
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isoxazoles as efficient alkyne amination reagents in divergent heterocycle synthesis
Xin-Qi Zhu
 *a,
Zhi-Xu Meng
a,
Bo Zhou
b,
Ming-Yu Teng
a and
Long-Wu Ye
*abc
*a,
Zhi-Xu Meng
a,
Bo Zhou
b,
Ming-Yu Teng
a and
Long-Wu Ye
*abc
aYunnan Key Laboratory of Modern Separation Analysis and Substance Transformation, College of Chemistry and Chemical Engineering, Yunnan Normal University, Kunming 650500, China. E-mail: xinqizhu@ynnu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, and College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: longwuye@xmu.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 13th February 2025
Abstract
During the past decades, the exploration of new alkyne amination reactions has attracted increasing attention due to the high efficiency in heterocycle synthesis. In addition to the well-established alkyne amination reagents (such as nitrogen ylides and azides), isoxazoles and their derivatives have been proven to be efficient amination reagents, especially the N,O-bifunctional reagents of alkynes, in the transition metal-catalyzed transformation of alkynes through metal carbene intermediates. Isoxazole derivatives have been extensively applied to the rapid synthesis of a diverse range of structurally complex N-containing molecules, especially the valuable N-heterocycles in atom-economic manner. In this review, we summarize the latest trends and developments of isoxazole-enabled alkyne amination reactions and their applications in divergent heterocycle synthesis, including amination of ynamides, amination of ynol ethers, amination of thioynol ethers, amination of electron-deficient alkynes, amination of unpolarized alkynes and asymmetric amination of alkynes. Finally, we list the current challenges and opportunities for potential breakthroughs in this field.
 Xin-Qi Zhu | Xin-Qi Zhu was born in 1993 in Ruijin, Jiangxi province of China. He received his BSc degree in chemistry from Nanchang University in 2015. He then obtained his PhD in 2020 from Xiamen University under the guidance of Prof. Long-Wu Ye. After postdoctoral research with Prof. Long-Wu Ye at Xiamen University and with Prof. Jieping Zhu at École Polytechnique Fédérale de Lausanne (EPFL), he joined Yunnan Normal University in 2023 as a full professor. His current research interests include the development of novel asymmetric organocatalyzed and transition-metal-catalyzed reactions. |
 Zhi-Xu Meng | Zhi-Xu Meng was born in Puan, Guizhou Province of China. She earned her BSc from Zunyi Normal University in 2022. She is currently a third-year graduate student in the group of Prof. Ming-Yu Teng of Yunnan Normal University. |
 Bo Zhou | Bo Zhou was born in 1992 in Lu’an, Anhui province of China. He received his BSc in chemistry from Wuhan University of Technology (2013). He then obtained his PhD in 2019 from Xiamen University under the guidance of Prof. Long-Wu Ye. After postdoctoral studies with Prof. Guangbin Dong at the University of Chicago, he joined Xiamen University in 2022 as an associate Professor. His current research interests include asymmetric synthesis based on alkynes and their further applications. |
 Ming-Yu Teng | Ming-Yu Teng was born in 1981 in Tongren, Guizhou province of China. He received his BSc in chemistry from Zhejiang University (2003). He then obtained his PhD in 2009 from Nanjing University, under the guidance of Prof. Yi Pan and Le-Yong Wang. After postdoctoral studies with Prof. You-Xuan Zheng and Bing-Cai Pan at Nanjing University, he joined Yunnan Normal University in 2013 as an assistant Professor. He was promoted to the associate Professor in 2016 and full Professor in 2024. His current research interests include the development of organic optical functional materials, transition-metal catalysis and heterocycle synthesis. |
 Long-Wu Ye | Long-Wu Ye was born in 1981 in Wenzhou, Zhejiang province of China. He received his BSc in chemistry from Zhejiang University (2003). He then obtained his PhD in 2008 from Shanghai Institute of Organic Chemistry with Prof. Yong Tang. After postdoctoral studies with Prof. Subhash Sinha at The Scripps Research Institute and with Prof. Liming Zhang at the University of California at Santa Barbara, he joined Xiamen University in 2011 as an associate Professor. He was promoted to a full Professor in 2012. His current research interests focus on the alkyne transformations and their applications in heterocycle synthesis. |
Key learning points(1) Overview of current developments in the isoxazoles as efficient alkyne amination reagents.(2) Strategies for the divergent synthesis of heterocycles employing isoxazoles and alkynes. (3) Important reaction types in amination of alkynes with isoxazoles. (4) α-Imino metal carbenes as the key intermediates in amination of alkynes with isoxazoles. (5) The current challenges and the future directions in amination of alkynes with isoxazoles. |
1. Introduction
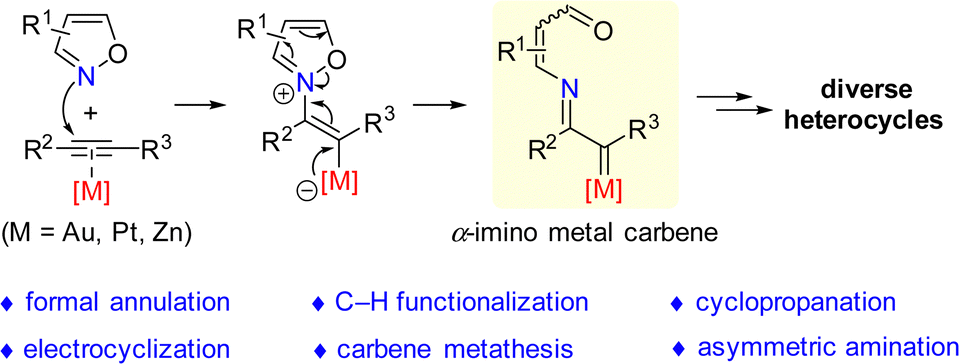
Transition metal-catalyzed alkyne amination reaction is a powerful methodology for the rapid assembly of valuable N-heterocycles from readily available alkynes via α-imino metal carbene intermediates.1 In the past decades, a variety of amination reagents, including azides,2 nitrogen ylides,3 sulfilimines,4 and others,5 have been demonstrated in the transition metal-catalyzed intermolecular and intramolecular alkyne amination reactions. In spite of the efficiency of these approaches, the limitation in atom-economy still exists.Isoxazoles and their derivatives (such as benzo[c]isoxazoles and benzo[d]isoxazoles) are important skeletons, which could be found in a broad range of natural products, pharmaceuticals and bioactive molecules.6 However, the reaction of isoxazole derivatives with alkynes was underexplored for a long duration probably due to the weak nucleophilicity of isoxazole derivatives. Compared with the above-mentioned alkyne amination reagents,2–5 the reaction of isoxazoles with alkynes exhibits unique advantages in heterocycle synthesis. Mechanistically, the labile N–O bonds of isoxazole make it a potential amination reagent in the amination reaction of alkynes, and no waste would be formed in the transformation. Therefore, the development of a new catalytic system and exploration into the electronic properties of alkyne species are important.
In 2015, Ye's group demonstrated a gold-catalyzed reaction of ynamides with isoxazoles, which represents the first example of alkyne amination reaction using isoxazole derivatives.7 This work provided a new strategy for the amination reaction of alkynes, offering an efficient and divergent method for the synthesis of a wide range of functionalized N-containing heterocycles through an atom-economic pathway. The typical reaction pathway of alkyne amination reaction with isoxazole derivatives is shown in Scheme 1. The nitrogen atom in the isoxazole derivative initially attacks the metal-activated triple bond to afford the vinyl metal intermediate, triggering the fragmentation of N–O bond to generate the key α-imino metal carbene intermediate. The α-imino metal carbene intermediate is regarded as a high electrophilic Fischer-type carbene complex, which could further undergo a series of transformations, such as formal annulation, C–H functionalization, cyclopropanation, electrocyclization, carbene metathesis and other asymmetric aminations, and lead to the assembly of diverse heterocycles.
 | ||
| Scheme 1 Reaction pathway for the amination of alkynes with isoxazoles. | ||
Considering the wide applications of isoxazoles in alkyne amination reactions, especially as the N,O-bifunctional reagents of alkynes, it is necessary to summarize the latest advances in the amination of alkynes with isoxazoles. During the past years, several relevant reviews on transition metal-catalyzed amination of alkynes have been demonstrated.8 However, the latest comprehensive summary on the amination of different types of alkynes with isoxazoles has not been reported.
The aim of this review is to provide latest trends and developments of alkyne amination reactions with isoxazoles in divergent heterocycle synthesis, which is organized by different types of alkynes, including amination of ynamides, amination of ynol ethers, amination of thioynol ethers, amination of electron-deficient alkynes, amination of unpolarized alkynes and asymmetric amination of alkynes. In general, ynamides, ynol ethers, and thioynol ethers are considered as electron-rich alkynes, which exhibit α-addition by isoxazoles in the alkyne amination reactions with isoxazoles. In contrast, electron-deficient alkynes, such as propiolates, alkynones and alkynyl sulfones, display β-addition by isoxazoles. The unpolarized alkynes demonstrate relatively poor chemoselectivity in the alkyne amination reactions with isoxazoles. We believe this review will promote the further development of heterocycle synthesis, alkyne chemistry and carbene chemistry.
2. Amination of ynamides
2.1. Formal annulation
Ynamides are special alkynes directly attached to the nitrogen atom containing an electron-withdrawing group (EWG), which have electron-rich triple bonds and tunable electronic properties. Ynamides have been proven to be versatile synthons in organic synthesis.9 Since the first report on formal annulation of ynamides with isoxazoles by Ye's group,7 a series of formal annulation reactions between ynamides and isoxazoles have been developed, such as [3+2], [4+2], [4+3], [5+1] and [5+2] annulations, leading to the construction of diverse N-heterocycles (Scheme 2).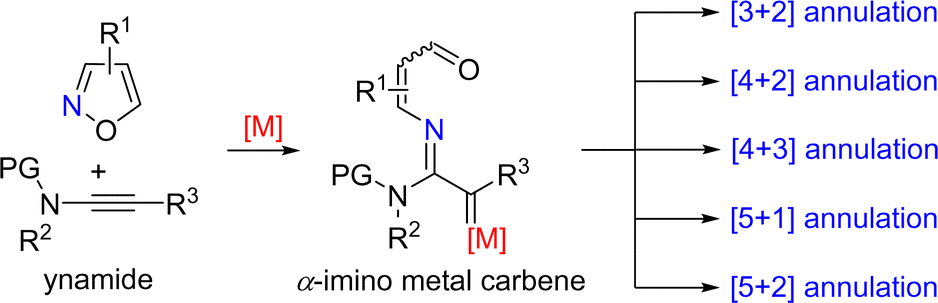 | ||
| Scheme 2 Formal annulation reactions of ynamides with isoxazoles. | ||
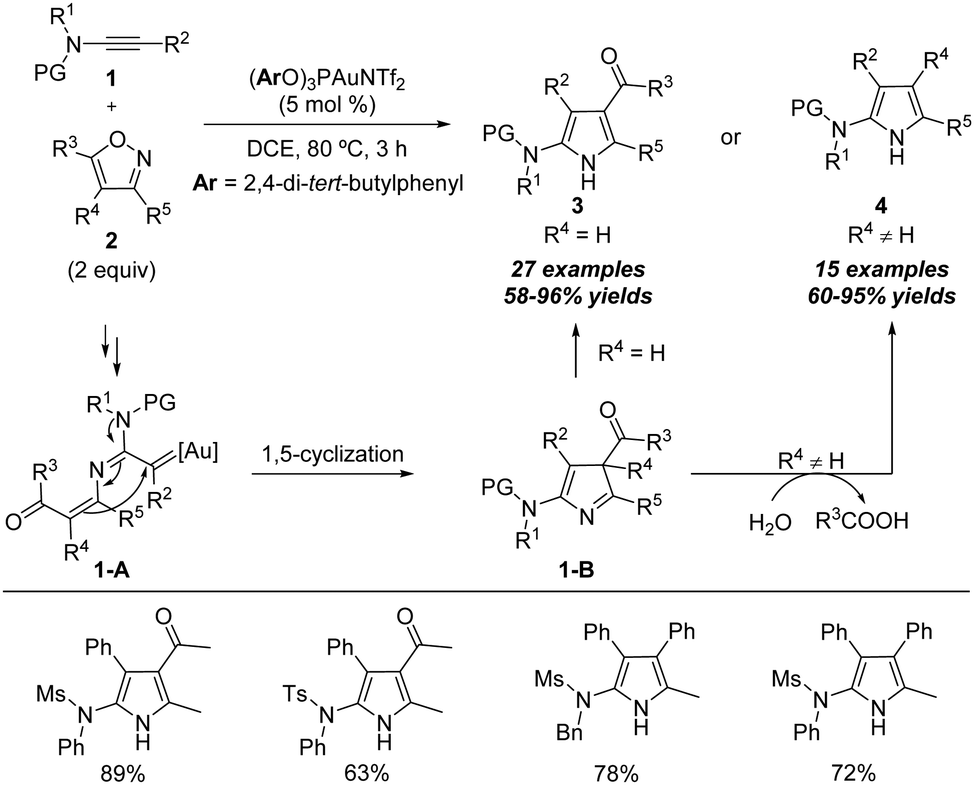 | ||
| Scheme 3 Gold-catalyzed formal [3+2] annulation of ynamides 1 with isoxazoles 2. | ||
The proposed mechanism of the above gold-catalyzed [3+2] annulation of ynamides with isoxazoles is shown below. First, nucleophilic attack of isoxazole 2 onto the gold-activated triple bond of ynamides and N–O bond cleavage lead to the formation of the α-imino gold carbene intermediate 1-A. Subsequently, a 1,5-cyclization of 1-A occurs to form the 3H-pyrrole intermediate 1-B and releases the gold catalyst. Then 3H-pyrrole 1-B readily isomerizes into the 1H-pyrrole 3 via a 1,3-H shift when the disubstituted isoxazole is used as the substrate. Alternatively, for the trisubstituted isoxazole substrate, a water-assisted deacylative aromatization of 1-B occurs to afford the final 1H-pyrrole 4.
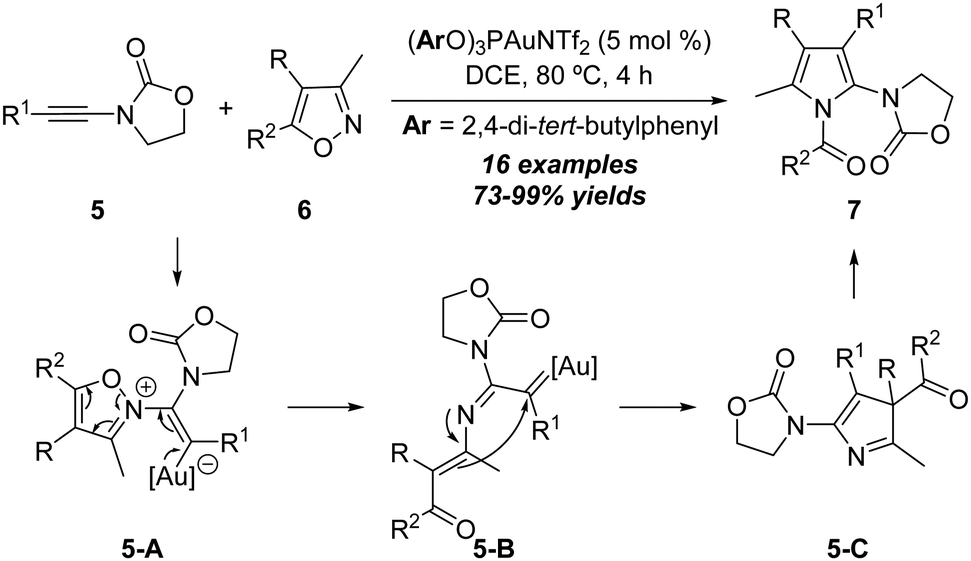
In 2015, the same group disclosed another interesting gold-catalyzed formal [3+2] annulation between ynamides and isoxazoles.10 The treatment of oxazolinone-type ynamides 5 with isoxazoles 6 in the presence of the (ArO)3PAuNTf2 (Ar = 2,4-di-tert-butylphenyl) catalyst afforded a series of N-acetylpyrroles 7 in 73–99% yields (Scheme 4). Substrates bearing both electron-donating and -withdrawing groups on the aryl substituents could be well tolerated in this [3+2] annulation. The reaction starts with the coordination of the gold catalyst with ynamide 5, which is attacked by isoxazole 6 to form the vinyl gold intermediate 5-A. Then, ring opening of isoxazole moieties in 5-A happens to deliver the α-imino gold carbene intermediate 5-B which can be trapped by the intramolecular enamine moiety to form 3H-pyrrole intermediate 5-C. Finally, an intermolecular nucleophilic attack of 5-C by another intermediate 5-C occurs to provide the desired product 7.
 | ||
| Scheme 4 Gold-catalyzed [3+2] annulation of ynamides 5 with isoxazoles 6. | ||
Similarly, benzo[c]isoxazoles were also developed as amination reagents. In 2016, Hashmi and co-workers reported a gold-catalyzed formal [3+2] annulation of ynamides 8 with anthranils (benzo[c]isoxazoles) 9 by employing IPrAuCl/AgNTf2 as the catalyst, affording a variety of 7-acylindoles 10 in generally good yields (Scheme 5).11 Interestingly, unpolarized alkynes 11 were also proven as suitable substrates in this annulation, and the corresponding 7-acylindoles 12 were produced in 50–74% yields.
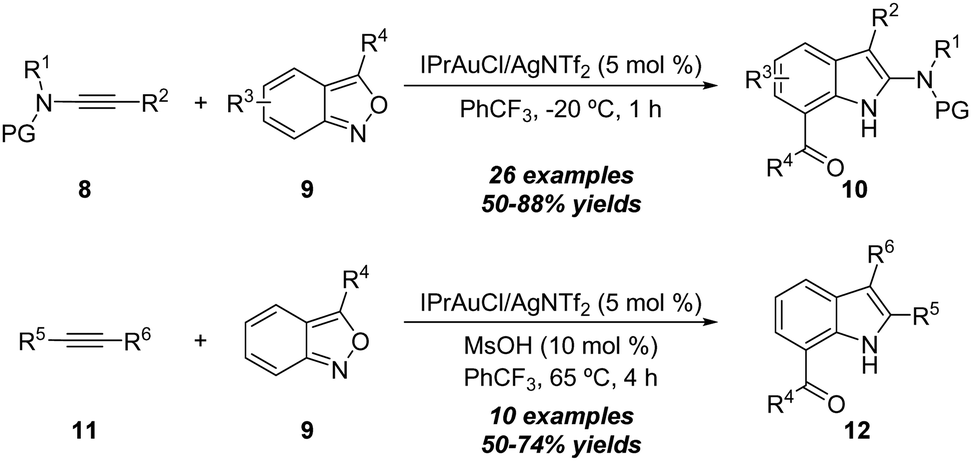 | ||
| Scheme 5 Gold-catalyzed formal [3+2] annulation of ynamides 8 or unpolarized alkynes 11 with anthranils 9. | ||
In 2020, the same group reported another related gold-catalyzed formal [3+2] annulation/acyl migration of ynamides 13 with anthranils 14 by employing PicAuCl2 as the catalyst, furnishing the 6-acylindoles 15 or 5-acylindoles 16 in moderate to excellent yields via selective 1,4- or 1,3-acyl migration (Scheme 6).12 Based on the control experiments and density functional theory (DFT) calculations, the mechanism for this selective 1,4- or 1,3-acyl migration is proposed. Initially the α-imino gold carbene intermediate 13-A is generated from nucleophilic N-attack of anthranil and N–O bond cleavage. After intramolecular cyclization, the consecutive 1,2-acyl shift and anthranil-assisted deprotonation/deauration/protonation process occur to furnish 6-acylindole 15.
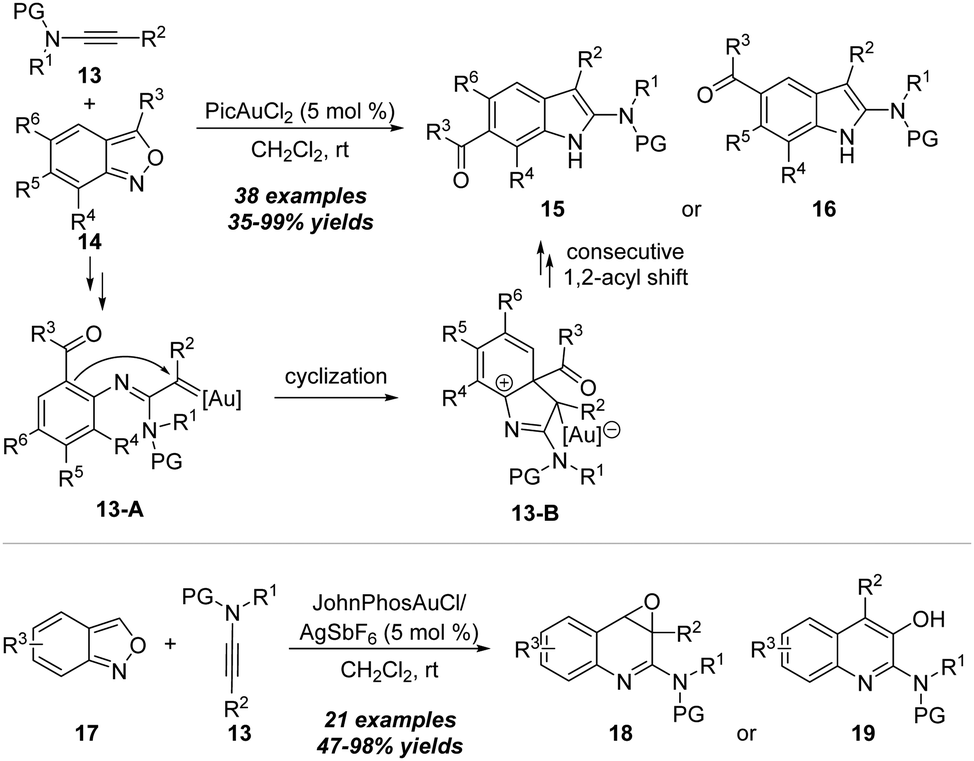 | ||
| Scheme 6 Gold-catalyzed formal [3+2] annulation of ynamides 13 with anthranils 14 or 17. | ||
In the same work, the switchable synthesis of quinoline oxides 18 and quinolines 19 from gold-catalyzed amination of ynamides 13 with anthranils 17 was demonstrated through 1,7-cyclization. With 5 mol% JohnPhosAuCl and AgSbF6 in dichloromethane at room temperature, the reaction proceeded smoothly with various functional groups, including electron-withdrawing and electron-donating groups on the aromatic ring, leading to the preparation of a range of quinoline oxides 18 and the rearranged quinoline products 19 in moderate to excellent yields.
Recently, a unique gold-catalyzed formal [3+2] annulation of yndiamides 20 with isoxazoles 21 was demonstrated by Anderson group for the direct synthesis of polysubstituted diaminopyrroles 22 (Scheme 7).13 By using (ArO)3PAuNTf2 (Ar = 2,4-di-tert-butylphenyl) as the catalyst and 3 Å MS as the additive in DCE, a series of functionalized diaminopyrroles 22 were obtained in moderate to good yields. This methodology tolerates a wide range of aryl and alkyl isoxazoles, as well as various symmetrical yndiamides. It is worth noting that the diaminopyrroles could be obtained in good regioselectivities when employing unsymmetrical yndiamide substrates. The reaction starts with the addition of isoxazole 21 onto gold-activated yndiamides to afford the vinyl gold intermediate 20-A. Subsequent ring opening provides α-imino gold carbene intermediate 20-B, which can undergo intramolecular cyclization to produce iminium intermediate 20-C. Next, 20-C undergoes protodeauration to afford 3H-pyrrole 20-D. Finally, diaminopyrrole 22 forms via double 1,5-H migration.
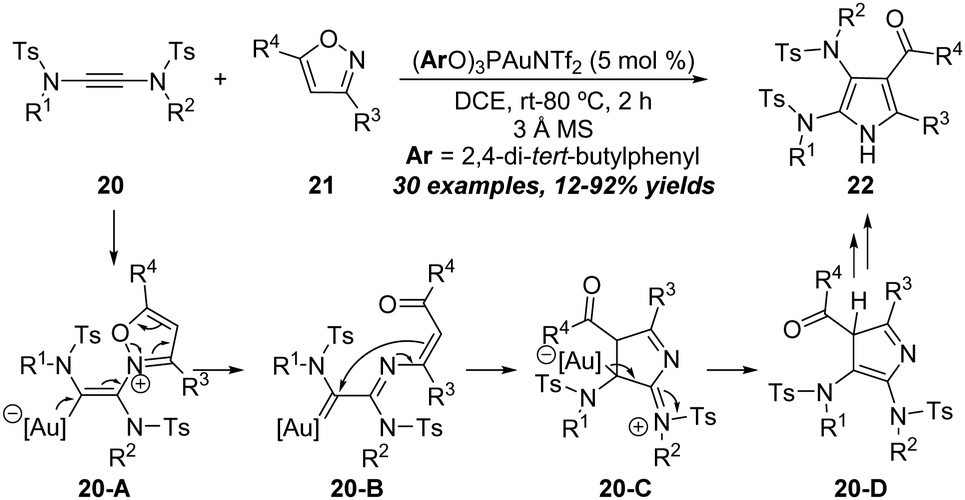 | ||
| Scheme 7 Gold-catalyzed formal [3+2] annulation of yndiamides 20 with isoxazoles 21. | ||
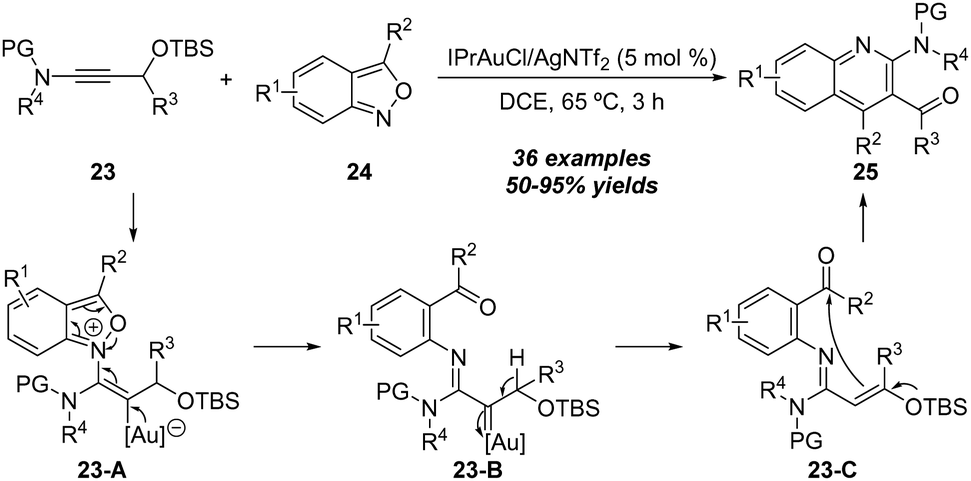 | ||
| Scheme 8 Gold-catalyzed [4+2] annulation of propargylic silyl ethers 23 with anthranils 24. | ||
In 2018, Hashmi and co-workers realized another gold-catalyzed [4+2] annulation of ynamides with isoxazoles (Scheme 9).15 In the presence of 5 mol% KAuBr4 catalyst, the reaction proceeded at −20 °C for 2 h, and was then heated to 40 °C. The formal [4+2] annulation of N-benzyl ynamides 26 with anthranils 27 led to the desired quinoline-based polyazaheterocycles 28 in 24–72% yields. The plausible mechanism involves the formation of α-imino gold carbene intermediate 26-B, followed by the subsequent N-benzyl C–H insertion of gold carbene, and enamine-aldehyde addition/elimination to generate the expected quinoline-based polyazaheterocycle 28.
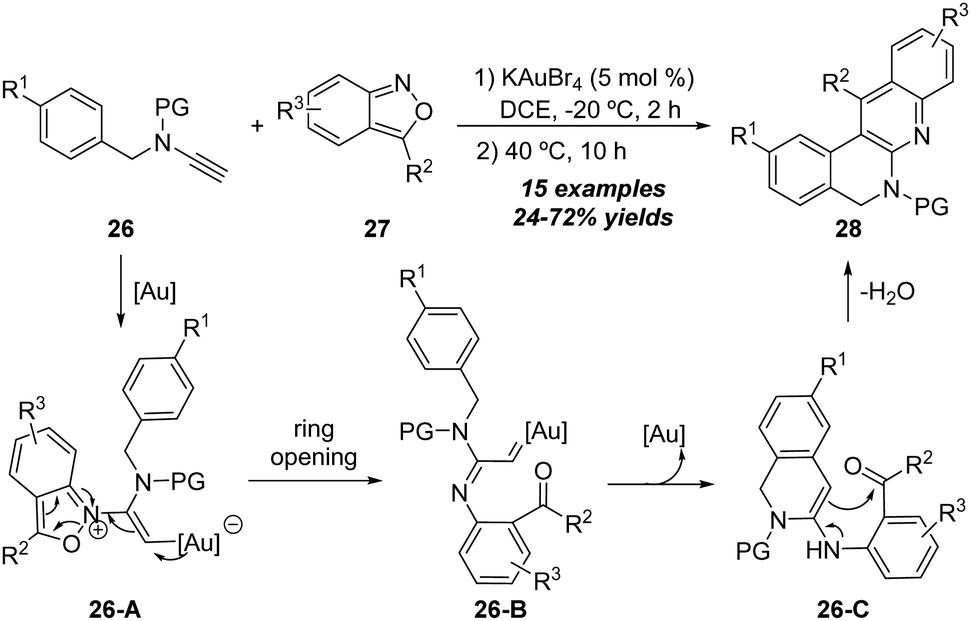 | ||
| Scheme 9 Gold-catalyzed [4+2] annulation of N-benzyl ynamides 26 and anthranils 27. | ||
In the same report, the authors demonstrated a gold-catalyzed [4+2] annulation of N-furanylmethyl ynamides with anthranils. As shown in Scheme 10, 2-amino pyrroles 30 were initially obtained under the optimized reaction conditions from the amination of N-furanylmethyl ynamides 29 with anthranils 27, which could be readily transformed into thermodynamically more stable pyrrolo[2,3-b]quinolines 31. Mechanistically, the α-imino gold carbene 29-B is first generated from nucleophilic addition and the fragment of N–O bonds. Then 29-B is captured by N-furanylmethylene species to generate spiro intermediate 29-C, followed by a C–O cleavage of the furan ring (29-C) to form the imine intermediate 29-D, which can be transformed into 2-amino pyrrole 30 via aromatization. Finally, a Friedel–Crafts type cyclization of 2-amino pyrrole 30 affords the desired pyrrolo[2,3-b]quinoline 31.
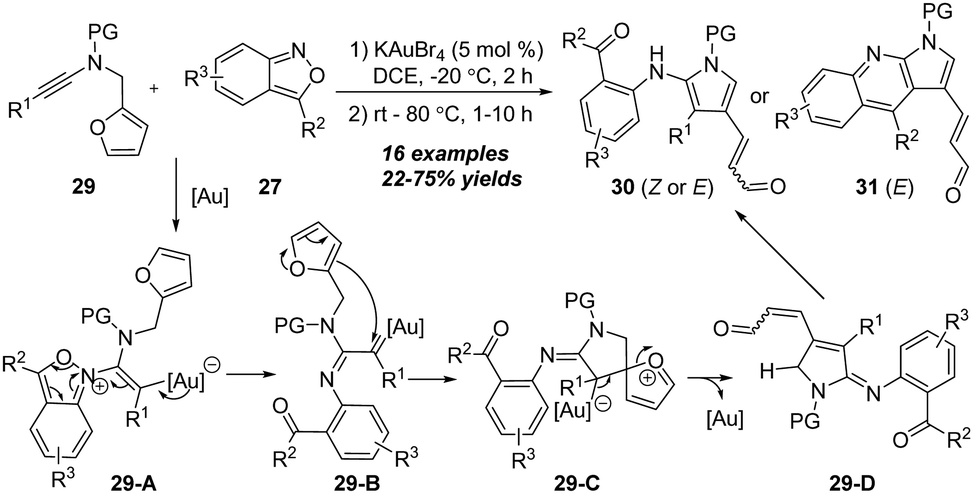 | ||
| Scheme 10 Gold-catalyzed [4+2] annulation of N-furanylmethyl ynamides 29 with anthranils 27. | ||
By using a similar strategy, in 2018, Liu and co-workers disclosed a gold-catalyzed tandem [4+2] annulation of N-aryl ynamides 32 with anthranils 33, delivering diverse 6H-indolo[2,3-b]quinoline derivatives 34 in 19–88% yields (Scheme 11).16 Similar to the above report,15 the key N-phenyl C–H insertion of gold carbene and the enamine-aldehyde addition/elimination were involved in this [4+2] annulation.
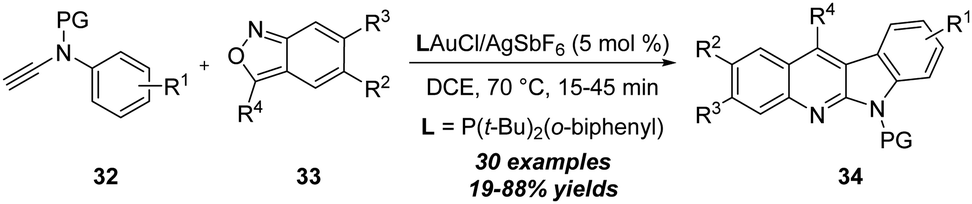 | ||
| Scheme 11 Gold-catalyzed [4+2] annulation of N-aryl ynamides 32 with anthranils 33. | ||
Later in 2019, the same group further reported that this formal [4+2] cycloaddition strategy was also applicable to the reaction of N-propargyl ynamides 35 with anthranils 36 under gold catalysis.17 As outlined in Scheme 12, the reaction of N-propargyl ynamides 35 with anthranils 36 took place efficiently in the presence of 5 mol% of AuCl3, and various pyrrolo[2,3-b]quinolines 37 could be produced in 31–91% yields after the treatment of 20 mol% TsOH. The reaction of N-propargyl ynamide 35 and anthranil 36 first generates α-imino gold carbene intermediate 35-A, which is trapped by the alkyne moiety via carbene/alkyne metathesis, affording vinyl cation intermediate 35-B. After that, vinyl cation 35-B is trapped by water, followed by elimination of the gold catalyst, providing intermediate 35-C. Further oxidation/intramolecular cyclization/elimination occurs to deliver pyrrolo[2,3-b]quinoline 37.
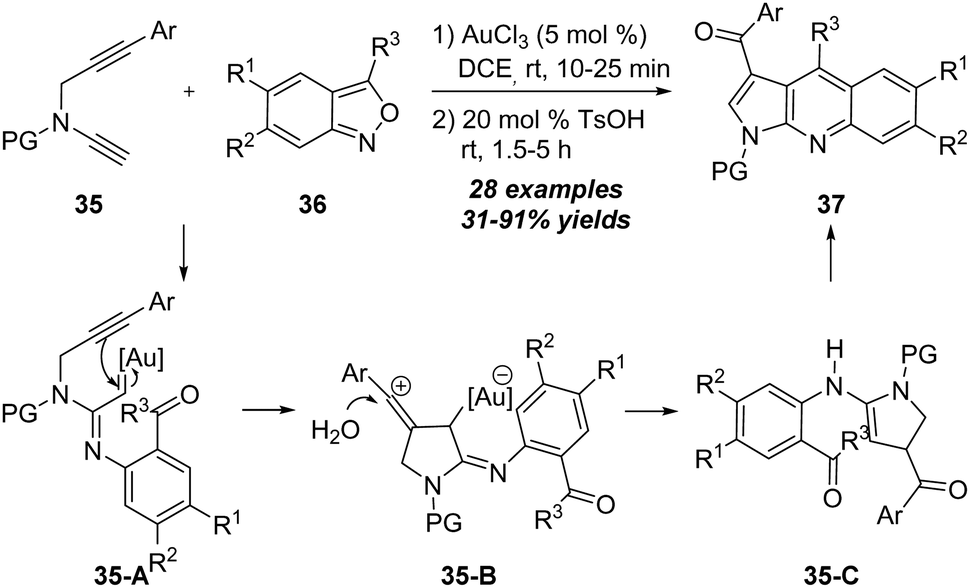 | ||
| Scheme 12 Gold-catalyzed tandem [4+2] annulation of N-propargyl ynamides 35 with anthranils 36. | ||
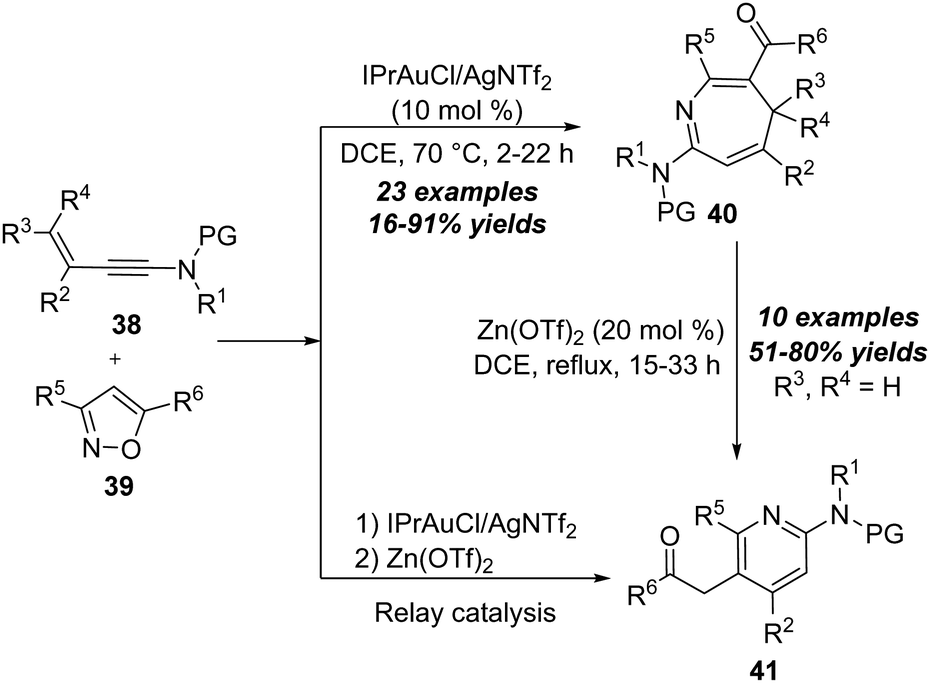 | ||
| Scheme 13 Gold-catalyzed formal [4+3] annulation of 3-en-1-ynamides 38 with isoxazoles 39. | ||
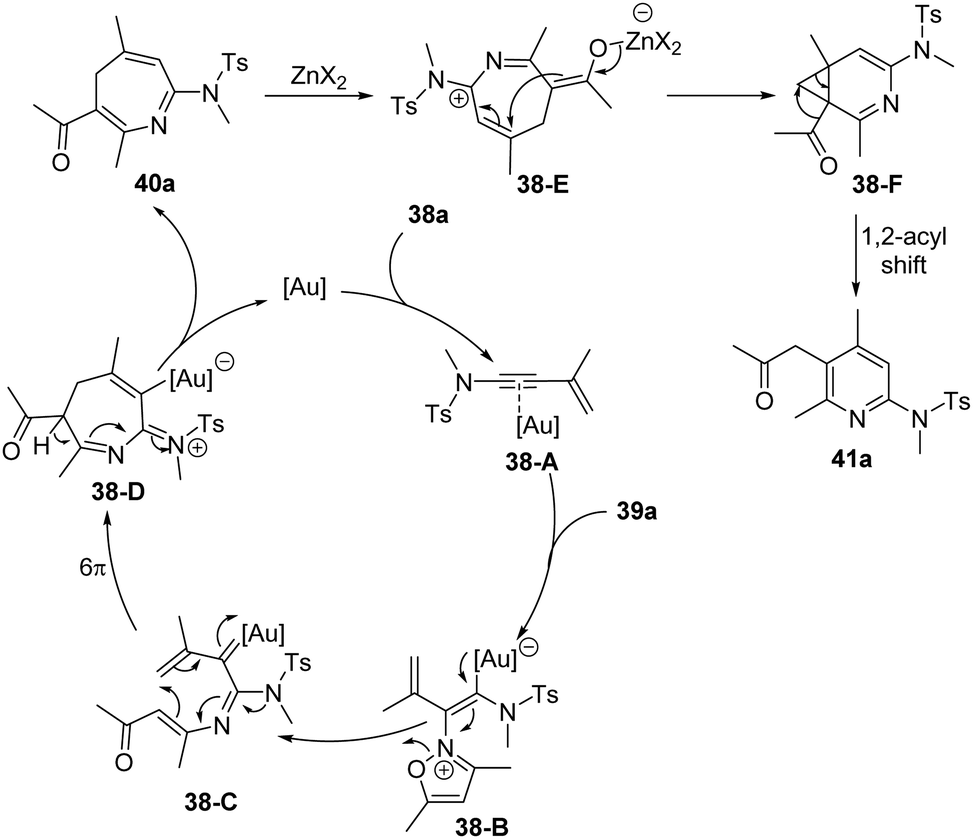
A proposed mechanism for this formal [4+3] annulation is depicted in Scheme 14. The reaction begins with coordination of the gold catalyst with ynamide 38a, followed by nucleophilic addition with isoxazole 39a, delivering the vinyl gold intermediate 38-B. Subsequent ring-opening forms the α-imino gold carbene 38-C. Then, a heptatrienyl cation type 6π electrocyclization occurs to produce seven-membered iminium intermediate 38-D. Subsequent elimination and protodeauration of 38-D furnish 4H-azepine 40a with the regeneration of the gold catalyst. Further treatment of 4H-azepine 40a with the zinc catalyst generates 2-azapentadienyl cation 38-E, that undergoes an intramolecular cyclization to afford cyclopropane intermediate 38-F. Finally, a 1,2-acyl shift of 38-F takes place to yield 2-amino pyridine 41a.
 | ||
| Scheme 14 Proposed mechanism for gold-catalyzed formal [4+3] annulation of 3-en-1-ynamides 38 with isoxazoles 39. | ||
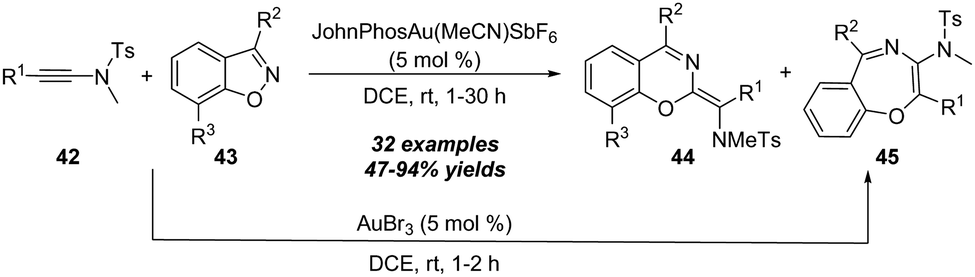 | ||
| Scheme 15 Gold-catalyzed formal [5+1]/[5+2] annulation of ynamides 42 with benzo[d]isoxazoles 43. | ||
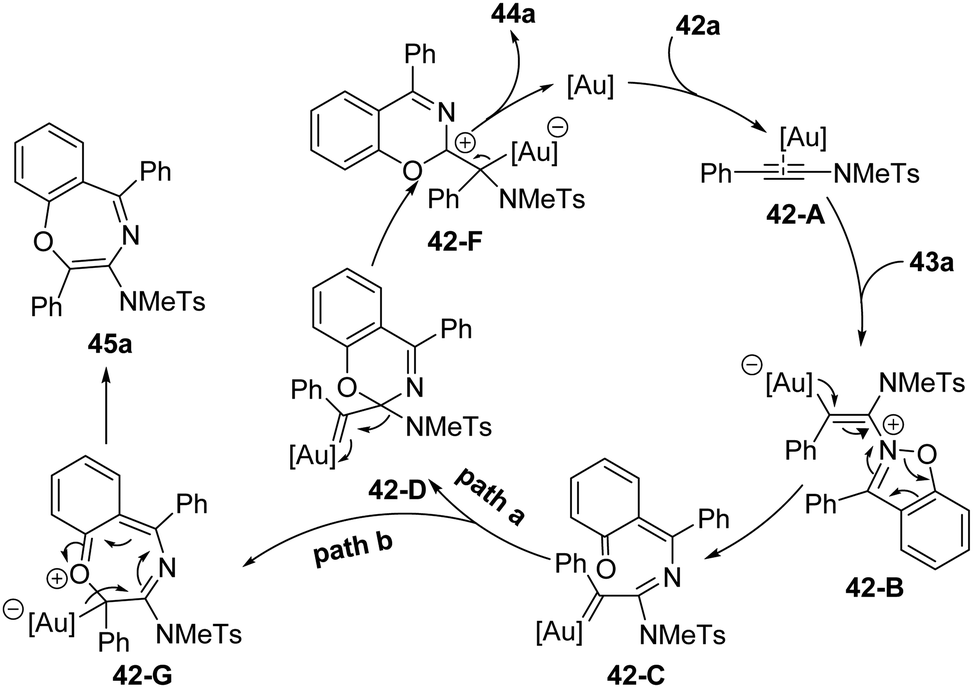
The proposed mechanism for this gold-catalyzed cascade formal [5+1]/[5+2] annulation is depicted in Scheme 16. The reaction commences with the coordination of the gold catalyst with ynamide 42a followed by nucleophilic attack, furnishing vinyl gold intermediate 42-B. A N–O bond fragment of isoxazole moiety leads to α-imino gold carbene intermediate 42-C. Then, there are two possible pathways for the intermediate 42-C. In path a, 6-membered intermediate 42-D is produced through a 6π electrocyclization. Subsequent 1,2-amino migration of 42-D followed by the elimination of the gold catalyst provides the 2H-benzo[e][1,3]oxazine 44a. Alternatively, in path b, a 1,7-cyclization via O-attack to gold carbene 42-C occurs to deliver 7-membered intermediate 42-G. Finally, the elimination of the gold catalyst yields the final benzo[f][1,4]oxazepine 45a. Such a type of protocol on gold-catalyzed formal [5+1] annulation of ynamides with benzo[d]isoxazoles was also nicely demonstrated by Sahoo and co-workers in 2019.20
 | ||
| Scheme 16 Proposed mechanism for gold-catalyzed formal [5+1]/[5+2] annulation of ynamides 42 with benzo[d]isoxazoles 43. | ||
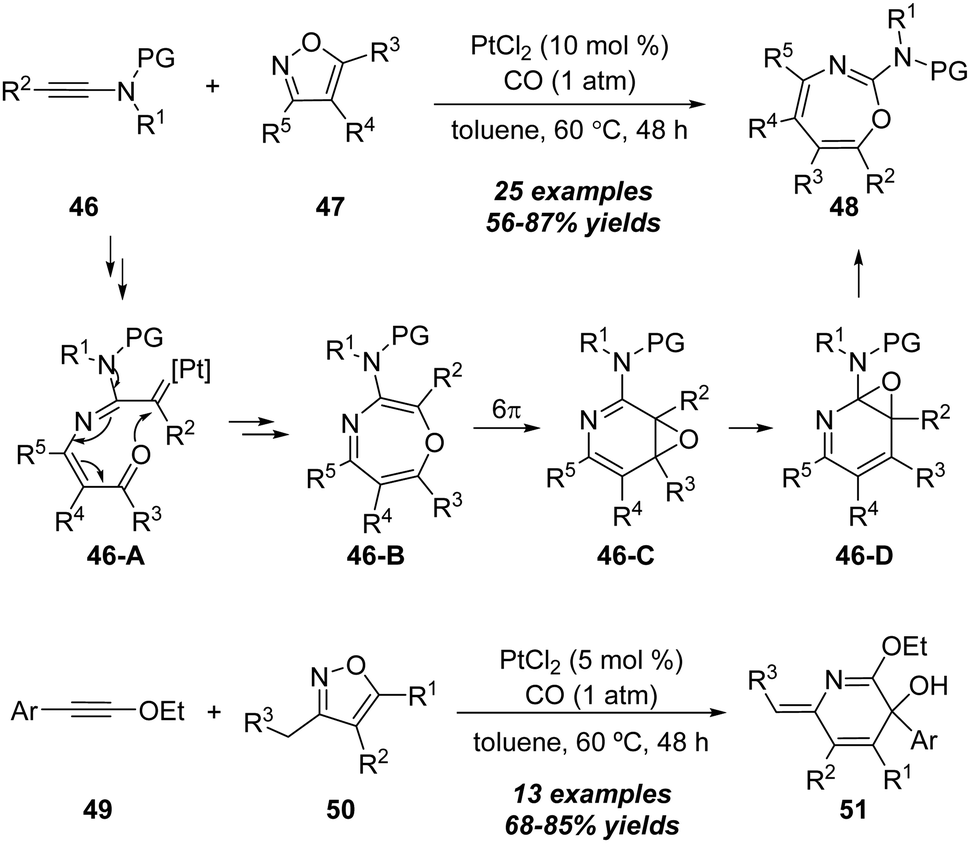 | ||
| Scheme 17 Platinum-catalyzed formal [5+2] annulation of alkynes with isoxazoles. | ||
In the same work, a selective platinum-catalyzed formal [4+2] annulation was also achieved when the ynol ethers 49 were used as substrates. As outlined in below, the reaction of ynol ethers 49 with various isoxazoles 50 in the presence of PtCl2 (5 mol%) under CO atmosphere in toluene led to the construction of 2,5-dihydropyridines 51 in generally moderate to excellent yields.
In 2018, Liu group reported gold-catalyzed formal [5+2] and [5+1] annulations between ynamides 52 and 1,2-benzisoxazoles 53 (Scheme 18).22 A series of 2H-benzo[e][1,3]oxazines 54 and benzo[f][1,4]oxazepines 55 could be selectively synthesized in high yields through the ligand-controlled chemoselectivity. By employing IPrAuCl/AgNTf2 as the catalyst, aryl-substituted ynamides underwent the formal [5+2] annulation via a 1,7-cyclization; however P(t-Bu)2(o-biphenyl)AuCl/AgNTf2 changed the chemoselectivity to a formal [5+1] annulation. Control experiments indicated that a 1,2-sulfonamide shift was involved in the formal [5+1] annulation process.
 | ||
| Scheme 18 Gold-catalyzed formal [5+2] and [5+1] annulations between ynamides 52 and 1,2-benzisoxazoles 53. | ||
In 2018, Wan and Hu reported an interesting Brønsted acid-catalyzed formal [5+2+1] annulation of ynamides and isoxazoles with water.23 As described in Scheme 19, a variety of oxygen-bridged tetrahydro-1,4-oxazepines 58 were isolated in generally moderate to good yields by employing ynamides 56, isoxazoles 57 and water using 15 mol% HNTf2 as the catalyst. It is worth noting that this protocol represents the first metal-free reaction of isoxazoles with alkynes.
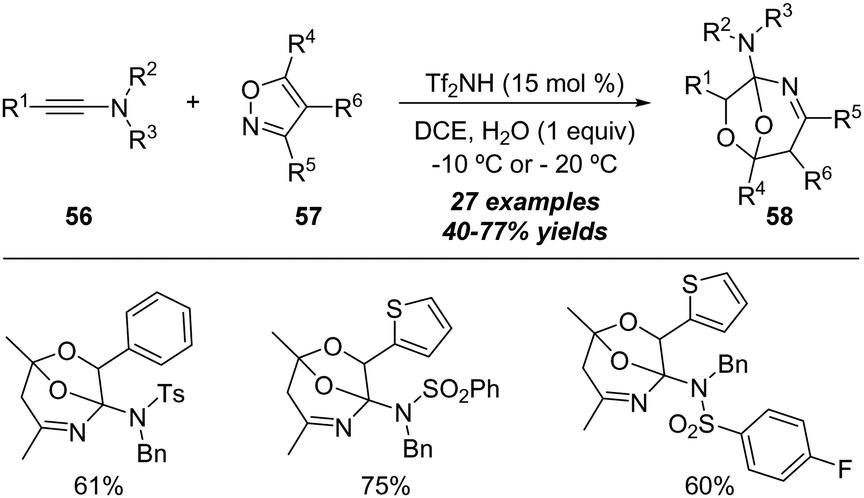 | ||
| Scheme 19 Brønsted acid-catalyzed formal [5+2+1] cycloaddition of ynamides 56 and isoxazoles 57 with water. | ||
The proposed mechanism is shown in Scheme 20. First, ynamide 56a reacts with HNTf2 to give keteniminium intermediate 56-A, which was attacked by isoxazole 57a to afford intermediate 56-B. Then carbocation 56-C is generated from ring-opening, and the carbocation 56-C may go through two pathways. For path a, a subsequent intramolecular 1,7-cyclization of 56-C delivers the seven-membered intermediate 56-D. Ultimately, the addition of H2O to the iminium 56-D and subsequent acid-catalyzed ketalization afford the bridged tetrahydro-1,4-oxazepine 58a. In path b, water may attack the carbonyl group of 56-C first to assist the intramolecular cyclization, affording the seven-membered intermediate 56-G. The desired product 58a forms by the acetal formation step with the regeneration of the catalyst.
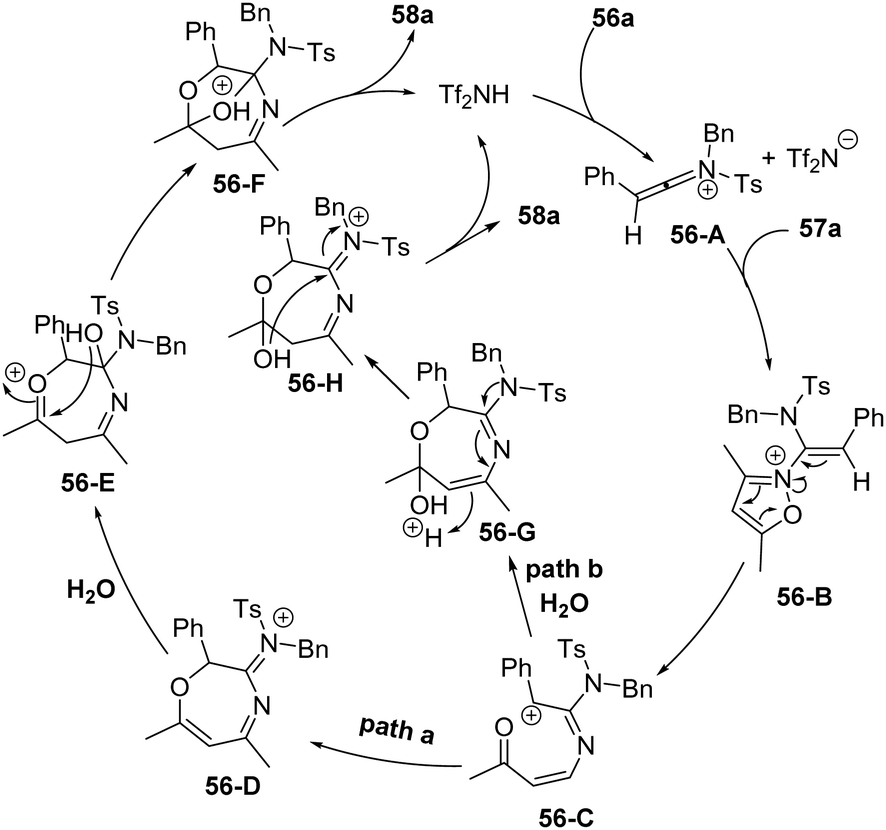 | ||
| Scheme 20 Proposed mechanism for Brønsted acid-catalyzed formal [5+2+1] cycloaddition of ynamides 56 and isoxazoles 57 with water. | ||
In 2021, Liu and co-workers reported another related gold-catalyzed formal [5+2] annulation of 1,3-diynamides 59 with anthranils 60 for the efficient synthesis of functionalized quiniline oxides 61.24 The [3+2] annulation adducts 7-formylindoles 62 were also generated as minor products. Interestingly, the furoquinoline derivatives 63 could be obtained from quiniline oxides 61 through a gold-catalyzed intramolecular cyclization/aromatization sequence (Scheme 21).
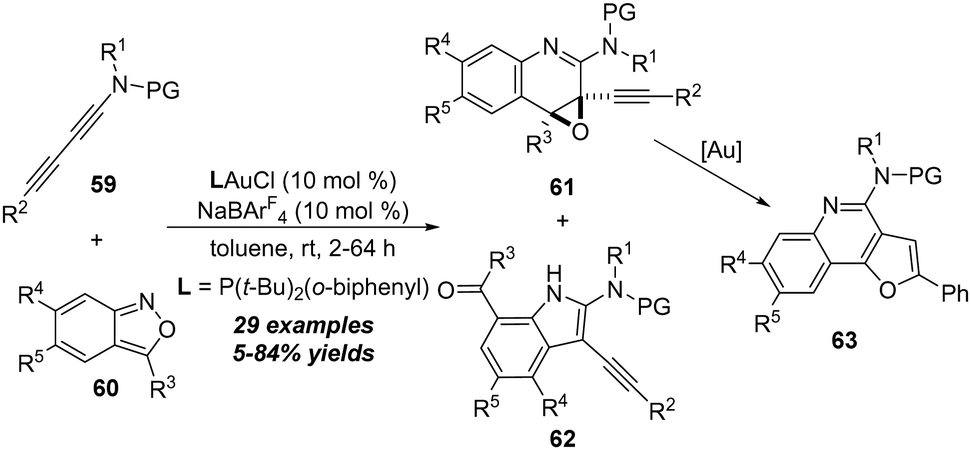 | ||
| Scheme 21 Gold-catalyzed formal [5+2] annulation of 1,3-diynamides 59 with anthranils 60. | ||
2.2. Cyclopropanation
In the amination of alkynes, the key metal carbene intermediates could also undergo cyclopropanation to give cyclopropane-fused heterocycles. In 2019, Hashmi and co-workers demonstrated a gold-catalyzed cyclopropanation between readily available N-allylynamides 65 and anthranils 64 (Scheme 22).25 This protocol features simple conditions, high efficiency, and excellent functional group compatibility. However, the alkene moieties on N-allylynamides are limited to di- and trisubstituted alkenes. The mechanism of this intermolecular tandem reaction is postulated to involve the formation of vinyl gold intermediate 64-A, followed by ring-opening of 64-A, leading to the formation of α-imino gold carbene 64-B. Finally, intramolecular cyclopropanation of 64-B delivers the corresponding 3-azabicyclo[3.1.0]hexane-2-imine 66.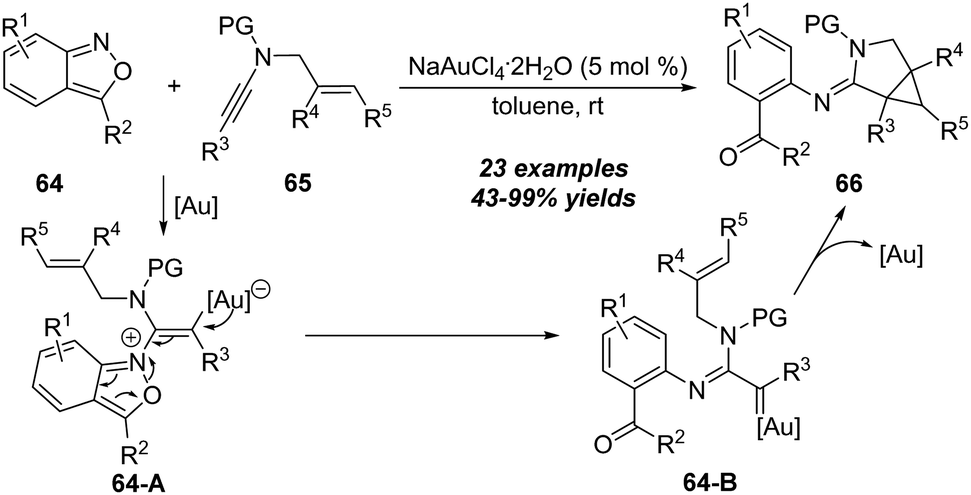 | ||
| Scheme 22 Gold-catalyzed cyclopropanation between N-allylynamides 65 and anthranils 64. | ||
3. Amination of ynol ethers
In 2018, Ye, Lu and co-workers reported a zinc-catalyzed [3+2] annulation reaction of ynol ethers 67 with 3,5-disubstituted isoxazoles 68 under mild reaction conditions, leading to the atom-economic synthesis of 2-alkoxyl 1H-pyrroles 69 in 62–96% yields (Scheme 23).26 Interestingly, the scope of the above zinc-catalyzed transformation could be extended to the reaction of ynol ether 67a with trisubstituted isoxazoles 70, and 3H-pyrroles 71 were selectively formed in moderate to excellent yields under similar reaction conditions. Notably, this protocol represents the first example of non-noble metal-catalyzed reaction of ynol ethers with isoxazoles. The reaction starts with the nucleophilic attack of isoxazole 68 on the zinc-activated ynol ether to afford the vinyl zinc intermediate 67-A, which is further transferred into the carbocation intermediate 67-B upon the cleavage of the N–O bond. Then, a 1,5-cyclization takes place to form intermediate 67-C, which undergoes deprotonation and 1,3-H shift to give the final product 69. Of note, the zinc-catalyzed reaction showed significant difference with platinum catalysis.21 In addition, theoretical calculations provided further evidence for the proposed reaction mechanism, especially for the distinct selectivity.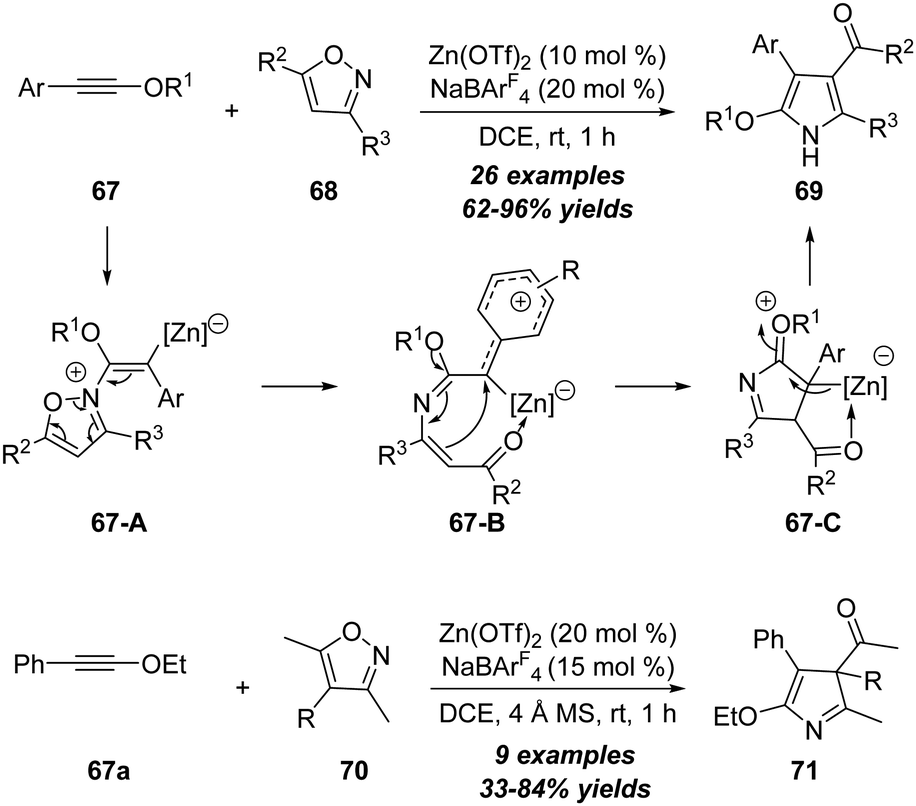 | ||
| Scheme 23 Zinc-catalyzed [3+2] annulation reaction of ynol ethers 67 with 3,5-disubstituted isoxazoles 68 or 70. | ||
In 2019, Liu's group further realized a gold-catalyzed annulation of ynol ethers 72 with anthranils 73, leading to the formation of benzofuro[2,3-b]quinolines and 6H-chromeno[3,4-b]quinolines 74 efficiently (Scheme 24).27 This new synthetic method employed readily available starting materials through a one-pot reaction for the synthesis of biologically significant benzofuro[2,3-b]quinolines. Mechanistically, the formation of the α-imino gold carbene intermediate and intramolecular C–H insertion are the key for this reaction. In the same work, the authors also assessed the scope of a one-pot transformation for the direct synthesis of 6H-chromeno[3,4-b]quinolines from a series of propargyl ethers 75 and anthranils 73. In the presence of 10 mol% of gold(I) and 20 mol% of zinc(II) as catalysts, the annulation delivered the desired 6H-chromeno[3,4-b]quinolines 76 in 18–84% yields.
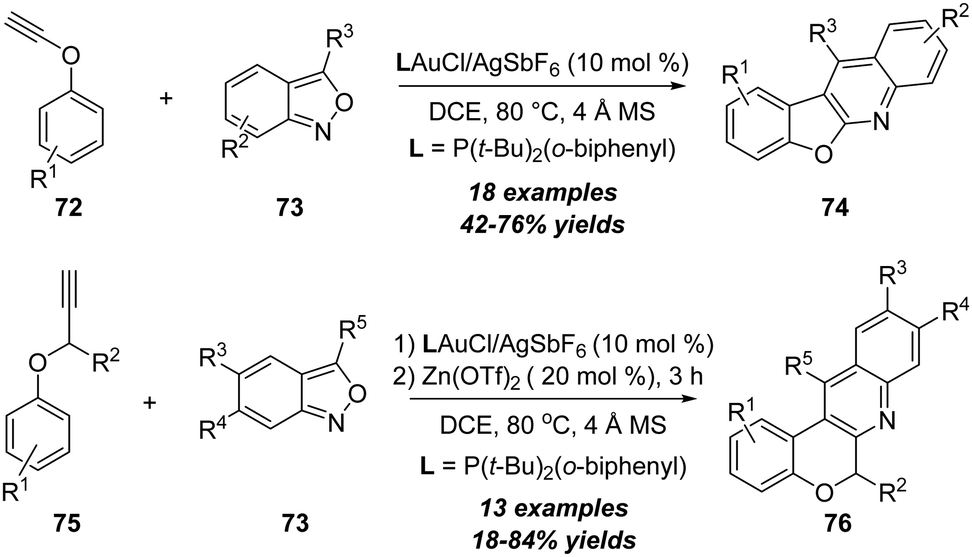 | ||
| Scheme 24 Gold-catalyzed annulation of ynol ethers 72 or 75 with anthranils 73. | ||
4. Amination of thioynol ethers
Different with ynamides and ynol ethers, the amination reaction of thioynol ethers demonstrated unique reactivities due to the special properties of sulfur atoms. In 2018, Ye, Lu and co-workers reported a novel zinc-catalyzed reaction of thioynol ethers 77 with isoxazoles 78, leading to the preparation of β-keto enamides 79 in moderate yields, which could be readily converted to diverse heterocycles (Scheme 25).28 The reaction proceeded through an unprecedented 1,2-sulfur migration based on thioynol ethers. Importantly, this protocol not only represents the first example of the non-noble metal-catalyzed reaction of isoxazoles with alkynes, but also represents the first non-noble metal-catalyzed alkyne amination by an N–X bond nucleophile (X = N or O). The reaction begins with the nucleophilic attack of isoxazole 78 onto the zinc-coordinated thioynol ether to provide a vinyl zinc intermediate 77-A, which can isomerize into the phenyl-stabilized carbocation intermediate 77-B upon cleavage of the isoxazole N–O bond. Followed by 1,2-sulfur migration to produce migrated intermediate 77-C. Through DFT calculations, it is found that 1,2-sulfur migration is kinetically favorable, which eventually furnishes β-keto enamide 79 via H2O addition, proton transfer and proton demetallization.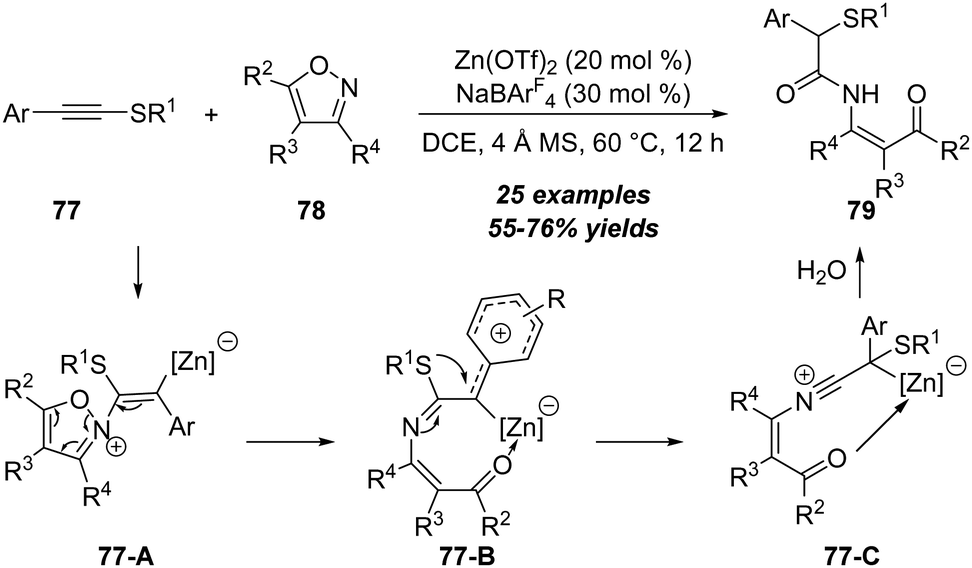 | ||
| Scheme 25 Zinc-catalyzed reaction of thioynol ethers 77 with isoxazoles 78. | ||
By employing the amination of thioynol ethers, Davies group extended this strategy to the synthesis of sulfenyl pyrroles 83 and indoles 84. In the presence of 5 mol% of gold(I) as the catalyst, the [3+2] annulation of alkynyl thioethers 80 with isoxazoles 81 or 82 led to the convergent and efficient synthesis of the corresponding sulfenylated pyrroles 83 or indoles 84 in moderate to excellent yields (Scheme 26).29 This reaction showed broad structural and functional group tolerance. Initial investigations into the annulation with trisubstituted isoxazoles also revealed the unique reactivity for alkynyl thioethers. Different from typical α-addition of alkynyl thioethers, the observed results and isotopic labelling studies matched with the β-selective addition pathway.
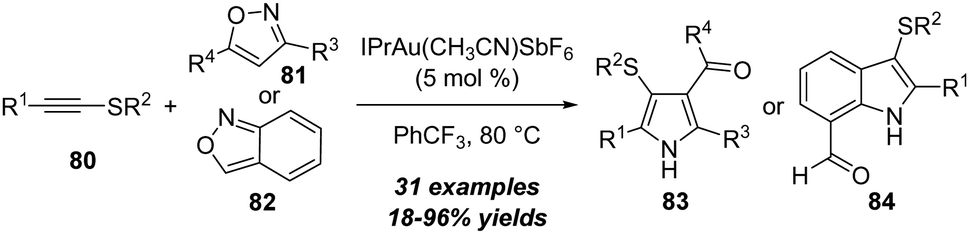 | ||
| Scheme 26 Gold-catalyzed formal [3+2] annulation of alkynyl thioethers 80 with isoxazoles 81 or 82. | ||
5. Amination of electron-deficient alkynes
Apart from the above electron-rich alkynes, electron-deficient alkynes were also suitable substrates to react with isoxazoles and led to the synthesis of heterocycles. In 2017, Liu group first disclosed a gold-catalyzed [4+1] annulation between propiolate derivatives 85 and isoxazoles 86, leading to facile synthesis of 2,4-dicarbonylpyrroles 87 in generally moderate to excellent yields (Scheme 27).30 Both aromatic and alkyl substituted propiolates 85 were suitable substrates to produce the target pyrrole products. The reaction mechanism involves the formation of α-oxo gold carbene intermediate 85-A, ring closure (85-B), intramolecular cyclization and ring-opening of 85-C.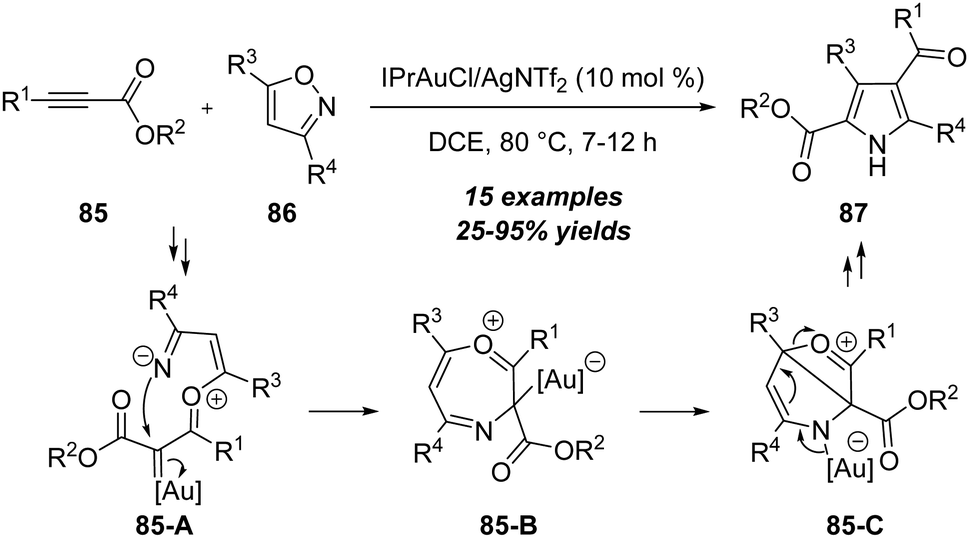 | ||
| Scheme 27 Gold-catalyzed [4+1] annulation between propiolate derivatives 85 and isoxazoles 86. | ||
In the same report, the authors found that the unsubstituted isoxazoles 88 could proceed through the N-attack onto propiolates 85 in the presence of 10 mol% IPrAuCl/AgNTf2, and led to the formation of 3,8-dicarbonylimidazo[1,2-a]pyridines 89 in 61–75% yields via a formal [2+2+1]/[4+2] annulation (Scheme 28).30 As shown below, a similar formal [5+2] annulation of propiolate 85 with isoxazole 88 first occurs to furnish the 7-membered intermediate 88-A through a N-attack. Further Diels–Alder reaction with another isoxazole 88 affords the bridged intermediate 88-B. An aromatization of intermediate 88-B forms α-imino gold carbene 88-C by the loss of water, which is attacked by the pyridine moiety and isomerized to afford the desired imidazo[1,2-a]pyridine 89.
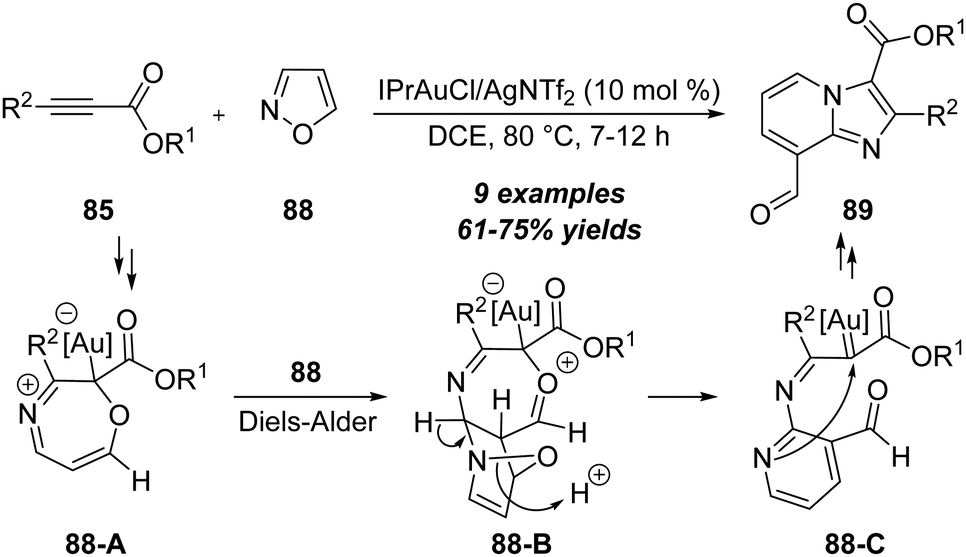 | ||
| Scheme 28 Gold-catalyzed formal [2+2+1]/[4+2] annulation. | ||
In the same year, Liu and co-workers extended the reaction to readily available anthranil substrates and developed a gold-catalyzed formal [4+2] annulation/cyclization cascade of propiolate derivatives 90 with anthranils 91, delivering quinoline oxides 92 in moderate to excellent yields with excellent diastereoselectivities (Scheme 29).31 Significantly, a novel relay catalysis using gold and zinc catalysts was also achieved to produce bridged oxygenated tetrahydroquinoline derivatives 93 in high yields with good to excellent diastereoselectivities. Mechanistically, the reaction of alkyne with anthranil first gives α-oxo gold carbene 90-A, followed by 1,7-cyclization, 6π electrocyclization, water attack and Payne rearrangement to produce expoxide 90-C. Finally, product 93 forms via ring-opening and lactonization.
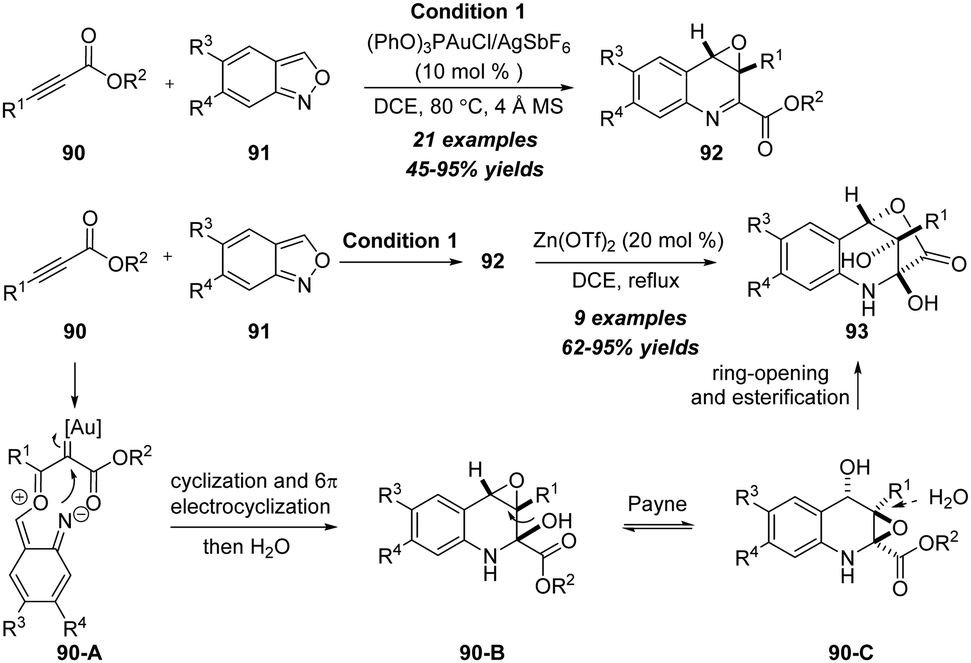 | ||
| Scheme 29 Gold-catalyzed formal [4+2] annulation/cyclization cascade of propiolate derivatives 90 with anthranils 91. | ||
Later in 2018, the same group demonstrated a gold-catalyzed formal [4+2] annulation between propiolates and 1,2-benzisoxazoles (Scheme 30).32 When using tert-butyl propiolates 94 as substrates, this reaction preferably underwent formal [4+2] annulation, providing 6H-1,3-oxazin-6-one derivatives 96 in generally high yields. Alternatively, a direct Michael-type addition occurred when using ethyl propiolates as substrates. In addition, gold-catalyzed Michael-type reactions of ethyl propiolates 97 with 1,2-benzisoxazoles 95 led to assembly of ethyl phenoxyacrylates 98 in 69–88% yields.
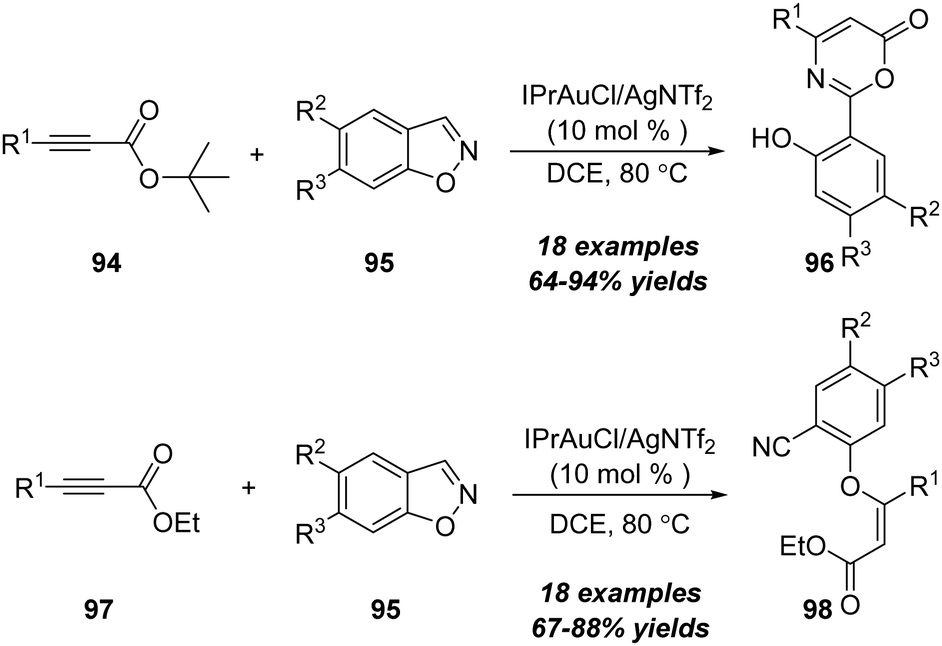 | ||
| Scheme 30 Gold-catalyzed formal [4+2] annulation between propiolates 94 or 97 and 1,2-benzisoxazoles 95. | ||
The proposed reaction mechanism of above gold-catalyzed formal [4+2] annulation is depicted in Scheme 31. First, nucleophilic attack of 1,2-benzisoxazole 95a onto gold-activated aryloxy alkyne 94 or 97 results in vinyl gold 94-A, which can be trapped by the ester group to form tricyclic intermediate 94-B. After the fragment of N–O bond of 94-B, there are two pathways depending on the substituents. For the ethyl propiolate 97, the 94-E underwent ring-opening to produce the nitrium intermediate 94-F. Then the dissociation of nitrium intermediate 94-F forms gold-coordinated alkyne species 94-H and 2-hydroxybenzonitrile 94-G, which further react to furnish the observed 3-phenoxyacrylate 96a. For the tert-butyl propiolate 94, a loss of the tert-butyl cation moiety in 93-C generates stable 6H-1,3-oxazin-6-one derivative. Subsequent deauration takes place to afford the desired 6H-1,3-oxazin-6-one 98a.
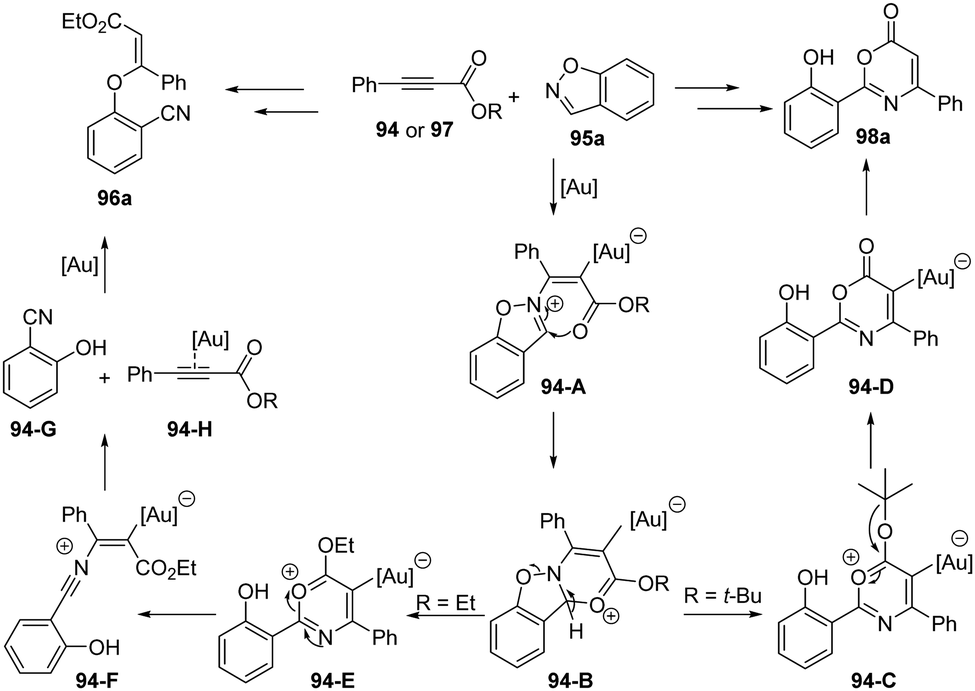 | ||
| Scheme 31 Plausible mechanism for gold-catalyzed formal [4+2] annulation between propiolates 94 and 1,2-benzisoxazoles 95. | ||
Another interesting protocol on gold-catalyzed cyclization/cascade skeletal rearrangement of o-cyanophenylalkynones 99 with 3-amino-benzo[d]-isoxazoles 100 was described by Liu and co-workers in 2021.33 As outlined in Scheme 32, this protocol provides an efficient approach for the construction of medium-sized benzolactones 101 in moderate to excellent yields.
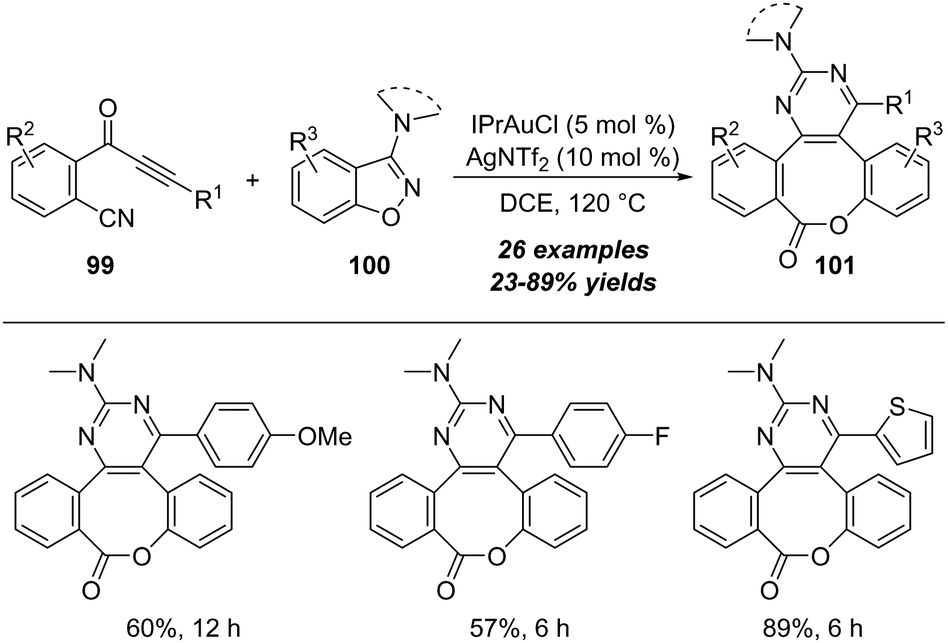 | ||
| Scheme 32 Gold-catalyzed cyclization/cascade skeletal rearrangement of o-cyanophenylalkynones 99 with 3-amino-benzo[d]-isoxazoles 100. | ||
In the proposed reaction mechanism (Scheme 33), an initial attack of benzo[d]isoxazole 100 onto gold-activated ynone affords vinyl gold intermediate 99-A, which undergoes ring-opening to yield the α-imino gold carbene intermediate 99-B. Then intramolecular cyclization of 99-B provides the cyclized intermediate 99-C. Subsequently, carbene transfer of 99-C via C–C bond cleavage followed by 1,2-aryl migration gives the 4-membered ring intermediate 99-E, which can be converted into 99-F after the elimination of the gold catalyst. Finally, a formal [2+2] cycloaddition of 99-F occurs to deliver fused ring intermediate 99-G, and the desired product 101 forms through ring expansion.
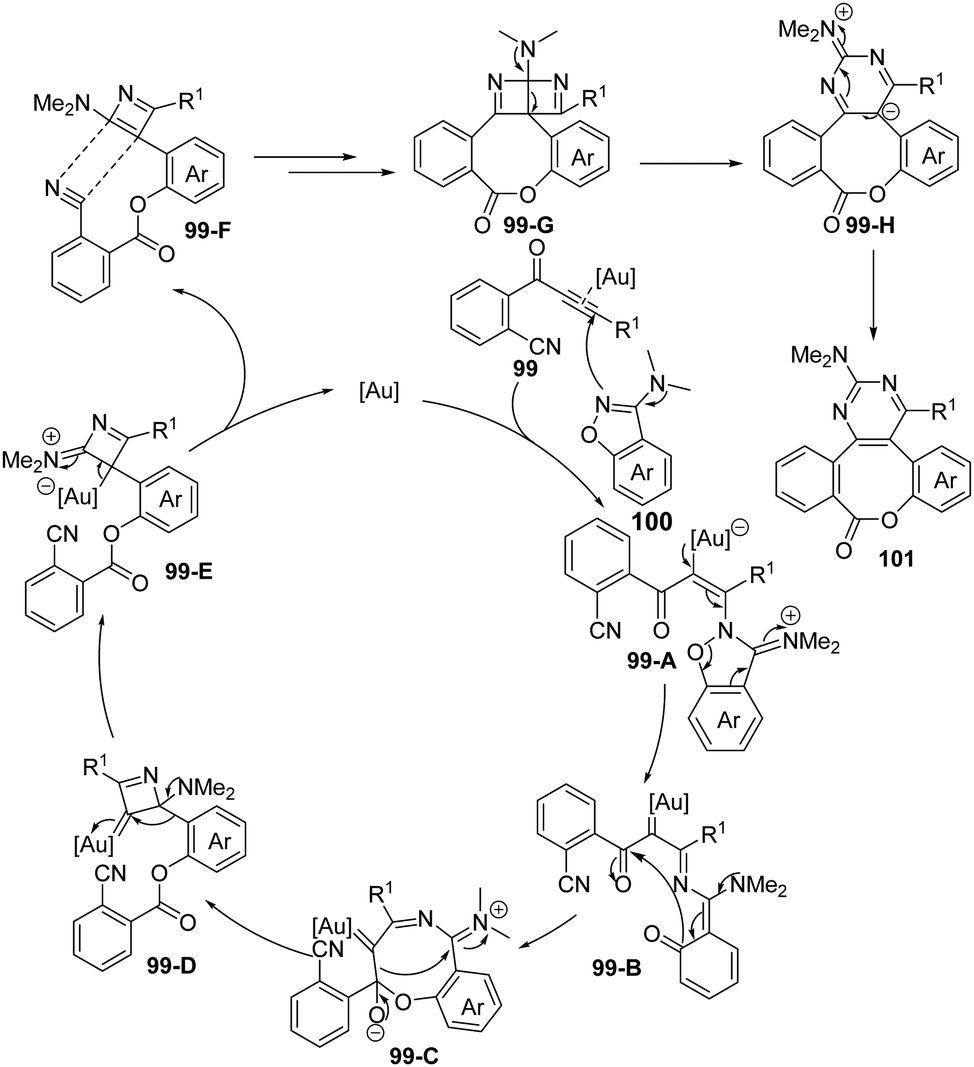 | ||
| Scheme 33 Proposed mechanism for gold-catalyzed cyclization/cascade skeletal rearrangement of o-cyanophenylalkynones 99 with 3-amino-benzo[d]-isoxazoles 100. | ||
Besides ynones, alkynyl sulfones were also employed as appropriate alkynes in the amination with isoxazoles. In 2022, Hashmi and co-workers reported a gold-catalyzed formal [4+2] annulation of alkynyl sulfones 102 with anthranils 103, for the assembly of diverse 3-hydroxyquinolines 104 in moderate to excellent yields (Scheme 34).34
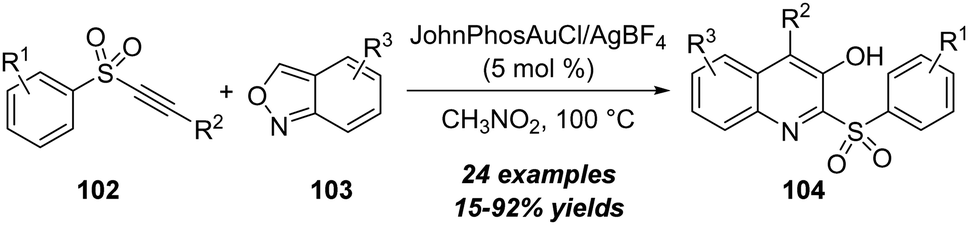 | ||
| Scheme 34 Gold-catalyzed formal [4+2] annulation of alkynyl sulfones 102 with anthranils 103. | ||
6. Amination of unpolarized alkynes
Apart from the above-mentioned polarized alkynes, some specific unpolarized alkynes could also react with isoxazole derivatives in the presence of metal catalysts. In 2018, Hashmi group made a seminal contribution in gold-catalyzed regiospecific C–H annulation of unpolarized alkynes with anthranils (Scheme 35).35 The gold-catalyzed reaction of o-ethynylbiaryls 105 with anthranils 106 produced diverse N-doped polycyclic aromatic hydrocarbons (PAHs) 107 in 20–70% yields. First, the gold-coordinated alkyne is regioselectively attacked by anthranil 106 to produce the α-imino gold carbene intermediate 105-B, which can be further trapped by the phenyl group through C–H functionalization and deliver the six-membered intermediate 105-C. Subsequent enamine–aldehyde addition/elimination of 105-C affords the final product 107.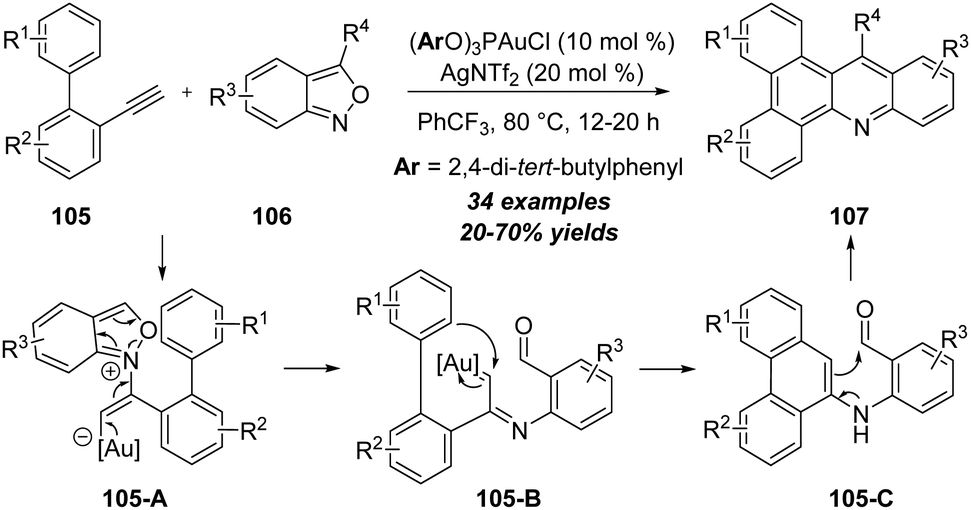 | ||
| Scheme 35 Gold-catalyzed regiospecific C–H annulation of unpolarized alkynes 105 with anthranils 106. | ||
In 2018, a gold-catalyzed [4+1] annulation of 1,4-diyn-3-ols with isoxazoles was disclosed by Liu's group.36 As shown in Scheme 36, in the presence of 10 mol% of IPrAuCl/AgOTf, the reaction of 1,4-diyn-3-ols 108 with isoxazoles 109 proceeded smoothly, allowing the efficient formation of multisubstituted pyrroles 110 in mostly good to excellent yields. Interestingly, the gold carbene-enabled 1,2-alkyne shift is the key for the transformation.
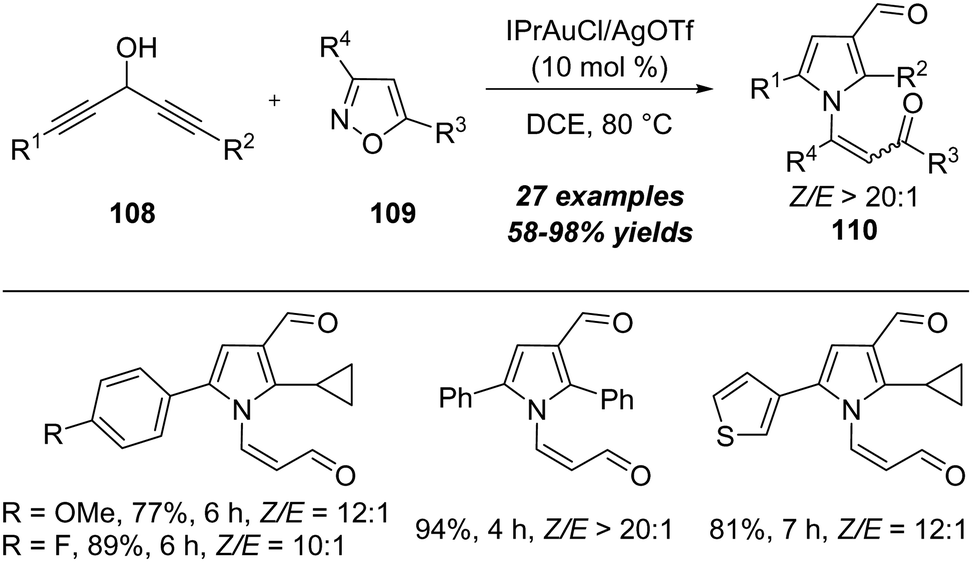 | ||
| Scheme 36 Gold-catalyzed [4+1] annulation between 1,4-diyn-3-ols 108 and isoxazoles 109. | ||
In 2019, the same group described an elegant protocol for the gold-catalyzed bicyclic annulations of 4-methoxy-1,2-dienyl-5-ynes 111 with isoxazoles 112.37 As shown in Scheme 37, it was found that the treatment of 4-methoxy-1,2-dienyl-5-ynes 111 with disubstituted isoxazoles 112 using IPrAuCl/AgNTf2 as the catalyst could afford various 8-formylindolizines 113 in moderate to excellent yields under mild reaction conditions. In addition, the use of non-substituted isoxazole 112a under the standard conditions led to the formation of the 7-formylindolizines 114. The reaction begins with the nucleophilic attack of alkyne moiety onto gold-activated allene to provide gold carbene intermediate 111-A, followed by the attack of isoxazole 112a to produce alkynyl intermediate 111-B through a ring-opening of the isoxazole. The subsequent N-attack and protodeauration take place to generate pyrrole intermediate 111-C, which undergoes a carbonyl-ene reaction and elimination/1,2-acyl migration to deliver the final product 114.
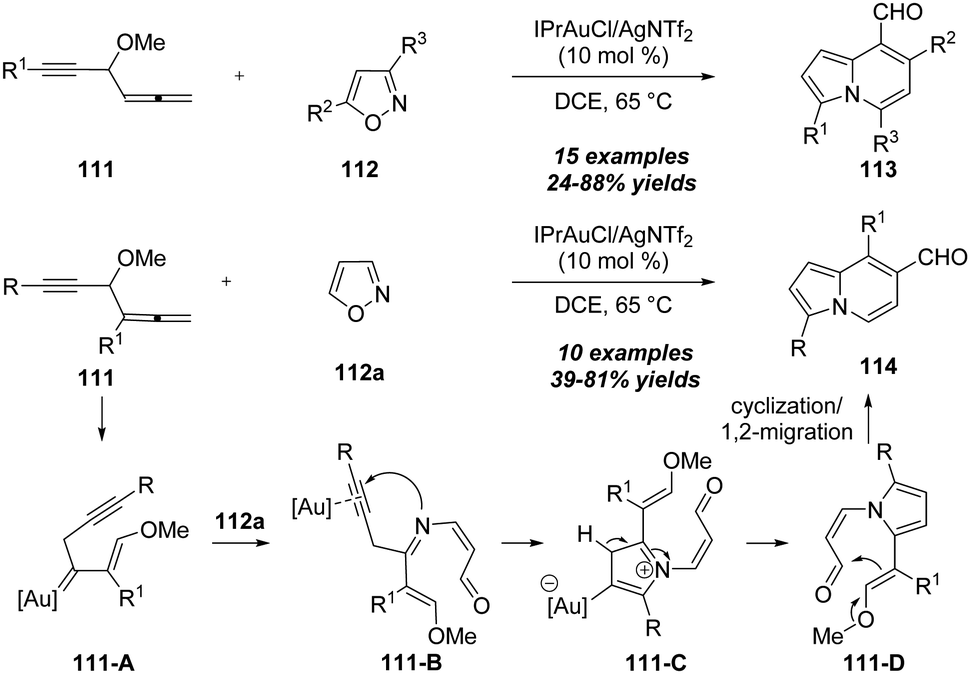 | ||
| Scheme 37 Gold-catalyzed bicyclic annulations of 4-methoxy-1,2-dienyl-5-ynes 111 with isoxazoles 112. | ||
By using a similar strategy, in 2019, Liu and co-workers disclosed the efficient construction of tetrahydro-1H-benzo[b]azepine derivatives from gold-catalyzed [4+3] annulation of 2-alkenyl-1-alkynylbenzenes 115 with anthranils 116.38 As shown in Scheme 38, terminal alkynes (R4 = H) reacted well to furnish benzo[b]azepine derivatives 117 via a novel skeletal rearrangement, while internal 1,5-enynes (R4 = alkyl) afforded products 118 in moderate to good yields without the rearrangement process.
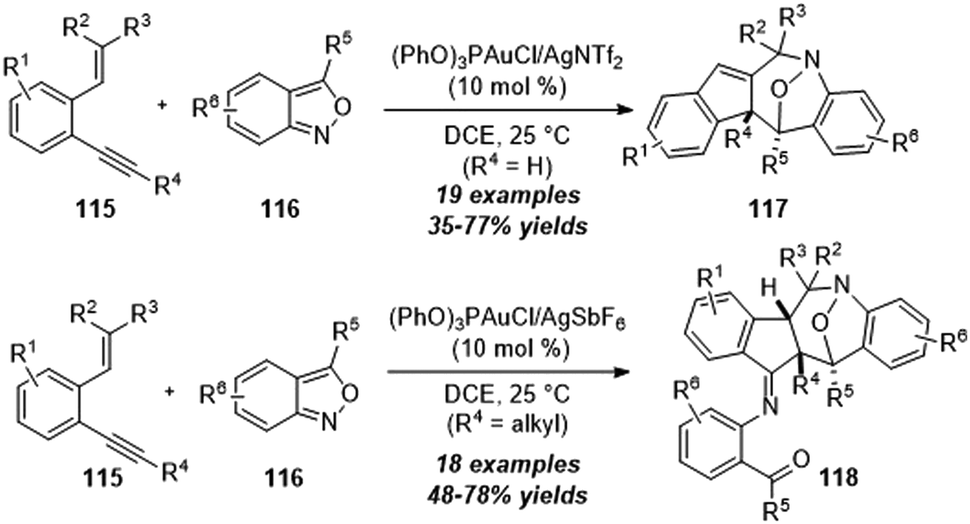 | ||
| Scheme 38 Gold-catalyzed [4+3] annulations of 2-alkenyl-1-alkynylbenzenes 115 with anthranils 116. | ||
In the same year, a gold-catalyzed 1,3-carbofunctionalization of vinyl propargyl esters 119 with anthranils 120 was achieved by Liu and co-workers, leading to the convenient synthesis of 1,3-dihydrobenzo[c]-isoxazoles 121 in 42–92% yields with excellent diastereoselectivities (Scheme 39).39 The proposed mechanism of gold-catalyzed 1,3-carbofunctionalization of vinyl propargyl esters 119 with anthranils 120 is shown below. First, a gold-assisted intramolecular cyclization of gold-activated alkyne takes place to afford vinyl gold intermediate 119-A. A formal [5+3] annulation of intermediate 119-A with anthranil 120 furnishes the 8-membered intermediate 119-B. Further ring-opening of the oxonium moiety yields benzylic cation intermediate 119-C. Finally, the desired product 121 is obtained through a further intramolecular cyclization.
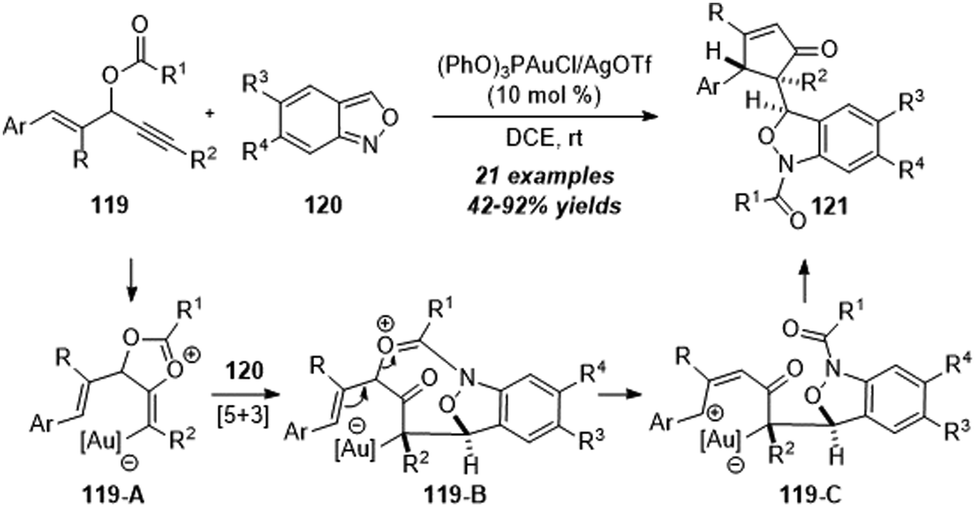 | ||
| Scheme 39 Gold-catalyzed 1,3-carbofunctionalization of vinyl propargyl esters 119 with anthranils 120. | ||
A related gold-catalyzed [4+1] annulation of 4-methoxy-1,2-dienyl-5-ynes 122 with anthranils 123 was also explored by Liu and co-workers in 2019, enabling the preparation of pyrroles 124 (Scheme 40).40 For the allenyl ester substrates 125, the formed pyrroles could further undergo intramolecular aldol reaction by the treatment of DBU, and produced diverse pyrrolo[1,2-a]quinoline derivatives 126.
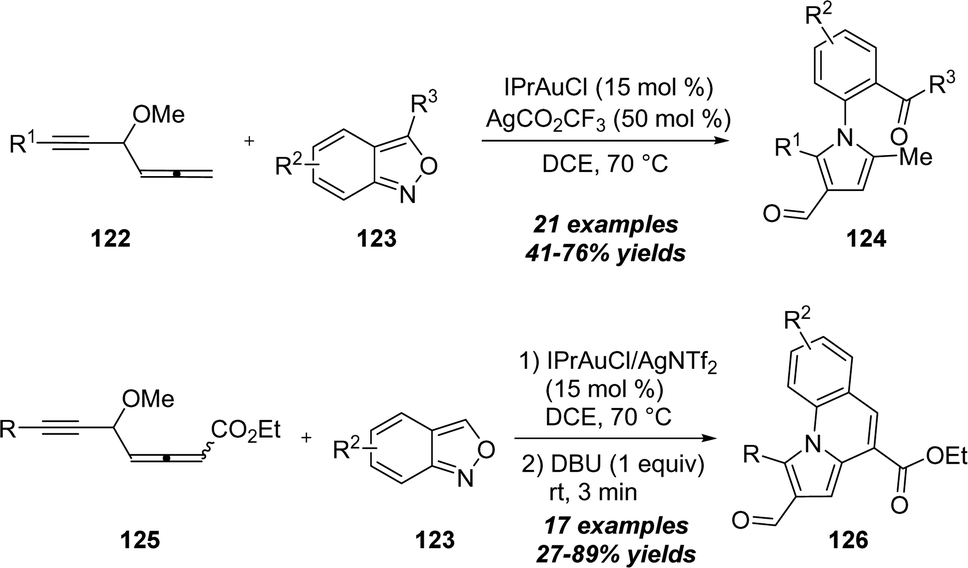 | ||
| Scheme 40 Gold-catalyzed [4+1] annulation of 4-methoxy-1,2-dienyl-5-ynes 122 or 125 with anthranils 123. | ||
In 2020, the same group reported a unique gold-catalyzed aminoaromatization of 1,5-diynes with anthranils.41 As described in Scheme 41, the aminoaromatization of 1,5-diynes 127 with anthranils 128 in the presence of 10 mol% (o-biphenyl)di-tert-butylphosphine gold chloride and AgSbF6 as the catalyst afforded the corresponding 1-amino-2-napthaldehyde derivatives 129 in generally moderate to good yields. Based on the control experiments, a proposed mechanism is demonstrated. First, α-imino gold carbene intermediate 127-B is generated from the gold-catalyzed addition of 1,5-diyne 127 with anthranil 128 through vinyl gold intermediate 127-A. Subsequent 1,2-H shift of gold carbene affords α,β-unsaturated 3-hydroxy-enimine 127-C. Finally, gold-catalyzed intramolecular cyclization, protodeauration and aromatization take place to produce the desired 1-amino-2-napthaldehyde 129.
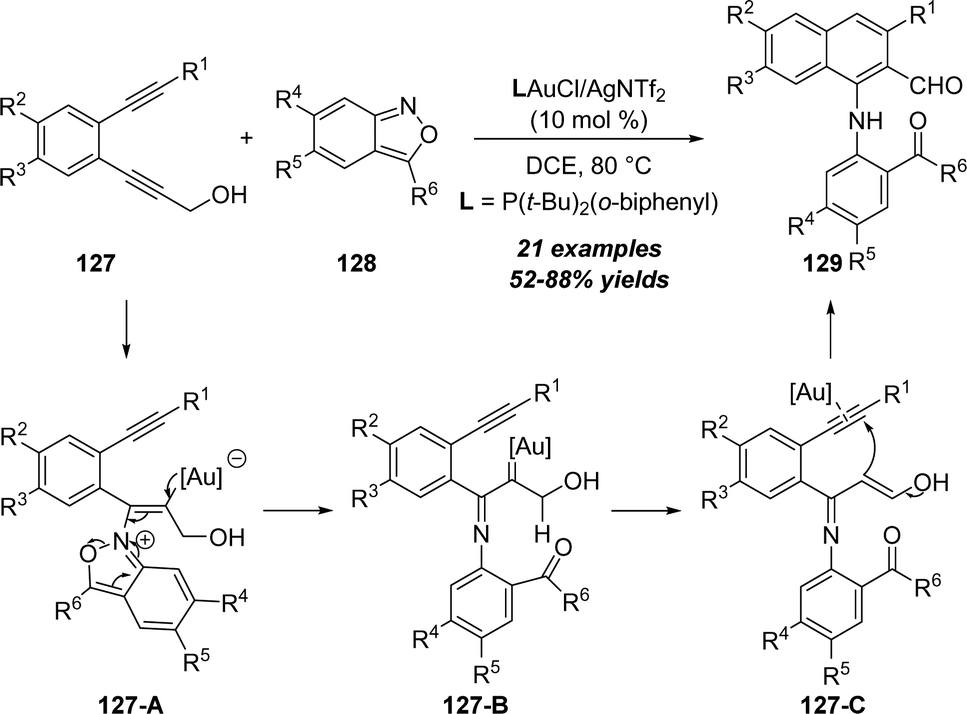 | ||
| Scheme 41 Gold-catalyzed aminoaromatization of 1,5-diynes 127 with anthranils 128. | ||
In 2017, Liang and co-workers disclosed a copper-catalyzed [4+2] annulation between propargylic alcohols 130 with benzo[d]isoxazoles 131 for the preparation of functionalized quinoline derivatives 132 in good yields under mild conditions. (Scheme 42).42 Notably, this protocol represents a rare example of non-noble-metal catalyzed reaction of unpolarized alkynes with isoxazole derivatives.
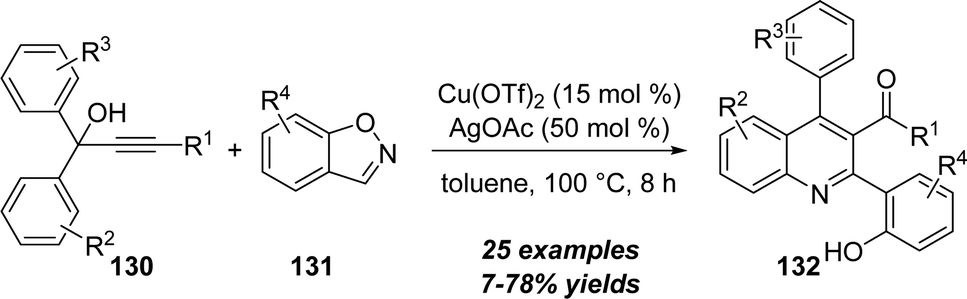 | ||
| Scheme 42 Copper-catalyzed [4+2] annulation of propargylic alcohols 130 and benzo[d]isoxazoles 131. | ||
By changing the structures of alkynes, Liu group also reported a gold-catalyzed formal [4+2] annulation of vinylallenes 133 or enyne acetates 136 with anthranils 134, providing an efficient method for the synthesis of various valuable 3,4-dihydroquinoline derivatives 135 (Scheme 43).43 Of note, this protocol involves the α-alkyl gold carbene intermediates and presents the alkyl C–H reactivity of α-alkyl gold carbenes with an external nucleophile.
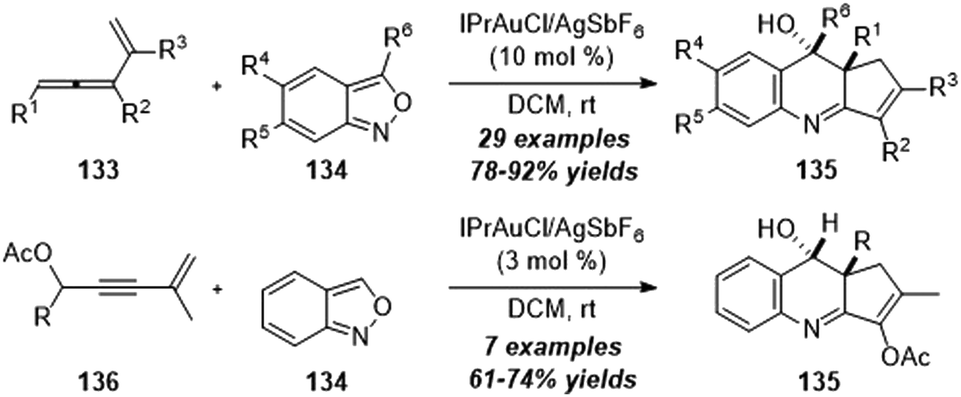 | ||
| Scheme 43 Gold-catalyzed formal [4+2] annulation of vinylallenes 133 or enyne acetates 136 with anthranils 134. | ||
7. Asymmetric amination of alkynes
Although significant advances have been achieved in the amination reaction of alkynes with isoxazoles, the catalytic asymmetric amination was not realized until in 2020 attribute to the impediment in the asymmetric alkyne functionalization.44 Ye and co-workers realized the first example of catalytic asymmetric reaction of isoxazoles with alkynes.45 As shown in Scheme 44, the functionalized chiral 2H-azepines 139 could be achieved in good yields with moderate to excellent enantioselectivities through a zinc-catalyzed enantioselective formal [4+3] annulation of enynol ethers 137 with isoxazoles 138. It is worth noting that this protocol not only represents the first asymmetric heptatrienyl cation type 6π electrocyclization, but also constitutes the first asymmetric reaction of ynol ethers. Based on experimental studies and DFT calculations. The plausible reaction mechanism is proposed based on the model substrates. The reaction commences with the nucleophilic attack of isoxazole 138 onto the zinc-activated ynol ether to afford vinyl zinc intermediate 137-A. Then, the N–O bond cleavage of isoxazole produces zinc-stabilized allyl cation species 137-B, which is transformed into 7-membered intermediate 137-C through a heptatrienyl cation type 6π electrocyclization. Notably, the 6π electrocyclization step is the enantiodetermining step. At Last, the deprotonation of intermediate 137-C delivers the desired chiral 2H-azepine 139.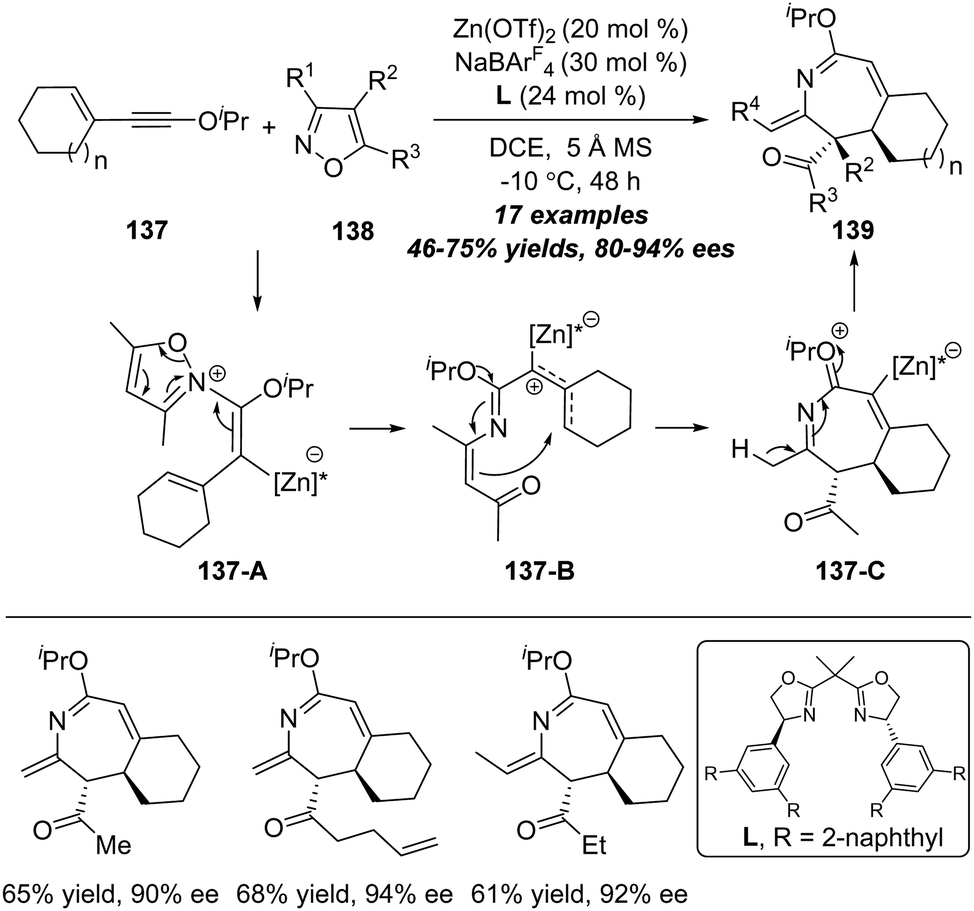 | ||
| Scheme 44 Zinc-catalyzed enantioselective formal [4+3] annulation of enynol ethers 137 with isoxazoles 138. | ||
In 2019, Liu and co-workers reported a gold-catalyzed asymmetric formal [4+3] annulation of enynones 140 with anthranils 141 by employing the chiral phosphine ligand, producing enantioenriched bridged epoxybenzoazepines 142 in moderate to excellent yields with 88–99% ees through an enantioselective dearomatization process (Scheme 45).46 The proposed mechanism for this gold-catalyzed asymmetric formal [4+3] annulation is shown below. Initially, the gold catalyzed cyclization and tautomerization of enynone 140 occur to provide the 3-furyl methyl cation intermediate 140-A which can serve as a 1,3-all carbon dipole. Then, nucleophilic addition of anthranil 141 onto 1,3-dipole 140-A affords oxonium ion intermediate 140-B, which could be the enantio-determining step. Moreover, 140-B undergoes further intramolecular cyclization to deliver the bridged intermediate 140-C. Finally, the desired chiral bridged epoxybenzoazepine 142 is obtained from aromatization with the regeneration of the gold catalyst.
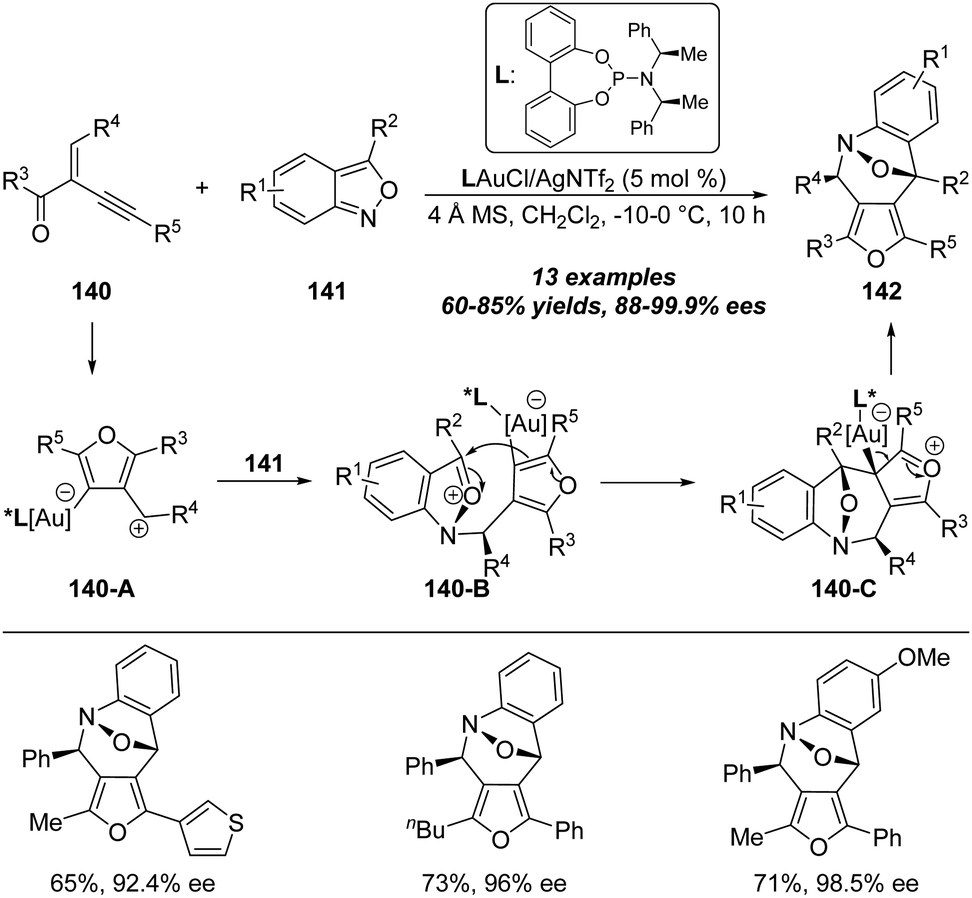 | ||
| Scheme 45 Gold-catalyzed asymmetric formal [4+3] annulation of enynones 140 with anthranils 141. | ||
8. Conclusion
In summary, isoxazoles and their derivatives, as the N,O-bifunctional reagents of alkynes, have attracted more and more attention during the past decades in the development of alkyne amination reactions, providing an efficient and divergent way for the synthesis of a wide range of functionalized heterocycles, especially for valuable N-containing heterocycles. Divergent metal carbene-mediated cascade aminations, including formal annulation, C–H functionalization, cyclopropanation, electrocyclization, carbene metathesis and other asymmetric aminations, could be achieved by exploiting the flexible properties of metal carbene intermediates.Despite these achievements, there are still considerable challenges that should arouse more research in the near future. Firstly, the developed amination reactions of alkynes with isoxazoles are mostly limited to transition-metal catalysis, especially for noble metal catalysts. Therefore, non-noble metal catalyzed and metal-free protocols need to be further developed for the goal of green synthesis. Secondly, most amination reactions require special alkyne substrates, such as electron-rich alkynes (such as ynamides, ynol ethers and thioynol ethers), electron-deficient alkynes, and conjugated alkynes. Thus, catalytic systems with higher efficiency and selectivity are still in great demand to uncover the amination of simple alkynes. Thirdly, the catalytic asymmetric amination reaction of isoxazoles with alkynes has been rarely demonstrated. Thereby, more types of asymmetric catalytic systems are needed. Finally, further synthetic applications in the synthesis of natural products, bioactive molecules and pharmaceuticals by using this strategy are highly desirable. We anticipate that the amination reactions of alkynes with isoxazoles summarized here will attract more research interest into this field. It is our expectation that the amination of alkynes could be developed as an efficient tool for the assembly of various functional molecules in organic synthesis and other related areas.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22101238, 22125108, and 22331004), the Yunnan Fundamental Research Project (202401CF070024) and Yunnan Normal University.Notes and references
- For selected reviews on α-imino metal carbenes, see: (a) M. Akter, K. Rupa and P. Anbarasan, Chem. Rev., 2022, 122, 13108 CrossRef CAS PubMed; (b) L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Chem. Rev., 2021, 121, 9039 CrossRef PubMed; (c) K. Pal and C. M. R. Volla, Chem. Rec., 2021, 21, 4032 CrossRef PubMed; (d) T. Wang and A. S. K. Hashmi, Chem. Rev., 2021, 121, 8948 CrossRef; (e) P. W. Davies, Chem. Rec., 2021, 21, 3964 CrossRef PubMed; (f) E. Aguilar and J. Santamaría, Org. Chem. Front., 2019, 6, 1513 RSC; (g) Y. Jiang, R. Sun, X.-Y. Tang and M. Shi, Chem. – Eur. J., 2016, 22, 17910 CrossRef; (h) P. W. Davies and M. Garzón, Asian J. Org. Chem., 2015, 4, 694 CrossRef; (i) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC; (j) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 CrossRef PubMed.
- For selected examples on amination reaction of alkynes based on azides, see: (a) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260 CrossRef PubMed; (b) C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838 CrossRef PubMed; (c) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354 CrossRef CAS PubMed; (d) B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8358 CrossRef CAS PubMed; (e) J. Matsuoka, Y. Matsuda, Y. Kawada, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2017, 56, 7444 CrossRef CAS; (f) W.-B. Shen, Q. Sun, L. Li, X. Liu, B. Zhou, J.-Z. Yan, X. Lu and L.-W. Ye, Nat. Commun., 2017, 8, 1748 CrossRef PubMed; (g) X. Liu, Z.-S. Wang, T.-Y. Zhai, C. Luo, Y.-P. Zhang, Y.-B. Chen, C. Deng, R.-S. Liu and L.-W. Ye, Angew. Chem., Int. Ed., 2020, 59, 17984 CrossRef CAS; (h) C.-Y. Weng, G.-Y. Zhu, B.-H. Zhu, P.-C. Qian, X.-Q. Zhu, J.-M. Zhou and L.-W. Ye, Org. Chem. Front., 2022, 9, 2773 RSC; (i) J.-M. Xie, Y.-L. Zhu, Y.-M. Fu, C.-F. Zhu, L.-J. Cheng, Y.-E. You, X. Wu and Y.-G. Li, Org. Lett., 2023, 25, 421 CrossRef CAS; (j) S. P. Sancheti, D. J. Mondal and N. T. Patil, ACS Catal., 2023, 13, 4391 CrossRef; (k) D. F. L. Rayo, A. Mansour, W. Wu, B. N. Bhawal and F. Gagosz, Angew. Chem., Int. Ed., 2023, 62, e202212893 CrossRef PubMed; (l) L. C. Greiner, N. Arichi, S. Inuki and H. Ohno, Angew. Chem., Int. Ed., 2023, 62, e202213653 CrossRef PubMed; (m) X. Liu, L.-G. Liu, C.-M. Chen, X. Li, Z. Xu, X. Lu, B. Zhou and L.-W. Ye, Angew. Chem., Int. Ed., 2023, 62, e202216923 CrossRef PubMed; (n) Y. Chen, Y.-H. Yan, B.-H. Zhu, F. Chen, L. Li and P.-C. Qian, Org. Lett., 2023, 25, 2063 CrossRef PubMed . For selected examples of intermolecular amination reaction of alkynes based on azides, see: ; (o) C. Shu, Y.-H. Wang, B. Zhou, X.-L. Li, Y.-F. Ping, X. Lu and L.-W. Ye, J. Am. Chem. Soc., 2015, 137, 9567 CrossRef PubMed; (p) C. Shu, Y.-H. Wang, C.-H. Shen, P.-P. Ruan, X. Lu and L.-W. Ye, Org. Lett., 2016, 18, 3254 CrossRef PubMed; (q) C. Shu, C.-H. Shen, Y.-H. Wang, L. Li, T. Li, X. Lu and L.-W. Ye, Org. Lett., 2016, 18, 4630 CrossRef PubMed; (r) B. Zhou, Y.-Q. Zhang, X. Liu and L.-W. Ye, Sci. Bull., 2017, 62, 1201 CrossRef PubMed; (s) C.-Y. Shi, J.-X. Gong, Z. Li, C. Shu, L.-W. Ye, Q. Sun,B. Zhou and X.-Q. Zhu, Chin. Chem. Lett., 2025, 36, 109895 CrossRef.
- For selected examples on amination reaction of alkynes based on nitrogen ylides, see: (a) R. J. Reddy, M. P. Ball-Jones and P. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 13310 CrossRef PubMed; (b) M. Garzón, E. M. Arce, R. J. Reddy and P. W. Davies, Adv. Synth. Catal., 2017, 359, 1837 CrossRef; (c) X. Zhang and Z. Geng, RSC Adv., 2016, 6, 62099 RSC; (d) A. D. Gillie, R. J. Reddy and P. W. Davies, Adv. Synth. Catal., 2016, 358, 226 CrossRef; (e) M. Garzón and P. W. Davies, Org. Lett., 2014, 16, 4850 CrossRef PubMed; (f) E. Chatzopoulou and P. W. Davies, Chem. Commun., 2013, 49, 8617 RSC; (g) P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem., Int. Ed., 2011, 50, 8931 CrossRef PubMed; (h) C. Li and L. Zhang, Org. Lett., 2011, 13, 1738 CrossRef PubMed.
- For selected examples on amination reaction of alkynes based on sulfilimines, see: (a) X. Tian, L. Song, M. Rudolph, F. Rominger, T. Oeser and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 3589 CrossRef PubMed; (b) X. Tian, L. Song, M. Rudolph, Q. Wang, X. Song, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 1598 CrossRef PubMed; (c) X. Tian, L. Song, C. Han, C. Zhang, Y. Wu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 2937 CrossRef PubMed; (d) X. Tian, L. Song, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 4327 CrossRef PubMed; (e) X. Tian, L. Song and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2020, 59, 12342 CrossRef PubMed; (f) A. V. Mackenroth, P. W. Antoni, F. Rominger, M. Rudolph and A. S. K. Hashmi, Org. Lett., 2023, 25, 2907 CrossRef PubMed; (g) Q. Sun, C. Hüßler, J. Kahle, A. V. Mackenroth, M. Rudolph, P. Krämer, T. Oeser and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2024, 63, e202313738 CrossRef PubMed.
- For other selected examples, see: (a) A. Prechter, G. Henrion, P. Faudot Dit Bel and F. Gagosz, Angew. Chem., Int. Ed., 2014, 53, 4959 Search PubMed; (b) L. Zhu, Y. Yu, Z. Mao and X. Huang, Org. Lett., 2015, 17, 30 CrossRef PubMed; (c) S. K. Pawar, R. L. Sahani and R.-S. Liu, Chem. – Eur. J., 2015, 21, 10843 CrossRef PubMed; (d) Z. Zeng, H. Jin, J. Xie, B. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2017, 19, 1020 CrossRef PubMed; (e) W. Xu, G. Wang, N. Sun and Y. Liu, Org. Lett., 2017, 19, 3307 CrossRef PubMed; (f) W. Xu, Y. Chen, A. Wang and Y. Liu, Org. Lett., 2019, 21, 7613 CrossRef PubMed.
- (a) G. C. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef PubMed; (b) J. Zhu, J. Mo, H.-Z. Lin, Y. Chen and H.-P. Sun, Bioorg. Med. Chem., 2018, 26, 3065 CrossRef PubMed; (c) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef PubMed; (d) J. Wang, Y. Wu, C. Ma, G. Fiorin, J. Wang, L. H. Pinto, R. A. Lamb, M. L. Klein and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1315 CrossRef PubMed; (e) A. A. Jensen, N. Plath, M. H. F. Pedersen, V. Isberg, J. Krall, P. Wellendorph, T. B. Stensbøl, D. E. Gloriam, P. Krogsgaard-Larsen and B. Frølund, J. Med. Chem., 2013, 56, 1211 CrossRef PubMed; (f) L.-F. Yu, J. B. Eaton, A. Fedolak, H.-K. Zhang, T. Hanania, D. Brunner, R. J. Lukas and A. P. Kozikowski, J. Med. Chem., 2012, 55, 9998 CrossRef CAS PubMed; (g) D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, S. Knapp, S. J. Conway and T. D. Heightman, J. Med. Chem., 2011, 54, 6761 CrossRef CAS PubMed; (h) C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061 CrossRef CAS PubMed; (i) N. S. Duncan, M. J. Campbell, D. S. Backos, C. Li, K. C. Rider, S. Stump, M. J. Weaver, M. P. Gajewski, H. D. Beall, P. Reigan and N. R. Natale, Bioorg. Med. Chem., 2022, 69, 116911 CrossRef CAS PubMed; (j) J. Li, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2020, 7, 2325 RSC.
- A.-H. Zhou, Q. He, C. Shu, Y.-F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L.-W. Ye, Chem. Sci., 2015, 6, 1265 RSC.
- (a) L. Li, T.-D. Tan, Y.-Q. Zhang, X. Liu and L.-W. Ye, Org. Biomol. Chem., 2017, 15, 8483 RSC; (b) R. L. Sahani, L.-W. Ye and R.-S. Liu, Adv. Organomet. Chem., 2020, 73, 195 CrossRef CAS; (c) X.-Q. Zhu, J.-J. He, B. Zhou and L.-W. Ye, Cell Rep. Phy. Sci., 2023, 4, 101645 CrossRef CAS.
- For selected reviews on ynamide chemistry, see: (a) L. Hu and J. Zhao, Acc. Chem. Res., 2024, 57, 855 CrossRef CAS PubMed; (b) Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Chem. Soc. Rev., 2021, 50, 2582 RSC; (c) C. C. Lynch, A. Sripada and C. Wolf, Chem. Soc. Rev., 2020, 49, 8543 RSC; (d) Y.-B. Chen, P.-C. Qian and L.-W. Ye, Chem. Soc. Rev., 2020, 49, 8897 RSC; (e) F.-L. Hong and L.-W. Ye, Acc. Chem. Res., 2020, 53, 2003 CrossRef CAS PubMed; (f) J. Luo, G.-S. Chen, S.-J. Chen, J.-S. Yu, Z.-D. Li and Y.-L. Liu, ACS Catal., 2020, 10, 13978 CrossRef CAS; (g) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393 CrossRef CAS; (h) G. Evano, C. Theunissen and M. Lecomte, Aldrichimica Acta, 2015, 48, 59 CAS; (i) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed.
- X.-Y. Xiao, A.-H. Zhou, C. Shu, F. Pan, T. Li and L.-W. Ye, Chem. – Asian J., 2015, 10, 1854 CrossRef CAS PubMed.
- H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794 CrossRef CAS PubMed.
- X. Tian, L. Song, K. Farshadfar, M. Rudolph, F. Rominger, T. Oeser, A. Ariafard and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2020, 59, 471 CrossRef CAS PubMed.
- Z. Tong, P. J. Smith, H. D. Pickford, K. E. Christensen and E. A. Anderson, Chem. – Eur. J., 2023, 29, e202302821 CrossRef CAS PubMed.
- H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 12688 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16549 CrossRef CAS PubMed.
- M.-H. Tsai, C.-Y. Wang, A. S. K. Raj and R.-S. Liu, Chem. Commun., 2018, 54, 10866 RSC.
- Y.-C. Hsu, S.-A. Hsieh and R.-S. Liu, Chem. – Eur. J., 2019, 25, 5288 CrossRef CAS PubMed.
- S. S. Giri and R.-S. Liu, Chem. Sci., 2018, 9, 2991 RSC.
- W. Xu, J. Zhao, X. Li and Y. Liu, J. Org. Chem., 2018, 83, 15470 CrossRef CAS PubMed.
- R. Vanjari, S. Dutta, B. Prabagar, V. Gandon and A. K. Sahoo, Chem. – Asian J., 2019, 14, 4828 CAS.
- W.-B. Shen, X.-Y. Xiao, Q. Sun, B. Zhou, X.-Q. Zhu, J.-Z. Yan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2017, 56, 605 CAS.
- P. D. Jadhav, X. Lu and R.-S. Liu, ACS Catal., 2018, 8, 9697 CAS.
- Y. Zhao, C. Wang, Y. Hu and B. Wan, Chem. Commun., 2018, 54, 3963 CAS.
- Y. B. Pandit, Y.-T. Jiang, J.-J. Jian, T.-C. Chen, T.-C. Kuo, M.-J. Cheng and R.-S. Liu, Org. Chem. Front., 2021, 8, 2563 CAS.
- L. Song, X. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. Commun., 2019, 55, 9007 CAS.
- X.-Q. Zhu, H. Yuan, Q. Sun, B. Zhou, X.-Q. Han, Z.-X. Zhang, X. Lu and L.-W. Ye, Green Chem., 2018, 20, 4287 CAS.
- M. D. Patil and R.-S. Liu, Org. Biomol. Chem., 2019, 17, 4452 RSC.
- X.-Q. Zhu, Q. Sun, Z.-X. Zhang, B. Zhou, P.-X. Xie, W.-B. Shen, X. Lu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2018, 54, 7435 RSC.
- P. E. Simm, P. Sekar, J. Richardson and P. W. Davies, ACS Catal., 2021, 11, 6357 CrossRef CAS PubMed.
- R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 1026 CrossRef CAS PubMed.
- R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736 CrossRef CAS PubMed.
- Y. B. Pandit, R. L. Sahani and R.-S. Liu, Org. Lett., 2018, 20, 6655 CrossRef CAS PubMed.
- A. Wang, X. Hu, X. Xie and Y. Liu, Adv. Synth. Catal., 2021, 363, 3769 CrossRef CAS.
- Y. Wu, C. Hu, T. Wang, L. Eberle and A. S. K. Hashmi, Adv. Synth. Catal., 2022, 364, 1233 CrossRef CAS.
- Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 6935 CAS.
- R. D. Kardile, B. S. Kale, P. Sharma and R.-S. Liu, Org. Lett., 2018, 20, 3806 CrossRef CAS PubMed.
- A. S. K. Raj, K.-C. Tan, L.-Y. Chen, M.-J. Cheng and R.-S. Liu, Chem. Sci., 2019, 10, 6437 Search PubMed.
- R. R. Singh, M. Skaria, L.-Y. Chen, M.-J. Cheng and R.-S. Liu, Chem. Sci., 2019, 10, 1201 RSC.
- M. Skaria, P. Sharma and R.-S. Liu, Org. Lett., 2019, 21, 2876 CAS.
- H.-C. Hsieh, K.-C. Tan, A. S. K. Raj and R.-S. Liu, Chem. Commun., 2019, 55, 1979 Search PubMed.
- Y. B. Pandita and R.-S. Liu, Adv. Synth. Catal., 2020, 362, 3183 Search PubMed.
- Y.-P. Han, X.-S. Li, Z. Sun, X.-Y. Zhu, M. Li, X.-R. Song and Y.-M. Liang, Adv. Synth. Catal., 2017, 359, 2735 CrossRef CAS.
- B. D. Mokar, P. D. Jadhav, Y. B. Pandit and R.-S. Liu, Chem. Sci., 2018, 9, 4488 CAS.
- For selected reviews on asymmetric alkyne functionalization, see: (a) G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060 CAS; (b) M. Bao, S. Zhou, W. Hu and X. Xu, Chin. Chem. Lett., 2022, 33, 4969 CrossRef CAS.
- X.-Q. Zhu, Z.-S. Wang, B.-S. Hou, H.-W. Zhang, C. Deng and L.-W. Ye, Angew. Chem., Int. Ed., 2020, 59, 1666 CrossRef CAS PubMed.
- R. D. Kardile, T.-H. Chao, M.-J. Cheng and R.-S. Liu, Angew. Chem., Int. Ed., 2020, 59, 10396 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |