
A passage from pincer complexes to rationally designed phosphine-free Co(III) catalysts supported by a pentadentate ligand for activation of alcohols: studies on sp3 C–H alkylation of 9H-fluorene and quinoline synthesis†
Prashant
Kukreti
,
Rahul
Chauhan
and
Kaushik
Ghosh
 *
*
Department of Chemistry, Indian Institute of Technology, Roorkee 247667, India. E-mail: kaushik.ghosh@cy.iitr.ac.in
First published on 14th May 2025
Abstract
In this study, a pentadentate carboxamide ligand (BPAPA-H = (2-(bis(pyridin-2-ylmethyl)amino)-N′-phenyl-N′-(pyridin-2-yl)acetohydrazide) is introduced and utilized for the synthesis of cobalt(III) catalysts [Co(III)(BPAPA)Cl]ClO4 (C1) and [Co(III)(BPAPA)Br]ClO4 (C2). These cobalt(III) catalysts are used for selective mono sp3 C–H alkylation of 9H-fluorene with cheap and abundant alcohols via the borrowing hydrogen (BH) approach, with the generation of ecologically benign water as a side product. Employing the existing methodology, 41 derivatives of mono sp3 C–H alkylated fluorene were produced utilizing different aromatic and aliphatic alcohols as alkylating agents, with isolated yields reaching as high as 97%. To investigate the catalytic potential in the synthesis of heterocycles, our optimized cobalt(III) catalyst facilitated the acceptorless dehydrogenative (AD) coupling of 2-aminobenzyl alcohol with aromatic ketones, resulting in the formation of 25 quinoline derivatives with yields reaching as high as 96%. The current methodology was also explored in the gram-scale synthesis of sp3 C–H alkylation of fluorene and quinoline synthesis for large-scale applications. A series of control experiments were carried out to explain and reveal the possible reaction mechanism and intermediates. The key intermediates involved in the catalytic cycle were characterized with the help of HRMS studies.
Introduction
The development of economically viable and eco-friendly methods from renewable sources for the formation of C–C and C–N bonds is a great area of research with considerable interest in sustainable organic synthesis.1–3 Alcohols are one such renewable resource that could easily be obtained from lignocellulose biomass by fermentation or catalytic reactions.4 Due to this, in recent years, borrowing hydrogen (BH) and acceptorless dehydrogenation (AD) reactions of alcohols catalysed by transition metals have received a lot of attention.5–7 These methods offer a straightforward and effective means of creating N-heterocycles and other significant compounds from easily accessible alcohols, with hydrogen and water serving as eco-friendly by-products.8,9 In the past few years, utilization of nonprecious base metal catalysts (Mn, Fe, Co, Ni) in BH and AD strategies has received significant attention in sustainable chemistry due to their high earth-abundance, eco-friendliness, and cost effectiveness in comparison with noble 4d metal catalysts (such as Ru, Rh or Pd).10–16The direct substitution of alcohols with various nucleophiles via a BH approach has recently attracted significant attention for C–C and C–N bond formation.17–20 In recent years, synthetic techniques for producing value-added substituted fluorene derivatives (9-monoalkylated and 9,9-dialkylated fluorenes) through selective alkylation of 9H-fluorenes have attracted considerable interest.21–24 Fluorene derivatives are well recognised for their biological and pharmacological properties.25–28 Several FDA-approved drugs containing a fluorene moiety exhibit various biological activities such as anticancer, antimalarial, antiarrhythmic, and neuromuscular blocking agents as shown in Fig. 1.29–32 Fluorene-based molecules are also utilized for biological applications such as lysosomal imaging,33–35 ATP sensing,36 protein binding,37–40 and β-amyloid inhibitors for the treatment of Alzheimer's disease.41
 | ||
| Fig. 1 Drug molecules based on fluorene and quinoline. | ||
In addition to their biological applications, π-conjugated polymers, such as polyfluorenes, are widely used in optoelectronic devices such as polymer light-emitting diodes (PLEDs), polymer solar cells, field effect transistors, and electrochromic devices.42–46 Fluorene-based probes are also developed for the detection of inorganic and organic molecules.47–50 Similarly, quinoline scaffolds are also found in many molecules with antibacterial, antifungal, antiviral, anti-protozoal, antimalarial, anticancer, and cardiovascular activities.51–54
Traditionally, sp3 C–H mono-alkylation of fluorene was achieved using an excess of alkyl halides in combination with strong bases like n-BuLi under severe conditions.55–58 Nevertheless, these approaches encounter various difficulties, such as yielding low to moderate outcomes and exhibiting poor selectivity, frequently resulting in unwanted dialkylation by-products. To overcome these challenges, it is essential to develop strategies for selective sp3 C–H monoalkylation that operate under mild conditions. In this regard, methods that employ transition metal-catalyzed borrowing hydrogen (BH) represent a sustainable and effective solution, allowing for the use of renewable alcohols as alkylating agents.59–75
The borrowing hydrogen approach was utilised for the first time by Gnanaprakasam and co-workers in 2020, demonstrating Ru(II) catalysed mono-alkylation of fluorene using alcohols.76 In 2022, Dai et al. utilized a spirocyclic NHC–Ir pincer complex for mono-alkylation of fluorene.77 There are only a few reports on the monoalkylation of fluorene catalysed by base metal (Mn, Ni, Cu, Zn) catalysts.78–83 Recently in 2024, Samanta and co-workers followed by Balaraman and co-workers developed Co(II) catalysts for monoalkylation of fluorene using an NNN pincer and an NN bidentate ligand.84,85 Utilizing a cobalt catalyst derived from a pincer PNP ligand, Zheng and co-workers reported quinoline synthesis by AD of alcohols for the first time in 2017.86 After that, various research groups developed cobalt-based pincer catalysts for the synthesis of the quinoline scaffold (Fig. 2b).87–95
 | ||
| Fig. 2 Previous reports on (a) sp3 C–H alkylation of fluorene and (b) quinoline synthesis by cobalt catalysts and (c) present report. | ||
Notably, the majority of cobalt-based catalysts described for BH and AD reactions are typically supported by bidentate or tridentate ligands, which usually lead to high-spin Co(II) complexes. However, it is well-known that ligands with strong π-accepting abilities, with a strong ligand field, are more efficient for alcohol dehydrogenation processes.92,96 This is especially important because the dehydrogenation of primary alcohols begins with the formation of a metal-alkoxide intermediate, produced through coordination with a π-donor alkoxide anion at the metal center. From a mechanistic point of view, previous reports have indicated the necessity of at least one labile group in the catalyst.10–16 In our earlier research on Ru(II) catalysts for BH and AD of alcohols, we found that a metal center featuring a five-coordinate structure along with one labile group can effectively function as a catalyst.97
In this manner, 3d metal complexes supported by pentadentate ligands could act as effective catalysts for organic transformations based on BH and AD. In this context, pentadentate mono-carboxamide ligands represent a promising ligand framework. These are one of the ligand frameworks that provide such a strong field environment, capable of making Co(III) low-spin complexes from Co(II) precursors.98–100 Nevertheless, based on our review of the literature, we found no reports of 3d metal catalysts derived from pentadentate ligands being used for the dehydrogenation of primary alcohols.
Keeping all these reports in mind, we have designed and synthesized a novel phosphine-free pentadentate mono-carboxamide ligand BPAPA-H and utilized it for the synthesis of versatile cobalt(III) catalysts [Co(III)(BPAPA)Br]ClO4 (C1) and [Co(III)(BPAPA)Br]ClO4 (C2). The catalysts were used for the selective sp3 C–H alkylation of fluorenes via the BH approach of alcohols and quinoline synthesis via the AD route of 2-aminobenzyl alcohols with aromatic ketones.
Results and discussion
In the current report, a pentadentate mono-carboxamide ligand (BPAPA-H) was synthesized via DCC & HOBT coupling of 2-(1-phenylhydrazinyl)pyridine and bis(pyridin-2-ylmethyl)glycine in an equimolar ratio in DMF. The ligand is characterized by 1H-NMR, 13C-NMR, FT-IR, UV-vis spectroscopy, and HRMS techniques (ESI,† Fig. S1–S5). The FT-IR spectrum of BPAPA-H displays a band at 1663 cm−1 for the (–CONH) amide group. The absorption spectrum of the ligand exhibits one main peak at 269 nm with a shoulder peak at 309 nm. BPAPA-H is then utilized to synthesize air-stable cobalt(III) complexes C1 and C2 in methanol as shown in Fig. 3a. The complexes are fully characterized by different spectroscopic techniques such as FT-IR, UV-vis, 1H NMR, 13C NMR, and HRMS (ESI,† Fig. S6–S15). The stretching frequencies of the amide (–CO–) group in C1 and C2 show a decline from 1663 cm−1 to 1648 cm−1 and 1647 cm−1, respectively, suggesting that the amide nitrogen coordinates with cobalt and that there is subsequent metal-to-ligand back donation into the π-antibonding orbital of the CONH moiety. The absorption spectra display two distinct peaks for C1 at 451 nm and 345 nm, whereas C2 shows absorption at 345 nm and 258 nm. The molecular mass of BPAPA-H, C1, and C2 was authenticated with the help of HRMS studies. In the HRMS spectrum, the peak at m/z 425.2091 corresponds to [BPAPA-H + H+], the peak at m/z 517.0957 to the [Co(BPAPA)Cl]+ cation, and the peak at m/z 561.0455 to the [Co(BPAPA)Br]+ cation (ESI,† Fig. S5, S10, and S15). | ||
| Fig. 3 (a) Synthetic scheme of [Co(III)(BPAPA)Cl]ClO4 and [Co(III)(BPAPA)Br]ClO4, (b) ORTEP diagram (25% probability level) of C1 (CCDC: 2356288), and (c) ORTEP diagram (25% probability level) of C2 (CCDC: 2356288). H atoms connected to C atoms and ClO4 are omitted for clarity. | ||
Crystal structures and description
The molecular structures of C1 and C2 are determined by single-crystal X-ray crystallography. Suitable size and good diffraction quality crystals of C1 and C2 were obtained by the ether diffusion method in methanol and acetonitrile, respectively. The ORTEP diagrams of C1 and C2 are shown in Fig. 3a and b. C1 and C2 were crystallized in the triclinic “P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ” space group, and a distorted octahedral geometry was found around the cobalt center. The N(7)–N(33) bond distance is 1.430(3) Å and the C(45)–O(9) bond distance is 1.238(3) Å in C1. The N(2)–N(8) bond distance is 1.416(3)Å and the C(17)–O(2) bond distance is 1.228(3) Å in C2. All selected bond lengths, angles, and structure parameters for C1 and C2 are depicted in the ESI† (Tables S1–S5).
” space group, and a distorted octahedral geometry was found around the cobalt center. The N(7)–N(33) bond distance is 1.430(3) Å and the C(45)–O(9) bond distance is 1.238(3) Å in C1. The N(2)–N(8) bond distance is 1.416(3)Å and the C(17)–O(2) bond distance is 1.228(3) Å in C2. All selected bond lengths, angles, and structure parameters for C1 and C2 are depicted in the ESI† (Tables S1–S5).
Selective sp3 C–H alkylation of fluorenes
In this study, we begin by optimizing reaction parameters for the mono-alkylation of fluorene (Table 1). The model substrates used in the study were fluorene (1a) and benzyl alcohol (2a). The influence of each reaction parameter (base, solvent, time, temperature, and catalyst mol%) was investigated, and several reactions were carried out to determine the best conditions. A detailed summary of the optimization of reaction parameters is presented in the ESI† (Tables S6–S11).|
|
|||||
|---|---|---|---|---|---|
| S. no | Catalyst (mol%) | Base (equivalent) | Temp. (°C) | Solvent | Yield (%) |
| 1 | C1 (1) | t BuOK 1.0 eq. | 100 | Toluene | 88 |
| 2 | BPAPA-H (1.0) | t BuOK 0.75 eq. | 100 | Toluene | NR |
| 3 | CoCl2·6H2O (1.0) | t BuOK 0.75 eq. | 100 | Toluene | NR |
| 4 | CoBr2 (1.0) | t BuOK 0.75 eq. | 100 | Toluene | NR |
| 5 | C1 (1.0) | t BuOK 0.75 eq. | 100 | Toluene | 88 |
| 6 | C2 (1.0) | t BuOK 0.75 eq. | 100 | Toluene | 94 |
| 7 | C1 (1.0) | KOH 0.75 eq. | 100 | Toluene | 81 |
| 8 | C1 (1.0) | Cs2CO3 0.75 eq. | 100 | Toluene | 62 |
| 9 | C1 (1.0) | t BuOK 0.75 eq. | 100 | Xylene | 76 |
| 10 | C1 (1.0) | t BuOK 0.75 eq. | 100 | DMF | NR |
| 11 | C1 (1.0) | t BuOK 0.5 eq. | 100 | Toluene | 71 |
| 12 | C1 (1.0) | t BuOK 1.0 eq. | 100 | Toluene | 94 |
| 13 | C1 (0.5) | t BuOK 0.75 eq. | 100 | Toluene | 53 |
| 14 | C1 (1.0) | t BuOK 0.75 eq. | 90 | Toluene | 78 |

As an initial attempt, we focused our study on the catalytic synthesis of sp3 C–H alkylated fluorene. For this, we performed a reaction with fluorene (1a, 0.5 mmol), benzyl alcohol (2a, 0.75 mmol), and C1 (1 mol%) with 1.0 equivalent tBuOK in toluene at 100 °C for 12 hours under an inert atmosphere of N2. This provides us an 88% yield of the respective mono-alkylated fluorene molecule 3a (Table 1, entry 1). Notably, efforts to carry out the reaction with just the ligand or CoCl2·6H2O or CoBr2 did not result in the production of the desired alkylated product 3a (Table 1, entries 2–4). Utilizing 0.75 equivalent tBuOK with catalysts C1 and C2 provides 88% and 94% yields of 3a, respectively. The reaction works effectively in non-polar solvents, producing 3a with 92%, 76%, and 67% yields using toluene, xylene, and benzene, respectively. On the other hand, 1,4-dioxane produced 3a in a 32% yield, but polar solvents such as DMSO, DMF, and t-amyl alcohol did not exhibit any product formation (ESI,† Table S7). Trace amounts of product 3a were formed when the reaction was performed using either the catalyst or the base alone (Table S6†). To figure out the optimal base for the reaction, several bases were examined (Table S8†). Strong bases such as tBuOK, KOH, and NaOH produced high yields of 3a with 94%, 81%, and 74%, respectively. The moderately strong base Cs2CO3 yielded 62%, whereas weaker bases like Na2CO3, K2CO3, and NaHCO3 showed no product formation. These findings suggest that a strong base is essential for the reaction to occur effectively. The maximum yield of 3a was achieved using 1 mol% C2 and 0.75 equivalents of tBuOK at 100 °C for 12 hours in toluene (Table 1, entry 6). The yield of 3a was not enhanced by raising the temperature beyond 100 °C, increasing the catalyst loading above 1 mol% and the base amount above 0.75 equivalents, and expanding the reaction period above 12 hours (ESI,† Tables S6, S9 and S10). However, a considerable drop in the yield of 3a was observed when the reaction parameters were lowered from their optimum values. After establishing the optimum reaction conditions, we move towards exploring the substrate scope for the current protocol.
Initially, several benzyl alcohols with various functional groups were used as alkylating partners to assess respective sp3 C–H alkylated fluorenes. Aromatic alcohols having electron-drawing groups at different positions, such as 4-F, 4-Cl, 4-Br, 4-CF3, 3-Cl, and 3-Br, have been explored as alkylation agents. The current methodology proceeds efficiently with these alcohols providing the respective alkylated fluorene derivatives (3b–3g) with 86–90% yields. Next, alcohol derivatives with electron-donating groups, such as 4-OMe, 4-tBu, 3-OMe, 2,4-dimethoxy, and [1,3]dioxole, were explored. All of these substrates provided the corresponding alkylated fluorene derivatives (3h–3l) in excellent yields of 91–96%. Furthermore, aromatic alcohols such as 4-Me, 4-Ph, 3-OPh, and 2-Me benzyl alcohols were examined, which provided admirable yields (86–92%) of the corresponding alkylated products (3m–3p). Additionally, the polycyclic aromatic alcohol 1-naphthylmethanol was used efficiently, yielding a high 95% yield of the alkylated fluorene 3q. The optimum conditions also showed good tolerance to secondary alcohols and alkylated product 3r was produced in an 88% yield utilizing 1-phenylethanol and 9H-fluorene.
Fluorene derivatives such as 2,7-dibromo fluorene and 2-bromo fluorene were also compatible and explored with various benzyl alcohols. With the current approach, the alkylated compounds (3s–3ab) were obtained in yields of 81–93% using 2,7-dibromofluorene and benzyl alcohol derivatives that contained both electron-donating and electron-withdrawing groups. Additionally, 2-bromofluorene exhibited significant reactivity with various benzyl alcohols yielding the alkylated compounds (3ac–3ah) with a range from 81% to 90% yields. The catalytic potential of C2 was also examined with aliphatic alcohols and showed good reactivity with them. Reactions with primary aliphatic alcohols, such as butanol, hexanol, octanol, decanol, and iso-amyl alcohol, produced the corresponding alkylated fluorenes (3ai–3am) with significant yields of 85–92%. Similarly, the secondary aliphatic alcohol cyclohexanol resulted in the alkylated product 3an with an 89% yield. The chemoselectivity of the methodology was also examined using 3,7-dimethyloct-6-en-1-ol, a primary alcohol bearing an internal double bond. The reaction selectively provides the monoalkylated fluorene derivative 3am in an 86% yield with excellent selectivity. The catalytic potential of C2 was further examined on a gram scale to figure out its potential for large-scale synthesis (Scheme 1). Notably, the methodology showed good results, affording gram-scale production of 3a and 3h with 81% and 84% yields, respectively.
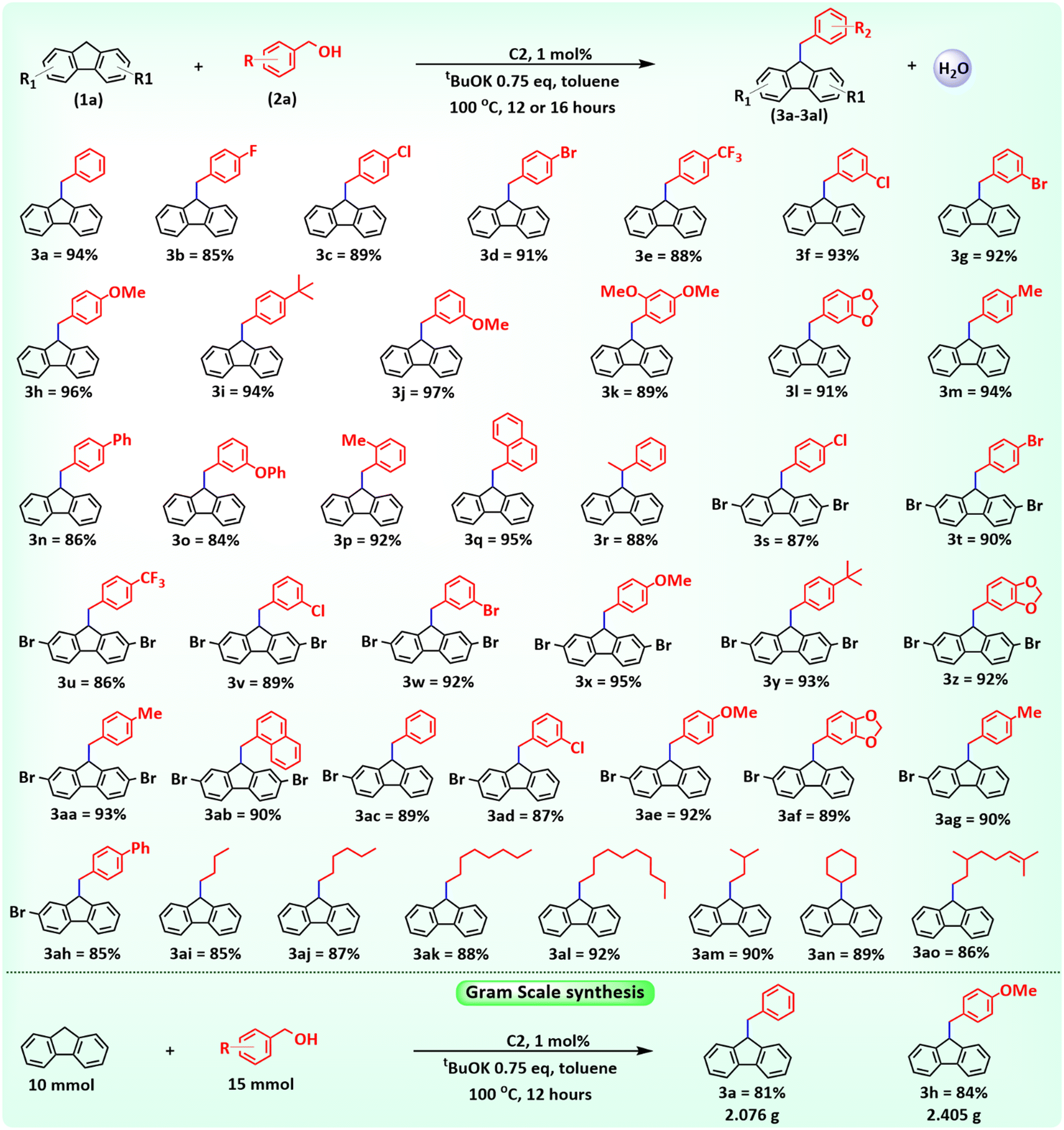 | ||
| Scheme 1 Scope of the sp3 C–H alkylation of 9H-fluorene. Reaction conditions: fluorene derivative 1a (0.5 mmol), benzyl alcohol derivative 2a (0.6 mmol), tBuOK (0.75 equivalent), temperature 100 °C under a N2 atmosphere, and time 12 hours for aromatic alcohols, and 16 hours for aliphatic alcohols under an inert atmosphere of N2. Isolated yields after column chromatography. | ||
Quinoline synthesis
To further expand the catalytic potential of C2 for N-heterocyclic synthesis, C2 was also utilized for quinoline synthesis via AD cyclization of 2-aminobenzyl alcohol (4a) and acetophenone (5a). Initially, a comprehensive analysis of reaction parameters such as the base, solvent, catalyst loading, temperature, and reaction time was carried out (ESI,† Tables S12–S17). Screening several bases revealed that tBuOK was the most efficient base to facilitate the dehydrogenative cyclization of 4a and 5a (ESI,† Table S13). Toluene has been found to be the most effective solvent for quinoline synthesis 6a (ESI,† Table S14). Lowering the base amount below 0.75 equivalents and the catalyst loading below 1 mol% reduced the production of 6a (ESI,† Tables S12 and S15). Reactions were performed at different time periods and temperatures, which demonstrates that an optimal reaction time of 12 hours at 100 °C provides the highest yield of 6a (ESI,† Tables S16 and S17). Raising the temperature above 100 °C, using catalyst amounts greater than 1 mol%, increasing the base quantity beyond 0.75 equivalents, or prolonging the reaction time past 12 hours did not enhance the yield of 6a. Conducting the reactions only with the ligand or CoCl2·6H2O or CoBr2 resulted in no product formation (ESI,† Table S12).Under the optimized conditions, 2-aminobenzyl alcohol was first coupled with a variety of substituted acetophenone derivatives to explore the scope of the current protocol. C2 exhibits remarkable catalytic efficiency with acetophenone derivatives having electron-drawing groups like 4-F, 4-Cl, 4-CF3, 4-CN, and 2-Cl (Scheme 2, 6b–6f), with 81–94% yields. Next, 2-aminobenzyl alcohol was reacted with acetophenone derivatives having electron-donating groups at various positions, including 4-OMe, 4-Me, 3-OMe, 3-Me, and 2-OMe. C2 demonstrated outstanding catalytic efficiency, producing the respective quinoline molecules (Scheme 2, 6g–6k) with 92–96% yields. Aromatic aliphatic ketones, such as propiophenone and butyrophenone, were also investigated, providing the quinoline derivatives 6l and 6m with 91% and 93% yields, respectively. Aromatic cyclic ketones, which include α-tetralone and 6-OMe-α-tetralone, produce the respective compounds 6n and 6o with high yields of 90 and 91%, respectively. Heteroaryl ketones such as 2-acetylthiophene, 2-acetylpyridine, 3-acetylpyridine, and 4-acetylpyridine were well tolerated utilizing the present strategy. These heteroaryl ketones provide their corresponding quinolines 6p–6s with 87–90% yields, respectively. Polycyclic aromatic ketones such as 2-acetyl naphthalene delivered the quinoline derivative 6t with a significant 94% yield. 2-Amino-5-chlorobenzyl alcohol was also explored with various aromatic ketones with 83–86% yields of the respective quinoline derivatives 6u–6w. Aliphatic ketones, such as 4 methylcyclohexanone, yielded the quinoline derivative 6x with a remarkable yield of 82%. The aliphatic amino alcohol, 3-aminopropan-1-ol, has a lower reactivity than aromatic amino alcohols, yielding the respective product 6y with a modest yield of 52%. Gram scale synthesis of 6j and 6p was also performed using C2, which resulted in 82% and 80% yields on a gram scale level (Scheme 2).
 | ||
| Scheme 2 Reaction conditions and stoichiometry: 2-amino benzyl alcohol (4a, 0.5 mmol), acetophenone derivative (5a, 0.5 mmol), temperature: 100 °C; base: 0.75 equivalent, and time 12 h under an inert atmosphere of N2. Isolated yields after column chromatography | ||
Control experiments
To explain the reaction pathway and possible intermediates involved in the sp3 C–H alkylation of 9H-fluorene, a series of control experiments were conducted (Scheme 3). Using the optimized reaction parameters, 4-methoxy benzyl alcohol with C2 provides the corresponding aldehyde 2b with a 92% yield. The formation of H2 during AD of benzyl alcohol and quinoline synthesis was confirmed via the reduction of styrene to ethyl benzene in the presence of 10% Pd–C in THF (Scheme 3a). The GC-MS analysis of the reaction mixture revealed the formation of the hydrogenation product ethylbenzene (shown in ESI† Fig. S19). Control experiments in the presence of radical quenchers (TEMPO) were also performed to figure out the radical or non-radical nature of the AD of benzyl alcohol by C2. In the presence of TEMPO, no major difference in the yield of respective aldehyde 2b was observed, explaining a non-radical pathway (Scheme 3b). The reaction of 2b with fluorene only in the presence of a base yields an alkenylated product 4b (Scheme 3c). Notably, 4b and 4-methoxy benzyl alcohol provide the respective alkylated fluorene 3h with an 87% yield in the presence of C2 (Scheme 3d). In the absence of C2, no formation of the corresponding sp3 C–H alkylated fluorene 3h was observed (Scheme 3e). In the presence of TEMPO, using standard reaction parameters, fluorene and 4-methoxy benzyl alcohol provide the respective alkylated products with no major difference in the yield (Scheme 3f). This experiment reveals the non-radical nature of the overall reaction. A mercury poisoning test was performed to explain the homogeneous nature of the catalytic reaction (Scheme 3g). No significant difference in the yield of product 3h was observed in the presence of 1 drop Hg, which explains the homogeneous nature of the reaction. | ||
| Scheme 3 Control experiments for mechanistic investigation (a–g) sp3 C–H alkylation of 9H-fluorene, (h–j) quinoline synthesis, (k and l) involvement of Co-hydride intermediate, (m and n) effect of air on the reactions, (o and p) GC-MS and (q) HRMS of the intermediates. | ||
A few control experiments were performed to figure out the reaction pathway for quinoline synthesis. Under optimized reaction conditions, only in the presence of a base, no formation of quinoline molecule 6h was observed from 2-aminobenzylalcohol and acetophenone (Scheme 3h). Utilizing optimized reaction parameters in the presence of C2, 2-aminobenzylalcohol, and acetophenone with 1 equivalent TEMPO did not show a significant change in the yield of quinoline molecule 6h (Scheme 3f). This experiment explains the non-radical nature of the overall reaction. A mercury poisoning test was also performed, which explains the homogeneous nature of the catalytic reaction (Scheme 3j).
Additionally, a few more control experiments were also performed to detect the potent intermediates during catalysis. Metal-hydride is one of the key intermediates involved in the organic transformation based on BH and AD of alcohols. A number of attempts were also performed to characterize Co(III)-hydride through 1H NMR studies. However, we were unable to characterize a Co(III)-hydride via1H NMR. The reason for this could be the less stable nature of these types of intermediates. To confirm the involvement of metal-hydride as a potent intermediate, control experiments with NaBH4 while lowering the base equivalents were performed. This results in the formation of desired products with no significant change in their yield, confirming the involvement of metal-hydride as a key intermediate (Scheme 3k and l).101,102 Performing the reactions in air led to a significant decrease in the yields of monoalkylated fluorene 3h and quinoline 6h (Scheme 3m and n), likely due to the high sensitivity of in situ formed metal-hydride in the presence of moisture and oxygen from air. The in situ formation of 9-benzylidene-9H-fluorene and (E)-3-(2-aminophenyl)-1-(p-tolyl)prop-2-en-1-one during the catalytic cycle was also confirmed through GC-MS studies (Scheme 3o and p). In the GC-MS spectrum, the m/z value of 284 and 237 stands for 9-benzylidene-9H-fluorene and (E)-3-(2-aminophenyl)-1-(p-tolyl)prop-en-1-one, respectively (ESI,† Fig. S18 and S20). HRMS studies were also performed to characterize the cobalt(III) intermediates. The cobalt hydride and cobalt-alkoxy intermediates of catalyst C2 were characterized by HRMS with an m/z value of 493.1292 for [(BPAPA)Co–H]+ and an m/z value of 589.1751 for [(BPAPA)Co–OCH2Ph]+, respectively (ESI,† Fig. S21 and S22).
Probable mechanism
Following that, we try to figure out the plausible reaction mechanism for the sp3 C–H alkylation of 9H-fluorene by a Co(III) catalyst. A proposed reaction mechanism for sp3 C–H alkylation of 9H-fluorene is outlined in Fig. 4a. The catalytic process begins with the dehydrogenation of primary alcohols to the corresponding aldehyde, catalysed by a Co(III) catalyst. Firstly, alcohols deprotonate in the presence of a base, producing an alkoxide anion, which reacts with the Co(III) catalyst C2 to form a cobalt(III)-alkoxy intermediate (C2A), which upon β-H elimination forms a cobalt hydride species (C2B) and aldehyde. Now the aldehyde reacts with a fluorene molecule in the presence of a base to produce 9-benzylidene-9H-fluorene. Then 9-benzylidene-9H-fluorene coordinates with intermediate C2B and by migratory insertion of hydride produces intermediate C2C, which reacts with another benzyl alcohol to produce a sp3 C–H alkylated fluorene molecule.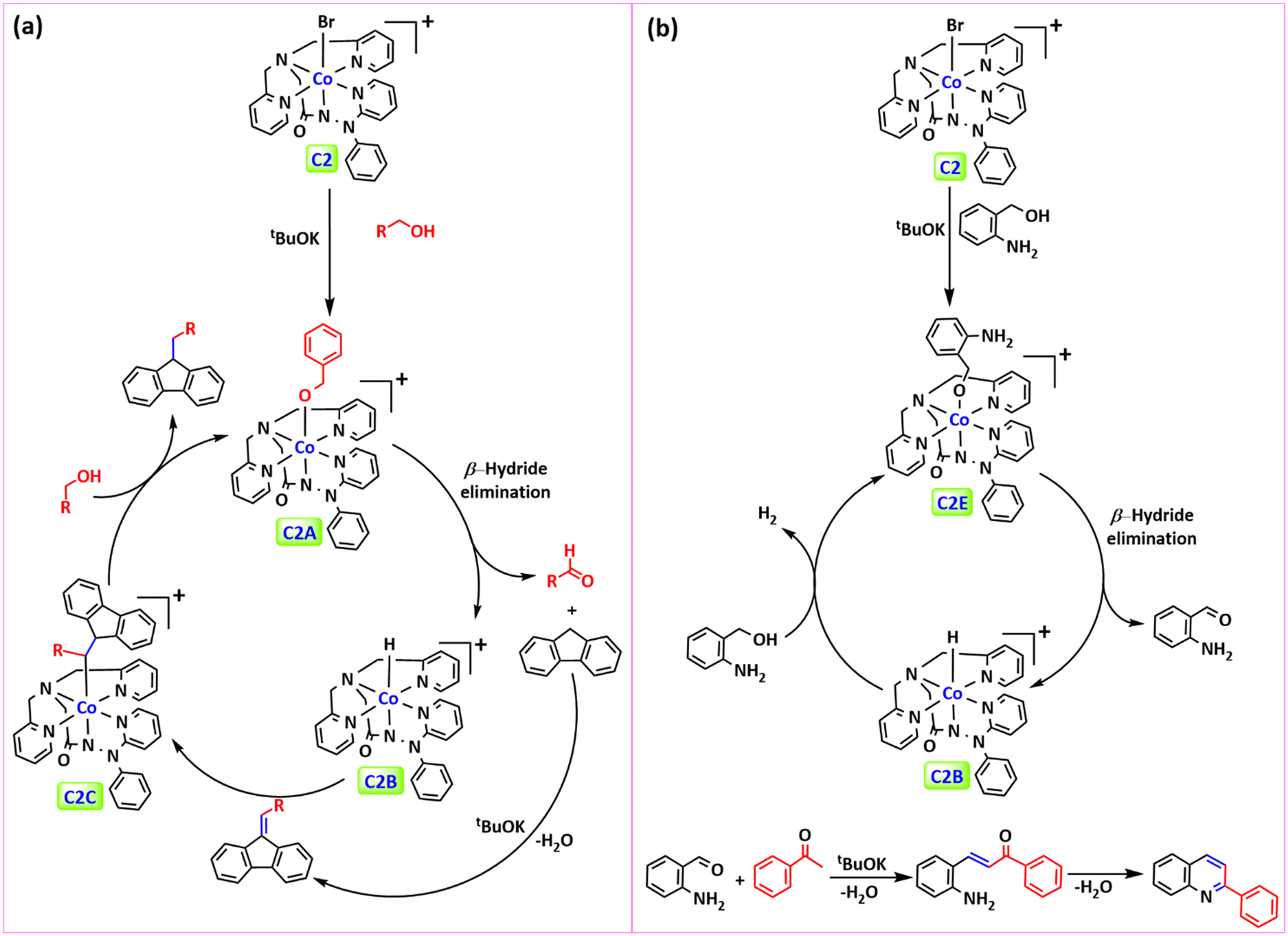 | ||
| Fig. 4 Proposed reaction pathway: (a) sp3 C–H mono-alkylation of 9H-fluorene and (b) quinoline synthesis. | ||
A possible pathway for the synthesis of quinoline is depicted in Fig. 4b. In the first step, when a base is present, 2-aminobenzyl alcohol is deprotonated to produce the corresponding alkoxide anion. This alkoxide then interacts with the Co(III) complex, resulting in the formation of intermediate C2E. After the elimination of β-hydride from intermediate C2E, intermediate C2B is generated, resulting in the release of 2-aminobenzaldehyde. When another molecule of 2-aminobenzyl alcohol reacts with intermediate C2B, it converts back to intermediate C2E and produces H2 gas. Finally, 2-aminobenzaldehyde reacts with acetophenone in a basic environment, leading to the formation of the amino-α-β unsaturated carbonyl compound, which provides a quinoline molecule after the intramolecular condensation reaction.
Conclusion
In summary, we have reported two versatile phosphine-free, air-stable, base metal Co(III) catalysts (C1 & C2) supported by a novel pentadentate mono-carboxamide ligand. These catalysts enabled the first Co(III) catalysed sp3 C–H alkylation of 9H-fluorene using greener biomass-derived alcohols as coupling partners. In addition, both catalysts efficiently catalyse quinoline synthesis via dehydrogenative coupling of 2-amino benzyl alcohol with aromatic ketones. C2 exhibited appreciable functional group tolerance and high catalytic efficiency to produce sp3 C–H alkylated fluorenes and quinoline derivatives having yields up to 93%. The catalytic efficiency of C2 was also explored for the gram-scale reactions for large-scale applications. Several control experiments were performed, which showed the involvement of Co-hydride as a key intermediate in the reaction. Based on our literature survey, we have developed the first base metal catalyst derived from a pentadentate ligand for BH and AD-based reactions. We firmly believe that this type of strategy can open up new route maps for the development of new sustainable base metal catalysts for BH and AD-based organic transformations.Data availability
All data are incorporated into the main manuscript and the ESI;† crystallographic data were deposited with the CCDC. Original data files are made available on request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
KG gratefully thanks CSIR, New Delhi CSR-2225-CMD/23-24 for financial assistance. PK shows his appreciation to IIT Roorkee, and RC is thankful to CSIR New Delhi for fellowships. We are grateful to DST-FIST for providing a single-crystal XRD facility under the grant code SR/FST/CS-II/2018/72(C) and to the Central Instrumentation Facility, IIT Roorkee, for access to instrumentation.Notes and references
- X. Chen, F. Xiao and W. M. He, Org. Chem. Front., 2021, 8, 5206–5228 RSC.
- J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Org. Chem. Front., 2021, 8, 384–414 RSC.
- S. Pimparkar, A. Koodan, S. Maiti, N. S. Ahmed, M. M. M. Mostafa and D. Maiti, Chem. Commun., 2021, 57, 2210–2232 RSC.
- M. Maji, D. Panja, I. Borthakur and S. Kundu, Org. Chem. Front., 2021, 8, 2673–2709 RSC.
- M. Trincado, J. Bösken and H. Grützmacher, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS.
- Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1761–1779 CrossRef.
- A. Cook and S. G. Newman, Chem. Rev., 2024, 124, 6078–6144 CrossRef CAS.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- S. Budweg, K. Junge and M. Beller, Catal. Sci. Technol., 2020, 10, 3825–3842 RSC.
- R. Ferraccioli, Curr. Org. Chem., 2018, 22, 1875–1892 CrossRef CAS.
- V. Vermaak, H. C. M. Vosloo and A. J. Swarts, Coord. Chem. Rev., 2024, 507, 215716 CrossRef CAS.
- P. Chandra, T. Ghosh, N. Choudhary, A. Mohammad and S. M. Mobin, Coord. Chem. Rev., 2020, 411, 213241 CrossRef CAS.
- I. Borthakur, A. Sau and S. Kundu, Coord. Chem. Rev., 2022, 451, 214257 CrossRef CAS.
- M. Subaramanian, G. Sivakumar and E. Balaraman, Org. Biomol. Chem., 2021, 19, 4213–4227 RSC.
- T. K. Roy, R. Babu, G. Sivakumar, V. Gupta and E. Balaraman, Catal. Sci. Technol., 2024, 14, 2064–2089 RSC.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS.
- N. Joly, S. Gaillard, A. Poater and J. L. Renaud, Org. Chem. Front., 2024, 7278–7317 RSC.
- F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef CAS PubMed.
- A. S. Das, A. R. Nair, A. Sreekumar and A. Sivan, ChemistrySelect, 2022, 7, e202201097 CrossRef CAS.
- X. Lei, M. Thomaidi, G. K. Angeli, A. Dömling and C. G. Neochoritis, Synlett, 2022, 33, 155–160 CrossRef CAS.
- Shivani, A. Mishra, P. Kaur and K. Singh, ACS Omega, 2022, 7, 39045–39060 CrossRef CAS PubMed.
- Y. V. Svyaschenko, A. Orthaber and S. Ott, Chem. – Eur. J., 2016, 22, 4247–4255 CrossRef CAS PubMed.
- S. R. Choi, M. A. Larson, S. H. Hinrichs and P. Narayanasamy, Bioorg. Med. Chem. Lett., 2016, 26, 1997–1999 CrossRef CAS PubMed.
- C. T. Yen, C. C. Wu, J. C. Lee, S. L. Chen, S. L. Morris-Natschke, P. W. Hsieh and Y. C. Wu, Eur. J. Med. Chem., 2010, 45, 2494–2502 CrossRef CAS PubMed.
- C. C. Lee, D. M. Chang, K. F. Huang, C. L. Chen, T. C. Chen, Y. Lo, J. H. Guh and H. S. Huang, Bioorg. Med. Chem., 2013, 21, 7125–7133 CrossRef CAS PubMed.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- D. Zhou, W. Tuo, H. Hu, J. Xu, H. Chen, Z. Rao, Y. Xiao, X. Hu and P. Liu, Eur. J. Med. Chem., 2013, 64, 432–441 CrossRef CAS.
- R. M. Burch, M. Weitzberg, N. Blok, R. Muhlhauser, D. Martin, S. G. Farmer, J. M. Bator, J. R. Connor, C. Ko, W. Kuhn, B. A. McMillan, M. Raynor, B. G. Shearer, C. Tiffany and D. E. Wilkins, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 355–359 CrossRef CAS PubMed.
- M. D. Hopkins, G. L. Ozmer, R. C. Witt, Z. C. Brandeburg, D. A. Rogers, C. E. Keating, P. L. Petcoff, R. J. Sheaff and A. A. Lamar, Org. Biomol. Chem., 2021, 19, 1133–1144 RSC.
- E. M. Hussein, M. S. Malik, R. I. Alsantali, B. H. Asghar, M. Morad, M. A. Ansari, Q. M. S. Jamal, A. A. Alsimaree, A. N. Abdalla, A. A. S. Al-Qarni, R. S. Jassas, H. M. Altass and S. A. Ahmed, J. Mol. Struct., 2021, 1246, 131232 CrossRef CAS.
- J. Shaya, P. R. Corridon, B. Al-Omari, A. Aoudi, A. Shunnar, M. I. H. Mohideen, A. Qurashi, B. Y. Michel and A. Burger, J. Photochem. Photobiol., C, 2022, 52, 100529 CrossRef CAS.
- A. L. Capodilupo, V. Vergaro, E. Fabiano, M. De Giorgi, F. Baldassarre, A. Cardone, A. Maggiore, V. Maiorano, D. Sanvitto, G. Gigli and G. Ciccarella, J. Mater. Chem. B, 2015, 3, 3315–3323 RSC.
- S. Yao, H. Y. Ahn, X. Wang, J. Fu, E. W. Van Stryland, D. J. Hagan and K. D. Belfield, J. Org. Chem., 2010, 75, 3965–3974 CrossRef CAS PubMed.
- R. Ye, Q. Cui, C. Yao, R. Liu and L. Li, Phys. Chem. Chem. Phys., 2017, 19, 31306–31315 RSC.
- G. Kumar, R. Agarwal and S. Swaminathan, Chem. Commun., 2012, 48, 2412–2414 RSC.
- S. Lee, C. Esteva-Font, P. W. Phuan, M. O. Anderson and A. S. Verkman, MedChemComm, 2015, 6, 1278–1284 RSC.
- Q. Xia, Z. Wang, W. Wan, H. Feng, R. Sun, B. Jing, Y. Ge and Y. Liu, Chem. Commun., 2023, 59, 10008–10011 RSC.
- P. J. Perry, M. A. Read, R. T. Davies, S. M. Gowan, A. P. Reszka, A. A. Wood, L. R. Kelland and S. Neidle, J. Med. Chem., 1999, 42, 2679–2684 CrossRef CAS PubMed.
- A. Pasieka, D. Panek, P. Zaręba, E. Sługocka, N. Gucwa, A. Espargaró, G. Latacz, N. Khan, A. Bucki, R. Sabaté, A. Więckowska and B. Malawska, Bioorg. Med. Chem., 2023, 88–89, 117333 CrossRef CAS.
- I. Reche, I. Gallardo and G. Guirado, ChemElectroChem, 2014, 1, 2104–2109 CrossRef CAS.
- C. R. Hao, X. Zong, Y. Cheng, M. Zhao, M. Luo, Y. Zhang and S. Xue, Sustainable Energy Fuels, 2021, 5, 5548–5556 RSC.
- A. Goel, S. Chaurasia, M. Dixit, V. Kumar, S. Prakash, B. Jena, J. K. Verma, M. Jain, R. S. Anand and S. S. Manoharan, Org. Lett., 2009, 11, 1289–1292 CrossRef CAS PubMed.
- S. Liu, K. Zhang, J. Lu, J. Zhang, H. L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2013, 135, 15326–15329 CrossRef CAS PubMed.
- P. Mäkinen, F. Fasulo, M. Liu, G. K. Grandhi, D. Conelli, B. Al-Anesi, H. Ali-Löytty, K. Lahtonen, S. Toikkonen, G. P. Suranna, A. B. Muñoz-García, M. Pavone, R. Grisorio and P. Vivo, Chem. Mater., 2023, 35, 2975–2987 CrossRef.
- K. Shah, E. Hassan, F. Ahmed, I. Anis, M. Rabnawaz and M. R. Shah, Ecotoxicol. Environ. Saf., 2017, 141, 25–29 CrossRef CAS PubMed.
- R. Dahiwadkar, H. Kumar and S. Kanvah, J. Photochem. Photobiol., A, 2022, 427, 1–8 CrossRef.
- R. Batool, N. Riaz, H. M. Junaid, M. T. Waseem, Z. A. Khan, S. Nawazish, U. Farooq, C. Yu and S. A. Shahzad, ACS Omega, 2022, 7, 1057–1070 CrossRef CAS PubMed.
- L. Li, Q. Chen, Z. Niu, X. Zhou, T. Yang and W. Huang, J. Mater. Chem. C, 2016, 4, 1900–1905 RSC.
- R. Kumar, A. Thakur, Sachin, D. Chandra, A. K. Dhiman, P. K. Verma and U. Sharma, Coord. Chem. Rev., 2024, 499, 215453 CrossRef CAS.
- S. Mukherjee and M. Pal, Drug Discovery Today, 2013, 18, 389–398 CrossRef CAS.
- A. Uddin, M. Chawla, I. Irfan, S. Mahajan, S. Singh and M. Abid, RSC Med. Chem., 2021, 12, 24–42 RSC.
- A. Saxena, S. Majee, D. Ray and B. Saha, Bioorg. Med. Chem., 2024, 103, 117681 CrossRef CAS PubMed.
- Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416–7417 CrossRef CAS.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946–953 CrossRef CAS PubMed.
- C. Xia and R. C. Advincula, Macromolecules, 2001, 34, 6922–6928 CrossRef CAS.
- M. Ranger and M. Leclerc, Chem. Commun., 1997, 3, 1597–1598 RSC.
- S. Bansal and B. Punji, ChemCatChem, 2024, 16, e202301558 CrossRef CAS.
- S. Bansal, A. Biswas, A. Kundu, M. Adhikari, S. Singh and D. Adhikari, ACS Catal., 2024, 14, 8087–8095 CrossRef CAS.
- M. Jafarzadeh, S. H. Sobhani, K. Gajewski and E. Kianmehr, Org. Biomol. Chem., 2022, 20, 7713–7745 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- V. Yadav, E. Balaraman and S. B. Mhaske, Adv. Synth. Catal., 2021, 363, 4430–4439 CrossRef CAS.
- A. Mishra, A. D. Dwivedi, S. Shee and S. Kundu, Chem. Commun., 2019, 56, 249–252 RSC.
- A. Mandal, M. Pradhan, C. Mitra, S. Nandi, B. Sadhu and S. Kundu, ACS Catal., 2024, 706–718 Search PubMed.
- P. Chakraborty, N. Garg, E. Manoury, R. Poli and B. Sundararaju, ACS Catal., 2020, 10, 8023–8031 CrossRef CAS.
- S. N. R. Donthireddy and A. Rit, Chem. – Eur. J., 2024, 30, 1–9 CrossRef PubMed.
- N. Wang, Y. Li, Z. Chen, C. Zhao and Z. Ke, ACS Catal., 2024, 18032–18044 CrossRef CAS.
- A. Mondal, R. Kumar, A. K. Suresh, M. K. Sahoo and E. Balaraman, Catal. Sci. Technol., 2023, 13, 5745–5756 RSC.
- M. Siddique, B. Boity and A. Rit, Catal. Sci. Technol., 2023, 13, 7172–7180 RSC.
- A. K. Bains, A. Biswas, A. Kundu and D. Adhikari, Adv. Synth. Catal., 2022, 364, 2815–2821 CrossRef CAS.
- A. K. Bains, A. Biswas and D. Adhikari, Adv. Synth. Catal., 2022, 364, 47–52 CrossRef CAS.
- A. K. Bains, A. Biswas and D. Adhikari, Chem. Commun., 2020, 56, 15442–15445 RSC.
- A. Mondal, H. J. Phukan, D. Pal, S. Kumar, M. Roy and D. Srimani, Chem. – Eur. J., 2024, 30, e202303315 CrossRef CAS PubMed.
- A. Samanta, A. Chaubey, D. Pal, K. Majhi and D. Srimani, Chem. Commun., 2024, 10398–10401 RSC.
- M. A. Shaikh, S. G. Agalave, A. S. Ubale and B. Gnanaprakasam, J. Org. Chem., 2020, 85, 2277–2290 CrossRef CAS PubMed.
- K. L. Dai, Q. L. Chen, W. P. Xie, K. Lu, Z. B. Yan, M. Peng, C. K. Li, Y. Q. Tu and T. M. Ding, Angew. Chem., Int. Ed., 2022, 61, e202206446 CrossRef CAS PubMed.
- S. Pal, A. K. Guin, S. Chakraborty and N. D. Paul, ChemCatChem, 2024, 16, e202400026 CrossRef CAS.
- A. Mondal, R. Sharma, D. Pal and D. Srimani, Chem. Commun., 2021, 57, 10363–10366 RSC.
- A. Biswas, A. K. Bains and D. Adhikari, Catal. Sci. Technol., 2022, 12, 4211–4216 RSC.
- R. Sharma, A. Mondal, A. Samanta and D. Srimani, Catal. Sci. Technol., 2023, 13, 611–617 RSC.
- S. Sahoo, S. Manna and A. Rit, Chem. Sci., 2024, 15, 5238–5247 RSC.
- R. Saha, S. K. Maharana, N. C. Jana and B. Bagh, Chem. Commun., 2024, 10144–10147 RSC.
- N. Kamal and S. Samanta, J. Org. Chem., 2024, 89, 1910–1926 CrossRef PubMed.
- R. Kumar, S. R. Padhy and E. Balaraman, J. Org. Chem., 2024, 89, 15103–15116 CrossRef CAS PubMed.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS PubMed.
- S. Mondal, S. Pal, S. Khanra, S. Chakraborty and N. D. Paul, Eur. J. Inorg. Chem., 2023, 26, 1–13 Search PubMed.
- B. Goswami, M. Khatua, R. Chatterjee, N. Kamal and S. Samanta, Organometallics, 2023, 42, 1854–1868 CrossRef CAS.
- M. Khatua, B. Goswami, A. Devi, N. Kamal, S. Hans and S. Samanta, Inorg. Chem., 2024, 63, 9786–9800 CrossRef CAS PubMed.
- S. Kumari, A. Maji, R. Chauhan, V. K. Chaudhary, T. Kush, M. Joshi, A. R. Choudhury and K. Ghosh, Eur. J. Org. Chem., 2024, 27, e202400051 CrossRef CAS.
- H. Tian, W. Xue, J. Wu, Z. Yang, H. Lu and C. Tang, Org. Chem. Front., 2022, 9, 4554–4560 RSC.
- A. Singh, A. Maji, M. Joshi, A. R. Choudhury and K. Ghosh, Dalton Trans., 2021, 50, 8567–8587 RSC.
- S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2017, 54, 90–93 RSC.
- S. Shee, K. Ganguli, K. Jana and S. Kundu, Chem. Commun., 2018, 54, 6883–6886 RSC.
- S. Q. Zhang, B. Guo, Z. Xu, H. X. Li, H. Y. Li and J. P. Lang, Tetrahedron, 2019, 75, 1–13 CrossRef.
- R. Saini, P. Kukreti, R. Chauhan, A. Panwar and K. Ghosh, Dalton Trans., 2025, 5838–5848 RSC.
- A. Maji, A. Singh, N. Singh and K. Ghosh, ChemCatChem, 2020, 12, 3108–3125 CrossRef CAS.
- C. K. Sams, F. Somoza, I. Bernal and H. Toftlund, Inorg. Chim. Acta, 2001, 318, 45–52 CrossRef CAS.
- R. K. Afshar, R. Bhalla, J. M. Rowland, M. M. Olmstead and P. K. Mascharak, Inorg. Chim. Acta, 2006, 359, 4105–4113 CrossRef CAS.
- S. Biswas, S. Bose, J. Debgupta, P. Das and A. N. Biswas, Dalton Trans., 2020, 49, 7155–7165 RSC.
- A. Maji, S. Gupta, M. Maji and S. Kundu, J. Org. Chem., 2022, 87, 8351–8367 CrossRef CAS PubMed.
- J. Rana, P. Nagarasu, M. Subaramanian, A. Mondal, V. Madhu and E. Balaraman, Organometallics, 2021, 40, 627–634 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2429425 and 2356288. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cy00422e |
| This journal is © The Royal Society of Chemistry 2025 |