
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous catalytic hydrogenation of N-benzyl nicotinamide: a comparative study with nicotinamide adenine dinucleotide†
Makoto
Hirano
 *a,
Wataru
Onodera
b,
Masazumi
Tamura
b and
Yutaka
Amao
*ac
*a,
Wataru
Onodera
b,
Masazumi
Tamura
b and
Yutaka
Amao
*ac
aGraduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka, 558-8585, Japan. E-mail: ss23240i@st.omu.ac.jp
bGraduate School of Engineering, Osaka Metropolitan University, 3-3-138, Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
cResearch Centre of Artificial Photosynthesis, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@omu.ac.jp
First published on 7th May 2025
Abstract
In this study, we report the hydrogenation of N-benzyl nicotinamide (BNA+), a structural mimic of the coenzyme nicotinamide adenine dinucleotide (NAD+) required for enzymatic reactions, to 1,4-dihydro-N-benzyl nicotinamide (1,4-BNAH) using a Pt/SiO2 catalyst. This reaction was performed under mild conditions (20 °C, 1 atm H2), with a view to future applications in enzymatic reactions. A production rate of 1,4-BNAH reaching 170 mmol-1,4-BNAH g-Pt−1 h−1 or 81 mmol-1,4-BNAH mmol-Pt per surface sites per h was achieved, which was approximately 6.8 times higher than that of 1,4-NADH (26 mmol-1,4-NADH g-Pt−1 h−1 or 12 mmol-1,4-NADH mmol-Pt per surface sites per h). Kinetic analysis revealed that both BNA+ and NAD+ hydrogenation were first-order in H2 pressure; however, the reaction order with respect to NAD+ concentration was 0.14, indicating strong adsorption on the Pt/SiO2 catalyst surface, whereas that for BNA+ was 0.56, suggesting weaker adsorption that contributes to the enhanced 1,4-BNAH production rate. Moreover, the Pt/SiO2 catalyst exhibited excellent recyclability, retaining its activity over five consecutive cycles without deactivation. This stability is attributed to the weaker adsorption of BNA+, mitigating catalyst fouling. Consequently, hydrogenation of BNA+ emerges as a promising strategy for cofactor regeneration in biocatalytic processes.
Introduction
Enzymes are catalysts with excellent features, such as high activity, selectivity and specificity, that allow them to perform even the most complex chemical processes under the mildest experimental and environmental conditions.1 Coenzymes, which are small organic molecules, activate enzymes and directly participate in enzymatic reactions,2 such as quinone coenzymes, vitamin coenzymes, and adenosine triphosphate.3 Notably, nicotinamide adenine dinucleotide (NAD), a type of vitamin coenzyme, serves as an electron carrier in various redox reactions and can exist in two states: oxidized (NAD+) and reduced (1,4-NADH). 1,4-NADH has the 4-position of the pyridine ring in a reduced state (Fig. 1(A)), with hydrogen at this position facilitating enzyme-mediated redox activation. In industrial processes, enzyme-catalyzed reactions often utilize 1,4-NADH, which is converted into NAD+. To regenerate 1,4-NADH within the reaction system, nonenzymatic methods have been developed for 1,4-NADH regeneration from NAD+ (Fig. 1(B)), including the use of reducing agents such as dithionite4 and NaBH4,5 homogeneous catalysis6–10 with metal complexes such as Ru, Rh, and Ir, and electrocatalytic11–13 and photocatalytic14–16 methods. Recently, the hydrogenation of NAD+ using supported metal catalysts has been considered an effective approach for 1,4-NADH regeneration17 because it enables the separation of the product and the catalyst; using H2, it reduces waste generated from sacrificial reducing agents and electron mediators, enhancing the E-factor (kg-waste per kg-1,4-NADH) and contributing to green chemistry.18 For instance, Wang et al. demonstrated 1,4-NADH regeneration by hydrogenating NAD+ with Pt/Al2O3 catalyst.19 However, the selectivity for 1,4-NADH production has been low, primarily owing to side reactions, such as reductions at the 6-position of the nicotinamide ring. To improve selectivity, a Pt/SiO2 catalyst doped with Sn improved selectivity from 30 to 90% during NAD+ hydrogenation.20 Table S1† summarizes the examples of 1,4-NADH production using H2 and heterogeneous catalysts.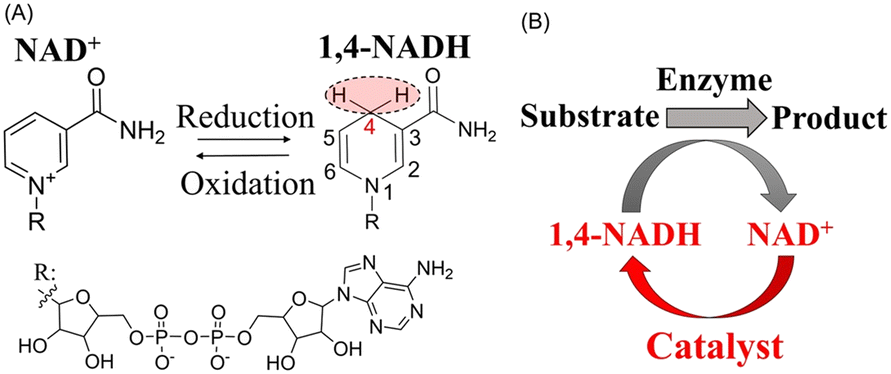 | ||
| Fig. 1 (A) Molecular structures of NAD+ and 1,4-NADH. (B) Catalytic regeneration of 1,4-NADH via NAD+ reduction. | ||
In addition to research on the regeneration of 1,4-NADH from NAD+, the development of synthetic nicotinamide cofactor mimics (mNADHs) has been explored21–23 to address the high-cost issue and low stability of 1,4-NADH.21–23 mNADHs can be synthesized relatively easily from nicotinamide and an appropriate alkyl-phenyl halide (e.g., benzyl chloride), allowing low-cost, one- or two-step synthesis.21 Because of its structural simplicity, stability, and applicability to enzyme reactions, 1,4-dihydro-N-benzyl nicotinamide (1,4-BNAH)—the reduced form of N-benzyl nicotinamide (BNA+), in which a benzyl group is attached to the nitrogen atom of the pyridine ring is a representative example of mNADHs (Fig. 2(A)).24–26 Notably, when the OYE3 enzyme from Saccharomyces pastorianus was used with 1,4-BNAH as a cofactor, higher enzymatic activity was observed compared with 1,4-NADH in the reduction of activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.27 Various methods for regenerating 1,4-BNAH from BNA+ have been reported (Fig. 2(B) and Table S2†), similar to 1,4-NADH regeneration from NAD+. In noncatalytic reactions, 1,4-BNAH can be produced using dithionite28 and NaBH4.29 As a catalytic reaction, the cobalt complex catalyst CoCl(DMG)2(py) achieved a production ratio of 1,4- to 1,6-isomers of approximately 80
C double bonds.27 Various methods for regenerating 1,4-BNAH from BNA+ have been reported (Fig. 2(B) and Table S2†), similar to 1,4-NADH regeneration from NAD+. In noncatalytic reactions, 1,4-BNAH can be produced using dithionite28 and NaBH4.29 As a catalytic reaction, the cobalt complex catalyst CoCl(DMG)2(py) achieved a production ratio of 1,4- to 1,6-isomers of approximately 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20–90:10 when using H2 as a reducing source.30 Moreover, rhodium complex catalyst, [Cp*Rh(Bpy)(H2O)]2+, known for its high selectivity in 1,4-NADH regeneration from NAD+, has been applied to the production of 1,4-BNAH with formate as a reducing source, achieving a high selectivity of >95%.31 Another approach involves systems that combine photo molecular catalysts,32 N-doped carbon nanodots (N-CD)33 or mesoporous indium tin oxide (ITO)34 with electron mediator [Cp*Rh(Bpy)(H2O)]2+ for 1,4-BNAH regeneration. However, these methods use homogeneous catalysts or dissolved electron mediators in reaction solution, making the purification of the product difficult. Further, the 1,4-BNAH production rate is approximately 1.5–2.5 times slower than that of 1,4-NADH, potentially because of the lower electron-withdrawing properties of the 1-benzyl group compared with the ribosyl group, thereby making the pyridine ring less capable of accepting electrons (Fig. S1†).34,35 Recently, bioinspired metal sulfide electrocatalyst, which directly generate 1,4-BNAH without using an electron mediator, have been reported, showing a high selectivity of 94%.36 Further discussions on the relative production rates of 1,4-NADH and 1,4-BNAH are anticipated in future studies. To our knowledge, there are no reported examples of heterogeneous catalysts for the hydrogenation of BNA+; for example, the enzyme biohybrid catalysts with immobilized platinum nanoparticles were tested with BNA+ instead of NAD+ but had difficulty forming nanoparticles.37 Therefore, exploring methods for the generation of 1,4-BNAH from BNA+ using heterogeneous catalysts with H2 is crucial. In this work, we investigated BNA+ hydrogenation to produce 1,4-BNAH using a metal-supported catalyst that facilitates both product separation and catalyst recycling (Fig. 2(C)). To facilitate future integration with enzymatic processes, the hydrogenation was performed under mild conditions (20 °C, 1 atm H2). We then compared the catalytic activities in the hydrogenation of NAD+ and BNA+ and performed kinetic studies, which offered new insights into how differences in molecular structure influence reactivity. Finally, the recyclability of the heterogeneous Pt/SiO2 catalyst was evaluated. Altogether, this investigation of NAD+ analogue hydrogenation using a heterogeneous catalyst lays essential groundwork for broadening the potential applications of these cofactors.
:20–90:10 when using H2 as a reducing source.30 Moreover, rhodium complex catalyst, [Cp*Rh(Bpy)(H2O)]2+, known for its high selectivity in 1,4-NADH regeneration from NAD+, has been applied to the production of 1,4-BNAH with formate as a reducing source, achieving a high selectivity of >95%.31 Another approach involves systems that combine photo molecular catalysts,32 N-doped carbon nanodots (N-CD)33 or mesoporous indium tin oxide (ITO)34 with electron mediator [Cp*Rh(Bpy)(H2O)]2+ for 1,4-BNAH regeneration. However, these methods use homogeneous catalysts or dissolved electron mediators in reaction solution, making the purification of the product difficult. Further, the 1,4-BNAH production rate is approximately 1.5–2.5 times slower than that of 1,4-NADH, potentially because of the lower electron-withdrawing properties of the 1-benzyl group compared with the ribosyl group, thereby making the pyridine ring less capable of accepting electrons (Fig. S1†).34,35 Recently, bioinspired metal sulfide electrocatalyst, which directly generate 1,4-BNAH without using an electron mediator, have been reported, showing a high selectivity of 94%.36 Further discussions on the relative production rates of 1,4-NADH and 1,4-BNAH are anticipated in future studies. To our knowledge, there are no reported examples of heterogeneous catalysts for the hydrogenation of BNA+; for example, the enzyme biohybrid catalysts with immobilized platinum nanoparticles were tested with BNA+ instead of NAD+ but had difficulty forming nanoparticles.37 Therefore, exploring methods for the generation of 1,4-BNAH from BNA+ using heterogeneous catalysts with H2 is crucial. In this work, we investigated BNA+ hydrogenation to produce 1,4-BNAH using a metal-supported catalyst that facilitates both product separation and catalyst recycling (Fig. 2(C)). To facilitate future integration with enzymatic processes, the hydrogenation was performed under mild conditions (20 °C, 1 atm H2). We then compared the catalytic activities in the hydrogenation of NAD+ and BNA+ and performed kinetic studies, which offered new insights into how differences in molecular structure influence reactivity. Finally, the recyclability of the heterogeneous Pt/SiO2 catalyst was evaluated. Altogether, this investigation of NAD+ analogue hydrogenation using a heterogeneous catalyst lays essential groundwork for broadening the potential applications of these cofactors.
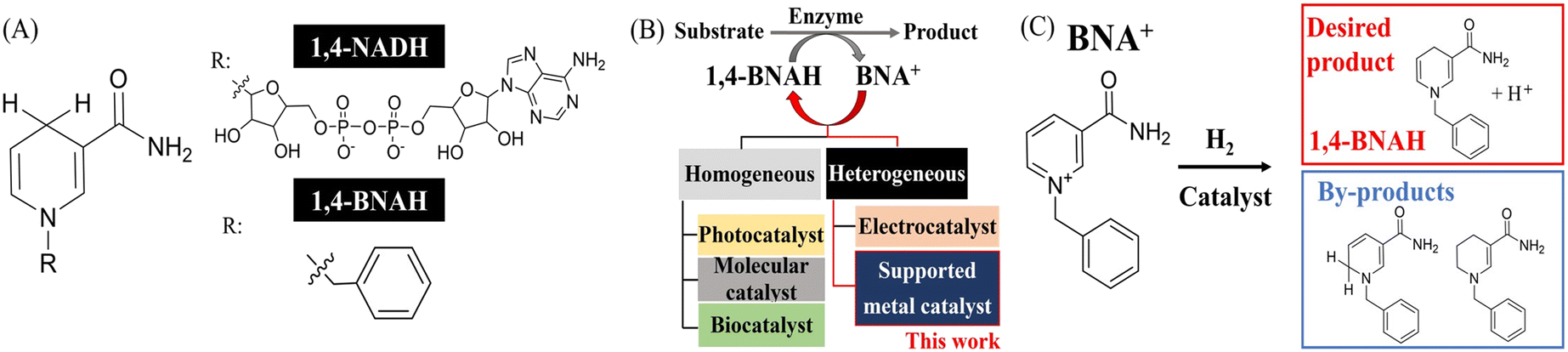 | ||
| Fig. 2 (A) Molecular structures of 1,4-NADH and 1,4-BNAH. (B) 1,4-BNAH regeneration using different catalyst types. (C) Hydrogenation of BNA+ to the desired product, 1,4-BNAH, with the production of other by-products. | ||
Experimental
Materials
Benzyl-3-carbamoylpyridin-1-ium bromide (BNA+, ≥95%, Combi-Blocks), 1-benzyl-1,4-dihydronicotinamide (1,4-BNAH, ≥95%, TCI), the reduced form of β-nicotinamide adenine dinucleotide (1,4-NADH, ≥95%, Oriental Yeast), the oxidized form of β-nicotinamide adenine dinucleotide (NAD+, ≥95%, Oriental Yeast), potassium dihydrogen phosphate (≥99%, Wako), dipotassium hydrogen phosphate (≥99%, Wako), tetraammineplatinum(II) nitrate [Pt(NH3)4](NO3)2 (Sigma-Aldrich), iridium(IV) nitrate [Ir(NO3)4HNO3] (Furuya Metal), nitrosyl ruthenium(III) nitrate [Ru(NO)(NO3)3] (Sigma-Aldrich), rhodium(III) nitrate [Rh(NO3)3] (Sigma-Aldrich), silica (SiO2) powder (CARiACT G-6, Fuji Silysia Chemical) with a pore size of 6 nm, particle size of 75–150 μm, and a surface area of 500 m2 g−1, methanol (≥99.7%, Wako), 1.0 mol L−1 Tris-HCl buffer (pH 8.8, Wako), 1,4-dioxane (super dehydrated, ≥99.5%, Wako), sucrose (Wako), and chloroform-d (≥99.8%, Wako), and hydrogen gas (≥99.999%) was provided by a YH-500 hydrogen gas generator (Scitem). A stock solution of 1,4-BNAH was prepared by dissolving it in acetonitrile, and the desired concentration was adjusted using 0.1 mol L−1 Tris-HCl buffer (pH 8.8). The purities of BNA+, NAD+ and 1,4-NADH were measured via nuclear magnetic resonance (NMR) using sucrose as an internal standard, whereas those of 1,4-BNAH were measured using 1,4-dioxane. Final concentrations were calculated by integrating the relevant proton signals in the 1H NMR spectra.Catalyst preparation
Silica-supported metal catalysts (Pt/SiO2, Ir/SiO2, Ru/SiO2, Rh/SiO2) were synthesized using the metal precursors. First, 1 g of SiO2 powder was dispersed in an aqueous solution containing the metal precursor. The volume and concentration of the precursor solution were adjusted to a 4 wt% metal loading for Ir/SiO2 catalyst, Ru/SiO2 catalyst, and Rh/SiO2 catalyst, and either 4, 1, or 0.5 wt% for Pt/SiO2 catalyst. A 2 mL aliquot of this solution was added to the silica and stirred at 80–90 °C for 2 h, forming a paste. The resulting mixture was dried overnight at 110 °C and calcined at 500 °C for 3 h at a heating rate of 10 °C min−1. For the H2 reduction pretreatment of Pt/SiO2, the catalyst was treated with H2 at 150 °C (heating rate of 10 °C min−1) for 1 hour, followed by passivation at room temperature in a 2% O2 atmosphere for 10 min.Catalyst characterization
X-ray diffraction (XRD) patterns were measured on a Rigaku MiniFlex600 diffractometer using Cu Kα radiation (λ = 0.154 nm, 40 kV, and 15 mA). The scanning range was 5–75° in 2θ with a step size of 0.02°, and the patterns were normalized to the integrated intensity of the SiO2 peaks. Hydrogen temperature-programmed reduction (H2-TPR) measurements were carried out using a Microtrac-BEL system equipped with a thermal conductivity detector (TCD). The flow gas was 5% H2/Ar at a flow rate of 30 mL min−1. Prior to the measurement, the sample was pre-treated at −30 °C for 30 min. The temperature was then ramped from −30 to 250 °C at a rate of 10 °C min−1.The number of surface Pt0 atoms on the Pt/SiO2 was estimated by CO pulse chemisorption. Before analysis, the catalyst was reduced under 5% H2 in He (30 mL min−1) at 150 °C for 30 min. Next, CO pulses (10% CO in He) were introduced at room temperature using a BELCAT instrument (MicrotracBEL). The average Pt particle size was calculated from the CO uptake, assuming a 1:1 stoichiometry between adsorbed CO and surface Pt atoms on spherical particles. Metal dispersion was determined by dividing the number of surface metal atoms (derived from CO uptake) by the total number of Pt atoms loaded onto the catalyst.
Hydrogenation
The hydrogenation of BNA+ under 1 atm H2 was carried out in a two neck-round bottom flask (25 or 300 mL) sealed with a butyl rubber cap and equipped with a magnetic stirrer. For experiments at H2 pressures of 2 and 4 bar, a custom-made 300 mL autoclave was used. A 0.1 mol L−1 Tris-HCl buffer solution (pH 8.8) containing BNA+ or NAD+ was placed into the flask, and the mixture was degassed by bubbling argon for 30 min. After degassing, the catalyst was introduced, and the headspace was purged with H2 (flow rate: 500 mL min−1) for 20 min. Next, the reaction was conducted in a water bath with stirring. Aliquots (0.1–0.2 mL) for sample were collected, filtered, and analyzed by high-performance liquid chromatography (HPLC) to determine the BNA+, NAD+, 1,4-BNAH, and 1,4-NADH concentrations. HPLC analysis was performed on a Hitachi Primaide system equipped with a TOSOH ODS column (4.6 × 250 mm2 and 5 μm). The mobile phase for BNA+ and 1,4-BNAH was a 1:1 (v/v) mixture of methanol and 100 mmol L−1 potassium phosphate buffer (pH 7). For NAD+ and 1,4-NADH, a 1:9 (v/v) mixture of methanol and the same buffer was used. Detection wavelengths were 265, 360, 260, and 340 nm for BNA+, 1,4-BNAH, NAD+, and 1,4-NADH, respectively (Fig. S2 and S3†).
Details of the calculation method of catalyst activity for conversion (%), yield (%), selectivity (%), and production rates (mmol-product per g-Pt per h or mmol-product mmol-Pt per surface sites per h) are provided in eqn (1)–(6), conversion (%), yield (%), and selectivity (%), the production rate of the 1,4-BNAH or 1,4-NADH per amount of Pt (mmol-product per g-Pt per h), the production rate per Pt surface site (mmol-product mmol-Pt per surface sites per h), and the total turnover number (total TON) are defined. The Pt surface sites are determined by CO chemisorption measurement. In these equations, the symbol “[-]” represents a millimolar concentration (mmol L−1), and “[-]0” denotes the initial concentration (mmol L−1) of BNA+ or NAD+. The production rates of 1,4-BNAH or 1,4-NADH were calculated at conversions below 25%.
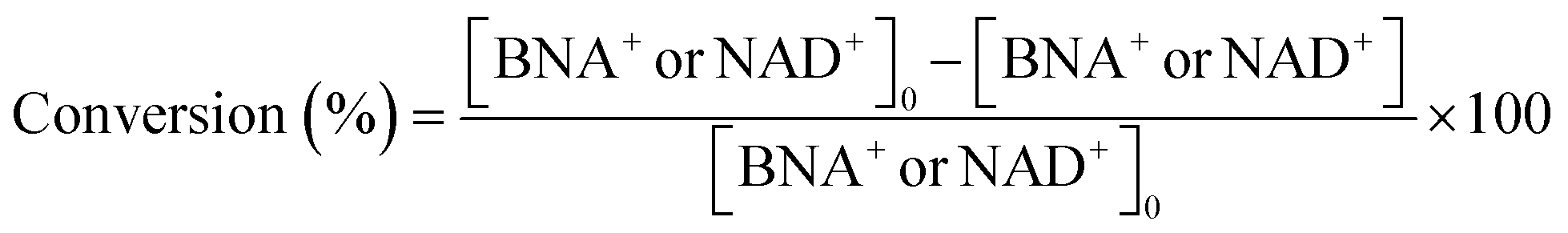 | (1) |
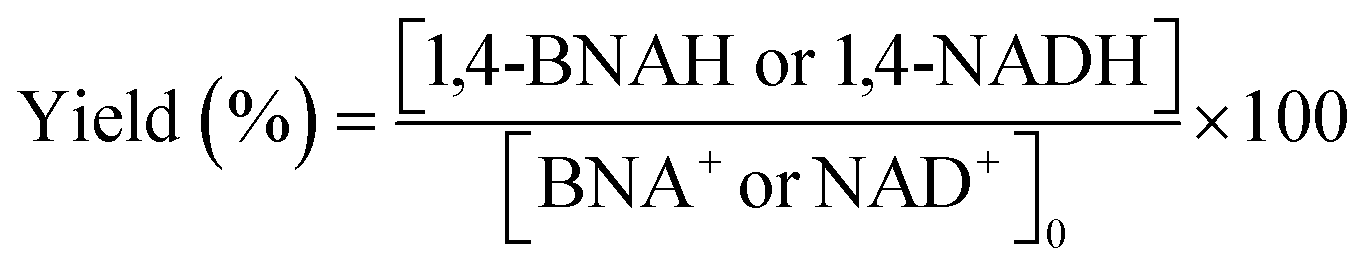 | (2) |
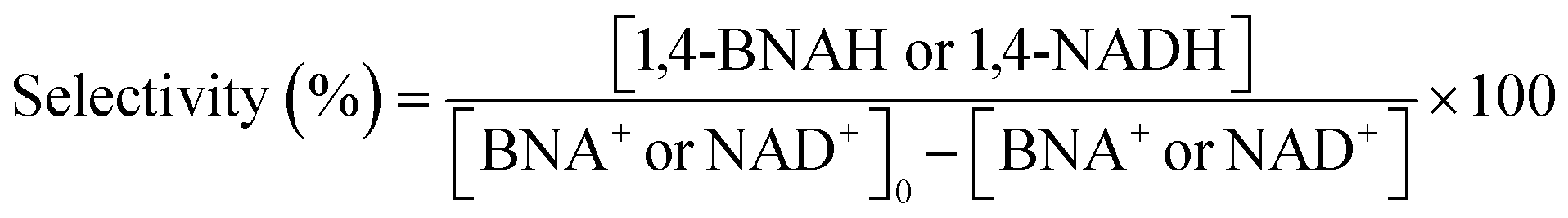 | (3) |
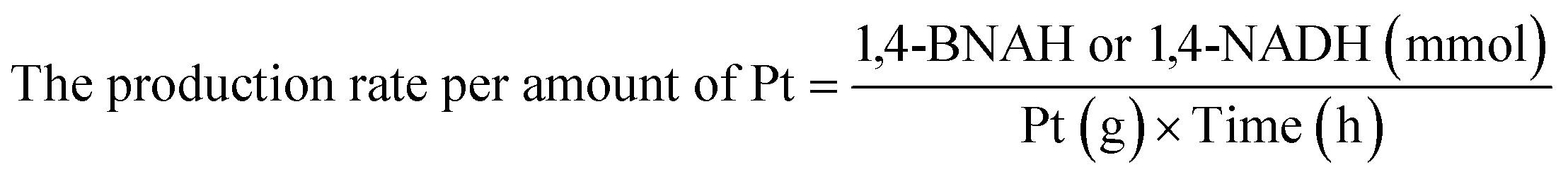 | (4) |
 | (5) |
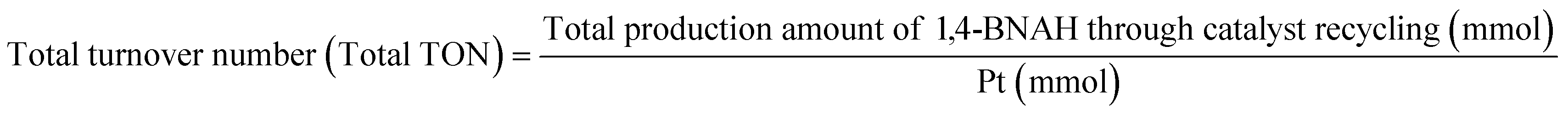 | (6) |
Catalyst recycling experiments used the same procedure for each reaction cycle. After 1 h of hydrogenation, the catalyst was recovered by centrifugation, washed with deionized water, and reused in subsequent runs. For product extraction and NMR analysis, BNA+ (1.7 mmol L−1) was reacted with the Pt/SiO2 (4 wt%, 20 mg) in 80 mL of Tris-HCl buffer (0.1 mol L−1, pH 8.8) at 20 °C and 1 atm H2 for 45 min. The reaction mixture was extracted three times with 30 mL of dichloromethane. The combined organic layers were washed with 80 mL of brine, dried over magnesium sulfate, and filtered. The filtrate was concentrated by rotary evaporation, and the residue was dissolved in deuterated chloroform. Quantitative 1H NMR spectra were obtained on a Bruker Avance-300 spectrometer with 128 scans, using 1,4-dioxane as an internal standard.
Results and discussion
Screening of supported noble metals for BNA+ hydrogenation
We investigated the effect of different noble metal species (Pt, Rh, Ir, Ru) supported in SiO2 for BNA+ hydrogenation. Based on Wang et al.'s work on 1,4-NADH regeneration using H2, in which Pt delivered the highest yields, we selected the metal species and support.38 In selecting the catalyst support, among SiO2, carbon, and MgO supports, the Pt/SiO2 catalyst had the highest production rate of 1,4-NADH, presumably because of the negative charge on SiO2 enhancing interactions with cationic NAD+.39 Thus, we chose SiO2 as the support in this study. Fig. 3 summarizes the screening test results of silica-supported noble metal catalysts for BNA+ hydrogenation. The Pt/SiO2 catalyst achieved conversion, yield, and selectivity values of 89, 30, and 34%, respectively; the Rh/SiO2 catalyst achieved 45, 3, and 7%; the Ir/SiO2 catalyst yielded 19, 6, and 32%; the Ru/SiO2 catalyst achieved 14.2% conversion and 14% yield. These results indicate that the Pt/SiO2 catalyst is appropriate for the hydrogenation of BNA+. During the reaction using the Pt/SiO2 catalyst, the solution gradually turned yellow (Fig. S4†). | ||
| Fig. 3 Hydrogenation of BNA+ (1.7 mmol L−1) in 20 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) at 20 °C and 1 atm H2 for 45 min using 5 mg of either 4 wt% Pt/SiO2 catalyst, 4 wt% Rh/SiO2 catalyst, 4 wt% Ir/SiO2 catalyst, 4 wt% Ru/SiO2 catalyst, or SiO2. | ||
Reaction solution analysis after BNA+ hydrogenation
To confirm the reaction products generated by the hydrogenation of BNA+ using the Pt/SiO2 catalyst, the reaction mixture was extracted with dichloromethane and analyzed by NMR, following a previously reported procedure.30 This analysis revealed three primary products: 1,4-BNAH (30% yield), its isomer 1,6-BNAH (12% yield), and the over-reduced species 1-benzyl-1,4,5,6-tetrahydropyridine-3-carboxamide (BTC, 10% yield). The resulting product distribution is shown in Fig. 4. Regarding the ratio of 1,4-BNAH to 1,6-BNAH produced by reduction, yielding a 1,4-BNAH:1,6-BNAH ratio of 72:28, which exceeds the 55:45 ratio reported with NaBH4 (ref. 29) and is comparable to the 80:20 ratio obtained with the homogeneous Co complex catalyst (CoCl(DMG)2(py))30 (Fig. S5†).
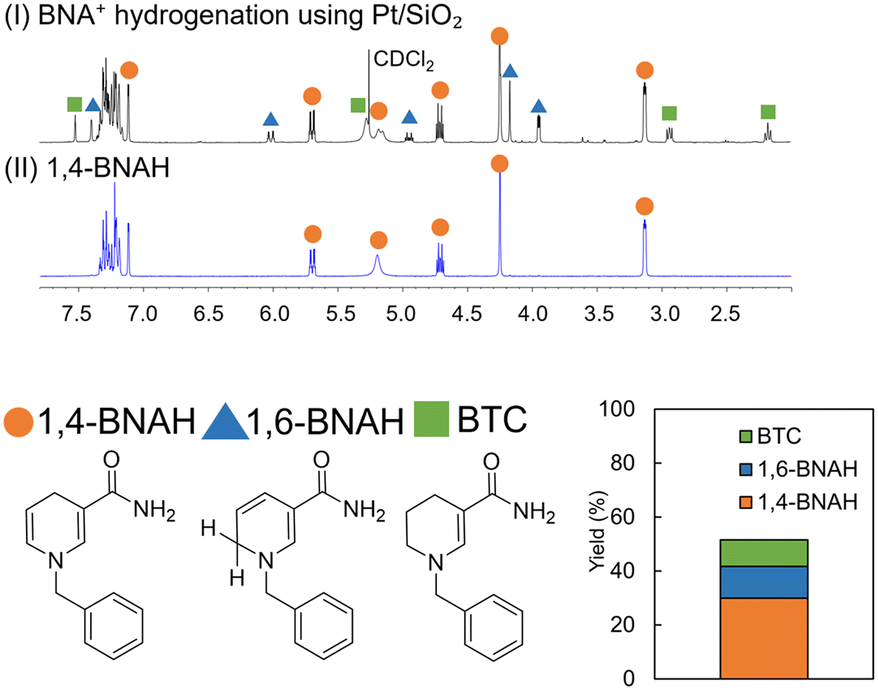 | ||
| Fig. 4 1H NMR spectra (in CDCl2) (I) Spectrum of the CH2Cl2 extracted solution obtained after the hydrogenation of BNA+ using a Pt/SiO2 catalyst. (II) Spectrum of the 1,4-BNAH standard compound. In the hydrogenation reaction, BNA+ (1.7 mmol L−1) was treated with Pt/SiO2 catalyst (4 wt%, 20 mg) in Tris-HCl buffer (0.1 mol L−1, pH 8.8, 80 mL) at 20 °C under 1 atm of H2 for 45 minutes. The NMR signals corresponding to 1,4-BNAH, 1,6-BNAH, and 1-benzyl-1,4,5,6-tetrahydropyridine-3-carboxamide (BTC) are marked with orange circles, blue triangles, and green squares, respectively. The relative composition of these products in the extracted solution is presented in the accompanying bar graph. | ||
Effect of H2 pretreatment on the Pt/SiO2 catalyst
H2-TPR measurements were performed to determine H2 pretreatment conditions of the Pt/SiO2 catalyst and the oxidation state of the Pt species. The H2 uptake calculated from the TPR profile matched the theoretical value for complete reduction of PtO2 (PtO2 + 2H2 → Pt + 2H2O), confirming that all Pt species are converted to the metallic ones at 130 °C (Fig. S6 and Table S3†). Accordingly, the catalyst was pretreated in flowing H2 at 150 °C, and the hydrogenation of BNA+ was tested with this H2-pretreated Pt/SiO2 catalyst. In these experiments, a 1.5 mmol L−1 BNA+ solution in 0.1 mol L−1 Tris-HCl buffer (pH 8.8) was reacted with 5 mg of either 4 wt% Pt/SiO2 catalyst or H2-pretreated 4 wt% Pt/SiO2 catalyst at 20 °C under 1 atm H2. As shown in Fig. 5A, the Pt/SiO2 catalyst reached conversion, yield, and selectivity of 93, 19, and 21%, respectively, after 60 min of reaction, whereas the H2-pretreated catalyst achieved values of 96, 22, and 23% at the same reaction time. Next, we compared the production rates of 1,4-BNAH in Fig. 5B. For the Pt/SiO2 catalyst without H2 pretreatment, the production rate was 68 mmol-1,4-BNAH g-Pt−1 h−1 (reaction time: 4.5 min; conversion: 17%; selectivity: 20%). In contrast, the H2-pretreated Pt/SiO2 catalyst exhibited a production rate of 117 mmol-1,4-BNAH g-Pt−1 h−1 (reaction time: 2 min; conversion: 11%; selectivity: 20%), indicating an approximately 1.7 fold increase in 1,4-BNAH production. Wang et al. reported that, compared to a catalyst without H2 pretreatment, applying H2 pretreatment to a Pt/Al2O3 catalyst for NAD+ hydrogenation increased the turnover frequency (TOF) of 1,4-NADH generation from 4 to 13 h−1, because of reduced Pt surfaces with increased H2 uptake capacity.19 A similar mechanism has potentially improved the 1,4-BNAH production rate in our system. These results indicate that H2 pretreatment of Pt/SiO2 catalyst to its metallic state does not alter product selectivity but enhances the production rate of 1,4-BNAH.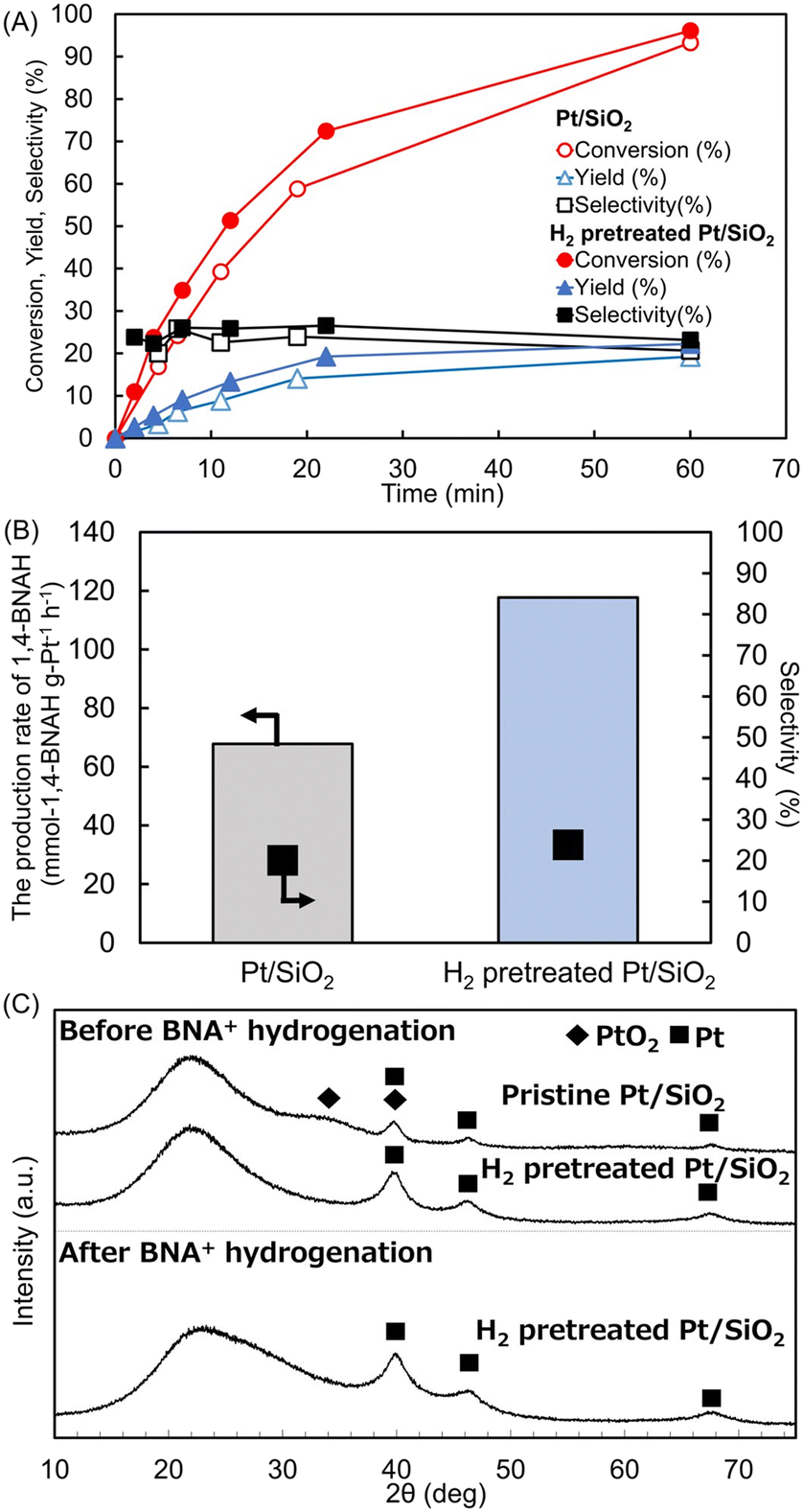 | ||
| Fig. 5 (A) Time course of BNA+ hydrogenation using Pt/SiO2 catalyst and H2-pretreated Pt/SiO2 catalyst. The reaction conditions: BNA+ (1.5 mmol L−1) in 20 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) with a 4 wt% Pt/SiO2 catalyst (5 mg) at 20 °C under 1 atm H2. (B) Comparison of the 1,4-BNAH production rates. (C) XRD patterns of the 4 wt% Pt/SiO2 catalyst comparing of pristine, without H2 pretreated, and H2 pretreated samples at before (upper) and after (lower) BNA+ hydrogenation. | ||
Fig. 5C presents the X-ray diffraction (XRD) patterns of the pristine Pt/SiO2 catalyst, the catalyst after H2 pretreatment, and the H2 pretreated catalyst following BNA+ hydrogenation. In the pristine Pt/SiO2 catalyst, reflections attributable to PtO2(100)40 are observed together with metallic Pt peaks. After H2 pretreatment, only metallic Pt reflections remain, confirming complete reduction of PtO2. Moreover, the diffraction pattern is unchanged after 45 min of BNA+ hydrogenation at 1 atm H2, indicating that Pt retains its metallic state throughout the reaction.
Effect of Pt loading amount
To investigate the effect of Pt loading on the Pt/SiO2 catalyst, 1 and 0.5 wt% Pt/SiO2 catalysts were prepared and compared with the 4 wt% Pt/SiO2 catalyst using all H2-pretreated samples. In the BNA+ hydrogenation reaction, the total Pt amount was standardized to that in 5 mg of the 4 wt% catalyst (0.2 mg Pt) by employing 20 mg of the 1 wt% catalyst and 40 mg of the 0.5 wt% catalyst. Time-course studies revealed that after 60 min, the 1 wt% Pt/SiO2 catalyst achieved 95% conversion, 26% yield, and 27% selectivity, while the 0.5 wt% Pt/SiO2 catalyst exhibited 90% conversion, 26% yield, and 29% selectivity (Fig. 6A and S7†). These results indicate that conversion, yield, and selectivity remained similar across different Pt loadings.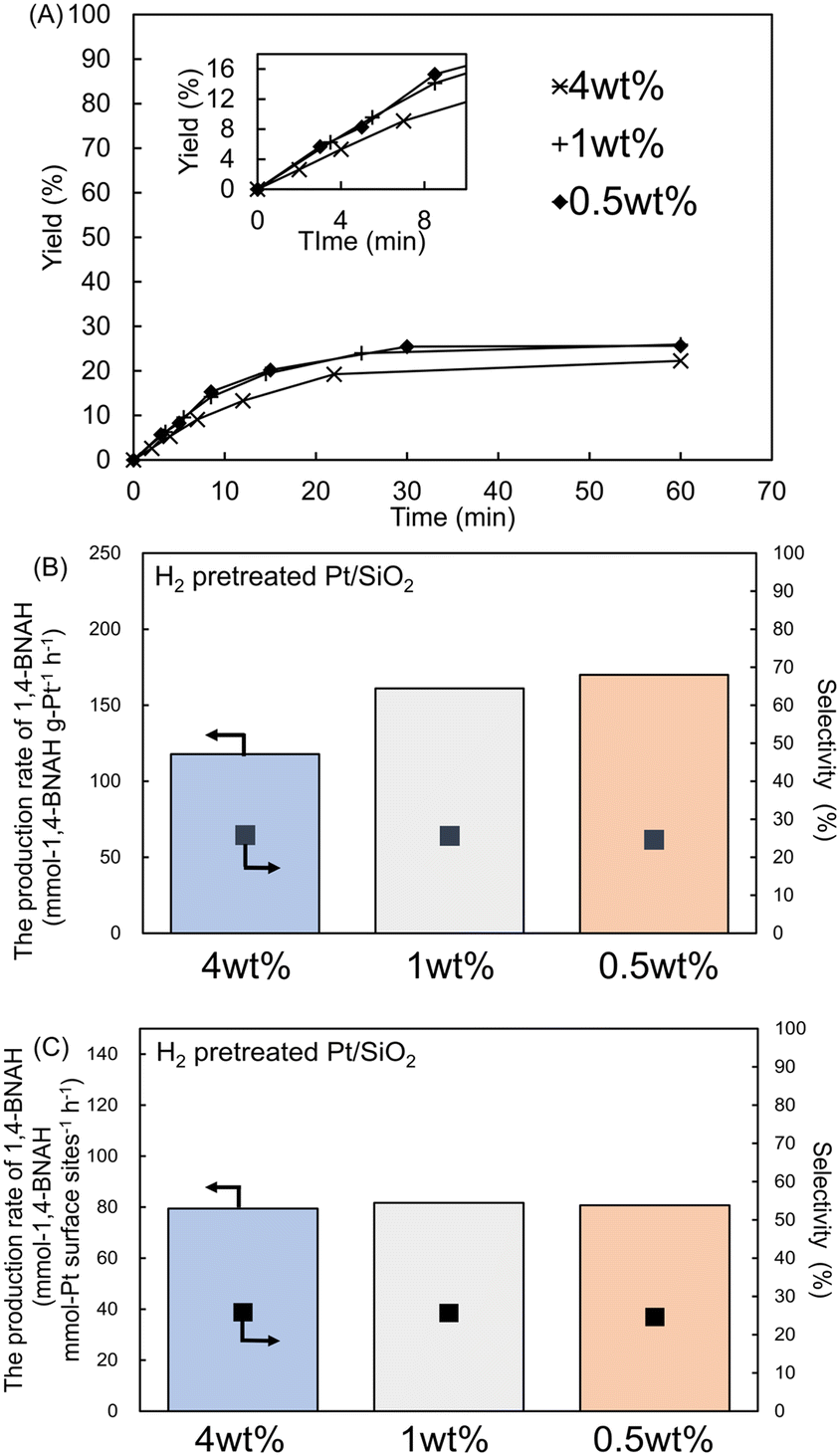 | ||
| Fig. 6 (A) Time course of yield in the hydrogenation of BNA+. Reaction conditions: BNA+ (1.5 mmol L−1) in 20 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) was reacted with H2-pretreated Pt/SiO2 catalysts (4 wt%: 5 mg, 1 wt%: 20 mg, 0.5 wt%: 40 mg) at 20 °C under 1 atm of H2. The inset shows an enlarged view of the yield change during the initial 0–10 minutes. (B) Comparison of the 1,4-BNAH production rate per total amount of Pt. (C) Comparison of the 1,4-BNAH production rate per surface Pt site. | ||
Notably, the 1 and 0.5 wt% Pt loadings exhibited a higher 1,4-BNAH production rate per amount of Pt than the 4 wt% loading. The 1 and 0.5 wt% Pt/SiO2 catalysts exhibited production rates of 161 mmol-1,4-BNAH g-Pt−1 h−1 (reaction time: 3.5 min; conversion: 24%; yield: 6.2% selectivity: 26%) and 170 mmol-1,4-BNAH g-Pt−1 h−1 (reaction time: 3 min; conversion: 23%; yield: 5.7% selectivity: 25%), respectively (Fig. 6B). Thus, reducing the Pt loading from 4 wt% to either 1 or 0.5 wt% resulted in a 1.4–1.5-fold increase in the production rate of 1,4-BNAH. Additionally, similar trends in 1,4-BNAH production rates were observed when the catalyst amount was varied from 5 to 10 mg for the 4, 1, and 0.5 wt% Pt/SiO2 catalysts (Fig. S8†).
CO pulse chemisorption analysis was performed to determine the number of surface Pt sites on the catalysts (Table S4†). Pt metal dispersion rates were 29, 38, and 41% for the 4, 1, and 0.5 wt% Pt/SiO2 catalysts, respectively. Using these dispersion rates, the production rate of 1,4-BNAH per Pt surface site was calculated to be 79, 81, and 82 mmol-1,4-BNAH−1 mmol-Pt per sites per h for the 4, 1, and 0.5 wt% catalysts, respectively (Fig. 6C and S9†). These findings suggest that the higher production rate arises from an increased number of Pt surface sites resulting from higher Pt dispersion. Particle sizes determined by CO pulse chemisorption were 3.9, 2.9, and 2.8 nm for the 4, 1, and 0.5 wt% catalysts, respectively (Table S4†). Furthermore, particle sizes calculated from the XRD patterns using the Scherrer equation were 3.7, 2.8, and 2.6 nm, respectively (Fig. S10†), and the values match those obtained from CO pulse chemisorption (Table S5†).
In summary, the Pt/SiO2 catalyst is a suitable metal species. H2 pretreatment of Pt/SiO2 improved the 1,4-BNAH production rate and lowering the Pt loading from 4 to 1 or 0.5 wt% increased metal dispersion and further accelerated the reaction. On other hand, the metal state and Pt particle size did not affect selectivity. In this study, the 0.5 wt% H2 pretreated Pt/SiO2 catalyst was chosen for BNA+ hydrogenation.
Comparison of 1,4-BNAH and 1,4-NADH production rates
Fig. 7(A) and (B) show the time courses for the hydrogenation of BNA+ and NAD+, each at 1.5 mmol L−1, using 40 mg of the H2 pretreated 0.5 wt% Pt/SiO2 catalyst in 20 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) at 20 °C under 1 atm H2. For BNA+ hydrogenation, 89% conversion, 29% yield, and 29% selectivity were achieved after 60 min. For the NAD+ hydrogenation, after 380 min the reaction achieved 98% conversion, a 28% yield, and 29% selectivity. Similar selectivity was obtained in both the BNA+ and NAD+ hydrogenation reactions. The yield and selectivity of the hydrogenation of NAD+ to 1,4-NADH using a Pt/SiO2 catalyst have been reported to be 23 and 20%, respectively.39 Next, the production rate of 1,4-NADH was determined to be 25 mmol-1,4-NADH g-Pt−1 h−1 and 12 mmol-1,4-NADH mmol Pt per sites per h (reaction time: 40 min; conversion: 14%; yield: 2.8% selectivity: 20%). NAD+ hydrogenation using Pt/Al2O3 catalyst reported a TOF of 13 h−1 for 1,4-NADH production, which is consistent with our results.19 Notably, hydrogenation of BNA+, the 1,4-BNAH production rate was determined to be 170 mmol-1,4-BNAH g-Pt−1 h−1 and 81 mmol-1,4-BNAH mmol-Pt per sites per h (reaction time: 3 min; conversion: 23%; yield: 5.7% selectivity: 25%). Therefore the production rate of 1,4-BNAH was approximately 6.8 times higher than that of 1,4-NADH (Fig. 7(C)); this observation contrasts with trends reported in comparative studies on the reduction of NAD+ and BNA+ to 1,4-NADH and 1,4-BNAH, respectively, such as photoelectrode reduction system via [Cp*Rh(Bpy)(H2O)]2+ as electron mediator. For instance, in a light-driven reaction using nitrogen-doped carbon dots (N-CD), TOFN-CD was 20.5 mmol g−1 h−1 for 1,4-NADH production and 8.26 mmol g−1 h−1 for 1,4-BNAH production (Fig. S1†).33 Similarly, in light-driven reactions using ITO, the production rates of 1,4-NADH and 1,4-BNAH were 0.47 and 0.33 mmol L−1 h−1, respectively;34 these studies indicate that the production rate of 1,4-NADH is 1.4–2.5 times higher than that of 1,4-BNAH, potentially owing to differences in their reduction potentials. On the other hand, in this study, the hydrogenation of BNA+ using the Pt/SiO2 catalyst resulted in a production rate of 1,4-BNAH, which is higher than that of 1,4-NADH.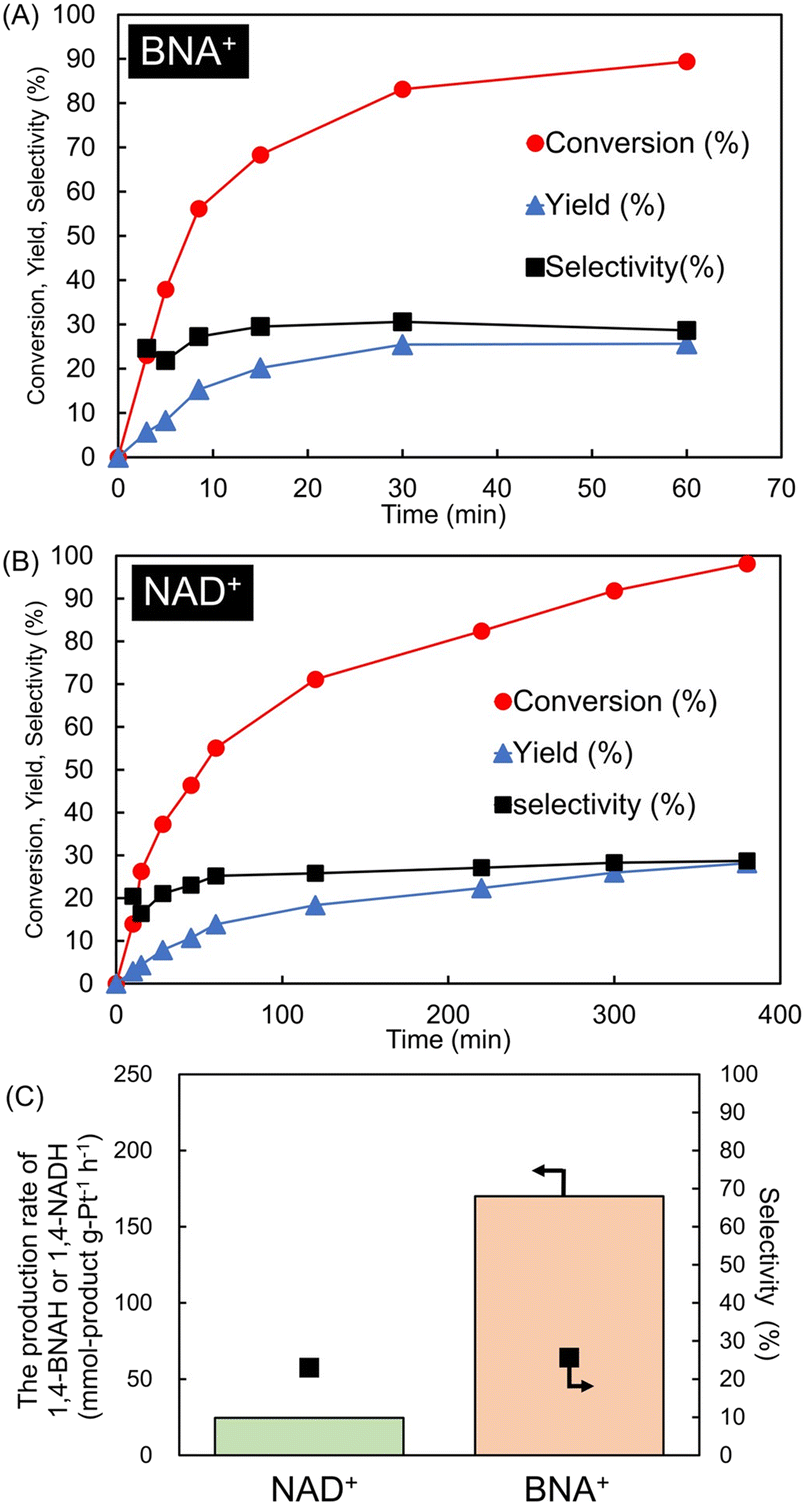 | ||
| Fig. 7 Time courses for the hydrogenation of BNA+ (A) and NAD+ (B), respectively, using H2-pretreated 0.5 wt% Pt/SiO2 catalysts (40 mg). Reaction condition: BNA+ or NAD+ 1.5 mmol L−1 in 20 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) at 20 °C under 1 atm H2. (C) Compares the production rates of 1,4-BNAH and 1,4-NADH per total Pt amount. | ||
Examination of reaction order
To investigate the reaction order of BNA+ and NAD+ hydrogenation, we examined the initial concentration dependence of NAD+ and BNA+ as well as the H2 pressure dependence in the reactions using the H2 pretreated 0.5 wt% Pt/SiO2 catalysts. By analyzing the production rates of 1,4-BNAH and 1,4-NADH, we determined the reaction orders regarding the initial concentrations of BNA+ and NAD+, as well as with respect to the H2 pressure. The production rate r (mmol-1,4-BNAH or 1,4-NADH g-Pt−1 h−1) was expressed using rate eqn (7). In this rate equation, the parameter k is the rate constant, and [BNA+ or NAD+] [1,4-BNAH or 1,4-NADH] denotes the concentration of substrate or product. The exponent a indicates the reaction order with BNA+ or NAD+. Similarly, [H2] represents the hydrogen concentration in the solution, and b is the reaction order with H2.| r = k[BNA+ or NAD+]a[H2]b | (7) |
We carried out experiments at 20 °C and 1 atm H2 using 1.8 mmol L−1 NAD+ or BNA+ under varying the Pt/SiO2 catalyst loading amount to 5, 10, and 40 mg. The results indicated that the production rates of 1,4-BNAH were 163, 157, and 148 mmol-1,4-BNAH g-Pt−1 h−1, respectively (Fig. S11-1†), whereas those of 1,4-NADH were 20, 19, and 21 mmol-1,4-NADH g-Pt−1 h−1, respectively (Fig. S11-2†). In all cases, the production rate increased proportionally with the catalyst loading amount, confirming that the catalytic reaction can be observed within the 5 to 40 mg range of Pt/SiO2 catalyst loading.
Using the 10 mg Pt/SiO2 catalyst under 1 atm H2 at 20 °C, hydrogenation was performed with different initial concentrations for each substrate, BNA+ and NAD+. For BNA+, initial concentrations of 1.0 and 0.4 mmol L−1 resulted in 1,4-BNAH production rates of 135 and 68 mmol-1,4-BNAH g-Pt−1 h−1, respectively (Fig. S12-1†). In contrast, for NAD+, initial concentrations of 0.8 and 0.3 mmol L−1 resulted in production rates of 18 and 15 mmol-1,4-NADH g-Pt−1 h−1, respectively (Fig. S12-2†). When the logarithm of the production rates was plotted against the logarithm of the initial substrate concentrations, which included the results from the 1.8 mmol L−1 substrate condition in the 10 mg Pt/SiO2 loading test, a linear relationship was observed (Fig. 8(A)). From the slopes of these plots, the reaction orders for BNA+ and NAD+ were determined to be 0.56 and 0.14, respectively.
 | ||
| Fig. 8 (A) log–log plots of the formation rate (r) of 1,4-BNAH or 1,4-NADH plotted against the logarithm of the substrate concentration in which circles and triangles represent BNA+ and NAD+, respectively. (B) log–log plots of the formation rate (r) of 1,4-BNAH or 1,4-NADH plotted against the logarithm of H2 pressure. (C) Illustration showing the adsorption of NAD+, BNA+, and H2 on a Pt/SiO2 catalyst. | ||
Next, the effect of H2 pressure was evaluated by varying the pressure to 2 and 4 bar under substrate concentration of 1.8 mmol L−1 for both BNA+ and NAD+ at 20 °C with the 10 mg Pt/SiO2 catalyst. The results indicated that the production rates of 1,4-BNAH were 297 and 675 mmol-1,4-BNAH g-Pt−1 h−1 at 2, and 4 bar, respectively (Fig. S13-1†), whereas those of 1,4-NADH were 47 and 88 mmol-1,4-NADH g-Pt−1 h−1 at 2 and 4 bar, respectively (Fig. S13-2†). When the logarithm of the formation rates was plotted against the logarithm of the H2 pressure, linear relationships were again observed. The reaction orders relative to H2 pressure were found to be 1.1 for the hydrogenation of both NAD+ and BNA+ (Fig. 8(B)).
Therefore, the lower reaction order of NAD+ compared with BNA+ suggests that NAD+ is strongly adsorbed on the Pt/SiO2 surface (Fig. 8C). In contrast, the suppression of nonspecific adsorption for BNA+ appears to accelerate the production of 1,4-BNAH relative to 1,4-NADH. The specific reaction mechanism and the substrate adsorption phenomena on the catalyst will be investigated in future studies.
The reusability of the catalyst was evaluated. Heterogeneous catalysts offer advantages over enzymes and homogeneous catalysts, considering they can be easily recovered by filtration. The reaction was scaled up to 100 mL using a 0.8 mmol L−1 BNA+ solution and 50 mg of the H2 pretreated 0.5 wt% Pt/SiO2 catalyst. After each 60 min reaction, the catalyst was recovered by centrifugation and decantation, then reintroduced into a fresh BNA+ solution for subsequent cycles. This process was repeated five times (Fig. 9). The results showed no changes in conversion, yield, or selectivity, confirming the stability of the catalytic activity. The repeated use of the catalyst resulted in a total turnover number of 119.
 | ||
| Fig. 9 The results of the catalyst recycling experiments. In these experiments, BNA+ (0.8 mmol L−1) was reacted in 100 mL of 0.1 mol L−1 Tris-HCl buffer (pH 8.8) using an H2-pretreated 0.5 wt% Pt/SiO2 catalyst (50 mg) at 20 °C under 1 atm H2. | ||
In contrast, the hydrogenation of NAD+ using a Pt/TiO2 catalyst reported a decline in yield during five catalyst recycling cycles.19 FT-IR spectroscopy detected two new additional peaks assigned to the vibrations of C–O and –CH2– groups, respectively, suggesting that the deactivation may be a result of the deposition of organic species, which probably arose from the fragment of NAD+.17 Based on the reaction order results, BNA+ appears to exhibit reduced adsorption onto the Pt surface compared to NAD+, therefore mitigating the accumulation of organic species on the catalyst. Consequently, the reusability of the catalyst is expected to be enhanced when employing BNA+ hydrogenation relative to NAD+ hydrogenation.
Conclusions
1,4-NADH, a key coenzyme, functions as a reductant in enzymatic reactions. A 1,4-NADH analogue, 1,4-BNAH, is of growing importance due to its applicability in enzymatic systems as well as its improved stability and cost-effectiveness. In this study, we investigated the hydrogenation of BNA+ to produce 1,4-BNAH using heterogeneous catalysts. As part of catalyst optimization, Rh, Pt, Ru, and Ir were supported on SiO2 and evaluated. Among them, Pt/SiO2 exhibited the highest catalytic activity. Furthermore, 1,4-BNAH could be synthesized under mild conditions (20 °C, 1 atm H2), highlighting the potential for integration with enzyme-catalyzed systems. It was also revealed that H2 pretreatment of the Pt/SiO2 catalyst enhanced the 1,4-BNAH formation rate. When comparing Pt loadings of 4, 1, and 0.5 wt%, the catalysts with 1 and 0.5 wt% Pt exhibited higher production rates than the 4 wt% catalyst, likely due to improved Pt dispersion. In addition, while product selectivity was similar for both BNA+ and NAD+ hydrogenation, the production rate of 1,4-BNAH reached 170 mmol g-Pt−1 h−1, approximately 6.8 times higher than that of 1,4-NADH. In contrast, photoelectrochemical reduction methods typically yield higher production rates for 1,4-NADH than for 1,4-BNAH, suggesting that the Pt/SiO2-catalyzed hydrogenation of BNA+ may offer a favorable approach for practical cofactor regeneration processes.Analysis of the reaction orders revealed that the reaction order concerning H2 pressure is almost first order for both substrates, whereas the reaction order concerning initial NAD+ concentration is 0.14. In contrast, the hydrogenation of BNA+ exhibited a reaction order of 0.56 concerning the initial BNA+ concentration. These results indicate that NAD+ is strongly adsorbed on the Pt/SiO2 while the adsorption of BNA+ is suppressed. This difference in adsorption characteristics likely contributes to the enhanced production rate observed for 1,4-BNAH compared to that for 1,4-NADH. The catalytic mechanism and the correlation between the molecular structure of substrate and its reactivity remain unclear at present, and further investigations are planned. On the other hand, the observation of high catalytic activity for BNA+ as an NAD+ analogue with a simpler structure than NAD+ is a highly significant achievement for broadening the applications of NAD+ analogues.
Finally, the Pt/SiO2 catalyst demonstrated reusability by maintaining its catalytic activity over five reaction cycles. This stability is attributed to the suppressed strong adsorption of BNA+ on the catalyst surface.
Our future work will focus on optimizing the catalyst to further improve reaction selectivity, as well as integrating BNA+ and other synthetic mNADHs with enzyme systems that utilize these cofactors. These advancements aim to establish an efficient cofactor regeneration process for biocatalytic applications.
Data availability
The authors confirm that the data supporting the findings of this manuscript are available within the article and its ESI.†Author contributions
Professor Yutaka Amao conceived and directed the project, supervised the research, and revised the manuscript. Professor Masazumi Tamura supervised the research and contributed to manuscript revisions. Makoto Hirano conducted the experiments and wrote the manuscript, and Wataru Onodera carried out the experimental work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grant-in-Aid for Specially Promoted Research (23H05404), Scientific Research (B) (22H01872), and (22H01871).Notes and references
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- A. Kirschning, Angew. Chem., Int. Ed., 2021, 60, 6242–6269 CrossRef CAS PubMed.
- B. J. Bryan Jones, D. W. Sneddon, W. Higgins and A. J. Lewis, Chem. Commun., 1972, 856–857 RSC.
- J. Kovár and H. Klukanová, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1984, 788, 98–109 CrossRef PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. Commun., 2025, 61, 3271–3282 RSC.
- F. Hollmann, B. Witholt and A. Schmid, J. Mol. Catal., 2003, 19, 167–176 Search PubMed.
- L. Tensi and A. Macchioni, ACS Catal., 2020, 10, 7945–7949 CrossRef CAS.
- M. M. Grau, M. Poizat, I. W. C. E. Arends and F. Hollmann, Appl. Organomet. Chem., 2010, 24, 380–385 CrossRef CAS.
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS.
- H. Ning, Y. Wu, C. Liu, Z. Zhao, Z. Li, J. Dai, P. Zhang, F. Li, L. Sun and F. Li, Angew. Chem., Int. Ed., 2025, e202503018 CAS.
- N. H. A. Besisa, K. S. Yoon, T. G. Noguchi, H. Kobayashi and M. Yamauchi, ACS Sustainable Chem. Eng., 2024, 12, 9874–9881 Search PubMed.
- H. K. Song, S. H. Lee, K. Won, J. H. Park, J. K. Kim, H. Lee, S. J. Moon, D. K. Kim and C. B. Park, Angew. Chem., Int. Ed., 2008, 47, 1749–1752 CrossRef CAS PubMed.
- T. Katagiri and Y. Amao, Sustain. Energy Fuels, 2022, 6, 2581–2592 Search PubMed.
- M. Higashi, T. Toyodome, K. Kano and Y. Amao, Electrochim. Acta, 2023, 460, 142590 CrossRef CAS.
- W. Dong, J. Tang, L. Zhao, F. Chen, L. Deng and M. Xian, Green Chem., 2020, 22, 2279–2287 RSC.
- M. Wang, X. Ren, M. Guo, J. Liu, H. Li and Q. Yang, ACS Sustainable Chem. Eng., 2021, 9, 6499–6506 CrossRef CAS.
- T. Saba, J. W. H. Burnett, J. Li, X. Wang, J. A. Anderson, P. N. Kechagiopoulos and X. Wang, Catal. Today, 2020, 339, 281–288 Search PubMed.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880–1886 Search PubMed.
- J. W. H. Burnett, J. Li, A. J. McCue, P. N. Kechagiopoulos, R. F. Howe and X. Wang, Green Chem., 2022, 24, 1451–1455 Search PubMed.
- I. Zachos, C. Nowak and V. Sieber, Curr. Opin. Chem. Biol., 2019, 49, 59–66 CrossRef CAS PubMed.
- C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2014, 4, 788–797 CrossRef CAS.
- F. Liu, L. He, S. Dong, J. Xuan, Q. Cui and Y. Feng, Molecules, 2023, 28, 5850 CrossRef CAS PubMed.
- T. Knaus, C. E. Paul, C. W. Levy, S. De Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033–1039 CrossRef CAS PubMed.
- C. Nowak, A. Pick, L. I. Csepei and V. Sieber, ChemBioChem, 2017, 18, 1944–1949 CrossRef CAS PubMed.
- C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernández, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180–183 Search PubMed.
- S. A. Löw, I. M. Löw, M. J. Weissenborn and B. Hauer, ChemCatChem, 2016, 8, 911–915 CrossRef.
- R. D. Chapman, R. A. O'brien and P. A. Kondracki, Tetrahedron, 1996, 52, 9655–9664 Search PubMed.
- T. Okamoto, S. Yamamoto, A. Ohno and S. Oka, Bull. Inst. Chem. Res., 1983, 61, 64–71 CAS.
- T. Okamoto, I. Yamamoto and S. Oka, J. Mol. Catal., 1987, 39, 219–223 CrossRef CAS.
- P. S. Wagenknecht, J. M. Penney and R. T. Hembre, Organometallics, 2003, 22, 1180–1182 CrossRef CAS.
- M. R. Schreier, B. Pfund, D. M. Steffen and O. S. Wenger, Inorg. Chem., 2023, 62, 7636–7643 CrossRef CAS PubMed.
- J. Kim, S. H. Lee, F. Tieves, D. S. Choi, F. Hollmann, C. E. Paul and C. B. Park, Angew. Chem., 2018, 130, 14021–14024 CrossRef.
- J. Kim, Y. Um, S. Han, T. Hilberath, Y. H. Kim, F. Hollmann and C. B. Park, ACS Appl. Mater. Interfaces, 2022, 14, 11465–11473 CrossRef CAS PubMed.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., 1999, 111, 1524–1527 CrossRef.
- S. Tian, S. M. Lu, T. Liu, F. Liu, C. Feng, X. Zhang, H. Zhang, C. Ding and C. Li, ChemCatChem, 2023, 15, e202300009 CrossRef CAS.
- H. A. Reeve, L. Lauterbach, O. Lenz and K. A. Vincent, ChemCatChem, 2015, 7, 3480–3487 CrossRef CAS PubMed.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- T. Saba, J. Li, J. W. H. Burnett, R. F. Howe, P. N. Kechagiopoulos and X. Wang, ACS Catal., 2021, 11, 283–289 Search PubMed.
- M. R. Gao, Z. Y. Lin, J. Jiang, C. H. Cui, Y. R. Zheng and S. H. Yu, Chem. – Eur. J., 2012, 18, 8423–8429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00202h |
| This journal is © The Royal Society of Chemistry 2025 |