
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polystyrene-bound AlCl3 – a catalyst for the solvent-free synthesis of aryl-substituted tetrazoles†
Max
Schmallegger
 *a,
Mathias
Wiech
a,
Sebastian
Soritz
b,
Miriam de J.
Velásquez-Hernández
a,
Brigitte
Bitschnau
a,
Heidrun
Gruber-Woelfler
b and
Georg
Gescheidt
*a
*a,
Mathias
Wiech
a,
Sebastian
Soritz
b,
Miriam de J.
Velásquez-Hernández
a,
Brigitte
Bitschnau
a,
Heidrun
Gruber-Woelfler
b and
Georg
Gescheidt
*a
aInstitute of Physical and Theoretical Chemistry, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: schmallegger@tugraz.at; g.gescheidt-demner@tugraz.at
bInstitute of Process and Particle Engineering, Graz University of Technology, Inffeldgasse 13, 8010 Graz, Austria
First published on 7th February 2025
Abstract
Tetrazole moieties are components of various pharmacologically active molecules. Several synthetic protocols for the synthesis of tetrazoles have been developed. Among those, the reaction of organic nitriles with azides catalyzed by Lewis acids (LAs) provides a convenient access. Nevertheless, generally rather harsh reaction conditions have to be utilized for such syntheses. We have developed a simple, solvent-free procedure which allows a convenient isolation of tetrazoles using a heterogeneous catalyst: we show that polystyrene/AlCl3 composites produce tetrazoles at reasonable yields and allow a simple work-up procedure. We have characterized the AlCl3/polystyrene composite (gas sorption, XRD, IR) and investigated its efficacy in the preparation of aryl-substituted tetrazoles. We also have evaluated MgCl2, CuCl2, and ZnCl2 as Lewis-acid catalysts, but they are clearly outperformed by AlCl3 correlating with the Lewis-acid strength on the Gutmann-Beckett scale.
Introduction
Tetrazole-containing compounds have been used in a wide range of applications: they are employed as ligands, in photography, as precursors for more complex heterocycles and in drug development.1–8 Several tetrazole-containing drugs with a variety of biological activities are available on the market, including antihypertensive, antimicrobial, antiviral, antiallergic, cytostatic, and nootropic functions.3,7,9 5-monosubstituted tetrazoles in particular have been widely used in pharmaceutics because they can serve as a bio-isosteric replacement for carboxylic acids, exhibiting similar biological activity and improved resistance to metabolic degradation.10,11A widely used approach for the synthesis of 5-monosubstituted tetrazoles is the diazotization of imidohydrazides.12 Alternatively, they can be synthesized from amines,13,14 amides,15 and aldoximes,16,17 or in multi-component reactions, such as the Ugi tetrazole6,18 reaction or the Passerini tetrazole reaction.19 However, the most commonly used synthetic route involves the reaction of inorganic azide salts or organic azides with nitriles.8,20,21 This approach was first introduced in 1958, utilizing sodium azide and ammonium chloride in dimethylformamide.22 The use of Lewis-acid (LA) catalysis with organometallic or organosilicon azides has been shown to be efficient.11,23,24 Sharpless and co-workers25–27 presented systematic studies, showing that the activation of the nitrile by an electron-withdrawing group is a prerequisite for an efficient reaction.26,28,29
While the use of Lewis acids as catalysts is a well-established approach, it still suffers from certain shortcomings. This includes low reaction yields, harsh reaction conditions, the use of expensive catalysts, time-consuming work-up procedures, or the need for high-boiling point solvents.12,30–33 Therefore, the development of more efficient and convenient methods for the synthesis of tetrazoles is desired.8,17,34,35
In this study, we evaluated AlCl3, MgCl2, CuCl2, and ZnCl2 as Lewis acid catalysts for the synthesis of 5-monosubstituted tetrazoles via a solvent-free reaction. We correlated their Lewis acid strength, as determined by the Gutmann–Beckett method, with tetrazole product yields. AlCl3 was found to be the most efficient catalyst under our experimental conditions. It has already been shown that immobilized AlCl3 (e.g., on polymers) is a well-suited heterogeneous catalyst.36–42 Based on the early work by Neckers et al.,43,44 we report on the feasibility of using AlCl3 supported on a custom-made porous polystyrene photopolymer45 matrix for the solvent-free synthesis of 5-aryl tetrazoles from trimethylsilyl azide and arylnitrile derivatives. The prepared composites are characterized and tested on nitriles presenting substituents with electron donating and -withdrawing abilities.
Experimental section
Materials
Nitriles (R–N3), aluminium chloride (AlCl3), and styrene were purchased from Sigma-Aldrich. Trimethyl silyl azide (TMS–N3) and divinylbenzene were purchased from TCI Chemicals. Zinc chloride (ZnCl2), copper chloride (CuCl2), magnesium chloride (MgCl2) and isopropanol (i-PrOH) were purchased from Roth. All substances were acquired in the highest purity available and used without further purification.Preparation of polymer-bound AlCl3
Porous polystyrene was prepared following the procedure described by Seeberger et al.45 2.8 ml styrene, 2.2 ml divinyl benzene, 5 ml methanol and 170 mg of the commercial photoinitiator “Irgacure 819” (bis(2,4,6-trimethylbenzoyl)-phenylphosphine oxide) were mixed in a 10 ml glass vial. The mixture was placed in an Anycubic Wash & Cure 2.0 device (λ = 405 nm) and irradiated for 60 minutes. The obtained monolith was washed with ethanol and dried at 80 °C for 2 hours. 500 mg of the polymer was then ground and placed in a round-bottomed flask. 1000 mg AlCl3 and 20 ml of CCl4 were added to the mixture.46 The polymer/AlCl3 ratio was rationalized by testing composites with different mass ratios (polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AlCl3 = 1:1 and 1:3, respectively), which show inferior catalytic activity (see Fig. S14†). The mixture was heated to 50 °C and stirred for one hour until a brown solid was obtained. The composite was filtered, washed three times with deionized water and ethanol, and dried at 120 °C for two hours.
:AlCl3 = 1:1 and 1:3, respectively), which show inferior catalytic activity (see Fig. S14†). The mixture was heated to 50 °C and stirred for one hour until a brown solid was obtained. The composite was filtered, washed three times with deionized water and ethanol, and dried at 120 °C for two hours.
Characterization of the polymer-bound AlCl3 catalyst
Synthesis of tetrazoles
000 rpm for 6 min, the supernatant was transferred to an NMR tube and measured directly.
Determination of reaction yield by 1H NMR spectroscopy
1H NMR spectra were recorded on a 200 MHz Bruker Avance DPX spectrometer. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS), using the residual non-deuterated solvent as an internal reference. The reaction yield was determined by comparing the integrals of characteristic signals of the product and educt (see ESI†).Determination of Lewis acid strength by 31P NMR
31P NMR spectra were recorded on a 200 MHz Bruker Avance DPX spectrometer. Chemical shifts (δ) are reported in ppm, relative to 85% H3PO4 in H2O. The experiments were conducted in MeOH-d4.Results and discussion
Lewis acid strength and catalytic performance
As a benchmark to screen the catalytic activity of Lewis acids in the synthesis of 5-aryltetrazoles, we reacted benzonitrile with trimethylsilyl azide in the presence of the readily available metal chlorides MgCl2, CuCl2, ZnCl2, and AlCl3 under solvent-free conditions, yielding 5-phenyltetrazole 3a (Scheme 1).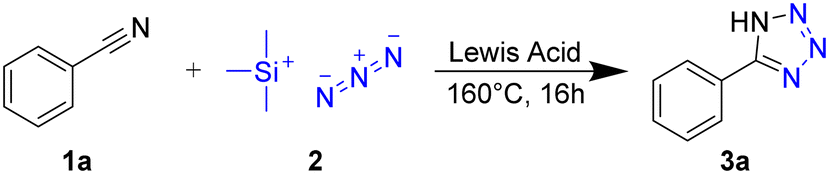 | ||
| Scheme 1 Synthesis of 5-phenyltetrazole (3a) from benzonitrile (1a) and TMS-N3 (2) in the presence of Lewis acids at elevated temperatures. | ||
The yield of 3a was the lowest for MgCl2 (10%), CuCl2 (55%), and ZnCl2 (81%), peaking with AlCl3 (96%, Fig. 1). These reactivities correlate with the Lewis-acid strength as determined by the NMR-based Gutmann–Beckett method49,50 for Lewis acidity within the limits of this approach:51 Here, the 31P resonance of triethylphosphine oxide shifts downfield as a result of de-shielding from coordination with Lewis acids. This effect correlates with the strength of the electron acceptor, i.e. the Lewis Acid. Accordingly, stronger Lewis acids give rise to a larger chemical shift perturbation Δδ31P (Scheme 2).51–53
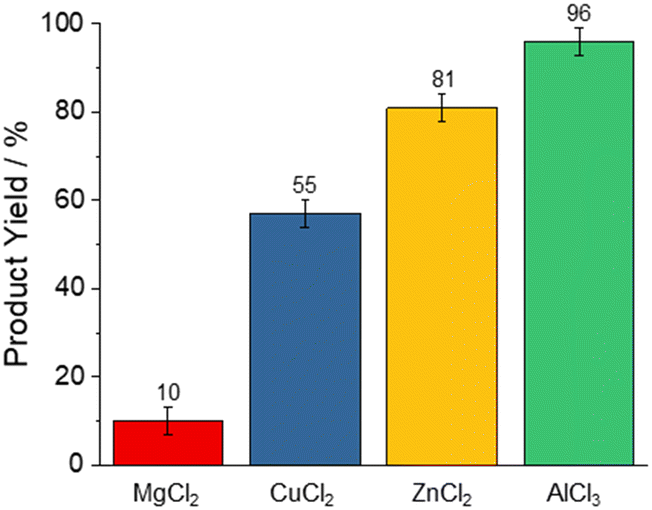 | ||
| Fig. 1 Yields of 5-phenyltetrazole with different Lewis acid catalysts at 160 °C for 16 h, as determined by 1H-NMR spectroscopy. | ||
 | ||
| Scheme 2 Interaction of triethylphosphine oxide with a Lewis acid, resulting in a change in chemical shift in the 31P NMR spectrum. | ||
The 31P chemical shifts of triethylphosphine in the presence of Lewis acids are presented in Table 1 (see ESI† for the corresponding spectra). Lewis acids that induce a higher chemical shift in the 31P NMR also lead to higher product formation when employed as catalysts. Based on these observations, we exclusively employed AlCl3 for further experiments.
| Lewis acid | δ 31P/ppm | Δδ31P/ppm | 3a yield/% |
|---|---|---|---|
| MgCl2 | 59.5 | 0.1 | 10 |
| CuCl2 | 62.7 | 3.3 | 55 |
| ZnCl2 | 64.3 | 4.9 | 81 |
| AlCl3 | 74.3 | 14.9 | 96 |
Preparation, performance and characterization of the heterogeneous catalyst
To produce a porous polymer support for AlCl3, we employed a straightforward photochemical pathway:45 a mixture of styrene, the cross-linker divinyl benzene, methanol, and the photoinitiator Irgacure 819 was prepared and irradiated for 60 min with 405 nm LEDs. The resulting polymer monolith was cleaned, dried, and ground into fine powder. In the second step, an AlCl3/CCl4 solution was added to the polymer (Scheme 3a, see Experimental section for details), yielding AlCl3@polystyrene (Scheme 3).54–56 The immobilization is distinguishable by a change in colour. The parent polystyrene is white, whereas the polystyrene–AlCl3 composite exhibits a yellow-brownish colour.54–56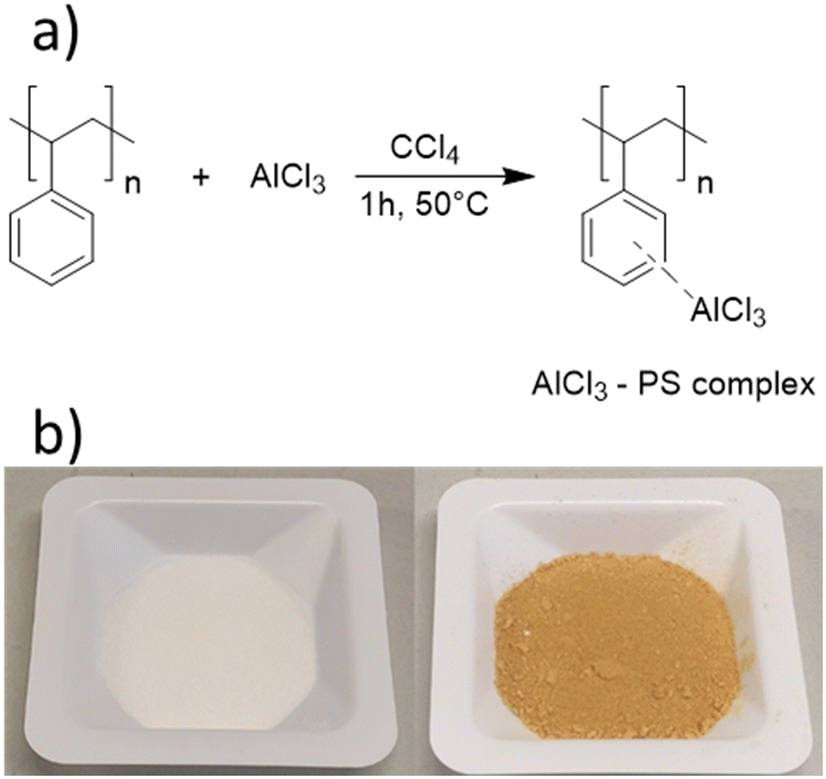 | ||
| Scheme 3 a) Reaction scheme for the immobilization of AlCl3 in a porous polystyrene matrix via crosslinking. b) Photograph of the porous polystyrene before (left) and after (right) immobilization of AlCl3. | ||
IR spectroscopy revealed new vibrational bands in the sample containing AlCl3. A strong band at approximately 1600 cm−1, characteristic of AlCl3, was detected. The broad band around 3300 cm−1 was attributed to hydroxyl groups from partially hydrolysed AlCl3. The corresponding spectra are shown in the ESI† (Fig. S9). Furthermore, X-ray powder diffraction (PXRD) measurements were performed. The AlCl3@polystyrene composite exhibited new reflexes compared to the blank polymer reference (Fig. 2). These are attributable to AlCl3·6(H2O) based on data from the ICSD database (ICSD-22071). This confirms, that upon immobilization and work-up, a substantial portion of intact AlCl3 is present in the composite. We also recorded PXRD patterns of the composite after employing it as heterogenous catalyst. Additional reflexes are detected after five consecutive reaction runs at 160 °C for 6 h (see below). The corresponding peaks reflect the formation of the hydrolysis products AlCl(OH)2·2(H2O) and Al(OH)3 due to the (partial) hydrolysis of immobilized AlCl3 (see Fig. S13†).57 This is indicative of a loss in the catalytic activity of the composite upon reuse.
 | ||
| Fig. 2 Normalized PXRD patterns of the polymer reference (green), the polystyrene/AlCl3 composite before (blue) and after 5 catalytic runs (red); signals stemming from hydrolysis products AlCl(OH)2·2(H2O) and Al(OH)3 are marked with an asterisk. The diffractogram of AlCl3·6(H2O) (ICSD-22071) is shown for comparison (black). | ||
For reactants to access the catalytically active species efficiently, it is critical that the composite provides a sufficiently high surface area. BET measurements revealed that the AlCl3–polymer composite is mesoporous, with a surface area of approximately 6 m2 g−1 (see Fig. S10†). The analysis further indicates, that the mesopores have an average pore size of 32 Å.
Using the AlCl3-containing polymer composite as a catalyst for the conversion of benzonitrile (1a) and trimethylsilyl azide (2) to the corresponding tetrazole 3a, yields of up to 79% were achieved (solvent-free, Scheme 1, 160 °C, 6 h; see Experimental section for details). We assessed the influence of reaction time on the NMR yield of 3a by stopping the reactions after 1 to 6 h (Fig. 3).
 | ||
| Fig. 3 Product yield of 5-phenyltetrazole as determined by 1H-NMR in presence of polystyrene-bound AlCl3 as heterogeneous catalyst (black) or after hot filtration after 1 hour (red). | ||
Longer reaction times did not significantly improve product yield. While yields obtained with the composite catalysts are generally lower than those obtained when employing AlCl3 directly (79% vs. 96%), the required time decreases considerably (6 h vs. 16 h). Furthermore, when comparing turnover number (TON) and turnover frequency (TOF), we observe almost identical values when employing the composite compared to AlCl3: for the conversion of 1a and 2 to the corresponding tetrazole 3a, a TON of 8.8 (compared to 9.6 for AlCl3) and a TOF of 1.46 h−1 (compared to 1.6 h−1 for AlCl3) is obtained. While higher TONs and TOFs for the catalytic tetrazole synthesis were reported58–60 the results still underpins the practicality of utilizing low-cost precursors along with a simple loading of AlCl3 leading to catalytically active composites.
Importantly, the work-up procedure becomes much more convenient: The AlCl3–polymer composite can be easily separated from the reaction product by filtration. However, filtration cannot remove Al species leached from the composites. To study the leaching of the catalyst, “hot filtration” tests were performed: the composite material with polymer-bound AlCl3 was filtered from the reaction mixture after 1 h. The samples were maintained at 160 °C for 1–5 h. For samples containing polymer-bound AlCl3, the product yield continued to increase with increasing time. In contrast, the yield remained constant for the sample in which the composite catalyst was removed via filtration (Fig. 3). Although the leaching of catalytically active Al species from the polymer cannot be entirely excluded, if it occurs, it is promptly transformed into a non-catalytically active species. This indicates that the polymer-bound AlCl3 truly acts as a heterogeneous catalyst.
Because the polymer composite can be easily separated from the reaction mixture, we tested the activity of the catalyst upon reuse. The composite was collected, washed, dried, and reused in subsequent reaction runs. For substrate 1a, a second run yielded 58% 3a, which gradually decreased to 40% in the fifth run (Fig. 4). As indicated by the PXRD results, this inactivation is a consequence of the hydrolysis of AlCl3 within the composite. Leaching of catalytically active species may also contribute to the loss of reactivity.
 | ||
| Fig. 4 1H-NMR determined yields of 5-phenyltetrazole with (reused) polystyrene-bound AlCl3 as heterogeneous catalyst. Conditions: 160 °C for 6 hours, solvent free. | ||
In addition to probing the persistence of the active species, the stability of the polymer support under experimental conditions was assessed. The NMR spectra recorded from the solutions extracted between the reaction runs showed no solubilized polymer components (see Fig. S3†), illustrating the stability of the support material.
To explore the versatility of our approach, we used nitriles 1b–f as additional starting compounds. These derivatives were chosen to represent steric (3c), electron-withdrawing (3d), and -donating (3b, 3e, 3f) effects (Scheme 4).
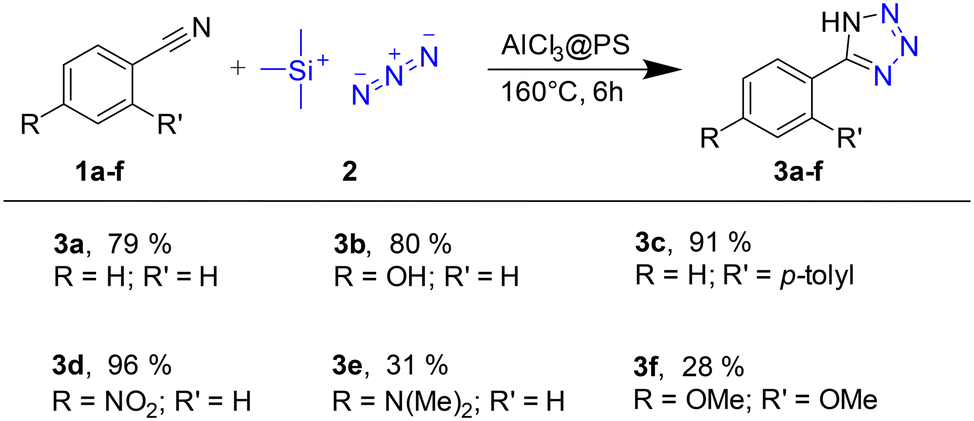 | ||
| Scheme 4 Scope of the solvent-free tetrazole synthesis; yields were determined by 1H-NMR. | ||
Except for 3e and 3f, which carry electron-donating substituents (4-dimethylamino or 2,4-dimethoxy groups, respectively), the yields of 3a–3d range from 79% to 96%. This is in line with less efficient conversions reported for analogous reactions showing that substrates with electron-withdrawing or weak electron donating groups provide higher reactivity.26,28,29
Conclusions
The embedding of AlCl3 into porous polystyrene results in a catalyst capable of producing 5-aryl tetrazoles in yields exceeding 80%. However, when the aromatic moiety contains electron-donating substituents, yields decrease to approximately 30%. The work-up process is straightforward, as the product can be extracted by washing the AlCl3/polystyrene composite with DMSO. Notably, this composite offers significantly shorter reaction times (6 hours compared to 16 hours). Although AlCl3 is partially hydrolysed during the reaction, we did not observe any leaching of catalytically active species, as confirmed by hot filtration experiments.Our study demonstrates that utilizing low-cost precursors along with a simple loading of AlCl3 leads to catalytically active composites. These composites not only provide efficient reaction times and good yields but also facilitate easy product isolation. Additionally, the use of DMSO as a solvent, instead of problematic high-boiling alternatives, underpins the practicality of this method.
Data availability
The data supporting this article have been included as part of the ESI† See DOI: https://doi.org/10.1039/D4CY01215A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NAWI Graz and the Austria Wirtschaftsservice aws (Prototype funding for Universities and Universities of Applied Sciences; Title: “Nanocomposites for an efficient synthesis of active pharmaceutical substances”) for financial support. M. W. and G. G. thank the FWF (Grant I5710 “Smart Inhibitors”) for financial support. The authors thank Prof. Paolo Falcaro for access to the facility for gas sorption measurements. The authors thank Flavius Schweiger, Jeremias Hajek and Arwin Samardzic for performing preliminary experiments.References
- C. X. Wei, M. Bian and G. H. Gong, Molecules, 2015, 20, 5528–5553 CrossRef CAS PubMed.
- L. M. T. Frija, A. Ismael and M. L. S. Cristiano, Molecules, 2010, 15, 3757–3774 CrossRef CAS PubMed.
- L. V. Myznikov, A. Hrabalek and G. I. Koldobskii, Chem. Heterocycl. Compd., 2007, 43, 1–9 CrossRef CAS.
- F. Lv, Y. Liu, J. Zou, D. Zhang and Z. Yao, Dyes Pigm., 2006, 68, 211–216 CrossRef CAS.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Am. Ethnol., 2008, 47, 2832–2835 CAS.
- O. I. Shmatova and V. G. Nenajdenko, J. Org. Chem., 2013, 78, 9214–9222 CrossRef CAS PubMed.
- E. A. Popova, R. E. Trifonov and A. Ostrovskii, Russ. Chem. Rev., 2019, 88, 644–676 CrossRef CAS.
- R. Vishwakarma, C. Gadipelly and L. K. Mannepalli, ChemistrySelect, 2022, 7, e202200706 CrossRef CAS.
- D. S. Wishart, C. Knox, A. C. Guo, S. Shrivastava, M. Hassanali, P. Stothard, Z. Chang and J. Woolsey, Nucleic Acids Res., 2006, 43, D668–D672 CrossRef PubMed.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- J. Roh, K. Vávrová and A. Hrabálek, Eur. J. Org. Chem., 2012, 31, 6101–6118 CrossRef.
- F. R. Benson, Chem. Rev., 1947, 41, 1–61 CrossRef CAS PubMed.
- Y. Joo and J. M. Shreeve, Org. Lett., 2008, 10, 4665–4667 CrossRef CAS PubMed.
- W. K. Su, Z. Hong, W. G. Shan and X. Zhang, Eur. J. Org. Chem., 2006, 12, 2723–2726 CrossRef.
- J. V. Duncia, J. B. Santella and M. E. Pierce, J. Org. Chem., 1991, 56, 2395–2400 CrossRef CAS.
- K. Ishihara, M. Kawashima, T. Shioiri and M. Matsugi, Synlett, 2016, 27, 2225–2228 CrossRef CAS.
- S. D. Guggilapu, S. K. Prajapti, A. Nagarsenkar, K. K. Gupta and B. N. Babu, Synlett, 2016, 27, 1241–1244 CrossRef CAS.
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970–2042 CrossRef CAS PubMed.
- I. Ugi and R. Meyr, Chem. Ber., 1961, 94, 2229–2233 CrossRef CAS.
- S. C. Sarngadharan, J. Aronson, C. Gelbaum, K. Griffith, J. Faris, A. B. Moihdeen, M. Patel, M. Malone, K. Richman, C. A. Eckert, C. L. Liotta and P. Pollet, Org. Process Res. Dev., 2022, 26, 1432–1441 CrossRef CAS.
- S. Swami, S. N. Sahu and R. Shrivastava, RSC Adv., 2021, 11, 39058–39086 RSC.
- W. G. Finnegan, R. A. Henry and R. Lofquist, J. Am. Chem. Soc., 1958, 80, 3908–3911 CrossRef CAS.
- F. Abrishami, M. Daryanavard and F. Nakhaei, J. Iran. Chem. Soc., 2023, 20, 1821–1829 CrossRef CAS.
- A. Babu and A. Sinha, ACS Omega, 2024, 9, 21626–21636 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210–12216 CrossRef CAS PubMed.
- L. Bosch and J. Vilarrasa, Am. Ethnol., 2007, 2, 3926–3930 Search PubMed.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- R. J. Herr, Bioorg. Med. Chem., 2002, 10, 3379–3393 CrossRef CAS PubMed.
- G. I. Koldobskii, V. A. Ostrovskii and V. S. Popavskii, Chem. Heterocycl. Compd., 1981, 17, 965–988 CrossRef.
- P. K. Kadaba, Synthesis, 1973, 2, 71–84 CrossRef.
- Y. Satoh and N. Marcopulos, Tetrahedron Lett., 1995, 36, 1759–1762 CrossRef CAS.
- D. Habibi, H. Nabavi and M. Nasrollahzadeh, J. Chem., 2013, 2012, 1–4 Search PubMed.
- S. Rostamizadeh, H. Ghaieni, R. Aryan and A. Amani, Chin. Chem. Lett., 2009, 20, 1311–1314 CrossRef CAS.
- A. Rahmatpour, Heteroat. Chem., 2012, 23, 472–477 CrossRef CAS.
- M. G. Mazzotta, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2342–2350 CrossRef CAS PubMed.
- A. El Maatougui, J. Azuaje, E. Sotelo, O. Caamaño and A. Coelho, ACS Comb. Sci., 2011, 13, 7–12 CrossRef CAS PubMed.
- K. Srirattnai, S. Damronglerd, S. Omi, S. Roengsumran, A. Petsom and G.-H. Ma, Tetrahedron Lett., 2002, 43, 4555–4557 CrossRef CAS.
- K. P. Borujeni and B. Tamami, Catal. Commun., 2007, 8, 1191–1196 Search PubMed.
- A. Rahmatpour, Appl. Organomet. Chem., 2011, 25, 585–590 Search PubMed.
- A. Rahmatpour and J. Aalaie, Heteroat. Chem., 2011, 22, 85–90 Search PubMed.
- D. C. Neckers, D. A. Kooistra and G. W. Green, J. Am. Chem. Soc., 1972, 94, 9284–9285 CrossRef CAS.
- E. C. Blossey, L. M. Turner and D. C. Neckers, Tetrahedron Lett., 1973, 21, 1823–1826 Search PubMed.
- F. R. Bou-Hamdan, K. Krüger, K. Tauer, D. T. McQuade and P. H. Seeberger, Aust. J. Chem., 2013, 66, 213–217 CrossRef CAS.
- J. F. Rabek and J. Lucki, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2537–2551 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- F. Ambroz, T. J. Macdonald, V. Martis and I. P. Parkin, Small Methods, 2018, 2, 1–17 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- P. Erdmann and L. Greb, Angew. Chem., 2022, 61, 1–8 Search PubMed.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- D. C. Neckers, D. A. Kooistra and G. W. Green, J. Am. Chem. Soc., 1972, 94, 9284–9285 CrossRef CAS.
- E. C. Blossey, L. M. Turner and D. C. Neckers, Tetrahedron Lett., 1973, 21, 1823–1826 CrossRef.
- J. F. Rabek and J. Lucki, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2537–2551 CrossRef CAS.
- A. D. V. Souza, C. C. Arruda, L. Fernandes, M. L. P. Antunes, P. K. Kiyohara and R. Salomão, J. Eur. Ceram. Soc., 2015, 35, 803–812 CrossRef CAS.
- M. A. Ashraf, Z. Liu, C. Li and D. Zhang, Appl. Organomet. Chem., 2021, 35, e6133 CrossRef CAS.
- N. Taherzad, L. Kafi-Ahmadi and A. Poursattar Marjani, Appl. Organomet. Chem., 2023, 37, e7089 CrossRef CAS.
- M. A. Jani and K. Bahrami, Appl. Organomet. Chem., 2020, 34, e6014 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01215a |
| This journal is © The Royal Society of Chemistry 2025 |