
Reactive carbon capture using saline water: evaluation of prospective sources, processes, and products
Anya
Dickinson-Cove
a,
Erika
La Plante
b,
Yiming
Liu
 c,
Dante
Simonetti
a,
Eric M. V.
Hoek
ae,
Gaurav
Sant
ad and
David
Jassby
*ad
c,
Dante
Simonetti
a,
Eric M. V.
Hoek
ae,
Gaurav
Sant
ad and
David
Jassby
*ad
aDepartment of Civil & Environmental Engineering, University of California, Los Angeles, California 90095, USA. E-mail: jassby@ucla.edu
bDepartment of Materials Science and Engineering, University of California, Davis, California 95616, USA
cDepartment of Civil and Environmental Engineering, Rice University, Houston, Texas 77251, USA
dInstitute for Carbon Management, University of California, Los Angeles, California 90095, USA
eEnergy Storage and Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, California 94272, USA
First published on 22nd November 2024
Abstract
Reactive carbon capture (RCC) processes involve the capture of carbon dioxide (CO2) and conversion to a value-added product using a single sorbent/reaction medium. Not only can RCC processes generate valuable byproducts that can reduce the cost of carbon capture, but RCC tends to have lower energy demand than processes involving the transfer of CO2 between the mediums used for capture and subsequent reactions. Saline water has been proposed as a potential medium for RCC due to it's relative abundance and low cost. Additionally, the composition and chemistry of many saline water sources: (1) elevates the CO2 content (as compared to atmospheric concentrations), (2) provides various cations that can form valuable products with CO2, and (3) enhances the kinetics of chemical reactions used to convert CO2 to stable byproducts. In addition to established industrial processes for converting CO2 into inert or valuable byproducts, we found 20 new processes and technologies that have been developed specifically to capture and convert CO2 using saline water. Both preexisting and emerging processes can be broadly classified as electrochemical or chemical titration processes. When assessing the potential viability of applying any of these processes for large scale carbon capture, several factors must be considered, such as the net carbon footprint of the process, the market size, location of customers and value of the end product, the energy demand and chemical costs of the process, and any other environmental impacts. The feasability of many emerging saline-based RCC processes is difficult to determine, as many technologies were tested using synthetic saline waters and/or concentrated CO2 sources. Notwithstanding the early stage of development of many saline-based RCC technologies, the major limitation to implementation of this approach to carbon capture is the mismatch in the scale of the markets for products of saline-based RCC and the scale of carbon capture needed to meet climate goals. However, because the products of many of the processes reviewed here are stable and non-hazardous, these technologies may also be used for carbon sequestration efforts where the products are managed as waste, in which case the carbon capture potential of these technologies can surpass the market-imposed limitations on RCC. Thus, the potential benefits of saline water-based RCC identified in this review encourage further study and development of these technologies.
 Anya Dickinson-Cove | Anya Dickinson-Cove is a PhD student in the Department of Civil and Environmental Engineering at the University of California, Los Angeles. She received her MS in Civil and Environmental Engineering from UCLA (2023) and a BSc in Environmental Engineering from California State Polytechnic University (2020). Anya's research focuses on waste recovery and valorization systems. |
 Erika La Plante | Dr. Erika La Plante is an Assistant Professor of Materials Science and Engineering at the University of California, Davis. Previously, she held positions at the University of Texas at Arlington and the University of California, Los Angeles. She obtained her PhD in Earth and Environmental Sciences with a focus in Geochemistry at the University of Illinois at Chicago and her BS in Geology from the University of the Philippines. Erika applies her expertise in the kinetics of low-temperature aqueous processes at mineral-fluid interfaces to address the many research questions in the fields of climate, sustainability, built environment, and energy. |
 Eric M. V. Hoek | Dr. Eric Hoek is a professor in the Department of Civil & Environmental Engineering at the University of California, Los Angeles and a faculty scientist in the Energy Storage & Distributed Resources Division at Lawrence Berkeley National Laboratory. Prof. Hoek's research explores the union of membrane technologies, nanomaterials and electrochemistry. Prof. Hoek, his advisees and collaborators have published over 160 peer-reviewed journal articles, 30 U.S. patents, and two books. He is also co-Editor-in-Chief of The Encyclopedia of Membrane Science & Technology, former Associate Editor of the Elsevier journal Desalination, and founding/former Editor-in-Chief of the Nature journal npj Clean Water. |
 Gaurav Sant | Dr. Gaurav Sant is a Professor and the Pritzker Endowed Chair in Sustainability at the Samueli School of Engineering at the University of California, Los Angeles. Gaurav has published over 200 publications and has an h-index of 55. He is also the Director of UCLA's Institute for Carbon Management (ICM), the Co-Founder of Equatic Inc., the Grand Prize Winner of the Temasek Foundation's 2021 Liveability Challenge, the Co-Founder of Concrete- AI Inc., the Co-Founder of Nextli Technologies Inc., and the Founder of CarbonBuilt Inc., a Grand Prize Winner of the 2021 NRG COSIA Carbon XPRIZE. |
 David Jassby | Dr. David Jassby is a professor in the Department of Civil and Environmental Engineering at the University of California, Los Angeles. He received his PhD in Civil and Environmental Engineering from Duke University (2011), an MS in Civil and Environmental Engineering from UC Davis (2005), and a BSc in Biology from Hebrew University (2002). David's research is primarily concerned with membrane separations, CO2 capture and sequestration, environmental chemistry and electrochemistry, resource recovery, and water treatment technologies. |
Introduction
Reducing atmospheric CO2 is a critical component of mitigating climate change, but reducing emissions alone will not be enough to meet climate goals set by the IPCC.1 In addition, historic carbon emissions will need to be removed from the atmosphere, and at a rapid pace. While there are existing biological and geological processes that can sequester atmospheric CO2, these processes are too slow to achieve the level of sequestration necessary. Thus, numerous processes have emerged, some of which enhance the naturally occurring carbon sequestration processes, and some of which are novel industrial processes. Among these processes are reactive carbon capture processes, which convert atmospheric CO2 to value-added products, making the processes more economically favourable. In reactive carbon capture, the reactions that convert the CO2 to the end product occur within the medium that captures the CO2. One medium that has been identified for reactive carbon capture is saline water, which often has an enhanced capacity to dissolve CO2 compared to freshwater, is widely available, and contains numerous chemical components that can react with dissolved CO2 to form valuable products. While a recent review by Mustafa et al.2 summarized technologies that can be used to capture carbon in reject brine, many of the technologies included in that review are not considered here as potential saline-based RCC processes, as they either (1) don’t capture carbon directly in the saline water stream but use a product from the saline water to subsequently capture carbon3–5 or (2) don’t generate a value-added product directly from the saline water.6,7 Here, we will review technologies that have been specifically developed to capture CO2 using saline water, as well as a number of technologies developed for other purposes that can also be applied to RCC.To begin, we will discuss the fundamental chemistry of carbon capture in saline water, and then review different saline water sources. Then, we will cover some of the fundamental chemistry of the conversion of dissolved CO2 to stable chemical products before discussing the variety of technologies that apply this chemistry. Additionally, we will discuss potential products of saline-based RCC, evaluate costs associated with energy and chemical demand, and consider the overall environmental impacts of these processes.
Saline water as a sorbent for CO2
Whereas the majority of conventional, non-biological carbon capture, utilization, and sequestration (CCUS) technologies involve dissolution of gaseous CO2 into a basic solvent, such as aqueous hydroxides or amines, or absorption with an amine-based solid sorbent,8 there are a growing number of technologies that capture CO2 in saline water.9 This includes saline-based reactive carbon capture processes (RCC), which use saline water as the medium to both capture and convert CO2 into a value-added product. As the adsorption/absorption of CO2 by any medium is often an equilibrium process, the CO2 capture capacity of both conventional and aqueous-based processes is in part determined by the concentration of CO2 in the gaseous phase in contact with the solvent and/or sorbent. As the concentration of CO2 in the gaseous feed stream targeted for capture decreases, the volume of the solvent or sorbent required to capture a given amount of CO2 increases. The concentration of CO2 in point source emissions such as flue gas ranges from 3–20%,10 but is only around 420 ppm (0.04%) in the atmosphere,11 which makes carbon capture and sequestration from ambient air using conventional technologies difficult due to the large volumes of air and sorbent that would be required. Still, there is a need for processes that can capture and sequester atmospheric CO2, as at least 40% of global CO2 emissions are from non-point or mobile sources, and because removing legacy CO2 emissions inherently requires treating atmospheric CO2.8,12 The abundance and low cost of saline water (in comparison to conventional solvents and sorbents) make it an attractive alternative medium for both CCU in general and RCC in specific.When gaseous CO2 dissolves into water, it undergoes a series of rapid acid/base reactions, which leads to the formation of different carbonate species (the distribution of which depends on solution pH and the presence of cations, such as calcium, that can form sparingly soluble species). Because of this, CO2 concentrations in seawater are at least 140 times larger compared to their atmospheric concentrations12 (Freshwater CO2 concentrations are much more variable, ranging from soft waters that contain very little CO2 to hard waters that can have more than double the CO2 concentration in seawater.13 However, the limited availability of freshwater and its value as drinking water makes using these sources for RCC less attractive than saline waters). The elevated concentration of CO2 in water creates an opportunity for more efficient capture and sequestration of CO2 from non-point sources. Thus, saline water-based CCU technologies, including RCC, have the potential to play a key role in achieving carbon sequestration equivalent to the 80% reduction in CO2 emissions called for by the IPCC.8 RCC technologies make up an important subset of CCU technologies as they directly convert captured carbon into valuable end products and have many potential applications for saline water-based CO2 capture.
Not only does saline water have the potential to capture large volumes of CO2 (atmospheric or otherwise) efficiently and inexpensively, but various constituents of saline water can be used to directly convert the dissolved CO2 into valuable chemical products. Potentially high-value materials in saline waters comprise salts of sodium (Na+), lithium (Li+), calcium (Ca2+), magnesium (Mg2+), barium (Ba2+), and strontium (Sr2+). Additionally, it has recently been demonstrated that some valuable organic products can be directly produced from carbonates and hydrogen present in saline water.14–16 Processes used to generate either inorganic or organic chemicals from carbonates in saline water can be classified as reactive carbon capture (RCC) processes, wherein the sorbent (saline water) serves both as the medium for carbon capture and conversion to a value-added product (Fig. 1). This type of carbon capture process is particularly attractive because it can eliminate the need for downstream CO2 handling, such as the burial of the concentrated gas in a geological formation, or transfer of captured carbon into another medium used for sequestration, thereby reducing energy demand, process complexity, and cost. An additional potential benefit of using saline water as the source of carbon sequestering cations is the reduction in energy/chemical demand compared to processes that use solid wastes/ores as the cation source for carbon capture.17
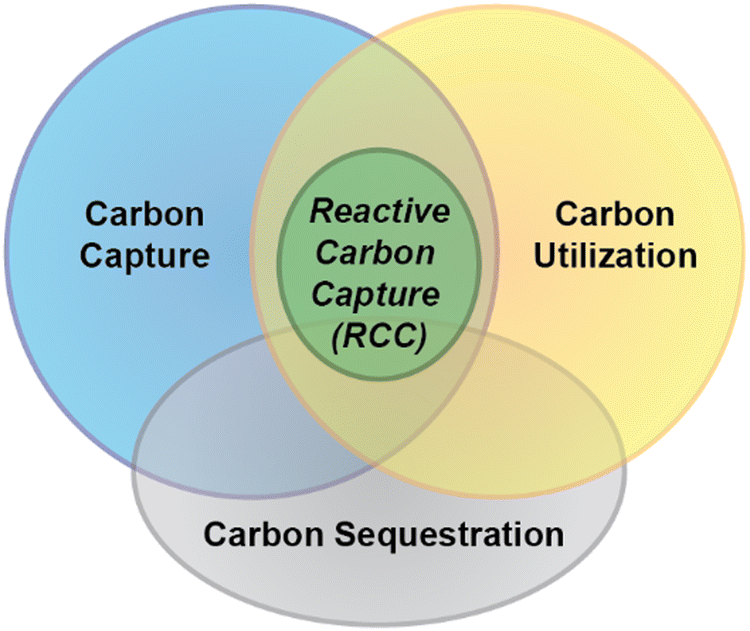 | ||
| Fig. 1 Reactive carbon capture processes involve the capture of gaseous CO2 and conversion to a value added product within the same medium (for instance, saline water). RCC is inherently a combination of carbon capture and utilization (sometimes referred to as “CCU”) but in some instances can also achieve sequestration when the end products are long term stable sinks for CO2. However, many RCC processes generate products that when used re-release the CO2. | ||
CO2 capture chemistry
To understand how saline water can be used to efficiently convert atmospheric CO2 into valuable products, the mechanisms for CO2 dissolution into water and precipitation out of water must be understood. When a gaseous mixture containing CO2 is in contact with liquid water, the gaseous CO2 will reach equilibrium with various carbonate species in the water.18 It should be noted that while the gas–liquid phase equilibrium of CO2 is governed by Henry's law at temperatures and concentrations relevant to most applications reviewed here, the equilibrium between the two phases is not well described by Henry's law for temperatures above 100 °C,19 or aqueous mol fractions (XCO2,(aq)) above 2% (∼44 g L−1).20 Water-based RCC technologies operating outside of these conditions would require more careful consideration of the CO2–water equilibrium.Several reactions govern the aqueous CO2 system, beginning with the rapid dissolution of CO2 into water:18
| CO2(g) ↔ CO2(aq), k = 1 × 1010 s−1, KH = 29.4 atm M−1 |
Note: all equilibrium constants presented in this section are for standard temperature (25 °C and 1 atm) unless otherwise noted. Given pure water at equilibrium with air at standard temperature and pressure and with a CO2 concentration of 420 ppmv, the concentration of CO2(aq) in water is approximately 1.42 × 10−5 M. This value is lower than the atmospheric concentration of CO2 (420 ppmv ≈ 1.71 × 10−5 M air), but because of the carbonate system, CO2(aq) rapidly transforms to the various carbonate species, which increases the total amount of CO2 that can be dissolved. Once dissolved, CO2 hydrates to form carbonic acid (H2CO3).21
| CO2(aq) + H2O ↔ H2CO3, k = 0.06 s−1 |
Carbonic acid then rapidly deprotonates to form bicarbonate and carbonate.18,21
| H2CO3 ↔ HCO3− + H+, k = 1.0 × 107 s−1, pKa = 6.352 |
| HCO3− ↔ CO3−2 + H+, k = 3.0 × 100 s−1, pKa = 10.329 |
The total sum of [CO2(aq)] + [H2CO3] + [HCO3−] + [CO32−] is known as the total dissolved inorganic carbon or “CT”. At a pH of approximately 5.85, the CT in pure water is equal to the concentration of CO2 in the atmosphere, while at pH >5.85, the CT in pure water is greater than to the concentration of CO2 in the atmosphere. For example, in pure water at neutral pH (7) in equilibrium with air, the formation of carbonic acid, bicarbonate, and carbonate increases the CT from 1.42 × 10−5 M to 8.59 × 10−5 M. As the pH of the water increases, the equilibrium of the above equations shifts further to the right, leading to an increase in CT.
Carbonate system in saline water
The dissolution of CO2 and formation of carbonate species occurs in all waters that are in contact with the atmosphere, such as seawater, surface water, and wastewater (Fig. 2). It should be noted that groundwater not in contact with the atmosphere but in contact with carbonate minerals can also contain carbonate species formed by mineral dissolution.22 The total dissolved inorganic carbon (“DIC”, analogous to Ct) in these waters is often higher than in pure water due to the alkalinity of saline waters, so the Ct can even further surpass the concentration of CO2 in air. The alkalinity shifts the carbonate system towards the formation of bicarbonate and carbonate, allowing for further dissolution of CO2 and effectively increasing the total inorganic carbon content in real waters compared to pure water. For example, in seawater, the CT is 2.3 × 10−3 M.23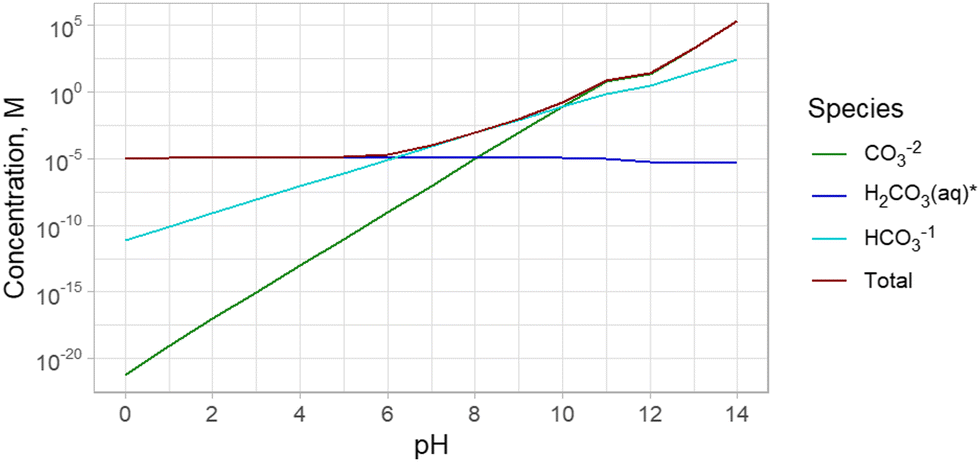 | ||
| Fig. 2 Concentration of carbonate species in seawater (modelled as 0.6 M NaCl) in contact with ambient atmosphere containing 400 ppm CO2. Note that H2CO3* indicates the total of dissolved CO2 and carbonic acid (H2CO3). At near neutral pH, the typical pH of seawater, the dominant carbonate species is HCO3−. Note that the region between pH 11 and 12 has a reduced rate of increase of CO32− concentration due to carbonate precipitation. | ||
The impact of salinity on the aqueous carbonate system can be understood by considering the influence of salinity on the ionic activity of the carbonate species and thus impacting the thermodynamic constants governing carbonate speciation. The equilibrium constants for the formation of HCO3− and CO32− are functions of the ionic activity (“{X}”) of the species, which is the product of concentration, and an activity coefficient (γ), which is a function of temperature and salinity. While the thermodynamic equilibrium constant (K°) is a function of ionic activity, it is common to use a distribution coefficient (K*) (which is a function of concentration) to describe equilibria.
| {X} = γ[X] |

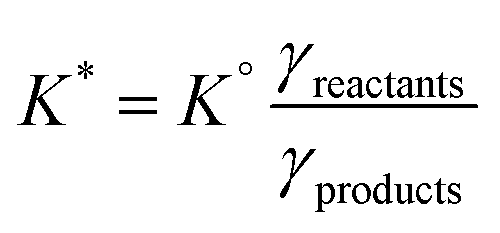
The activity coefficients can be calculated using a variety of models, many based off the Pitzer equations for the activity coefficient,24 which account for electrostatic interactions and ion pairing in complex solutions. The activity coefficient, and thus activity, of ionic species in water tends to decrease with increasing ionic strength, though this trend does not necessarily hold at high ionic strengths (>1 M).18 Using the activity coefficients determined for seawater at standard temperature (25 °C) with a salinity of 35 g L−1, the carbonate distribution coefficients (pK1* = 5.8401 and pK2* = 8.963624) are found to be lower than the constants for the carbonate system in freshwater (pK1* = 6.352 and pK2* = 10.32918). The lower pKa* values mean that equilibrium favors the formation bicarbonate and carbonate more in seawater as compared to pure water. While this is generally the case for saline waters, the exact value of the equilibrium constants will vary with composition, which should be considered in the design and optimization of saline RCC. As some RCC approaches involve increasing the ionic strength of a saline water either through chemical addition or concentration, more careful consideration of activity coefficients and their impact may be needed in designing these systems, as trends in ionic strength and activity can invert at high concentrations.18
The elevated CO2 content of saline waters, the relative abundance of these waters, and the ability to produce valuable end products makes RCC processes using saline water ideal for large-scale CCUS purposes. The elevated concentration of dissolved CO2 in water (compared to air) makes it easier (by reducing kinetic limitations) to drive chemical reactions25 that can transform dissolved CO2 into stable minerals (e.g., CaCO3) that can permanently sequester CO2. While solvents and sorbents used in conventional CCUS can have even greater CO2 capacity (further enhancing sequestration kinetics), these technologies require significant volumes of manufactured media to capture CO2, rather than using a natural resource such as saline water. Thus, saline water-based CO2 capture and sequestration can strike the balance between enhancing sequestration kinetics and utilizing an abundant, low-cost medium. In this review, we will cover recent efforts in this area of RCC in saline streams. Because these processes involve the handling of large volumes of water and the modification of various water quality parameters, there are many parallels between RCC and conventional water treatment processes. While these processes were not originally designed and optimized for RCC, we will discuss the extent of carbon capture currently achieved using these processes as well as how the chemistry and process design can be applied more expressly for RCC.
Though saline waters make an ideal medium for capturing and converting CO2 for the reasons listed above, it should be noted that not all carbonate solid formation results in carbon capture. For instance, precipitation of MgCO3 or CaCO3 from saline waters such as seawater, which have the majority of carbonates present as HCO3−, actually can cause a net release of CO2, as the divalent cation can sequester 2 mol CO2 as HCO3−/mol cation in the aqueous phase, but only sequesters 1 mol CO2 as a solid carbonate.26
| Ca2+ + 2HCO3− → CaCO3(s) + H2CO3(aq) → CaCO3(s) + H2O(l) + CO2(g) |
This can be addressed in one of two ways – adding alkalinity to the saline water source to retain the DIC,26 or by avoiding the formation of divalent carbonates and instead designing processes to generate bicarbonate solids such as NaHCO3. Generally, further discussion below of methods employing divalent carbonate formation for RCC all include some form of alkalinity addition.
Saline water sources
Just as the technologies used for gaseous carbon capture can differ for concentrated point sources versus atmospheric capture, the development of RCC technologies for carbon capture from saline water must take into consideration the composition of the saline water. Water with a total dissolved solids concentration exceeding 10 g L−1 is classified as saline;27 a variety of different water resources fall into this classification, including seawater, some surface and groundwater, and many waste streams and brines. (While some sources classify waters with TDS below 30 g L−1 as “brackish” and waters with TDS over 100 g L−1 as “brines”,27 here we will refer to saline waters as all waters with TDS over 10 g L−1 to encompass all potential source waters for saline RCC). The suitability of these sources for RCC applications varies according to the total salinity of the water, its alkalinity, the species of the dissolved solids capable of reacting with carbonates to form value-added products, and whether other constituents in the water can interfere with the RCC process.Another factor of seawater composition that impacts the design of RCC processes utilizing seawater is the ratio of carbonates relative to various carbonate-forming cationic species. Carbonates (primarily as HCO3−) are significantly less abundant (more than 250 times lower28) than the dominant anion in seawater, chloride, which serves as the counterion to the vast majority of the cations in seawater. As a result, there are more cations capable of forming solid carbonates than there are dissolved carbonates in seawater. This can be understood by considering the differing sources of ions in seawater. Ca, Mg, and many other metals found in seawater enter the oceans via dissolution of terrestrial minerals, which are then transported through runoff or river flow to the ocean.30 Meanwhile, DIC in seawater is primarily a result of CO2 dissolution across the gas/water interface between the ocean and the atmosphere.32 Since these are distinct processes, the amount of carbonate species present in seawater does not directly correlate to the amount of carbonate-forming cations, as would be the case if the ionic species were formed by the direct dissolution of a carbonate minerals. For instance, the concentration of Ca and Mg ions are roughly 5 and 25 times greater, respectively, than the concentration of carbonates (as HCO3−) on a mol basis.28 As a result, RCC processes can be designed to increase the concentration of carbonate species relative to the concentration of various cations, such as by contacting the seawater with a CO2-rich gaseous or aqueous stream, which can increase the potential amount of carbon capture beyond the amount of DIC naturally present in seawater, if sufficient alkalinity is present to facilitate additional CO2 dissolution into the water.
A final aspect of seawater composition that is relevant to RCC are constituents of the water that may interfere with RCC processes. These include ions that are not directly relevant for carbon capture, but which can impact the RCC process. For instance, sulfate (SO42−) is 13 times more concentrated in seawater than HCO3− on a mol basis,28 and forms solids with many ions present in seawater, such as CaSO4 (Ksp = 4.93 × 10−5), SrSO4 (Ksp = 3.8 × 10−7), and BaSO4 (Ksp = 1.1 × 10−10). (Ksp values for dissociation reaction of metal sulfates “XSO4” as shown below).
| XSO4 → X2+ + SO42− |
For RCC processes designed to generate CaCO3 (Ksp = 2.8 × 10−9) from seawater,33 the formation of CaSO4 is unlikely to consume Ca ions as its solubility product is over 104 times greater than CaCO3 (meaning it would require much greater concentrations of Ca and/or SO42− to precipitate than CaCO3). While the solubility products of SrSO4 and BaSO4 are much lower than CaSO4 (increasing their tendency to precipitate), Sr and Ba are present at much lower concentrations than other major cations (Na, Mg, Ca) in seawater,28 reducing the likelihood of formation of these solids. However, as many emerging RCC processes involve a concentration step to improve the kinetics of product formation, the impact of increased concentrations on the saturation of other precipitating species needs to be carefully considered in process design. This is particularly relevant for membrane-based RCC processes, as concentration polarization at the membrane/water interface can lead to precipitate formation (scaling), which results in significant losses in process efficiency.33 In other RCC processes, the major issue posed by the formation of non-carbonate precipitates is the reduced purity of the desired product.
Other seawater constituents beyond dissolved solids which can impact the viability of seawater-based RCC process include dissolved organic matter (known as “dissolved organic carbon”, “DOC”), as well as larger suspended organic and inorganic matter (known as “total suspended solids,” “TSS”). These constituents can contribute to clogging, surface fouling, and deterioration of system components, reducing efficiency and increasing operational costs. TSS are defined as aqueous constituents that are removed by a 0.45 μm filter, while DOC represents the organic fraction that passes through the filter. DOC concentrations vary throughout the ocean, typically ranging between 30–80 μM.34 DOC concentrations fall at the higher end of this spectrum in coastal waters,34 an important consideration in locating systems for seawater-based RCC, as organic matter can cause fouling on system surfaces, reducing process efficiency.35 Studies of organic fouling in seawater reverse-osmosis processes have shown that the formation of an organic foulant layer on membrane surfaces is exacerbated by the presence of divalent cations,36 which is particularly relevant to RCC processes that increase the concentrations of these ions to improve carbon-capture kinetics. Various pretreatment technologies, such as screening and filtration, are available to remove TSS and DOC from seawater35 prior to RCC processes sensitive to the presence of these constituents. In addition, drawing seawater from beach wells, rather than relying on an open ocean intake, has been shown to dramatically reduce DOC concentrations.37 However, the cost of pretreatment/beach well intake and any impacts on carbon capture capacity should be considered when evaluating the feasibility of using seawater for the RCC process.
Though the composition of some inland salt lakes may lend themselves to the chemistry of RCC processes, the actual availability of inland saline waters may limit their viability for meaningful carbon capture efforts. Even if all inland saline waters were available for RCC purposes, they account for only 0.9% of all water on Earth, whereas seawater accounts for 96.5%.39 However, only a much smaller portion of this already relatively small volume of available inland saline water would be likely to be usable, as these waters often host ecosystems that are sensitive to changes in volume and/or composition of the water body.38 Additionally, the presence of dissolved solids, dissolved organics, and suspended solids in surface waters poses similar challenges to RCC applications as described previously for seawater.
Some groundwater resources are rich in valuable elements,42 and there is precedent for extraction of saline groundwater to recover valuable constituents such as lithium43 in the form of lithium carbonate. A large percentage of global lithium production comes from the mining of brines in South America.44 In North America, there is a significant effort to extract lithium from lower-quality brines, such as produced water from oil and gas extraction and geothermal brines used for power generation.42 These North American brines are very rich in calcium. For example, Smackover brines (generated from the oil and gas fields of southern Arkansas) have calcium concentrations as high as 1 M, making them attractive for RCC.45 Importantly, these brines are already being brought to the surface – a byproduct of other industries, potentially reducing the overall cost of the RCC. The extent to which this type of reactive carbon capture could be applied to produce other products is likely an economic question, as the lesser value of other carbonate products may not be sufficient to justify the extraction of groundwater, which can be energy and cost intensive.
While it is difficult to quantify the volume of saline groundwaters worldwide, approximately 16% of the total land area on earth has underlying saline water at depths of 500 m or less,46 indicating that this is a potentially vast resource. (Note that groundwater exists at depths below 500 m, but is typically only extracted as a byproduct of oil and gas production,47 which is covered in the subsequent section on wastewater.) As with surface waters, understanding the source of salinity in these waters informs the chemistry of these waters and thus their suitability for RCC. The majority of these waters became saline either through evaporation of surface water, dissolution of formation minerals, a combination of evaporation and dissolution, or saline water was present at the time the aquifer geology was formed (“connate water”).46
Due to variations in the source of groundwater salinity, there is significant variation in the TDS of different groundwaters – for example, saline aquifers are reported to have salinities ranging from <50–340 g L−1 TDS in Israel,48 10–350 g L−1 TDS in China,49 and 10–300 g L−1 TDS in the United States.50 While the ionic composition of groundwater is also variable, major ions found in groundwater typically include Na+, Ca2+, Mg2+, Cl−, and SO4.2–14,17–51 As with saline surface waters, the overabundance of cations that can form carbonate precipitates relative to the DIC in groundwater indicates the potential for processes that can capture additional CO2 as solid minerals.
As NaCl is the major constituent of saline groundwaters (which typically have lower TSS and DOC contents compared to surface waters52), fewer process modifications and pretreatment steps may be needed to remove these constituents to enable the use of saline groundwater for RCC as compared to other potential saline water sources. However, as with other water sources, the presence of other precipitating ions may interfere with RCC purposes. Because groundwaters have varying ionic compositions, the potential for formation of competing precipitates such as CaSO4 should be evaluated on a source-by-source basis.
One additional constituent that can inhibit the utilization of these waters for RCC is naturally occurring radioactive materials (NORM) present in some groundwater. While the prevalence and distribution of NORM in groundwater is difficult to assess, monitoring of groundwater in the US has found many regions where groundwater contains radionuclides of radium and uranium at levels above 5 and 10 pCi L−1, respectively.53 Uranium is most mobile in low TDS, carbonate-rich groundwaters, whereas radium is more mobile in high TDS, chloride-dominant groundwaters,53 making radionuclides of radium more likely to occur in groundwaters targeted for RCC. Additionally, it should be noted that water treatment methods including electrodialysis and lime softening have been applied to remove up to 90% of radionuclides from groundwaters,53 capturing the NORM as a liquid concentrate or in a solid sludge, respectively. While this is useful for water treatment purposes, it raises concerns about RCC processes, which (a) use electrodialysis to concentrate carbonate-capturing cations, or (b) use precipitative softening to react cations with carbonates to produce solids. In both cases, the radionuclides could become concentrated in the product, rendering it radioactive, and complicating its further use or disposal.
Wastewater which typically contains Na+ and Ca2+ concentrations equal to or greater than the concentration of seawater (∼0.4 M Na and 0.01 M Ca28) include desalination brines, landfill leachates, flue gas desulfurization effluent, and wastewaters from the dairy and oil and gas industries.55 Similarly, desalination brines, flue gas desulfurization effluent, and wastewater from the oil and gas industry have typical Mg2+ concentrations equal to or greater than in seawater.55 While the Na+, Ca2+, and Mg2+ in less concentrated wastewater could still be used for RCC processes, it is useful to consider seawater as a gauge to determine which wastewater provides a similar or greater carbon capture potential than this low-cost, abundant saline water source. Bicarbonates found in oil and gas wastewater, desalination brines, and municipal wastewaters, and carbonates found in pharmaceutical wastes are all present at concentrations much lower than concentrations of carbonate-forming cations, presenting similar carbon capture potential as in other carbonate-depleted saline water sources reviewed here.
While some wastewaters have high salinity levels, which make them attractive for use in RCC, they can also have high levels of DOC and TSS, and may contain hazardous or toxic materials,57 making them unsuitable for reuse in RCC processes without additional pretreatment. DOC and TSS can be removed from saline wastewater using a variety of treatment methods, with filtration methods potentially being suitable here as they can reject larger particulates and organic compounds found in saline wastewater57 while allowing the passage of the ions that can be used in RCC. However, some hazardous contaminants in saline wastewater, such as heavy metals,58 cannot be easily separated from other salt ions during filtrations and would require more careful process design to ensure that any products generated during RCC would not be contaminated. Precipitative processes using elevated pH to generate Ca, or Mg solids would require such process design, as many heavy metals readily precipitate at lower pH than these solids.18
Despite some wastewater requiring pretreatment to make their composition suitable for RCC processes, there are still compelling cases for using at least some wastewater in RCC. Wastewater is unique amongst saline water sources for RCC in that many of these sources must already undergo some treatment processes prior to discharge or disposal, so additional processing to capture carbon may be integrated with these processes. For instance, one of the common treatment objectives for high-salinity wastewater is TDS reduction.54 This provides the opportunity to synergistically pair RCC processes that remove dissolved solids through the formation of solid carbonate species with desalination operations, effectively lowering the TDS and minimizing surface scaling while producing valuable carbonate solids. Some wastewater treatment processes, such as precipitative softening, already convert carbonates into solids,59 lending themselves to modification for RCC purposes. However, the extent to which RCC efforts can be integrated with existing treatment infrastructure will be determined by the ability to meet the required level of treatment (for all contaminants of concern, not just TDS) while also achieving a meaningful level of carbon capture.60
Beyond the impact of the saline water composition on carbon capture potential and energy demand, operational considerations such as equipment scaling/damage and product purity are impacted by the composition of the saline water source. Concerns related to the composition of different saline waters are discussed in the preceding sections. However, these concerns have not been addressed for many of the technologies discussed in this review. For technologies where these concerns were assessed, discussions of their impacts can be found in the following sections on each specific technology.
CO2 conversion processes
Various chemical processes can be used to capture carbon and generate valuable products using saline water. Here, we will discuss the underlying chemistry by which these processes convert carbon to a stable product. Additionally, we will review actual processes and technologies used to achieve saline-based RCC. The technologies and processes are divided into processes that are currently in development, as these are being optimized for CO2 capture, and established processes that were developed and optimized for purposes other than CO2 capture with saline water but have the potential to be optimized for RCC.CO2 conversion chemistry
Formation of products from the DIC in saline waters temporarily decreases the concentration of dissolved carbonates, allowing the water, once in contact with the air, to absorb more atmospheric CO2, driving the carbon capture process. In conditions where the bulk CO2 partial pressure is effectively constant (i.e. the atmosphere), the total amount of dissolved CO2 is also maintained at a constant concentration, following Henry's law. As such, when some of the dissolved CO2 converts to HCO3− and CO3− to maintain equilibrium, more CO2 will then dissolve, satisfying equilibrium with the bulk gas phase. If a saline water is maintained in a nonequilibrium state (i.e. carbonate solids are continuously precipitated and removed), such reactions can continue until available reactants in the saline water source are consumed through the product formation process. Thus, if the correct conditions (i.e. pH, temperature) are maintained, the carbon-capture potential of saline water can extend beyond its initial DIC concentration when additional gaseous CO2 is available to dissolve and form products (e.g., by bubbling air or other CO2-containing gas through the water) and is ultimately determined by the concentration of reactants other than CO2. While the carbon capture potential of saline water sources is ultimately limited by the availability of other reactants (i.e. carbonate-forming cations), the kinetics of carbon conversion processes would likely be limited by the DIC concentration, which is determined largely by the concentration of CO2 in the gaseous phase.18 Thus, contacting waters with gaseous streams containing elevated CO2 concentrations is a straightforward way to capture additional CO2 more efficiently in the aqueous phase and convert it to an end product. Again, it is imporant to note that in order to prevent equilibrium shifting back and re-releasing CO2, sufficient alkalinity must be present in the aqueous stream. This is particularly critical for systems removing divalent carbonate solids (CaCO3, MgCO3). Below we will summarize the chemical reactions that can effectively convert DIC, whether dissolved from the atmosphere or a CO2-concentrated gas, into stable products, thus enabling RCC.
The formation of precipitates is governed by equilibrium between the solid and dissolved ionic phases. For a solid precipitate to form, ion activity potential (IAP) of the species forming the solid must be greater than the solubility product (Ksp) of the solid.18 (The solubility product of various carbonate solids is listed below in Table 1)
For the reaction
| AaBb(solid) ↔ aA+ + bB−, |
| IAP = {A}a{B}b |
| Ksp = {A}eqa{B}eqb |
It should be noted that many solids will not begin to precipitate until the IAP greatly exceeds the Ksp (i.e. the solution becomes supersaturated with respect to this solid). For instance, seawater is already oversaturated with respect to both CaCO3 and MgCO3 by a factor of 2, but additional concentration or other processing is required to induce precipitation.71 Some processes can reduce the degree of oversaturation required for solid formation by reducing thermodynamic barriers to solid formation. However, even when oversaturated and thermodynamically favoured, some carbonates, such as MgCO3 are still extremely slow to form – see following discussion on the kinetics of carbonate formation. The dissociation constants for various carbonate solids (XCO3 or XHCO3) are included below in Table 1, for generic dissociation reactions shown below.
| XCO3 → X+2 + CO32− |
| XHCO3 → X+ + HCO3− |
As carbonate is rarely the sole anion present in saline waters, it is important to also consider other solids which can form from saline waters, as they may either reduce the efficiency of an RCC process or diminish the purity of the generated products, as noted in the discussion of competing ions present in saline water sources. For instance, many cations readily form solids with hydroxides.18 As hydroxide ion addition is used to increase alkalinity (and drive the carbonate system equilibrium towards elevated concentrations and CO3−2 speciation), it is important to assess the possibility that some this alkalinity may be consumed through the formation of relatively insoluble species such as Mg(OH)2.31 The dissociation constants for various hydroxide solids (XOH or X(OH)2) are included below in Table 2, for generic dissociation reactions shown below.
| XOH → X+ + OH− |
| X(OH)2 → X+ + 2OH− |
| Solid | K sp (STP) | Ref. |
|---|---|---|
| Ca(OH)2 | 5.5 × 10−6 | 68 |
| Fe(OH)2 | 4.87 × 10−17 | |
| Mg(OH)2 | 5.61 × 10−12 |
Another major consideration for RCC processes based upon precipitation is the rate of precipitate formation.

Should the rate of precipitation be lower than the rate of competing reactions, inhibition of production formation could occur. For instance, in many waters the rate of precipitation of Mg(OH)2 can be greater than the rate of precipitation of CaCO3.12 The formation of Mg(OH)2 reduces solution pH, shifting carbonate equilibrium away from CO32− and thus can inhibit the formation of CaCO3. In some waters, this can be prevented by maintaining solution pH above the pH needed for CaCO3 formation but below the pH for Mg(OH)2 formation – de Lannoy et al. achieved this separation by maintaining pH between 9.3–9.6 in synthetic seawater.12 However, in other waters, such as the synthetic seawater used by Xie et al., Mg(OH)2 precipitated first around a pH of 10, with CaCO3 precipitating later at a pH of 12.3.17 Again, this demonstrates that a thorough understanding of solution composition as well as the reaction kinetics relevant to precipitate formation aids in designing or optimizing RCC processes.
Even without the impact of competing ions, some carbonate minerals are still slow to form, such as MgCO3. For these kinetically hindered carbonates, increasing the temperature and/or pressure can enhance the rate of precipitation,72 but may incur an energetic cost that outweighs the carbon capture potential. Researchers have identified understanding and enhancing the kinetics of carbonate formation under ambient conditions as a critical topic for further study to aid the development of carbon capture technologies.72 It should be noted that at the time of this review, even within reviews focused on aqueous mineral carbonation, discussion of mineral carbonation kinetics primarily focused upon dissolution of Ca or Mg rich minerals, rather than on formation of mineral carbonates.73 RCC using saline water largely avoids these kinetic limitations by utilizing the ions already dissolved. Additionally, in the specific case of magnesium carbonates, several hydrated phases have more favourable kinetics and thus form more readily under ambient conditions (i.e. nesquehonite (MgCO3·3H2O) or hydromagnesite (4MgCO3·Mg(OH)2·4H2O)).72 Whether designing processes which produce more readily precipitated forms of a carbonate (either with or without post-processing to convert to the desired carbonate product) are viable for saline-based RCC is a techno-economic question, influenced by the relative value of the various phases of the carbonate and the energy demand required to generate the desired phase.
Overall, the formation of inorganic carbonate solids from saline solutions is at first a seemingly straightforward precipitation reaction. However, when considering complex saline water sources with varying chemical compositions, the concentration and solubility of each constituent must be considered. Carbonate-forming cations with low solubility and/or high concentrations are generally good candidates for carbon sequestration via precipitation. Additionally, carbonates with favourable precipitation kinetics under ambient conditions are desirable for RCC applications, as they can rapidly form without incurring additional energetic cost.
To convert CO2 into an organic product, the carbon must be reduced from a valence state of +4 to 0, requiring an energy source to provide electrons to the reaction.77
| nCO2 + nH2O + (4n)e− → CnH2nOn + O2n |
While many carbon capture technologies rely on autotrophic organisms to facilitate this reaction (i.e. through photosynthesis), this process can also be driven electrochemically, as reviewed later in this section.
Processes designed for reactive carbon capture
As capture and conversion of CO2 has emerged as an important objective towards mitigating climate change, focus has increased on RCC processes, including those that utilize saline water. To begin, we will discuss the technologies that have been specifically developed for carbon capture and conversion into a valuable product using saline water. As these saline-based RCC processes are an emerging carbon-capture approach, there are a limited number of technologies that currently fall into this category. However, many preexisting technologies and processes achieve some degree of CO2 capture via saline water, though this is not the express intent of these technologies. Because of the pressing need for CO2 capture and the massive potential benefits of saline-based RCC processes outlined in this review, it is worthwhile to discuss how these technologies that have not been specifically developed for saline-based RCC can be modified to optimize CO2 capture and utilization – see the subsequent section, “Reactive Carbon Capture Using Pre-Existing Technologies and Processes.”Though processes that use saline water to convert CO2 to a solid precipitate via chemical addition have existed long before interest in carbon capture, some aspects of these systems that are critical to optimizing carbon capture, such as the rate of carbonate formation and % conversion of cationic species to solids, have only recently been studied. For instance, De Vito et al.78 monitored the rate of precipitate formation in a system designed to produce carbonate solids by bubbling CO2 in a brine solution, finding that the concentration of the saline water impacted the carbonate formation rate and process. Their system provided pure CO2 bubbles to a synthetic MgCl2 brine at concentrations between 7 and 32 g L−1, achieving nearly 100% conversion of the Mg to carbonate precipitates (primarily nesquehonite, MgCO3·3H2O) in time frames ranging from 10 minutes to 30 days, respectively. This is an interesting result because typically increasing the concentration of the precipitate-forming species increases the rate of precipitation, whereas in this study, the lowest concentration solution achieved the most rapid carbonate formation. This suggests that the reaction may have been limited by CO2. If so, this has several implications for the development of precipitative processes for carbon capture – first, methods to improve CO2 dissolution (such as the use of microbubbles79) are likely to be important to achieving rapid and scalable saline-based RCC; second, RCC processes capturing CO2 from more dilute sources than the pure CO2 gas used by De Vito et al. will be even further rate-limited by the lower CO2 concentrations, again pointing to the need to enhance dissolution; and third, there may be instances in which the kinetic benefits of pre-concentrating the saline water prior to carbon capture provide little benefit, as the reaction is limited by the concentration of CO2 rather than by the concentration of cationic species.
Many emerging saline-based RCC processes that utilize chemical addition to induce precipitation, such as the one demonstrated by Bang et al.,79 feature novel approaches to adding CO2 to the saline water to enhance carbon capture. In their system, real brine from a seawater desalination facility was repeatedly cycled through pH adjustment with sodium hydroxide (NaOH) followed by contact with CO2 microbubbles and filtration to collect precipitates. Adding base to solutions has two impacts relevant to precipitative carbon capture, as previously discussed – first, it raises the pH, shifting the carbonate equilibrium towards formation of CO32−, thus improving the kinetics for carbonate precipitation, and second, it increases the concentration of OH−, which can increase the formation of hydroxide precipitates. The formation of hydroxide precipitates may seem counter to the objective of carbon capture, as it sequesters cations in hydroxide solids that may otherwise be able to form carbonate solids. However, Bang et al. found that in the instance of magnesium, the formation of hydroxide solids prior to contact with CO2 microbubbles facilitated the formation of magnesium carbonate, achieving 86% conversion of Mg in the brine to carbonate solids, along with 99% conversion of Ca. While this study did not consider the % conversion of CO2 to carbonate solids or the rate of CO2 capture, it provides a useful demonstration of the principles of precipitative CO2 capture and highlights how the interactions of different cationic species in real saline waters can impact the overall carbon capture potential of these processes.
Additionally, it should be noted that this process and the one demonstrated by de Vito et al. achieve net carbon capture by adding CO2 to a solution that is undersaturated with respect to carbonates. This is because these approaches use additional CO2 to drive the formation of carbonate solids rather than simply precipitating carbonates that were already present in the saline water and releasing CO2 in the process. (It should be noted than simply dissolving the CO2 to form aqueous HCO3− would result in greater CO2 capture (2 mols CO2 per mol Ca or Mg, versus 1 mol CO2 per mol when forming solids), but this approach does not yield saleable products and thus falls out of the purview of our discussion of RCC technologies).
While RCC processes achieving carbon capture by chemical titration like those demonstrated by De Vito et al. and Bang et al. seek to optimize the conversion of carbon to an end product (% CO2 converted) and the rate of conversion (CO2 converted/time), an additional optimization is likely required to make carbon capture via chemical addition viable from a carbon accounting perspective. Life cycle analyses completed by Beeftink et al. found that the amount of carbon sequestered during conventional precipitative processes like drinking water softening – 0.95 kg CO2eq per person per year, equivalent to 84 kg CO2eq per million gallons (MG) treated water – offset only about 20% of the carbon footprint of the energy and chemicals used for the process.80 However, their analysis showed that when considering downstream impacts of drinking water softening, such as improved household appliance efficiency and reduced scaling in pipes and appliances, precipitation processes had a net negative carbon footprint of −1.93 kg CO2eq per person per year or −170 kg CO2eq per MG treated water.80 Thus, if precipitation-based processes are to be applied for carbon sequestration purposes through RCC, the energy and chemical demand will need to be reduced or the process will need to be paired with applications where softening can improve downstream efficiency to maintain a carbon negative footprint. The main approach reported for reducing the carbon footprint of chemicals used in water softening is through electrochemical generation processes, covered in subsequent sections. Other novel approaches to improving the net carbon capture capacity of processes that can be applied for saline-water based RCC are covered below.
One way to reduce the energy demand of precipitative RCC processes is to reduce the thermodynamic barriers to precipitate formation. Lowering the saturation index (SI) required for precipitation to initiate means, for example, less energy needs to be spent to concentrate the saline water to the necessary SI. Burhenne et al.81 demonstrated the application of a bench-scale fluidized bed pellet reactor that precipitated CaCO3 from a feed of K2CO3 generated from atmospheric CO2. While K2CO3 is not an ideal representative of most saline waters (even after contact with concentrated CO2) due to the elevated solubility of K2CO3 compared to other carbonate salts, this process provides a critical demonstration of the impact of seeding material on carbonate precipitation, which can be applied to other carbonate species (e.g., CaCO3) precipitation from saline waters.
Another way to improve the thermodynamics of precipitate formation is by using a catalyst. Dindi et al. modified the Solvay process for producing NaHCO3 (discussed in further detail in following subsection) to utilize desalination brine as both the sorbent for CO2 capture and the source of Na+ ions for the process. In this process, an amine-based solvent is added to the brine prior to contact with CO2 in flue gas. As the CO2 dissolved into the brine, it forms carbamates with the amine, which can then react with sodium in the brine to form NaHCO3. This process provides an important advantage over more conventional amine-based CO2 capture processes, as the use of brine as the bulk sorbent solution allows for the removal of absorbed CO2 through precipitation, shifting equilibrium to allow further absorption of CO2. The authors evaluated multiple amine catalysts and ultimately found that the optimal CO2 absorption, salt removal, and NaHCO3 production were achieved using 30% 2-amino-,2-methyl-propanol (AMP) mixed with the brine. While the process was successfully demonstrated for synthetic brines at concentrations typical of desalination brines (0.85 M NaCl), as well as at both lower and higher concentrations (0.6–1.8 M NaCl), the authors noted that similar studies found that the increased ionic strength of the brine has been reported elsewhere to limit CO2 absorption capacity. Absorption capacity was not evaluated in this study but is an important factor for process design and viability, and further development of the process will likely require further evaluation of the impact of brine concentrations on this parameter. An additional part of the process developed by Dindi et al. that may require further refinement is the amine-catalyst recovery process – after evaluating distillation, amine-chloride precipitation, and ultra-high lime with aluminum (UHLA) recovery methods, they found that the UHLA process was the only viable process.82 While this process was able to recover the AMP catalyst, it reduces the CO2 sequestration potential of the process as it requires desorption of any CO2 that remains bound to the AMP prior to recovery, and it consumes lime (CaO), a chemical whose production often has a significant carbon footprint.83
Another material that has recently been studied for its ability to reduce the thermodynamic barrier to carbonate formation is carboxylated polystyrene (PS). Power et al. added carboxylated PS microspheres to batch reactors containing solutions of MgCl and NaHCO3, and demonstrated the formation of magnesite (MgCO3) after a 60 day reaction time without additional energy input.31 The study suggests that the carboxyl groups on the PS help to dehydrate the Mg ions, overcoming a critical energetic barrier to MgCO3 formation. This represents a major advancement from conventional methods of producing MgCO3 as it eliminates some of the energy demand to drive this kinetically unfavourable reaction. However, the extremely long reaction time makes this process less attractive for large scale implementation, so further process development would be needed to utilize this material as a catalyst in RCC processes. Additionally, the primary benefit to producing magnesite as opposed to more readily precipitated phases of magnesium carbonate (such as nesquehonite) presented by Power et al. is the stability of MgCO3, which is more stable than many of the other metastable phases of magnesium carbonate.31 For RCC purposes, which emphasize the generation of a valuable end product, the stability of the product is a lesser concern than the saleability of the product – see section “Products from Saline Water RCC” for discussion of the value of various forms of magnesium carbonate (note that within the broader context of CCS in which product value is not emphasized, the stability of the end sequestration product is a critical consideration). Whether this process to directly generate magnesite from solution provides benefits over methods to form other phases of magnesium carbonate or not, it is still critical to note that the significant increase in overall carbon capture potential from saline waters that could be realized by producing magnesium carbonates in addition to calcium and sodium carbonates merits further study of potential applications of this material for RCC purposes.
A different approach to increasing the CO2 absorption capacity and precipitate production from brines is mixing them with materials rich in carbonate-forming species. Soong et al.66 evaluated the CO2 sequestration capacity of brines produced during oil production mixed with fly ash, which can contain significant levels of calcium. Two process approaches were evaluated – mixing the ash with the brine and contacting the resultant slurry with CO2 gas, and contacting filtrate collected from the ash brine mixture with CO2 gas. For CO2 contact with ash-brine slurries of 10 wt% using fly ash from various sources, they demonstrated CO2 consumption of 0.06–0.32 mol L−1, and for CO2 contact with the slurry filtrates they demonstrated consumption of 0.38–0.55 mol L−1. Importantly, as the composition of the fly ash varied by source, the composition of recovered solids also varied, particularly when the fly ash was not separated from the brine after initial contact. Using this approach, different fly ash sources yielded 30–50% CaCO3 in the recovered solids. However, when the fly ash was separated from the brine prior to contact with CO2, the solids recovered contained above 90% CaCO3.66 This study demonstrates that while the addition of carbonate-forming species to brines can increase the CO2 sequestration capacity, careful process design such as the inclusion of an intermediate filtration step is essential to ensuring that the process can generate products (such as CaCO3) that are sufficiently pure for beneficial use. Additionally, this process was only demonstrated as a proof-of-concept using pure CO2 gas, and the CO2 sequestration capacity using ambient air was not evaluated, limiting the ability to assess this process as a stand-alone CO2 capture approach. However, as increasing the concentration of species such as Ca and Mg can shift the carbonate system equilibrium to allow for an increase in the total amount of dissolved carbonate species, contacting brines with fly ash or other Ca or Mg rich materials is likely to increase the carbon sequestration capacity for precipitative processes when compared to raw brines. This approach demonstrates a potential method to adding alkalinity in processes designed to precipitate CaCO3, a step that is essential to ensuring net carbon capture.
Some processes have combined multiple methods to optimize the amount of CO2 mineralized. For instance, Zhang et al.67 utilized both a catalytic material (nickel nanoparticles) and an additional source of carbonate-forming cations (blast furnace slag) to achieve 100% conversion of calcium in a synthetic desalination brine into calcium carbonate. While both the catalyst and added cations were shown to enhance the carbon capture in the plug flow precipitator used in the study as compared to system operating without catalyst or cation addition, there are major drawbacks to this approach, including a relatively slow carbon capture rate, contamination of the end material with the catalyst, and toxicity concerns related to the nickel nanoparticle used as catalyst. While studies of strategies to enhance carbon capture efficiency of different technologies can be illuminating, the overall carbon capture rate is a major design parameter governing whether a RCC technology can achieve a meaningful volume of carbon capture on a reasonable time scale.
Ultimately, many of the saline-based RCC processes using chemical titration approaches which have emerged in the past two decades are still at low levels of technical readiness. Most of the processes reviewed above have only been demonstrated at a bench scale, and often utilize synthetic saline waters and/or a concentrated CO2 source. Still, trends that indicate how chemical titration processes may be implemented to achieve efficient, scalable CO2 capture and conversion emerge in the literature, including: (1) enhancement of CO2 dissolution into saline water (via pH adjustment (e.g., alkalinity addition) and more efficient gas contacting) and (2) enhancement of the rate of carbonate formation (using seed materials, catalysts, or process design). Still, whether these can be implemented while also decreasing the overall energy intensity of this RCC approach to make it viable as a carbon-negative process remains to be seen.
Electrochemical processes
Many emerging RCC technologies rely on electrochemical reactions to drive the formation of the desired chemical products from carbonates in saline water. Many of these electrochemical processes have emerged as a potential means to reduce the carbon footprint associated with the other RCC processes by reducing required chemical addition. Under the umbrella of electrochemical RCC, some technologies utilize electrodialysis (ED) systems with configurations that allow ions in feed streams (usually saline water and a carbonate-rich stream) to be separated and then combined to generate desired products. Other novel applications of electrochemistry for RCC can involve direct reaction of carbonates on electrode surfaces to generate valuable chemical products. Both ED and other electrochemical processes often take advantage of the ability to separate H+ and OH− to generate the alkalinity, which is then used to shift equilibrium towards the formation of carbonate solids.Electrodialysis for the generation of inorganic products
Electrodialysis is a process in which a saline stream is fed through channels separated by ion exchange membranes in a “stack” between two electrodes, which cause the charged species within the saline stream to migrate across the membranes towards the electrode with an opposite charge. By orienting membranes that are only permeable to cationic or anionic species in a specific order within the electrode stack, the migration of ions can be stopped so that certain steams become concentrated with ions, while other streams become diluted, as is typical in desalination applications.84 Modifications of ED can capitalize on specific membrane properties and/or membrane stack configuration to separate the desired ions for carbon capture from other ions present in saline waters.85 See (Fig. 3) for a comparison of ED schematics covered in this review.While in theory ED water could be applied to simply concentrate saline waters in order to increase the saturation level of carbonate-forming species and thereby decrease the barrier to precipitate formation, there are several limitations to the actual viability of such applications. Indeed, no example of this approach was found at the time of this review. Though ED is a potentially lower-carbon approach to concentrating saline waters when compared to thermally driven concentration processes,86 it is often not as energy efficient as reverse osmosis.87 Multiple other factors may also limit the practicality of using ED to simply concentrate saline waters to enhance capture carbon. First, most saline waters already contain high concentrations of non-carbonate hardness (see previous discussion of saline water compositions), so processes that can specifically increase the concentration of the inorganic carbon species rather than simply increasing the concentration of all species may be more efficient. (Also note the distinction between processes that concentrate all species and processes that increase the concentration of cationic species, as the latter can lead to increased CO2 capture within the saline water when supplemental CO2 is provided, as previously discussed.) Another factor that may limit applications of ED for concentrating saline waters for RCC is the existence of a limiting concentration beyond which the concentrated stream cannot be increased.86 As the concentration gradient between the concentrate and dilute streams increases, osmosis and diffusion increase in the opposite direction of the electrically driven separation, limiting further transport of ions across the membrane. Though this can in part be overcome by increasing the current density of the ED system, the associated increase in energy consumption can become prohibitive.86 Finally, ED systems are more susceptible to scaling than systems such as RO that can also be used for concentrating saline water, which can pose major limitations in processes designed to produce carbonate solids.87
An additional barrier to the application of ED to saline-based RCC processes that produce carbonate solids is scaling within the membrane stack, which can greatly reduce the efficiency of the process. Scaling, the accumulation of precipitated solids on membrane surfaces, can be comprised of carbonate solids and/or other solids such as gypsum (CaSO4). While carbonate precipitation is ultimately desirable to collect the carbonate end products in RCC, if it occurs within the ED stack it can cause scale formation on membrane surfaces in the concentrated channels. Thus, RCC processes utilizing ED must be carefully designed and optimized to achieve precipitation outside of the membrane stack. Generally, this is addressed by (1) operating the system such that concentration polarization at the membrane surfaces is minimized by convective mixing within the channel86 and (2) operating at concentrations below the required saturation index for precipitation. The latter approach means additional steps would be required after ED to induce precipitation and collect the desired carbonate solids. Here, the use of seeds or catalysts outside the stack to allow precipitation at lower saturation index has been investigated.65
Instead of using ED to concentrate saline waters used in RCC, many processes utilize unique membranes and/or stack configurations to combine target ions from two separate feed streams to generate a product stream containing the ionic species of the desired product (i.e. NaHCO3, CaCO3, etc.). Some systems have an additional product stream of another valuable, non-carbonate chemical. The feed streams are typically saline water that provides the desired cations, and a stream with high CT. Some of these systems also have stack configurations that allow transport of alkali (as OH−) to the product stream. Additionally, the use of bipolar membranes to generate acid and alkalinity in separate streams within and ED stack can reduce the reagent demand in processes that consume alkali.65 Below, we will provide a summary of the configuration of 4 RCC processes using ED in this manner, as this is the defining feature that enables these systems to capture CO2.
One application of ED for saline-based RCC was demonstrated by Dara et al.,62 who designed an electrodialysis system to use saline water and carbonic acid generated by dissolved CO2 in deionized water to produce sodium bicarbonate (NaHCO3) and hydrochloric acid (HCl). Their system featured 4 channels between a Ti mesh cathode and a Pt/Ir-coated Ti anode separated by ion exchange membranes: a feed stream containing carbonic acid separated by an AEM from the first product stream, which was in turn separated by a CEM from a feed stream containing NaCl, which was separated by an AEM on the other side from the second product stream (Fig. 3). The electrodes were oriented such that anions and cations would move out of the feed streams and into the product streams: HCO3− and Cl− moved toward the cathode into the first and second product streams, respectively, while Na+ and H+ moved toward the anode into the first and second product streams, respectively. This system design effectively created two product streams – concentrated NaHCO3 and concentrated HCl – both of which are useful for various industrial applications (see discussion in following section). Additionally, the system effectively desalinated the saline feed stream, demonstrating additional benefits for the treatment of saline wastewater. A major drawback of this system is the low conductivity of the carbonic acid stream, which was attributed to low rate of diffusion of the carbonate species.62 This study was an early-stage proof of concept and did not evaluate the amount of carbon capture potential of the system. Additionally, only a synthetic saline stream of 1 M NaCl was investigated, whereas most real saline waters contain a variety of ionic species, which may hinder the ability of the system to produce pure chemical products and/or reduce the system efficiency due to membrane scaling. However, it may be possible to use ion-selective membranes within the ED stack to prevent ions besides Na and Cl from entering/leaving the saline stream, better ensuring the purity of the produced chemical streams.85 Also, it should be noted that as the CO2 captured in this system is initially captured in DI water, this may not technically be considered as “saline-based RCC,” but it may be possible to use of saline water to initially capture the CO2 and generate the carbonic acid stream.
Indeed, other applications of ED have taken this approach. Mustafa et al.61 designed an electrodialysis system which used carbonates from a saline stream to generate NaHCO3 and Na2CO3, capitalizing on the increased CO2 content of saline waters and eliminating the need to dissolve additional CO2 into the source water. In this system, alternating CEMs and AEMs created four chambers between the anode and cathode compartments of the electrodialysis stack – a chamber to collect produced acid, a chamber fed with a NaCl brine, a chamber that collected NaHCO3 and Na2CO3 (we will refer to this as the “product stream”), and a chamber fed with a saline, carbonate rich stream (Fig. 3). During optimization trials of the system, increasing voltage and CO2 concentration led to greater CO2 uptake into the product stream, while the impact of the concentration of the brine stream had a more nuanced impact on CO2 uptake. Uptake increased at a brine concentration of 0.75 M NaCl, after which the uptake declined, which the authors attributed to loss of system efficiency due to scaling caused by formation of precipitate within the ED stack. This highlights scaling as a critical issue for systems designed to concentrate precipitating species such as sodium carbonate – determining and maintaining an optimal concentration that is low enough to prevent precipitation within the system, while still high enough to improve the kinetics of downstream precipitative product recovery. Another unique approach to carbon capture employed in the study by Mustafa et al. was the dehydration of the NaHCO3/Na2CO3 stream using freeze-drying rather than heating, effectively preventing CO2 evolution. While they found that the solids produced by freeze-drying had a 15% greater NaHCO3 content compared to the liquid product solution, the solids produced this way were still far from pure, consisting of only 55% NaHCO3, with much of the remainder being Na2CO3.62 Depending on the intended use case of the produced solids, this purity level may not be sufficient for RCC purposes, and further process modification may be needed to achieve a substantially pure carbonate or bicarbonate product.
ED systems for saline-based RCC have also been used to produce solids other than sodium carbonate and bicarbonate. While sodium is the most abundant cation in most saline waters, sodium carbonates are more soluble than many other carbonate solids (see previous section on inorganic product formation from aqueous carbonates). The lower solubility of other carbonate solids makes them attractive for saline-based RCC processes, and it can also exacerbate the impact of scaling within an ED stack. As such, the design of ED systems intended to produce lower solubility carbonates such as CaCO3 may include additional features or modifications to overcome this.
Zhao et al.65 recently designed an electrodialysis stack with bipolar membranes (BPMED), which when used in tandem with a crystallizer achieved sequestration of carbon as CaCO3 without significant membrane fouling. Their system was comprised of a series of groups of bipolar membranes and AEMs between a cathode and an anode. Each group of membranes formed 3 distinct channels, beginning with a bipolar membrane nearest to the cathode, followed by a channel fed with alkali solution, and AEM, a channel fed with seawater, another AEM, a channel fed with acid solution, and another bipolar membrane (Fig. 3). The bipolar membranes were oriented such that the positive face was in contact with the alkali channel, where OH− generated by water hydrolysis in the BPM collects, while the negative face within contact with the acid channel, where H+ generated by hydrolysis collected. The AEMs enclosing the seawater channel prevented Ca and Mg from depositing on the bipolar membranes, effectively preventing fouling on these surfaces. This allowed for continued generation of alkalinity in the form of OH−, which was able to transport across the AEM into the seawater stream, where it helped maintain a sufficiently high pH to allow for CaCO3 precipitation when the seawater stream was subsequently circulated through a crystallizer. The major benefits of this system are the reduced reagent demand for seawater softening and the ability to use a membrane-based system to soften the seawater without major efficiency losses due to fouling.65
Another approach to reducing the impact of scaling on BPMs used to generate alkali in saline based RCC processes is to demineralize water that is contacted with the BPMs, as discussed by de Lannoy et al.12 While this approach may seem counterintuitive in a system in which the ultimate objective is to produce carbonate minerals from the saline feed water, their proposed system demineralizes only a small portion (not quantified) of saline water stream that was then fed to a BPMED system to generate acid and/or base (Fig. 3). The acid or base is then used to shift the pH of the remaining saline water stream to convert the DIC to either gaseous CO2 or solid CaCO3, respectively. This system is distinct from the other ED applications reviewed here for several reasons. First, by only using a demineralized fraction of the seawater for acid/base generation, they reduce scaling within the ED stack, potentially increasing the efficiency and reducing the need for membrane cleaning or replacement.12 Second, the ability of the process to generate two different products out of the dissolved carbonates is unique and makes the system adaptable to market demands (though the low current value of pure CO2 gas75 means this is not a likely candidate for RCC purposes). While these differences between the systems developed by de Lannoy et al. and others may be beneficial for carbon capture applications, other differences indicate potential downsides of these systems. For instance, unlike the systems developed by Dara et al. and Mustafa et al., which utilized the most abundant cation in most saline waters (Na+) to capture DIC as solid carbonates, this proposed system utilizes Ca+ ions to generate carbonate solids. Furthermore, while tests of the base addition step of their system demonstrated complete DIC removal from the seawater, it should be noted that this is an underutilization of the Ca+2 ions in seawater, which outnumber DIC on a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol basis. However, methods utilized in other processes reviewed here to increase the carbonate concentration in seawater could be used to ensure that the entire carbon capture capacity of the system is realized.
:1 mol basis. However, methods utilized in other processes reviewed here to increase the carbonate concentration in seawater could be used to ensure that the entire carbon capture capacity of the system is realized.
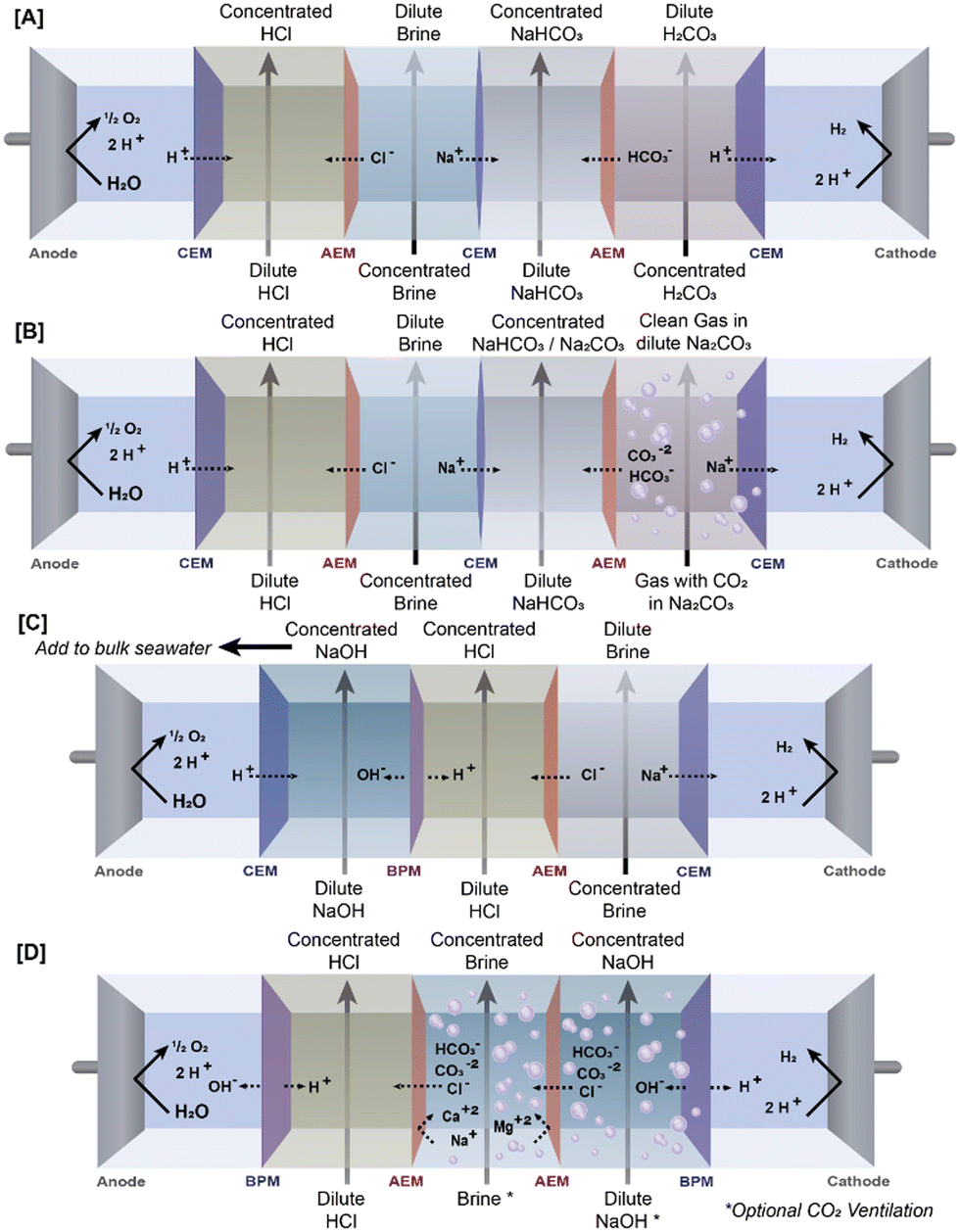 | ||
| Fig. 3 Comparison of ED stack schematics from (A) Dara et al., (B) Mustafa et al., (C) de Lannoy et al., and Zhao et al. | ||
Xie et al.17 demonstrated a membrane electrolysis process that increase the pH of synthetic seawater, which was then bubbled with CO2 gas to generate CaCO3, along with Mg(OH)2 (which was later converted to Mg5(CO3)4(OH)2·4H2O (hydromagnesite)). In their system, the seawater was fed to the cathode chamber, where water was split by the cathode to produce OH−, thus maintaining an elevated pH to facilitate precipitation. When the pH of the cell reached 10.1, Mg(OH)2 precipitated and was filtered out of the solution. Subsequently, CO2 was bubbled into the cell, allowing CaCO3 to precipitate. The Mg(OH)2 was converted into hydromagnesite by heating a suspension of the Mg(OH)2 with bubbled CO2. This system converted over 99% of both the Mg+2 and Ca+2 into carbonate solids.17 A major benefit of the system developed by Xie et al. is the production of pure, separate calcium and magnesium solids, which was achieved through careful control of the system pH and timing of the CO2 addition. This eliminates any need for additional costly product purification processes. Additionally, the anion exchange membrane used in their system primarily serves to separate acid generated at the anode and alkali generated at the cathode, rather than to allow selective transport of ions between feed and product streams. As such, reductions in flux due to scaling on the surface of the membrane are likely less of a concern in such systems, and scaling was not discussed as an issue. However, further study is required before it can be concluded that the carbonate production by membrane electrolysis systems is less impacted by scaling, but if this is the case, this would be a major benefit of such systems, particularly when compared to ED.
While systems such as those demonstrated by Xie et al. effectively convert Ca and Mg in seawater to stable carbonates, RCC systems utilizing sodium to capture CO2 may have a greater overall carbon capture capacity, as sodium is typically more abundant in saline waters (see previous section on saline water composition). While a major benefit of processes that generate calcium and magnesium carbonates is the durability of these solids (as this enables their use for long-term carbon sequestration), the durability of sodium carbonates is generally less discussed in relation to carbon capture processes. Instead, the focus is on producing NaHCO3 and Na2CO3 for consumption, so the long-term durability of these products is less of a concern than the ability to generate them efficiently. An additional benefit to processes using sodium to convert CO2 to solids is that the formation of NaHCO3 avoids the need to add alkalinity to prevent the net release of carbon when forming carbonates with divalent cations. While ED systems have been demonstrated as a way to combine Na from seawater with carbonates, the low mobility of bicarbonate and carbonate ions can limit the efficiency of these systems and prevent them from reaching the full carbon capture potential provided by the sodium ions.62 As such, other electrochemical systems with geometries that do not require the transport of bicarbonate or carbonate across membrane interfaces may be an approach to reach the full carbon capture potential more efficiently.
A prototype system developed by Park et al.64 used a two chamber system separated by a ceramic membrane to electrolytically convert seawater first to NaOH and then to NaHCO3. In this system, the anode is within an inner chamber created by the ceramic membrane, which allows separation between the H+ generated at the anode and OH− generated at the cathode. Unlike the membranes used within ED systems, the ceramic membrane allows transport of both cations and anions, so Cl− ions move across the membrane towards the anode and Na+ moves across the membrane towards the cathode, thus producing HCl in the inner anode chamber and NaOH in the outer cathode chamber. The NaOH solution generated via electrolysis was subsequently bubbled gaseous CO2, forming aqueous NaHCO3. This eliminated the need to transport bicarbonate/carbonate ions across the membrane interface and was achieved at ambient temperature and pressure. It should be noted that in this study, the ultimate end product was CaCO3, which was produced by adding CaCl2 to the NaHCO3 solution. By using NaHCO3 as an intermediate product and adding supplemental Ca, their system surpassed the carbon capture capacity of forming CaCO3 directly from seawater, while still taking advantage of the solubility reduced solubility of CaCO3 compared to NaHCO3 and NaCO3. The precipitation of CaCO3 was also achieved without increasing temperature or pressure, which can be major energetic demands in conventional CaCO3 production processes. However, the actual amount of CO2 captured was not quantified, so an objective comparison of the energy savings of this process cannot be made at this time. Still, the system developed by Park et al. demonstrates how the chemistry of a saline water can be manipulated using electrochemical approaches to enhance the uptake of CO2 and facilitate more thermodynamically favourable formation of the desired carbonate products. The extent to which such a system can be considered as an RCC process depends largely on the net carbon footprint of the process, and whether it can be applied to capture carbon from non-concentrated CO2 sources.
Conversion of aqueous carbonates to organic products was also demonstrated by Zhang et al., who modified a membrane electrolyzer to allow for the generation of formates (KHCO2 and NaHCO2) from KHCO3 and NaHCO3 solutions with 97% and 93% carbon conversion efficiency, respectively.15 The ability of their system to achieve this efficiency relied upon maintaining bicarbonate as the dominant carbonate species, which means that careful control of pH was required. The bicarbonate reacts with H+ generated by the anode and passes across a CEM to form CO2* within the cathode compartment, where it can then be reduced on the cathode surface to produce formate. They were able to achieve this through the use of a near-neutral anolyte solution, reactor design, and control of CO2 partial pressure in the reactor headspace.15 Importantly, the presence of Cl−, SO42−, and I− were shown to have no impact on the conversion efficiency of this system when present at concentrations equal to or less than the concentration of bicarbonate.15 However, further testing with solutions containing concentrations of these anions that are more representative of the composition of saline waters (where non-bicarbonate anions typically dominate) (e.g., those tested in the study by Nakata et al.14) are needed to further demonstrate the applicability of this technology for saline-based RCC. Discussion of this type of system often involves using a hydroxide solution to dissolve carbonates (Sullivan et al.) but does not thoroughly investigate the use of saline waters as the carbon capture medium.
Another system similar to the one developed by Zhang et al., but which generates CO rather than formate from an aqueous bicarbonate stream, was recently demonstrated Li et al.88 CO is primarily used as a feedstock to generate organic chemicals,89 and for that reason we chose to include discussion of this system in this section. In their electrolyzer, a bipolar membrane was oriented so that H+ are generated within the compartment being fed a bicarbonate solution. As in the system demonstrated by Zhang et al., this leads to the generation of CO2*. This compartment is bound on the other side by an Ag-coated carbon support that catalyses the conversion of CO2 to CO (rather than allowing the CO2 to reach the cathode where it could be converted to formate, similar to Zhang et al.'s system). While the researchers highlight the ability to generate CO as the major benefit of this system,88 it should be noted that this system was only able to achieve a maximum of 83% carbon conversion (lower than that demonstrated by Zhang et al.). An additional shortcoming of this system is that it has only been demonstrated with a KHCO3 solution, and thus further study is needed to determine if this can be applied for RCC from real saline waters. However, the flexibility of the system developed by Zhang et al. to operate with a sodium-based solution and with other anions present indicates that there may be potential for the system developed by Li et al. to be applied for solutions more representative to real saline waters.
Reactive carbon capture using pre-existing technologies
While current research on saline based RCC is focused on optimizing the amount of carbon capture, many processes using similar chemistries (i.e. carbonate solid precipitation) have long been employed in a variety of industries but were optimized for metrics other than carbon capture. In some instances, the processes are designed specifically to produce a desired carbonate product (such as the Solvay process), whereas in other instances the production of valuable carbonates is a byproduct of another process (such as water softening). Understanding these preexisting processes for converting salinity into valuable carbonates provides important insight into not only the chemistry of carbonate solid production, but also into the implementation of such processes at larger scales than much of the current CCU research.| NH3(g) + NaClaq + CO2(g) → NH4Cl + NaHCO3(s) |
At least one quarter (though likely much more) of the world's soda ash is produced using processes similar to the Solvay process, accounting for at least 14 million metric tonnes annually91 (much of the remainder is mined from natural deposits). Though originally developed as a purely production process, growing interest in carbon sequestration has led to the study of this process for carbon sequestration purposes. In 2019, Palitsakun et al. demonstrated a Solvay process utilizing synthetic brines at concentrations intended to model seawater and desalination brine, and demonstrated up to 100% capture of a pure CO2 stream using ammonia gas as the reaction intermediate.63 The Solvay process has been demonstrated using various CO2 sources, including diesel exhaust (4.8% CO2) and flue gas from a natural gas power plant (10% CO2), capturing 99 and 98% of the CO2, respectively,92 indicating the robustness of the process for CO2 capture from a variety of sources. While the overall reaction chemistry of the Solvay process involves the precipitation of a carbonate solid from a saline solution, the conventional Solvay process has an associated carbon footprint of 1.61–2.29 tonnes CO2 emitted/ton NaHCO3 produced,4 so it cannot be classified as a saline water-based RCC process. The carbon footprint of the Solvay process is largely associated with the energy intensive catalyst and solid recovery steps,93 which typically require heat and result in 1.48–2.04 tonnes of indirect CO2 emissions per tonne NaHCO3 produced.4 However, the increased interest in carbon capture and utilization processes has motivated recent studies of modifications to the Solvay process, which can decrease the energy demand and thus make it viable as a CCU process.
A common approach to modifying the Solvay process is to replace ammonia as a catalyst, which can reduce the energy demand associated with its regeneration. For instance, El Naas et al. demonstrated a modification of the Solvay process which used lime (CaO) instead of ammonia to maintain the pH required for NaHCO3 to precipitate.
| CaO(s) + H2O → Ca(OH)2(aq) |
| Ca(OH)2(aq) + 2NaCl(aq) + 2CO2(g) → CaCl2 + 2NaHCO3(s) |
This process produced NaHCO3 at a 30% energy savings compared to the conventional Solvay process, half of which was due to the elimination of the ammonia recovery step.93 It should be noted that lime can have a high associated carbon footprint when produced using conventional methods (calcining CaCO3), and while the authors did not assess the carbon footprint of their process versus the conventional Solvay process, it is possible that some of the reduction in carbon footprint from the lower energy demand may be offset by carbon footprint of the process chemicals.
Precipitative water softening processes often use a pH shift toward basic conditions to shift the carbonate equilibrium towards CO32− formation, improving the kinetics of CaCO3 precipitation. The pH shift can be induced by adding chemicals, such as NaOH, or through electrochemical generation of alkalinity. While the latter approach has the benefit of reducing the chemical demands of the softening system, quenching of OH− alkalinity produced at the cathode in electrochemical systems by H+ ions produced by the anode can significantly reduce the efficiency of electrochemical softening processes. To that end, Ba et al.94 developed an integrated electrolysis-microfiltration-ion exchange (IEMI) system for water softening, which features a porous, tubular anode oriented such that protons generated at its surface can be drawn through the anode and out of the electrolytic cell, preventing quenching of alkalinity generated in cell. This novel system design allowed for higher pH to be reached in the electrolytic cell (effluent pH 11.9) than in conventional electrolytic cells (effluent pH 11) using ion exchange membranes to separate the OH− and H+. The electrolytic cell with the porous anode achieved production of CaCO3 (1.9 kW h kg−1 CaCO3), which was as or more efficient than a conventional (CEM separated) electrolytic cell (1.4–11.2 kW h kg−1 CaCO3) with the same current density (18 mA cm−2) applied and hardness removal efficiency achieved (∼65%) in each cell. In the system developed by Ba et al., the majority of the calcium hardness was removed in a crystallizer fitted with a microfilter, with the ion exchange column receiving the filtrate from the crystallizer used to primarily remove remaining magnesium hardness. While the addition of the ion exchange column helped improve the overall hardness removal of the IEMI system to above 90%,94 it bears less relevance to reactive carbon capture objectives. However, the novel use of a porous anode to abstract protons from the electrolytic cell, enabling more energy efficient production of CaCO3 is directly applicable to electrolytic systems designed to capture carbonate from saline waters and produce CaCO3. While the CO2 sequestration capacity of the system was not directly analyzed or optimized in the study by Ba et al., they did investigate the impacts of increasing the HCO3− concentration in the synthetic brine fed to the electrolytic cell, and found that the removal of calcium hardness increased with increasing HCO3− alkalinity from 100 to 400 mg L−1 as CaCO3.94
Management of precipitation softening sludges includes collecting and dewatering the precipitates. During the initial collection of precipitates, the solids will act as a slurry due to high water content, whereas after dewatering they must be handled as a sludge. Solids may initially be concentrated by methods such as gravity thickening, followed by dewatering using either drying beds or mechanical dewatering such as centrifugation or filtration, sometimes followed by further volume reduction through pelletization or recalcination, before offtake of the solids.59 While methods like recalcination which re-release the CO2 from the precipitate are not appropriate for CCUS purposes, many of the processes used in management of softening sludges can be applied to water based CCUS.
At water treatment facilities, precipitated solids are usually initially separated from water by gravity. Clarifiers settle solids formed during water softening to produce a slurry with 15–20% (wt/wt) solids content, which can be further dewatered in thickeners to reach solids contents of 25–30%.95 A major parameter for clarifier design is the solids settling velocity, as the clarifier area must be large enough to maintain an overflow rate below the settling velocity in order to retain the solids.96 The composition of solids formed during carbonate precipitation can significantly impact the settling velocity and thus the footprint required for solids management. For instance, the Ca:Mg ratio in softening sludges is correlated to their dewaterability during settling, with high ratios (indicative of less Mg, often in the form of Mg(OH)2) indicating improved settleability. Interestingly, supplementary CO2 can re-dissolve the Mg in settled sludge, providing as much as a 3-fold reduction in area required to settle the sludge,97 and providing an additional opportunity to sequester carbon (as bicarbonate ions). Other lightweight components within softening or coagulation sludges, such as aluminum hydroxide flocs and organics, also reduce settleability.95 The settleability of solids formed in water based CCUS processes using gravity separation methods is a key design parameter and should be carefully considered. As such, the extensive knowledge base on solids settleability and gravity settler design within the water/wastewater treatment (W/WWT) industry can be applied to aid CCUS efforts.
An additional design parameter for solids management is the desired end solids content. It is known that higher solids content must be achieved for handleability of softening sludges (>50%), though the exact solids content required for handleability varies and often must be determined empirically.97 As such, water treatment facilities usually employ an additional dewatering step after gravity separation to produce solids which can be easily transported – saline water-based RCC processes which produce carbonate solids would likely also need to employ additional dewatering, if these solids are meant to be used by another entity or buried. Examples of such dewatering steps include filter presses,98 centrifuges,95 and thermal drying.99 The energy demand for solids dewatering should be a major consideration in the design of RCC processes. The carbon footprint of the energy required to dewater solids generated from water softening can effectively cancel out the carbon capture achieved by generating the solids.80
Another important but easily overlooked consideration for the dewatering of precipitated carbonate solids is the management of the effluents produced during dewatering. Many water and wastewater treatment facilities dispose of the effluents by discharge or by returning them to the plant's headworks.95 However, this option may not be viable for saline water-based RCC processes which are not co-located with a W/WWT facility. Additionally, the composition of these effluents can vary depending on the carbonate precipitation method and the chemical composition of the influent water source, which may prohibit direct discharge of the effluents in cases where they contain elevated concentrations of regulated pollutants. Additionally, effluents from dewatering of lime softening sludge can have elevated pH and calcium concentration – as high as pH 12.24 and 380 mg L−1 Ca95 – due to dissolution of unreacted lime from the softening sludge. For RCC purposes, ensuring maximal conversion of available calcium to carbonate solids is critical for efficient sequestration, so steps to further react calcium in dewater effluents may be employed.
Summary of recent studies of potential saline water-based RCC technologies
Table 3 summarizes each of the RCC processes reviewed in the section of this review focused on CO2 conversion processes and products. Qualitative features, including the technology type, saline water source, CO2 source, products, and concurrent processes, as well as quantitative features including CO2 capture and energy consumption are reported as they were discussed in each respective article. The technology stage, benefits, and limitations are based upon information within the reports as well as assessment by the authors of this review.| Paper | Technology | Saline water source | CO2 source | CO2 captured | Energy demand | Products | Concurrent processes | Technology Stage | Benefits | Limitations |
|---|---|---|---|---|---|---|---|---|---|---|
| Dara et al. 201762 | Electrodialysis | Synthetic (1 M NaCl) | Pure CO2 gas, dissolved in DI water | Not reported | Not reported | HCl NaHCO3 | Desalination | Proof of concept | Simultaneous CO2 capture, water treatment, a chemical production | Current limited by low conductivity of carbonic acid stream (attributed to low rate of diffusion of carbonate species in the stream and low mobility of the species) |
| Dara et al. 2019 | Gas-Fed electrodialysis | Synthetic (1 M NaCl) | CO2 gas (0–50% in O2 gas) | Not reported | Not reported | HCl NaHCO4 | Desalination | Proof of concept | Utilization of sodium (more abundant) for RCC | Low conductivity of carbonate stream potentially elevates energy demand/slows reaction |
| Zhao et al. 202065 | BMED with crystallizer | Synthetic seawater | CO2 gas, dissolved in alkali water | Not reported | 0.89 kW h kg−1 CO2 | CaCO3 | Seawater softening | Bench scale | Prevention of membrane fouling | Utilization of divalent cation (Ca) for carbon capture requires alkalinity addition for net CO2 capture |
| Nakata et al.14 | Electrolysis with BDD cathode | Seawater and NaCl solution | CO2 gas dissolved in solution | Not reported | Not reported | Formaldehyde formic acid | None | Proof of concept | Chemical production, reduced H2 generation | Lower efficiency due to impurities in seawater, lower solubility of CO2 in seawater vs methanol |
| Islam et al.76 | Sonication | Synthetic seawater and NaCl Solution | CO2 gas and Flue gas | Not reported | Not reported | Methane, ethane, ethylene | None | Proof of concept | Organic chemical production | Additional purification steps needed for product recovery |
| Dindi et al.82 | Amine-catalyzed precipitation | Synthetic desalination brine | Synthetic flue gas (15% CO2) | Not reported | Not reported | NaHCO3 | Desalination | Proof of concept/bench scale | Utilization of sodium (more abundant) for RCC | Use of lime, which can have a large carbon footprint when produced by calcining limestone |
| Soong et al.66 | Carbonate precipitation | Oriskany sandstone aquifer brine | Pure CO2 gas | 0.031–0.273 mol CO2 per h | Not reported | CaCO3 | None | Proof of concept | Use of waste products (produced water brine and fly ash) | Not all available Ca consumed NaCl impurity in solids collected from 2 step method solids in 1 step method in slurry with fly ash, inhibiting recovery for beneficial use |
| Mustafa et al.61 | Electrodialysis | Synthetic (NaCl solutions) | Synthetic (NaCO3 solution) | 1.9 mol CO2 per h | 11.2 kW h kg−1 CO2 | HCl NaHCO3 Na2CO3 | Desalination | Bench scale | Use of waste products (alkaline and NaCl brines) | Optimal conditions for CO2 removal differ from optimal conditions for brine desalination |
| Power et al.31 | Enhanced precipitation | Synthetic (NaHCO3/MgCl solution) | Synthetic (NaHCO3/MgCl solution) | Not reported | Not reported | MgCO3 | None | Proof of concept | Room temperature magnesite formation | Long reaction time (60 days), Utilization of divalent cation (Mg) for carbon capture requires alkalinity addition for net CO2 capture |
| Taniguchi et al.100 | Electrodialysis | Synthetic | Flue gas | 9900 kmol h−1 CO2 (proposed) | 0.225 (ED), 0.525 (overall) kW h kg−1 CO2 | CO2 gas | None | Described systems | Low energy consumption | CO2 absorption capacity of potassium solution likely higher than natural saline waters; energy demand is theoretical, and system has not been demonstrated |
| Lannoy et al.12 | Bipolar membrane electrodialysis | Seawater | DIC in seawater | 20 kmol CO2 per h (proposed) | 2.72-3.85 kW h kg−1 CO2 | CaCO3 or CO2 gas | None | Described prototype based on bench scale | System flexibility to generate solid or gaseous process depending on BPMED product used | Utilization of divalent cation (Ca) for carbon capture requires alkalinity addition for net CO2 capture |
| Ba et al.94 | Integrated electrolysis microfiltration ion exchange system | Synthetic industrial waste water | HCO3− alkalinity in water source | Not reported | Not reported | CaCO3 | Water softening | Bench scale | Use of membrane-less ED system with tubular electrode reduces capital and O&M costs associated with fragile IEMs, makes ED system tolerant to higher salinity influent | Demonstrated for low salinity influent which limits CO2 capture potential, utilization of divalent cation (Ca) for carbon capture requires alkalinity addition for net CO2 capture |
| el Naas et al.93 | Modified solvay process | Multi-stage flash desalination brine | Synthetic flue gas (10% CO2) | 1.25 mol CO2 per h | 0.773 kW h kg−1 CO2 | NaHCO3, CaCl | None | Bench scale | Eliminates use of ammonia in Solvay process, reducing associated energy demand for separation and regeneration | Use of lime, which can have a large carbon footprint when produced by calcining limestone; utilization of divalent cations (Ca,Mg) for carbon capture requires alkalinity addition for net CO2 capture |
| Xie et al.17 | Membrane electrolysis | Synthetic seawater | Pure CO2 gas | 0.078 mol CO2 per h | 1.22 kWh per kg CO2 | MgCO3, CaCO3, HCl | HCl generation | Bench scale | Separation of magnesium and calcium solids, oxidation of H2 at the anode rather than Cl | Involves H2 gas management (produced at cathode, supplied to anode); utilization of divalent cations (Ca,Mg) for carbon capture requires alkalinity addition for net CO2 capture |
| Palitsakun et al.63 | Modified solvay process | Synthetic brine (NaCl) | Pure CO2 gas | 2.5 mol CO2 per h | Not reported | NaHCO3, Na2CO3 | None | Bench scale | Ammonia is less expensive and toxic than other amine catalysts | Incomplete recovery of ammonia, solid composition mixed between NaHCO3 and Na2CO3 |
| Zhang et al.67 | Continuous plug flow reactor with nickel nanoparticle catalyst | Synthetic desalination brine | Pure CO2 gas | 0.036 mol CO2 per h | Not reported | CaCO3, MgCO3 (various) | None | Bench scale | Carbonic acid formation accelerated by nickel NPs | Separation of catalyst and precipitate not discussed, toxicity of nickel nanoparticle; utilization of divalent cations (Ca,Mg) for carbon capture requires alkalinity addition for net CO2 capture |
| Bang et al.79 | Precipitation with microbubbles | RO desalination brine | Pure CO2 Gas | Not reported | Not reported | CaCO3, MgCO3 (various) | None | Proof of concept | Formation of both magnesium and calcium carbonates | Significant chemical consumption (NaOH); Utilization of divalent cations (Ca,Mg) for carbon capture requires alkalinity addition for net CO2 capture |
| Chaalal et al.92 | Modified solvay process | Ammonated seawater | 10% CO2 in methane; diesel exhaust (4.8% CO2) | 0.162 mol CO2 per h | Not reported | NaHCO3, Na2CO3 | Brine desalination | Bench scale | Applicable to multiple CO2 containing gas streams | Ammonia regeneration can be energy/cost intensive |
| Park et al.64 | Membrane electrolysis | Synthetic brine (NaCl) | Pure CO2 Gas | Not reported | Not reported | CaCO3, HCl | None | Proof of concept | More rapid formation of CaCO3 compared to natural processes at room temp/pressure | Requires addition of carbonate forming species; utilization of divalent cations (Ca) for carbon capture requires alkalinity addition for net CO2 capture |
| De Vito et al.78 | Enhanced precipitation | Synthetic industrial waste water (MgCl) | Pure CO2 gas | 0.002–1.64 mol CO2 per h | Not reported | MgCO3*3H2O, MgCO3*5H2O | None | Pilot scale | Formation of more readily precipitated magnesium carbonates at room temperature, pressure | Only demonstrated with pure MgCl |
| Zhang et al.15 | Membrane electrolysis | KHCO3 and NaHCO3 solutions | KHCO3 and NaHCO3 solutions | Not reported | Not reported | Formate solids | None | Bench scale | Demonstrated with K and Na, and performance sustained in presence of other anions | Careful gas and pH management required |
| Li et al.88 | Bipolar membrane electrolysis | KHCO3 solutions | KHCO3 solutions | Not reported | Not reported | CO | None | Bench scale | Generation of CO feedstock for organic products | Only demonstrated with pure KHCO3 |
Products of saline water-based RCC
Here, we evaluate the products that are formed by current saline-based processes, as well as discuss a selection of other products, which while not currently generated at scale, have been or could be feasibility produced using a saline-based RCC process. To evaluate each product, we will consider the advantages posed by replacing conventional production methods with RCC processes, current and prospective demand for the product, and the carbon footprint of use cases for each product. It is important to consider whether the end use of the solids generated during RCC will re-release the captured carbon in evaluating the life cycle impact of the RCC process. While end uses which re-release the CO2 generally increase the carbon footprint, in instances where RCC-generated chemicals can replace chemicals generated using more carbon-intensive processes, the overall carbon footprint of the end use can still be reduced.When assessing the value of potential products of saline water-based RCC, average market prices of the various products are considered. However, there is likely to be a trade-off between the purity of products from RCC, the energy required, and the market price of different product purities. Each RCC process should be individually evaluated to determine the product purity which is the most cost- and carbon- effective.
The economic benefit of generating valuable products rather than simply sequestering carbon is understood to be a major driver of this trend toward CCU processes. Reactive carbon capture processes fall into the classification of carbon capture and utilization (CCU) technologies – processes that convert the captured carbon into a valuable product. The volume of carbon capture by CCU technologies, currently between 10–15 MT CO2 per year, exceeds the volume of carbon capture by technologies solely developed to sequester carbon (CCS), and is continuing to grow at a more rapid rate compared to CCS.75 However, not all CCU processes generate the same products – for instance, some non-saline based RCC processes use biological mechanisms to generate complex organic products – so it is essential to understand the value of the products generated by each respective process when comparing CCU approaches and evaluating their potential for large-scale implementation.
Carbonate solids
The majority of saline water-based RCC processes generate inorganic products, often in the form of carbonate solids. Once carbonate solids have been collected and dewatered, they can be sold as valuable chemical products. A wide range of carbonate solids can be generated, depending on which cation(s) are present in the saline water source and the method of carbonate formation employed.Generally, solids can be produced at a larger volume with lower quality, or at a smaller volume with higher quality (i.e. more pure).97 Both methods can be employed for saline-water based RCC. The advent of ion-selective aqueous separation processes, such as ion-selective electrodialysis or adsorption, creates the potential to extract the more valuable elements from saline streams to produce valuable, high purity products. However, higher energy demand and/or lower production volume of high purity products can reduce the carbon capture potential of such RCC processes. Potential trade-offs between the value of the generated products and the amount of CO2 captured create a compelling case for integrated RCC processes capable of utilizing the dominant cations in saline waters (Na, Ca, Mg) to provide the majority of carbon capture, while utilizing other cations (i.e. Li) to produce valuable end products. Thus, we will include a selection of carbonate solids produced from cations which are present low levels in saline water in our analysis below, in the instances where these carbonates have compelling value.
Many of the solids which can potentially be produced using saline-based RCC have end uses which may result in the re-release of the CO2 captured in the solids. In these instances, carbon sequestration is not achieved, but net emissions reductions are still possible when these products are used in lieu of carbonates produced from carbon emitting processes.
Currently, these carbonates are produced by mining geological deposits or are extracted from saline brines.95 Much of NaHCO3 and Na2CO3 production currently utilizes the Solvay process, which combines NaCl-rich brines with an ammonia catalyst and a CO2 source to generate NaHCO3, which can be converted to Na2CO3via heating.2 This process can be CO2-intensive due to the energy demand for heating and catalyst recovery (see previous discussion of Solvay process).
The end use of NaHCO3 and Na2CO3 determines whether RCC processes that produce these minerals are net carbon negative or simply carbon neutral. Na2CO3 is primarily used as a feedstock within the chemical industry, and is used in chemical processes in various other industries, whereas NaHCO3 is primarily produced as a consumer good.96 In either case, the solid is typically dissolved and/or reacted, which means the carbon is not permanently sequestered. Still, replacing Na2CO3 and NaHCO3 produced by conventional processes with large carbon footprints with Na2CO3 or NaHCO3 produced by RCC can reduce the overall carbon footprint of these end uses.
The global production of high-grade CaCO3 was 114 MT per year as of 2013, and the price ranges between $30–350 per metric tonne.75 It is estimated that by using carbon capture methods to generate CaCO3 to meet this demand, 50 MT per year of CO2 could be captured and utilized.75 However, much larger amounts of lower-grade calcium carbonate minerals, such as limestone and dolomite (CaMgCO3, see discussion below on complex inorganic carbonates), are produced and consumed annually. Over 54 and 901 MT per year of dolomite and limestone, respectively, are consumed in the United States alone,101 with similar rates of consumption worldwide. These minerals are much less valuable than high purity CaCO3, with prices just above $10 per metric tonne in 2018.98 However, as annual consumption of these minerals is significantly greater than of high-grade CaCO3, using RCC processes to meet the demand for calcium carbonate minerals may significantly increase the amount of carbon capture and utilization as compared to the carbon capture potential for high-grade CaCO3 production alone.
In assessing the potential demand for CaCO3 produced from RCC processes as a replacement for CaCO3 generated from conventional mineral extraction processes, the end use case must be considered. For instance, based on reported data on limestone and dolomite consumption by use by the USGS,101 it can be estimated that at least 12% of limestone and 6% of dolomite is used as coarse crushed aggregate stone, a product that cannot easily be generated from the CaCO3 produced by saline-based RCC processes. However, other processes may be more suited to utilize CaCO3 generated from saline-based RCC. For instance, cement manufacturing accounts for 10% of limestone consumption in the U.S.,101 and there is evidence that CaCO3 generated from precipitative carbon capture processes can be substituted for limestone in cement production.102,103 This use case is particularly attractive as it provides a stable medium for long-term CO2 storage. Other processes such as lime production, which accounts for roughly 4% of limestone consumption,101 re-release the CO2 from the calcium carbonate, making them unsuitable for carbon sequestration efforts.
| CaCO3 → CaO + CO2 |
However, if the CaCO3 used in these processes is replaced with RCC-generated CaCO3, new CO2 emissions can be prevented, effectively “closing the loop” for carbon in the process.
Current examples of how lower-grade carbonate solids are valorised can be found in the management of softening-sludges generated in the water treatment industry. Many municipalities generate large volumes of carbonate minerals as a byproduct of lime softening, and then sell the carbonate-rich sludges to offset sludge management costs.98 As municipalities often produce more softening sludge than there is demand for lime within their region (the cost of transporting softening sludges long distances to meet additional demand for lime production can become prohibitive), other applications such as flue gas SOx scrubbing and wastewater pH control have previously been investigated.104 While the dissolution of carbonates into acidic wastewaters does not directly lead to release of CO2 gas, this approach may not be viewed as suitable for long-term, permanent CO2 storage. The use of lime sludges for flue gas scrubbing does result in release of CO2 gas,104 again making this disposal approach unsuitable for long term sequestration.
| SO2 + CaCO3 → CaSO3 + CO2 |
Again, in instances where RCC-generated CaCO3 is used in in flue gas scrubbing in place of CaCO3 generated using more carbon intensive methods, these applications may still result in net reductions in CO2 emissions.
Though the production of MgCO3 can require more process steps than for Na2CO3 or CaCO3, it's chemical value and relative abundance still make it an attractive product for RCC. Saline-based RCC processes may prove advantageous over other carbon capture technologies which generate MgCO3 as it avoids the energy costs associated with converting magnesium to a form with readily reacts with CO2. Some carbon-capture processes generate MgCO3 by crushing and then dissolved Mg-rich minerals, but these two steps are associated with a high energy demand, corresponding to a net production of 1.3 kg CO2eq generated for every kg of CO2 consumed in MgCO3 production.104 Saline based processes utilize Mg already dissolved in the source water, so the energy expense of solubilizing the minerals is effectively eliminated. Many other magnesium compounds, such as MgO (magnesia) and Mg(OH)2, are already produced from aqueous resources to avoid the costs of mineral processing.106
Similar to CaCO3, the end use of MgCO3 informs the overall carbon capture potential of RCC processes. For instance, MgCO3 is used to produce magnesia, but this process releases the CO2 from the mineral.106
| MgCO3 → MgO + CO2 |
As with lime production, the release of CO2 prevents RCC processes with this end use from being truly carbon negative but enables carbon neutrality by preventing release of CO2 from newly extracted MgCO3.
000 times less than the concentration of Na.28 However, geographic scarcity of lithium and the increasing demand for lithium carbonate for battery production42 makes this carbonate significantly more valuable than other carbonate products of saline-based RCC. Therefore, we include discussion of Li2CO3 as a potential RCC product, as producing even a small quantity of this carbonate can significantly impact the economics of RCC.
While Li2CO3 is predominantly used for battery applications, it is also used for glassmaking and other industrial processes,108 as well as for medical applications.109 In 2018, the worldwide production of Li2CO3 was over 95000 metric tons per year.42 The price of lithium carbonate has generally increased over the past decade, and was $37000 per metric tonne in 2023.44
As with other carbonates, it is vital to compare Li2CO3 production from RCC to current/conventional production method. Lithium carbonate is typically produced by adding soda ash (Na2CO3) to Li-rich brines, or by reacting concentrate generated from lithium-rich ores with Na2CO3 and CaCO3.110 A thorough TEA-LCA of the production of lithium carbonate via both conventional methods has been completed by Kelly et al., finding that brine production has a significantly lower carbon footprint than ore production of Li2CO3 (3 vs. 20 tonnes CO2eq/ton Li2CO3, respectively). They also found that for brine-based production, the soda ash was the most significant contributor to the carbon footprint for Li2CO3 production, while for ore-based production, the energy demand was the most significant contributor.110 These findings have multiple important implications for RCC processes. First, saline water-based RCC processes for Li2CO3 production would be similar to brine-based processes, and thus are likely to also have a lower carbon footprint than ore-based processes (depending on the energy consumption needed to concentrate the saline water source). Second, as previously discussed, soda ash can be generated using RCC processes, so further reductions of the overall carbon footprint of Li2CO3 production may be possible.
The end use of lithium carbonate also informs the carbon footprint of RCC processes generating Li2CO3. The primary use of Li2CO3 is for batteries – more specifically, for generation of cathode materials. Li2CO3 is directly used to generate cathode materials such as lithium (manganese and/or nickel and/or cobalt) oxides, releasing the CO2 stored in the solid in the process.110 It should be noted that the electrolyte in most lithium ion batteries consists of a non-carbonate lithium salt dissolved in an organic carbonate solvent,111 so Li2CO3 is only sometimes used indirectly for the generation of the non-carbonate salt, a process that would also release the CO2.112 Thus, as with other end uses that release the CO2 from the carbonate product, RCC-generated Li2CO3 can only provide CO2 emission reductions when used in lieu of Li2CO3 generated using conventional methods.
While no accurate assessment of global SrCO3 consumption could be found at the time of this review, the demand can be indirectly estimated using global celestite production as a proxy, as the majority of celestite is processed to produce SrCO3.118 Global celestite production was 219000 metric tonnes in 2018,118 roughly equating to 104000 tonnes of strontium. The average price of strontium carbonate was just below $900 per metric tonne in 2018.118
Strontium is less abundant in many saline waters compared to cations Na, Ca, and Mg, but at almost 8 mg L−1 in seawater,28 it is more abundant than most other cations. The value of SrCO3 is higher than carbonates produced using the more abundant cations, but like barium, the value may not be sufficiently high to justify targeted recovery, as the relatively low concentration of Sr limits the total CO2 capture capacity of saline water-based RCC processes producing this mineral.
Organic products
Few saline water-based processes reported at the time of this review can generate organic products. However, as many organic chemicals are currently produced from fossil fuels, which typically have high associated carbon footprints, alternative methods of producing hydrocarbons are highly compelling. Current approaches can produce simple hydrocarbons including formaldehyde and formic acid, which can be used as feedstocks for the generation of more complex and valuable hydrocarbons.119 The value ($ per ton) of the organic products reviewed here is greater than the value of many of the carbonate salts which can be produced from the most abundant cations in saline waters (NaHCO3/Na2CO3, MgCO3, CaCO3), and the global demand is on a similar scale to the demand for aforementioned inorganic products. However, the costs associated with producing these organic products using saline water may be greater than the costs of producing these organic products from fossil fuel sources, which would reduce the viability of RCC processes generating these simple organic products.000 tonnes per year,121 and the price of formic acid was $700–800 per metric tonne as of 2014.120 (It should be noted that the production capacity is determined by the capacity of formic acid production facilities and thus is likely an overestimate of the demand but provides a scale of reference for demand nonetheless).
000 bcf in 2022.123 These hydrocarbons are primarily produced either directly or as a byproduct of fossil fuel extraction; methane can also be produced through biological processes.122 When methane is used as a fuel, CO2 and any un-combusted methane (which is an even more potent greenhouse gas) are released to the atmosphere, so as with other RCC products, replacing conventional production with RCC only serves to reduce the carbon footprint rather than offer long term storage. As biological production of methane is already a widely used lower-carbon method,122 there may be little incentive to use RCC processes to generate this product.
Summary of product values and market sizes
The product values, market size, and market value of each product is summarized in Fig. 4. The products formed using the cations that are most abundant in most saline waters (Na, Ca, and Mg) are the least valuable, while LiCO3 is significantly more valuable than other products. However, the market size for both CaCO3 and Na2CO3 are much larger than the other products (with the exception of formaldehyde). As a result, the market value of CaCO3 and Na2CO3 outweighs the value of products from less abundant cations in seawater. The high demand for these products makes saline-based RCC processes more economically attractive, so long as the solids can be produced at a low cost (due to the low value of the products). Additional techno-economic analysis of saline water-based RCC processes could be valuable. As previously noted, some of these processes can produce multiple mineral carbonate products. Additionally, there could be the potential to combine processes that are used to generate different mineral carbonate products. Saline-based RCC processes that generate multiple mineral carbonates could allow capitalization on both the large market size of products such as CaCO3 and Na2CO3, as well as the high value of products like Li2CO3. However, little analysis of systems producing multiple mineral carbonates can be found at present. Such analysis could provide insights into the feasibility of saline-based RCC processes. It may also highlight the potential benefits of using real saline waters, which contain multiple carbonate-forming cationic species, as feedstocks for RCC.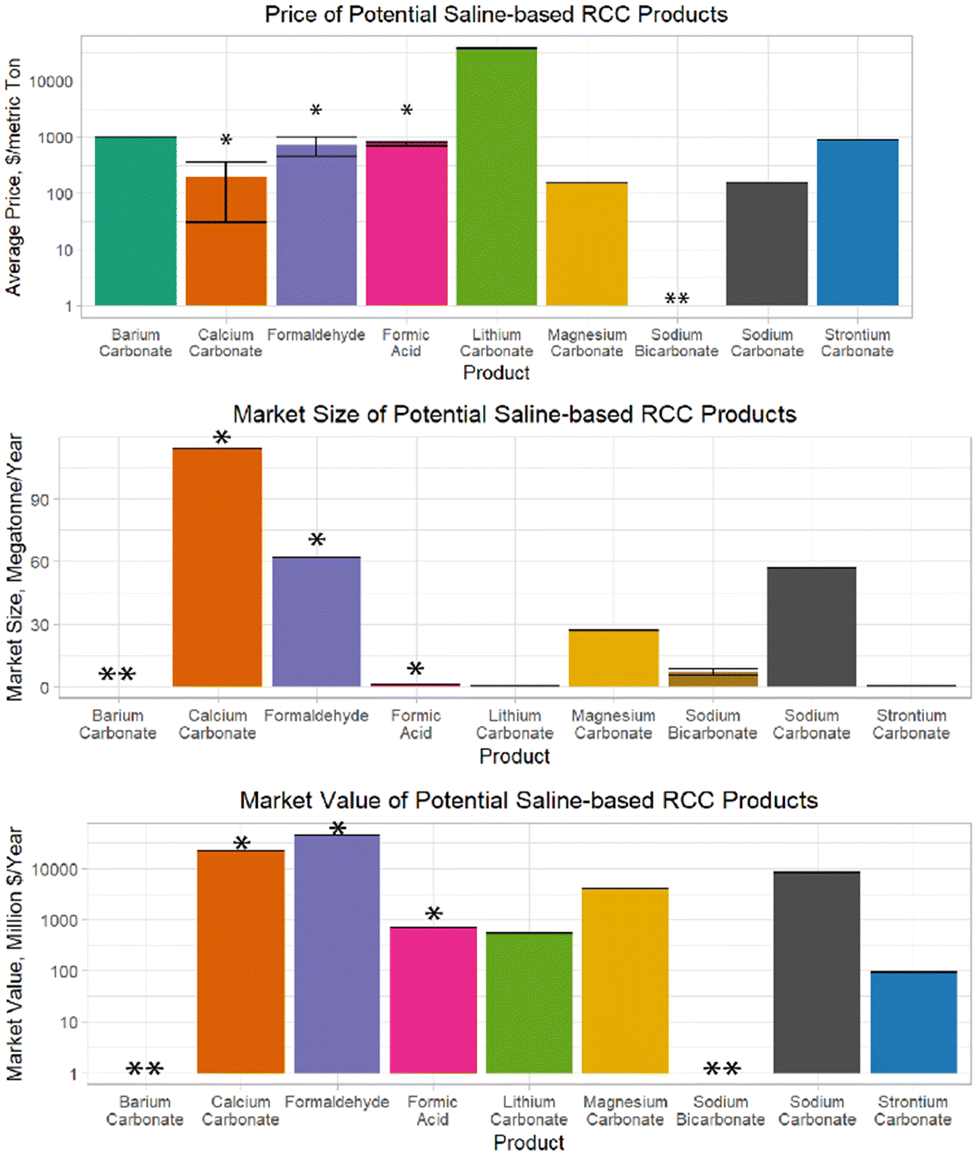 | ||
| Fig. 4 The price per tonne, market size (megatonnes per year), and market value (million $ per year) of potential products of saline-based RCC. Note that all values are from 2018, unless indicated with a single asterisk (CaCO3 data from 2013, formaldehyde from 2019, and formic Acid from 2014). Double asterisks indicate missing data. Note that price and market value are plotted on a logarithmic scale. | ||
It is also important to contextualize the market size of the potential products of saline-based RCC. A thorough techno-economic analysis by Shokrollahi et al. compared 34 CCU products and ranked CaCO3 product as the most favorable product in terms of environmental impact and immediate applicability, and second most favorable in terms of economic potential (Syngas produced from CO2 ranked first in this category.).124 (Note – this analysis only considered Mg, Ca, and Na carbonates). This highlights the outsize potential of saline-based RCC processes capable of producing CaCO3 within the field of carbon utilization.
Costs of saline-water based RCC
One of the primary purposes of saline water based RCC processes is to offset the cost of carbon capture with the value of the generated products. The cost of carbon capture varies widely between different technologies both in terms of capital and operation and maintenance (O&M) costs. Major differences in O&M costs emerge due to energy and chemical demands of the different processes, the saline water source used, and any associated waste handling. The overall cost of different RCC processes is also impacted by the value of the products generated – a process that has greater energy or chemical cost but produces a significantly more valuable product may have a lower overall cost per tonne of carbon captured than processes that are more energy or chemical efficient, but that generate less valuable products.At the time of this review, few technologies reviewed here have been analyzed to determine the cost per tonne of CO2 captured. Processes using electrolytically generated alkalinity to induce CaCO3 precipitation, such as that proposed by de Lannoy et al., are estimated to cost $300–600 per ton net CO2 captured.12 This value reflects the cost per net capture of CO2, which was determined using technoeconomic/lifecycle analyses that considered factors such as the energy demand and associated carbon footprint.125 However, many emerging CO2 capture technologies that are actively being commercialized are reporting roadmaps to an eventual cost <$100 per ton net CO2 captured, the threshold considered as commercially viable.126 This value does not include any offsets to costs from the sale of the CaCO3 generated in the process. However, at a market price of between $30–350 per ton CaCO3 (equivalent to $13–150 per ton CO2), such processes may not be profitable.
A techno-economic review of CCUS technologies published in 2022 noted that systems that convert CO2 to chemical products such as mineral carbonates are still in the development stage (technical readiness level 6).127 In addition, the quantification of CO2 capture costs and the potential of CCU technologies (which include saline-based RCC) is uniquely complicated amongst CCUS technologies as it requires analysis of complex market dynamics.127 The high energy intensity of many CCU processes has been implicated in limiting the implementation of these technologies, again emphasizing the need to (a) quantify and (b) reduce the energy demand for RCC technologies, as discussed in previous sections of this review. Additionally, at the date of this publication, no techno-economic analysis of RCC processes that produce high-value carbonates such as lithium in addition to large-market size products CaCO3 and Na2CO3 could be found.
Aspects of the carbon capture process that most significantly impact the cost of carbon capture include the electrical demand and cost of electricity, chemical demand, and required equipment for the process.12 Thus, to provide a general evaluation of the cost of carbon capture with saline based RCC technologies reviewed here, we will discuss the energy costs, chemical costs, and relevant equipment costs of each. The equipment costs summarized here include only equipment which falls outside the purview of typical chemical processing equipment (pipes and fittings, tanks, mixers, pumps, etc.) and which may significantly impact overall cost of the technology due to either the amount required, the frequency of replacement, or relatively high price of the equipment itself. This includes equipment such as ion exchange membranes, where the number and frequency of membrane replacement required for a model RCC process had an outsized impact on overall process cost,12 and electrodes – the cost of electrodes is often the main capital expense for electrochemical processes, as electrodes are often made with expensive materials such as iridium and platinum. Yet, an often-overlooked aspect of many CCUS processes (at least in the scientific literature) is the cost of the balance of plant – the valves, control hardware and software, power source, etc. Often, these costs can be significant and should not be neglected.
When comparing the costs of RCC processes, it is critical to present the costs in terms of $ per ton net CO2 captured, as this normalizes the costs to the amount of CO2 capture, which can vary greatly between different technologies. Additionally, this value is normalized to the net amount of carbon captured, so processes which can convert a large amount of CO2 and/or are low cost but have large associated carbon emissions are fairly represented. Because it is crucial to evaluate the cost of RCC technologies in this way, we will also include discussion of the carbon footprint in this section. It should be understood that reducing the carbon footprint of any RCC process increases its cost effectiveness in $ per ton net CO2 capture.
Summary of energy, chemical, and relevant equipment requirements for saline-based RCC processes
Though most saline-based RCC technologies are at early stages of development and thus typically have not been thoroughly economically evaluated, the costs of various technologies can be assessed by comparing energy, chemical, and capital infrastructure demands. Table 4 summarizes the energy costs, and also identifies chemical inputs and major equipment which contributes to capital costs. While the energy costs are a direct quantification, the other two categories are qualitative, as many of these technologies are at an early stage of development and thorough economic analyses of the process costs are not available at the time of this review.| Paper | Energy costs (kW h kg−1 CO2) | Chemical Inputs | Major Equipment |
|---|---|---|---|
| Dara et al. 201762 | 4.8 | Sulfuric acid in electrode compartments | Pt/IR coated mesh electrodes, PC-cell IEMs |
| Dara et al. 2019128 | CO2 conversion not reported | Titanium & graphite electrodes, membranes (PC-cell, Fumatech) | |
| Zhao et al. 202065 | 0.89 | NaNO3 in electrode chambers | Electrodes, YDS BPM, IEM |
| Nakata et al.14 | CO2 conversion not reported | Electrode (BDD, Pt), membranes (Nafion) | |
| Islam et al.76 | Energy demand not reported | Hydrogen gas | Sonicator |
| Dindi et al.82 | Energy demand not reported | Amine catalyst (2-amino, 2-methyl propanol (AMP) (∼30% amine regeneration demonstrated) | |
| Soong et al.66 | Energy demand not reported | Fly ash (waste material) | |
| Mustafa et al.61 | 11.18181818 | Ti/PT/Ir electrodes, IEMs (PC Cell) | |
| Power et al.31 | Energy demand not reported | Carboxylated polystyrene catalyst | |
| Taniguchi et al.100 | 0.225 | ED system (electrodes, membranes) | |
| De Lannoy et al.12 | 2.72–3.85 | ED system (electrodes, Neosepta/Selemion membranes), Membrane contactor systems for degassing | |
| Ba et al.94 | 4.318181818 | Ti mesh electrodes, MF Membranes, IX Resin | |
| Galvez Martos104 | Energy demand not reported | NaOH | |
| El-Naas93 | 0.732277778 | CaO | Bubble contact reactor |
| Xie et al.17 | 1.222904492 | Hydrogen gas | Gas diffusion anode, nickel foam cathode, AEM |
| Palitsakun et al.63 | Energy demand not reported | NH3 | |
| Zhang et al.67 | Energy demand not reported | Furnace slag, NaOH | Nickel nanoparticle catalyst material |
| Bang et al.79 | Energy demand not reported | NaOH | |
| Park et al.64 | CO2 capture not reported | CaCl2 | Ceramic membrane |
| De Vito et al.78 | Energy demand not reported | NH3 | |
| Liu et al.6 | Energy demand not reported | Buffer (HBO3, Tris) |
Energy demand
The energy cost of saline-water based RCC processes includes energy needed to move the saline water (pumping), the energy demand of the process itself (including the energy to pretreat the water and to drive the chemical processes), and the energy demand for any auxiliary operations (e.g., waste handling). Here, we will discuss the energy demand of RCC processes (including pretreatment), as this is a more consistent metric to compare technologies than energy cost, which can vary by location, power source, and time. However, it is important to note when using energy demand as the comparative metric that while energy costs generally scale with the energy demand, technologies requiring other energy sources (i.e. fuels used in thermally driven processes) may result in difference costs compared to technologies requiring electricity for the same energy demand. It should also be noted that the energy cost of saline-based RCC processes can vary based upon the product formed. As previously discussed, some carbonates (i.e. CaCO3, NaHCO3) readily precipitate under ambient conditions, whereas other carbonates (MgCO3) require additional energy to drive precipitation.31Pairing saline water-based RCC processes with other processes already handling large volumes of saline water is a potential way to reduce the costs acquiring the saline water. Eisaman et al. evaluated the impact of co-locating a seawater-based carbon capture process with power facilities using seawater for cooling, and with desalination facilities.125 This effectively reduces costs of conveying water by reusing water (i.e. reject brine or waste cooling water), which has already been collected and conveyed to a facility. They found that co-location with a power plant would reduce the costs by 33%, while co-location with a desalination plant would reduce costs by 55%. The lower cost reduction associated with power plant colocation was attributed to the need for additional treatment and pumping to increase the concentration of the seawater to a concentration comparable to a desalination brine.
In electrically driven saline-based RCC processes that involve the electrolysis of water to generate alkalinity as OH−, there is a base energy demand associated with this reaction. For processes that utilize bipolar membranes, the energy consumption associated with electrolysis is around 600–700 kW h per ton NaOH,130 though in practice the energy consumption is greater due to resistance caused by the ED stack. Processes that utilize conventional electrolysis via a cathode and anode typically have even greater energy consumption, due to overpotentials associated with the generation of O2 and H2 gas at the electrodes.130 The base energy demand associated with direct electrolysis is around 1400 kWh per ton NaOH,131 though again actual energy demand is much greater.
The energy demand of RCC processes utilizing ED is also strongly impacted by the current efficiency of the ED system. Systems that use ED to generate strong acid and/or base suffer from reduced current efficiency as the concentration of the generated acid/base increases.12 As the concentration of acid/base used in RCC can impact the carbon capture efficiency,12 it is important to assess the trade-off between increased reaction efficiency and increased energy demand with concentration. Additionally, approaches to preventing or reducing losses of current efficiency during operation are an important lever to reducing energy demand in electrochemical RCC processes. Zhao et al. found that combining their ED system with a seeded crystallizer reduced the energy cost per kg CO2 sequestered, as circulating the concentrate stream through the seeded crystallizer prevented scaling from occurring within the membrane stack, thus preventing losses in efficiency.65
It should be noted that the energy demand of electrically driven CO2 capture processes can extend beyond the energy required to drive the process, particularly in instances where the electrically driven process serves primarily to concentrate the carbonates. For instance, Taniguchi et al. determined that the energy demand for an ED system intended to concentrate carbonates captured in a potassium solution was 0.225 kW h kg−1 CO2, but the overall energy demand nearly doubled when solution circulation and CO2 recovery from the concentrate were considered.100 While this study considered vacuum stripping to produce CO2 gas as the recovery method (as opposed to conversion to a valuable product), it is useful in demonstrating the magnitude of energy consumption directly associated with electrically driven CO2 capture processes with energy associated with other components of potential RCC processes.
Because approaches to saline-based RCC often involve shifting pH with base to induce CO2 dissolution or precipitation, many electrochemical processes have been proposed to provide in situ generation of alkalinity/acidity (see previous section). This approach can reduce the chemical costs by producing base/acid directly from the saline water stream, instead incurring the expense of the energy required for electrolysis. Additionally, in instances where the electricity used to drive the process is provided by a low-carbon source (solar, wind, etc.), the carbon footprint of the chemicals required for the RCC process can be reduced.
While electrochemistry provides a promising solution for reducing the chemical costs of bases/acids used in RCC processes, regeneration methods provide the key to reducing costs associated with catalysts used in RCC. Being able to regenerate the catalyst can not only reduce the cost per tonne CO2 captured, increasing the efficiency of regeneration can help reduce the carbon footprint. Many factors impact regenerability, including (but not limited to) the catalyst solubility and boiling point. There can be a trade-off between the CO2 capture potential and ease of regeneration for catalysts, as demonstrated by Dindi et al. in their investigation of alkanolamines as an alternative catalyst for ammonia in the Solvay process.82 They found that while the alkanolamine catalyst 2-amino, 2-methyl propanol (AMP) increased the conversion of NaCl to NaHCO3, the distillation process used to effectively recover ammonia from the Solvay process only achieved 20% recovery of the AMP catalyst (due to higher boiling point of AMP), though slightly higher recovery of 30% was achieved after recovery process modification.82 Designing RCC processes to utilize more easily recovered or regenerated catalysts can improve the project economics and carbon capture potential.
The chemical demands of saline-water based RCC processes can extend beyond the chemicals directly used in the process to include chemicals used to pre-treat the water source. As previously discussed, many saline water sources (i.e. seawater) contain other constituents (TSS, DOC) that must be removed prior to RCC processes. While some pretreatment methods such as filtration require relatively little chemical inputs, other processes such as coagulation and flocculation can be chemically intensive. For instance, Beeftink et al. estimated that coagulation prior to softening consumed 4.72 g of FeCl3 per m3.80 While the amount of required coagulant will vary according to the composition of the source water and the type of coagulant used (i.e. alum, FeCl, etc.), this estimate illustrates that there can be non-negligible chemical inputs required for pretreatment to enable RCC using real saline waters. Chemical demands of pretreatment can be reduced by using more energy intensive processes such as electrocoagulation or filtration (MF, UF, NF).
Most chemicals used in saline-based RCC processes have an associated carbon footprint, whether they are generated as part of the process (as is typical in electrically driven processes) or purchased as a feedstock (typical to chemical titration processes). In the electrically driven processes reviewed here, the energy demand reported for each technology includes the energy required to generate these products. However, chemical titration processes often don’t report the energy demand or carbon footprint associated with the chemical feedstocks they require, so we will briefly discuss the carbon footprint of some common chemical feedstocks for saline based RCC here.
One of the most common feedstocks for these processes is NaOH, which alone can contribute more than 1 kg CO2 generated/kg CO2 captured in carbon capture processes utilizing NaOH evaluated by Medina-Martos et al.132 This study assumed that the chlor-alkali process is used to produce NaOH, and found that using 100% renewable energy to drive this process (as opposed to a typical mix of 30% renewable energy sources used today) significantly reduced the carbon footprint of the process, in some cases allowing for a net negative carbon footprint.132
Another feedstock to chemical titration processes is lime (CaO), which is commonly used in softening processes to produce carbonate solids but has not been as extensively applied to carbon-capture purposes. Lime is typically produced by heating limestone (CaCO3), requiring a large amount of thermal energy. This has an associated carbon footprint of 1-1.8 kg CO2 generated/kg CaO produced.133 (Note that lime softening can capture between 1–2 mols CO2 per mol of lime added, so this would equate to 1.3–4.6 kg CO2 generated/kg CO2 captured). The higher CO2 generation per amount of CO2 captured provides a clear demonstration of why processes using NaOH as a source of alkalinity are the focus of development for saline based RCC.
Though the chemical and energy demands of saline water based RCC processes (particularly when considering energy for conveyance and pretreatment) may exceed the allowable demand to remain net carbon-negative when using power generated using coal or natural gas, there are still ways that these processes can effectively provide carbon capture. The limit to the energy demand for a net-carbon negative RCC process increases significantly as the carbon footprint of the power source decreases to near 0 for sources such as solar, wind, and nuclear. Additionally, as previously discussed, processes that electrochemically generate the required chemicals can reduce the carbon footprint of the process. To achieve meaningful carbon capture with saline based RCC, it is important to (1) carefully plan RCC deployment to utilize low-carbon energy, and (2) continue to improve the energy efficiency of the RCC processes. More comprehensive analysis of the carbon footprint of proposed saline-based RCC facilities that include both carbon associated with the energy demand for water conveyance and pretreatment and carbon associated with chemical inputs to the process is necessary to assess the actual carbon capture potential of these technologies.
In order to compare the energy demands and other costs of RCC processes to those of CO2 conversion processes in sequential CCU, it is necessary to clearly delineate the costs of carbon capture and costs of carbon utilization in both steps. For saline-based RCC, it is important to note that several of the technologies reviewed here used a source of concentrated CO2 in their proof-of-concept and bench-scale studies. It can thus be understood that these technologies could be paired with any of the aforementioned carbon capture processes to act as sequential RCC, and the energy and costs of the processes would be the sum of the capture and conversion processes (thus increasing the energy demand range to 0.5–13 kW h kg−1 CO2). This is why a major point of this review is the potential to use saline water sources which can capture CO2 from the atmosphere and then be directly converted into an end product, as this carbon source eliminates the carbon capture step required for sequential CCU. As previously noted, a critical step in the development of saline-based RCC technologies is study of the impacts of using real saline waters on the energy demand and costs of these processes. Notwithstanding the impacts of the carbon source on the energy demand of saline-based RCC processes and sequential CCU processes, a direct comparison between the energy demands and costs solely related to CO2 conversion would illuminate the potential benefits and drawbacks of these processes. However, though many reviews of carbon conversion processes exist,134–136 there has not been comprehensive quantification of the energy demands and costs of these processes at the time of this review, making such comparisons difficult.
Environmental impacts
While the purpose of RCC processes is to positively impact the environment by reducing atmospheric CO2, it is important to not overlook other potential environmental impacts these processes could have when implemented at scale. Two of the primary potential impacts – water pollution and ecological damage – as well as methods to mitigate these impacts are discussed in this section.Wastewater management
The environmental impacts of saline-water based RCC processes must also include any impacts of waste generated by the process. While saline-water RCC processes can generally be compared to water softening processes, in the case of RCC, the generated solids are the desired product, and the treated water is a byproduct (whereas softened water is the desired product and solids are treated as wastes). Thus, different waste management approaches may need to be applied to saline based RCC. However, the waste management objective to reduce the amount of waste ultimately discharged to the environment can still be applied to saline-based RCC processes.Waste diversion to beneficial reuse is a broadly employed strategy that aligns with many RCC processes. In some instances, saline water is softened and/or desalinated during RCC, and such water can be considered as a desired product when diverted for potable or non-potable beneficial reuse. Depending on the quality of the saline water used for RCC, the resulting wastewater may need to be diverted to a water treatment or reclamation facility prior to reuse.
Additionally, it should be noted that while wastewater streams produced by RCC processes may be desalinated due to the removal of ionic species through formations of carbonates, this cannot always be assumed to be the case. For instance, many of the electrochemical approaches reviewed here effectively concentrate the saline stream, so even after removal of carbonate forming species, other ionic species may remain in the water at increased concentrations. In these instances, RCC wastewater would requiring additional treatment prior to beneficial use or discharge. Another consideration for the management of RCC wastewater is that the wastewater may exceed the volume of water that can be beneficially reused, and thus would need to be disposed.
Discharging of wastewater can be achieved by either discharge to the environment, deep well injection, or evaporation. Discharging to the environment often requires that the wastewater is of suitable quality. A water quality standard that may prevent discharge of waters used in RCC to the environment is salinity limits. While there have not been nationwide limits on salinity for discharge, many local water quality authorities have set limits or guidelines, generally limiting discharge concentrations to around the TDS limits for water use (500 mg L−1 in drinking water, 1000 mg L−1 for other uses137). However, as the discharge limits for salinity are determined on a case-by-case basis, there are instances in which the discharge limit may be as high as the US EPA's recommendation of 40 g L−1 TDS (slightly above the salinity of seawater).138 Another water quality marker which should be considered when releasing wastewater from RCC processes to the environment is the dissolved CO2 content (DIC). While generally less stringently regulated than salinity, releasing CO2-depleted water into aquatic environments can cause temporary, localized pH shifts which could harm ecosystems. Though waters released to the environment would eventually re-equilibrate with the atmosphere, in regions such as the mixed surface layer of the ocean, this process can take up to 1 year.12
Ecosystem impacts
RCC within the carbon capture landscape
While there are several promising aspects of saline-based RCC, it is essential to compare the potential of this approach to the amount of carbon capture required to mitigate climate change. In their 2013 review of the prospects of CCU technologies, Aresta et al. highlight that while chemical production consumes approximately 200 megatonnes CO2/year, this makes up less than 1% of anthropogenic CO2 emissions, which are on the scale of 32000 megatonnes per year.136 The production of inorganic carbonates, the primary output of many of the RCC processes reviewed here, accounts for only about a quarter of the CO2 consumption associated with chemical production,136 further highlighting the mismatch between the potential supply and demand. It is also important to consider the capacity of other CCU/CCS approaches. For instance, urea production makes up over half of the 200 megatonnes per year of CO2 capture associated with chemical production.136 This is the only carbon utilization pathway identified by Aresta et al. that has a greater carbon capture potential than inorganic carbonate production pathways (such as saline-based RCC). Processes that have greater carbon capture potential typically involve storage/sequestration of CO2, such as CO2-enhanced oil and gas recovery, ocean storage, and injection into deep saline formations (which can include in situ mineralization). Numerous in-depth reviews of various CCUS approaches have been published.2,127,139
Even though it may seem intuitive to compare the capacity of CCU and CCS processes, the “capacity” of CCU processes is determined by market demand, while the “capacity” of CCS is determined by physical availability. The IEA identifies processes that involve carbon storage as critical to reaching climate goals and indicates that there is more than sufficient storage capacity to achieve these goals – while 94 Gt of carbon storage is needed by 2050 to meet the 2 °C target, there is between 2000–20000 Gt of geological carbon storage potential in the United States alone.140 While saline-based RCC (as well as all other CCU approaches) may not suffice to fully achieve the carbon capture needed to meet climate goals due to the mismatch between the demand for CO2-derived products and the amount of CO2 requiring removal, it should be emphasized that this failure is related not to any technical shortcomings of RCC but instead is a result of the framework of producing saleable products to reduce the cost of carbon capture. Therefore, while there is certainly space for RCC systems to generate carbon-neutral or negative chemical products as part of carbon capture efforts, these technologies need not be restricted to implementation in instances where the products can be sold, but may also be used to generate products that can provide long term storage/sequestration of CO2 (e.g., ex situ mineral carbonation). Two major barriers have been identified for these types of processes – the kinetics of carbonate formation, and potential environmental impacts associated with the mineral source.141 The first concern is addressed by many of the technologies reviewed here, which use various approaches to enhance the rate of carbonate formation. However, the ability of saline-based RCC technologies to address the second barrier is equally, if not more, important. As discussed previously, using saline water as the source of carbonate-forming cations can avoid the need to mine and dissolve carbonate-forming minerals. Additionally, for processes that ultimately generate solid carbonates, there is an added benefit of the ease of accounting for the stored carbon (which is far more difficult in many other CCUS approaches). A 2022 techno-economic review of CCUS technologies suggested a similar approach – integrating CCU technologies (such as RCC) with CCS systems to increase the carbon capture potential of these approaches. Here, saline-based RCC technologies may provide potential benefit over other CCU approaches that generate products that are less easily stored or disposed of. Inorganic carbonates have the longest CO2 storage times of the CCU products considered in Shokrollahi et al.'s techno-economic analysis of CCU processes and products.124
Conclusions
Several major trends emerged in our review of the existing literature on saline water-based RCC processes. Many studies focused on methods to improve the kinetics of carbonate formation, either by concentrating the saline water stream, using catalysts, or optimizing the CO2 transfer efficiency. These approaches allowed for carbon capture at ambient temperatures and pressures, which has the potential to greatly reduce the energy demand compared to processes requiring elevated temperatures/pressures. Additionally, nearly all of the studies reviewed involved careful control of the reaction pH to produce the desired products, which falls in line with Liu et al.'s assessment that pH was the single most important factor in determining carbon capture potential of precipitative processes.6Interestingly, many studies focused on concentrating the saline water stream, whereas only a few discussed other approaches to enhancing CO2 dissolution, though at least one study demonstrated that the precipitation reaction was limited by CO2. This may in part be due to the fact that many of the studies reviewed used pure CO2 or a gas stream containing CO2 at concentrations above those found in the atmosphere. While this can simplify bench-scale analysis, it limits the applicability of many of the methods reviewed here to carbon capture from ambient air. Further study of methods to enhance gas transfer into saline waters in RCC processes would be useful, particularly in investigating the CO2 capture efficiency of these processes from more realistic CO2 sources. While it is in theory feasible to provide pure CO2 produced from another carbon capture method with many of the technologies reviewed here, this such approaches cannot be classified as RCC, and would eliminate the energy savings from avoiding CO2 gas processing, a major benefit of RCC processes.
Another limit to the existing studies of saline based RCC processes is the use of synthetic saline water sources, which are often pure NaCl (or other chloride salt) solutions. Again, while using these solutions is helpful to quantify the carbon capture potential of the process, they fall short of estimating the carbon capture potential of the process using real saline water, in which the presence of competing constituents can impact both the carbon capture efficiency and the purity of the end product. Further work investigating how these processes can be used to produce carbonate products of sufficient purity for beneficial use would be useful in demonstrating the actual viability of these technologies for RCC.
Overall, the development of saline based RCC processes is promising based on the underlying chemistry but limited in actual demonstration and evaluation. Only one of the reviewed studies provided a cost estimate of the process, and nearly one third provided no quantified information on the carbon capture potential of their respective RCC process. Most technologies reviewed here were bench scale operations. However, as discussed, the existence of processes utilizing similar chemistries in the chemical manufacturing and water treatment industries provides invaluable examples of how these technologies can be applied at scale.
Finally, we highlight that there is a significant mismatch between the scale of the markets for products of saline-based RCC processes and the scale of carbon capture needed to meet climate goals. Fortunately, because many of the products of saline-based RCC processes are stable and non-hazardous, there is the potential to apply these technologies at larger scales, although this will require treating the products as a waste rather than a value-added product (once market demands have been met). As the primary objective of RCC is to reduce atmospheric CO2, the demonstrated carbon capture potential should encourage further study and development of these technologies, even in instances where the market demand for the associated products is limited.
Author contributions
Anya Dickinson-Cove: investigation, writing – original draft, writing – review & editing, data curation, visualization. Dr. David Jassby: supervision, funding acquisition, writing – review & editing. Dr. Erika La Plante: writing – review & editing. Dr. Eric M. V. Hoek: writing – review & editing. Dr. Yiming Liu: investigation, writing – original draft. Dr. Gaurav Sant: conceptualization.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Department of Energy ARPA-E MINER program.Notes and references
- Ipcc, AR5 Climate Change 2014: Mitigation of Climate Change, 2014.
- J. Mustafa, A. A. H. I. Mourad, A. H. Al-Marzouqi and M. H. El-Naas, Desalination, 2020, 483, 114386 CrossRef CAS
.
- D. Kang, Y. Yoo and J. Park, Desalination, 2020, 473, 114147 CrossRef
- J. H. Lee, D. W. Lee, C. Kwak, K. Kang and J. H. Lee, Ind. Eng. Chem. Res., 2019, 58, 15533–15541 CrossRef CAS
- L. Lu, Y. Fang, Z. Huang, Y. Huang and Z. J. Ren, Chem. Eng. J., 2016, 306, 330–335 CrossRef CAS
- Q. Liu and M. M. Maroto-Valer, Fuel Process. Technol., 2012, 98, 6–13 CrossRef CAS
- P. C. De Carvalho Pinto, T. R. Da Silva, F. M. Linhares, F. V. De Andrade, M. M. De Oliveira Carvalho and G. M. De Lima, Clean Technol. Environ. Policy, 2016, 18, 1123–1139 CrossRef CAS
- Ipcc, AR6 Climate Change 2022: Mitigation of Climate Change, 2022.
- J.-P. Gattuso, A. K. Magnan, L. Bopp, W. W. L. Cheung, C. M. Duarte, J. Hinkel, E. McLeod, F. Micheli, A. Oschlies, P. Williamson, R. Billé, V. I. Chalastani, R. D. Gates, J.-O. Irisson, J. J. Middelburg, H.-O. Pörtner and G. H. Rau, Front. Mar. Sci., 2018, 5, 337 CrossRef
- B. Metz, O. Davidson, H. C. Connick, M. Loos and L. A. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, 2005.
- R. Baciocchi, G. Storti and M. Mazzotti, Chem. Eng. Process., 2006, 45, 1047–1058 CrossRef CAS
- C.-F. Lannoy, M. D. Eisaman, A. Jose, S. D. Karnitz, R. W. DeVaul, K. Hannun and J. L. B. Rivest, Int. J. Greenhouse Gas Control, 2018, 70, 243–253 CrossRef
-
B. I. McNeil and K. Matsumoto, Fish Physiology, Elsevier, 2019, vol. 37, pp. 1–32 Search PubMed
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS
- Z. Zhang, D. Xi, Z. Ren and J. Li, Cell Rep. Phys. Sci., 2023, 4, 101662 CrossRef CAS
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS
- H. Xie, T. Liu, Z. Hou, Y. Wang, J. Wang, L. Tang, W. Jiang and Y. He, Environ. Earth Sci., 2015, 73, 6881–6890 CrossRef CAS
-
M. M. Benjamin, Water Chemistry, 2nd edn, 2015 Search PubMed
- J. J. Carroll and A. E. Mather, J. Solution Chem., 1992, 21, 607–621 CrossRef CAS
- L. W. Diamond and N. N. Akinfiev, Fluid Phase Equilib., 2003, 208, 265–290 CrossRef CAS
-
W. Stumm and J. J. Morgan, Aquatic chemistry: chemical equilibria and rates in natural waters, Wiley, New York, 3rd edn, 1996 Search PubMed
- F. J. Pearson, Jr. and B. B. Hanshaw, Isotope Hydrology, International Atomic Energy Agency (IAEA), 1970.
- E. Halewood, K. Opalk, L. Custals, M. Carey, D. A. Hansell and C. A. Carlson, Front. Mar. Sci., 2022, 9, 1061646 CrossRef
- F. J. Millero, T. B. Graham, F. Huang, H. Bustos-Serrano and D. Pierrot, Mar. Chem., 2006, 100, 80–94 CrossRef CAS
-
J. Crittenden, R. R. Trussel, D. W. Hand, K. J. Howe and G. Tchobanoglous, MWH's Water Treatment: Principles and Design, Wiley Online Books, 3rd edn, 2012 Search PubMed
- P. Renforth and G. Henderson, Rev. Geophys., 2017, 55, 636–674 CrossRef
-
R. A. Freeze and J. A. Cherry, Groundwater, Prentice-Hall, Englewood Cliffs, N.J, 1979 Search PubMed
-
M. D. Angelis, in Water Encyclopedia, ed. J. H. Lehr and J. Keeley, Wiley, 1st edn, 2004, pp. 159–160 Search PubMed
- M. Thamminidi, Physical Geography, PMF IAS, 2023.
- M. Lebrato, D. Garbe-Schönberg, M. N. Müller, S. Blanco-Ameijeiras, R. A. Feely, L. Lorenzoni, J.-C. Molinero, K. Bremer, D. O. B. Jones, D. Iglesias-Rodriguez, D. Greeley, M. D. Lamare, A. Paulmier, M. Graco, J. Cartes, J. Barcelos, E. Ramos, A. De Lara, R. Sanchez-Leal, P. Jimenez, F. E. Paparazzo, S. E. Hartman, U. Westernströer, M. Küter, R. Benavides, A. F. Da Silva, S. Bell, C. Payne, S. Olafsdottir, K. Robinson, L. M. Jantunen, A. Korablev, R. J. Webster, E. M. Jones, O. Gilg, P. Bailly Du Bois, J. Beldowski, C. Ashjian, N. D. Yahia, B. Twining, X.-G. Chen, L.-C. Tseng, J.-S. Hwang, H.-U. Dahms and A. Oschlies, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22281–22292 CrossRef CAS PubMed
- I. Power, P. Kenward, G. Dipple and M. Raudsepp, Cryst. Growth Des., 2017, 17(11), 5652–5659 CrossRef CAS
- T. DeVries, Annu. Rev. Environ. Resour., 2022, 47, 317–341 CrossRef
- A. Matin, F. Rahman, H. Z. Shafi and S. M. Zubair, Desalination, 2019, 455, 135–157 CrossRef CAS
-
T. Dittmar and A. Stubbins, Treatise on Geochemistry, Elsevier, 2014, pp. 125–156 Search PubMed
- A. H. A. Dehwah, S. Li, S. Al-Mashharawi, H. Winters and T. M. Missimer, Desalination, 2015, 360, 19–27 CrossRef CAS
- W. Yin, X. Li, S. R. Suwarno, E. R. Cornelissen and T. H. Chong, Water Res., 2019, 159, 385–396 CrossRef CAS PubMed
- W. M. Last, Geosci. J., 2002, 6, 347–369 CrossRef
- W. D. Williams, Environ. Conserv., 2002, 29, 154–167 CrossRef
- P. H. Gleick and I. Shiklomanov, Water in the 21st century, 105 edn, 1993, 104.
- C. M. White, B. R. Strazisar, E. J. Granite, J. S. Hoffman and H. W. Pennline, J. Air Waste Manage. Assoc., 2003, 53, 645–715 CrossRef CAS
- G. Zhang, C. Taberner, L. Cartwright and T. Xu, SPE J., 2011, 16, 959–967 CrossRef CAS
- B. Jaskula, Minerals Yearbook - Lithium, U.S. Geological Survey, 2018.
- M. L. Vera, W. R. Torres, C. I. Galli, A. Chagnes and V. Flexer, Nat. Rev. Earth Environ., 2023, 4, 149–165 CrossRef CAS
- B. Jaskula, Mineral Commodity Summaries, January 2023 - Lithium, U.S. Geological Survey, 2023.
- R. Darvari, J.-P. Nicot, B. R. Scanlon, J. R. Kyle, B. A. Elliott and K. Uhlman, J. Geochem. Explor., 2024, 257, 107363 CrossRef CAS
- F. Van Weert, J. Van der Gun and J. Reckman, Global overview of saline groundwater occurrence and genesis, International Groundwater Resources Assessment Centre, 2009.
- B. R. Scanlon, R. C. Reedy and J. P. Nicot, Environ. Sci. Technol., 2014, 48, 12386–12393 CrossRef CAS
- Y. Yechieli and O. Sivan, Hydrogeol. J., 2011, 19, 71–81 CrossRef CAS
- Y. Li and Z. Pang, Greenhouse Gases, 2017, 7, 53–64 CrossRef CAS
- J. Androwski, A. Springer, T. Acker and M. Manone, J. Am. Water Resour. Assoc., 2011, 47, 93–102 CrossRef CAS
- C. Li, X. Gao, S. Li and J. Bundschuh, Environ. Sci. Pollut. Res., 2020, 27, 41157–41174 CrossRef CAS PubMed
- W. M. Alley, Ground water and surface water: a single resource, U.S. Geological Survey, Denver, Colo, 1998.
- O. S. Zapecza and Z. Szabo, Groundwater Quality: Hydrologic Conditions and Events, 1988, 50–57.
- P. Sahu, J. Water Reuse Desalin., 2021, 11, 33–65 CrossRef CAS
- A. Panagopoulos, Environ. Sci. Pollut. Res., 2022, 29, 23736–23749 CrossRef CAS
-
E. M. V. Hoek, J. Wang, T. D. Hancock, A. Edalat, S. Bhattacharjee and D. Jassby, Oil & produced water management, Morgan & Claypool Publishers, San Rafael, CA, 2021 Search PubMed
- D. Ariono, M. Purwasasmita and I. G. Wenten, J. Eng. Technol. Sci., 2016, 48, 367–387 CrossRef CAS
-
S. Nallakukkala and B. Lal, in Gas Hydrate in Water Treatment, ed. B. Lal and S. Nallakukkala, Wiley, 1st edn, 2022, pp. 269–287 Search PubMed
- C. Gabrielli, G. Maurin, H. Francy-Chausson, P. Thery, T. T. M. Tran and M. Tlili, Desalination, 2006, 201, 150–163 CrossRef CAS
- L. Lu, J. S. Guest, C. A. Peters, X. Zhu, G. H. Rau and Z. J. Ren, Nat. Sustainability, 2018, 1, 750–758 CrossRef
- J. Mustafa, A. H. Al-Marzouqi, N. Ghasem, M. H. El-Naas and B. Van Der Bruggen, Desalination, 2023, 548, 116263 CrossRef CAS
- S. Dara, M. Lindstrom, J. English, A. Bonakdarpour, B. Wetton and D. P. Wilkinson, J. CO2 Util., 2017, 19, 177–184 CrossRef CAS
- S. Palitsakun, A. Seubsai and K. Sudsakorn, Songklanakarin J. Sci. Technol., 2019, 41, 984–991 CAS
- H. Park, J. Lee, J. Han, S. Park, J. Park and B. Min, Energies, 2015, 8, 8704–8715 CrossRef CAS
- Y. Zhao, J. Wang, Z. Ji, J. Liu, X. Guo and J. Yuan, Chem. Eng. J., 2020, 381, 122542 CrossRef CAS
- Y. Soong, D. L. Fauth, B. H. Howard, J. R. Jones, D. K. Harrison, A. L. Goodman, M. L. Gray and E. A. Frommell, Energy Convers. Manage., 2006, 47, 1676–1685 CrossRef CAS
- N. Zhang, R. M. Santos, S. M. Smith and L. Šiller, Chem. Eng. J., 2019, 377, 120479 CrossRef CAS
-
J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill Education, 17th edn, 2017 Search PubMed
- L. H. Lalasari, M. K. Widowati, N. C. Natasha, E. Sulistiyono and A. B. Prasetyo, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 176, 012040 Search PubMed
- J. W. Ball and D. K. Nordstrom, WATEQ4F -- User's manual with revised thermodynamic data base and test cases for calculating speciation of major, trace and redox elements in natural waters, Report 90-129, USGS, 1991.
- E. C. La Plante, D. A. Simonetti, J. Wang, A. Al-Turki, X. Chen, D. Jassby and G. N. Sant, ACS Sustainable Chem. Eng., 2021, 9, 1073–1089 CrossRef CAS
- H. S. Santos, H. Nguyen, F. Venâncio, D. Ramteke, R. Zevenhoven and P. Kinnunen, Inorg. Chem. Front., 2023, 10, 2507–2546 RSC
- S. P. Veetil and M. Hitch, Int. J. Environ. Sci. Technol., 2020, 17, 4359–4380 CrossRef CAS
- M. Erans, E. S. Sanz-Pérez, D. P. Hanak, Z. Clulow, D. M. Reiner and G. A. Mutch, Energy Environ. Sci., 2022, 15, 1360–1405 RSC
- P. Tcvetkov, A. Cherepovitsyn and S. Fedoseev, Sustainability, 2019, 11, 5834 CrossRef CAS
- M. H. Islam, O. S. Burheim, J.-Y. Hihn and B. G. POllet, Ultrason. Sonochem., 2021, 73, 105474 CrossRef CAS PubMed
-
J. J. Middelburg, Marine Carbon Biogeochemistry: A Primer for Earth System Scientists, Springer International Publishing, Cham, 2019 Search PubMed
- C. De Vito, V. Ferrini, S. Mignardi, M. Cagnetti and F. Leccese, Period. Mineral., 2012, 81, 333–344 Search PubMed
- J.-H. Bang, Y. Yoo, S.-W. Lee, K. Song and S. Chae, Minerals, 2017, 7, 207 CrossRef
- M. Beeftink, B. Hofs, O. Kramer, I. Odegard and A. Wal, J. Cleaner Prod., 2021, 278, 123925 CrossRef CAS
- L. Burhenne, C. Giacomin, T. Follett, J. Ritchie, J. S. J. McCahill and W. Mérida, J. Environ. Chem. Eng., 2017, 5, 5968–5977 CrossRef CAS
- A. Dindi, D. V. Quang and M. R. M. Abu-Zahra, Appl. Energy, 2015, 154, 298–308 CrossRef CAS
- Y. Shan, Z. Liu and D. Guan, Appl. Energy, 2016, 166, 245–252 CrossRef CAS
- E. Jones, M. Qadir, M. T. H. Vliet, V. Smakhtin and S.-M. Kang, Sci. Total Environ., 2019, 657, 1343–1356 CrossRef CAS PubMed
- J. Mustafa, N. Ghasem, M. H. El-Naas, B. V. D. Bruggen and A. H. Al-Marzouqi, J. Cleaner Prod., 2024, 438, 140578 CrossRef CAS
- B. Sun, M. Zhang, S. Huang, J. Wang and X. Zhang, Desalination, 2021, 498, 114793 CrossRef CAS
- S. K. Patel, P. M. Biesheuvel and M. Elimelech, ACS EST Eng., 2021, 1, 851–864 CrossRef CAS
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS
-
R. Pierantozzi, Kirk-Othmer Encyclopedia of Chemical Technology, ed. O. Kirk, Wiley, 1st edn, 2000 Search PubMed
-
S. E. Manahan, Green Chemistry and the Ten Commandments of Sustainability, ChemChar Research, Columbia, MO, 3rd edn, 2011 Search PubMed
- W. Bolen, Mineral commodity summaries 2022 - Soda Ash, U.S. Geological Survey, 2022.
- O. Chaalal and M. M. Hossain, Environ. Prog. Sustainable Energy, 2016, 35, 250–256 CrossRef CAS
- M. H. El-Naas, A. F. Mohammad, M. I. Suleiman, M. Al Musharfy and A. H. Al-Marzouqi, Desalination, 2017, 411, 69–75 CrossRef CAS
- X. Ba, J. Chen, X. Wang, H. Xu, J. Sun, Y. Qi, Y. Li, J. Wang and B. Jiang, Desalination, 2023, 553, 116481 CrossRef CAS
- H. Zhou, D. W. Smith and S. J. Stanley, Can. J. Civ. Eng., 1992, 19, 794–805 CrossRef
-
G. Tchobanoglous, F. L. Burton and H. D. Stensel, Wastewater Engineering: Treatment and Reuse, McGraw Hill Companies, Inc., New York, USA, 5th edn, 2014 Search PubMed
- R. Committee, Am. Water Works Assoc., 1981, 73, 600–608 CrossRef
- J. H. Leeuwen, D. Whie, R. Baker and C. Jones, Land Contam. Reclam., 2011, 18, 393–415 Search PubMed
- H. T. Campion, J. AWWA, 1934, 26, 488–494 CrossRef CAS
- I. Taniguchi and T. Yamada, Energy Procedia, 2017, 114, 1615–1620 CrossRef CAS
- J. Willet, Mineral commodity summaries 2022 - Crushed Stone, U.S. Geological Survey, 2018.
- C. W. Hargis, I. A. Chen, M. Devenney, M. J. Fernandez, R. J. Gilliam and R. P. Thatcher, Materials, 2021, 14, 2709 CrossRef CAS PubMed
- L. McDonald, F. Glasser and M. Imbabi, Materials, 2019, 12, 554 CrossRef CAS PubMed
- J.-L. Galvez-Martos, A. Elhoweris, J. Morrison and Y. Al-horr, J. CO2 Util., 2018, 27, 161–169 CrossRef CAS
- W.-L. Tan, A. L. Ahmad, C. P. Leo and S. S. Lam, J. CO2 Util., 2020, 42, 101333 CrossRef CAS
- A. M. Merrill, Minerals Yearbook - Magnesium Compounds, U.S. Geological Survey, 2018.
- M. S. Diallo, M. R. Kotte and M. Cho, Environ. Sci. Technol., 2015, 49, 9390–9399 CrossRef CAS PubMed
-
E. P. Comer, Lithium Needs and Resources, Elsevier, 1978, pp. 237–240 Search PubMed
- S. A. Barroilhet and S. N. Ghaemi, Acta Psychiatr. Scand., 2020, 142, 161–172 CrossRef CAS
- J. C. Kelly, M. Wang, Q. Dai and O. Winjobi, Resour., Conserv. Recycl., 2021, 174, 105762 CrossRef CAS
- T. G. Goonan, Lithium use in batteries, Report 1371, U.S. Geological Survey, 2012.
- N. Susarla and S. Ahmed, Ind. Eng. Chem. Res., 2019, 58, 3754–3766 CrossRef CAS
- S. Park, J.-H. Bang, K. Song, C. W. Jeon and J. Park, Chem. Eng. J., 2016, 284, 1251–1258 CrossRef CAS
- Ö. Z. Mete, A. V. Subhas, H. H. Kim, A. G. Dunlea, L. M. Whitmore, A. M. Shiller, M. Gilbert, W. D. Leavitt and T. J. Horner, Earth Syst. Sci. Data, 2023, 15, 4023–4045 CrossRef
- M. McRae, Minerals Yearbook - Barite, U.S. Geological Survey, 2018.
- C. A. Johnson, N. Piatik and M. M. Miller, Critical Mineral Resources of the United States—Economic and Environmental Geology and Prospects for Future Supply, Chapter D, U.S. Geological Survey, 2017.
- A. Hatfield, Mineral Commodity Summaries, January 2024 - Strontium, U.S. Geological Survey, 2024.
- S. A. Singerling and J. Ober, Minerals Yearbook - Strontium, U.S. Geological Survey, 2018.
-
A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 1–34 Search PubMed
- D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed
-
J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 1st edn, 2016, pp. 1–22 Search PubMed
-
R. Schmidt, K. Griesbaum, A. Behr, D. Biedenkapp, H.-W. Voges, D. Garbe, C. Paetz, G. Collin, D. Mayer and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–74 Search PubMed
- U. S. E. I. Administration, 2022.
- M. Shokrollahi, N. Teymouri and P. Navarri, J. Cleaner Prod., 2024, 434, 139620 CrossRef CAS
- M. D. Eisaman, J. L. B. Rivest, S. D. Karnitz, C.-F. De Lannoy, A. Jose, R. W. DeVaul and K. Hannun, Int. J. Greenhouse Gas Control, 2018, 70, 254–261 CrossRef CAS
- K. S. Lackner and H. Azarabadi, Ind. Eng. Chem. Res., 2021, 60, 8196–8208 CrossRef CAS
- W. Y. Hong, Carbon Capture Sci. Technol., 2022, 3, 100044 CrossRef CAS
- S. Dara, A. Bonakdarpour, M. Ho, R. Govindarajan and D. P. Wilkinson, React. Chem. Eng., 2019, 4(1), 141–150 RSC
- C. WateReuse Association Desalination, Seawater Desalination Power Consumption, WateReuse Association.
- G. Pourcelly, Russ. J. Electrochem., 2002, 38, 919–926 CrossRef CAS
- A. Kumar, F. Du and J. H. Lienhard, ACS Energy Lett., 2021, 6, 3563–3566 CrossRef CAS
- E. Medina-Martos, J.-L. Gálvez-Martos, J. Almarza, C. Lirio, D. Iribarren, A. Valente and J. Dufour, J. CO2 Util., 2022, 60, 101991 CrossRef CAS
- M. Simoni, M. D. Wilkes, S. Brown, J. L. Provis, H. Kinoshita and T. Hanein, Renewable Sustainable Energy Rev., 2022, 168, 112765 CrossRef CAS
- A. Saravanan, P. Senthil Kumar, D.-V. N. Vo, S. Jeevanantham, V. Bhuvaneswari, V. Anantha Narayanan, P. R. Yaashikaa, S. Swetha and B. Reshma, Chem. Eng. Sci., 2021, 236, 116515 CrossRef CAS
- A. Rafiee, K. Rajab Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS
- C. Alaska Department of Envrionmental, Water Quality Standards, Chapter 70, Report 18 AAC 70, 2023.
- P. Southern, California Coastal Water Research, Management of Brine Discharges to Coastal Waters Recommendations of a Science Advisory Panel, Report 694, 2012.
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS
- Iea, 20 years of carbon capture and storage, 2016.
- A. A. Olajire, J. Pet. Sci. Eng., 2013, 109, 364–392 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |