwar, ChemCatChem, 2018, 10, 881–906 CrossRef CAS
- X. Duan, T. Li, X. Jiang, X. Liu, L. Xin, H. Yang, Y. Kuang and X. Sun, Mater. Rep.: Energy, 2022, 100146 CAS
- A. M. Harzandi, S. Shadman, M. Ha, C. W. Myung, D. Y. Kim, H. J. Park, S. Sultan, W. S. Noh, W. Lee, P. Thangavel, W. J. Byun, S. Hun Lee, J. N. Tiwari, T. J. Shin, J. H. Park, Z. Lee, J. S. Lee and K. S. Kim, Appl. Catal., B, 2020, 270, 118896 CrossRef CAS
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS
- A. Kumar, V. Q. Bui, J. Lee, L. Wang, A. R. Jadhav, X. Liu, X. Shao, Y. Liu, J. Yu, Y. Hwang, H. T. D. Bui, S. Ajmal, M. G. Kim, S. G. Kim, G. S. Park, Y. Kawazoe and H. Lee, Nat. Commun., 2021, 12, 6766 CrossRef CAS PubMed
- M. Li, H. Zhu, Q. Yuan, T. Li, M. Wang, P. Zhang, Y. Zhao, D. Qin, W. Guo, B. Liu, X. Yang, Y. Liu and Y. Pan, Adv. Funct. Mater., 2022, 33, 2210867 CrossRef
- W. Fang, Y. Wu, S. Xin, Y. Hu, J. Dang, M. Li, B. Chen, H. Zhao and Z. Li, Chem. Eng. J., 2023, 468, 143605 CrossRef CAS
- L. Huo, C. Jin, J. Tang, X. Xu, K. Jiang, L. Shang, Y. Li, J. Zhang, L. Zhu, J. Chu and Z. Hu, ACS Appl. Energy Mater., 2022, 5, 15136–15145 CrossRef CAS
- M. Luo, J. Cai, J. Zou, Z. Jiang, G. Wang and X. Kang, J. Mater. Chem. A, 2021, 9, 14941–14947 RSC
- Q.-Q. Yan, D.-X. Wu, S.-Q. Chu, Z.-Q. Chen, Y. Lin, M.-X. Chen, J. Zhang, X.-J. Wu and H.-W. Liang, Nat. Commun., 2019, 10, 4977 CrossRef CAS PubMed
- P. Zhou, N. Li, Y. Chao, W. Zhang, F. Lv, K. Wang, W. Yang, P. Gao and S. Guo, Angew. Chem., Int. Ed., 2019, 58, 14184–14188 CrossRef CAS PubMed
- X. Li, P. Shen, Y. Luo, Y. Li, Y. Guo, H. Zhang and K. Chu, Angew. Chem., 2022, 134, e202205923 CrossRef
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS PubMed
- Z. W. Chen, J. Li, P. Ou, J. E. Huang, Z. Wen, L. Chen, X. Yao, G. Cai, C. C. Yang and C. V. Singh, Nat. Commun., 2024, 15, 359 CrossRef CAS PubMed
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang and C. Zhao, Energy Environ. Sci., 2018, 11, 3375–3379 RSC
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi and Z. Jiang, Angew. Chem., 2019, 131, 2648–2652 CrossRef
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed
- G. Sun, Z.-J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li and S. C. Purdy, Nat. Commun., 2018, 9, 4454 CrossRef PubMed
- A. E. Baber, H. L. Tierney, T. J. Lawton and E. C. H. Sykes, ChemCatChem, 2011, 3, 607–614 CrossRef CAS
- C. H. Chen, D. Wu, Z. Li, R. Zhang, C. G. Kuai, X. R. Zhao, C. K. Dong, S. Z. Qiao, H. Liu and X. W. Du, Adv. Energy Mater., 2019, 9, 1803913 CrossRef
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed
- J. Park, S. Lee, H. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed
- X. Yin, H. Wang, S. Tang, X. Lu, M. Shu, R. Si and T. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed
- F. Li, G. F. Han, Y. Bu, S. Chen, I. Ahmad, H. Y. Jeong, Z. Fu, Y. Lu and J. B. Baek, Nano Energy, 2022, 93, 106819 CrossRef CAS
- W. Peng, J. Han, Y.-R. Lu, M. Luo, T.-S. Chan, M. Peng and Y. Tan, ACS Nano, 2022, 16, 4116–4125 CrossRef CAS PubMed
- S. Yuan, Z. Pu, H. Zhou, J. Yu, I. S. Amiinu, J. Zhu, Q. Liang, J. Yang, D. He and Z. Hu, Nano Energy, 2019, 59, 472–480 CrossRef CAS
- S. Jiao, M. Kong, Z. Hu, S. Zhou, X. Xu and L. Liu, Small, 2022, 18, 2105129 CrossRef CAS PubMed
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, Z. Wang, Z. Hu, Y. Zhao and H.-W. Li, Nano Energy, 2021, 88, 106221 CrossRef CAS
- Y. Zhu, M. Klingenhof, C. Gao, T. Koketsu, G. Weiser, Y. Pi, S. Liu, L. Sui, J. Hou and J. Li, Nat. Commun., 2024, 15, 1447 CrossRef CAS PubMed
- C. Yue, C. Feng, G. Sun, N. Liu, H. Hao, W. Bao, X. Zhang, F. Sun, C. Zhang, J. Bi, Y. Zhou, H. C. Chen, Y. Pan, D. Sun and Y. Lu, Energy Environ. Sci., 2024, 17, 5227–5240 RSC
- Y. Zhu, X. Zhu, L. Bu, Q. Shao, Y. Li, Z. Hu, C. Chen, C. Pao, S. Yang and X. Huang, Adv. Funct. Mater., 2020, 30, 2004310 CrossRef CAS
- K. Wang, J. Cao, X. Yang, X. Sang, S. Yao, R. Xiang, B. Yang, Z. Li, T. O’Carroll and Q. Zhang, Adv. Funct. Mater., 2023, 33, 2212321 CrossRef CAS
- L. Wang, M. Ma, C. Zhang, H. Chang, Y. Zhang, L. Li, H. Chen and S. Peng, Angew. Chem., 2024, 136, e202317220 CrossRef
- R. Liu, Z. Gong, M. Yan, G. Ye and H. Fei, Nano Res, 2023, 16, 256–263 CrossRef CAS
- C. Zhou, J. Shi, Z. Dong, L. Zeng, Y. Chen, Y. Han, L. Li, W. Zhang, Q. Zhang and L. Gu, Nat. Commun., 2024, 15, 6741 CrossRef CAS PubMed
- Z. Chen, X. Li, J. Zhao, S. Zhang, J. Wang, H. Zhang, J. Zhang, Q. Dong, W. Zhang and W. Hu, Angew. Chem., Int. Ed., 2023, 62, e202308686 CrossRef CAS PubMed
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang and G.-Y. Shi, Nat. Commun., 2021, 12, 3021 CrossRef PubMed
- T. Zhang, J. Jin, J. Chen, Y. Fang, X. Han, J. Chen, Y. Li, Y. Wang, J. Liu and L. Wang, Nat. Commun., 2022, 13, 6875 Search PubMed
- C. Tsounis, B. Subhash, P. V. Kumar, N. M. Bedford, Y. Zhao, J. Shenoy, Z. Ma, D. Zhang, C. Y. Toe and S. Cheong, Adv. Funct. Mater., 2022, 32, 2203067 CrossRef CAS
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS
- D. Wang, H. Li, N. Du and W. Hou, Adv. Funct. Mater., 2021, 31, 2009770 CrossRef CAS
- Y. Wang, L. Chen, Z. Mao, L. Peng, R. Xiang, X. Tang, J. Deng, Z. Wei and Q. Liao, Sci. Bull., 2019, 64, 1095–1102 CrossRef CAS PubMed
- B. Cao, M. Hu, Y. Cheng, P. Jing, B. Liu, B. Zhou, X. Wang, R. Gao, X. Sun and Y. Du, NPG Asia Mater., 2021, 13, 1 CrossRef CAS
- Q. Wang, Z. L. Zhao, S. Dong, D. He, M. J. Lawrence, S. Han, C. Cai, S. Xiang, P. Rodriguez, B. Xiang, Z. Wang, Y. Liang and M. Gu, Nano Energy, 2018, 53, 458–467 CrossRef CAS
- L. Wang, X. Liu, L. Cao, W. Zhang, T. Chen, Y. Lin, H. Wang, Y. Wang and T. Yao, J. Phys. Chem. Lett., 2020, 11, 6691–6696 CrossRef CAS PubMed
- J. Yu, J. Li, C.-Y. Xu, Q. Li, Q. Liu, J. Liu, R. Chen, J. Zhu and J. Wang, Nano Energy, 2022, 98, 107266 CrossRef CAS
- B. Liu, Y. Cheng, B. Cao, M. Hu, P. Jing, R. Gao, Y. Du, J. Zhang and J. Liu, Appl. Catal., B, 2021, 298, 120630 CrossRef CAS
- Y. Li, Y. Peng, W. Dong, X. Jiang, L. Lu, D. Yang, L.-C. Hsu, W. Li, B. Su and A. Lei, J. Am. Chem. Soc., 2024, 146, 14194–14202 CrossRef CAS PubMed
- T. Li, T. Lu, X. Li, L. Xu, Y. Zhang, Z. Tian, J. Yang, H. Pang, Y. Tang and J. Xue, ACS Nano, 2021, 15, 20032–20041 CrossRef CAS PubMed
- Y. Gu, B. Xi, R. Wei, Q. Fu, Y. Qain and S. Xiong, Nano Lett, 2020, 20, 8375–8383 Search PubMed
- Z. Zhang, X. Zhao, S. Xi, L. Zhang, Z. Chen, Z. Zeng, M. Huang, H. Yang, B. Liu and S. J. Pennycook, Adv. Energy Mater., 2020, 10, 2002896 Search PubMed
- W. Luo, Y. Wang, L. Luo, S. Gong, M. Wei, Y. Li, X. Gan, Y. Zhao, Z. Zhu and Z. Li, ACS Catal., 2022, 12, 1167–1179 Search PubMed
- S. Sultan, M. H. Diorizky, M. Ha, J. N. Tiwari, H. Choi, N. K. Dang, P. Thangavel, J. H. Lee, H. Y. Jeong and H. S. Shin, J. Mater. Chem. A, 2021, 9, 10326–10334 RSC
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan and Y. Qin, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed
- M. R. Kandel, U. N. Pan, D. R. Paudel, P. P. Dhakal, N. H. Kim and J. H. Lee, Composites, Part B, 2022, 239, 109992 CrossRef CAS
- L. Cao, X. Wang, C. Yang, J. Lu, X. Shi, H. Zhu and H.-P. Liang, ACS Sustainable Chem. Eng., 2020, 9, 189–196 Search PubMed
- C. Chen, D. Wu, Z. Li, R. Zhang, C. Kuai, X. Zhao, C. Dong, S. Qiao, H. Liu and X. Du, Adv. Energy Mater., 2019, 9, 1803913 CrossRef
- M. Li, H. Zhu, Q. Yuan, T. Li, M. Wang, P. Zhang, Y. Zhao, D. Qin, W. Guo and B. Liu, Adv. Funct. Mater., 2023, 33, 2210867 Search PubMed
- Y. Chen, J. Li, N. Wang, Y. Zhou, J. Zheng and W. Chu, Chem. Eng. J., 2022, 448, 137611 Search PubMed
- M. A. Z. G. Sial, M. Mateen, R. Naz, M. Abbas, N. Abbas, S. H. Talib, M. R. S. A. Janjua and M. Qamar, Int. J. Hydrogen Energy, 2024, 51, 540–549 CrossRef CAS
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y. R. Lu, T. S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed
- K. L. Zhou, C. Wang, Z. Wang, C. B. Han, Q. Zhang, X. Ke, J. Liu and H. Wang, Energy Environ. Sci., 2020, 13, 3082–3092 RSC
- M. Yi, N. Li, B. Lu, L. Li, Z. Zhu and J. Zhang, Energy Storage Mater, 2021, 42, 418–429 CrossRef
- J. Zhu and S. Mu, Adv. Funct. Mater., 2020, 30, 2001097 CrossRef CAS
- L. Zhao, J. Zhu, Y. Zheng, M. Xiao, R. Gao, Z. Zhang, G. Wen, H. Dou, Y. Deng and A. Yu, Adv. Energy Mater., 2022, 12, 2102665 CrossRef CAS
- J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 15298–15312 CrossRef CAS PubMed
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat Energy, 2016, 1, 1–9 Search PubMed
- S. Fang, X. Zhu, X. Liu, J. Gu, W. Liu, D. Wang, W. Zhang, Y. Lin, J. Lu, S. Wei, Y. Li and T. Yao, Nat. Commun., 2020, 11, 1029 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef
- P. Li, G. Zhao, P. Cui, N. Cheng, M. Lao, X. Xu, S. X. Dou and W. Sun, Nano Energy, 2021, 83, 105850 Search PubMed
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 Search PubMed
- X. Wang, Q. He, L. Song, M. Jaroniec, Y. Zheng and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 13635–13640 RSC
- A. Pei, G. Li, L. Zhu, Z. Huang, J. Ye, Y. C. Chang, S. M. Osman, C. W. Pao, Q. Gao, B. H. Chen and R. Luque, Adv. Funct. Mater., 2022, 32, 2208587 CrossRef CAS
- M. Qin, J. Chen, X. Zheng, M. Qi, R. Yang, S. Mao and Y. Wang, Appl. Catal., B, 2022, 316, 121602 CrossRef CAS
- Y. Pan, Z. Liu, M. Wang, C. Zhang, C. Chen and Y. Lin, Sci. China Mater., 2020, 63, 921–948 CrossRef CAS
- Y. N. Regmi, G. R. Waetzig, K. D. Duffee, S. M. Schmuecker, J. M. Thode and B. M. Leonard, J. Mater. Chem. A, 2015, 3, 10085–10091 RSC
- N. H. Ahmad Junaidi, W. Y. Wong, K. S. Loh, S. Rahman and W. R. W. Daud, Int. J. Energy Res., 2021, 45, 15760–15782 CrossRef CAS
- J. Yang, B. Chen, X. Liu, W. Liu, Z. Li, J. Dong, W. Chen, W. Yan, T. Yao, X. Duan, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 9495–9500 CrossRef CAS PubMed
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 1–6 Search PubMed
- M. K. Oudenhuijzen, J. H. Bitter and D. C. Koningsberger, J Phys Chem B, 2001, 105, 4616–4622 CrossRef CAS
- F. H. Saadi, A. I. Carim, W. S. Drisdell, S. Gul, J. H. Baricuatro, J. Yano, M. P. Soriaga and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 12927–12930 CrossRef CAS PubMed
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby and P. Zelenay, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed
- H. Li, H. Zhu, Z. Zhuang, S. Lu, F. Duan and M. Du, Sustainable Energy Fuels, 2020, 4, 996–1011 RSC
- J. Hwang, S. H. Noh and B. Han, Appl. Surf. Sci., 2019, 471, 545–552 CrossRef CAS
- S. Tang, X. Zhou, T. Liu, S. Zhang, T. Yang, Y. Luo, E. Sharman and J. Jiang, J. Mater. Chem. A, 2019, 7, 26261–26265 RSC
- Y.-Q. Su, Y. Wang, J.-X. Liu, I. A. W. Filot, K. Alexopoulos, L. Zhang, V. Muravev, B. Zijlstra, D. G. Vlachos and E. J. M. Hensen, ACS Catal., 2019, 9, 3289–3297 CrossRef CAS
- X. Zhang, J. Wang, J. Wang, J. Wang, C. Wang and C. Lu, J. Phys. Chem. Lett., 2021, 12, 11135–11142 CrossRef CAS PubMed
- S. Ghosh, S. R. Kadam, S. Kolatkar, A. Neyman, C. Singh, A. N. Enyashin, R. Bar-Ziv and M. Bar-Sadan, ACS Appl. Mater. Interfaces, 2021, 14, 581–589 CrossRef PubMed
- R. Sundararaman, W. A. Goddard and T. A. Arias, J. Chem. Phys., 2017, 146, 114104 CrossRef PubMed
- D. Kim, J. Shi and Y. Liu, J. Am. Chem. Soc., 2018, 140, 9127–9131 CrossRef CAS PubMed
- K. B. Oldham, J. Electroanal.
Chem., 2008, 613, 131–138 CrossRef CAS
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef
- M. Gajdoš, A. Eichler and J. Hafner, J. Phys.:Condens. Matter, 2004, 16, 1141 CrossRef
| This journal is © The Royal Society of Chemistry 2025 |