
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation and characterization of the dimetal decacarbonyl dication [Ru2(CO)10]2+ and the metal-only Lewis-pair [Ag{Ru(CO)5}2]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) †
†
Malte
Sellin
 a,
Jörg
Grunenberg
b and
Ingo
Krossing
*a
a,
Jörg
Grunenberg
b and
Ingo
Krossing
*a
aInstitut für Anorganische und Analytische Chemie und Freiburger Materialforschungszentrum (FMF), Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bInstitut für Organische Chemie; Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
First published on 6th December 2024
Abstract
The reaction of Ag+ with Ru3(CO)12 in a CO atmosphere under concommittant irradiation with UV-light yields a salt of the metal-only Lewis-pair [Ag{Ru(CO)5}2]+. Switching the silver cation for a more process-selective deelectronator yields a salt of the homoleptic transition metal carbonyl cation [Ru2(CO)10]2+, which fills the gap between the known cations [Ru(CO)6]2+ and [Ru3(CO)14]2+. The amount of π-backdonation in this series was studied by a combination of vibrational spectroscopy and computed relaxed force constants.
Introduction
Carbon monoxide (CO) is an excellent π-acceptor and a moderate σ-donor ligand. With this combination, transition metal carbonyls (TMCs) are amongst the most fundamental complex classes in organometallic chemistry.1,2 Their electronic properties are ideal for the stabilisation of unusually low formal oxidation states of transition metals down to −IV.3By contrast, the stabilization of metal cations exclusively by carbon monoxide ligands needs the strict absence of nucleophiles. Therefore, the first homoleptic transition metal carbonyl cation (TMCC) – [Mn(CO)6]+ – was not isolated until 1961 by Fischer et al.4 Even after that, little progress on this kind of compounds was made until the 1990s, when the groups of Willner and Aubke introduced superacidic systems such as HF/SbF5 or FSO3H/SbF5 to isolate various closed-shell TMCCs up to the oxidation state + III.5–7 Most of these TMCCs are “non-classical” carbonyl complexes in which σ-donation is stronger than π-backdonation, yielding positively polarized (non-classical) carbonyl ligands, which have a higher force-constant than uncoordinated carbon monoxide.2,6,8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡
‡
Over the last years, our group prepared9 several missing TMCCs by the reaction of binary TMCs with deelectronators (also known as one-electron oxidants)§ such as [NO]+, Ag+ or synergistic Ag+/0.5 I2 yielding the open-shell TMCCs [Ni(CO)4]˙+,10 [M(CO)6]˙+ (M = Cr, Mo, W)11 and the heptacarbonyls [M(CO)7]+ (M = Nb, Ta).12 However, often other oxidation paths (e.g. halonium addition) were observed besides the desired deelectronation.§ Thus, we developed selective deelectronation reagents first on the basis of fluorinated ammoniumyl,13 then aromatic radical cations,14–17e.g. [anthraceneHal]˙+ (anthraceneHal = C14F8Cl2) with a formal potential of +1.42 V vs. ferrocene. Using these reagents, even strongly Lewis-basic TMCs like W(CO)6 and Fe(CO)5 can be selectively deelectronated and isolated as their respective radical-cation salts.15,18,19
Attempts to isolate the heavier homologues of the radical cation [Fe(CO)5]˙+ by the reaction of the respective trimetal dodecacarbonyls with [anthraceneHal]˙+ under carbon monoxide atmosphere yielded the clustered TMCCs [M3(CO)14]2+ (M = Ru, Os) instead.14 These complexes are members of the even smaller family of homomultinuclear TMCCs, with the only other examples being [Pt2(CO)6]2+20 and [Hg2(CO)2]2+.21 By contrast, a series of heteromultinuclear TMCCs forms by combining TMCs with coinage metals and yields the class of metal-only Lewis pair (MOLP)22 cations [M{TMC}2]+, e.g. [M{Fe(CO)5}2]+, [Ag{M′3(CO)12}2]+ (M = Cu, Ag, Au; M′ = Ru, Os; Fig. 1).14,23–27
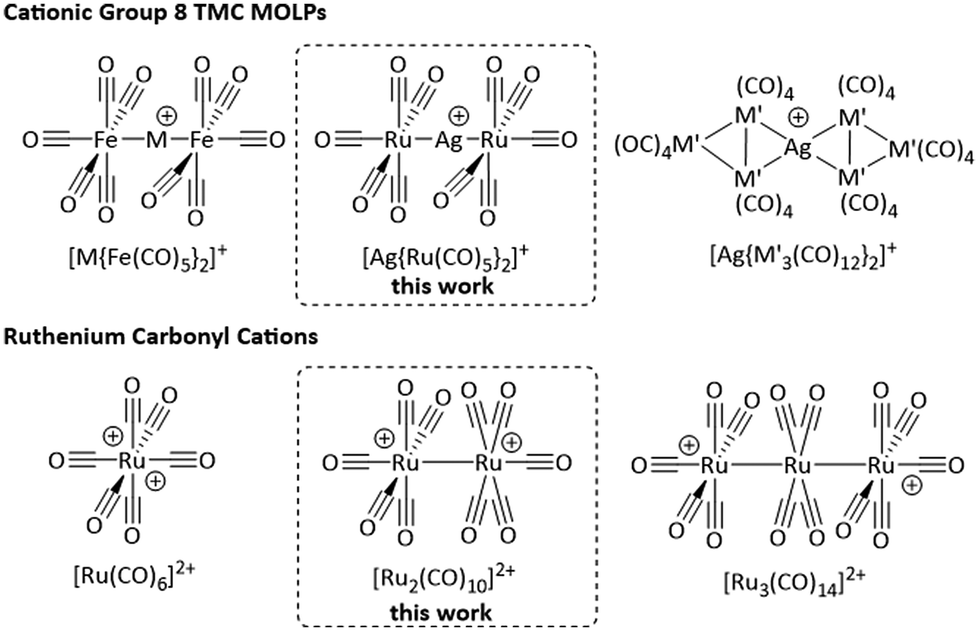 | ||
| Fig. 1 Overview to the cationic group 8 TMC metal-only Lewis pairs (MOLPs; M = Cu, Ag, Au; M′ = Ru, Os) and the ruthenium carbonyl cations in comparison to the MOLP based TMCC [Ag{Ru(CO)5}2]+ and the homodinuclear TMCC [Ru2(CO)10]2+ from this work. | ||
Results and discussion
Herein, we report the light-induced reaction of the cluster Ru3(CO)12 to the metal-only Lewis-pair [Ag{Ru(CO)5}2]+ and the all-RuI transition metal carbonyl cation [Ru2(CO)10]2+, filling the gap between superelectrophilic [Ru(CO)6]2+ and the [Ru3(CO)14]2+ cluster.14,28Synthesis and characterisation of [Ag{Ru(CO)5}2]+
We previously reported that the silver(I) cation reacts with Ru3(CO)12 as a Lewis-acid instead as an oxidant and forms [Ag{Ru3(CO)12}2]+.14 Repeating the same reaction with an excess of silver(I) under irradiation with UV-light (370 nm) and carbon monoxide pressure (3 bar), the silver(I) cation still did not react as an oxidation reagent, but rather formed [Ag{Ru(CO)5}2]+ (eqn (1); [F{Al(ORF)3}2]− counterion).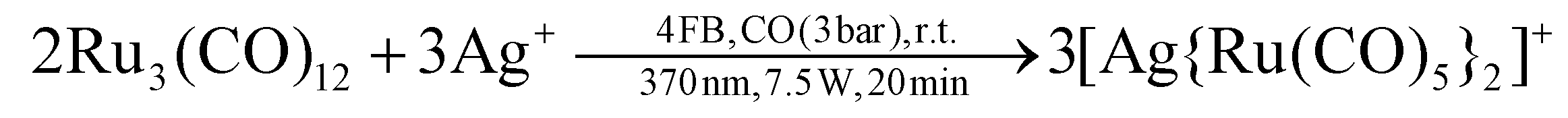 | (1) |
Layering the colourless 4FB solution (4FB = 1,2,3,4-tetrafluorobenzene) with n-pentane led to the formation of a 89% yield of colourless plates suitable for single crystal X-ray diffraction studies of [Ag{Ru(CO)5}2]+[F{Al(ORF)3}2]− (RF = C(CF3)3; Fig. 2). Related to complexes between iron pentacarbonyl and metal cations [M{Fe(CO)5}2]x+ (M = Cu+, Ag+, Au+, Hg2+), the silver(I) cation binds directly to the metal centre of the pentacarbonyl complex.24–27,29 In comparison to the nearly linear Fe–M–Fe cores, the Ru–Ag–Ru axis is with 171.7° significantly bent, which could be a consequence of weaker secondary Ag⋯C contacts due to the larger Ag–Ru distance. Still, the Ag–Ru distances are with 2.682(2) Å in [Ag{Ru(CO)5}2]+ significantly shorter than in [Ag{Ru3(CO)12}2]+ (2.887(1) Å).14 This bending also leads to a lowering of the symmetry and a splitting/doubling of all IR bands in comparison to the optimized D4h symmetric structure.
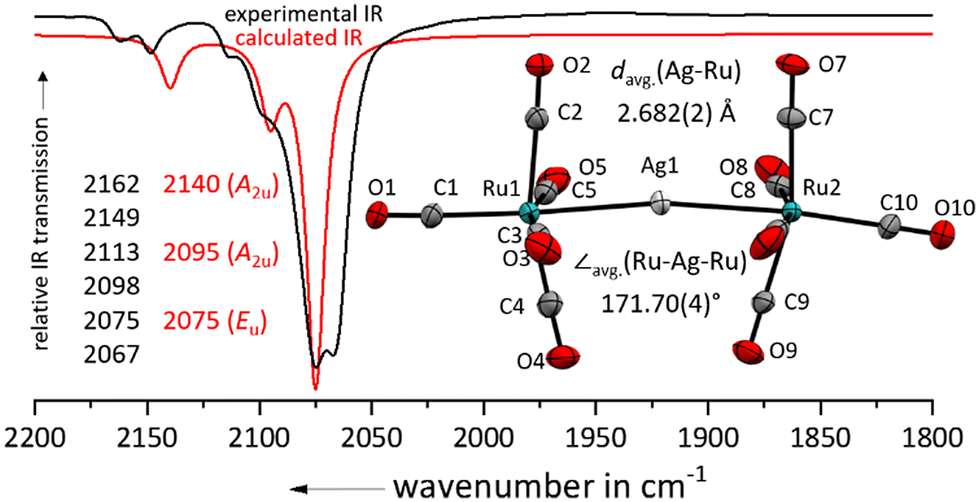 | ||
| Fig. 2 Experimental ATR-IR spectrum of [Ag{Ru(CO)5}2]+[F{Al(ORF)3}2]− (black line) in comparison with the and calculated IR spectrum of [Ag{Ru(CO)5}2]+ (red line, B3LYP(D3-BJ)/def2-TZVPP, scaled by 0.968 according to Duncan et al.31). Note the doubling of all lines by symmetry lowering induced by bending. Solid-state structure of the complex cation in [Ru2(CO)10]2+([F{Al(ORF)3}2]−)2·(4FB)2. Ellipsoids depicted at 50% probability; colour code: silver – light grey, ruthenium – turquoise, oxygen – red, carbon – light grey. | ||
We may speculate that the [Ag{Ru(CO)5}2]+ cation could serve in the future as a synthon for the highly unstable Ru(CO)5, as the Fe(CO)5 in the closely related [Ag{Fe(CO)5}2]+ can be transmetalated to copper using CuBr.29
Synthesis and characterisation of [Ru2(CO)10]2+
Since we aimed for a homonuclear TMCC, we switched to halogenated arenium radical cations as more selective deelectronators. Initial attempts to generate the [Ru2(CO)10]2+ dication by the reaction of Ru3(CO)12¶ with an excess of the deelectronator [anthraceneHal]˙+ under carbon monoxide atmosphere only yielded [Ru3(CO)14]2+, unreacted [anthraceneHal]˙+ and [Ru2(CO)10]2+ as a side-product. Even exchanging [anthraceneHal]˙+ for the far more powerful [naphthaleneF]˙+ deelectronation reagent (naphthaleneF = C10F8; +2.00 V vs. Fc+/0)15,16 led to the same result.However, when a 4FB solution of Ru3(CO)12 and [anthraceneHal]˙+[WCA]− (WCA = [Al(ORF)4]−, [F{Al(ORF)3}2]−) is irradiated with UV-light (370 nm) under carbon monoxide pressure (3 bar), the respective [Ru2(CO)10]2+([WCA]−)2 complex salts forms as a major product besides some minor unknown side-products in 66% overall yield (eqn (2); [Al(ORF)4]−/[F{Al(ORF)3}2]− counterion).||
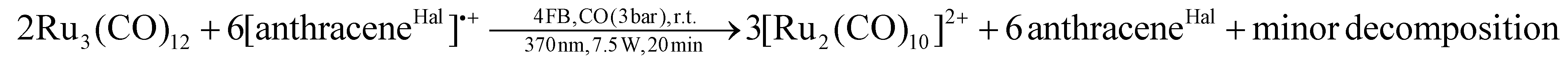 | (2) |
Layering the solution with n-pentane led to the formation of colourless needles suitable for single crystal X-ray diffraction studies. The solid-state structure of the salt [Ru2(CO)10]2+([F{Al(ORF)3}2]−)2·(4FB)2 shows, that the geometry of the complex dication is close to the expected D4d symmetry (Fig. 3) with a torsion angle between the two sets of equatorial carbonyl ligands of only 36° compared to the ideal 45°.
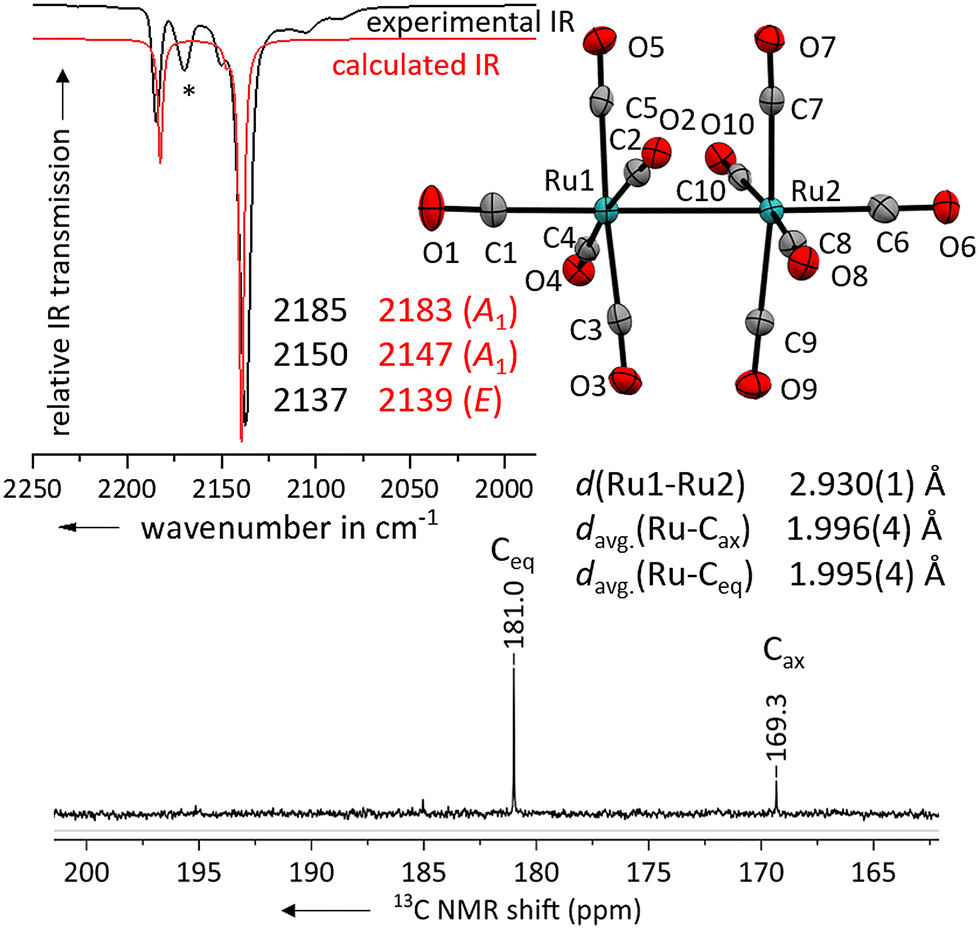 | ||
| Fig. 3 Experimental ATR-IR spectrum of [Ru2(CO)10]2+([Al(ORF)4]−)2 (black line, impurity marked with (*)) in comparison with the calculated IR spectrum of [Ru2(CO)10]2+ (red line, B3LYP(D3-BJ)/def2-TZVPP, scaled by 0.968 according to Duncan et al.31). Solid-state structure of the complex cation in [Ru2(CO)10]2+([F{Al(ORF)3}2]−)2·(4FB)2. Ellipsoids depicted at 50% probability; colour code: ruthenium – turquoise, oxygen – red, carbon – grey. Carbonyl region of the 13C NMR spectrum of [Ru2(CO)10]2+([F{Al(ORF)3}2]−)2 in 4FB. | ||
The Ru–Ru bond distance of [Ru2(CO)10]2+ is with 2.930(1) Å slightly longer than the one in the [Ru3(CO)14]2+ cluster with 2.891(1) Å and that in neutral Ru3(CO)12 at 2.844(2) Å.30 The average C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O distances of the axial carbonyl ligands are to some extent longer than the ones of the equatorial carbonyl ligands. However, as the elongation of the CO bond upon π-backdonation is comparably low, this effect is not significant in relation to the higher errors in the scXRD.
O distances of the axial carbonyl ligands are to some extent longer than the ones of the equatorial carbonyl ligands. However, as the elongation of the CO bond upon π-backdonation is comparably low, this effect is not significant in relation to the higher errors in the scXRD.
To further investigate the electronic properties of the carbonyl ligands, we measured a 13C NMR spectrum of [Ru2(CO)10]2+ ([F{Al(ORF)3}2]−)2 in 4FB. The shift of the 13C peak is well suited to compare the electronic situation between different carbonyl ligands bound to the same transition metal, but not in between TMCCs having different transition metals.9 The peak for the axial carbonyl ligands is with 169.3 ppm already close to the one observed in [Ru(CO)6]2+ (168.8 ppm).7 This indicates, that the axial carbonyl ligands in [Ru2(CO)10]2+ are positively polarized (non-classical). The equatorial carbonyl ligands are more electron-rich with a 13C NMR shift of 181.0 ppm and electronically in-between the axial and equatorial carbonyl ligands of [Ru3(CO)14]2+ (171.6 & 185.1 ppm).14
The three bands assigned to the CO-stretching vibrations of [Ru2(CO)10]2+ are in excellent agreement to the DFT-calculated values (B3LYP(D3-BJ)/def2-TZVPP, scaled by 0.968 according to Duncan et al.,31Fig. 3). Unfortunately, no Raman spectrum could be obtained due to the strong fluorescence induced by minor (photo-)decomposition products.
Computational investigations on [Ru2(CO)10]2+
As a computational measure, which directly distinguishes between classical and non-classical interactions of carbonyl ligands with the metal, we calculated the QTAIM charges (B3LYP(D3-BJ)/def2-TZVPP) of Ru(CO)5 and the ruthenium carbonyl cations. The QTAIM charges are in line with the 13C NMR shifts throughout the entire series from Ru(CO)5 to [Ru(CO)6]2+: with the increasing positive partial charge of the carbonyl ligand, the 13C NMR shift is more high-field. Both the equatorial and axial carbonyl ligands are positively charged, thus making the complex a non-classical TMCC. As already observed in the [Ru3(CO)14]2+ cluster, the interaction of the metal centre with the axial carbonyl ligands are less classical than the equatorial ones (decrease of the π-backdonation). In contrast to the relatively small differences of the 13C NMR shifts of the axial carbonyl ligand of [Ru2(CO)10]2+, the carbonyl ligands in [Ru(CO)6]2+ are nearly twice as positively charged (Fig. 4 and Table 1).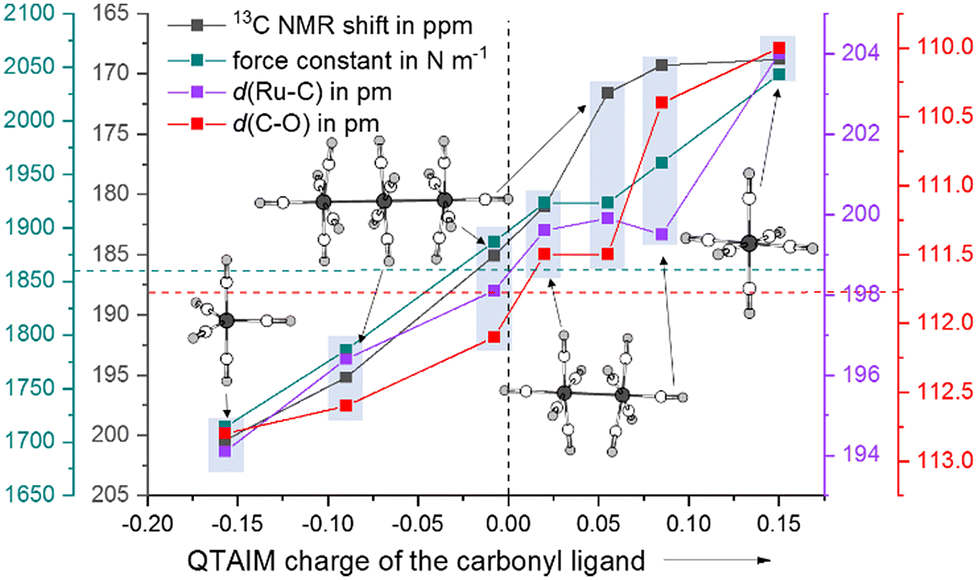 | ||
| Fig. 4 Comparison of the experimental data and calculated force-constants of the carbonyl ligands of Ru(CO)5, [Ru3(CO)14]2+, [Ru2(CO)10]2+ and [Ru(CO)6]2+ as a function on their QTAIM charges. The dotted lines are showing the data of free carbon monoxide. | ||
| [Ru3(CO)14]2+ | [Ru2(CO)10]2+ | [Ru(CO)6]2+ | |
|---|---|---|---|
| ṽ(CO) (IR) in cm−1 | 2173 | 2185 | 2198 |
| 2139 | 2150 | ||
| 2114 | 2137 | ||
| 2058 | |||
| δ 13CCO in ppm | 171.5 (ax) | 169.3 (ax) | 168.8 |
| 185.0 (eq) | 181.0 (eq) | ||
| 195.1 (cen) | |||
| QTAIM charge | +0.055 (ax) | +0.085 (ax) | +0.150 |
| −0.008 (eq) | +0.020 (eq) | ||
| −0.090 (cen) | |||
| k CO in N m−1 | 1.923 (ax) | 1.961 (ax) | 2.043 |
| 1.887 (eq) | 1.923 (eq) | ||
| 1.786 (cen) |
As we were not able to collect a Raman spectrum of the [Ru2(CO)10]2+ complex, the determination of the force-constants from the experimental data would have been inaccurate and would have relied on multiple assumptions. Therefore, we instead computationally determined the full set of relaxed force-constants for the ruthenium carbonyl series using the TPSSH/def2-TZVPP level of theory. Since the values of complete matrix of relaxed force constants do not depend on both, the chosen coordinate system and any assumption concerning the coupling terms, they can be directly used as unique bond strength descriptors.32 As expected, both the equatorial and the axial sets of carbonyl ligands in [Ru2(CO)10]2+ have force constants (1.923 & 1.961 N m−1) higher than the one of uncoordinated carbon monoxide (1.856 N m−1).
Energetics of the dimerization of [Ru(CO)5]˙+ to [Ru2(CO)10]2+
While the [Ru2(CO)10]2+ dimer is expected to be preferred against the monomeric 17 VE radical cation [Ru(CO)5]˙+ according to the isolobal principle, this result stands in contrast to its lighter homologue [Fe(CO)5]˙+,18 which exists as the open-shell monomer under the same conditions. In agreement with this observation, DFT-calculations (B3LYP(D3-BJ)/def2-TZVPP) show, that the dimerization of [M(CO)5]˙+ to [M2(CO)10]2+ is strongly endergonic in the gas-phase for both iron and ruthenium (Δr,(g)G° = + 289.0 kJ mol−1 (Fe)/+171.6 kJ mol−1 (Ru)) due to the high Coulomb repulsion between the cationic monomers. However, the situation is very different, when the solvation of 4FB is considered, giving Δr,(4FB)G° = +101.8 kJ mol−1 (Fe)/−9.8 kJ mol−1 (Ru) upon inclusion of a COSMO-RS solvation model,33 explaining both the open-shell nature of [Fe(CO)5]˙+ and the dimeric form of [Ru2(CO)10]2+ (eqn (3)).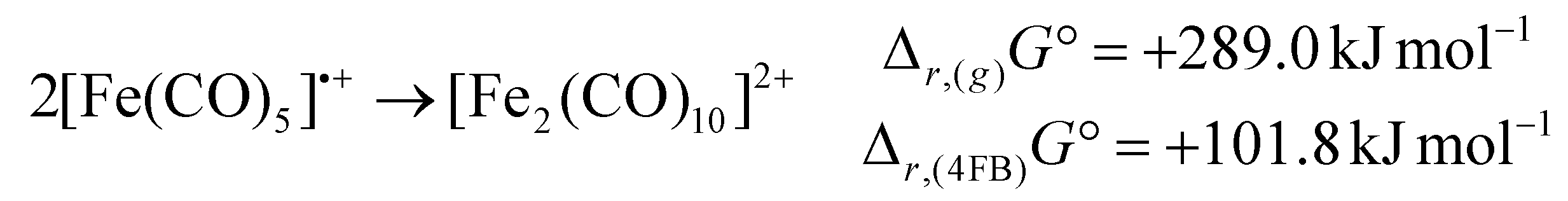 | (3a) |
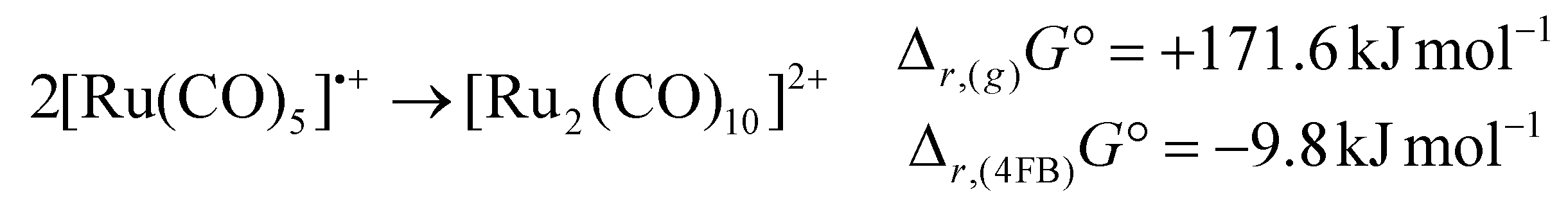 | (3b) |
Conclusions
In summary, we report the isolation and characterization of the MOLP-based TMCC [Ag{Ru(CO)5}2]+ through the reaction of Ru3(CO)12 and Ag(I) under CO pressure and irradiation with UV-light. Switching the silver(I) cation in this reaction for [anthraceneHal]˙+, the TMCC [Ru2(CO)10]2+ forms. This complex is the missing link between [Ru(CO)6]2+ and [Ru3(CO)14]2+. Additionally, this complex can be seen as the dimerized heavier homologue of the recently prepared [Fe(CO)5]˙+.18 Both experimental and quantum-chemical investigations show, that the axial carbonyl ligands are almost as electron-poor as the carbonyl ligands in the superelectrophilic [Ru(CO)6]2+ and the relevant trends of this new series are plotted in Fig. 4.Experimental
General prodedures
All manipulations were carried out by using standard Schlenk technique or a nitrogen filled glovebox (O2/H2O < 0.1 ppm). All the reactions were performed in Schlenk tubes with grease free PTFE-valves. The solvent 1,2,3,4-tetrafluorobenzene (4FB, C6F4H2, from fluorochem) was stirred a few days over calcium hydride (CaH2) and distilled. The distillate was stirred over Ag+[Al(ORF)4]− and condensed to remove traces of less fluorinated benzenes. This leads to a minor contamination of RFOH (≪1%), which does not affect the reactions. n-Pentane was dried using a Grubbs apparatus. 4FB, and n-pentane were stored over 3 Å molar sieves. Octafluoronaphthalene (ABCR), 9,10-dichlorooctafluoroanthrace (Sigma Aldrich) and triruthenium dodecacarbonyl (Sigma Aldrich) were bought from commercial sources. [NO]+[Al(ORF)4]−,34 Ag+[F{Al(ORF)3}2]−, [NO]+[F{Al(ORF)3}2]−35 and [naphthaleneF]+·[F{Al(ORF)3}2]−15 were prepared according to literature procedures.Vibrational spectroscopy
FTIR spectra were recorded inside a glovebox with a Bruker ALPHA equipped with QuickSnap Eco ATR module and ZnSe crystal. The spectra were measured at RT in the range of 4000–550 cm−1 with 32 scans and a resolution of 4 cm−1. The data were processed with the Bruker OPUS 7.5 software package. The intensities are reported as follows: ≥0.8 = very strong (vs), ≥0.6 = strong (s), ≥0.4 = medium (m), ≥0.2 = weak (w), <0.2 = very weak (vw). The data were processed with the Bruker OPUS 7.5 software package. The graphical representations were created with ORIGINPRO 2021.NMR spectroscopy
NMR spectra were recorded at RT on a Bruker Avance DPX 200 MHz. The samples were dissolved in 4FB (0.6 ml) in a 5 mm thick-walled NMR tube with J. Young PTFE valve. The spectra were calibrated by using the 1H signal of the solvent 4FB (δ = 6.97 ppm, rel. to tetramethylsilane). The field corrections of other nuclei were adjusted accordingly. The MestReNova software package was used for measuring, processing and creation of the graphical representations of the spectra. 1H and 13C NMR spectra are referenced against TMS and 19F NMR spectra against CFCl3.Single crystal X-ray diffraction
The data were collected on a Bruker D8 VENTURE dual wavelength Mo/Cu three-circle diffractometer with a microfocus sealed X-ray tube using mirror optics as monochromator and a Bruker PHOTON III detector. Single crystals were selected at RT in PFPE oil JC 1800 (Sunoit Performance Material Science), mounted on CryoLoops with a diameter of 0.1 to 0.2 mm and shock-cooled using an Oxford Cryostream 800 low temperature device. The data were gathered at 100(2) K using Mo Kα radiation (λ = 0.71073 Å). All data were integrated with SAINT (version 8.38A) and a multi-scan absorption correction using SADABS or TWINABS was applied. The structures were solved by direct methods using SHELXT36 and refined by full-matrix least-squares methods against F2 by SHELXL-2018/337 using the GUI software ShelXle.38 Disordered moieties were refined using bond lengths restraints and displacement parameter restraints and were modelled with the program DSR.39 The gathered data were finalized with the tool FinalCif.40 The graphical representations of the crystal structures were generated with Mercury (version 4.0).41 Crystallographic data for the structures reported in this paper have been deposited with the CCDC (2390017 & 2390232†).42Computational details
Geometry optimizations were performed with the TURBOMOLE software43 (v7.2 or v7.5) using the DFT functionals B3LYP44 with the def2-TZVPP45 basis set, the resolution-of-identity (RI) approximation,46 dispersion correction (D3-BJ),47 a fine integration grid (m4) and the default SCF convergence criteria (10−6 a.u.). All structures were checked for proper spin occupancies and imaginary frequencies with the integrated EIGER and AOFORCE48 modules. IR and Raman spectra were simulated at B3LYP(D3BJ)/def2-TZVPP level with a scaling factor of 0.9657,49 for transition metal carbonyls, the specialized scaling factor of 0.968 was used.31 Gibbs free energies of solvation were calculated with the COSMO-RS model50 at the BP86(D3)/def2-TZVPD//BP86(D3)/def-TZVP level of theory using the fine cavity construction algorithm ($cosmo_isorad) and the CosmoThermX software. Relaxed force constants were calculated as diagonal elements of the compliance matrix on the TPSSH/def2-TZVPP level of theory. Evaluating several modern DFT methods the TPSSH functional, additional relying on the electron kinetic energy density, seems to evenly describe the electronic coupling in organometallic compounds.51 Transformation of the cartesian DFT force constant matrix into internal coordinates was achieved using the freely available COMPLIANCE 3.0.2. code. Provided that a Cartesian Hessian matrix is available as input, all internal force constants for arbitrary atom–atom pairs, as well as their couplings, can be calculated intuitively using a graphical interface.52UV-setup
The Samples were irradiated with a custom-built UV-lamp. A UV-LED strip (370 nm, 47 mW per LED, ca. 2.8 m) was glued in a spiral form in a PVC tube with a diameter of 10 cm (Fig. 5). This LED setup has a power of 40 W and a light intensity equivalent to ca. 7.5 W. For irradiation durations of over a few minutes, a cooling system is required. This set-up was placed on a stirring plate and the Schlenk-tube inside the lamp, to allow uniform irradiation. | ||
| Fig. 5 Photograph of the UV-setup. | ||
Synthetic procedures
ATR IR (ZnSe) ṽ/cm−1 = 2162 (vw), 2148 (vw), 2113 (vw), 2098 (sh), 2075 (w), 2067 (w), 1524 (vw), 1508 (vw), 1354 (vw), 1300 (vw), 1276 (w), 1266 (w), 1240 (vs), 1214 (vs), 1177 (w), 1052 (vw), 973 (vs), 863 (vw), 826 (vw), 811 (vw), 760 (vw), 750 (vw), 727 (s), 684 (vw), 637 (vw), 578 (vw), 567 (w).
ATR IR (ZnSe) ṽ/cm−1 = 2184 (vw), 2170 (vw), 2150 (vw), 2137 (m), 2105 (vw), 2088 (vw), 1513 (vw), 1447 (vw), 1353 (vw), 1296 (w), 1272 (w), 1264 (w), 1240 (m), 1207 (vs), 1190 (w), 1176 (w), 1126 (vw), 1089 (vw), 1080 (vw), 1068 (vw), 1046 (vw), 967 (vs), 844 (vw), 834 (vw), 756 (vw), 744 (vw), 726 (s), 712 (vw), 682 (vw), 627 (vw), 566 (w), 558 (w).
1H-NMR (400 MHz, 4FB, 298 K): only solvent and minor impurities.
13C-NMR (50 MHz, 4FB, 298 K): δ = 181.0 (s, 8C, [Ru2(CO)10]2+ equatorial), 169.3 (s, 2C, [Ru2(CO)10]2+ axial), 120.7 (q, 36C, anion – OC(CF3)3), 78.4 (m, 12C, anion – OC(CF3)3) ppm.
19F-NMR (188 MHz, 4FB, 298 K): δ = −76.2 (s, 108F, anion – OC(CF3)3), −184.8 (s, 2F, anion – Al-F-Al) ppm.
Author contributions
MS performed all the synthetic and analytical work and wrote the manuscript together with IK. JG performed the DFT calculations for the determination of the force constants and added these parts to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for [Ag{Ru(CO)5}2]+[F{Al(ORF)3}2]− and [Ru2(CO)10]2+([F{Al(ORF)3}2]−)2·(4FB)2 has been deposited at the CCDC 2390232 and 2390017.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 350173756 and the ERC Advanced Grant InnoChem (101052935). M. S. is grateful for a PhD fellowship from the “Fonds der Chemischen Industrie FCI”.References
- E. W. Abel and F. G. A. Stone, Q. Rev., Chem. Soc., 1970, 24, 498 CAS.
- A. J. Lupinetti, S. H. Strauss and G. Frenking, in Progress in Inorganic Chemistry 2001, pp. 1–112 Search PubMed.
- (a) J. E. Ellis and R. A. Faltynek, J. Am. Chem. Soc., 1977, 99, 1801 CAS; (b) J. E. Ellis, K. L. Fjare and T. G. Hay, J. Am. Chem. Soc., 1981, 103, 6100 CAS; (c) J. T. Lin, G. P. Hagen and J. E. Ellis, J. Am. Chem. Soc., 1983, 105, 2296 CAS.
- E. O. Fischer, K. Fichtel and K. Öfele, Chem. Ber., 1962, 95, 249 CrossRef CAS.
- (a) C. Bach, H. Willner, F. Aubke, C. Wang, S. J. Rettig and J. Trotter, Angew. Chem., Int. Ed. Engl., 1996, 35, 1974 CrossRef CAS; (b) E. Bernhardt, B. Bley, R. Wartchow, H. Willner, E. Bill, P. Kuhn, I. H. T. Sham, M. Bodenbinder, R. Bröchler and F. Aubke, J. Am. Chem. Soc., 1999, 121, 7188 CrossRef CAS.
- H. Willner and F. Aubke, Angew. Chem., Int. Ed. Engl., 1997, 36, 2402 CrossRef CAS.
- E. Bernhardt, C. Bach, B. Bley, R. Wartchow, U. Westphal, I. H. T. Sham, B. von Ahsen, C. Wang, H. Willner, R. C. Thompson and F. Aubke, Inorg. Chem., 2005, 44, 4189 CrossRef CAS PubMed.
- (a) A. J. Lupinetti, G. Frenking and S. H. Strauss, Angew. Chem., Int. Ed., 1998, 37, 2113 CrossRef CAS; (b) H. Willner and F. Aubke, Chem. – Eur. J., 2003, 9, 1668 CrossRef CAS; (c) H. Willner and F. Aubke, Organometallics, 2003, 22, 3612 CrossRef CAS.
- M. Sellin and I. Krossing, Acc. Chem. Res., 2023, 56, 2776 CrossRef CAS.
- M. Schmitt, M. Mayländer, J. Goost, S. Richert and I. Krossing, Angew. Chem., Int. Ed., 2021, 60, 14800 CrossRef CAS PubMed.
- (a) J. Bohnenberger, W. Feuerstein, D. Himmel, M. Daub, F. Breher and I. Krossing, Nat. Commun., 2019, 10, 624 CrossRef CAS PubMed; (b) J. Bohnenberger, M. Schmitt, W. Feuerstein, I. Krummenacher, B. Butschke, J. Czajka, P. J. Malinowski, F. Breher and I. Krossing, Chem. Sci., 2020, 11, 3592 RSC.
- W. Unkrig, M. Schmitt, D. Kratzert, D. Himmel and I. Krossing, Nat. Chem., 2020, 12, 647 CrossRef CAS.
- M. Schorpp, T. Heizmann, M. Schmucker, S. Rein, S. Weber and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 9453 CrossRef CAS PubMed.
- M. Sellin, C. Friedmann, M. Mayländer, S. Richert and I. Krossing, Chem. Sci., 2022, 13, 9147 RSC.
- M. Sellin, J. Willrett, D. Röhner, T. Heizmann, J. Fischer, M. Seiler, C. Holzmann, T. A. Engesser, V. Radtke and I. Krossing, Angew. Chem., Int. Ed., 2024, e202406742 CAS.
- C. Armbruster, M. Sellin, M. Seiler, T. Würz, F. Oesten, M. Schmucker, T. Sterbak, J. Fischer, V. Radtke, J. Hunger and I. Krossing, Nat. Commun., 2024, 15, 6721 CrossRef CAS.
- M. Sellin, M. Seiler, M. Mayländer, K. Kloiber, V. Radke, S. Weber, S. Richert and I. Krossing, Chem. – Eur. J., 2023, 29, e202300909 CrossRef CAS.
- J. M. Rall, M. Schorpp, M. Keilwerth, M. Mayländer, C. Friedmann, M. Daub, S. Richert, K. Meyer and I. Krossing, Angew. Chem., Int. Ed., 2022, e202204080 CAS.
- J. M. Rall, L. Nork, T. A. Engesser, M. Mayländer, S. Weber, S. Richert and I. Krossing, Chem. – Eur. J., 2024, 30, e202400105 CrossRef CAS PubMed.
- (a) Q. Xu, B. T. Heaton, C. Jacob, K. Mogi, Y. Ichihashi, Y. Souma, K. Kanamori and T. Eguchi, J. Am. Chem. Soc., 2000, 122, 6862 CrossRef CAS; (b) Q. Xu, Y. Souma, B. T. Heaton, C. Jacob and K. Kanamori, Angew. Chem., Int. Ed., 2000, 39, 208 CrossRef CAS.
- M. Bodenbinder, G. Balzer-Jöllenbeck, H. Willner, R. J. Batchelor, F. W. B. Einstein, C. Wang and F. Aubke, Inorg. Chem., 1996, 35, 82 CrossRef CAS.
- (a) J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS; (b) B. R. Barnett, C. E. Moore, P. Chandrasekaran, S. Sproules, A. L. Rheingold, S. DeBeer and J. S. Figueroa, Chem. Sci., 2015, 6, 7169 RSC.
- (a) M. Sellin, M. Seiler and I. Krossing, Chem. – Eur. J., 2023, e202300908 CrossRef CAS PubMed; (b) J. Bohnenberger, D. Kratzert, S. Pan, G. Frenking and I. Krossing, Chem. – Eur. J., 2020, 26, 17203 CrossRef CAS.
- P. J. Malinowski and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 13460 CrossRef CAS.
- S. M. Rupf, S. Pan, A. L. Moshtaha, G. Frenking and M. Malischewski, J. Am. Chem. Soc., 2023, 145(28), 15353–15359 CrossRef CAS PubMed.
- G. Wang, Y. S. Ceylan, T. R. Cundari and H. V. R. Dias, J. Am. Chem. Soc., 2017, 139, 14292 CrossRef CAS PubMed.
- S. Pan, S. M. N. V. T. Gorantla, D. Parasar, H. V. R. Dias and G. Frenking, Chem. – Eur. J., 2021, 27, 6936 CrossRef CAS.
- C. Wang, B. Bley, G. Balzer-Jöllenbeck, A. R. Lewis, S. C. Siu, H. Willner and F. Aubke, J. Chem. Soc., Chem. Commun., 1995, 2071 RSC.
- V. Zhuravlev and P. J. Malinowski, Eur. J. Inorg. Chem., 2023, 26, e202300177 CrossRef CAS.
- C. Slebodnick, J. Zhao, R. Angel, B. E. Hanson, Y. Song, Z. Liu and R. J. Hemley, Inorg. Chem., 2004, 43, 5245 CrossRef CAS PubMed.
- M. K. Assefa, J. L. Devera, A. D. Brathwaite, J. D. Mosley and M. A. Duncan, Chem. Phys. Lett., 2015, 640, 175 CrossRef CAS.
- (a) L. H. Jones and B. I. Swanson, Acc. Chem. Res., 1976, 9, 128 CrossRef CAS; (b) J. J. Turner and J. A. Timney, J. Mol. Spectrosc., 2022, 387, 111662 CrossRef CAS; (c) K. Brandhorst and J. Grunenberg, Chem. Soc. Rev., 2008, 37, 1558 RSC.
- C. C. Pye, T. Ziegler, E. van Lenthe and J. N. Louwen, Can. J. Chem., 2009, 87, 790 CrossRef CAS.
- P. J. Malinowski, T. Jaroń, M. Domańska, J. M. Slattery, M. Schmitt and I. Krossing, Dalton Trans., 2020, 49, 7766 RSC.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- (a) D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933 CrossRef CAS; (b) D. Kratzert and I. Krossing, J. Appl. Crystallogr., 2018, 51, 928 CrossRef CAS.
- FinalCif, https://www.xs3.uni-freiburg.de/research/finalcif.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS.
- (a) M. von Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746 CrossRef CAS; (b) O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B Condens. Matter, 1988, 37, 785 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136 CrossRef CAS; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC; (c) R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 5119 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511 CrossRef CAS.
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, J. Phys. Chem. A, 2015, 119, 1701 CrossRef CAS PubMed.
- (a) A. Klamt, J. Phys. Chem., 1995, 99, 2224 CrossRef CAS; (b) A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074 CrossRef CAS.
- (a) K. P. Jensen, Inorg. Chem., 2008, 47, 10357 CrossRef CAS PubMed; (b) J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- K. Brandhorst and J. Grunenberg, J. Chem. Phys., 2010, 132, 184101 CrossRef.
- The IUPAC Compendium of Chemical Terminology, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019 Search PubMed.
- W. Manchot and W. J. Manchot, Z. Anorg. Allg. Chem., 1936, 226, 385 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2390232 and 2390017. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03364g |
| ‡ We use the subdivision of (non-)classical not for the entire complex, but separated for the interaction between the transition metal and each spectroscopically independent carbonyl ligand.9,14 |
| § The deelectronation is a special-case of the oxidation reaction and describes ‘a complete net removal of one or more electrons from a molecular entity’ (https://doi.org/10.1351/goldbook.O04362).53 |
| ¶ Monomeric Ru(CO)5 was not used, because it is only obtainable by a high-pressure carbonylation of ruthenium or Ru3(CO)12. Due to its rapid loss of CO to form Ru2(CO)9 and finally Ru3(CO)12, it is not commercially available.54 |
| || Using an excess of Ru3(CO)12 under the same conditions unfortunately does not yield higher clusters such as [Ru4(CO)18]2+, but [Ru3(CO)14]2+ and polymeric {Ru(CO)4}. |
| This journal is © The Royal Society of Chemistry 2025 |