
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precursor reaction pathway leading to BiFeO3 formation: insights from text-mining and chemical reaction network analyses†
Viktoriia
Baibakova
 a,
Kevin
Cruse
a,
Michael G.
Taylor
b,
Carolin M.
Sutter-Fella
a,
Gerbrand
Ceder
a,
Anubhav
Jain
a and
Samuel M.
Blau
*a
a,
Kevin
Cruse
a,
Michael G.
Taylor
b,
Carolin M.
Sutter-Fella
a,
Gerbrand
Ceder
a,
Anubhav
Jain
a and
Samuel M.
Blau
*a
aLawrence Berkeley National Laboratory, 1 Cyclotron Rd, Berkeley, CA 94720, USA. E-mail: smblau@lbl.gov
bTheoretical Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
First published on 27th May 2025
Abstract
BiFeO3 (BFO) is a next-generation non-toxic multiferroic material with applications in sensors, memory devices, and spintronics, where its crystallinity and crystal structure directly influence its functional properties. Designing sol–gel syntheses that result in phase-pure BFO remains a challenge due to the complex interactions between metal complexes in the precursor solution. Here, we combine text-mined data and chemical reaction network (CRN) analysis to obtain novel insight into BFO sol–gel precursor chemistry. We perform text-mining analysis of 340 synthesis recipes with the emphasis on phase-pure BFO and identify trends in the use of precursor materials, including that nitrates are the preferred metal salts, 2-methoxyethanol (2 ME) is the dominant solvent, and adding citric acid as a chelating agent frequently leads to phase-pure BFO. Our CRN analysis reveals that the thermodynamically favored reaction mechanism between bismuth nitrate and 2ME interaction involves partial solvation followed by dimerization, contradicting assumptions in previous literature. We suggest that further oligomerization, facilitated by nitrite ion bridging, is critical for achieving the pure BFO phase.
1 Introduction
Bismuth ferrite (BiFeO3, or BFO) is a well-studied multiferroic material with significant applications in sensors, memory devices, and spintronics.1,2 Perovskite BFO thin films have attracted considerable interest as multiferroic perovskites and a semiconductor alternative to lead-based perovskite solar cells. The crystallinity and crystal structure of BFO are key factors influencing its functional properties, which highlights the importance of the synthesis process.2,3 Several methods, including sol–gel, hydrothermal, and solid-state techniques, have been employed to synthesize BFO.4–9 Among these, sol–gel synthesis is particularly advantageous for growing single-phase films due to its ability to achieve uniform composition at the molecular level.10 In this process, precursor materials are mixed in solution, followed by deposition, spin-coating, and temperature treatment.1,7–9However, challenges remain in designing sol–gel protocols to achieve target outcomes.10,11 Achieving a pure crystalline BFO phase is a topic of ongoing investigation, as even small changes in sol–gel protocol conditions can lead to different structures.12,13 Variations in precursors, ratios, pH, temperature, time, pressure, atmosphere, and other factors can produce a wide range of compounds, often resulting in BFO with impurity phases or the formation of an amorphous phase.7–9,11 While most parameters can be numerically analyzed and sequentially optimized to identify cutoff values,3,11 selecting precursor materials for sol–gel BFO synthesis is not straightforward due to the complex underlying chemical interactions within the precursor solution.12,14 For example, although the most common sol–gel BFO precursor solution, comprising nitrate salts and 2ME, has been experimentally studied,1,14 the specific reaction pathways involved have yet to be explored from a computational perspective.
To add insight into the impact of precursors on BFO phase formation and the reactions occurring in the precursor solution, we propose the combined use of text-mining15,16 and Chemical Reaction Network (CRN) methods.17–19 Our approach is outlined in Fig. 1. Text-mining facilitates the extraction of synthesis protocols and phase outcomes from scientific literature, providing a broad perspective on experimental trends.11 By systematically analyzing 340 sol–gel synthesis recipes targeting phase-pure BFO thin films, we identified key trends in the choice of metal precursors, solvents, and additives as critical factors influencing outcome phase. This text-mining analysis further informs the selection of systems for CRN modeling20 to explore the underlying chemical reaction pathways at a molecular level. Our study investigates the detailed reaction pathways and intermediate structures involved in BFO synthesis, modeling the energetics and viability of various synthesis routes. Our findings highlight oligomerization and dimerization as dominant mechanisms, challenging the previously suggested hypothesis of a gradual nitrite-to-solvent replacement pathway. Based on these insights, we propose optimizing precursor materials by focusing on nitrate salts, selecting solvents stabilizing de-nitrated complexes, and avoiding surfactants.
 | ||
| Fig. 1 Graphical abstract. To study the precursor solution, we used (a) text mining and (b) chemical reaction networks (CRNs). (c) Integrating text mining with CRNs provides insights into how precursor materials influence the reaction pathway. We hypothesize that nitrate salts and solvents that stabilize de-nitrated complexes promote dimerization. The dimerization suggests that surfactants obstruct the reaction, and our text-mining analysis supports this claim. | ||
This paper consists of the following parts. The Methods section describes in-detail the text-mining techniques and CRN analysis used to extract and model the synthesis data, as well as assumptions made regarding structural representation. In the Results section, we present the findings from the text-mining analysis and CRN simulations, including trends in precursor selection and reaction pathways. We emphasize the oligomerization pathway in the sol–gel process. In the Discussion section, we interpret our findings in the context of existing studies, explaining that nitrate salts and solvents that stabilize de-nitrated complexes can enhance oligomerization while surfactants tend to inhibit it, and explore the potential future implications of these results.
2 Methods
We used data-driven methods, text-mining and CRN, to study the molecular reactions at play in the precursor solution for BFO thin films. The general workflow is outlined in Fig. 1.2.1 Text-mining
We used text mining to study the diversity of the precursor solution composition and establish the link between precursor materials and the resulting phase (Fig. 1a). We selected 178 scientific publications reporting sol–gel synthesis of BFO and targeted phase-pure perovskite thin films through a multi-step process. A keyword search of an in-house database containing nearly 5 million materials science articles published between 2000 and 2020 identified 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 196 papers discussing impurity phase formation. Chemical Named Entity Recognition was then used to extract relevant materials, narrowing the focus to BFO. Further filtering based on synthesis method and relevance to sol–gel-derived BFO thin films resulted in 121 suitable articles. To supplement our dataset with articles published after 2020, we scraped Web of Science, filtering for papers on sol–gel synthesis targeting BFO. This search yielded 57 additional papers. Publication selection is described in detail in our accompanying paper.11
196 papers discussing impurity phase formation. Chemical Named Entity Recognition was then used to extract relevant materials, narrowing the focus to BFO. Further filtering based on synthesis method and relevance to sol–gel-derived BFO thin films resulted in 121 suitable articles. To supplement our dataset with articles published after 2020, we scraped Web of Science, filtering for papers on sol–gel synthesis targeting BFO. This search yielded 57 additional papers. Publication selection is described in detail in our accompanying paper.11
Next, we manually extracted synthesis parameters and outcomes for 340 synthesis descriptions. Due to the complexity and variability of the synthesis descriptions and the limited automated capabilities at the time, we chose human-driven literature extraction which enabled us to capture nuanced insight that programmatic text-mining could not yet achieve. We extracted and tabulated all synthesis parameters mentioned in the experimental sections of selected papers, such as temperature, pH, time, concentration, etc., and performed a comprehensive programmatic analysis in our accompanying paper.11 However, in this work, we focus on precursor materials and output phases. To structure the data, we classified all materials (according to the claims of the authors and in a consistent way) into the following roles: metal source, solvent, chelating agent, dehydrating agent, or surfactant. We then conducted a statistical analysis to identify the most frequently used materials in each category, labeling less frequent materials as “other”. See ESI† for the full list of materials and details of the analysis.
2.2 Assumptions about metal complexes
To describe the chemical reaction pathway for the BFO system using a data-driven CRN approach, we made several assumptions about metal complexes. We focus solely on Bi due to its similar chemical behavior with Fe14 and to avoid computational complexity associated with Fe's incomplete d-shell. The Bi-metal ion core forms ionic and coordination bonds with inorganic (nitrite ion, nitric acid) and organic (2MEdehydr ion, 2ME) ligands. We assume a neutral precursor solution with Bi3+ charge, resulting in complexes with three ionic ligands and varying numbers of nonionic ligands. Ligands can be mono- or bi-dentate, influencing coordination geometry. Based on data from the Crystal Structure Database21,22 (see ESI†), Bi3+ ions coordinated with oxygen exhibit ligand-dependent coordination with coordination number (CN) ranging from 4 to 9 and the median CN of 6. Thus, we do not limit the CN but set an initial coordination of 6 for bismuth nitrate. Water ligands were excluded as they tend to form hydrogen bonds with organic ligands rather than coordinate with Bi-metal centers, see ESI.†2.3 Chemical reaction network
A Chemical Reaction Network (CRN) is a mathematical framework used to model and analyze chemical reactions, particularly in complex systems where experimental observations are difficult. CRNs allow for systematic exploration of chemical interaction spaces, enabling the identification of feasible reaction pathways and providing predictions of reaction outcomes.17,20,23 A CRN consists of two main elements: a dataset of chemical species (including reactants, reaction intermediates, and products) and the reactions between them, characterized by parameters such as free energy changes and activation barriers. Thus, the reaction pathway is broken down into elementary reaction steps occurring between intermediate species with minimal changes in structure and composition in each elementary step. This allows CRNs to model the entire reaction landscape in detail and identify energetically favorable pathways.24 The method is highly parallelizable and compatible with modern computational architectures, offering resource-efficient solutions for large-scale chemical systems and high-throughput studies.19,25To study the chemical reaction space of bismuth nitrate dissolved in 2ME, we built a CRN (Fig. 1b) and used a BEP-type approximation to relate the Gibbs free energy to the activation energy as the reaction driving force. We started by preparing a dataset of species for CRN including partial ligand exchanges: we represented reactant molecules as molecular graphs using SCINE Molassmebler26 and iteratively generated intermediate species toward the product (step 1). For each graph, we generated 3D conformers and pre-optimized them using Architector27 and TBLite28 (step 2). We selected three lowest energy conformers, optimized them and calculated thermodynamic potentials using QChem29 and QuAcc30 (step 3). We compiled this information into a comprehensive dataset of species and generated a dataset of reactions between them with High Performance Reaction Generation (HiPRGen)31 (step 4). Lastly, we produced reaction pathway trajectories with Reaction Network Monte Carlo (RNMC)31 (step 5). See more details on each step below. To handle structures with multiple Bi centers and bridging ligands, we performed steps 1 and 3 of the pipeline.
A chemical reaction network is defined by a set of species and reactions, and each run of RNMC produces a trajectory by selecting a reaction at every step; although no exhaustive method can address the vast space of possible complexes and reaction paths, the CRN approach captures as many possibilities as feasible, surpasses previous methods, and provides new insights.24
We converted Molassembler graphs into Architector's input format: dictionaries listing the metal core, coordination number (CN), and ligands in SMILES notation with bonding site indexes (ligands listed in ESI†). We ran Architector with force-field pre-optimization, the solvent set to “octanol” (approximating 2ME by dielectric constant), and requested 20 total metal-center symmetries. We used the Sella optimizer (from the Architector developer branch), which, although requiring 2–10 CPU node-hours per graph, yielded configurations more consistent with the input molecular graph and lower in energy compared to the default LBFGS optimizer.33 We also set Architector to perform geometry relaxation with GFN2-xTB34 and selected the 3 lowest-energy conformers based on xTB energy values for further DFT calculations. While satisfactory for this task, this method lacks support for oligomeric complexes; dimer structures were handled using SCINE Molassembler.
Overall, we collected a species dataset with the following data: relaxed 3D molecule, energy, entropy, and enthalpy. We note that during the geometry relaxations some bonds were not preserved, and the molecule graphs were updated accordingly.
The species dataset for the CRN is illustrated in Fig. 2. The fragmentation-recombination loop generated 809 unique molecular graphs, which we reduced to 110 graph dictionaries with ligand-based filtering as described earlier. Adding graph dictionaries with higher Bi CN enhanced the dataset for the potential product study (P). Architector produced a total of 1900 pre-optimized structures. Running DFT optimization of the three lowest energy conformers resulted in 231 molecules with unique molecular graphs. The optimized structures exhibit CN ranging from 4 to 10 with a median of 6. The ratio of nitric to 2ME ionic ligands indicates a trend toward more structures with 2ME ligands, consistent with the expected reaction direction. The study of potential products resulted in seven 3D conformers with CN of 7 and 8, and free energy analysis demonstrated that adding a fourth neutral 2ME ligand to Bi coordinated with three 2ME ions is not thermodynamically beneficial (see ESI†), corroborating earlier claims on similar structures.39
 | ||
| Fig. 2 Data flow in the generated CRN dataset follows the approach outlined in steps 1–3 described in the methods section. The dataset used in HiPRGen comprises 231 metal complexes with coordination numbers (CN) ranging from 4 to 10 and various ratios of nitrite to 2ME among the ionic ligands. ‘P’ denotes product structures. | ||
:100. We run 100 trajectories of 10000 steps each using GMC, Gillespie's next reaction simulator.40
2.4 Dimer study
To find the dimer lowest energy configuration, we followed steps 1–3 from the CRN box in Fig. 1 with several adjustments. We started with the molecule graph for the dimer-ready structure: [Bi3+(NO−3)2(2ME−)] determined in CRN analysis and connected two such graphs in order to form the dimer molecule graph using SCINE Molassembler. We built two different dimer molecular graphs, bridging with one and two nitrite ions and zero total charge, see ESI† for molecular graphs. Then, since Architector does not support multi metal complexes, we used SCINE Molassembler to generate 50 3D conformers and randomly chose 10 for each dimer graph. We followed with opt-freq-sp calculations in Q-Chem at the same level of theory as in original CRN step 3, and 17 dimer conformers converged. We chose the conformer with the lowest Gibbs free energy at room temperature, see ESI† for the full list of conformer energies and the lowest energy conformer XYZ.3 Results
3.1 Text-mining
To study the diversity of precursor materials and establish a connection with the resulting BFO material phases in reported experiments, we text-mined 340 synthesis recipes targeting phase-pure BFO and analyzed the collected dataset (see the results of the text-mining analysis in Fig. 3). We categorized the resulting phases into three groups: pure crystalline phase (73%), impure crystalline phase (24%), and amorphous phase (3%). Metal precursor nitrates are used in 94% of the recipes, while the remaining 6% use other salts, which yield all three phase outcomes. Thus, we could not observe a statistically significant correlation between salt choice and phase outcome from our data. Next, we incorporated solvents into the resulting material phase analysis. Specifically, 2ME is used in 73% of recipes, and ethylene glycol is used in 16% of the recipes. There are 50 recipes in our dataset that use only metal salts and solvents without other additives, and in 90% of those cases, they result in pure crystalline phase formation. These findings suggest that 2ME is the preferred solvent and is optimal for achieving a pure-phase crystalline BFO when no additional materials are included. | ||
| Fig. 3 (a) Precursor composition diagram from text-mined dataset. Vertical bars represent quantitative distribution of materials used in recipes. (b) Phase outcomes for different precursor materials. Solvent with no other additives leads to phase-pure crystalline BFO in 90% of cases. Addition of citric acid leads to a pure crystalline phase in 94% of cases. Acetic acid derivatives yield phase-pure BFO in 68% of cases. Overall portion of recipes reporting pure crystalline phase is 73%. | ||
Chelating agent citric acid, used in 12% of the recipes, maintains high phase-pure crystalline BFO yield: among 40 recipes with salts, solvent, and citric acid (no additional materials), 94% result in phase-pure BFO. In contrast, other chelators such as more common acetic acid or nitric acid (used in 34% and 5% of recipes respectively), show less consistent results. Among 190 recipes that use acetic acid, glacial acetic acid or acetic anhydride (which forms acetic acid after interacting with water), only 68% lead to phase-pure BFO, while 29% yield impure crystalline phase. Among 18 recipes using nitric acid, only 55% yield pure phase, and 45% result in impurities formation. These findings suggest that citric acid is a more reliable chelating agent for pure BFO synthesis compared to acetic or nitric acid. Contrary to the expectations that surfactants enhance phase-pure BFO formation,9,41 our analysis shows that their inclusion decreases the likelihood of achieving pure crystalline BFO phase: the fraction of recipes using surfactants and reporting phase-pure, impure, and amorphous phases is 66%, 31%, and 3% (with the overall recipes outcome phases fractions being 73%, 24%, and 3%), indicating that their role in phase purity enhancement is not straightforward.
To summarize out text-mining analysis, for researchers aiming to synthesize phase-pure BFO, we recommend using nitrate precursors, 2ME as the solvent, and citric acid as the chelating agent, while avoiding unnecessary additives that may introduce complexity without clear benefits.10 Motivated by text-mining results, we selected the most common precursor, bismuth nitrate dissolved in 2ME, and performed a computational analysis of its reaction pathways. We apply CRN approach to investigate the reaction path in detail in the context of observations from text-mining.
3.2 Chemical reaction network
The CRN analysis of bismuth nitrate dissolved in 2ME, as illustrated in Fig. 4, distills the complex CRN down to a handful of critical reactions, capturing both expected and unexpected pathways. Previous literature1,14 suggests that the complete replacement of nitrite ligands with 2ME ligands, as shown in Reaction (1), occurs before the formation of any oligomeric species on the way towards crystallization:| [Bi3+(NO−3)3] + 3·2ME → [Bi3+(2ME−dehydr)3] + 3HNO3 | (1) |
 | ||
| Fig. 4 Reaction path energy profile based on CRN analysis. The full solvation path is expected based on previous hypotheses but is found to be highly thermodynamically unfavorable. In contrast, the novel dimer path revealed by our analysis is thermodynamically favorable, suggesting a more likely route towards crystallization. | ||
Surprisingly, our calculations show that this overall reaction is endergonic, with a change in Gibbs free energy of ΔG(1) = +0.89 eV. However, the beginning of the path is energetically favorable. We call the exergonic part of the pathway “partial solvation”, in which one nitrite ligand is replaced by one solvent ligand (see ESI†), as shown in Reaction (2):
| [Bi3+(NO−3)3] + 1·2ME → [Bi3+(NO−3)2(2ME−dehydr)] + HNO3 | (2) |
In the CRN, Reaction (2) is composed of three elementary steps: solvation (3), H swap (4), acid detachment (5) as described with Reactions (3)–(5):
Solvation:
| [Bi(NO−3)3] + (2ME) → [Bi(NO−3)3(2ME)] | (3) |
H swap:
| [Bi(NO−3)3(2ME)] → [Bi(NO−3)2(2ME−dehydr)(HNO3)] | (4) |
Acid detachment:
| [Bi(NO−3)2(2ME−dehydr)(HNO3)] → [Bi(NO−3)2(2ME−dehydr)] + HNO3 | (5) |
The process begins with the solvation of the initial bismuth nitrate metal complex, where a neutral 2ME solvent molecule attaches to the complex. The attachment of 2ME is energetically favorable, with a solvation free energy of ΔG(3) = −0.22, indicating good solubility, as expected. CRN analysis reveals that the optimized complex, [Bi(NO−3)3(2ME)], has a CN of 8, with 2ME forming two Bi–O bonds. This bi-dentate interaction significantly contributes to the stability of the complex. Following the initial solvation, a proton transfer, i.e. H swap, occurs from the hydroxyl group (–OH) of 2ME to a nitrite ligand. This step has an associated free energy change of ΔG(4) = +0.38 eV. Despite being energetically unfavorable (endergonic), the overall free energy change relative to the initial state is ΔG = +0.16, which is five times smaller than ΔG(1). Further, we know that heat is required to drive this process forward, so the presence of an intermediate endergonic step is unsurprising. Subsequently, the byproduct nitric acid detaches from the bismuth core with ΔG(5) < 0, leading to the formation of a new complex, [Bi(NO−3)2(2ME−dehydr)], referred to as the “dimer-ready structure” for reasons that will be clarified below. This structure is characterized by the replacement of one nitrite ligand with one solvent ligand, making it the final stable complex on the expected solvation pathway that is energetically lower than the initial state, see ESI† for the reaction report. After the formation of the “dimer-ready structure,” the full solvation pathway continues with the substitution of the remaining two nitrite ligands with solvent molecules. However, these subsequent steps are increasingly endergonic, with significant energy costs, making the overall pathway thermodynamically unfavorable and thus highly unlikely to occur.
However, as illustrated in Fig. 4, our CRN analysis reveals that dimerization after partial solvation is exergonic, presenting a novel alternative mechanistic route towards crystallization that is vastly more thermodynamically favorable than the expected full solvation path. This “dimer path” can be described as two “dimer-ready structures” joining together as denoted by Reaction (6):
| 2[Bi(NO−3)2(2ME−dehydr)] → [Bi2(NO−3)4(2ME−dehydr)2] | (6) |
4 Discussion
Through text mining with the emphasis on phase-pure BFO, we examined what precursor materials experimentalists select based on expected behaviors in the solution, specifically the selection of metal salt, solvent, chelating agent, dehydrating agent, and surfactant. Solvents, typically volatile long organic molecules, are expected to help with forming a dense, stable polymeric precursor.1,14 Chelating agents, or chelators, are organic acids which should possess specific ligands that have high affinity and can bind/carry metal ions and are claimed to increase gel viscosity and encourage the formation of oligomeric structures.1,44 Dehydrating agents are employed to remove excess water by bounding it, thus promoting homogeneity of the final gel.1 The studies that employ surfactants commonly motivate it by the assumption that they improve phase purity by increasing gel homogeneity and promoting chemical reactions in it.1In our chosen precursor of study, Bi nitrate with 2ME as motivated by text mining, the reaction path was previously claimed to proceed as gradual replacement of nitrites with solvent ligands within a single-metal complex.1,14 There have been several experimental efforts where the BFO precursor was studied with Fourier Transform Infrared Spectroscopy (FTIR) for the bond presence before and after the reaction.14 The analysis indicated Bi–nitrite bonds before the reaction and Bi–2ME bonds after. The possible reaction pathway was described as the complete substitution of nitrite ligands with solvent for each Bi complex via gradual ligand replacement.1,14 This route is what we call “full solvation” and is described by Reaction (1).
We contradict an existing hypothesis that the chemical reaction in BFO precursor occurs as a gradual replacement of nitrite ligands with 2ME ligands within one complex. We find this reaction to be overall endergonic with ΔG(1) = +0.89 eV, making it energetically unfavorable. The estimated temperature for this reaction to proceed, from the formula T = ΔHrxn/ΔSrxn, is 300 °C, see ESI.†. However, while it was previously experimentally demonstrated that the precursor requires heating for bonds to change, the reported temperature was much lower – about 90 °C.14 Further, our CRN analysis reveals that every step of the “full solvation” route after the initial 2ME-nitrite swap is energetically unfavorable.
We instead hypothesize that the chemical reaction in the BFO precursor occurs through dimerization with both nitrite and solvent ligands coordinating Bi metal cores. Our CRN analysis revealed the first step of the oligomerization chain: formation of a dimer. The dimer configuration we report exhibits asymmetrical bridges involving both nitrite ions and solvent ions, consistent with a previous study which found that Bi complexes form dimeric structures with asymmetrically bridging ligands and additional coordination sites partially occupied by solvent molecules.43 This aspect of solvent involvement aligns with the subproducts we identified by the CRN. Hence, selecting the right precursor materials and their treatment is crucial for successful oligomerization. Returning to our findings, since we established dimerization as the preferential pathway in the precursor, we hypothesize that additives may lead to deviations from this route and result in impurity phase formation in BFO synthesis.
It was previously suggested that oligomer formation determines the structural motifs of the resultant BFO phase,14 and our results are consistent with this statement. In the BFO crystal, Bi–O–Bi bonds exhibit a rhombohedral arrangement with oxygen ions fourfold coordinated with metals. This motif can already be observed in the dimer structure as illustrated in Fig. 5. Two “dimer-ready structures” are linked with Bi–nitrite–Bi and Bi–2MEdehydr–Bi bonds with oxygens following a similar rhombohedral pattern. We expect that as more Bi complexes participate, oligomerization proceeds, forming spatial arrangement of bridges to bring oxygen ions to a fourfold coordination state and BFO phase seeding.
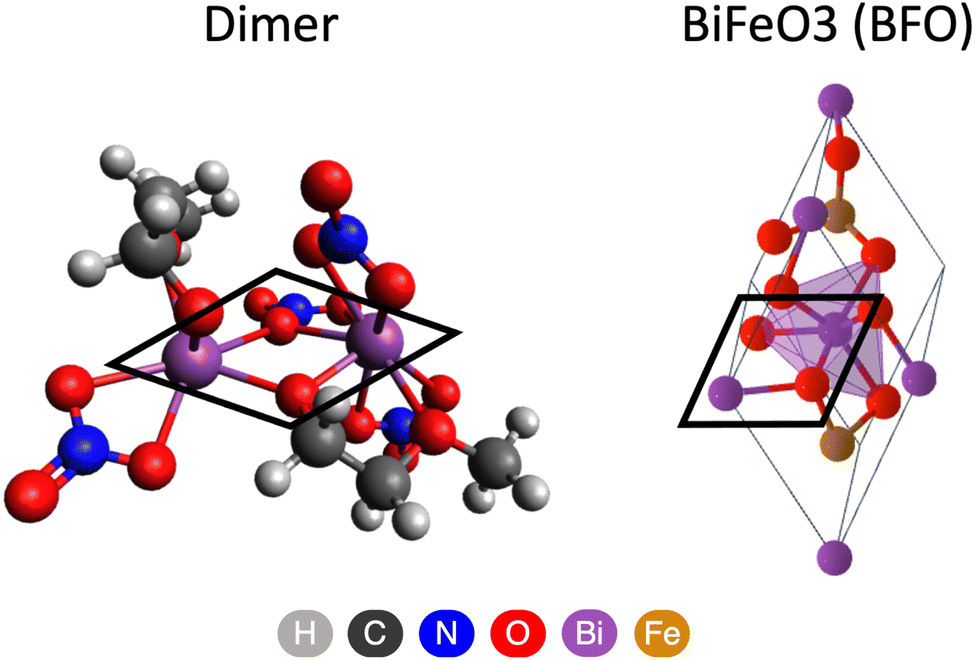 | ||
| Fig. 5 Comparing rhombohedral motifs forming in the dimer structure during bridging and persisting in the BFO crystal. | ||
To strengthen the connection between oligomerization and the final phase structure, we compare our findings to related studies reporting molecular dynamics (MD) simulations that illustrated the bridging process and the formation of rhombohedral crystal structural motifs. MD simulations showed that [Bi6O4(OH)4]6+ complexes in DMSO solution formed dimers via nitrite ion bridges and oligomerized further into [Bi6O4(OH)4](NO3)6 clusters.22,42,45,46 During the bridging process, two complexes were linked with rapidly formed temporary Bi–nitrite–Bi bonds with 1–3 nitrite bridges. Then these bonds quickly rearranged into Bi–O–Bi rhombohedral motifs with fourfold coordinated oxygen ions by Bi, characteristic to Bi2O3 crystals.42 These observations align with our findings and support our assertion that dimerization is the reaction pathway.
Next, we speculate on the potential implications of oligomerization being the favored pathway for the pure phase BiFeO3 formation, and particularly the effect of specific additive types in this process. We highlight that the following discussion is speculative and represents our attempt to connect all evidence reported in the literature, emphasizing that further research is needed. Our text-mining analysis reveals that only 2ME, ethylene glycol, and citric acid consistently lead to a pure phase (90 to 94% pure phase formation) while the addition of acetic acid derivatives, nitric acid, and surfactants, on average, decreasing the frequency of pure phase formation (66 to 68%). Previous studies suggest that impurity or amorphous phase formation may occur when competing chemical interactions are present within the precursor system.1 Building on this, we hypothesize that the mentioned additives may initiate side processes, diverting from oligomerization — a preferential reaction pathway within the precursor solution, as shown in our CRN analysis.
Our results suggest that an increase in nitrite ions would favor oligomerization, as the driving force behind this process has been attributed to the presence of free nitrite ions in solution.42 Surprisingly, our text-mining analysis showed that, in contrast to using pure nitrate salt, the addition of nitric acid decreases the formation of a pure phase. The reasons for this are unclear but may be related to excess nitrite competing with the solvent's ability to stabilize de-nitrated complexes. Previous MD simulations have shown that the ability of solvents to interact with under-coordinated Bi ions after nitrite ions depart is important for oligomerization.42,45 See more discussion in ESI.†
The approach used in this work has several limitations. Extracting synthesis recipes from natural texts is a challenging task. A synthesis process is typically described in a concise manner, frequently containing missing steps and experimental parameters.11,15 Also, the protocol complexity is not always explicitly described. To obtain a high-quality dataset, we extracted the synthesis recipes manually, which limited its size and influenced the range of conclusions we could derive. Advancements in automated text processing methods, such as large language models (LLMs), have effectively increased the volume of retrieved synthesis recipes; however, challenges remain in handling the versatility of features.47
The CRN method used in this work has several limitations, including: a minor trade-off in accuracy due to semi-empirical methods to manage the computational demand of data preparation with advanced structures like metal complexes; the use of the PCM solvation model; an inability to consider trimers due to the exponential increase in configurational conformers; and the inability to capture the complete chemical reaction pathway from individual molecules to final crystal formation. The primary limitation for this particular work is that we did not model Fe, due to its complex d-shell structure, which is not accurately accounted for by the semi-empirical methods used. We hypothesize that the Fe mechanism is similar to that of Bi based on experimental evidence. In the future, we hope these limitations will be overcome as methods progress.
5 Conclusions
This study presents a text-mining analysis of 340 sol–gel synthesis recipes for BFO precursor solution and crystallinity outcomes. Our analysis identified nitrates as the predominant metal salts and 2ME as the dominant solvent, with citric acid frequently leading to phase-pure BFO. The use of acetic acid and nitric acid as chelators resulted in variable phase purity, highlighting the complexity introduced by additional precursor materials.The CRN analysis uncovered the initial parts of the pathway involved in the formation of BFO. The study found that the commonly proposed full solvation route, involving the complete replacement of nitrite ligands with 2ME ligands, is energetically unfavorable. Instead, a more plausible mechanism involves partial solvation followed by dimerization. Our findings explain existing studies, proposing that oligomerization is facilitated by nitrite ion bridging. This pathway aligns with experimental observations and suggests that oligomerization plays an important role in BFO phase seeding.
This work demonstrates the power of combining text-mining with CRN analysis to uncover the underlying mechanisms of complex synthesis processes.
This study has several future implications. Subsequent studies should investigate the role of different solvents and chelating agents in the synthesis of BFO, focusing on their impact on the reaction pathways and final crystal structures. The method described in this work can be applied to study synthesis of other complex oxides, enhancing their crystallinity and phase purity. Implementation of advanced text processing methods and CRN expansion would help to scale up to larger datasets and investigate more complex systems.
Data availability
The code for dataset generation and data analysis described in the article is publicly available and can be found at Zenodo at https://doi.org/10.5281/zenodo.15389508. The code for building a chemical reaction network via HiPRGen and the dataset used in the paper are publicly available and can be found at Zenodo at https://doi.org/10.5281/zenodo.15389626.Author contributions
V. B.: conceptualization, methodology, software, investigation, data curation, writing – original draft, visualization. K. C.: data curation, formal analysis, writing – review and editing. M. T.: conceptualization, writing – review and editing C. S.-F.: conceptualization, supervision, writing – review and editing. G. C.: supervision, funding acquisition, writing – review and editing. A. J.: conceptualization, supervision, funding acquisition, project administration, writing – review and editing. S. B.: conceptualization, methodology, supervision, funding acquisition, project administration, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded and intellectually led by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under Contract No. DE-AC02-05-CH11231 (D2S2 program KCD2S2). We acknowledge support by the Center for High Precision Patterning Science (CHiPPS), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences at Lawrence Berkeley National Laboratory under contract DE-AC02-05CH11231. Computational resources were provided by the National Energy Research Scientific Computing Center (NERSC) and by the Lawrencium computational cluster resource provided by the IT Division at the Lawrence Berkeley National Laboratory. The Molecular Foundry supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 is acknowledged. We acknowledge U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry Program (KC0302031) under contract number E3M2 and the LANL's Director's Postdoc Fellowship (LANL-LDRD, 20210966PRD4) (M.G.T.). We thank Andrew Rosen for support with QuAcc and for other valuable discussions.Notes and references
- Q. Zhang, D. Sando and V. Nagarajan, J. Mater. Chem. C, 2016, 4, 4092–4124 RSC.
- J. Wang, J. Neaton, H. Zheng, V. Nagarajan, S. Ogale, B. Liu, D. Viehland, V. Vaithyanathan, D. Schlom and U. Waghmare, et al. , science, 2003, 299, 1719–1722 CrossRef CAS PubMed.
- P. Biswas, C. Thirmal, S. Pal and P. Murugavel, J. Appl. Phys., 2018, 123(2), 024101 CrossRef.
- S. M. Selbach, M.-A. Einarsrud and T. Grande, Chem. Mater., 2009, 21, 169–173 CrossRef CAS.
- S. Pillai, D. Bhuwal, T. Shripathi and V. Shelke, J. Mater. Sci.: Mater. Electron., 2013, 24, 2950–2955 CrossRef CAS.
- F. Gheorghiu, M. Calugaru, A. Ianculescu, V. Musteata and L. Mitoseriu, Solid State Sci., 2013, 23, 79–87 CrossRef CAS.
- Y. Ma, W. Xing, J. Chen, Y. Bai, S. Zhao and H. Zhang, Appl. Phys. A:Mater. Sci. Process., 2016, 122, 1–9 CrossRef.
- S. Gupta, M. Tomar and V. Gupta, J. Mater. Sci., 2014, 49, 5997–6006 CrossRef CAS.
- C. Anthony Raj, M. Muneeswaran, P. Jegatheesan, N. Giridharan, V. Sivakumar and G. Senguttuvan, J. Mater. Sci.: Mater. Electron., 2013, 24, 4148–4154 CrossRef.
- M. Abdelsamie, K. Hong, K. Cruse, C. J. Bartel, V. Baibakova, A. Trewartha, A. Jain, G. Ceder and C. M. Sutter-Fella, Matter, 2023, 6(12), 4291–4305 CrossRef CAS.
- K. Cruse, V. Baibakova, M. Abdelsamie, K. Hong, C. J. Bartel, A. Trewartha, A. Jain, C. M. Sutter-Fella and G. Ceder, Chem. Mater., 2023, 36, 772–785 CrossRef PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- D. Navas, S. Fuentes, A. Castro-Alvarez and E. Chavez-Angel, Gels, 2021, 7, 275 CrossRef CAS PubMed.
- Q. Zhang, N. Valanoor and O. Standard, J. Mater. Chem. C, 2015, 3, 582–595 RSC.
- K. Cruse, A. Trewartha, S. Lee, Z. Wang, H. Huo, T. He, O. Kononova, A. Jain and G. Ceder, Sci. Data, 2022, 9, 234 CrossRef PubMed.
- O. Kononova, H. Huo, T. He, Z. Rong, T. Botari, W. Sun, V. Tshitoyan and G. Ceder, Sci. Data, 2019, 6, 1–11 CrossRef PubMed.
- M. J. McDermott, S. S. Dwaraknath and K. A. Persson, Nat. Commun., 2021, 12, 3097 CrossRef CAS PubMed.
- X. Xie, E. W. Clark Spotte-Smith, M. Wen, H. D. Patel, S. M. Blau and K. A. Persson, J. Am. Chem. Soc., 2021, 143, 13245–13258 CrossRef CAS PubMed.
- S. M. Blau, H. D. Patel, E. W. C. Spotte-Smith, X. Xie, S. Dwaraknath and K. A. Persson, Chem. Sci., 2021, 12, 4931–4939 RSC.
- O. N. Temkin, A. V. Zeigarnik and D. Bonchev, Chemical reaction networks: a graph-theoretical approach, CRC Press, 2020 Search PubMed.
- A. Vaitkus, A. Merkys, T. Sander, M. Quirós, P. A. Thiessen, E. E. Bolton and S. Gražulis, J. Cheminf., 2023, 15, 123 Search PubMed.
- J. Näslund, I. Persson and M. Sandström, Inorg. Chem., 2000, 39, 4012–4021 CrossRef PubMed.
- J.-T. Zhang, H.-Y. Wang, X. Zhang, F. Zhang and Y.-L. Guo, Catal. Sci. Technol., 2016, 6, 6637–6643 RSC.
- M. Wen, E. W. C. Spotte-Smith, S. M. Blau, M. J. McDermott, A. S. Krishnapriyan and K. A. Persson, Nat. Comput. Sci., 2023, 3, 12–24 CrossRef PubMed.
- M. Aykol, J. H. Montoya and J. Hummelshøj, J. Am. Chem. Soc., 2021, 143, 9244–9259 CrossRef CAS PubMed.
- J.-G. Sobez and M. Reiher, J. Chem. Inf. Model., 2020, 60, 3884–3900 CrossRef CAS PubMed.
- M. G. Taylor, D. J. Burrill, J. Janssen, E. R. Batista, D. Perez and P. Yang, Nat. Commun., 2023, 14, 2786 CrossRef CAS PubMed.
- Light-weight tight-binding framework, https://www.github.com/tblite/tblite.
- E. Epifanovsky, A. T. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons and A. L. Dempwolff, et al. , J. Chem. Phys., 2021, 155(8), 084801 CrossRef CAS PubMed.
- A. S. Rosen, QuAcc – The Quantum Accelerator, Zenodo, 2023 Search PubMed.
- D. Barter, E. W. C. Spotte-Smith, N. S. Redkar, A. Khanwale, S. Dwaraknath, K. A. Persson and S. M. Blau, Digital Discovery, 2023, 2, 123–137 RSC.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- C. Zhu, R. H. Byrd, P. Lu and J. Nocedal, ACM Trans. Math. Softw., 1997, 23, 550–560 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- Avogadro an open-source molecular builder and visualization tool. Version 1.2.0, http://www.avogadro.cc/.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144(21), 214110 CrossRef PubMed.
- A. Hellweg and D. Rappoport, Phys. Chem. Chem. Phys., 2015, 17, 1010–1017 RSC.
- B. Mennucci, J. Tomasi, R. Cammi, J. Cheeseman, M. Frisch, F. Devlin, S. Gabriel and P. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- P. A. Williams, A. C. Jones, M. J. Crosbie, P. J. Wright, J. F. Bickley, A. Steiner, H. O. Davies, T. J. Leedham and G. W. Critchlow, Chem. Vap. Deposition, 2001, 7, 205–209 CrossRef CAS.
- D. T. Gillespie, J. Comput. Phys., 1976, 22, 403–434 CrossRef CAS.
- Z. Liu, H. Liu, G. Du, J. Zhang and K. Yao, J. Appl. Phys., 2006, 100(4), 044110 CrossRef.
- M. Walther and D. Zahn, Eur. J. Inorg. Chem., 2015, 2015, 1178–1181 CrossRef CAS.
- M. Mehring, Coord. Chem. Rev., 2007, 251, 974–1006 CrossRef CAS.
- C. V. Thompson and R. Carel, J. Mech. Phys. Solids, 1996, 44, 657–673 CrossRef CAS.
- M. Walther and D. Zahn, Chem. Phys. Lett., 2018, 691, 87–90 CrossRef CAS.
- M. Weber, M. Schlesinger, M. Walther, D. Zahn, C. A. Schalley and M. Mehring, Z. Kristallogr. Cryst. Mater., 2017, 232, 185–207 CrossRef CAS.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Nat. Mach. Intell., 2024, 1–11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dd00160a |
| This journal is © The Royal Society of Chemistry 2025 |