
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccentuating the ambient curing behavior of geopolymers: metamodel-guided optimization for fast-curing geopolymers with high flexural strength†
Kyungwon
Kim‡
 ab,
Hyejeong
Song‡
cd,
Sanghun
Lee
d,
Hyeongkyu
Cho
d,
Hyung Mi
Lim
a and
Hyunseok
Ko
*d
ab,
Hyejeong
Song‡
cd,
Sanghun
Lee
d,
Hyeongkyu
Cho
d,
Hyung Mi
Lim
a and
Hyunseok
Ko
*d
aDivision of Aerospace Convergence Materials Center, Korea Institute of Ceramic Engineering and Technology, Jinju, Gyeongsang-namdo 52851, Republic of Korea
bSchool of Convergence Science, Pusan National University, Busan, 46241, Republic of Korea
cDepartment of Materials Science and Engineering, Korea University, Seoul, 02841, Republic of Korea
dDivision of Low Carbon Energy and Materials Digitalization, Korea Institute of Ceramic Engineering and Technology, Jinj, Gyeongsang-namdo 5285, Republic of Korea. E-mail: hko@kicet.re.kr
First published on 27th January 2025
Abstract
A geopolymer, consisting of –Si–O–Al– covalent bonds in a polymeric network, has a simple manufacturing process with low CO2 emissions and excellent high-temperature performance, making it a promising modern refractory material. In particular, owing to its low-temperature and fast-curing conditions, geopolymers can be used for practical on-site applications. However, the properties of geopolymers are significantly dependent on the composition and content of various additives, and this complexity limits our understanding of the composition to a narrow scope. In this study, we investigated the optimal composition designed for fast and low-temperature curing geopolymers with additives, including Ca(OH)2, fumed silica, and chopped carbon fiber. A multivariate compositional optimization was systematically conducted using design of experiments and metamodeling. By utilizing the metamodel, we successfully developed an optimized geopolymer composition with only 45 sets of experiments. The flexural strength obtained was 27.83 MPa, the highest recorded value for a bulk fast-curing geopolymer to date. Furthermore, the curing speed was modulated to be swift at ambient conditions, achieving 98% of the full strength in 6 days at 20 °C (whereas it typically takes 1 to 4 weeks at 40 °C). We also investigated how superior strength could be achieved while curing at low temperatures for a short duration. It turned out that fumed silica slowed down the growth of the Ca compound, balancing two different effects stemming from Ca ions: strength degradation and rapid curing. The developed geopolymer is expected to be widely used in applications that require rapid curing at room temperature, such as external cement replacements for fire spread prevention structures, acid-exposed environments, or repair and finishing materials.
1 Introduction
With the increasing concerns over the greenhouse effect and global warming, one of the greatest challenges is diminishing CO2 production in human activities. Among various sources, cement, the second most used substance by humans after water, accounts for approximately one tenth of the total CO2 production.1 To address such concerns, a geopolymer (GP) has drawn great attention as an alternative structural material. A geopolymer is produced through a simple manufacturing process with low CO2 emissions (70–80% reduction2,3), and it offers good mechanical properties, heat resistance, and excellent acid resistance compared to ordinary Portland cements.4,5 The geopolymer-zeolite composites, derived from municipal solid waste incineration fly ash, exhibited enhanced performance in CO2 adsorption. This study explores the potential for simultaneous waste recycling and greenhouse gas reduction.6Fig. 1a schematizes the geopolymerization process from raw materials to cured GP. Geopolymers are produced by forming polymeric covalent bonds between alkali polysilicates and aluminosilicate oxides (e.g., slag, fly ash, and metakaolin7), constituting Si–O–Al bonds. Oligomers, small molecules such as sialate and sialate-siloxo, are initially formed through the alkaline depolymerization process of kaolinite, and a polymeric 3D network is formed with bridging alkali metal ions during the curing process.8 The GP reaction and its synthesis occur at low temperatures below 100 °C, which is advantageous for oxide materials, empowering its accessibility. More intriguingly, the functionality of GPs can be modulated by additives and reinforcing materials, making them versatile for various applications. Of particular note, this inorganic material is beneficial when it cures fast at room temperature, as it can be utilized for primary crack sealants,9 fire finishing materials, and other applications as construction materials, such as for soundproofing10 and as a moisture-absorbent material through pores and foam formation.11–15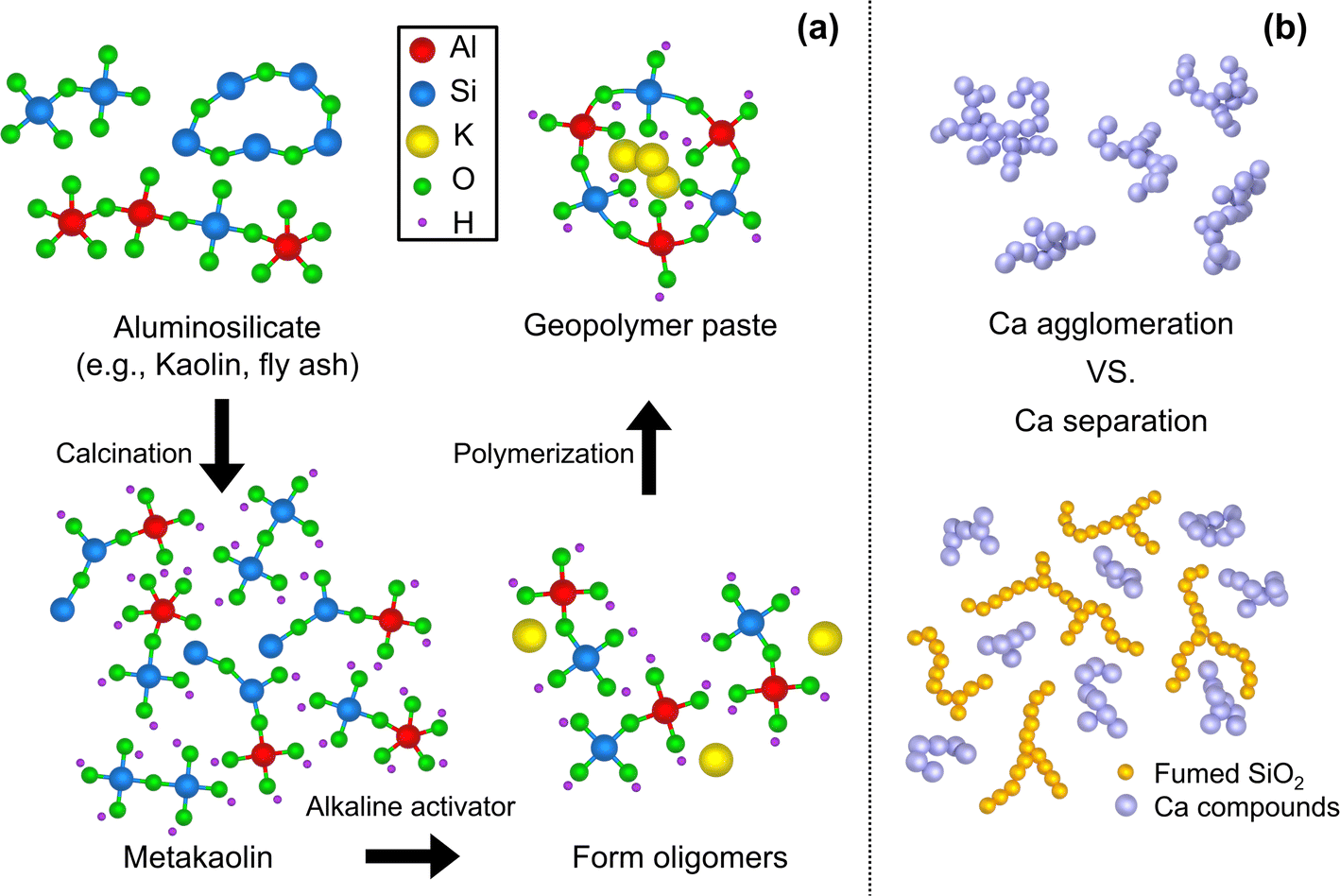 | ||
| Fig. 1 (a) Schematic of the geopolymerization reaction. The purpose of calcination heat treatment is to modify the chemical and physical properties of aluminosilicate raw materials. The aluminosilicate source is then dissolved in alkaline activators, forming oligomers. These oligomers undergo condensation, known as polymerization, leading to the formation of covalently bonded, non-crystalline (amorphous) geopolymer networks. (b) Illustration of the role of f-SiO2 in GPs. f-SiO2, with its high surface area, is expected to prevent the aggregation and growth of Ca compounds in the geopolymer. | ||
If the ability of GPs to cure rapidly under ambient conditions is further developed, they can be utilized in various applications. One approach to achieve low temperature and rapid synthesis is to employ calcium cations—a simple but widely used method in the cement industry. Studies have been performed on the mechanical strength and degradation mechanism of metakaolin-based geopolymers mixed with ordinary Portland cement, and cured at high temperature and high relative humidity.16 The Ca cations act as a charge balancing ions in the GP binder, along with metal cations in the activator,17 enabling rapid polymerization of the network (e.g., tobermorite (Ca5Si6O16(OH)2·4H2O), gehlenite (Ca2Al(AlSiO7)), and jennite (Ca9Si6O18(OH)6·8H2O)).18,19 Such applications are often accomplished by using raw materials that already contain CaO,20–22 or by adding calcium compounds (e.g., CaCO3 and Ca(OH)2). For instance, it has been found that the setting time was shortened from 15 hours to less than 1 hour with the addition of 0.4 mol Ca(OH)2,23 and the curing time was reduced from 20 hours to 4 hours when 4 wt% of CaO was incorporated.24 There have also been literature studies on the development and application scope of machine learning models to predict trends in the compressive strength of calcium-based geopolymers.25
While the addition of Ca compounds allows for more rapid curing at low temperature, it requires sacrificial decreases in both mechanical strength and workability.26 To compensate for these drawbacks, additives are often used in the geopolymer mixture.27 One example is nano-silica (SiO2), which increases the mechanical strength by condensing the material (compact structure by filling voids) and boosting the polymerization processes (due to the high surface area to volume ratio).28,29 Silica fume enhances the mechanical strength of geopolymers by contributing to the densification of the matrix, thereby improving its binding properties and overall durability.30 Fibers are also often used to complement the strength. The dispersed fibers can lead to a crosslinking effect,31 which boosts the strength of GP with their intrinsic high strength.32 It has been shown that the addition of chopped carbon fibers (denoted as Cf (all the abbreviations are listed in ESI, Table S6†)) in GP increased its flexural strength by four-fold.33 More intriguingly, synergetic effects have been reported when nano-silica and fibers are used at the same time; the flexural modulus increased by over 20% when nano-silica was added with 1–2 vol% of PVA fiber.34 A compact microstructure and improved workability have also been observed when both fiber and nanoparticles are simultaneously utilized, as the condensation is boosted at the fiber/matrix interface with the help of nano-silica.35,36 This infers that a synergistic effect is expected on the property and workability of GP, which is suitable for high-speed curing conditions.
As illustrated, additives are prevalently employed in the development of GPs. It should be pointed out that the characteristics of cured GPs are greatly affected by the mixture of starting materials: the molar concentration of the raw material, ratio of the alkali activators (i.e., alkali hydroxide to alkali silicate), and ratio of aluminosilicate to alkali activator and other additives. To date, research studies have been conducted on a wide range of chemical compositions.37,38 However, an understanding of the compositional effect is not only limited to a narrow range, but is also controversial. For instance, studies on the effect of sodium-based activators revealed opposing opinions. One study stressed that a decrease of the silicate solution in the alkali activator augmented the compressive strength.39 Conversely, another study reported on the observation of over 20% abated compressive strengths with low amounts of silicate solution.40 With the inclusion of multiple additives, the control of variables and consequential properties becomes even more sophisticated.41
Despite the complex interplay between parameters and material properties, the practical applications of geopolymers remain limited. However, recent studies have shown growing interest in utilizing statistical optimization and modeling techniques to better predict and optimize the properties of geopolymers. To enhance the material properties, Taguchi modeling was utilized to reach the target strength,42 and an optimization was performed on the fiber-reinforced, fly ash-based geopolymer foams, resulting in compressive strengths ranging from 0.02 to 0.45 MPa.43 Additionally, Design of Experiments (DOE) was applied to formulate mixture compositions, achieving compressive strengths of 18.9 MPa at 7 days and 22.3 MPa at 28 days in geopolymer concrete.44 Through mixture design and Response Surface Methodology (RSM), a model with a high R-square coefficient was validated, optimizing the compressive strength up to 35.31 MPa. By combining the effects of SiO2/Al2O3, H2O/Na2O, and the water-to-solid ratio, the compressive strength was maximized while preventing efflorescence, with metakaolin as the sole precursor achieving 53 MPa at 7 days.45 Techniques, such as artificial neural networks,15 regression analysis, and response surface methodology,46 have been actively employed to enhance the performance prediction and mix design optimization of geopolymer materials. There are cases where metamodels have been applied not only in geopolymers, but also across other fields of engineering. Studies were also performed using metamodels to improve the noise and vibrational performance of the Permanent magnet synchronous motors.47Via injection molding process variables and optimally determining the design variables efficiently, we adopted metamodel-based design optimization.48
Metamodeling techniques have been used as powerful tools in engineering to explore high-dimensional design spaces, making them particularly advantageous for design optimization. In particular, the Radial Basis Function (RBF) model is a promising approach for predicting properties influenced by multivariate parameters because it successfully captures the nonlinear relationship between variables and response values. Recent studies have utilized RBF models to predict the friction coefficient on textured and porous surfaces,49 estimate the mechanical and physical properties of wood composite materials,50 and forecast the wear rate of aluminum alloys.51 However, due to the controversial coactions of parameters and material traits, the practical applications of geopolymers remain limited.
In this study, by carrying out a statistical optimization, we have developed a balanced admixture GP that can rapidly cure at room temperature (RT) and is mechanically robust. A metamodel, or a surrogate model, based on systematically collected experimental data for the predefined design of experiments, is utilized to efficiently explore the multi-dimension parametric space. As a result, the developed GP cures at RT and established 80% of the saturated strength in 3 days, with an outstanding mechanical strength of 27.8 MPa. Moreover, the effect of additives, particularly the synergetic effect between fumed silica (a nanostructured silica that is a universal thickening agent with high surface area, is hereafter denoted as f-SiO2) and Ca precursor, on the mechanical performance was investigated.
2 Method
2.1 Geopolymer raw materials and pretreatments
Geopolymers condense and harden during the chemical reaction induced by the activator. Fig. 1b schematically shows the role of f-SiO2, along with Ca cations, used as curing accelerators during this process. When the aggregated Ca compounds become large, it becomes susceptible to micro cracks. The f-SiO2 with a high surface area has a significant effect in constituting an activated C–A–S–H gel, and becomes more consolidated as the calcium/silicate ratio decreases.52 Accordingly, it is expected that the relatively high content of Si will slow the Ca particle aggregation. In this study, f-SiO2 was expected to adjust the Ca compound growth rate to balance the boost of the curing speed and degradation of the mechanical strength.Metakaolin (MK) is used as the base aluminosilicate for GPs, which can be achieved by calcining the commercially available kaolin chemical (H2Al2Si2O8·H2O, DJ 5041-1400, Daejung Chemicals & Metals Co., Ltd, S. Korea). The kaolinite was calcined at 800 °C for 4 hours.53 X-ray fluorescence (ZSX Primus 4, Rigaku Co., Ltd, Japan) analysis was performed (ESI, Fig. S1(a)†), and showed that MK is mainly composed of Al2O3 (44 wt%) and SiO2 (53 wt%), and <0.5 wt% of other elements such as CaO, Fe2O3, MgO, and K2O. The average particle size of MK was measured as 10.49 μm, as illustrated in Fig. 2a, where the size distribution is measured with a particle size analyzer (LA-960, Horiba Co., Ltd, Japan). X-ray diffraction (Smart Lab, Rigaku, Japan) analysis was carried out to confirm the crystallographic changes, as shown in Fig. 2b. The main characteristics of the kaolin peaks indicate the presence of kaolinite (Al2Si2O5(OH)4; ICDD: The International Centre for Diffraction Data No. 14-0164 or 29-1488) and quartzite (SiO2; ICDD No. 46-1045 or 33-1161). However, after calcination, these peaks become negligible in MK. It can be seen that the amorphous phase of aluminosilicate, which is the starting structure for GP, is constituted. After geopolymerization (Fig. 2b), it was confirmed that the amorphous phase was maintained in the cured GP.
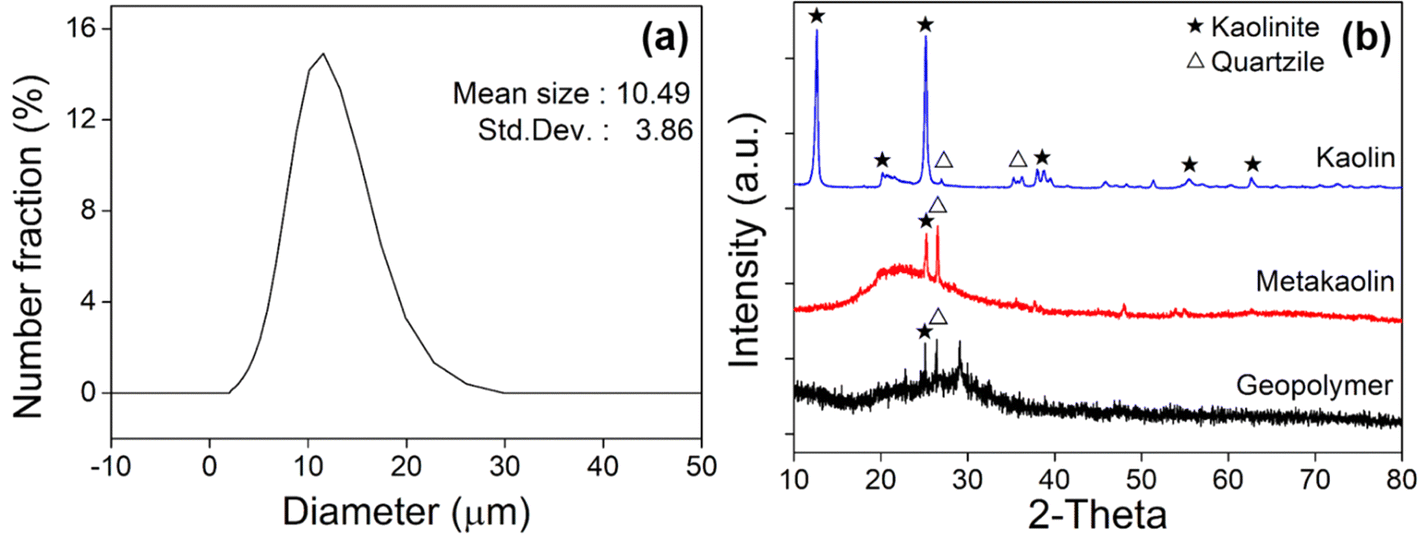 | ||
| Fig. 2 (a) Particle size distribution of metakaolin powder. (b) XRD results of kaolin, metakaolin, and cured geopolymer. | ||
As for the alkali activator, potassium hydroxide (KOH pellets, DJ 6597-4405, Daejung Chemicals & Metals Co., Ltd, S. Korea) and potassium silicate (K2SiO3, DJ 6617-4405, Daejung Chemicals & Metals Co., Ltd, S. Korea) were used. The potassium-based activators are chosen to achieve a denser structure54 and increased strength,55,56 as K+ offers high reactivity with the aluminosilicate materials.57 It was used by first dissolving KOH pellets in distilled water, and then adding potassium silicate solution. Calcium hydroxide (Ca(OH)2, DJ 2511-4400, Daejung Chemicals & Metals Co., Ltd, S. Korea) was used to enable fast curing. For structural additives, chopped Cf (C118-3K, HD fiber Co., Ltd, S. Korea) with a length of 4 ± 1 mm and f-SiO2 (Konasil 112945-52-5, OCI Co., Ltd, S. Korea) were utilized.
2.2 Experimental procedures
The powders (MK and Ca(OH)2) and dispersed solutions were prepared separately. The dispersed phase was prepared with the following sequence. Firstly, f-SiO2 was dispersed in a mixture of KOH and K2SiO3, and mixed for 5 min with sonication. The basicity of the solution was carefully maintained at pH > 10 for a homogenous dispersion of f-SiO2 particles.31 The chopped Cf was then added, followed by ultrasonication (SONICS Vibracell-VCX750, Sonics & Materials, Inc., USA) for 15 min. It should be noted that if Cf is not well-dispersed (i.e., entangled), there will be poor bonding between the GP matrix and fibers, inevitably lowering the mechanical strength. After securing a homogenized solution with f-SiO2 and Cf, MK and Ca(OH)2 were added. The mixture underwent a final mixing process in a planetary centrifugal mixer (DAC 150.1 FVZ-K, UniNanoTech Co., Ltd, USA) at 2000 rpm for 1 min. At this stage, the hardening is stimulated instantaneously by the addition of Ca (the surface is hardened within 30 minutes); thus, it is important to work quickly with the material. After being casted in a silicone mold, specimens were dried at RT for 1 day, and further dried as demolded, for up to 7 days at RT for evaluations.2.3 Physical property evaluation and observation
Mechanical strengths, namely flexural strength (the maximum stress a material can withstand in 3-point bending, σf) and compressive strength (the maximum uniaxial compressive stress a material can withstand, σc), were measured using a universal testing machine (5900 series, Instron Co., Ltd, USA). For σf, silicone molds (ASTM D790) with a dimension of 3.20 × 12.70 × 125 mm3 were used, as depicted in ESI, Fig. S1 (b).† The actual sample size had an error range of ±0.10 mm due to shrinkage. For σc, cylindrical specimens with a diameter of 25 mm and height of 25 mm were used, in accordance with ASTM C39. To investigate the microstructure and distribution of the chemical species, field emission scanning electron microscopy (JSM 7610F, JEOL, Japan) with energy-dispersive X-ray spectroscopy (JSM 7610F, JEOL, Japan) was used. Thermal gravimetric analysis measurement (1/LF/1100, Mettler Toledo, USA) was performed in the temperature range of 25–900 °C at a scanning rate of 0.5 min s−1 to investigate the high-temperature characteristics. To test for heat resistance, the non-combustible material test (Non-Comb 2005, FESTEC Co., Ltd, S. Korea) was performed.For the design of experiments (DOE) and metamodel analysis, we utilized PIAnO (Process Integration, Automation and Optimization, PIDOTECH Inc., S. Korea) and IDAES (Institute for the Design of Advanced Energy Systems, Institute for the Design of Advanced Energy Systems Integrated Platform, U.S.). The DOEs were designed with appropriately varied levels for each iteration in order to maximize σf and simultaneously minimize the curing time. The following variables were used as the input parameters: molar ratios of starting materials SiO2/Al2O3, K2O/Al2O3, K2O/SiO2, and H2O/K2O, and contents of activator (KOH and K2SiO3), and contents of additives (f-SiO2). Throughout the paper, the contents of components (e.g., KOH) are presented as a weight ratio to MK (WKOH = weight of KOH/weight of MK). As an additional example, the content of f-SiO2 is expressed as a weight ratio to MK (Wf-SiO2 = weight of f-SiO2/weight of MK). The weight ratio notation for the content of components used together is provided in ESI Table S6.† For each sampling point, we have taken an arithmetic average of 10 measurements for statistical stability, and the standard deviation was found to be less than 5% for all measurements. The metamodel for optimization design was performed using RBF interpolation, which effectively guides the prediction of material properties by interpolating data points based on their spatial relationships. The input parameters were alkali activators (KOH and K2SiO3) and H2O. Gaussian and multiquadric RBFs were employed as basis functions for modeling σf and curing time, respectively. Through Leave-One-Out Cross Validation, the regularization and shape parameters were selected to be the best ones.
3 Results & discussion
3.1 Design of the optimized composition
While various types of raw materials are used for geopolymer productions, the differences in the starting materials and their contents have a huge influence on the properties. Furthermore, some admixtures are often applied to achieve a fast-curing GP with reliable mechanical performances. Thus, these extensions enlarge the complexity in searching for an optimal design. Fig. 3 summarizes the design parameters available in the literature,17,32,58–76 as well as the measurements of σf and σc (maximum is taken when multiple measurements are presented in a single reference). Detailed information on the chemical composition is summarized in ESI Table S1.† It can be seen that σf and σc are distributed without any remarkable dependency on the chemical composition across wide ranges. Not surprisingly, the mechanical strength represented by the condensation of the microstructure cannot be solely understood by a single variant; it is a complex result of multiple variables. Based on the literature range, we carried out a preliminary DOE experiment (gray area in Fig. 3) to reduce the wide range of multivariants. As a result, we narrowed the parametric search space, as shaded in red in Fig. 3. The molar ratio ranges explored throughout the DOE are SiO2/Al2O3 = 2.8–4, K2O/Al2O3 = 0.87–2, K2O/SiO2 = 0.3–0.7, and H2O/K2O = 6.5–9.1. Up to a Ca content of 2.5 g per batch, the flexural strength increased (Fig. S2(a)†). However, beyond this amount, the formulation was not possible due to abrupt curing. Therefore, for subsequent DOE, this value was consistently maintained.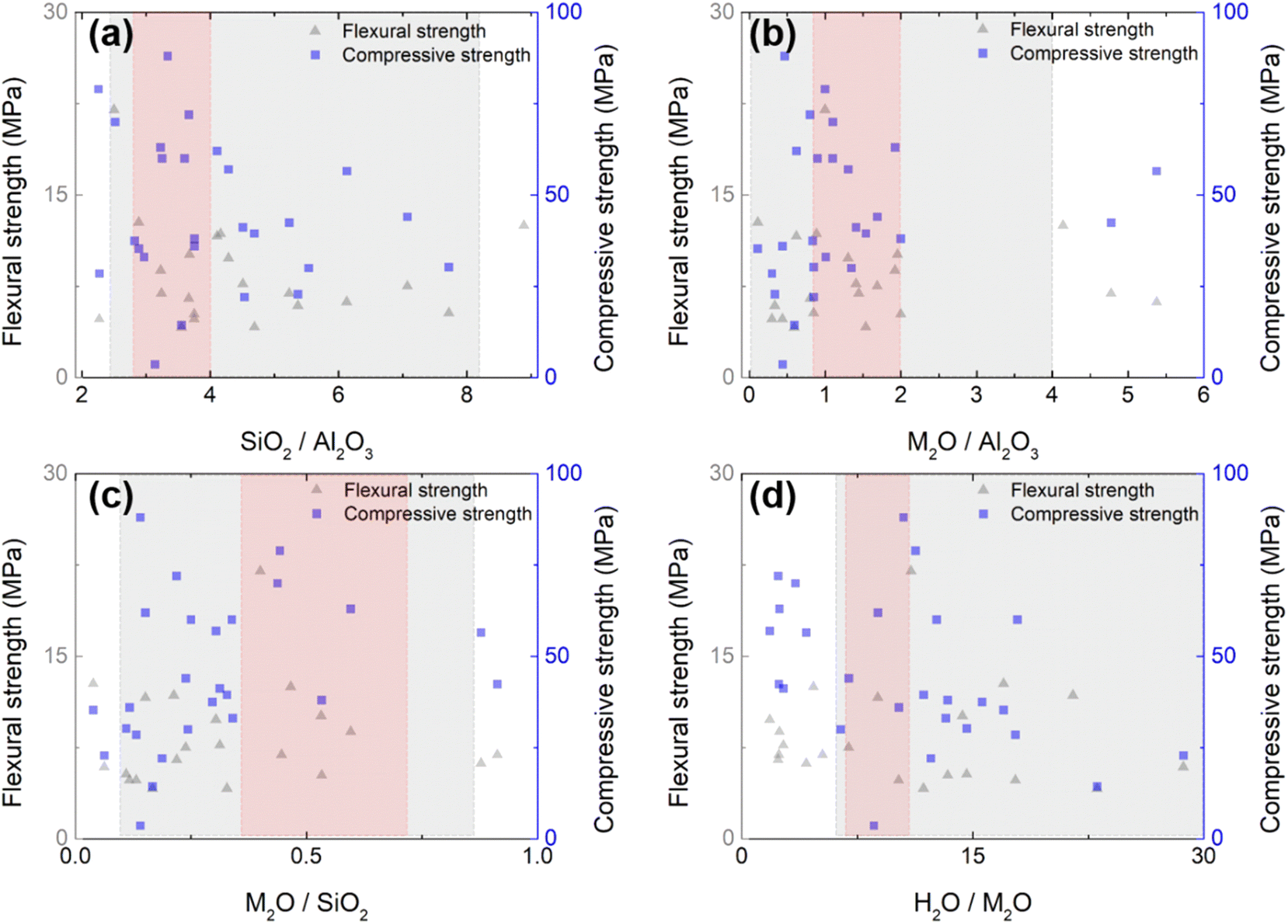 | ||
| Fig. 3 Flexural strength and compressive strength values according to the ratio of synthetic raw materials, including alkali activator and binder: (a) SiO2/Al2O3, (c) Na2O/SiO2, (b) Na2O/Al2O3, and (d) H2O/Na2O. The shaded grey area represents the range of our preliminary tests based on literature, while the red area represents the range explored in the DOEs of this study. | ||
In these ranges of variables, we sampled the first experiment-points for the 1st DOE (D1), and the details of the sampled points are shown in ESI Table S2.† From the sampling point analysis, we discovered that the factors imposing the most significance on the flexural strength development are f-SiO2 and alkali activators. The addition of an optimized proportion of silica fume was shown to enhance the microstructure and improve the compressive strength by 25%.77 The distribution of σf varies depending on the f-SiO2 content, and the maximum strength value of σf was observed at Wf-SiO2. To strategically predict the maximum σf, the data with f-SiO2 fixed at 0.03 were trained in the RBF model to investigate the influence of alkali activators. In Fig. 4a, it is clear that the σf increases as the amount of K2SiO3 increases (the contour becomes brighter). Conversely, the response surface for σf was relatively flat with changes in KOH. This indicates that further exploration to achieve the optimal σf is necessary.
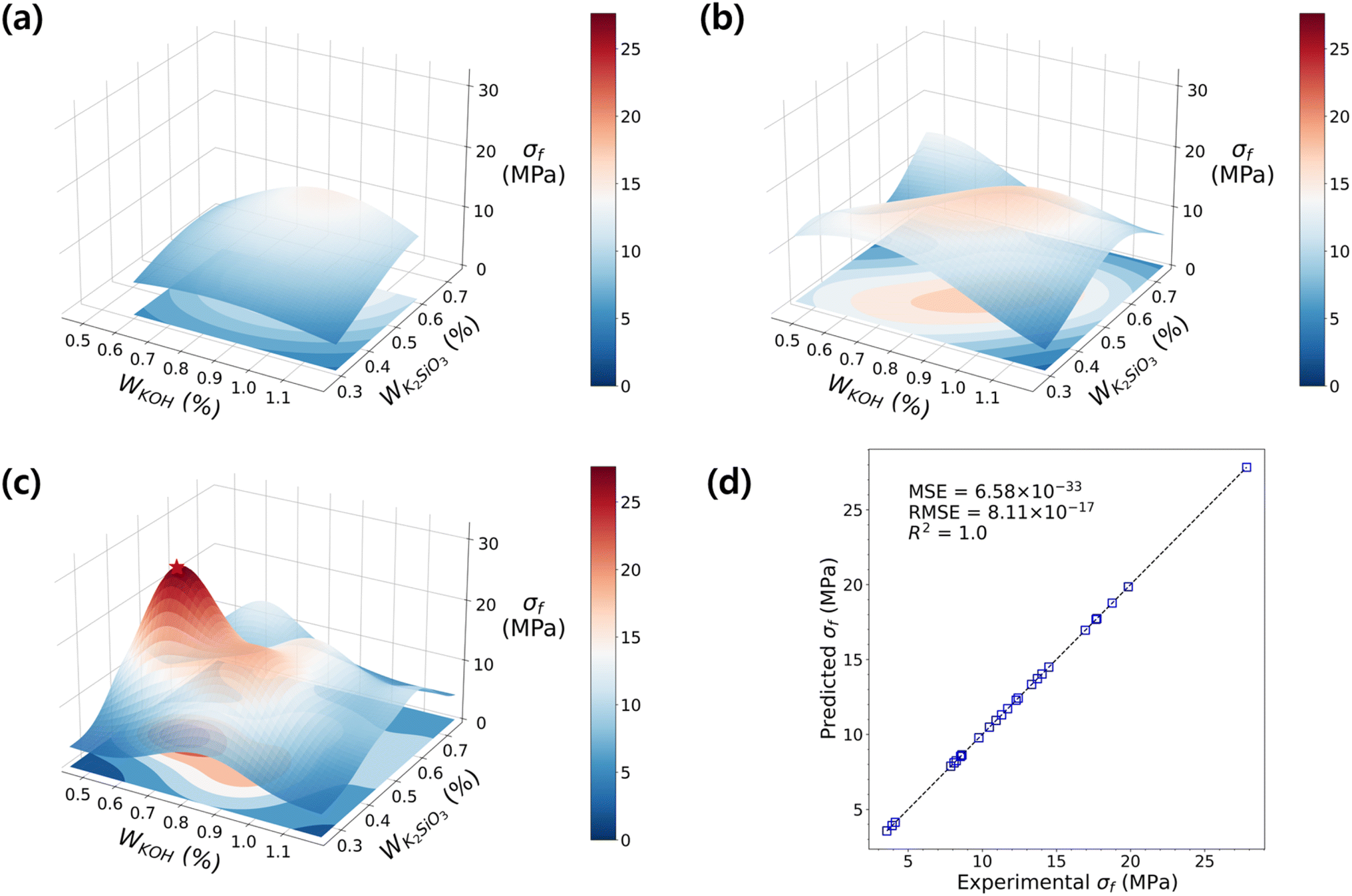 | ||
| Fig. 4 The RBF models for flexural strength of GPs with H2O fixed at 0. The development of the RBF model as DOE sets proceed: with the accumulated dataset of (a) D1 only, (b) D1 and D2, and (c) D1–D3. (d) The parity plot shows the distribution of experimental vs. predicted flexural strength, with the y = x line represented by dashed lines. The RBF model is fitted with R2 = 1.0. | ||
The second DOE, D2, was performed in a wider parametric space with reduced KOH and increased K2SiO3 content to explore the influence of KOH, which was not fully explored in D1, and the effect of increasing K2SiO3 on σf. The information on the sampled points is shown in ESI Table S3.†Fig. 4b shows the surface plot for the accumulated experimental dataset of D1 and D2. The model predicts the existence of an experiment point with greater strength than those predicted in D1. While the gentle response surface suggests that an optimized design regime has not been found yet, it provides a direction towards achieving the optimal σf; it infers that the parametric space of sampling approaches the vertex of the function.
Consequently, the σf increased in D3 compared to those from D1 and D2, suggesting the advance in DOE sequence proceeded successfully (attached as ESI, Table S4†). At the highest strength regime (Fig. 4c), the σf reached up to 27.83 MPa, which matches the metamodel result with a difference of only 0.22 MPa. The optimized mixing formulation for the highest flexural strength GP is found to be Wcf = 0.005, WKOH = 0.60, WK2SiO3 = 0.45, and WCa(OH)2 = 0.03. The shape of the response surface exhibits multiple curvatures, and shows a non-linear increasing trend depending on the ratio of alkali activators. Such trends, as shown in Fig. 4d, adequately explain the relationship with alka and renumli activators. Furthermore, they serve as a more effective guide for finding optimal points faster than linear models (Fig. S3†), which are typically known to be effective in data-driven modeling with limited data.
Similar to the process of finding the maximum flexural strength, we explored the optimal curing time within the sampled DOE using the RBF model. The terminology of “curing time” is often ambiguous, as there are various definitions to quantify the curing speed. Herein, we define two terms for the curing time: firstly, the surface curing time (tsc) is the temporal point at which there is no seepage when using a cotton swab to touch the surface; secondly, the final curing time is the duration to reach its maximum strength after the initial mixing and placement. Unconsolidated specimens from D1 and parts of D2 were excluded from analysis due to data abnormalities. In the response surface of D2 shown in Fig. 5a, it was observed that tsc decreased as K2SiO3 decreased. The influence of KOH is still minimal, but there is a possibility of an optimal tsc near the low values on the surface. Nonetheless, to ascertain this, further exploration in the surrounding region is required. Fig. 5b investigates the range of D3, where K2SiO3 is low and KOH is widely distributed, revealing a decreased tsc compared to D2. The response surface exhibits more dynamic changes than before, aligning with the direction indicated by the response surface in D2 towards the minimum point. This again supports the notion that our multi-step optimizations of co-variants proceeded in a correct direction, and the sampling points in D3 gradually approach the global minimum as the DOE progresses.
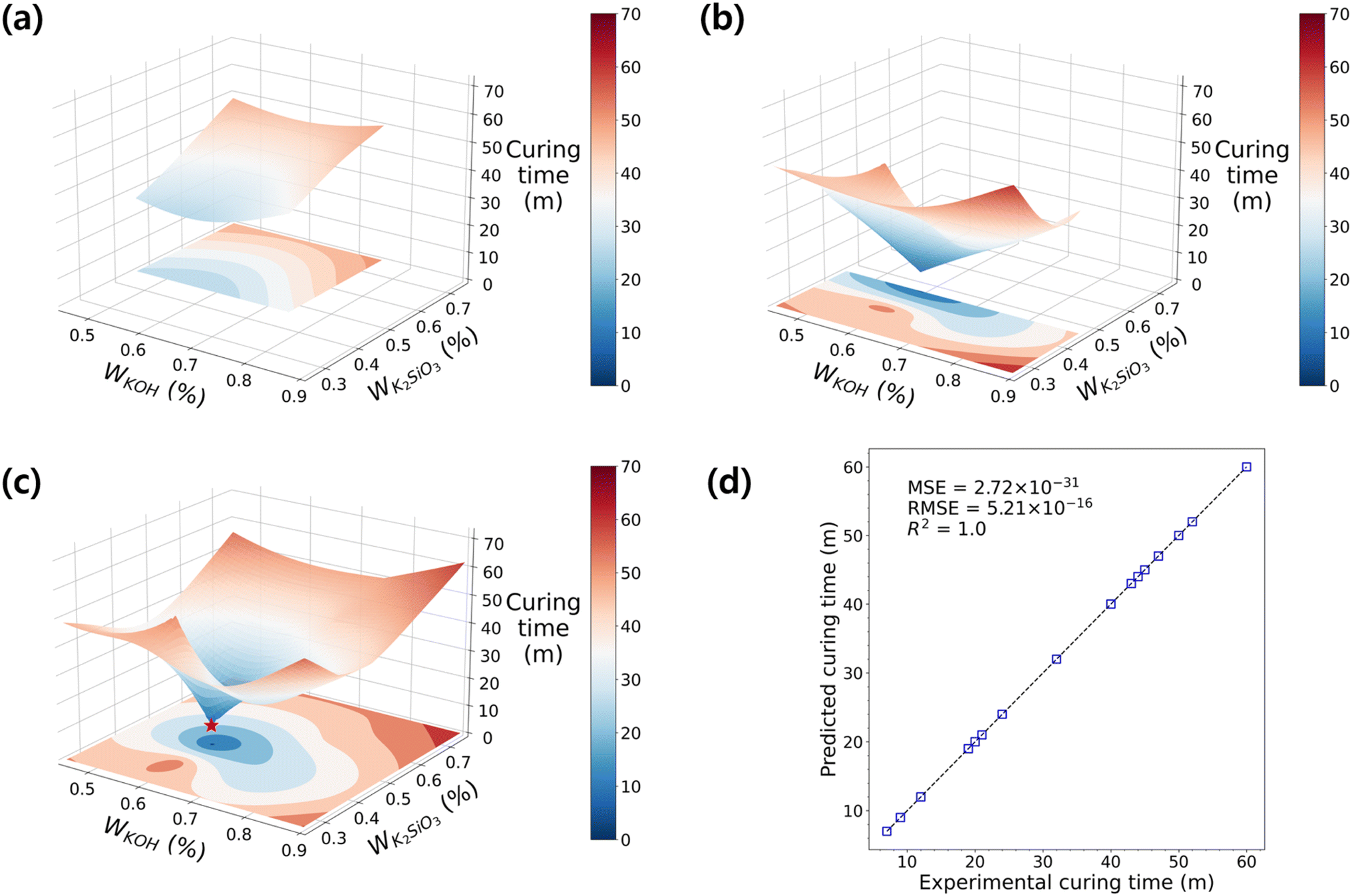 | ||
| Fig. 5 The RBF models for curing time of GPs with H2O fixed at 0. The development of the RBF model as DOE sets proceed: with the accumulated dataset of (a) D2 only, (b) D3 only, and (c) D2 and D3. (d) The parity plot shows the distribution of experimental vs. predicted curing time, with the y = x line represented by dashed lines. The RBF model is fitted with R2 = 1.0. | ||
Accordingly, by analyzing the cumulative data from D2 and D3, the optimal tsc of 7 minutes was achieved (Fig. 5c). Interestingly, the optimized mixing formulation for the RT fast curing of GP coincided with the experiment points exhibiting the highest flexural strength in Fig. 4c. Furthermore, the response surface in Fig. 5c displayed an inverse pattern compared to Fig. 4c. This suggests a strong negative correlation between the solidification rate and strength, indicating the successful attainment of a high-strength geopolymer that cures rapidly at RT. The detailed information regarding the correlations between data is shown in ESI Fig. S4.†
3.2 Synergistic effect of Ca compounds and fumed SiO2
Through the D1 and D2 experiments, we conducted flexural property tests that allowed for quick drying and easy measurements to effectively narrow down a wide range of parameters as a preliminary step to establish ambient temperature and rapid-curing geopolymer conditions. Subsequently, we conducted performance evaluations of both σf and σc in D3 to assess their potential applicability in structural applications. For the optimized GP, both σf and σc are measured to assess its performance for the structural material. In the case of the flexural strength, the highest value was obtained regardless of the raw material or process (as shown in Fig. 3). In the case of the compressive strength, it was ranked in the top tier with 55 MPa in the values of the literature studies investigated. Furthermore, it was confirmed that the strength was increased by 20% or more compared to a similar study using the metakaolin-based potassium alkali activator.78 The results of the specimens cured at ambient temperature for metakaolin-based geopolymers were compared in Table 1. The flexural strength was approximately 85% higher than the minimum value (4.10 MPa) across all drying conditions, and about 20% higher than the maximum flexural strength (22.0 MPa). The compressive strength was 55.12 MPa, which is 13% lower than the maximum value of 63 MPa reported in the literature. Clearly, a strengthening effect is observed with the formulation in this study, and the cause for the drastic augmentation in the strength needs to be discussed.| Author | Curing temperature (°C) | Curing conditions | Curing time (days) | Flexural strength (MPa) | Compressive strength (MPa) |
|---|---|---|---|---|---|
| This work | RT (room temperature) | RT | 80%: 3 days | 27.83 | 55.12 |
| 90%: 6 days | |||||
| Peigang He58 | RT | Sealed at 70 °C for 24 h | 3 | 22.0 | — |
| Kalaiyarrasi54 | RT | RT | 28 | 12.75 | 35.3 |
| Cyriaque Rodrigue Kaze79 | 90 | Oven for 24 h | 28 | 12.5 | — |
| Ali Nazari61 | 70 | Oven for 24 h | 27 | 11.8 | — |
| Pavel Rovnaník62 | RT | RT | 28 | 11.6 | 62 |
| Rashidah Mohamed Hamidi63 | 60 | Oven for 24 h | 1 | 10.11 | — |
| Peng Zhang64 | RT | 20 °C, 95% humidity | 28 | 9.8 | 57 |
| Huang Ji Zhuang65 | RT | 20 °C, 95% humidity demolded after 6 days | 35 | 8.8 | 63 |
| Harun Tanyildizi67 | RT | 28 | 7.5 | 44 | |
| Shilang Xu68 | RT | 28 | 6.9 | 42.4 | |
| A. Natali32 | RT | 7 | 6.9 | — | |
| F. N. Okoye17 | 100 | Oven for 72 h | 28 | 6.5 | 72 |
| M. Sofi69 | RT | Chamber at 30–35 °C for 24 h | 28 | 6.2 | 56.5 |
| Xueying Li71 | RT | Film-sealed curing | 28 | 5.3 | 30.23 |
| Yao Jun Zhang72 | RT | Chamber at 20 °C, 99% humidity | 28 | 5.2 | 38 |
| Hafez E. Elyamany73 | RT | Oven at various temperatures | 7 | 4.8 | 36 |
| A. Aboulayt74 | 40 | Oven at 12 h | 0.5 | 4.8 | 28.5 |
| Tanakorn Phoo-ngernkham75 | RT | Chamber at 23 °C for 24 h | 90 | 4.13 | 39.4 |
| Piotr Prochon76 | 65 | RT for 4 h, oven for 4 h | 3 | 4.10 | 14.3 |
One possible explanation for the high strength is the lowered viscosities of the alkali activators. As shown in Fig. 4, the strength increased when the amount of alkali hydroxide solution increased and that of the alkali silicate decreased. They are both sources for K+ cations, and these ions play a charge-balancing role in geopolymers, which fortify the geopolymeric network. Recent studies argued that the choice of alkali activator has minimal impact on the compressive strength, while the concentration plays a more significant role.77,80 While there are reports that the potassium silicates have higher reactivity,81 the viscous nature of the silicates increases the viscosity of the aluminosilicate gels, thus lowering the castability. Furthermore, f-SiO2, which plays a crosslinking role, is a particle with a high surface area and porosity, and has hygroscopicity. The presence of f-SiO2 further increases the viscosity of GP, resulting in poor fluidity. Nonetheless, the outstanding mechanical strengths cannot be solely explained by the alkali activators, which would have been achieved in previous studies; there must be a synergetic effect of the additives used in this study.
As stated, the presence or absence of additives can significantly influence the mechanical strength; hence, we have further analyzed the effect of additives in GP. By applying the optimized formula (Fig. 4), the following specimens are compared: GPOFC = optimized composition with f-SiO2 and Cf, GPOC = optimized composition without f-SiO2, GPOF = optimized composition without Cf, and GPO = optimized composition without Cf and f-SiO2. The σf of the GPOFC sample in Fig. 6a is as high as 27.82 MPa. The GPOFC rapidly fails without deformation when the applied stress reaches the maximum value. The σc of GPOFC (Fig. 6b) records the maximum stress of 55.12 MPa. On the other hand, GPOC showed a similar trend but with lower strength values of 22.56 and 48.19 MPa for σf and σc, respectively. Similarly, σf and σc decreased when Cf was removed from GPOFC (i.e., GPOF). When neither Cf nor f-SiO2 were added (i.e., GPO), the σf and σc are found to have the lowest values. The chemical bonding process forms strong interparticle bonds, enhancing the crack resistance under bending and leading to higher flexural strength. However, the compressive strength, while benefiting from these bonds, is more affected by porosity and microstructural flaws, as localized stress concentrations at weak points can reduce the overall strength.82 The isotropic distribution of pores uniformly impacts the compressive properties. However, there is a less uniform impact on the flexural properties owing to differences in stress distribution during bending.83 The results clearly show that the mechanical strength is improved with the presence of f-SiO2; however, the improvement remains questionable.
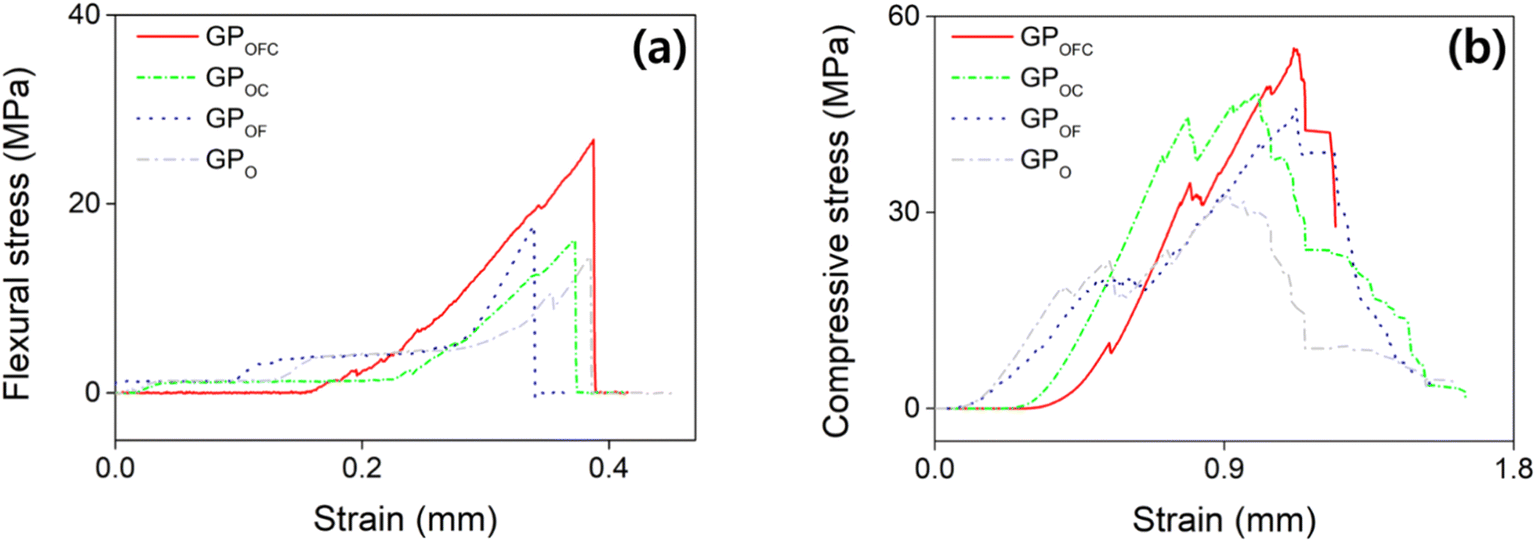 | ||
| Fig. 6 Representative stress–strain curves of GPOFC (red), GPOC (green), GPOF (blue), and GPO (gray) for (a) flexural test and (b) compressive test. | ||
To understand the strength mechanism of f-SiO2, microstructural analysis was carried out on the fracture surfaces of three specimens: the initial composition listed in D1 (D1-26 in ESI Table S2†), GPOFC, and GPOC. For the three specimens, the surface microstructures are exhibited in Fig. 7a–f. A distinctive surface roughness can be observed for GPOC, which is the only specimen without f-SiO2. f-SiO2 is known to form crosslinking interactions in gel binders, and it is likely that it helps in the densification of GP binders.84 The interpretation on the degree of densification from surface microscope characterization is consistent with the strength measurements, where the σf of GPOC is found to be significantly low (22.56 MPa). To confirm the effect of f-SiO2 on the distribution on the chemical species, EDS analysis is presented in Fig. 7g–i and S5.† Of particular note, the distribution of Ca ions showed drastic differences across the three specimens (Fig. 7g–i). The size of the Ca compound in GPIFC (Fig. 7g) is measured as 46.1 ± 5.6 μm, whereas that in GPOFC (Fig. 7h) is found to be 17.1 ± 5.8 μm. The pre-optimized specimen with the initial fumed silica and carbon fiber contents is referred to as GPIFC. It can be deduced that through the series of DOE optimization, the increased mechanical strengths resulted from the changes in the Ca compound distribution, becoming smaller and more sparse. There are also studies in the literature where the analysis results indicate “small doses of calcium hydroxide (up to 2% of mix weight) accelerate [the] setting and can enhance [the] compressive strength, with [the] C–S–H gel forming early in the reaction”, supporting this discussion.85 Meanwhile, the size of the Ca compound in GPOC (Fig. 7i) was 88.3 ± 4.9 μm; this size is 5 times greater than that for GPOFC when f-SiO2 was absent. Such difference provides sound evidence that the presence of f-SiO2 significantly affects the growth of the Ca compounds by impeding their growth. The limited growth of the Ca compound slows down the curing process for GP, allowing for sufficient time to undergo the steady geopolymerization reaction between the alkali activators and MK. Based on the analysis presented in this section, it is reasonable to believe that this homogeneous and steady reaction results in denser solidification of GP, and results in higher mechanical strengths in turn—the optimized amount of alkaline activators and the existence of f-SiO2 facilitate the manipulation of the GP reaction rate.
 | ||
| Fig. 7 (a–f) Cross-sectional images and (g–i) EDS analysis of geopolymers: (I) GPIFC, (II) GPOFC and (III) GPOC. (a–c) and (d–f) show surfaces at different magnifications. In (g–i), Ca elements are indicated in red, and the size of Ca compounds is estimated. | ||
3.3 Remarks on fast-curing geopolymer
As illustrated earlier, the manufacturing process of geopolymers is rather simple and curable under ambient conditions. With the excellent heat resistance and compatibility with other materials, geopolymers can be a powerful tool as a finishing material to prevent the spread of fires. The applicability becomes versatile, especially when they rapidly cure, so that it can be used on complicated cases (e.g., rough walls, inner cracks). However, it has been a challenge to find a balance between the curing speed and strength; typically, the strength is weaker when the curing time is shortened in the cement materials. In the following section, we compare our developed GP with the existing literature, in view of the strengths, curing time and temperature.In Fig. 8a, the σf is plotted against σc from the literature studies, depicting the distribution of the mechanical strength of GPs. The σc of GP w/Cement ranged from 30 to 60 MPa, while have relatively low σf. On the other hand, the single GP has a wide distribution of σc and a relatively high σf (>7.5 MPa) than the GP w/Cement. Thus, it can be seen that the GP in our study had the highest σf and a fairly high σc. Aside from the excellent mechanical characteristics, the greatest advantage is that this ceramic material can be cured at RT without heat treatment over a few days. The σf is compared in terms of their final curing time and temperature in Fig. 8b and c, respectively. The final curing time of the GP in this study was 6 days, which is fast compared to most of the other literature studies (excluding the subsequent drying process), where the curing time takes more than 10 days. A comparison with other references has been presented in Table 1. Surprisingly, we noticed that the optimized chemical formula can exhibit excellent early-stage strength. For the optimized case of D3-7, it only took 3 days and 6 days to reveal 80% and >98% of the saturated flexural strength, respectively (Fig. S2(b)†). By comparing the σf values from other studies where GPs were cured at RT, an overwhelming strength can be confirmed for the GPOFC in this study. In summary, the fast-curing geopolymer was formulated to realize mechanical strength while solidifying rapidly at RT, which is advantageous for applications of ceramic materials.
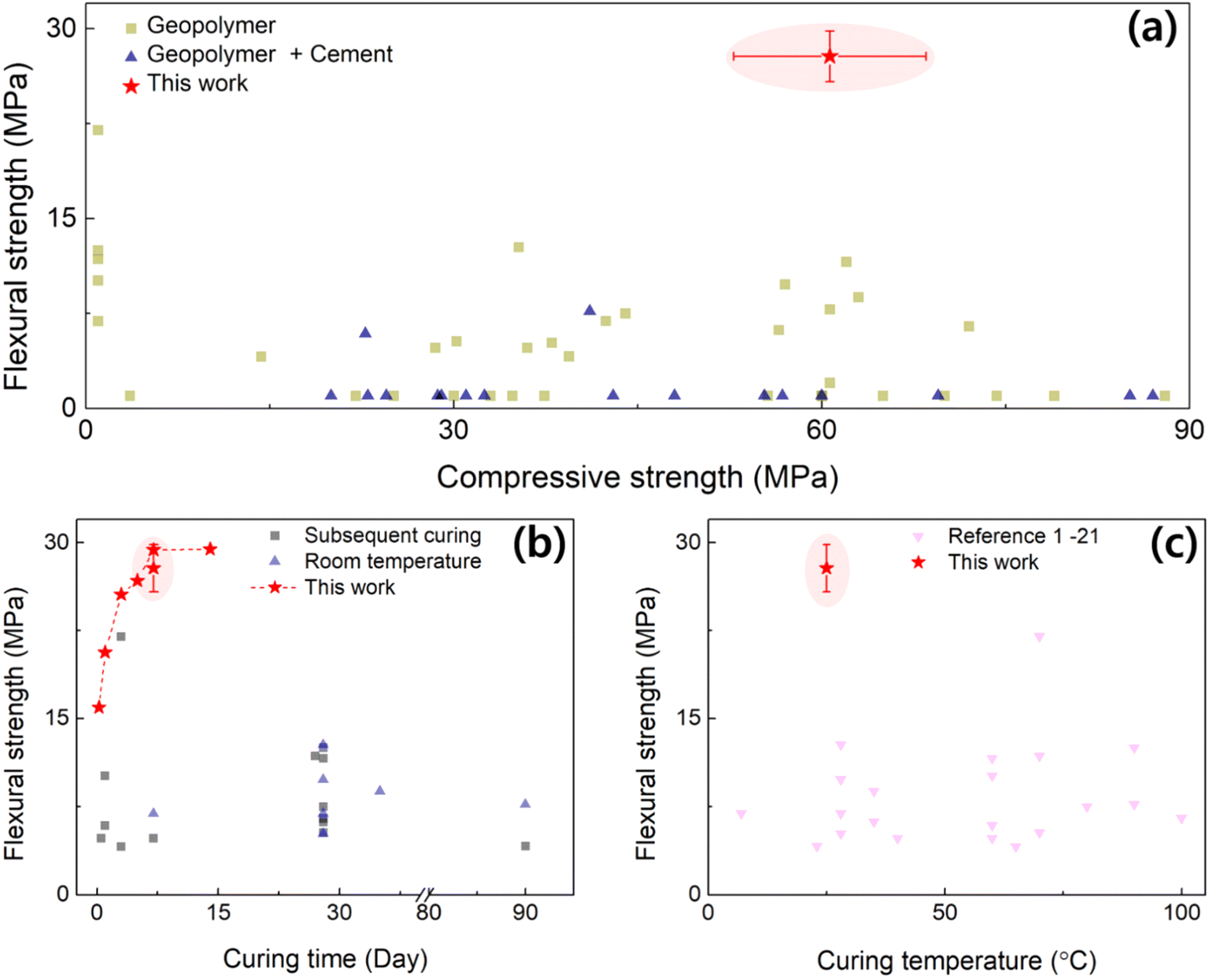 | ||
| Fig. 8 (a) Comparison of flexural strength and compressive strength with literature values. Literature values are classified into two categories: geopolymer alone and geopolymer combined with Portland cements, which are noted as Geopolymer and Geopolymer + cement, respectively. (b) Flexural strength plotted against curing time for groups with RT curing and subsequent curing. (c) Flexural strength as a function of curing temperature, comparing the present study with literature values. | ||
The fast-curing type GP is expected to be used as a non-combustible finishing material composite material that is structurally stable in fire resistance performance due to its rapid curing, free molding, and excellent non-combustibility performance. From the thermogravimetric analysis (TGA) in ESI Fig. S6,† the mass reduction is found to be 11.9% up to 900 °C, showing excellent high temperature resistance. Furthermore, the developed GP was tested for non-combustibility, according to ISO 1182. For fire spread prevention structure, the requirements at 750 °C are as follows: (a) a weight loss of 30% or less, and (b) an internal core temperature increase of 20 °C or less. The developed GP showed a maximum reduction of 11.9% up to 900 °C and an increase of 2.4 °C, respectively, fulfilling the requirements for the heat-resistant finishing materials. In addition, the foaming of GP can be applied to various applications, such as an insulator and soundproofing materials; the foaming rate and strength can be balanced by controlling the Ca precursors and f-SiO2.
4 Conclusion
A series of DOE was carried out to build a metamodel to interpret the effect of process variables on the properties of GPs, and it was used to optimize the design of multivariate fast-curing geopolymers with various additives, including f-SiO2, Cf, and Ca(OH)2. The optimal design targeted the flexural strength and curing time of the geopolymers. The major findings and perspectives are summarized, as follows:(1) The optimized geopolymer showed an outstanding flexural strength of 27.83 MPa and a rapid surface curing time of 7 minutes. The fast curing was achieved at room temperature within 7 minutes. This remarkable curing behavior occurs entirely at room temperature, without the need for elevated curing conditions. The high strength is astonishing, as it is achieved within a few days (takes 3 days to reach 80%, and 6 days to reach >98% of the full strength, whereas typical cements require several weeks) at room temperature (whereas high-strength GP in the literature used higher curing temperatures).
(2) The experimental data-driven statistical designing has successfully optimized the multi-variant problem for the targeted properties. Based on the DOE setup, we have been guided by metamodels to progress towards the optimal design. We found that the combination of DOE and metamodeling provided more efficient data-driven optimization compared to applying DOE alone, and such optimization scheme is relatively new in the field of geopolymers. Starting from powdered materials with potential experimental uncertainties, we effectively applied data-driven optimization techniques combining DOE and metamodeling. This success sets a valuable example for similar research efforts, showcasing the potential of a systematic approach to develop specialized materials.
(3) The synergistic effect of fumed SiO2 and carbon fiber on the curing characteristics and strengthening is elucidated, and will provide a design principle for future fast curing geopolymers. The presence of f-SiO2 adjusts the growth rate of calcium precursors, balancing two different effects originated from the calcium precursor, which are strength degradation and rapid curing. Such mechanistic insights can inform design rules within the field of fast-curing geopolymers.
The developed geopolymer is expected to be widely used in applications with its exceptional properties. Besides the high-temperature resistance, they can be freely casted owing to its rapid hardening cure-behavior. Some possible applications include repairing or finishing materials for buildings and constructions, fire-prevention materials (flame-retardant foam pads, heat shields, and fire-resistance fillers), and lightweight insulating foam materials by its pore-forming characteristics.
Data availability
The data and analysis scripts supporting this study are available in the public GitHub repository at https://github.com/shj0113/Metamodel-RBF.Author contributions
Conceptualization: Hyunseok Ko, Kyungwon Kim. Data curation: Kyungwon Kim, Sanghun Lee. Formal analysis: Hyunseok Ko, Hyejeong Song. Funding acquisition: Hyunseok Ko, Hyung Mi Lim. Investigation: Kyungwon Kim, Sanghun Lee. Methodology: Hyunseok Ko, Hyejeong Song. Project administration: Hyunseok Ko, Kyungwon Kim. Resources: Hyunseok Ko, Hyejeong Song. Software: Hyunseok Ko, Hyejeong Song. Supervision: Hyunseok Ko. Validation: Hyunseok Ko. Visualization: Hyunseok Ko, Kyungwon Kim, Hyejeong Song, Sanghun Lee. Writing – Original Draft: Hyunseok Ko, Kyungwon Kim. Writing – Review & Editing: Hyunseok Ko, Kyungwon Kim, Hyejeong Song.Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.Acknowledgements
This paper was supported by the Basic R&D program (KFB23001-0-01) funded by the Korea Institute of Ceramic Engineering and Technology (KICET, South Korea) and the Virtual Engineering Platform Project (P0022336), funded by the Ministry of Trade, Industry & Energy (MoTIE, South Korea).References
- E. Benhelal, E. Shamsaei and M. I. Rashid, J. Environ. Sci., 2021, 104, 84–101 CrossRef CAS PubMed.
- M. I. Khan, K. Azizli, S. Sufian and Z. Man, Ceram. Int., 2015, 41, 2794–2805 CrossRef.
- K. Komnitsas, D. Zaharaki, A. Vlachou, G. Bartzas and M. Galetakis, Adv. Powder Technol., 2015, 26, 368–376 CrossRef CAS.
- N. Lee and H.-K. Lee, Cem. Concr. Compos., 2016, 72, 168–179 CrossRef CAS.
- P. Thirumakal, M. Nasvi and K. Sinthulan, SN Appl. Sci., 2020, 2, 1–17 Search PubMed.
- Y. Wang, L. Chen, S. Li and Z. Zhang, Sep. Purif. Technol., 2025, 354, 129114, DOI:10.1016/j.seppur.2024.129114.
- P. Zhang, Z. Gao, J. Wang, J. Guo, S. Hu and Y. Ling, J. Cleaner Prod., 2020, 270, 122389, DOI:10.1016/j.jclepro.2020.122389.
- M. Albitar, P. Visintin, M. Mohamed Ali and M. Drechsler, KSCE J. Civ. Eng., 2015, 19, 1445–1455 CrossRef.
- A. Shalbafan, J. Welling and J. Hasch, Holzforschung, 2016, 70, 755–761 CrossRef CAS.
- C. Leiva, Y. Luna-Galiano, C. Arenas, B. Alonso-Fariñas and C. Fernández-Pereira, Waste Manage., 2019, 95, 504–512 CrossRef CAS.
- J. Feng, R. Zhang, L. Gong, Y. Li, W. Cao and X. Cheng, Mater. Des., 2015, 65, 529–533 CrossRef CAS.
- R. M. Novais, M. Seabra and J. Labrincha, J. Cleaner Prod., 2017, 143, 1114–1122 CrossRef CAS.
- T. Kovářík, J. Hájek, M. Pola, D. Rieger, M. Svoboda, J. Beneš, P. Šutta, K. Deshmukh and V. Jandová, Mater. Des., 2021, 198, 109355, DOI:10.1016/j.matdes.2020.109355.
- K. W. Kim, H. M. Lim, S.-Y. Yoon and H. Ko, Minerals, 2022, 12, 821, DOI:10.3390/min12070821.
- A. A. Shahmansouri, M. Yazdani, S. Ghanbari, H. A. Bengar, A. Jafari and H. F. Ghatte, J. Cleaner Prod., 2021, 279, 123697, DOI:10.1016/j.jclepro.2020.123697.
- X. Miao, X. Pang, S. Li, H. Wei, J. Yin and X. Kong, Chin. J. Chem. Eng., 2023, 60, 118–130 CrossRef CAS.
- F. Okoye, J. Durgaprasad and N. Singh, Ceram. Int., 2016, 42, 3000–3006 CrossRef CAS.
- G. Xu, J. Zhong and X. Shi, Fuel, 2018, 226, 644–657 CrossRef CAS.
- C. B. Cheah, W. K. Part and M. Ramli, Constr. Build. Mater., 2015, 88, 41–55 CrossRef.
- P. Topark-Ngarm, P. Chindaprasirt and V. Sata, J. Mater. Civ. Eng., 2015, 27, 04014198, DOI:10.1061/(ASCE)MT.1943-5533.0001157.
- E. Diaz, E. Allouche and S. Eklund, Fuel, 2010, 89, 992–996 CrossRef CAS.
- P. Chindaprasirt, P. De Silva, K. Sagoe-Crentsil and S. Hanjitsuwan, J. Mater. Sci., 2012, 47, 4876–4883 CrossRef CAS.
- C. Xu, S. Andre and J. Leslie, Int. J. Lab. Hematol., 2016, 38, 42–49 CrossRef.
- B. Kim, presented in part at the International Conference on Alkali Activated Materials and Geopolymers: Versatile Materials Offering High Performance and Low Emissions, Tomar, June, 2018 Search PubMed.
- W. Huo, Z. Zhu, H. Sun, B. Ma and L. Yang, J. Cleaner Prod., 2022, 380, 135159, DOI:10.1016/j.jclepro.2022.135159.
- H. Khater, J. Mater. Civ. Eng., 2012, 24, 92–101 CrossRef CAS.
- Z. Zidi, M. Ltifi, Z. B. Ayadi, L. E. Mir and X. Nóvoa, J. Asian Ceram. Soc., 2020, 8, 1–9 CrossRef.
- S. A. Khan and N. J. Zoeller, J. Rheol., 1993, 37, 1225–1235 CrossRef CAS.
- T. J. Kang, K. H. Hong and M. R. Yoo, Fibers Polym., 2010, 11, 719–724 CrossRef CAS.
- L. G. Baltazar, Crystals, 2022, 12, 288, DOI:10.3390/cryst12020288.
- H. Ko, H.-S. Lee and H. M. Lim, J. Asian Ceram. Soc., 2020, 8, 1285–1295 CrossRef.
- A. Natali, S. Manzi and M. Bignozzi, Procedia Eng., 2011, 21, 1124–1131 CrossRef CAS.
- T. Lin, D. Jia, P. He, M. Wang and D. Liang, Mater. Sci. Eng. A, 2008, 497, 181–185 CrossRef.
- H. Assaedi, T. Alomayri, A. Siddika, F. Shaikh, H. Alamri, S. Subaer and I.-M. Low, Materials, 2019, 12, 3624, DOI:10.3390/ma12213624.
- Y. Ling, P. Zhang, J. Wang and Y. Chen, Constr. Build. Mater., 2019, 229, 117068, DOI:10.1016/j.conbuildmat.2019.117068.
- J. Yu, M. Zhang, G. Li, J. Meng and C. K. Leung, Constr. Build. Mater., 2020, 239, 117853, DOI:10.1016/j.conbuildmat.2019.117853.
- P. Duxson, S. W. Mallicoat, G. C. Lukey, W. M. Kriven and J. S. Van Deventer, Colloids Surf., A, 2007, 292, 8–20 CrossRef CAS.
- Z. Yunsheng, S. Wei and L. Zongjin, Appl. Clay Sci., 2010, 47, 271–275 CrossRef.
- M. M. Al Bakri Abdullah, H. Kamarudin, K. N. Ismail, M. Bnhussain, Y. Zarina and A. Rafiza, Adv. Mater. Res., 2012, 341, 189–193 Search PubMed.
- D. Bondar, C. Lynsdale, N. B. Milestone, N. Hassani and A. A. Ramezanianpour, Cem. Concr. Compos., 2011, 33, 251–260 CrossRef CAS.
- C. Ferone, F. Colangelo, G. Roviello, D. Asprone, C. Menna, A. Balsamo, A. Prota, R. Cioffi and G. Manfredi, Materials, 2013, 6, 1920–1939 CrossRef CAS PubMed.
- P. Arjun Raj, D. Sarath, P. Nagarajan and B. S. Thomas, Iran. J. Sci. Technol., Trans. Civ. Eng., 2024, 1–24 Search PubMed.
- K. Walbrück, S. Witzleben and D. Stephan, Heliyon, 2024, 10, e35947, DOI:10.1016/j.heliyon.2024.e35947.
- A. Driouich, S. A. El Hassani, N. H. Sor, Z. Zmirli, M. A. O. Mydin, A. Aziz, A. F. Deifalla and H. Chaair, Results Eng., 2023, 20, 101573, DOI:10.1016/j.rineng.2023.101573.
- T. A. de Carvalho, F. Gaspar, A. C. Marques and A. Mateus, Constr. Build. Mater., 2024, 412, 134846, DOI:10.1016/j.conbuildmat.2023.134846.
- A. S. Kurzekar, U. Waghe, K. Ansari, A. Dabhade, T. Biswas, S. Algburi, M. A. Khan, E. Althaqafi, S. Islam and J. Palanisamy, Case Stud. Constr. Mater., 2024, e03826, DOI:10.1016/j.cscm.2024.e03826.
- S.-e. Kim and Y.-m. You, Appl. Sci., 2022, 12, 1625, DOI:10.3390/app12031625.
- G.-J. Kang, C.-H. Park and D.-H. Choi, J. Mech. Sci. Technol., 2016, 30, 1723–1732 CrossRef.
- G. Boidi, M. R. Da Silva, F. J. Profito and I. F. Machado, Surf. Topogr.:Metrol. Prop., 2020, 8, 044002, DOI:10.1088/2051-672X/abae13.
- A. İ. Kaya, M. İlkuçar and A. Çifci, Erzincan University Journal of Science and Technology, 2019, 12, 116–123 Search PubMed.
- A. Ashwin, R. H. Lakshman, C. C. Swaroop, M. Vignesh, R. V. Vignesh and R. Padmanaban, 2019, 561, 012046, DOI:10.1088/1757-899X/561/1/012046.
- J. Wang, T. Zhou, D. Xu, Z. Zhou, P. Du, N. Xie, X. Cheng and Y. Liu, Constr. Build. Mater., 2018, 167, 381–390 CrossRef CAS.
- M. Wang, Geopolymerization Mechanism of Aluminosilicate Geopolymer and Microstructure and Properties of Fly Ash Cenosphere/Geopolymer Composite, Harbin Institute of Technology, 2011 Search PubMed.
- N. Li, C. Shi, Q. Wang, Z. Zhang and Z. Ou, Mater. Struct., 2017, 50, 1–11 CrossRef CAS.
- K. El Hafid and M. Hajjaji, Constr. Build. Mater., 2018, 159, 598–609 CrossRef CAS.
- L. Gomez-Zamorano, E. Vega-Cordero and L. Struble, Constr. Build. Mater., 2016, 115, 269–276 CrossRef CAS.
- M. H. Fasihnikoutalab, S. Pourakbar, R. J. Ball and B. K. Huat, Int. J. Geosynth. Ground Eng., 2017, 3, 1–10 CrossRef.
- P. He, M. Wang, S. Fu, D. Jia, S. Yan, J. Yuan, J. Xu, P. Wang and Y. Zhou, Ceram. Int., 2016, 42, 14416–14422 CrossRef CAS.
- A. Kalaiyarrasi, P. Partheeban and V. Muthupandi, Int. J. Appl. Eng. Res., 2018, 13, 11466–11470 Search PubMed.
- T. da Silva Rocha, D. P. Dias, F. C. C. França, R. R. de Salles Guerra and L. R. d. C. de Oliveira, Constr. Build. Mater., 2018, 178, 453–461 CrossRef.
- A. Nazari, A. Maghsoudpour and J. G. Sanjayan, Constr. Build. Mater., 2015, 76, 207–213 CrossRef.
- P. Rovnaník, Constr. Build. Mater., 2010, 24, 1176–1183 CrossRef.
- R. M. Hamidi, Z. Man and K. A. Azizli, Procedia Eng., 2016, 148, 189–193 CrossRef CAS.
- P. Zhang, K. Wang, J. Wang, J. Guo, S. Hu and Y. Ling, Ceram. Int., 2020, 46, 20027–20037 CrossRef CAS.
- H. J. Zhuang, H. Y. Zhang and H. Xu, Procedia Eng., 2017, 210, 126–131 CrossRef CAS.
- P. Nath and P. K. Sarker, Constr. Build. Mater., 2017, 130, 22–31 CrossRef CAS.
- H. Tanyildizi and Y. Yonar, Constr. Build. Mater., 2016, 126, 381–387 CrossRef CAS.
- S. Xu, M. A. Malik, Z. Qi, B. Huang, Q. Li and M. Sarkar, Constr. Build. Mater., 2018, 164, 238–245 CrossRef CAS.
- M. Sofi, J. Van Deventer, P. Mendis and G. Lukey, Cem. Concr. Res., 2007, 37, 251–257 CrossRef CAS.
- C. Atiş, E. Görür, O. Karahan, C. Bilim, S. İlkentapar and E. Luga, Constr. Build. Mater., 2015, 96, 673–678 CrossRef.
- X. Li, X. Ma, S. Zhang and E. Zheng, Marterials, 2013, 6, 1485–1495 CrossRef CAS PubMed.
- Y. J. Zhang, S. Li, D. L. Xu, B. Q. Wang, G. M. Xu, D. F. Yang, N. Wang, H. C. Liu and Y. C. Wang, J. Mater. Sci., 2010, 45, 1189–1192 CrossRef CAS.
- H. E. Elyamany, M. Abd Elmoaty and A. M. Elshaboury, Constr. Build. Mater., 2018, 187, 974–983 CrossRef CAS.
- A. Aboulayt, M. Riahi, M. O. Touhami, H. Hannache, M. Gomina and R. Moussa, Adv. Powder Technol., 2017, 28, 2393–2401 CrossRef CAS.
- T. Phoo-ngernkham, P. Chindaprasirt, V. Sata, S. Hanjitsuwan and S. Hatanaka, Mater. Des., 2014, 55, 58–65 CrossRef CAS.
- P. Prochon, Z. Zhao, L. Courard, T. Piotrowski, F. Michel and A. Garbacz, Materials, 2020, 13, 1033, DOI:10.3390/ma13051033.
- T. Revathi, R. Jeyalakshmi and N. Rajamane, Mater. Today: Proc., 2018, 5, 8727–8734 CAS.
- D. Hardjito, presented in part at the 3rd Asian Concrete Federation International Conference, Ho Chi Minh City, November, 2008 Search PubMed.
- C. R. Kaze, A. Adesina and G. L. L.-Nana, J. Build. Eng., 2021, 43, 103229, DOI:10.1016/j.jobe.2021.103229.
- Y. Zhang, J. Chen and J. Xia, Materials, 2022, 16, 181, DOI:10.3390/ma16010181.
- H. Xu and J. Van Deventer, Min., Metall., Explor., 2002, 19, 209–214 CAS.
- S.-E. Bendaoudi, M. Bounazef and A. Djeffal, J. Mech. Behav. Mater., 2018, 27, 20180018, DOI:10.1515/jmbm-2018-0018.
- J. Seuba, S. Deville, C. Guizard and A. J. Stevenson, Sci. Rep., 2016, 6, 24326, DOI:10.1038/srep24326.
- Z. Xu, H. Yu, X. Sun, M. Zhao and D. Hui, J. Build. Eng., 2024, 97, 110930, DOI:10.1016/j.jobe.2024.110930.
- B. Kim, S. Lee, C.-M. Chon and S. Cho, Materials, 2021, 15, 194, DOI:10.3390/ma15010194.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed analysis of the experiments, metamodel, and analysis. See DOI: https://doi.org/10.1039/d4dd00217b |
| ‡ Co-first authorship. |
| This journal is © The Royal Society of Chemistry 2025 |