
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphology-dependent Li+ ion dynamics in X-ray amorphous and crystalline Li3PS4 prepared by solvent-assisted synthesis†
Jonas
Spychala
 a,
Christoph
Mandl
a,
Katharina
Hogrefe
a,
H. Martin R.
Wilkening
*ab and
Bernhard
Gadermaier
*a
a,
Christoph
Mandl
a,
Katharina
Hogrefe
a,
H. Martin R.
Wilkening
*ab and
Bernhard
Gadermaier
*a
aGraz University of Technology, Institute of Chemistry and Technology of Materials (NAWI Graz), Stremayrgasse 9, 8010 Graz, Austria
bAlistore – ERI European Research Institute, CNRS, Hub de l'Energie, Rue Baudelocque, F-80039 Amiens, France
First published on 4th November 2024
Abstract
Solid-state electrolytes with high ionic conductivity will be crucial for future energy storage systems. Among many possible materials, thiophosphates offer both favourable mechanical properties and fast ionic transport. β-Li3PS4, as a member of the thiophosphate family, has gained recent attention, due to its remarkable increase in Li+ ionic conductivity when prepared via solvent-assisted synthesis. Despite earlier studies, the lithium ion migration processes causing the increased conductivity remain, however, still uncertain. Here, we study both long-range cation transport and local Li+ jump processes by broadband impedance spectroscopy and nuclear magnetic resonance (NMR), respectively. In particular, we focus on the comparison between mechanochemical and solvent-assisted synthesis to determine the origin of the increased ionic conductivity observed in the latter. Our measurements reproduce the previously reported high ionic conductivity and reveal that synthesis conditions significantly affect the Arrhenius pre-exponential factor governing ionic conductivity. Diffusion-controlled 7Li (and 31P) NMR spin relaxation rates confirm rapid, anisotropic lithium ion hopping that is characterized by timescale-dependent activation energies Ea ranging from 0.40 eV (long-range transport, as also seen by conductivity spectroscopy) to values down to 0.09 eV (local barriers).
1 Introduction
The transformation towards an ecologically sustainable society necessitates energy storage devices at various scales, from hand-held devices to large stationary grid storage.1 To realize such a wide range of applications, further advancements in battery technology are urgently needed.2 Though lithium-ion batteries (LIBs) have become the standard solution for high-energy density storage, open questions remain concerning safety and cycleability.Since their commercialization in the 1990s, conventional LIBs have taken advantage of liquid electrolytes, which are flammable, and in many cases are also highly toxic or at least hazardous when released into the environment. Over the last years, solid-state batteries3 have gained much attention because they might offer improved safety and increased energy density. In such types of batteries, the flammable liquids are replaced with solids or even ceramic electrolytes that possess extraordinary Li+ conduction properties. In this way one would not only improve device safety but also open the door to further improvements in energy density as solid electrolytes might enable the use of Li metal anodes.4 If acting as a negative electrode material, the use of Li metal would lead, in the ideal case of materials combination, to a considerable increase in gravimetric energy density.5
Such a projection remains, however, also a bit theoretical, as current solid electrolytes are not always able to reliably produce ionic conductivities that are comparable to their liquid counterparts. Among the most promising materials in this regard we find oxides,3,6 that is, garnet-type materials7,8 including high-entropy variants,9 phosphates, sulfides and even halides.10–16 In particular, oxides and phosphates have shown to suffer from high interface resistance in combination with unfavourable mechanical properties such as increased hardness and/or brittleness. Consequently, softer materials from the sulfide and thiophosphate groups showing very high conductivities might prove advantageous.17,18 Their mechanical properties make them highly suitable to be used in solid-state batteries because the resistive issues at the interfaces can be greatly reduced.
The large family of thiophosphates comprises many different structure variants such as Li10GeP2S12,19,20 lithium argyrodites Li6PS5X (X = Br, Cl, I),11,21–23 Li3PS4,24–28 Li4PS4I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29,30 and even completely amorphous ones such as mixtures of the binaries Li2S and P2S5.31–33 In particular, amorphous and glassy materials in the Li2S–P2S5 compositional space have shown very high room temperature ionic conductivities reaching values as high as 1 mS cm−1, which sensitively depend on the exact Li2S–P2S5 ratio.34 Moreover, these materials are reported to be compatible with many active materials.35–37 Over the last decade, research has also focused on crystalline compounds within the Li2S–P2S5 phase diagram such as Li3PS4 and Li7P3S11 as the key players in this research area.38 Recent research also evaluates recycling strategies of Li3PS4-based batteries.39
29,30 and even completely amorphous ones such as mixtures of the binaries Li2S and P2S5.31–33 In particular, amorphous and glassy materials in the Li2S–P2S5 compositional space have shown very high room temperature ionic conductivities reaching values as high as 1 mS cm−1, which sensitively depend on the exact Li2S–P2S5 ratio.34 Moreover, these materials are reported to be compatible with many active materials.35–37 Over the last decade, research has also focused on crystalline compounds within the Li2S–P2S5 phase diagram such as Li3PS4 and Li7P3S11 as the key players in this research area.38 Recent research also evaluates recycling strategies of Li3PS4-based batteries.39
Li3PS4 prepared by mechanochemical synthesis, either using planetary or shaker ball mills, exhibits in many cases ionic conductivities below 10−7 S cm−1, far from sufficient for battery applications, especially if the γ-form of Li3PS4 is considered.24,40 Liu et al.41 were, however, able to prepare a sample with vastly increased ionic conductivity of approximately 0.1 mS cm−1 through precipitation from a tetrahydrofuran (THF) suspension. This remarkable improvement is mainly achieved by stabilizing the high-temperature β-phase of Li3PS4 at room temperature, possibly due to the surface morphology of the precipitated particles.41 The exact origin of the increased conductivity in this sample, as well as the underlying transport mechanism in this and similarly prepared samples,42 remain, however, still uncertain. Importantly, in a recent study, Raman spectroscopy and X-ray diffraction showed the presence of solvent residuals in these samples.43 These residuals form a separate Li3PS4• THF phase making most samples multiphase materials.43 Such a phase composition may significantly influence both the crystallinity and the overall (long-range) ionic conductivity.
Recent theoretical studies have demonstrated the anisotropic nature of lithium-ion migration in both the α modification and the β form of Li3PS4 as a contributing factor to the increased conductivity of β-Li3PS4 compared to that of its γ-phase.44,45 The crystal structure has been well explored by previous studies, and powerful calculations have been used to shed light on the dynamic processes. However, considering these studies, strong conclusive evidence for a single, prevailing diffusion mechanism is still lacking a bit. Our own recent study on mechanically prepared nanocrystalline Li3PS4 points to a zig-zag, channel-like (low-dimensional) diffusion pathway that governs diffusion-induced NMR spin relaxation.25
The present study aims at shedding light at the origins of enhanced ion conductivity in Li3PS4 samples prepared by solvent-assisted methods. In this regard it is also important to understand the changes of dynamic paraments as a function of crystallinity or morphology of these samples. Here, we use a combination of conductivity spectroscopy and diffusion-induced 7Li NMR to directly compare the results of samples either prepared by (mechanochemical) solid-state synthesis and solvent-assisted approaches.
2 Experimental
2.1 Solvent-assisted sample preparations
We prepared multiple variations of β-Li3PS4 for this study via a solvent-assisted synthesis route adopted from Liu et al.41 Stoichiometric amounts of Li2S (Alfa Aesar, 99.9%) and P2S5 (Honeywell Fluka, 99%) were manually homogenized in an agate mortar, before suspension in 30 ml tetrahydrofuran (THF, Alfa Aesar, 99.8%, anhydrous). The suspension was left to stir at room temperature for 24 h. Afterwards, the suspension was centrifuged for 4 min at 1100 rpm using a Heraeus Megafuge 10, then decanted, and finally re-suspended in 10 ml THF. This washing process was repeated three times; it seems to be necessary to obtain rather pure Li3PS4. After the final purification step, the remaining solvent was decanted and the wet product was set to dry for 24 h at 80 °C under a dynamic vacuum in a glass oven (Büchi B-585). The resulting β-Li3PS4 precursor will be called LPS-80. The batch was divided and heat-treated, again under dynamic vacuum, at 120 °C and 170 °C, respectively. The resulting samples, consisting of transparent crystals, will be referred to as LPS-120 and LPS-170 according to the highest temperature of the heat-treatment step. As a reference, we use a sample prepared via a mechanochemical synthesis route, which will be called LPS-solid. Information on the synthesis and characterization of this reference sample has been published earlier.25 Unless stated otherwise, all synthesis steps were carried out in glove boxes with an inert argon atmosphere (H2O and O2 ≤ 1 ppm) The solvents used were dried by means of molecular sieves (3 Å; solvent-to-sieve ratio of 10:1) for at least 48 h.
2.2 X-ray diffractometry to study the macroscopic crystal structure
X-ray powder diffraction (XRPD) measurements were performed with a Rigaku MiniFlex 600 diffractometer (θ–2θ Bragg–Brentano geometry, Cu Kα) with a D/teX Ultra silicon strip detector. Diffractogramms, were recorded in a 2θ range from 10° to 80° using a step size of 0.01° and a scanning rate of 2.5° min−1. The sample was protected from any contact with moisture and air through the use of an airtight sample holder equipped with a Kapton foil window. Baseline correction and Rietveld refinement was carried out with Malvern Panalytical X'PertHighScorePlus. Reference patterns for phase analysis were taken from the Inorganic Crystal Structures Database (ICSD).2.3 Impedance spectroscopy to measure the macroscopic ionic transport properties
For the variable-temperature impedance measurements, the β-Li3PS4 samples were pressed into pellets of 5 mm diameter by using a uniaxial Specac Mini Pellet press. We equipped the resulting pellets with Au electrodes in a vacuum with a LEICA EM SCD050 sputter coater. An Au layer with a thickness of 50 nm on each side of the pellet ensured good electrical contact. Subsequently, impedance experiments were carried out with a NovoControl Concept 80 broadband dielectric spectrometer that can cover a frequency range from 10−2 to 107 Hz. The temperature inside the airtight, home-built sample chamber, which was placed into the impedance cell (ZGS, Novocontrol), was controlled with a QUATRO cryosystem (Novocontrol) and varied from −80 °C to 160 °C in 20 K steps. The measurement chamber was continuously flushed with a stream of freshly evaporated N2 gas.2.4 MAS and time-domain NMR measurements
| Mρ (tlock) ∝ exp(−(tlock/T1ρ(ω1, T))k) with 0 < k ≤ 1 | (1) |
For laboratory-frame SLR NMR we have:
| M(tdelay) ∝ 1 − exp(−(tdelay/T1(ω0, T))k′) with 0 < k′ ≤ 1 | (2) |
1/T1ρ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) R1ρ denotes the spin-lock relaxation rate that depends on both absolute temperature T and the spin-locking frequency ω1; similarly, 1/T1R1 recorded in the laboratory frame of reference depends on T and the angular Larmor frequency ω0. The is orders of magnitude larger than ω1 and takes values in the MHz rather than in the kHz range as in the case of spin-lock R1ρ SLR NMR. The stretching exponents k and k′ were allowed to adopt values ranging between 0 and 1. In general, for 3D ion conductors, the rates follow a temperature dependence dictated by the so-called BPP model introduced by Bloembergen, Purcell and Pound which was developed for isotropic, uncorrelated motions.46 Models for spatially restricted diffusivity (2D and the limiting case for 1D) have been introduced by Richards47 and Sholl.48
R1ρ denotes the spin-lock relaxation rate that depends on both absolute temperature T and the spin-locking frequency ω1; similarly, 1/T1R1 recorded in the laboratory frame of reference depends on T and the angular Larmor frequency ω0. The is orders of magnitude larger than ω1 and takes values in the MHz rather than in the kHz range as in the case of spin-lock R1ρ SLR NMR. The stretching exponents k and k′ were allowed to adopt values ranging between 0 and 1. In general, for 3D ion conductors, the rates follow a temperature dependence dictated by the so-called BPP model introduced by Bloembergen, Purcell and Pound which was developed for isotropic, uncorrelated motions.46 Models for spatially restricted diffusivity (2D and the limiting case for 1D) have been introduced by Richards47 and Sholl.48
3 Results and discussion
3.1 Structural characterization
For structural analysis and phase identification, we recorded XRPD patterns of all three samples. Fig. 1a shows the corresponding patterns of the dried precursor LPS-80 (black) and the samples annealed at 120 °C, LPS-120 (blue), and 170 °C, LPS-170 (red). The dried sample, LPS-80, shows no identifiable reflections but appears completely amorphous, which is well in line with previous reports indicating that the precipitate is a fine, non-crystalline powder. Annealing at 120 °C hardly influences the atomic long-range order, as shown by the XRPD pattern of LPS-120, which is indistinguishable from that of the dried precursor. However, we cannot rule out the formation of X-ray invisible nanocrystalline regions embedded in an amorphous matrix as has been suggested and verified earlier by Tatsumisago and co-workers for the 75Li2S·25P2S5 system.49 As shown below, 31P and 6Li MAS NMR spectra do not change much up to 120 °C. Here, only annealing at 170 °C, that is, above the crystallization temperature of 140 °C,41 induces crystallization that is clearly seen in XRPD. Rietveld refinement of the LPS-170 pattern indicates that the main phase is β-Li3PS4 (ICSD: 180319) and that very small amounts of impurities of Li4P2S6 and unreacted Li2S are present (see Fig. S1†). | ||
Fig. 1 (a) XRPD patterns of the β-Li3PS4 precursor (bottom) and the samples annealed at 120 °C (middle) and 170 °C (top), respectively. Vertical bars shown at the very bottom indicate the reference pattern (ICSD: 180319) of β-Li3PS4. Values in brackets refer to the hkl indices. The inset shows the corresponding 1H MAS NMR lines. (b) 31P MAS NMR spectra (25 kHz spinning speed) of the samples annealed at the indicated temperatures 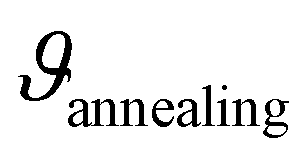 . For comparison, the corresponding spectrum of the mechanochemically prepared β-Li3PS4 sample is shown in grey.25 (c) 6Li MAS NMR spectra (25 kHz) for the same samples as in (b); again, the sample obtained via the solid-state mechanochemical approach is indicated in grey.25 . For comparison, the corresponding spectrum of the mechanochemically prepared β-Li3PS4 sample is shown in grey.25 (c) 6Li MAS NMR spectra (25 kHz) for the same samples as in (b); again, the sample obtained via the solid-state mechanochemical approach is indicated in grey.25 | ||
According to Mercier et al.,50 the crystal structure of β-Li3PS4 is well described by the Pnma space group with the lattice parameters a = 13.066(3) Å, b = 8.015(2) Å and c = 6.101(2) Å, as determined by synchrotron XRPD. Additionally, neutron diffraction data indicate a high degree of disorder in the lithium sublattice.51 β-Li3PS4 has three different lithium positions Li1 (8d), centred in the LiS4 tetrahedron, Li2 (4b) as part of an LiS6 octahedron and Li3 (4c). While the Li1 site is fully occupied, the other sites, Li2 and Li3, show occupancies of 0.7 and 0.3, respectively.
To gain further insight into the local structure and the extent of (site) disorder in the samples, we employed high-resolution 31P and 6Li MAS NMR to probe site-specific environments. The corresponding NMR spectra are shown in Fig. 1b and c, respectively. The 31P NMR spectra reveal a broad line for LPS-80 and LPS-120, centred around a chemical shift of 83.7 ppm. This resonance can be identified with 31P of the PS43− tetrahedra, in accordance with previous 31P MAS measurements and Raman spectroscopy data.43 Its chemical shift is comparable to that reported for 31P in amorphous samples, see, e.g., Marchini et al.43 A line width of ≈9 ppm (≈1.8 kHz) points to a large distribution of local chemical environments indicative of significant local disorder and/or polyhedra distortions.
Compared to LPS-80 and LPS-120, for crystalline LPS-170, we notice a considerably narrowed MAS NMR line with a width of 2 ppm (0.4 kHz). This decrease in line width can be explained by the higher extent of local ordering because of the increasing degree of crystallinity as already found by XRPD, see above. The absence of a broad 31P NMR line as seen for LPS-120, shows that LPS-170 is to be regarded as a sample with a low amount of amorphous material. Line narrowing is accompanied by a change in isotropic NMR chemical shift, which slightly increases to 86.2 ppm. This shift points to a change in the dominant local environment, sensed by the 31P nuclei. It is consistent with findings usually met for crystalline β-Li3PS4.43,51 In contrast to other studies on β-Li3PS4 samples prepared by liquid-based synthesis, we do not observe multiple 31P MAS NMR lines which have been earlier reported and associated with a rather high THF content.52 In our case, either most of the THF was removed during the vacuum drying process or the remaining THF did not lead to distinct coordination environments. To qualitatively assess the THF content remaining after the drying step, all samples were additionally investigated by 1H MAS NMR. The corresponding NMR spectra are shown in Fig. 1a (see inset); they are composed of three distinct, closely spaced 1H lines corresponding to the three different coordination environments of Li3PS4• 3THF, see Gobet et al.52 LPS-170 shows, however, a significantly lowered intensity in two of the THF lines indeed confirming a reduced THF content for this sample. Likely atomic rearrangements during crystallization enhanced the release of THF during the drying period.
Finally, 6Li MAS NMR spectra were recorded to collect information on the local magnetic environments to which Li is subjected in the three samples. As is shown in Fig. 1c, we observe only a single line for LPS-80 and LPS-120 at a chemical shift of 0.27 ppm. As observed by XRPD and 31P MAS NMR, the local and long-range order is very similar in these two samples. This shift increases to 0.62 ppm for LPS-170, reflecting the onset of crystallization as verified by XRPD. In previous studies, multiple 6Li NMR lines were detected and assigned to distinct Li sites. In all three samples, a single 6Li line is observed representing a population-weighted average chemical shift. The chemical environment of the Li-positions may be too similar to be resolved or fast exchange between the positions causes coalescence. While the chemical shift observed in LPS-170 resembles that of the LPS-solid sample, or LPS-80 and LPS-120, the chemical shift has no clear analogue in the literature, which could be explained by the local disorder of these samples.
3.2 Electrical conductivity
We used conductivity spectroscopy to study long-range ion transport in β-Li3PS4. Fig. 2a shows the real part of the complex ionic conductivity as σ′T vs. 1/T of all three samples. For each temperature, the ionic conductivity was extracted from the frequency-independent plateaus of the conductivity isotherms, hence σ′(ν → 0) can be identified with the direct current (DC) conductivity σDC (see Fig. 3a and Fig. S2†).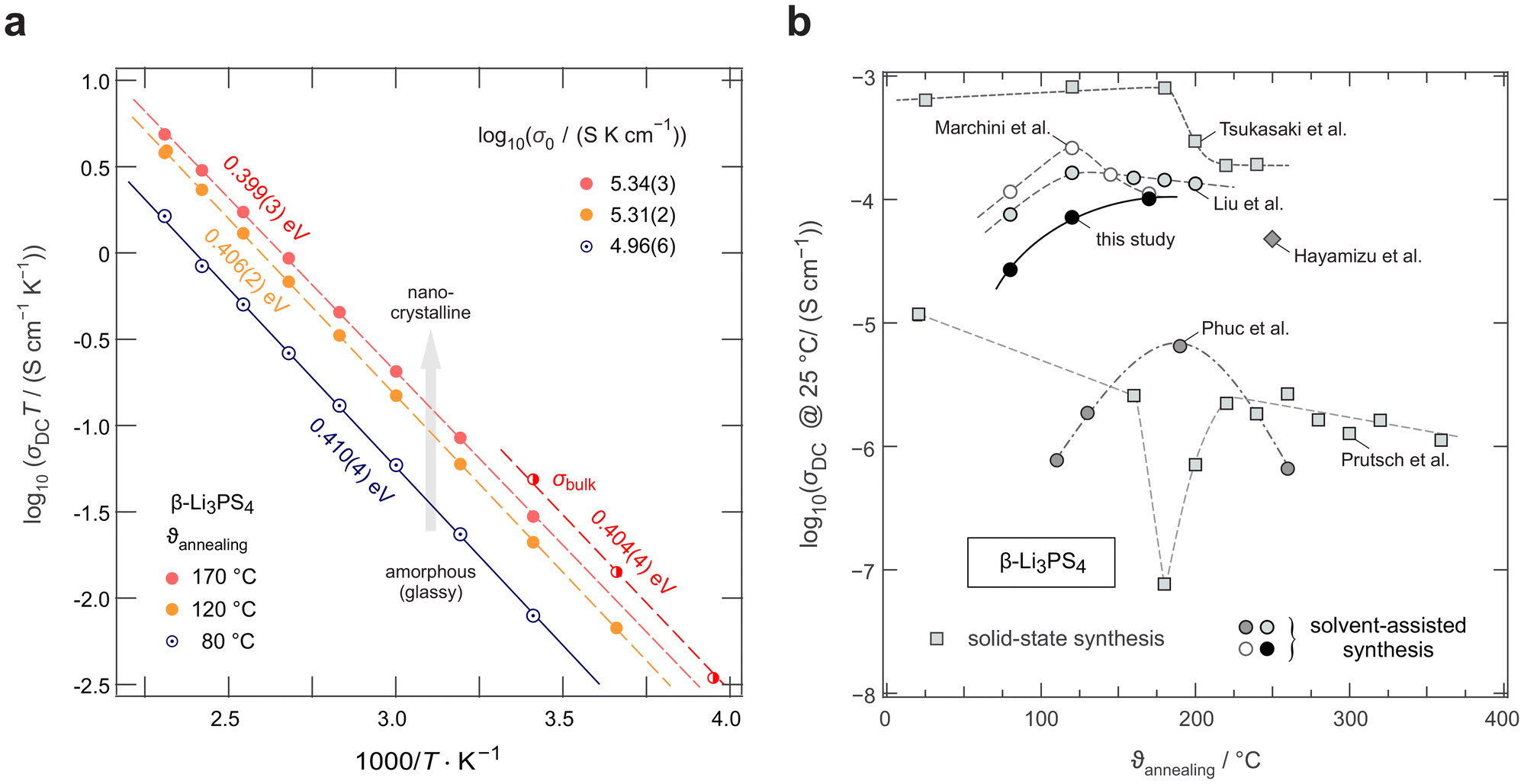 | ||
| Fig. 2 (a) Change of ionic conductivity, plotted as σDCT vs. 1/T, to determine Arrhenius activation energies and pre-exponential factors (σ0) of the β-Li3PS4 samples. Values in eV indicate activation energies. Log(σ0) values are shown in the upper part. While total conductivities are represented by filled symbols, half-filled symbols show the bulk conductivities referring to plateau II, see Fig. 3a. The latter separation is possible for the sample annealed at 170 °C. (b) Comparison of the overall (total) room temperature (25 °C) ionic conductivity of the β-Li3PS4 samples synthesized either via mechanochemical routes25–27 or solvent-assisted methods28,41,43 against their respective annealing temperatures. | ||
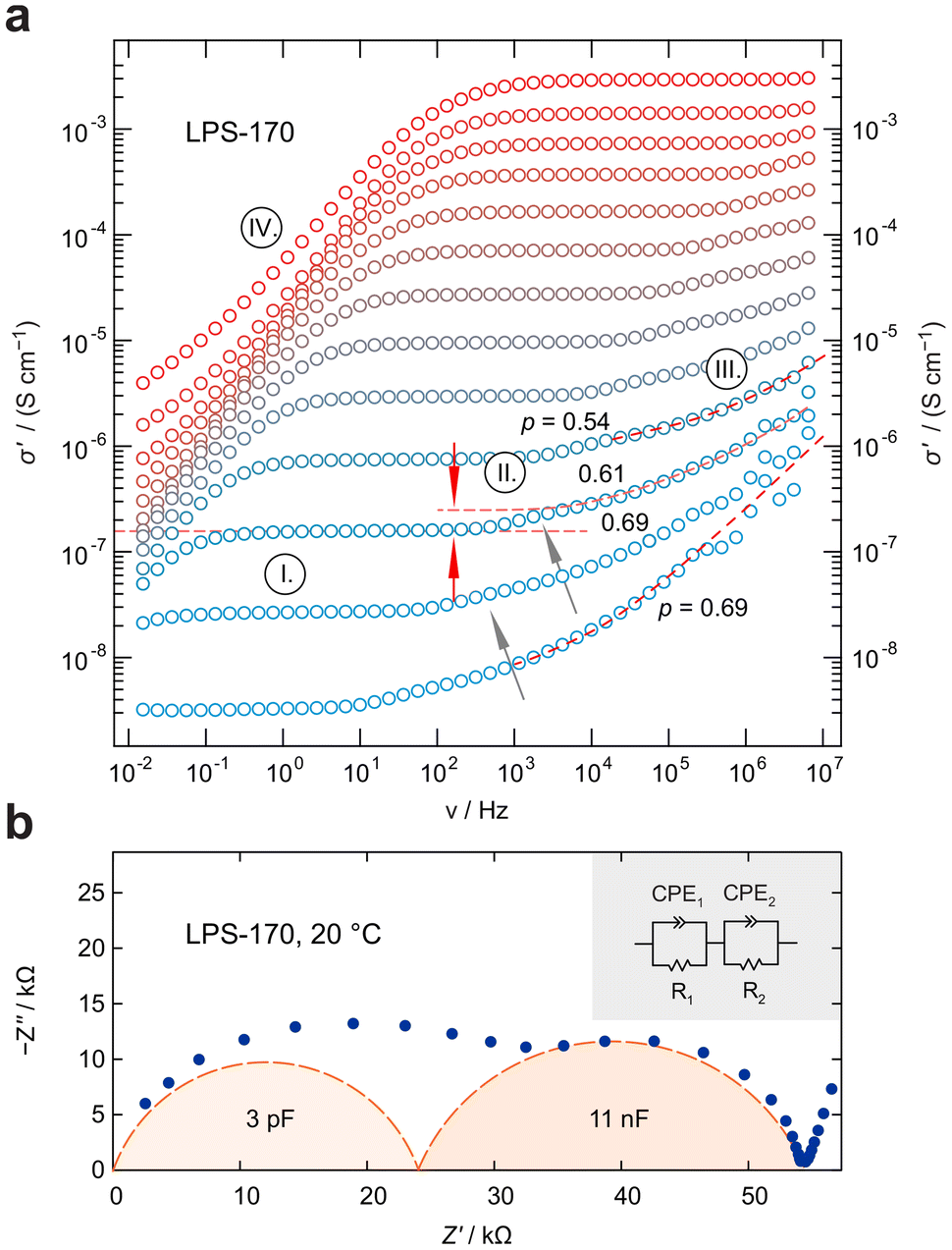 | ||
| Fig. 3 (a) Conductivity isotherms of LPS-170 recorded from −80 °C to 160 °C. The curves are composed of several regions. I shows the overall (DC) conductivity plateau influenced by grain boundary effects. It passes into a shallow dispersive region that finally reaches another plateau (II), which we assign to intragrain Li+ dynamics. The latter plateau is connected to region III, which is the main dispersive part of the isotherms. IV represents electrode polarization due to accumulated charge carriers in regions in front of the ion-blocking electrode. Dashed lines in the dispersive regions show Jonscher fits that yield the exponents indicated. (b) Nyquist plot (complex plane representation of the impedance) of data belonging to LPS-170, recorded at 20 °C. The two depressed semicircles mirror the two conductivity plateaus seen in (a). While capacitances in the order of a few pF are typically found for an electrical bulk response, values in the nF range do, however, point to the influence of grain boundary (g.b.) regions on electrical relaxation. | ||
While only a single plateau is observed in the amorphous samples, the crystalline LPS-170 sample shows two plateaus (see Fig. 3a). Using an appropriate equivalent circuit to simulate the data, we extracted capacitances of 3 pF for the more conductive process at higher frequencies and 11 nF for the less conductive one appearing at lower frequencies (see Fig. 3b). Considering these values, we ascribe the response with a higher conductivity to electrical relaxation in the bulk crystalline regions of the LPS-170 sample. A value of 11 nF indicates processes influenced by g.b. regions or amorphous fractions covering the crystallites.
To gain insight into the dimensionality of ionic transport, we analyzed the dispersive regimes of the conductivity isotherms with Jonscher's power law: σ ∝ νp.53 As pointed out by Sidebottom,54 the exponent p can be used as a first indication of the underlying dimensionality of the transport process in solids. Here, p adopts values ranging from 0.69 to 0.54 possibly suggesting that low-dimensional or at least anisotropic transport processes governing the isotherms at sufficiently low T. However, the Jonscher exponents are at best indicators and should not be interpreted as conclusive evidence for low dimensional charge carrier movements. For nanocrystalline, mechanosynthesised β-Li3PS4 studied by some of us earlier, exponents range from 0.5 to 0.35 and excellently agree with NMR results revealing quasi-1D (zig-zag) diffusion pathways in mechanosynthesised β-Li3PS4.25
To determine activation energies Ea for Li+ conduction in the three samples, we used linear fits to analyse the data shown in the Arrhenius plot of Fig. 2a. All three samples show very similar activation energies, Ea ≈ 0.4 eV and agree well with values reported in previous studies.43,55 While Ea does not change much, the increase in ionic conductivity upon annealing is driven through a rise of the associated Arrhenius pre-exponential factor. The highest ionic conductivity is found for LPS-170 characterized by 0.1 mS cm−1 at room temperature. However, reducing the annealing temperature to only 120 °C does not change the conductivity drastically. This finding suggests that the onset of long-range crystallographic ordering, which might form favourable transport pathways is not the only reason to explain facile transport in this system. Surface morphology effects or the formation of conductive (nano-)crystalline–amorphous interfacial regions might be considered as well to explain the changes detected, see below.
Interestingly, in the case of mechanochemically synthesized β-Li3PS4, amorphous samples often exhibit higher conductivities than their crystalline counterparts.25 In other cases, strongly disordered regions or defects have been shown to block long-range ion transport.20 In the present case, a possible explanation could be the morphology of the β-Li3PS4 particles resulting from solvent-assisted synthesis.41 Recent theoretical studies have shown that the migration pathways with the lowest activation energies are highly anisotropic and are constrained to the a-c plane. On the other hand, inter-plane migration is hindered by higher energy barriers across the PS43− bottleneck, as described by several studies.44,45 If solvent-assisted β-Li3PS4 forms surface structures with preferred orientations along the a-c plane, this behaviour might explain the increased ionic conductivity compared to that seen for samples that have been prepared by mechanochemical means.
To further illustrate the impact of synthesis conditions, that is, annealing temperature  , in Fig. 2b, we compare ionic conductivities from samples prepared by mechanochemistry and obtained by solvent-assisted synthesis. If plotted as a function of
, in Fig. 2b, we compare ionic conductivities from samples prepared by mechanochemistry and obtained by solvent-assisted synthesis. If plotted as a function of  , we observe a broad distribution of reported ionic conductivities for β-Li3PS4 independent of the synthesis method. Our analysis shows that no clear correlation between annealing temperature and ionic conductivity seems to exist, which indicates that phase composition, morphology, crystallinity, and possibly, interfaces all contribute to the overall ionic conductivity of Li3PS4.43 Importantly, the activation energies in most samples (Fig. 2) are remarkably similar and range from 0.35 eV to 0.47 eV. We think that the change in activation energy alone cannot explain the strong variation in conductivity. Instead, we suggest that, as mentioned above, changes of the Arrhenius pre-exponential factor, which is governed by, e.g., the charge carrier density and characteristic attempt frequencies,56 determines the final conductivity to a non-negligible extent. This influence is also seen when comparing the Arrhenius lines in Fig. 2a. Remarkably, as mentioned above, the samples annealed at 120 °C and 170 °C do not show a huge difference in ionic conductivity although 31P MAS NMR and XRPD show clear differences in local and long-range structures. Again, the formation of interconnected X-ray invisible nanocrystals49 or further interfacial/surface effects enabling a network of through-going bulk-type transport might explain this feature, which cannot be simply explained by the degree of crystallinity.
, we observe a broad distribution of reported ionic conductivities for β-Li3PS4 independent of the synthesis method. Our analysis shows that no clear correlation between annealing temperature and ionic conductivity seems to exist, which indicates that phase composition, morphology, crystallinity, and possibly, interfaces all contribute to the overall ionic conductivity of Li3PS4.43 Importantly, the activation energies in most samples (Fig. 2) are remarkably similar and range from 0.35 eV to 0.47 eV. We think that the change in activation energy alone cannot explain the strong variation in conductivity. Instead, we suggest that, as mentioned above, changes of the Arrhenius pre-exponential factor, which is governed by, e.g., the charge carrier density and characteristic attempt frequencies,56 determines the final conductivity to a non-negligible extent. This influence is also seen when comparing the Arrhenius lines in Fig. 2a. Remarkably, as mentioned above, the samples annealed at 120 °C and 170 °C do not show a huge difference in ionic conductivity although 31P MAS NMR and XRPD show clear differences in local and long-range structures. Again, the formation of interconnected X-ray invisible nanocrystals49 or further interfacial/surface effects enabling a network of through-going bulk-type transport might explain this feature, which cannot be simply explained by the degree of crystallinity.
Further analysis of the impedance data of the LPS-170 sample shown in Fig. 4, reveals a discrepancy between conductivity measurements and hopping processes identified by plotting the impedance loss angle as a function of frequency. The loss angle, in agreement with the location curve in the Nyquist representation (Fig. 3b), displays two distinct maxima representing two electrical (relaxation) processes. As can be seen in Fig. 4a, these processes differ in their respective apex frequencies by more than two orders of magnitude irrespective of temperature. At first glance, one would expect a similar difference to emerge also in the conductivity spectra. Indeed, as mentioned above, the conductivity isotherms are composed of two plateaus. The associated change in ionic conductivity is, however, significantly lower than one magnitude. This marked difference might be explained by a change in the number density of charge carriers Nc participating in each process. Indeed, Nc determining the interfacial grain boundary (g.b.) process, which itself depends on the (unknown) extent and geometry of the g.b. regions, is expected to be different as compared to that governing intragrain transport.
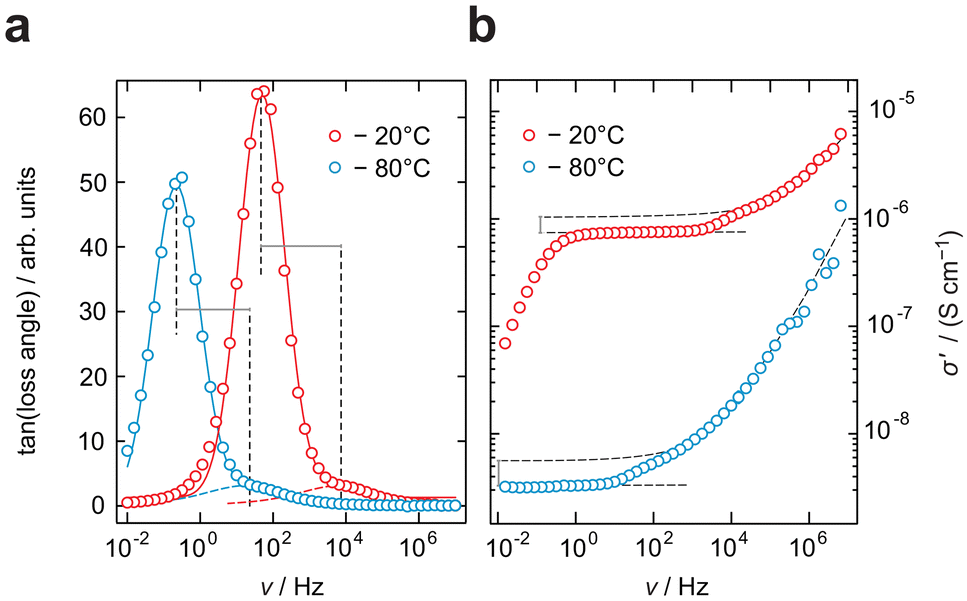 | ||
| Fig. 4 Isotherms of LPS-170 recorded at −20 °C and −80 °C. (a) Tangens of the loss angle against frequency. Two processes are visible with their respective frequencies of maximum loss differing by more than two orders of magnitude. (b) Conductivity isotherms for the same temperatures as depicted in (a). The two electrical processes seen in (a) result, however, in a much lower difference in ionic conductivity. Obviously, the two processes refer to distinct charge carrier densities determining the ionic conductivity of each plateau seen in the respective isotherms. | ||
3.3 Elementary steps of ion hopping probed by NMR
To understand ion hopping seen by NMR and how it connects to long-range ionic transport probed by electrical conductivity measurements, we recorded variable-temperature 7Li and 31P NMR SLR rates57 in both the laboratory (R1) and the rotating frame of reference (R1ρ). Here, we compare results on crystalline LPS-170 with those from our earlier study focussing on mechanochemically prepared nanocrystalline Li3PS4 (LPS-solid).25Fig. 5 shows the temperature-dependent 7Li and 31P NMR relaxation rates of these two samples.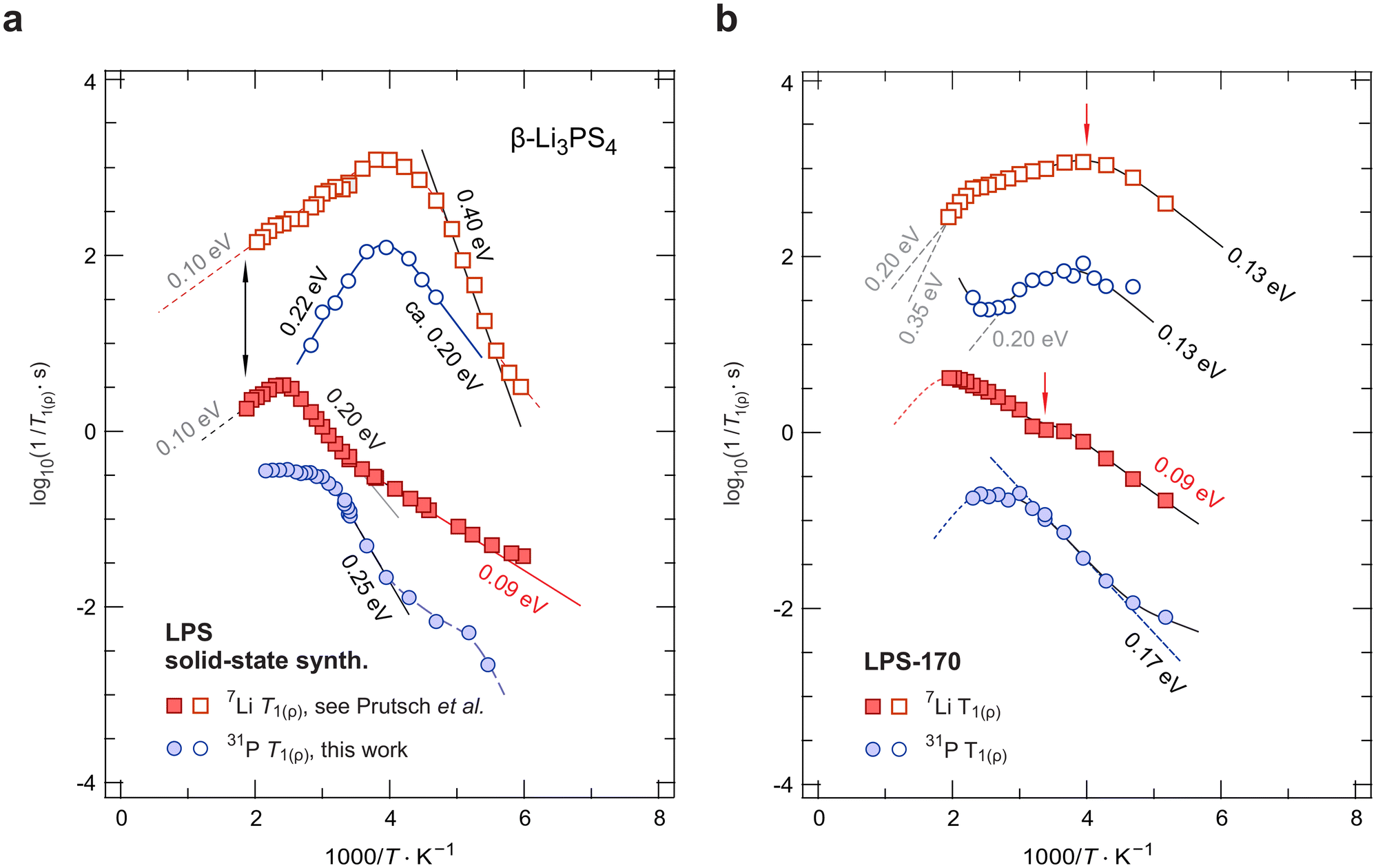 | ||
| Fig. 5 7Li and 31P NMR SLR and spin-lock rates of (a) mechanochemically prepared β-Li3PS4 obtained after an annealing step25 and (b) solvent-assisted β-Li3PS4 prepared in this work. Straight lines (dashed or solid) reflect linear fits used to analyse the slopes (in eV) of the flanks of the diffusion-induced relaxation rate peaks. See text for further details. | ||
To shed further light on Li+ diffusivity in β-Li3PS4, we performed 31P SLR NMR, since the P spins act as helpful local probes that sense magnetic fluctuations in their direct vicinity. These fluctuations can also originate from Li+ hopping processes; hence, 31P SLR NMR is able to indirectly sense the relaxation of the mobile cations. In addition, 31P SLR NMR would be directly affected by any rotational jump processes of the PS43− units themselves.
When coming from low temperatures, the 31P NMR SLR R1 rates (Fig. 5a) seem to strongly depend on temperature. As soon as 7Li R1 enters the flank with 0.09 eV, the 31P NMR rates adopt the same temperature dependence until they pass into a regime that is governed by an activation energy of 0.25 eV, see above. The local maxima seen in 31P SLR NMR might indicate the presence of rotational jump process the PS43− units are involved in. However, such motions are predicted by theory to take place at much higher T.33,45
The corresponding diffusion-induced spin-lock 31P NMR rates R1ρ pass through a peak located at a temperature of 250 K. They sense dynamic processes with correlation rates in the kHz range. The rate peak detected is almost symmetric with activation energies of 0.20 eV and 0.22 eV, respectively (see Fig. 5a). The very similar activation energies observed on the low-T flank of the 31P R1 and R1ρ peaks suggest that, on the average, the same overall process is being sensed by the 31P spins. However, 31P relaxation NMR is unable to see the quasi-1D diffusion process; instead the rates on the high-T side are additionally controlled by further motional processes in β-Li3PS4, which might include rotational jump processes33,45 On the other hand, the 31P NMR R1ρ(1/T) peak appears at the same temperature (250 K) as the corresponding spin-lock 7Li NMR peak. Hence, we conclude that the fluctuations produced by the rapid Li+ translational motions dictate at least the position on the temperature scale rather than the flanks of this peak.
The corresponding spin-lock 7Li NMR rates R1ρ of LPS-170 can be described by two overlapping relaxation peaks located at 250 K and 400 K, respectively. For the peak at low temperatures, an activation energy of approximately 0.13 eV describes the temperature dependence of the rates on the low-T side. This flank runs almost in parallel to the flank sensed by R1 measurements. Obviously, as in the LPS-solid sample, the Li+ ions in β-Li3PS4 have access to hopping pathways with extremely low barriers. At higher T, the rates seem to finally pass into a flank suggesting a steeper slope as compared to the situation in LPS-solid. Activation energies ranging from 0.20 eV to even 0.35 eV seem to characterize this flank. The upper value would be in agreement with that reported by Zhang et al.45 on the basis of ab initio molecular dynamics (AIMD). In summary, we see that the increase in conductivity as found for the solvent-assisted LPS sample, is reflected by 7Li SLR NMR flanks with rather low activation energies. Here, we attribute these low barriers to the rapid quasi-1D (or 2D, within the a-c plane) hopping processes. Consequently, higher ionic conductivities would be expected for morphologies with dominant a-c orientation, such as β-Li3PS4 nano-flakes.60
The same reduction in activation energies is also seen in 31P SLR NMR; the corresponding rates are also included in Fig. 5b. For the rates R1(1/T), we find a single rate peak at temperatures lower than 500 K, whose low-T flank is given by 0.17 eV instead of 0.25 eV as found for LPS-solid, see Fig. 5a. Presumably, this single peak is the result of an average of spin-fluctuations sensed by the 31P spins. In agreement with the decrease in Ea found, only a shallow peak is seen in spin-lock 31P SLR NMR. Again, activation energies of approximately 0.13 eV describe the partly accessible flanks. Most likely, the high-T flanks of the 31P NMR SLR rates R1 and R1ρ do not coincide at higher T, again pointing to strongly anisotropic Li+ diffusivity in LPS-170.
As a final remark, we also observe an increase of the 31P NMR R1ρ relaxation rates at temperatures above 390 K hinting at another NMR flank caused by slower magnetic spin-fluctuations. Considering the temperature range, the start of such a flank would be in agreement with the expectation for a typical high-temperature rotor phase as predicted by Zhang et al.45 Here, at least for the LPS-170 sample, we do not find strong evidence that at lower temperatures fast Li+ hopping is temporally coupled to any rotational jump processes of the PS43− units. For comparison, for the mechanosynthesized, solid-state sample,25 the presence of low-temperature PS43− rotational events, such as large-angle rotational jumps and small-angle wiggling, and their interrelation with Li+ translational dynamics cannot be excluded, see above.
3.4 7Li NMR spin–lattice relaxation modes
For the LPS-170 sample, we found at least two distinct 7Li NMR R1 relaxation modes, which are likely connected to several intragrain diffusion processes. The same two processes have been observed for LPS-solid, as discussed earlier.25 As mentioned above, the one seen in R1 NMR at low temperatures has been interpreted as being governed by the migration activation energy of Li+ hopping along the vacancy-rich zig-zag pathway. The second relaxation mode appearing at elevated T might be the same process but determined by the overall activation energy of this diffusion process, see above. For comparison with our earlier analysis interpreted in terms of quasi-1D diffusion, we re-analysed the 7Li NMR R1(1/T) behaviour with a sum of two relaxation rate peaks that both take 1D diffusion into account.61 For this purpose, we used a spectral density function J that at least contains the limiting cases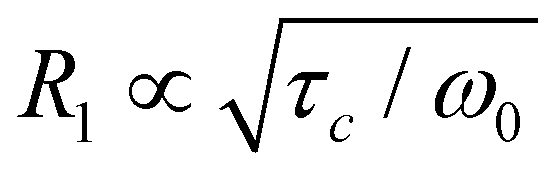 if ω0τc ≪ 1 and R1 ∝ τ−1cω−20 if ω0τc ≫ 1 (or R1 ∝ τ1−βcω−β0 with 1 < β ≤ 2).61–63 If the overall relaxation behaviour is interpreted as a superposition of two 1D peaks (see Fig. S3†), such an analysis yields an even higher activation energy (0.35 eV) for the peak seen at increased T. For comparison, this value of 0.35 eV, which is also close to that seen by spin-lock 7Li R1 NMR (0.40 eV, Fig. 5a), would be in agreement with those predicted by earlier maximum entropy method calculations and AIMD simulations.45,51
if ω0τc ≪ 1 and R1 ∝ τ−1cω−20 if ω0τc ≫ 1 (or R1 ∝ τ1−βcω−β0 with 1 < β ≤ 2).61–63 If the overall relaxation behaviour is interpreted as a superposition of two 1D peaks (see Fig. S3†), such an analysis yields an even higher activation energy (0.35 eV) for the peak seen at increased T. For comparison, this value of 0.35 eV, which is also close to that seen by spin-lock 7Li R1 NMR (0.40 eV, Fig. 5a), would be in agreement with those predicted by earlier maximum entropy method calculations and AIMD simulations.45,51
4 Conclusions
We measured ionic conductivities of β-Li3PS4, which has been prepared by solvent-assisted synthesis with subsequent annealing steps. Upon annealing, a clear increase in long-range ionic transport is observed, which is mainly due to a change of the Arrhenius pre-exponential factor rather than to a significant reduction in activation energy as deduced from conductivity spectroscopy. This change in dynamic parameters, governing the underlying Arrhenius relation, finally results in a conductivity of 0.1 mS cm−1 at room temperature. For comparison, such a high value compares well with results from Liu et al.41 who reported on a high ionic conductivity in nanoporous β-Li3PS4 synthesized by solvent-assisted approaches.In addition to conductivity measurements, we probed Li ion dynamics by diffusion-induced NMR relaxation. NMR, being directly sensitive to the magnetic-dipolar and/or electric quadrupolar spin fluctuations sensed by the diffusing Li ions, clearly points to a series of (length-scale dependent) activation energies governing overall ion dynamics in β-Li3PS4. Importantly, 7Li SLR NMR measurements sense rapid site-to-site Li+ hopping processes in the sample that has been annealed at elevated temperatures. The corresponding NMR activation energies of this sample turned out to be clearly lower than those determining Li+ hopping in a reference sample prepared by mechanochemical means and subsequent annealing. This lowering of hopping barriers probed by NMR agrees with the increase in Li+ dynamics detected by conductivity measurements.
Here, we only observe a slight increase in conductivity when comparing the X-ray amorphous sample annealed at 120 °C with the crystalline one obtained after annealing at 170 °C. Hence, the resulting conductivity of these two samples is hardly affected by the degree of overall crystallinity seen by XRPD. This observation is in strong contrast to earlier studies that report on significant changes.28 Here, we think that a single parameter, such as the extent of crystallinity, is not sufficient to fully describe the dynamic picture governing ionic conductivity. Instead, we conclude that long-range ion transport is affected by multiple factors including interfacial composition, surface morphology, the presence of nanocrystalline regions and phase composition, in particular.
Finally, 31P SLR NMR supports our conclusions drawn from 7Li SLR NMR. As compared to a recent study using maximum entropy methods and AIMD calculations,45 we could not find a strong influence of polyanion rotations on translational Li+ dynamics for the annealed sample LPS-170, at least for the low-temperature regime covered here. 31P SLR NMR of the sample prepared by solid-state synthesis shows that the 31P spins do not only sense the magnetic fluctuations produced by the 7Li spins in their direct vicinity. Instead, the different activation energies seen for 31P as compared to 7Li as well as the shift and appearance of (local) 31P SLR NMR rate maxima towards lower temperatures points to additional relaxation mechanism the 31P spins are involved in. These additional sources for longitudinal relaxation might indeed originate from rotational jump processes of the PS43− units.
Author contributions
Jonas Spychala: investigation (NMR), formal analysis, writing – original review & editing. Christoph Mandl: resources (sample preparation), formal analysis, investigation. Katharina Hogrefe: investigation (NMR). H. Martin R. Wilkening: supervision, writing – review & editing. Bernhard Gadermaier: conceptualization, supervision, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.Acknowledgements
We thank the Österreichische Forschungsförderungsgesellschaft mbH (FFG) for financial support in the frame of the CERES project (No. F44905). In addition, we thank the DFG for financial support (NMR and impedance spectrometers of the former research unit 1277).References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- R. H. Brugge, A. K. O. Hekselman, A. Cavallaro, F. M. Pesci, R. J. Chater, J. A. Kilner and A. Aguadero, Chem. Mater., 2018, 30, 3704–3713 CrossRef CAS.
- M. P. Stockham, B. Dong and P. R. Slater, J. Solid State Chem., 2022, 308, 122944 CrossRef CAS.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- C. Yu, F. Zhao, J. Luo, L. Zhang and X. Sun, Nano Energy, 2021, 83, 105858 CrossRef CAS.
- J. Lau, R. H. DeBlock, D. M. Butts, D. S. Ashby, C. S. Choi and B. S. Dunn, Adv. Energy Mater., 2018, 8, 1800933 CrossRef.
- S. Wang, Q. Bai, A. M. Nolan, Y. Liu, S. Gong, Q. Sun and Y. Mo, Angew. Chem., Int. Ed., 2019, 58, 8039–8043 CrossRef CAS.
- S. Ohno, A. Banik, G. F. Dewald, M. A. Kraft, T. Krauskopf, N. Minafra, P. Till, M. Weiss and W. G. Zeier, Prog. Energy, 2020, 2, 022001 CrossRef.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 855–856 CrossRef PubMed.
- I. Hanghofer, B. Gadermaier, A. Wilkening, D. Rettenwander and H. M. R. Wilkening, Dalton Trans., 2019, 48, 9376–9387 RSC.
- C. Yu, F. Zhao, J. Luo, L. Zhang and X. Sun, Nano Energy, 2021, 83, 105858 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS.
- L. Schweiger, K. Hogrefe, B. Gadermaier, J. L. M. Rupp and H. M. R. Wilkening, J. Am. Chem. Soc., 2022, 144, 9597–9609 CrossRef CAS PubMed.
- I. Hanghofer, M. Brinek, S. L. Eisbacher, B. Bitschnau, M. Volck, V. Hennige, I. Hanzu, D. Rettenwander and H. M. Wilkening, Phys. Chem. Chem. Phys., 2019, 21, 8489–8507 RSC.
- P. Adeli, J. D. Bazak, K. H. Park, I. Kochetkov, A. Huq, G. R. Goward and L. F. Nazar, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS PubMed.
- N. J. J. de Klerk, I. Rosłoń and M. Wagemaker, Chem. Mater., 2016, 28, 7955–7963 CrossRef CAS.
- M. Tachez, J. Malugani, R. Mercier and G. Robert, Solid State Ionics, 1984, 14, 181–185 CrossRef CAS.
- D. Prutsch, B. Gadermaier, H. Brandstätter, V. Pregartner, B. Stanje, D. Wohlmuth, V. Epp, D. Rettenwander, I. Hanzu and H. M. R. Wilkening, Chem. Mater., 2018, 30, 7575–7586 CrossRef CAS.
- K. Hayamizu, Y. Aihara, T. Watanabe, T. Yamada, S. Ito and N. Machida, Solid State Ionics, 2016, 285, 51–58 CrossRef CAS.
- H. Tsukasaki, S. Mori, S. Shiotani and H. Yamamura, Solid State Ionics, 2018, 317, 122–126 CrossRef CAS.
- N. H. H. Phuc, K. Morikawa, M. Totani, H. Muto and A. Matsuda, Solid State Ionics, 2016, 285, 2–5 CrossRef CAS.
- S. J. Sedlmaier, S. Indris, C. Dietrich, M. Yavuz, C. Dräger, F. von Seggern, H. Sommer and J. Janek, Chem. Mater., 2017, 29, 1830–1835 CrossRef CAS.
- A. Jodlbauer, J. Spychala, K. Hogrefe, B. Gadermaier and H. M. R. Wilkening, Chem. Mater., 2024, 1648–1664 CrossRef CAS.
- A. Hayashi, S. Hama, H. Morimoto, M. Tatsumisago and T. Minami, J. Am. Ceram. Soc., 2001, 84, 477–479 CrossRef CAS.
- F. Mizuno, A. Hayashi, K. Tadanaga and M. Tatsumisago, Adv. Mater., 2005, 17, 918–921 CrossRef CAS.
- J. G. Smith and D. J. Siegel, Nat. Commun., 2020, 11, 1483 CrossRef CAS.
- M. Tatsumisago and A. Hayashi, Solid State Ionics, 2012, 225, 342–345 CrossRef CAS.
- J. Trevey, J. S. Jang, Y. S. Jung, C. R. Stoldt and S.-H. Lee, Electrochem. Commun., 2009, 11, 1830–1833 CrossRef CAS.
- B. Gao, R. Jalem and Y. Tateyama, ACS Appl. Mater. Interfaces, 2021, 13, 11765–11773 CrossRef CAS.
- D. Xie, S. Chen, Z. Zhang, J. Ren, L. Yao, L. Wu, X. Yao and X. Xu, J. Power Sources, 2018, 389, 140–147 CrossRef CAS.
- A. Hayashi, K. Minami, S. Ujiie and M. Tatsumisago, J. Non-Cryst. Solids, 2010, 356, 2670–2673 CrossRef CAS.
- K. Wissel, A. Haben, K. Küster, U. Starke, R. Kautenburger, W. Ensinger and O. Clemens, Adv. Energy Sustainability Res., 2024, 5, 2300280 CrossRef CAS.
- K. Homma, M. Yonemura, T. Kobayashi, M. Nagao, M. Hirayama and R. Kanno, Solid State Ionics, 2011, 182, 53–58 CrossRef CAS.
- Z. Liu, W. Fu, E. A. Payzant, X. Yu, Z. Wu, N. J. Dudney, J. Kiggans, K. Hong, A. J. Rondinone and C. Liang, J. Am. Chem. Soc., 2013, 135, 975–978 CrossRef CAS PubMed.
- K. Wissel, L. M. Riegger, C. Schneider, A. I. Waidha, T. Famprikis, Y. Ikeda, B. Grabowski, R. E. Dinnebier, B. V. Lotsch, J. Janek, W. Ensinger and O. Clemens, ACS Appl. Energy Mater., 2023, 6, 7790–7802 CrossRef CAS.
- F. Marchini, B. Porcheron, G. Rousse, L. Albero Blanquer, L. Droguet, D. Foix, T. Koç, M. Deschamps and J. M. Tarascon, Adv. Energy Mater., 2021, 11, 2101111 CrossRef CAS.
- Y. Chen, L. Cai, Z. Liu, C. R. dela Cruz, C. Liang and K. An, Appl. Phys. Lett., 2015, 107, 013904 CrossRef.
- Z. Zhang, H. Li, K. Kaup, L. Zhou, P.-N. Roy and L. F. Nazar, Matter, 2020, 2, 1667–1684 CrossRef.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- P. M. Richards, Solid State Commun., 1978, 25, 1019–1021 CrossRef.
- C. A. Sholl, J. Phys. C: Solid State Phys., 1981, 14, 447–464 CrossRef CAS.
- H. Tsukasaki, S. Mori, H. Morimoto, A. Hayashi and M. Tatsumisago, Sci. Rep., 2017, 7, 4142 CrossRef.
- R. Mercier, J.-P. Malugani, B. Fahys, G. Robert and J. Douglade, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1887–1890 CrossRef.
- H. Stöffler, T. Zinkevich, M. Yavuz, A. Senyshyn, J. Kulisch, P. Hartmann, T. Adermann, S. Randau, F. H. Richter, J. Janek, S. Indris and H. Ehrenberg, J. Phys. Chem. C, 2018, 122, 15954–15965 CrossRef.
- M. Gobet, S. Greenbaum, G. Sahu and C. Liang, Chem. Mater., 2014, 26, 3558–3564 CrossRef CAS.
- A. K. Jonscher, Nature, 1977, 267, 673–679 CrossRef CAS.
- D. L. Sidebottom, Phys. Rev. Lett., 1999, 83, 983–986 CrossRef CAS.
- F. N. Forrester, J. A. Quirk, T. Famprikis and J. A. Dawson, Chem. Mater., 2022, 34, 10561–10571 CrossRef CAS PubMed.
- S. Breuer and M. Wilkening, Dalton Trans., 2018, 47, 4105–4117 RSC.
- F. Preishuber-Pflügl and M. Wilkening, Dalton Trans., 2016, 45, 8675–8687 RSC.
- Y. Yang, Q. Wu, Y. Cui, Y. Chen, S. Shi, R.-Z. Wang and H. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25229–25242 CrossRef CAS PubMed.
- M.-S. Lim and S.-H. Jhi, Curr. Appl. Phys., 2018, 18, 541–545 CrossRef.
- H. Wang, Z. D. Hood, Y. Xia and C. Liang, J. Mater. Chem. A, 2016, 4, 8091–8096 RSC.
- P. M. Richards, Topics in Current Physics, Springer, 1979, ch. 20, vol. 15, p. 813 Search PubMed.
- K. Volgmann, V. Epp, J. Langer, B. Stanje, J. Heine, S. Nakhal, M. Lerch, M. Wilkening and P. Heitjans, Z. Phys. Chem., 2017, 231, 1215–1241 CrossRef CAS.
- F. Stainer and H. M. R. Wilkening, Phys. Rev. B, 2024, 109, 174304 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld refinement, conductivity isotherms, analysis of NMR relaxation data with a 1D model. See DOI: https://doi.org/10.1039/d4dt02636e |
| This journal is © The Royal Society of Chemistry 2025 |