
Bicyclic (alkyl)(amino)carbenes (BICAACs): synthesis, characteristics, and applications
Ankita
Sharma
a,
Unnikrishnan
Nair K
ab and
Subrata
Kundu
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110 016, India. E-mail: subrata@iitd.ac.in
bDepartment of Chemistry, Ashoka University, Sonepat, Haryana-131029, India
First published on 8th November 2024
Abstract
Carbenes in general and isolable NHCs (N-heterocyclic carbenes) in particular have been useful ligands in recent years. The emergence of CAACs [cyclic(alkyl)(amino)carbenes], BICAACs [bicyclic(alkyl)(amino)carbenes], and many other carbenes has marked revolutionary milestones in this field. These carbenes possess an intriguing blend of highly electrophilic and nucleophilic characteristics, owing to their remarkably narrow HOMO–LUMO energy gap. The isolation and characterization of these carbenes hold significance not only due to their fascinating electronic properties but have demonstrated their prowess across various domains, including isolation of transition metal complexes, medicinal applications, catalysis, and radical stabilization. While the chemistry of 5-membered NHCs and CAACs has been extensively explored, the investigation of BICAACs has just begun. This review covers the synthesis, characterization, and reactivity of BICAACs and outlines the diverse applications of BICAACs in organometallic chemistry, metal-free catalysis, and main-group chemistry.
 Ankita Sharma | Ankita Sharma was born in Noida, Uttar Pradesh, India. She did her graduation in Chemistry (Hons) in 2018 from Miranda House, University of Delhi, and Master's from the Indian Institute of Technology Delhi in 2021. Currently she is pursuing her Ph.D. under the supervision of Dr Subrata Kundu at the Indian Institute of Technology Delhi. |
 Unnikrishnan Nair K | Unnikrishnan Nair K completed his M.Sc. in Analytical Chemistry from the University of Kerala in 2021. In December 2022, he joined Dr Subrata Kundu's research group at the Indian Institute of Technology Delhi as a project assistant. Presently, he serves as a project associate in the research group of Dr Munmun Ghosh at Ashoka University, Haryana. |
 Subrata Kundu | Subrata Kundu was born in Bankura, West Bengal, India. He received his Ph.D. degree in 2015 from the Indian Institute of Technology Kanpur under the guidance of Prof. V. Chandrasekhar and Prof. Sandeep Verma. After a couple of post-doctoral positions in the research groups of Prof. H. W. Roesky and Prof. Ian Manners, in December 2019, Subrata joined IIT Delhi as an assistant professor. Presently, he is working in the area of low-valent main-group chemistry. |
1. Introduction
Carbenes were thought to be laboratory curiosities until the first isolable carbene, {bis(diisopropylamino)phosphino}(trimethylsilyl) carbene, was prepared by Bertrand et al. in 1988.1 This remarkable compound was successfully isolated in liquid form and stabilized through favourable interactions with adjacent heteroatoms [P and Si], resulting from double-bond formation.1 In 1991, Arduengo et al. reported an isolable singlet carbene, which was characterized by the single crystal XRD technique for the first time.2 This pivotal achievement has paved the way for the isolation of many other types of singlet carbenes.3 Among the isolated carbenes, the so-called N-heterocyclic carbenes (NHCs) have found applications in medicine and materials science.4–6 In 2005, Bertrand et al. reported a new family of stable five-membered carbenes [cyclic(alkyl)(amino)carbenes (CAAC)], which result from the replacement of one of the two amino substituents of classical N-heterocyclic carbenes (NHCs) by a quaternary carbon atom.7 CAACs exhibit superior electrophilic and nucleophilic characteristics compared to NHCs.8Later, in 2017, Bertrand's group introduced bicyclic(alkyl)(amino)carbenes (BICAACs) with a bicyclo[2.2.2]octane framework.9 Furthermore, BICAACs have superior σ-donating and π-accepting characteristics in comparison with CAACs and consequently NHCs. Quantum chemical calculations reveal that among the carbene ligands whose chemistry has been explored to a decent extent, BICAACs have high energy HOMOs (higher nucleophilicity and more σ-donating) and low energy LUMOs (higher electrophilicity and more π-accepting). The singlet–triplet energy gaps (ΔES/T) in BICAACs are smaller than those in NHCs and slightly smaller than those in 5-membered CAACs (Fig. 1).9
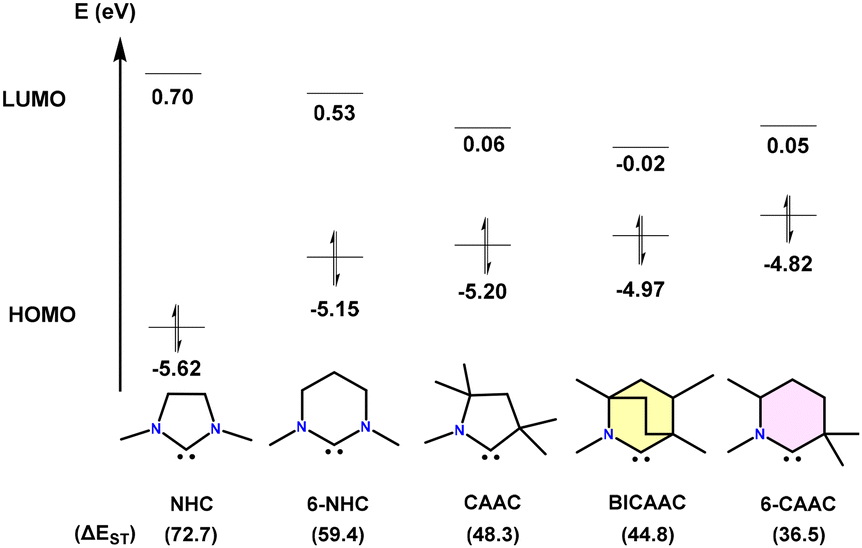 | ||
| Fig. 1 HOMO–LUMO gap (eV) and ΔES/T (kcal mol−1) of NHC, 6-NHC, CAAC, BICAAC, and 6-CAAC (calculated at the B3LYP/def2-TZVPP level of theory). | ||
Today, carbenes are among the most powerful tools in chemistry, used in materials science, catalysis, and the isolation of compounds in their lower oxidation states.19–23 Furthermore, in numerous instances, it has been observed that CAACs and BICAACs are very effective in stabilizing paramagnetic species, activating small molecules, and forming enthalpically strong bonds due to their superior electronic and steric characteristics.19 Comparative studies of the reactivity of several stable carbenes have been conducted in recent years, and it has been found that, frequently, CAACs and BICAACs exhibit different reactivity from NHCs. Furthermore, CAAC and BICAAC coordinated catalysts frequently outperformed their NHC-coordinated catalyst counterparts.19a–k
The chemistry of NHCs and five-membered CAACs is extensively covered in numerous reviews and books; therefore, we have not included CAAC-related works in this review.3,24–31 Since the chemistry of BICAACs is relatively new and lacks a dedicated review despite its reported applicability on various occasions, we focus on reports related to BICAACs. Additionally, the relevant properties of IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene), CAACs and 6-CAACs (6-CAAC = six-membered cyclic(alkyl)(amino)carbene) have been included on a few occasions for comparative purposes. The content of this review has been divided into the following sections: (1) Introduction, (2) Synthesis of BICAACs, (3) Evaluation of the electronic properties of BICAACs, (4) BICAAC metal complexes, (4) BICAACs as metal-free catalysts, (5) BICAAC-supported main-group compounds, and (6) Conclusions.
Various notations have been used in the literature to depict the bond between BICAACs and a central atom. In this review, we have chosen to use an arrow for consistency. However, in cases where significant π-back donation occurs, we followed the literature and used a double bond.
2. Synthesis of BICAACs
In 2017, Bertrand and co-workers reported the synthesis of BICAACs, which is presented in Scheme 1.9 The reaction of trivertal and 2,6-diisopropylaniline under acidic conditions formed an imine overnight. This imine, when treated with nBuLi at −78 °C and then alkylated with methyl chloride, produced an alkylated trivertal imine. Cyclization of the alkylated trivertal imine under anhydrous acidic conditions, followed by anion exchange with sodium tetrafluoroborate, provided the iminium salts [1a-H][BF4] and [1b-H][BF4]. Deprotonation of [1a-H][BF4] using KHMDS generated a free BICAAC (1a). The free carbene 1a was found to be stable in the solid state when stored under an inert atmosphere. Moreover, it was structurally characterized using single crystal X-ray diffraction. The yield of BICAACs for multi-gram synthesis from the primary starting materials varies from 15 to 33%. The 13C{1H} NMR spectrum of 1a showed a singlet at 334 ppm, which is deshielded compared to CAAC (320 ppm).7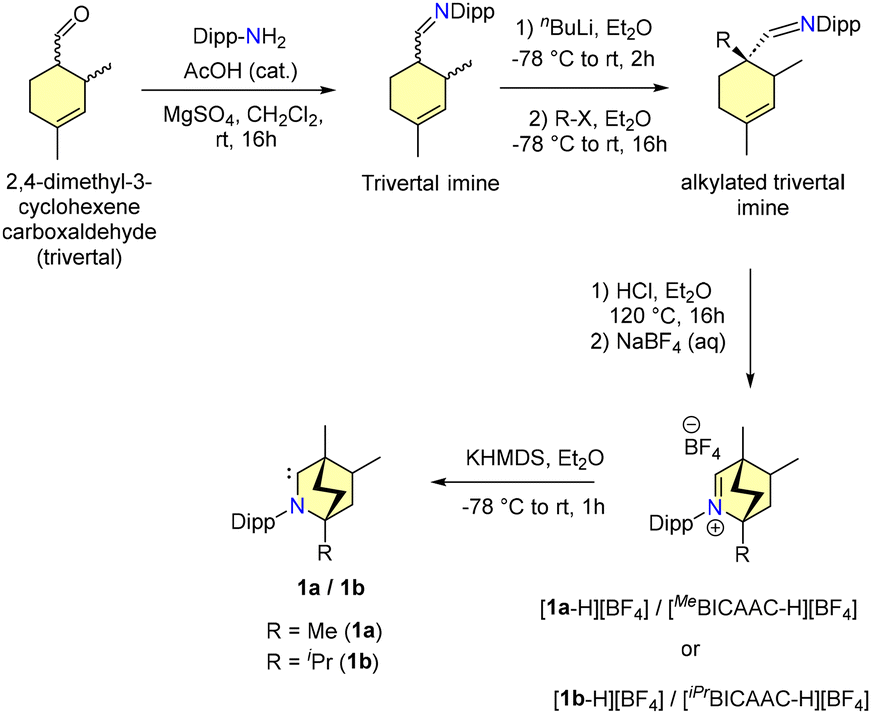 | ||
| Scheme 1 Synthesis of a BICAAC. | ||
3. Evaluation of the electronic properties of BICAACs
As discussed in the Introduction, the BICAAC's highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) have higher and lower energies, respectively, than CAACs and imidazole-based carbenes. This is also demonstrated by a considerable decrease in the singlet–triplet gap from 49.2 kcal mol−1 for CAACs to 45.7 kcal mol−1 for BICAACs for the DFT optimized model compounds as discussed in Fig. 1.26 BICAACs have higher σ-donation and π-acceptance than CAACs, possibly due to their broader carbene bond angle (106.9° for CAAC vs. 110.2° for BICAAC). The 6-CAAC has a 117.9° carbene bond angle.10 It should be noted that increasing the carbene bond angle of NHCs leads to higher p-character in both the HOMO and LUMO, resulting in a smaller ΔEST.32Tolman electronic parameters (TEPs) provide valuable information for comparing the electron-donating abilities of carbenes. TEPs were developed for the evaluation of the electron-donating ability of phosphines in phosphine coordinated metal-carbonyl complexes (by measuring the infrared stretching frequencies of carbonyl ligands) and were also used to determine the overall electron-donating ability of carbene ligands. Accordingly, TEPs were obtained from various square planar complexes, [(carbene)Rh(CO)2Cl]. It was found that the overall electron donation ability of Ad6-CAAC is better than that of MeBICAAC. However, MeBICAAC demonstrates superior donor ability to iPrNHC, cyCAAC, and acyclic diaminocarbene (iPrADCs) (Fig. 2).8–14
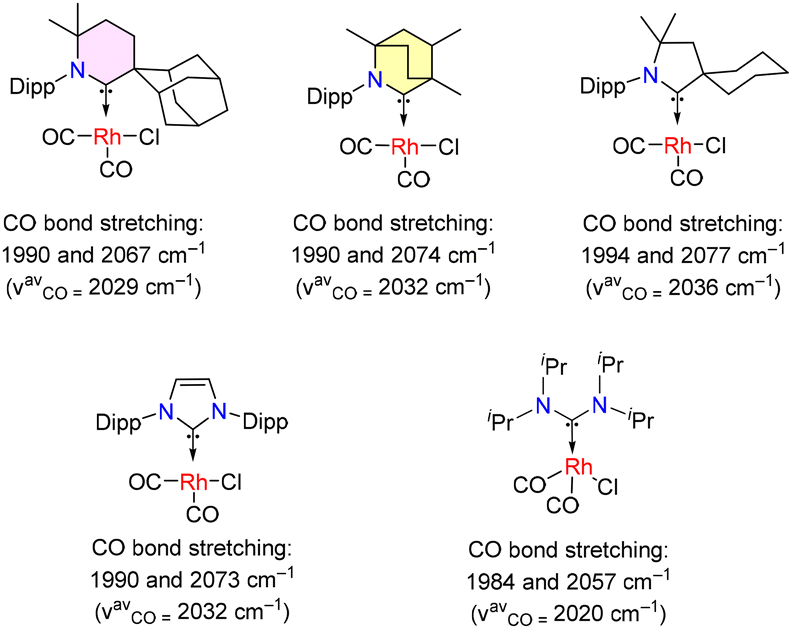 | ||
| Fig. 2 C–O stretching frequencies of carbene coordinated Rh(CO)2Cl complexes. | ||
Later, Jazzar and Bertrand et al. reported an excellent strategy to determine the basicity of carbenes by a proton exchange reaction between a free carbene and a carbene conjugate acid.15 In this reaction, the equilibrium will move in the direction of the more basic carbene, forming the corresponding conjugate acid, which could be monitored by 13C{1H} NMR.
Carbenes were found to be best placed in the sequence 6-CAAC > BICAAC > CAAC > NHC in strong connection with their theoretically expected basicity after completing a few strategic reactions (Scheme 2).
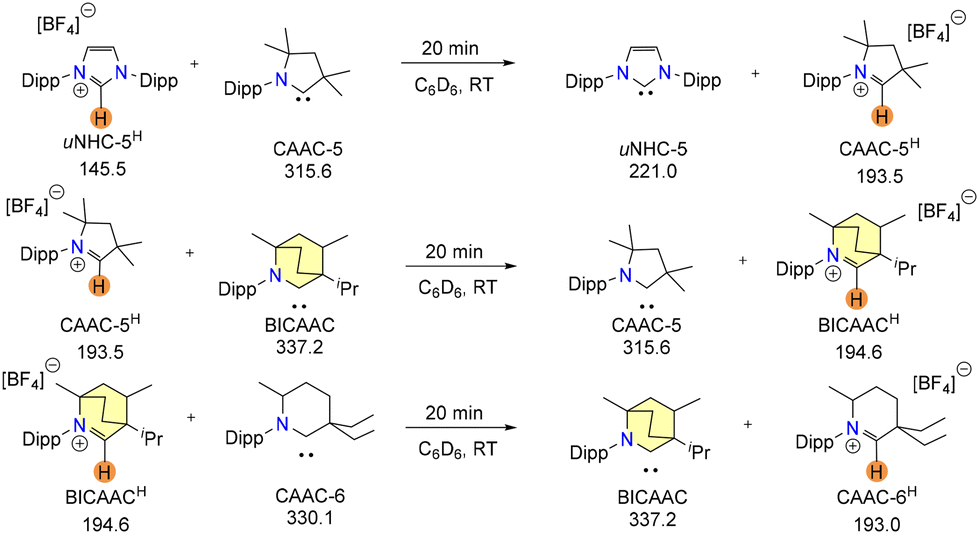 | ||
| Scheme 2 Proton exchange reactions between a carbene and carbene conjugate acid. 13C{1H} chemical shifts of the carbenic carbon in ppm units have been mentioned. | ||
Comparing the 31P{1H} NMR signals of carbene–phenylphosphinidene adducts and the 77Se{1H} NMR chemical shift of carbene–selenium adducts has proved to be an effective method for determining a carbene's π-accepting characteristics.16,17 The second technique is beneficial since the 77Se{1H} NMR scale is wider (Δ > 850 ppm), and the carbene selenium adduct is simple to generate. However, it should be kept in mind that 77Se is poisonous, and 77Se{1H} NMR is not as readily accessible as 31P NMR.17 Additionally, the presence of non-classical hydrogen bonds (NCHB) in carbene–selenium adducts results in noticeable downfield shifts in 77Se{1H} NMR spectra, which disturbs the Se scale and makes it challenging to examine the π-accepting characteristics of the carbene family. Furthermore, it is important to note that the choice of NMR solvent for recording the 77Se{1H} NMR spectrum is highly significant. A substantial change in the chemical shift (30 ppm) was found for the compound (iPr2N)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se when comparing CDCl3 with acetone-d6.33 In order to determine the π-acidity of carbene ligands, it is thus far more realistic to use the 31P chemical shifts of the carbene–phosphinidene adducts. It is evident that an increase in the carbene's π-accepting ability favors the back donation of the phosphorus atom's lone pair to the vacant p-orbital of the carbene center. As a result, the 31P NMR chemical shift of carbene–phosphinidene adducts should offer a simple approach for assessing the carbene's π-accepting properties: the more π-accepting the carbene is, the further downfield the phosphorus nucleus's chemical shift will be. The 31P{1H} and 77Se{1H} chemical shifts indicate the following order [6-CAAC > BICAAC > CAAC > NHC] of the π-accepting properties of the carbene ligands (Fig. 3).8–11
Se when comparing CDCl3 with acetone-d6.33 In order to determine the π-acidity of carbene ligands, it is thus far more realistic to use the 31P chemical shifts of the carbene–phosphinidene adducts. It is evident that an increase in the carbene's π-accepting ability favors the back donation of the phosphorus atom's lone pair to the vacant p-orbital of the carbene center. As a result, the 31P NMR chemical shift of carbene–phosphinidene adducts should offer a simple approach for assessing the carbene's π-accepting properties: the more π-accepting the carbene is, the further downfield the phosphorus nucleus's chemical shift will be. The 31P{1H} and 77Se{1H} chemical shifts indicate the following order [6-CAAC > BICAAC > CAAC > NHC] of the π-accepting properties of the carbene ligands (Fig. 3).8–11
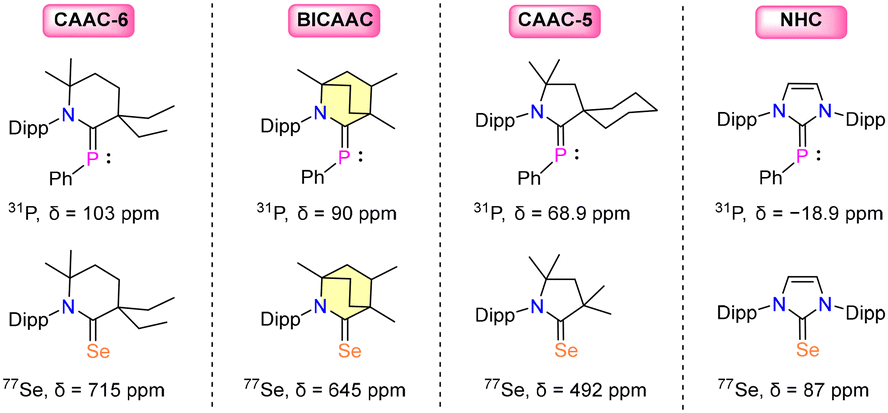 | ||
| Fig. 3 31P{1H} and 77Se{1H} NMR chemical shifts of carbene·(PPh) and carbene·(Se) adducts. | ||
Mondal and coworkers developed a methodology to establish the σ-donating and π-accepting properties of 5-membered CAACs using 15N NMR.18 This method describes that if a carbene exclusively functions as a σ-donor with species E, the formally unoccupied 2pπ(C) orbital is mainly stabilized by nitrogen π-donation. This results in nitrogen being electron deficient and a low-field 15N NMR signal. Conversely, when the carbene interacts with σ accepting and π-donating substances, the latter will also donate electrons to the empty 2pπ(C) orbital. This will result in a weaker π-donation by the nitrogen and a higher field 15N signal.18 Unfortunately, this method of exploring the electronic properties of BICAACs has yet to be explored.
4. BICAAC metal complexes
Coinage metal–carbene complexes have applications in medicinal chemistry due to their cytotoxic properties34a–c and are interesting candidates for application in OLEDs due to their unique photophysical properties.34d,e Several carbene–Au/Cu complexes are explored as metal catalysts.35–37 Along with the synthesis of BICAACs (Scheme 1), Bertrand et al. also reported the synthesis of BICAAC complexes of coinage metals in the same paper.9A homoleptic, bis-carbene complex [(MeBICAAC)2Cu][CuBr2] {2[CuBr2]} was isolated from the reaction of free carbene 1a with CuBr(SMe2) (Scheme 3a). Complex 2[CuBr2] exists as a mixture of diastereomers. In a similar reaction, a heteroleptic covalent, mono-carbene complex (iPrBICAAC)CuBr (3) was obtained using a bulky iPrBICAAC (1b). A heteroleptic gold complex (MeBICAAC)AuPh (4) was synthesized by the reaction of the free carbene (1a) with AuPh(PMe3) in THF (Scheme 3). Later, in 2020, Singh et al. also reported similar types of BICAAC coinage-metal complexes (Scheme 3b).38a Heteroleptic complexes (MeBICAAC)CuCl (5) and (MeBICAAC)CuI (6) were formed as the major products with some minor quantities of 2[CuI2] in the reaction of copper halides (CuCl and CuI) and 1a in equimolar amounts in THF (Scheme 3). It is known that Group-11 carbene metal complexes of type (carbene)MX (X = halides) slowly convert into [(carbene)2M]+[MX2]−.38b Similarly, an ionic gold complex 7[AuCl2] was formed in the reaction of equimolar amounts of AuCl(SMe2) and MeBICAAC in toluene and the formation of 7[AuCl2] proceeds through the disproportion of intermediate mono-carbene gold chloride.38a Homoleptic complex 2[PF6] can be synthesized exclusively through the reaction of a THF solution of 5, MeBICAAC, and KPF6 in equimolar concentrations for 10 h (Scheme 3b). It is important to note that, unlike BICAACs, no ionic complex formation was seen during the equimolecular interaction of cyCAAC and CuCl in THF, which solely produced a colorless complex [(cyCAAC)CuCl].38 Single crystal analysis of 2[CuBr2] shows a C–Cu–C bond angle of 180.0° but in 3 and 5, the C–Cu–X bond angles are 176.6° and 176.5°, respectively. In 2+ and 7+, an inversion center is located at the metal in both complexes. The positioning of the sterically demanding Dipp groups within the crystal units leads to infrequent (sp3)C–H⋯Cu/(sp3)C–H⋯Au intermolecular or intramolecular anagostic and pre-agostic binding interactions. The Ccarbene–Cu bond distance in 5 is 1.889(5) Å, which is close to the Ccarbene–Cu distance of 1.888(3) found in [(cyCAAC)CuCl]. The solution UV-Vis absorption spectra display bands between 312 nm and 364 nm, attributed to (σ + X)-π* charge transfer in complexes 5 and 6, and metal to ligand charge transfer (MLCT) transitions in 2[PF6] and 7[AuCl2]. However, complexes 5, 6, 2[PF6] and 7[AuCl2] exhibit broad absorption bands between 250 and 640 nm in the solid state. These unexpected bands may be caused by the mixing of intra-ligand interactions with certain weak non-covalent interactions.38
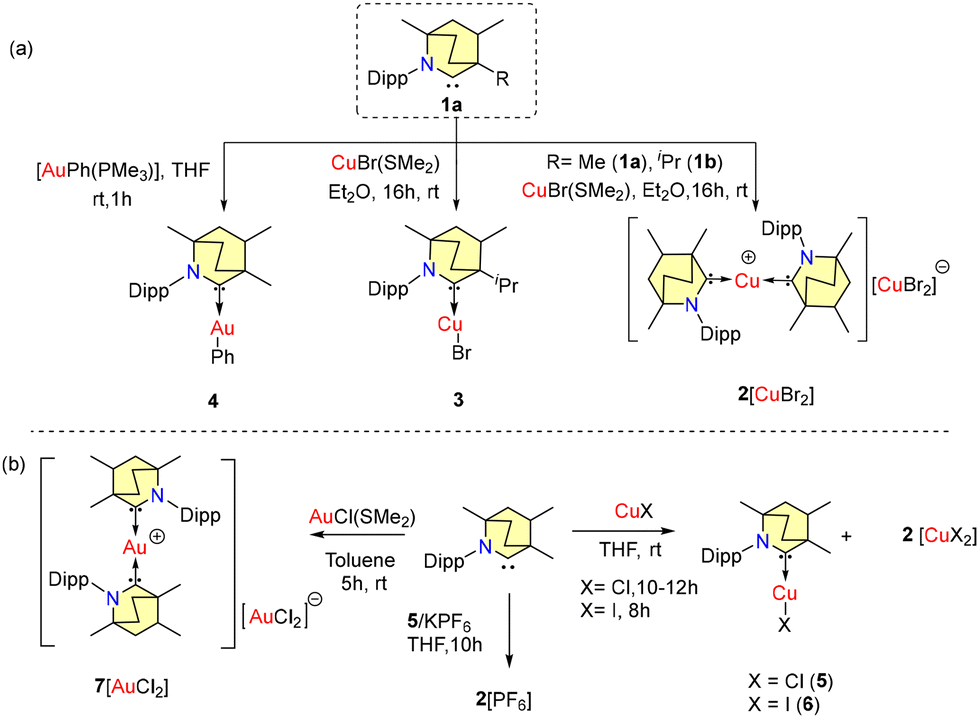 | ||
| Scheme 3 Synthesis of Cu/Au-BICAAC complexes. | ||
In 2018, Bertrand et al. reported that (BICAAC)CuBH4 (8a) could catalytically convert CO2 to formate in synergy with the Lewis pair DBU/BCF (TON-118) (Scheme 4).39 Complex 8a was found to exhibit higher activity compared to the reduction achieved solely with the Lewis pair DBU/BCF (TON-66) at 100 °C. The synthesis of catalyst 8a is given in Scheme 4b. The characterisation of 8a was done by single crystal X-ray diffraction. The 1H NMR analysis of 8a showed a broad quartet peak with a chemical shift of 54 ppm for B–H hydrogens with 1JB–H = 81.51 Hz.
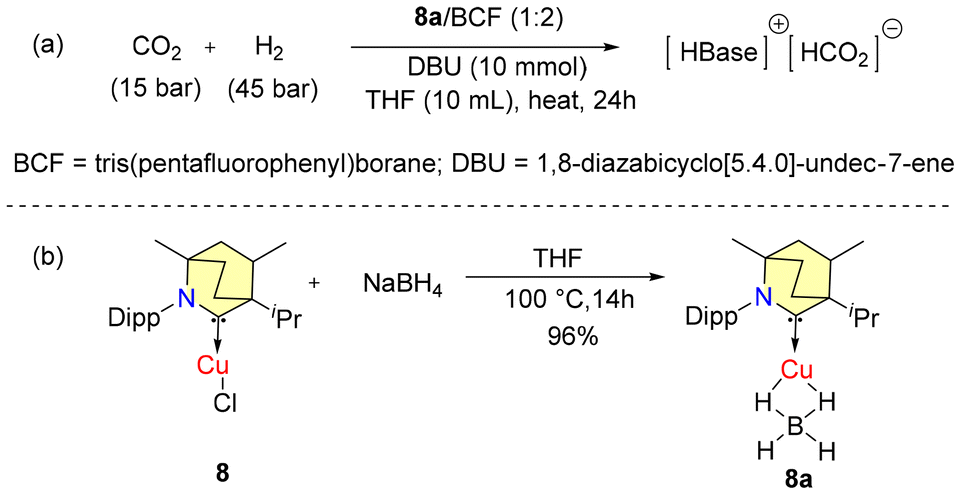 | ||
| Scheme 4 Reduction of CO2 to formic acid using 8a. | ||
However, the efficiency of 8a was found to be lower than that of the corresponding catalytic systems based on Et2CAAC (TON-305).39 The reduction of carbon dioxide to formic acid using copper hydrides as catalysts was not feasible because of the low stability, catalyst deactivation, and the inability of copper to activate H2.
In 2020, Bochmann and coworkers investigated the photophysical properties of BICAAC-coinage metal amides.40a Metal complexes 8–13 are synthesized according to Scheme 5a. All complexes were found to be stable in air and solvent media.
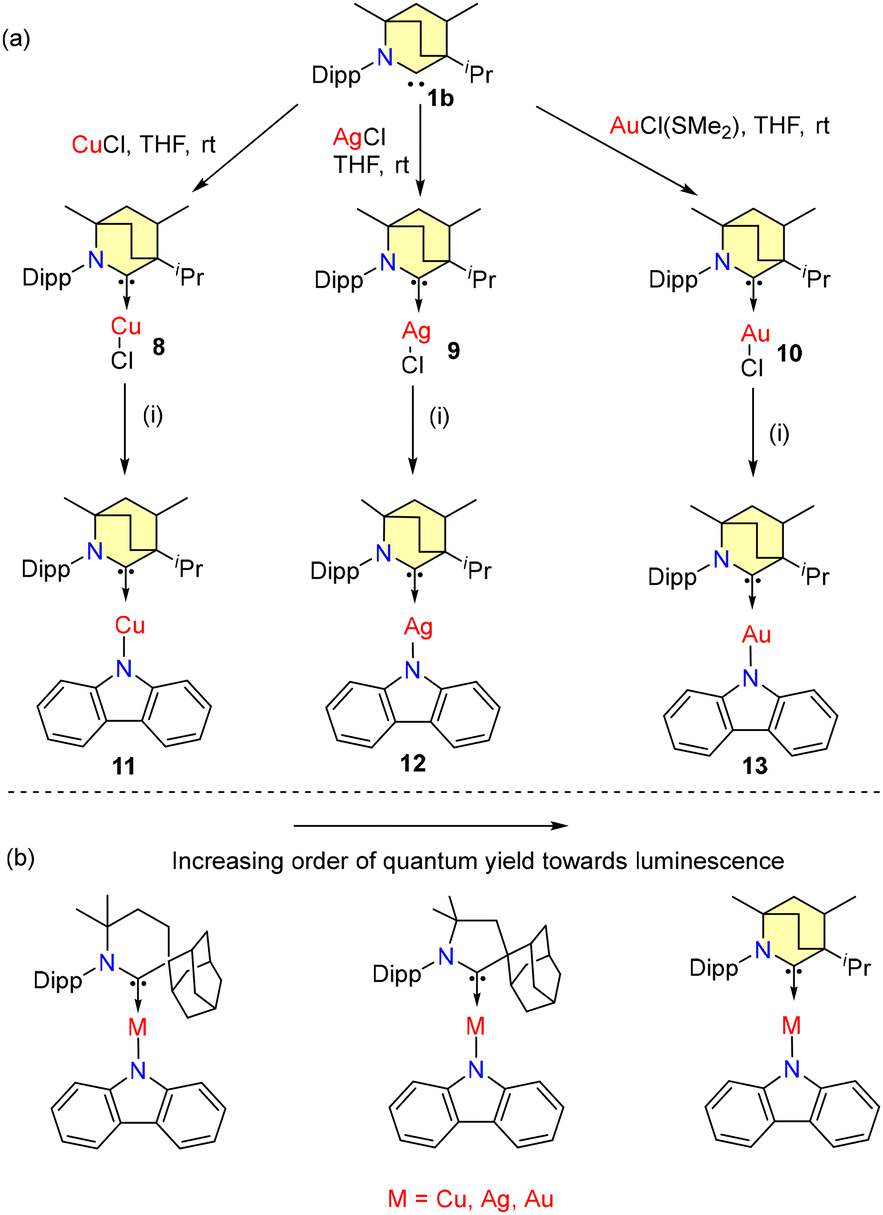 | ||
| Scheme 5 (a) Synthesis of BICAAC-metal amides: (i) carbazole, tBuOK, THF, and 3 h. (b) Relative photoluminescence properties. | ||
Notably, all the amide complexes 11–13 showed high stability upon irradiation with hard UV light at 290 nm. Among them, the gold complex 13 showed the highest stability in soft UV light. 11 showed green luminescence and 100% quantum yield (ΦPL) in the solution phase. Complexes 12 and 13 showed a quantum yield of 82% and 100%, respectively (Scheme 5b).40b
The CAAC-6 ligated metal carbazolate complex exhibited yellow luminescence and a low ΦPL (3.6% to 21.8%). In 2022, Cui et al. investigated the luminescence mechanism of 11 by conducting DFT/multireference configuration interaction studies, which revealed thermally activated delayed fluorescence (TADF).41
A highly selective protoboration of substituted terminal alkynes utilizing CAAC or BICAAC-supported copper catalysts was reported by the Bertrand group in 2021.42 Numerous aryl substrates, such as substituted aryl alkynes, alkyl alkynes, 2-ethynylnaphthalene, and heterocycles, produced the desired Markovnikov product with superior yields (51%–94%), showcasing a remarkable degree of α-selectivity (Scheme 6). In protoboration of alkyl substituted terminal alkynes (Et2CAAC)CuCl, (Et26-CAAC)CuCl and (CHMePhBICAAC)CuCl (8b) exhibited high conversion (yield) with comparable selectivity, which is different from the result obtained with (NHC)CuCl catalysts (Scheme 7). The catalytic activities of (iPrBICAAC)CuCl (8) and (CHMePhBICAAC)CuCl (8b) towards protosilylation of 5-phenyl-1-pentyne showed comparable α-selectivity, while a lower yield (50%) was obtained with 8b compared to 8 (99%) under the same reaction conditions (Scheme 7). Later in 2022, the same group reported highly α-selective methylboration of 5-phenyl-1-pentyne with 8 and 8b (Scheme 8).43
 | ||
| Scheme 6 Relative selectivity of protoboration using (carbene)CuCl as the catalyst. | ||
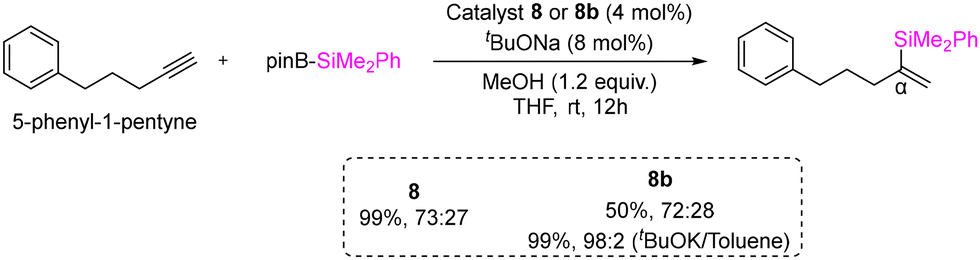 | ||
| Scheme 7 Protosilylation of 5-phenyl-1-pentyne. | ||
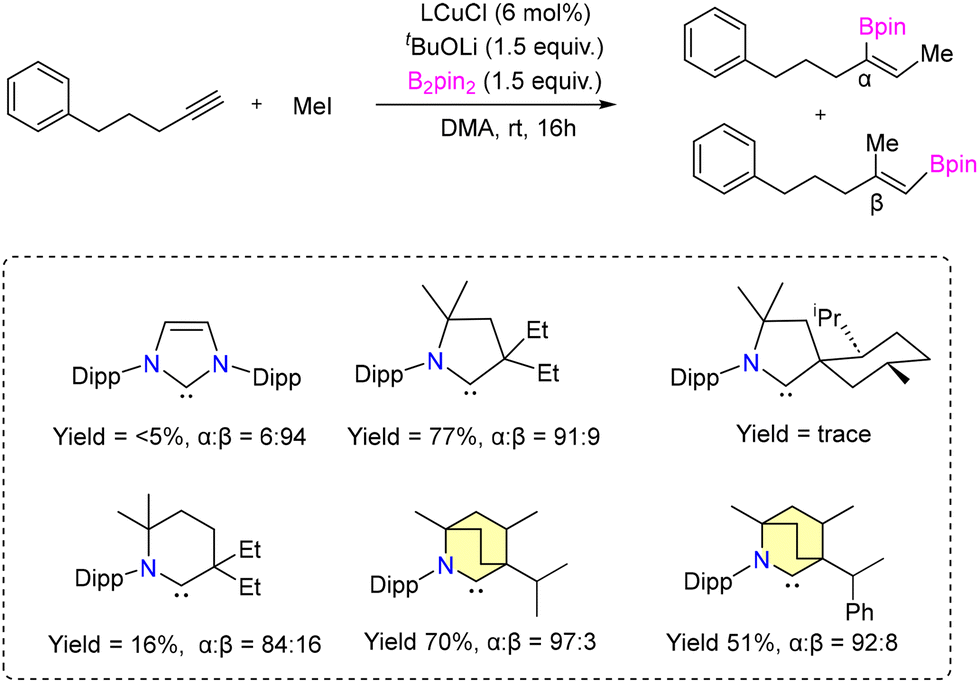 | ||
| Scheme 8 α-Selective methylboration of 5-phenyl-1-pentyne. | ||
No allylboration was observed. The study revealed that the presence of Lewis basic functional groups tends to a reduction in the catalytic efficacy of 8b. Interestingly, Et2CAAC-copper catalysts outperformed BICAAC-based catalysts in methylboration and allylboration with a broader substrate scope, selectivity, and product yield.43 The Bertrand group reported hydroamination and hydrohydrazination of alkynes catalyzed by BICAAC-gold catalysts in 2020.44 Hydroamination was conducted with optimized catalyst conditions of 2.5 mol% of 10/KBArF in benzene (Scheme 9). Remarkably, catalyst 10 demonstrated superior performance compared to previously known catalysts based on NHC and PPh3 due to better steric and electronic properties. Various substrates like aryl-substituted terminal alkynes reacted efficiently with aniline derivatives at 20 °C to afford the corresponding products in a very high quantitative yield. However, with alkyl substituted terminal alkynes, higher temperature was favored for better conversions. Similarly, internal alkynes reacted slowly and demanded higher temperatures and longer reaction times. Meanwhile, the conversion of low-boiling alkynes like ethoxy alkyne was achieved even at room temperature. Notably, sterically crowded anilines (Mesityl-NH2 and Dipp-NH2), electron-poor perfluoro anilines (C6F5-NH2) and dichloroanilines (C6H3Cl2-NH2) also afforded very high yields (Scheme 9a).44 Complex 10 efficiently catalyzed the hydrohydrazination of alkynes, where 1,1-disubstituted hydrazines were found to be less reactive (Scheme 9b). In the same report, they found that carbene–π alkyne cationic gold complexes are pivotal intermediates in both hydroamination and hydrohydrazination reactions. Notably, compounds 10 and 10a engage in the formation of cationic adducts, namely 14[BF4] and 14a[BF4], when reacting with n-hexyne (Scheme 10). It was noteworthy that 14[BF4] remained stable in air for up to four days, while 14a[BF4] decomposed after two days, yielding inactive bis-ligated cationic gold complex 7[BF4] and Au(0) nanoparticles, as depicted in Scheme 10.44
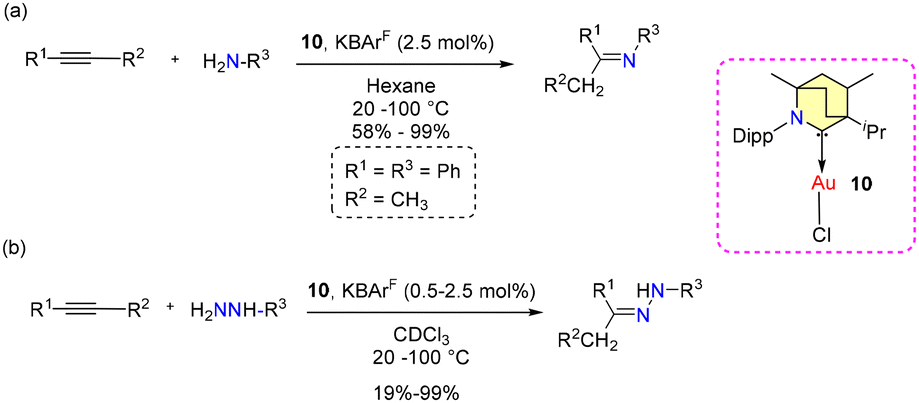 | ||
| Scheme 9 (a) Hydroamination and (b) hydrohydrazination using 10. | ||
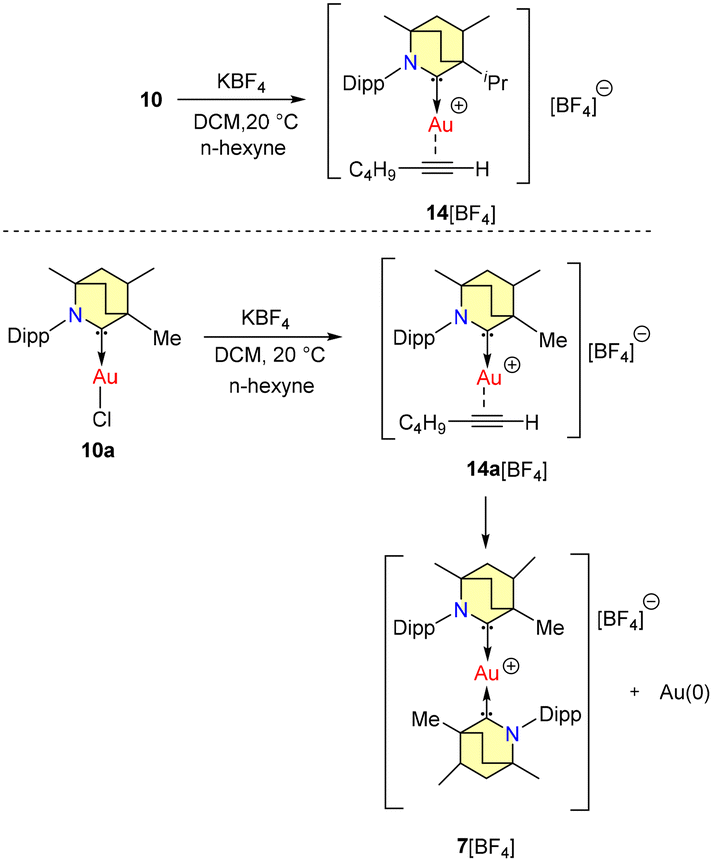 | ||
| Scheme 10 Formation of cationic adducts of BICAAC supported Au(I) and nhexyne. | ||
To confirm the existence of the active catalyst, gold cationic complexes of diphenylacetylene 15[BArF] and 15[BF4] were synthesized (Scheme 11) and the molecular structure of 15[BArF] was confirmed by single crystal X-ray diffraction studies.
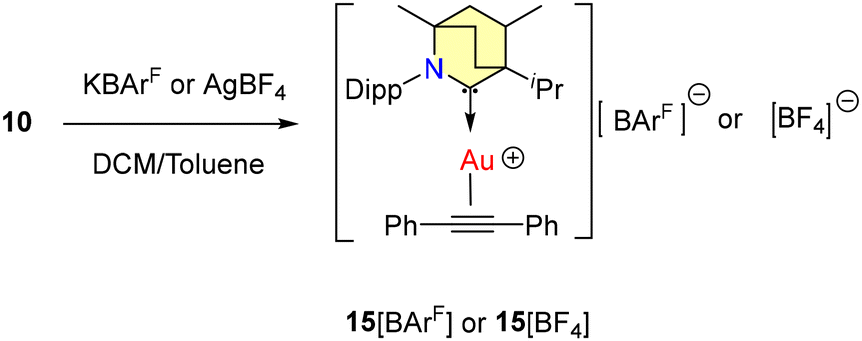 | ||
| Scheme 11 Formation of cationic complexes of diphenylacetylene. | ||
The interaction between p-tolyl-acetylene and 10 yielded 16[BArF], which then underwent subsequent reactions with p-toluidine and tosyl hydrazine, resulting in the anticipated products. Intriguingly, this process also led to the formation of Werner adducts, specifically 17[BArF] and 18[BArF], as illustrated in Scheme 12. Compounds 17[BArF] and 18[BArF] were structurally characterized by single-crystal X-ray diffraction.
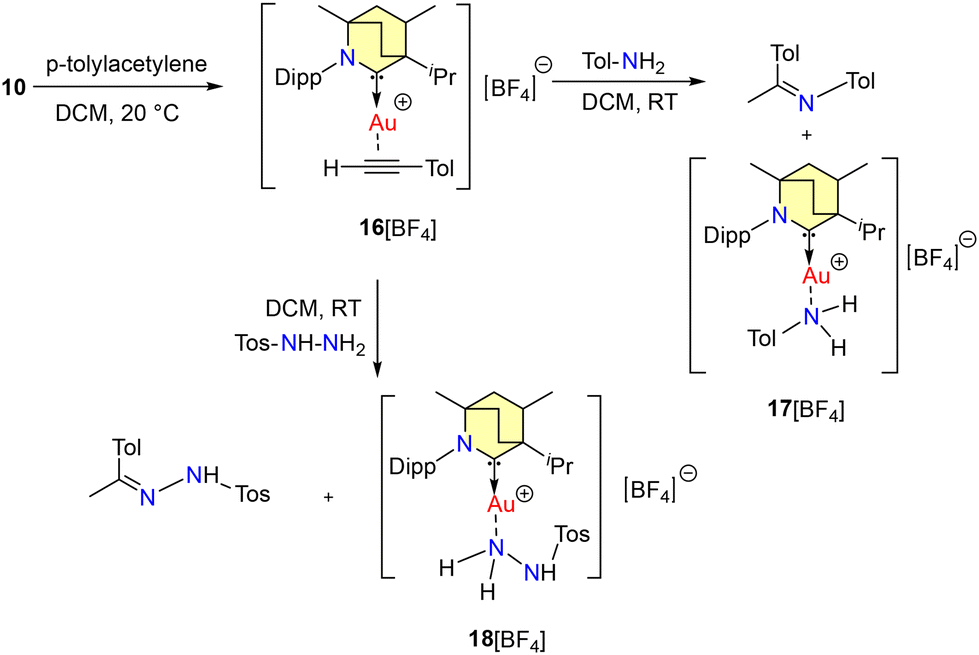 | ||
| Scheme 12 Formation of Werner adducts. | ||
In 2022, Singh et al. documented Negishi cross-coupling reactions catalyzed by BICAAC-nickel complexes.45 By combining free carbene 1a with the corresponding nickel halides in THF at room temperature, a mixture of rotamers (19–21a) was generated (Scheme 13).
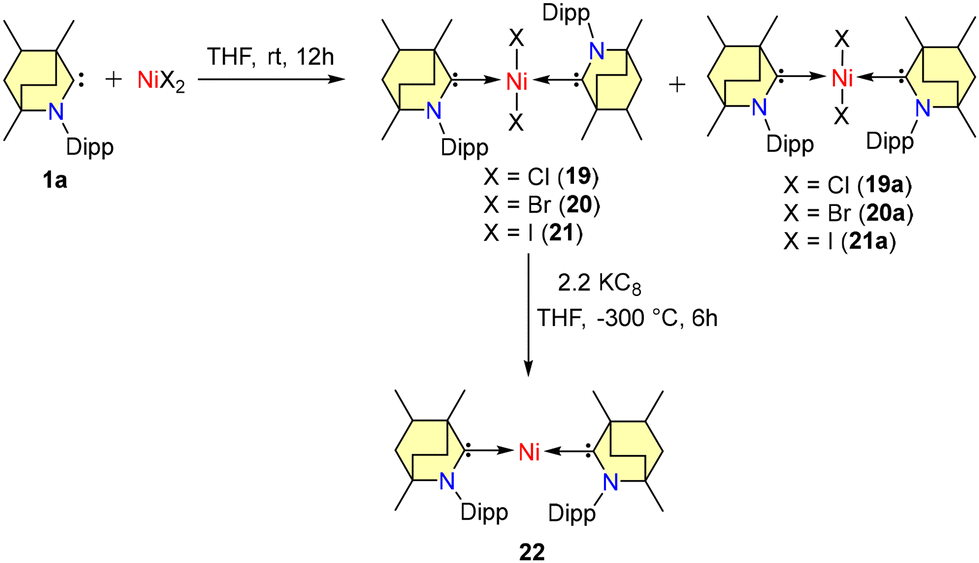 | ||
| Scheme 13 Synthesis of Ni-BICAAC complexes. | ||
In the case of complexes 19–21, the Dipp group was positioned in a trans-anti configuration, leading to a square planar geometry, while a trans–syn orientation was observed in 19a–21a. Remarkably, these rotamers remained non-interconvertible within the temperature range of −60 to 60 °C. The subsequent reduction of compounds 19–21 with KC8 led to the formation of air and moisture-sensitive, zero-valent nickel complex 22 (Scheme 13). Efforts to synthesize compound 22 from the combination of 1 and Ni(COD)2 resulted in suboptimal yield and slower conversion rates. Notably, unlike the case of CAACs, the stoichiometric interaction of BICAACs and NiBr2 formed four-coordinated [(MeBICAAC)NiBr2] in lower yield and the formation of [NiBr(μ-Br)(BICAAC)]2 was not observed.46 Furthermore, complexes 19–22 were employed for the Negishi cross-coupling reaction of various aryl halides with both fluorinated and non-fluorinated organozinc reagents. These reactions yielded moderate to excellent yields without the use of any additives (Scheme 14).
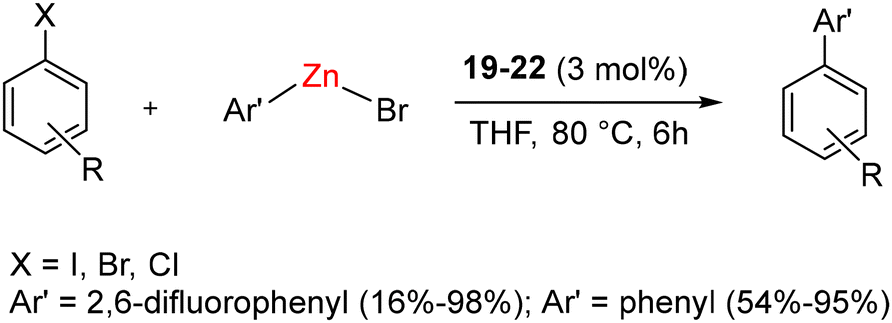 | ||
| Scheme 14 Negishi cross coupling using 22. | ||
Mechanistic investigations using UV-Vis studies, HRMS measurements, and controlled stoichiometric reactions provided insights into the catalytic process. Based on the findings, the authors proposed a Ni(I)/Ni(III) catalytic cycle for 19–21, while a Ni(0)/Ni(II) catalytic cycle was proposed for the Ni(0) complex 22 (Scheme 15). The complex 19 is reusable, stable, and thermally robust and showed the highest catalytic activity among 19–22.
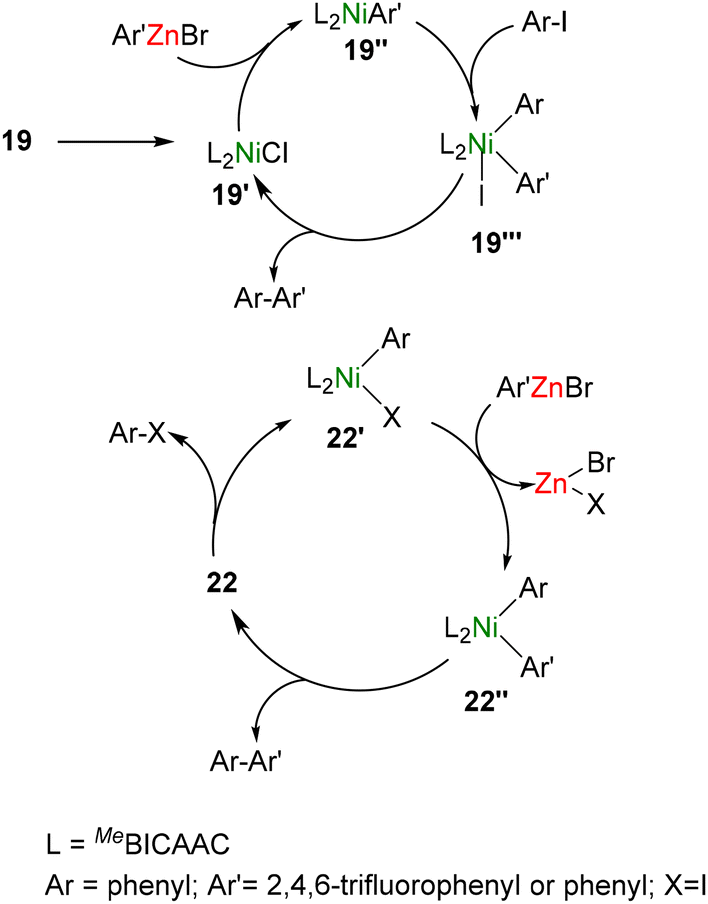 | ||
| Scheme 15 Mechanistic pathway of Negishi coupling with 19 and 22. | ||
In the year 2021, Mandal et al. reported the synthesis and reactivity of a water stable zero-valent zinc complex (BICAAC)2Zn (24) (Scheme 16).47a The process involved the reaction of BICAACMe (1a) with anhydrous zinc chloride in equimolar proportions in toluene at a temperature of 25 °C, resulting in the formation of dimeric complex 23. Subsequently, this complex was subjected to reduction by KC8 to form 24 (Scheme 16).
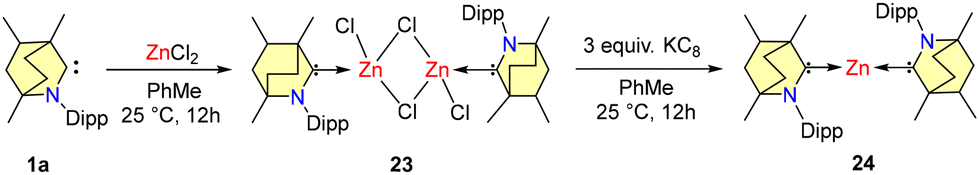 | ||
| Scheme 16 Synthesis of 23 and 24. | ||
Compound 24 can be stored under an inert atmosphere at room temperature indefinitely. Furthermore, compound 24 is stable in degassed water. However, compound 24 gradually decomposes into unidentified colorless solids after a few hours of exposure to air.47a
The closed shell triplet and singlet states of compound 24 are both more energetic than the singlet biradicaloid state. The ground state spin value being zero is a result of antiferromagnetic interaction, which arises from the weak superexchange between the unpaired electrons situated on the carbene carbon atoms and the filled d-orbitals of zinc (Fig. 4a). Furthermore, the LUMO–SOMO gap of 24 is lower than that of the similar zero valent zinc complex (CAAC)2Zn (Fig. 4b).47b
 | ||
| Fig. 4 (a) Singlet biradicaloid state of 24 and (b) LUMO–SOMO gap of (CAAC)2Zn and (BICAAC)2Zn. | ||
In contrast to compound 24, which was unable to activate H2, (CAAC)2Zn reacted with H2 to afford CAACH2 while depositing zinc metal. Compound 24 was capable of forming a zwitterionic adduct 25 upon interaction with CO2 and can reduce trityl chloride to Gomberg's triphenyl methyl radical much more efficiently than metallic zinc (Scheme 17).
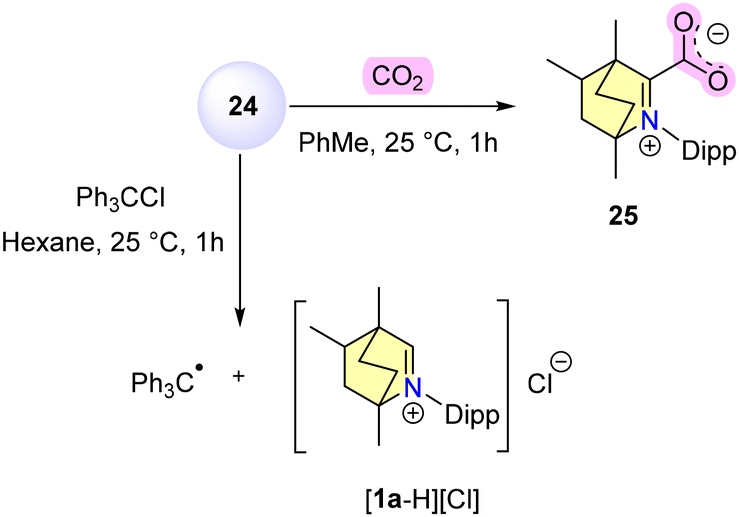 | ||
| Scheme 17 CO2 activation and reduction of triphenylmethylchloride with 24. | ||
The conversion of non-biodegradable polymers, such as polyethylene (PE), into chemical feedstocks represents a sustainable chemical transformation. The utilization of olefin metathesis within the realm of green chemistry, particularly in the context of sustainable catalysis, is an emerging trend. In 2022, Tuba et al. introduced temperature-activated BICAAC-Ru metathesis catalysts that exhibit high selectivity compared to the well-established second-generation Hoveyda–Grubbs and CAAC-Ru catalysts.48 The synthesis of mono-carbene BICAAC ruthenium complexes 26–29 involves a phosphene–carbene ligand exchange, utilizing in situ generated free carbenes 1b–e, with the first-generation Hoveyda–Grubbs catalyst (HG1), as depicted in Scheme 18. Additionally, bis-carbene complexes 30–31 are synthesized using first-generation Grubbs catalysts (G1), as outlined in Scheme 19.
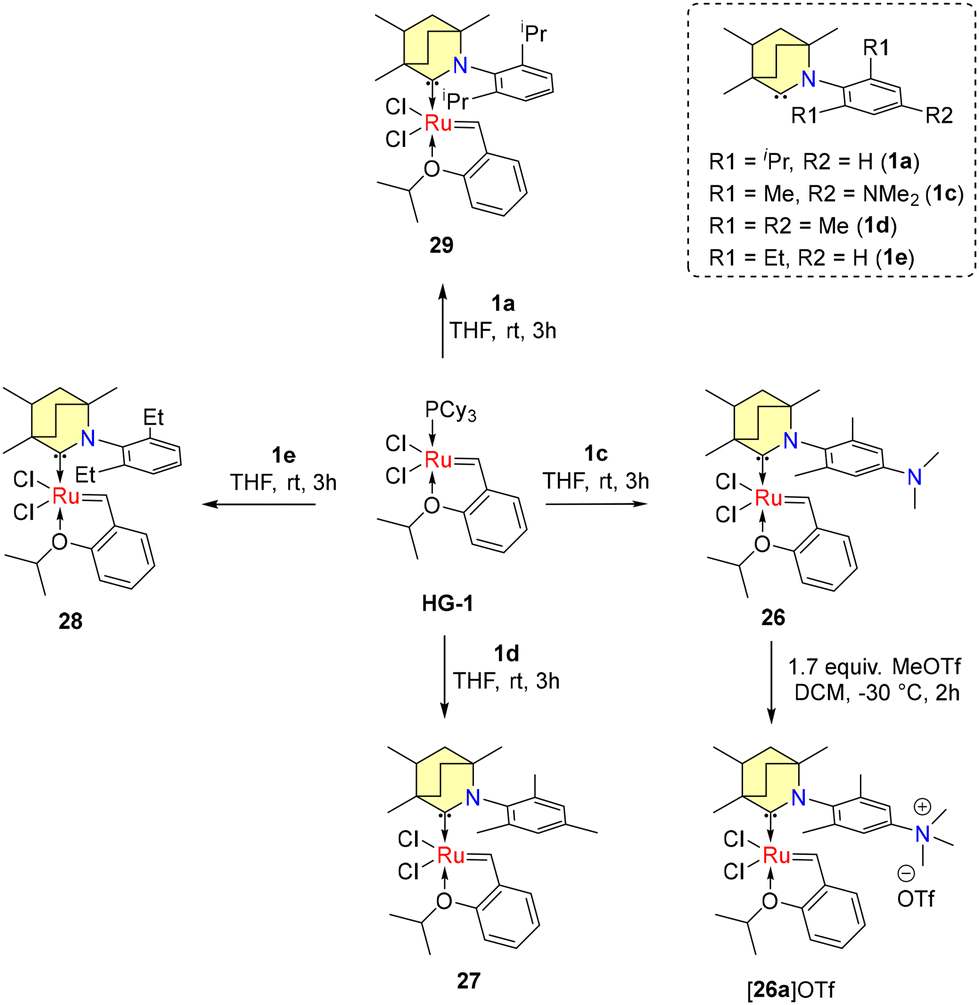 | ||
| Scheme 18 Synthesis of mono-carbene ruthenium complexes. | ||
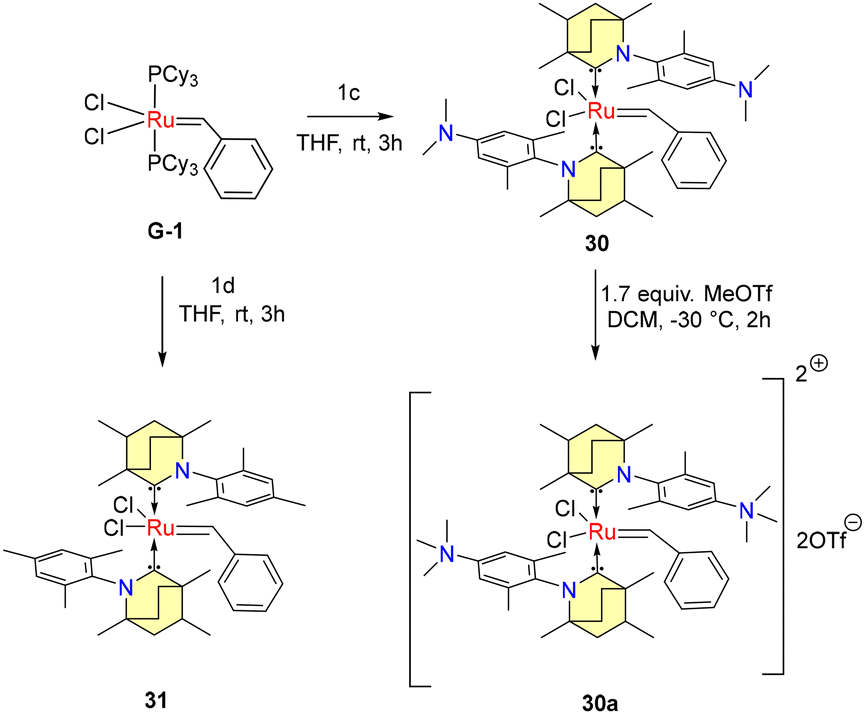 | ||
| Scheme 19 Synthesis of bis-carbene ruthenium complexes. | ||
While mono-carbene complexes 26–29 are stable in air, the bis-carbene complexes 30–31 can be purified in air but have a shorter shelf-life imparted by the strong trans-effect of the BICAAC ligands. The ionic methylated complex 26a[OTf] could be isolated, while 30a[OTf]2 was more sensitive. Remarkably high yields were achieved at 75 °C in the context of ring-closing metathesis reactions involving diallyl diethyl malonate with compounds 26–28 as well as 31. However, no reaction was observed with 26a[OTf] under similar conditions (Scheme 20a). For the isomerization metathesis reaction of 1-octadecene with 26/[RuH] (as part of a dual catalyst system) in the presence of 99.995% pure ethylene gas, selective production of propylene is achieved with a remarkable turnover number (TON) of 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 after a reaction time of 48 hours (Scheme 20b). Later, Turczel et al. reported metathesis reactions catalyzed by amberlyst-15/36 supported 26 having efficiency better than CAAC-based systems, as higher yields were obtained at lower catalyst loading.49
000 after a reaction time of 48 hours (Scheme 20b). Later, Turczel et al. reported metathesis reactions catalyzed by amberlyst-15/36 supported 26 having efficiency better than CAAC-based systems, as higher yields were obtained at lower catalyst loading.49
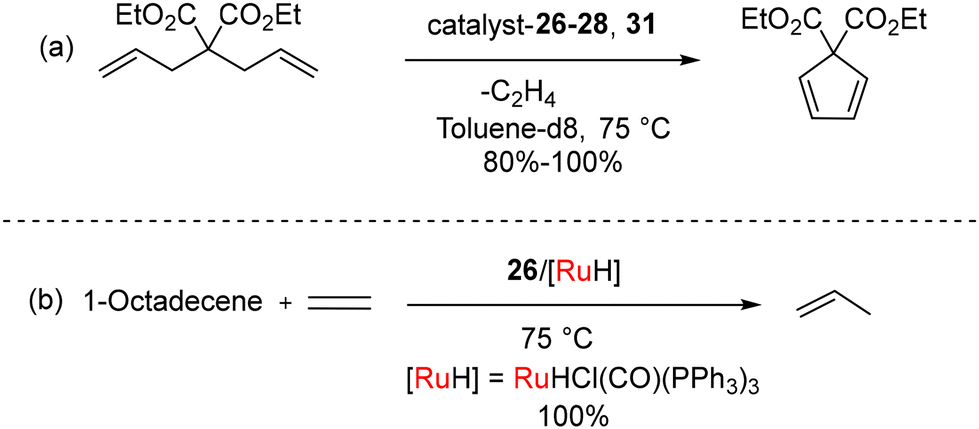 | ||
| Scheme 20 (a) Ring closing metathesis and (b) ISOMET reactions catalyzed by ruthenium complexes. | ||
In 2021, Singh et al. reported homogeneous cross-coupling reactions catalyzed by a palladium-BICAAC complex, conducted under open air conditions.50 The catalyst 32 was synthesized in accordance with Scheme 21.
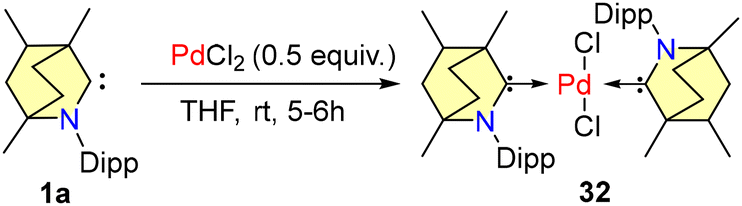 | ||
| Scheme 21 Synthesis of 32. | ||
The activity of the catalyst 32 was found to be better in comparison with tetracyclic-NHC-based palladium complexes. With less catalyst loading, the complex 32 exhibited higher activity than tetracyclic-NHC-based palladium complexes.51,52
In the context of Heck–Mizoroki cross-coupling reactions involving aryl bromides featuring electron-withdrawing substituents, the use of catalyst 32 led to impressive yields ranging from 71% to 83%, as illustrated in Scheme 22a.
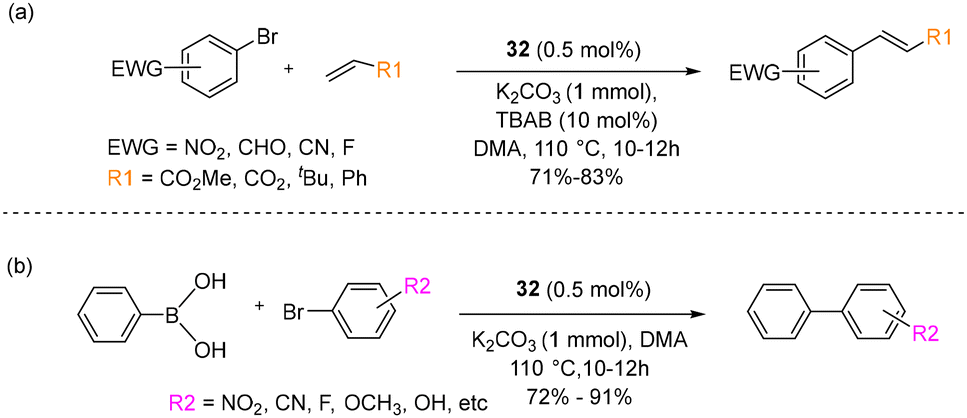 | ||
| Scheme 22 (a) Heck–Mizoroki and (b) Suzuki–Miyaura cross-coupling reactions using 32. | ||
Moreover, tert-butylacrylate and styrene as coupling partners yielded the formation of the respective products. Furthermore, they extended their investigation to the activity of the catalyst in Suzuki–Miyaura cross-coupling reactions, as depicted in Scheme 22b. Diverse substrates were explored, including aryl bromides featuring electron-withdrawing groups (EWGs), electron-donating groups (EDGs), and bromofuraldehyde, all yielding substantial yields ranging from 72% to 91%. Notably, following the completion of the reaction, catalyst 32 gradually degrades into inactive palladium nanoparticles. However, the absence of a considerable effect of mercury on the reaction medium suggested the homogeneous nature of the reaction.
Subsequently, in the year 2022, Tuba et al. disclosed findings regarding Mizoroki–Heck coupling reactions catalyzed by air and moisture resistant Pd-PEPPSI (PEPPSI = pyridine enhanced precatalyst preparation stabilization and initiation) complexes, supported by BICAAC ligands.53 Bispyridine-PdCl2 complexes 33a–b were produced by reacting PdCl2 with pyridine in a 1:3 molar ratio in methanol (Scheme 23). The reaction with equimolar amounts of free carbenes and palladium(II)-chloride-bis-pyridine complexes in a 1,4-dioxane medium at 80 °C for 2 hours formed the corresponding PEPPSI complexes (34a–e) (Scheme 23).
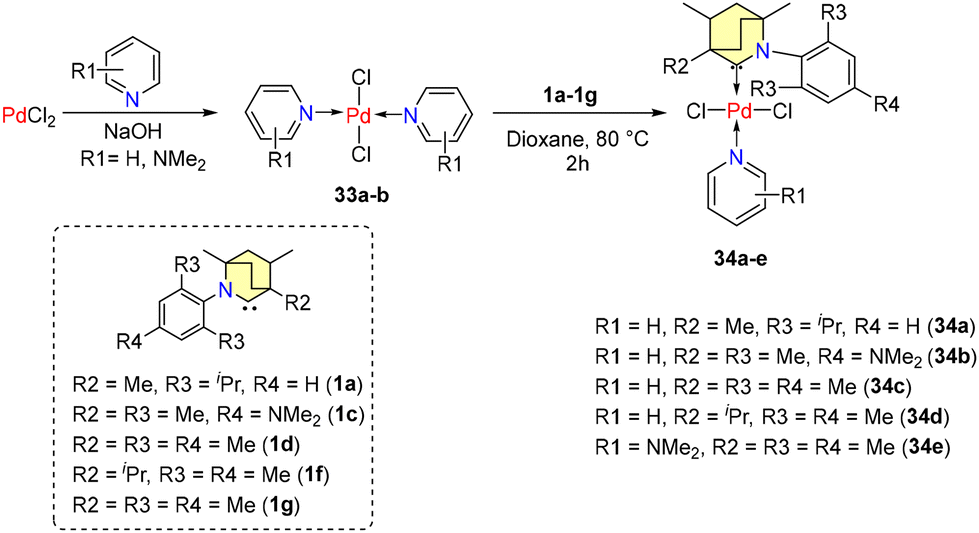 | ||
| Scheme 23 Synthesis of Pd-PEPPSI complexes. | ||
Crystal structure analysis showed that 34a and 34b are crystalized as racemates with enantio-pairs in the lattice with square planar coordination around Pd(II). Similar catalytic activity is shown by complexes (34a–e) in coupling reactions except for 34c, which has an isopropyl group adjacent to the carbene carbon atom, which hindered the activity. Moreover, the other substituents of the catalysts did not show any significant impact on efficiency. Coupling of various olefins like styrene, 1-dodecane, 1-octadecene, hepta-1,6-dien-4-ol, and 9-decene-1-ol with substituted aryl bromides yielded excellent conversions (95%–100%). Longer-chain olefins potentially underwent double bond isomerization, possibly catalyzed by intermediate metal hydrides. Poor yields were the result of substrates that were sterically hindered (Scheme 24). Notably, it was discovered that air reduced the catalytic efficiency.
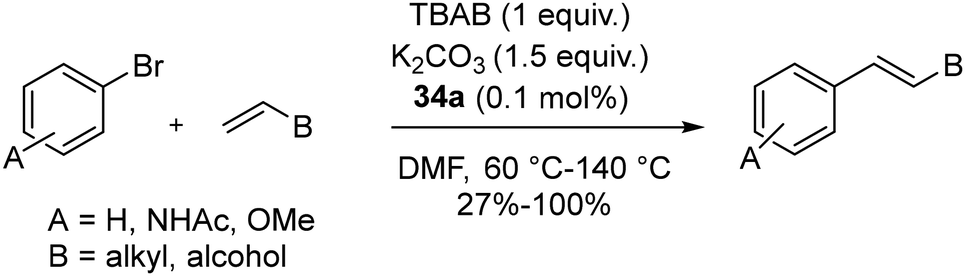 | ||
| Scheme 24 Mizoroki–Heck coupling catalyzed by 34a. | ||
Very recently in 2024, Sanjay Singh and coworkers reported bench-stable BICAAC derived iridium complexes exhibiting remarkable catalytic activity in transfer hydrogenation and hydrosilylation reactions.54 Complex 35 was synthesized using 0.5 equivalents of [IrCl(COD)]2 along with a free BICAAC (Scheme 25). The synthesized complex (35) was further treated with AgSbF6 to generate the corresponding iridium cation (35a), which demonstrated reactivity with acetonitrile, a Lewis base, forming the adduct (35b) at room temperature (Scheme 25).
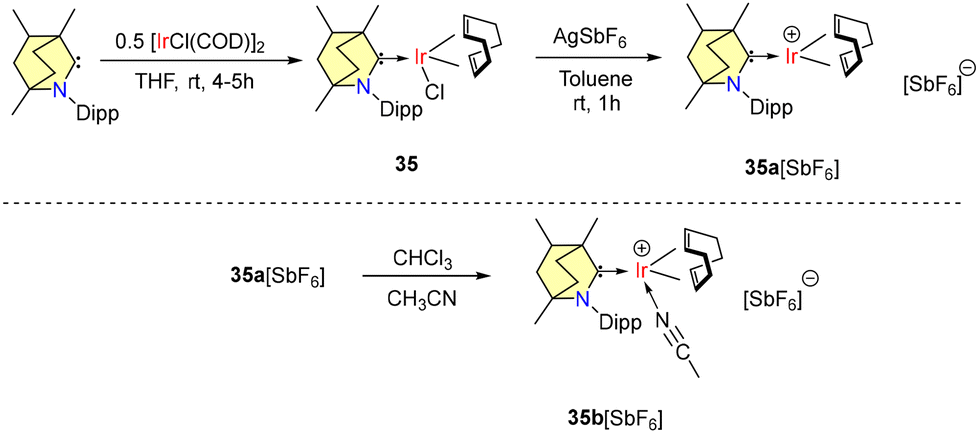 | ||
| Scheme 25 Synthesis of BICAAC-Ir complexes and their cationic reactivity. | ||
Subsequent studies demonstrated that these BICAAC-Ir complexes can function as catalysts for the transfer hydrogenation of aldehydes, ketones, and imines, using iPrOH as the hydrogen source (Scheme 26).54 These findings motivated further evaluation of the catalyst's general applicability in hydrosilylation reactions. Both electron-rich and electron-poor aldehydes underwent successful hydrosilylation with Et3SiH as the reducing agent with catalyst 35.
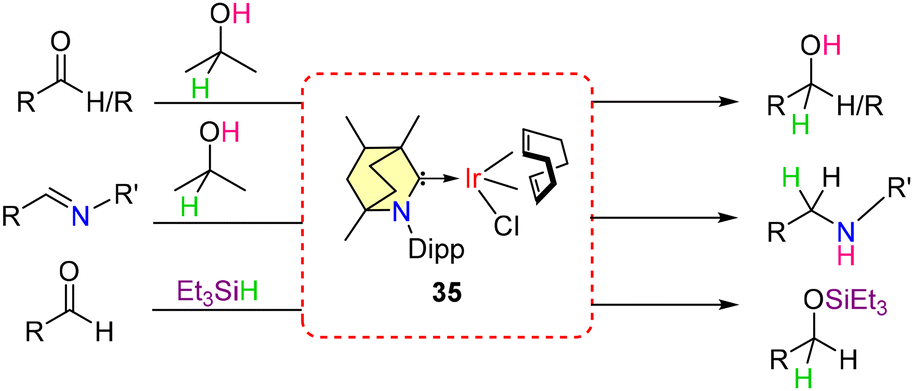 | ||
| Scheme 26 Transfer hydrogenation and hydrosilylation reactions of complex 35. | ||
5. BICAACs as metal-free catalysts
The first theoretical study on the activation of enthalpically strong bonds by BICAAC ligands was reported by Phukan et al. in 2018.55 It was found that BICAACs can activate N–H bonds via either an electrophilic pathway or a nucleophilic pathway. Conversely, the activation of P–H bonds occurs exclusively through a nucleophilic pathway, and Si–H bonds are activated through a hydride transfer pathway.55Tiwari et al. conducted computational studies on SO2 activation by several modelled BICAACs employing Density Functional Theory (DFT) methods.56 Later in 2019,57 Koley et al. theoretically predicted the electronic nature of carbene–alane and carbene–borane adducts using density functional theory. They reported that Ccarbene–boron bonds are slightly covalent in nature. Notably, the interaction energies, ΔEint, and dissociation energy De of the BICAAC·BH3 adduct surpassed those of all other carbene–borane adducts.
Similar patterns were observed with the BICAAC-AlH3 adduct as well. In AlH3 adducts, the electron density of Ccarbene–Al bonds is polarised towards the carbon center. Transition state energy analysis further unveiled that the tendency of BICAACs to form adducts with small boron hydrides, BH3, and insertion products with more hydridic reagents like B2Pin was thermodynamically favored.
In 2019, Singh et al. reported experimental results on B–H bond activation and borane adduct formation utilizing BICAAC ligands.58 The synthesis of borane adducts (36a–g) was carried out following Scheme 27. Among them, 36a demonstrated air stability for a limited duration, whereas the other adducts exhibited high sensitivity to moisture and oxygen. Notably, the reaction of a BICAAC with bulky B(C6F5)3 produced the BICAAC·B(C6F5)3 adduct (36g) as the major product. The production of BICAAC-borane adducts, [BICAAC·BH3] (36a), [BICAAC·BHCl2] (36b), [BICAAC·BH2Cl] (36c), [BICAAC·BF3] (36d), [BICAAC·BCl3] (36e), and [BICAAC·BBr3] (36f), is consistent with NHC and CAAC findings. However, reactions of the BICAAC with more hydridic boranes (9-BBN and catecholborane) that allow the activation of the B–H bond to create [BICAAC(H)-9-BBN] (37a) and [BICAAC(H)-Bcat] (37b) show that the BICAAC has reactivity akin to CAACs (Scheme 28). This reaction proceeded through the insertion of the carbene's carbon center into the B–H bonds, leading to the formation of air-sensitive complexes 37a and 37b (Scheme 28). The boron center has a tetrahedral geometry in 36a–g and a trigonal planar arrangement in 37a–b. The thermodynamic viability of adduct and insertion product formation was further substantiated by quantum computational intrinsic reaction coordinate (IRC) calculations. (NHC)·BF3, (CAAC)BF3, and (BICAAC)BF3 showed similar chemical shifts in 11B{1H} and 19F{1H} NMR (Scheme 24), which did not reflect the electronic and steric properties of the carbenes.59–61
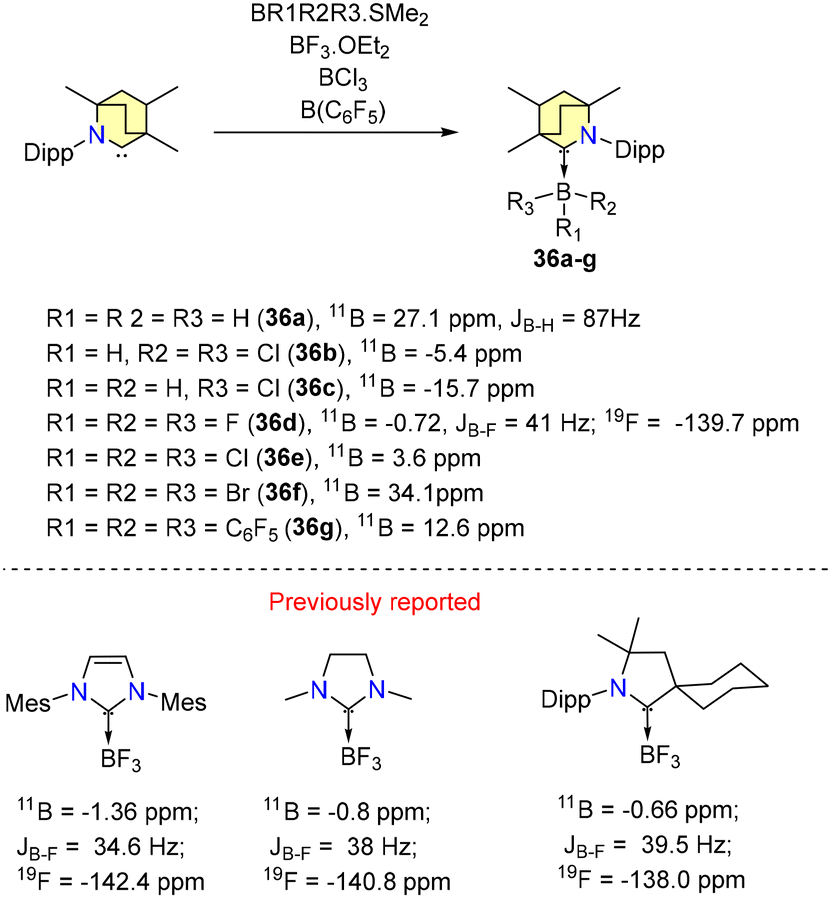 | ||
| Scheme 27 Synthesis of BICAAC-borane adducts and comparison of the chemical shifts of (carbene)·BF3 adducts in 11B{1H} and 19F{1H} NMR. | ||
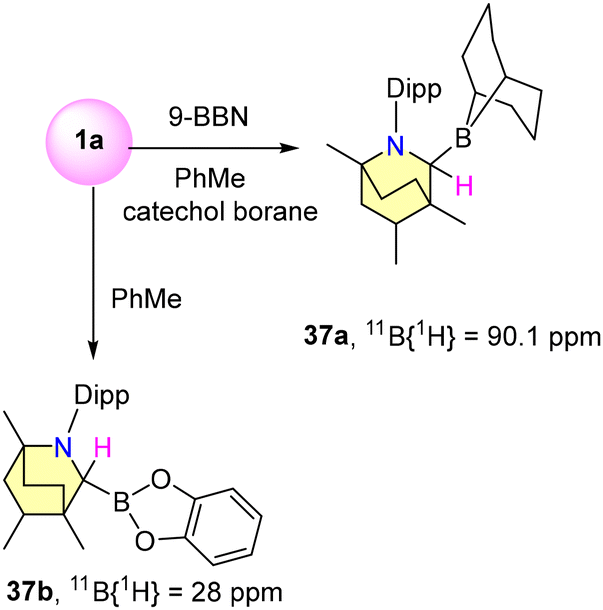 | ||
| Scheme 28 Formation of B–H insertion products (37a–b). | ||
Mandal et al. reported BICAAC catalyzed silylative dehydration of 2-methyl benzamide to their corresponding nitrile using PhSiH3 as a silylative reagent with a yield of 73% in 12 h (Scheme 29).62 Interestingly, extending the reaction time (24 h) resulted in a proportional increase in the product yield (86%). Further investigation revealed that NHCs, such as saturated IPr and IPr, demonstrated much reduced catalytic activity (48% and 14%) under identical conditions. The authors noted that the higher nucleophilicity of the BICAAC was essential for the efficiency of this reaction.
 | ||
| Scheme 29 Silylative dehydration of primary amides to nitriles using 1a. | ||
In the year 2020, the Bertrand research group unveiled a novel approach for activating CO molecules using a BICAAC and CAAC.63 Their investigation revealed that when free carbene 1a reacts with CO in deuterated benzene under ambient conditions, an immediate formation of amino ketene is triggered (Scheme 30). The resulting blue crystals of compound 38 were isolated from dry pentane, with a distinct CO stretching vibration (νstr co) noticeable at 2080 cm−1. Further experimentation showed that compound 38 readily interacts with equimolar amounts of 9,10-phenanthrenequinone (Quin), culminating in the synthesis of spirolactone 39a with a 74% yield. Subjecting a deuterated benzene solution of 39a to 4 atm CO at 80 °C produced black residues alongside carbonylated quinone 40 (Scheme 30).
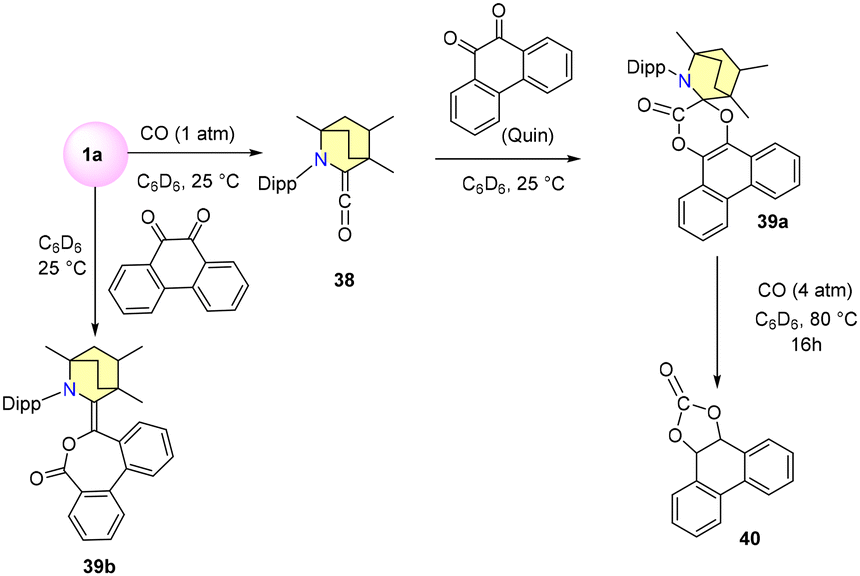 | ||
| Scheme 30 Reactivity of the (BICAAC)CO adduct. | ||
Additionally, the reaction of 39a with elemental sulfur (S8) yielded compounds 39c and 40. Intriguingly, the interaction of compound 40 with 1a regenerated 39a, thus confirming the reversible nature of the reductive elimination process (Scheme 31). In order to comprehend the mechanism and investigate potential pathways, ethylene carbonate 41 was selected as the desired substrate.63 At room temperature, it was noted that 1a readily combines with 41 to form a diastereomeric mixture, specifically a 76:24 ratio of 42a and 42b (Scheme 32).
 | ||
| Scheme 31 Reductive elimination at the BICAAC centre. | ||
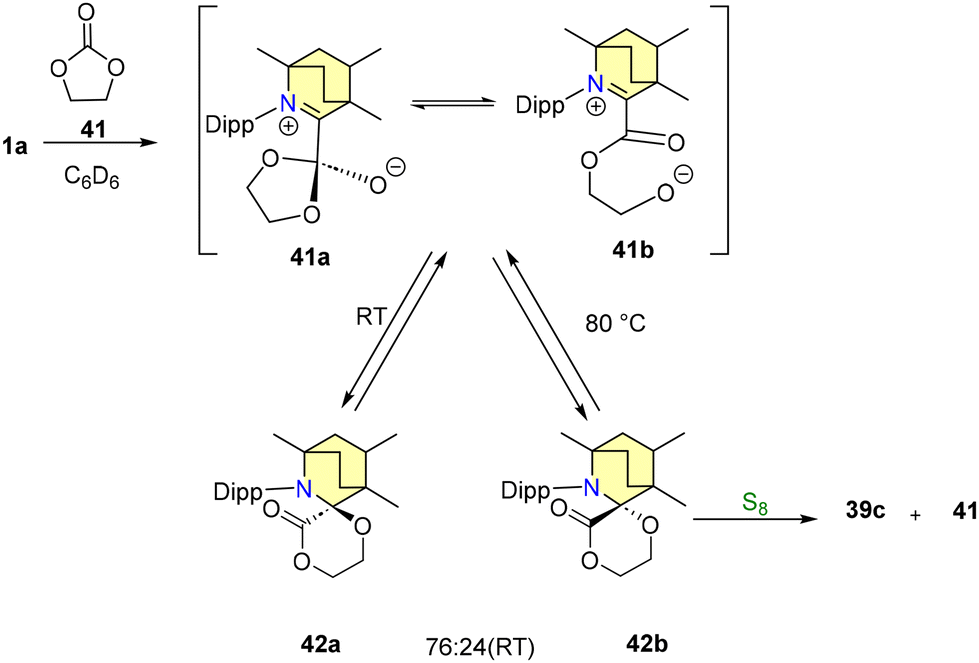 | ||
| Scheme 32 Formation of 42a–b. | ||
This transformation proceeds via the intermediates 41a and 41b. Interestingly, when the reaction temperature is elevated to 80 °C, the proportion of product formation experiences a reversal, as indicated in Scheme 32. Notably, efforts to employ 1a as an organic catalyst were unsuccessful, primarily due to the emergence of an inactive adduct 39b (Scheme 30). The bulky CAAC ligand catalytically facilitated the same carbonylation of an o-quinone into a cyclic carbonate.63
In another report, Mandal et al. have reported BICAAC catalyzed solvent-free, gram-scale reduction of nitriles to amine hydrochloride salts utilizing pinacolborane as the reducing agent.64 However, the authors did not mention anything on the possibility of hidden boron catalysis.65
This approach encompasses a diverse substrate scope, which includes the reduction of electron-donating and withdrawing group substituted aromatic nitriles, halogen-substituted benzonitriles, and heterocyclic nitriles, as well as cyclic and acyclic aliphatic nitriles, with moderate to good yields (Scheme 33). However, the observations reflect that ortho-substituted benzonitriles afforded unsatisfactory yields. Mechanistic insights revealed that the B–H insertion product 43 serves as a hydride donor during the reduction process (Scheme 34). This intermediate formation was validated through computational analyses and deuterium labelled (DBPin) experiments (Scheme 34). Similar results have also been reported by Sen et al. in 2022 using a six-membered NHC, which also supports this insertion reaction.66
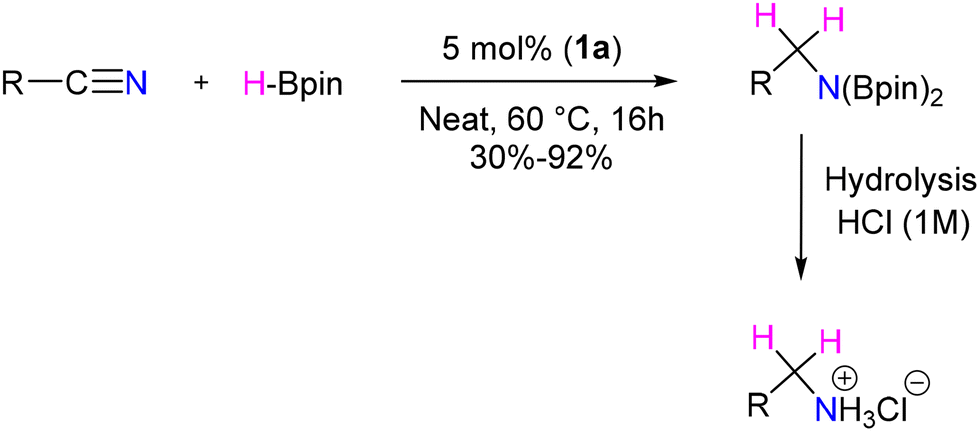 | ||
| Scheme 33 BICAAC catalyzed reduction of nitriles. | ||
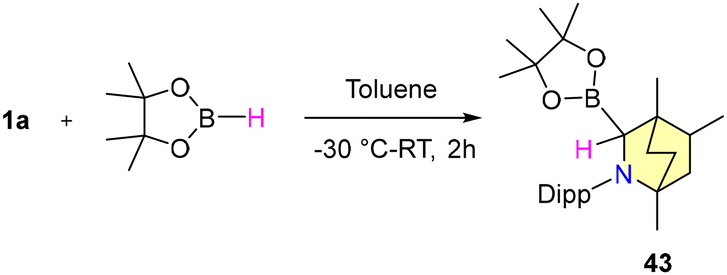 | ||
| Scheme 34 Reaction of HBpin with a BICAAC. | ||
In another report, Mandal et al. demonstrated a BICAAC as a catalyst for the N-methylation of primary amides using CO2 as the carbon source and H-Bpin as the reducing agent.67 The reaction exhibited robust tolerance towards various amides containing electron-donating, electron-rich, and heterocyclic groups, as well as biologically active molecules with amide moieties (Scheme 35).
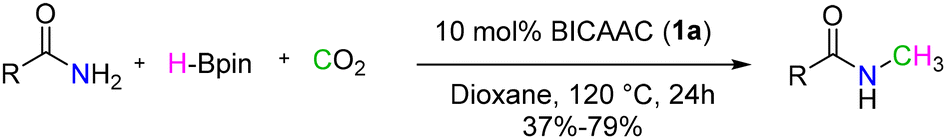 | ||
| Scheme 35 N-Methylation of primary amides. | ||
Mechanistic investigations have unveiled that the insertion product 43 and the adduct 45 possess the capability to catalyze N-methylation, likely attributable to their interconversion and the potential formation of intermediate 47 (Scheme 36). The role of 43 involves activating the amide bonds to generate N-borylated amide 48. On the other hand, adduct 45 interacts with H-Bpin, undergoing hydride transfer to establish transition state 46, which subsequently evolves into intermediate 47. The progression of 47 leads to the formation of N-borylated acetamide 49, followed by consecutive transformations resulting in aminal 51 and 50 (Bpin)2O. The transformation continues as 51 is further subjected to reduction by H-Bpin, ultimately yielding the desired N-methylated amide 52 (Scheme 36). The proposed catalytic cycle pathway finds support through controlled experiments.
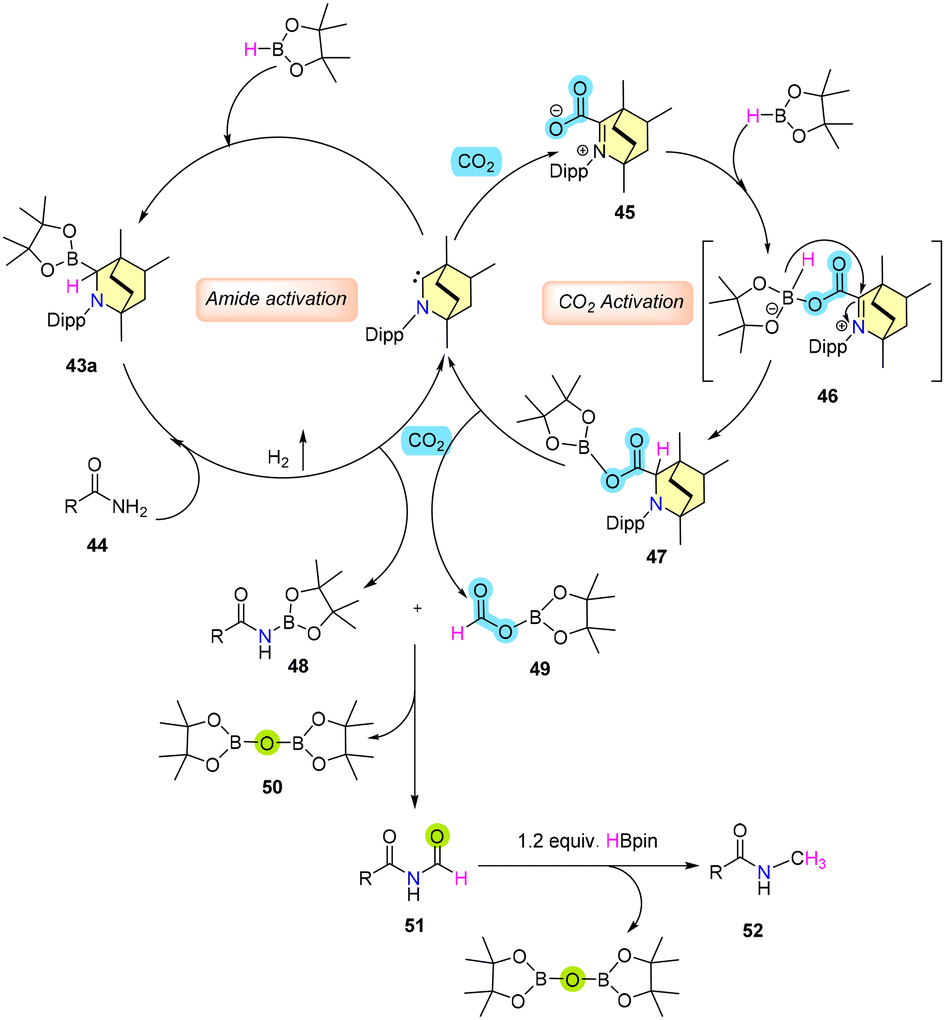 | ||
| Scheme 36 Mechanistic pathway for N-methylation of primary amides. | ||
In 2024, Singh and coworkers exploited the ambiphilic properties of the BICAAC for the dehydrogenation of alcohols and it was reported that the activation pathway closely resembles the conventional metal–ligand cooperative activation of a substrate (Scheme 37a).
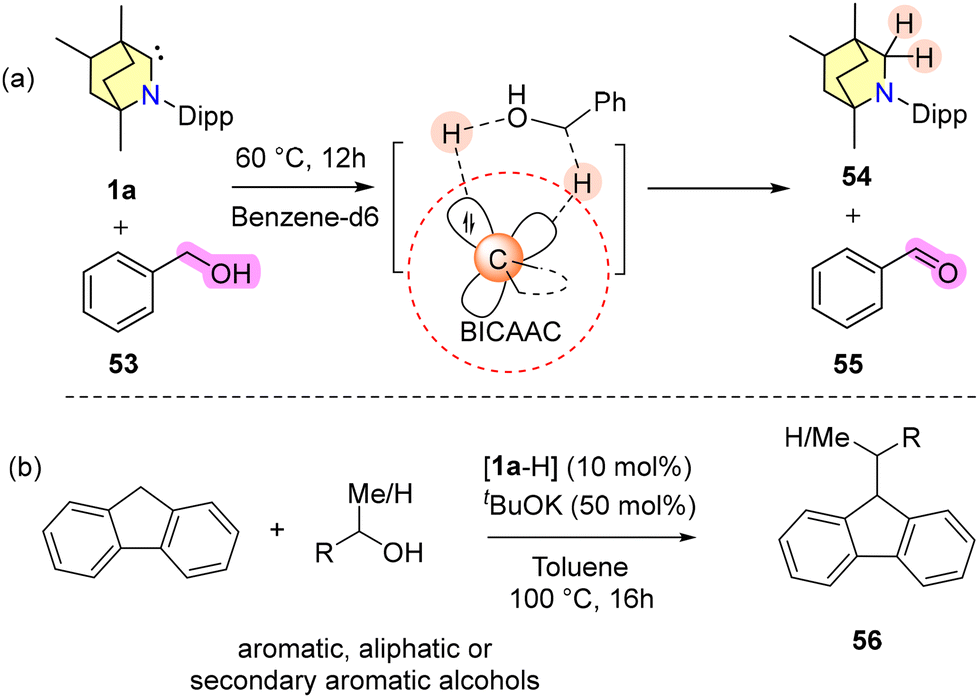 | ||
| Scheme 37 (a) Dehydrogenation of alcohol using a BICAAC through the ambiphilic action of the BICAAC and (b) BICAAC catalysed fluorene alkylation. | ||
A stoichiometric reaction of benzyl alcohol and a BICAAC at 60 °C resulted in the formation of (BICAAC)H2 (54) and benzaldehyde in 60% yield (Scheme 37a).68 Furthermore, they have used a BICAAC as a catalyst for the alkylation of fluorene at its 9-position using various alcohols as the alkyl source (Scheme 37b).
6. BICAAC-supported main-group compounds
In 2023, our group reported water and air-stable glyoxal radicals stabilised by a BICAAC (Scheme 38a).69 The synthesis of the radical 57[BF4] is given in Scheme 38. Interestingly, 57[BF4] was found to be stable under acidic and basic conditions as well as in H2O2, thiophenol and bovine serum. 57[BF4] showed excellent thermal stability (stable up to 200 °C). Notably, UV spectral analysis indicated that the BICAAC stabilized radical cation 57[BF4] exhibited stability towards acids and bases (Scheme 38b).69a Similar glyoxal radical ions were also reported with NHC ligands by Lee et al. in 2021 (Scheme 38b).69b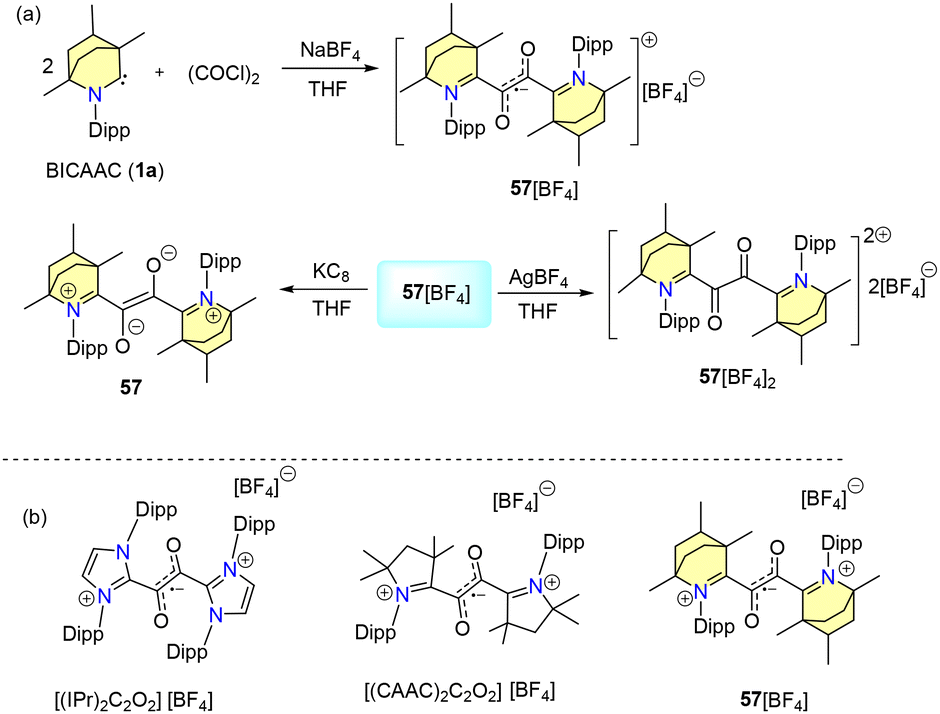 | ||
| Scheme 38 (a) Synthesis of a glyoxal radical cation using a BICAAC. (b) Examples of related radical ions. | ||
The DFT study revealed that the spin density of 57 has been delocalised over the central glyoxal unit, along with negligible contributions from the carbene groups.
Furthermore, cyclic-voltammetric investigations exhibited reversible one-electron oxidation and reduction phenomena of 57[BF4], which were verified by chemical synthesis (Scheme 38a).
In 2024, Mandal et al. carried out an extensive electrochemical study of compound 57[BF4] using cyclic voltammetric techniques.70 Based on the outcome of this study, they proposed its potential application in symmetric H-cell cycling (Scheme 39).70
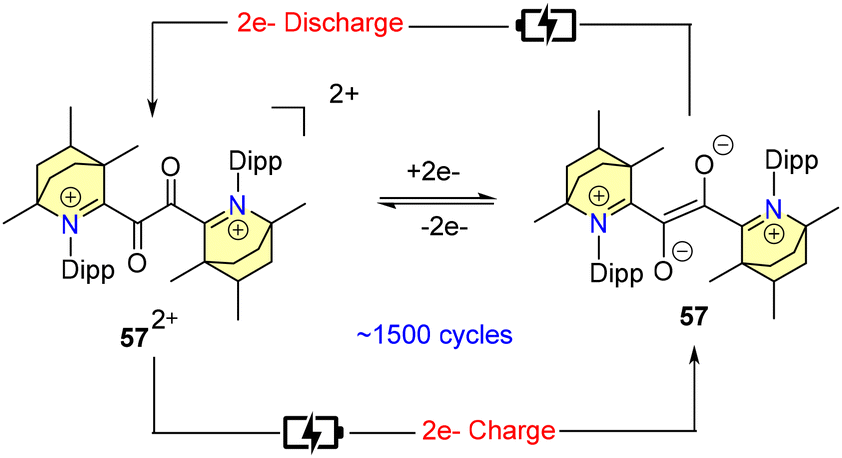 | ||
| Scheme 39 Schematic representation of symmetric H-cell cycling based on 57[BF4]. | ||
Sanjay Singh and co-workers in 2024 demonstrated that reacting BH2Cl and KC8 in the presence of a free BICAAC affords diborane complex 60 (Scheme 40).71a Similarly, a reaction of BHCl2 and 2 equiv. of KC8 in the presence of a BICAAC affords diborene 61 (Scheme 40).71a Both 60 and 61 are highly sensitive to air and moisture. Compound 60 exhibits a signal at δ = −16.4 ppm in 11B{1H} NMR spectroscopy, whereas it is further downfield shifted in 61 at 42.6 ppm. The 1H NMR spectrum displays a broad signal at 5 ppm corresponding to the BH protons of 61. The frontier molecular orbital diagram analysis indicates that diborene 61 has the lowest HOMO–LUMO gap in comparison with its NHC analogue.71b
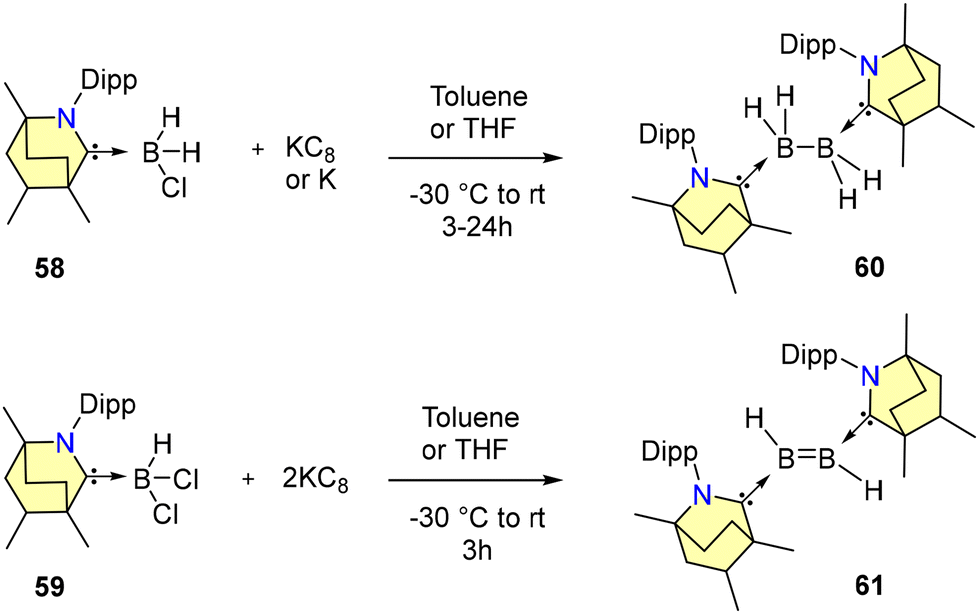 | ||
| Scheme 40 Synthesis of BICAAC-diborane (60) and diborene (61) complexes. | ||
The diborene 61 forms complexes with coinage metal halides (CuCl, AgBr, and CuI) (Scheme 41). It was observed that BB coordinated to two units of CuCl in a η2-coordination mode (62). In contrast, η1-coordinating complexes of Ag and Cu were formed when 61 was reacted with AgBr and CuI, yielding solid compounds 63 and 64, respectively. Compound 63 remains stable under inert conditions and below −15 °C.
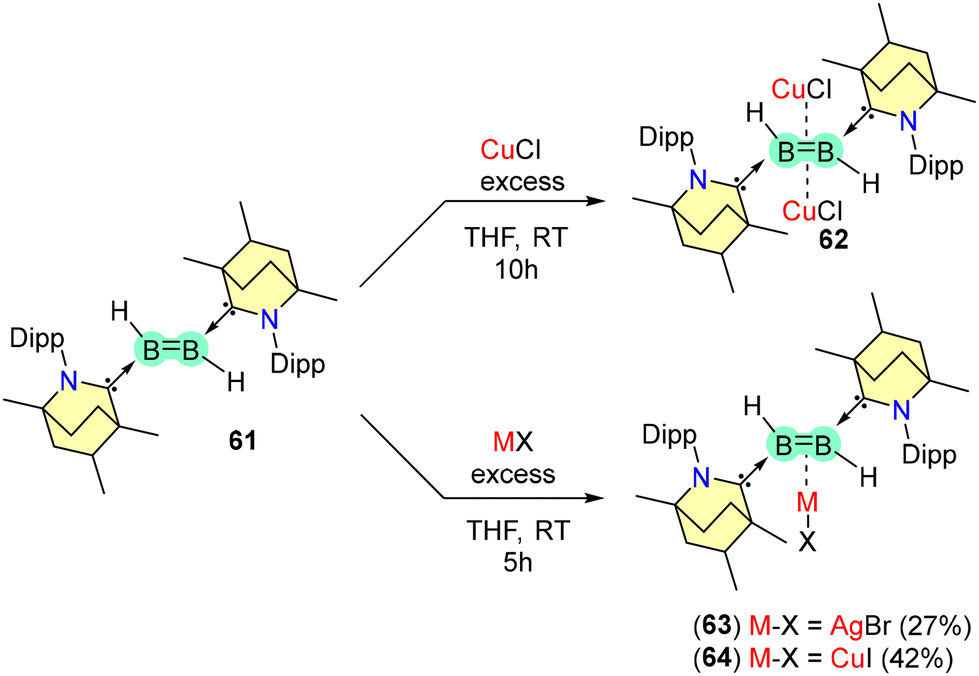 | ||
| Scheme 41 Coinage metal complexes of BICAAC-diborene. | ||
Only a few BICAAC complexes of heavier main group elements have been reported to date. Among them, very recently, Singh and co-workers reported the synthesis of low-valent stable silicon complexes (65–68) derived from BICAACs and Si(IV) precursors (Schemes 42 and 43).72 Starting with 1 equiv. of SiCl4 and a free BICAAC, compound 65 was synthesised with a yield of 76% (Scheme 42). Compound 65 exhibited a highly upfield-shifted signal at −106.8 ppm in the 29Si NMR spectrum, compared to its precursor (SiCl4 = −18 ppm). (MeBICAAC)SiCl4 (65) was thermally stable up to 196–198 °C.
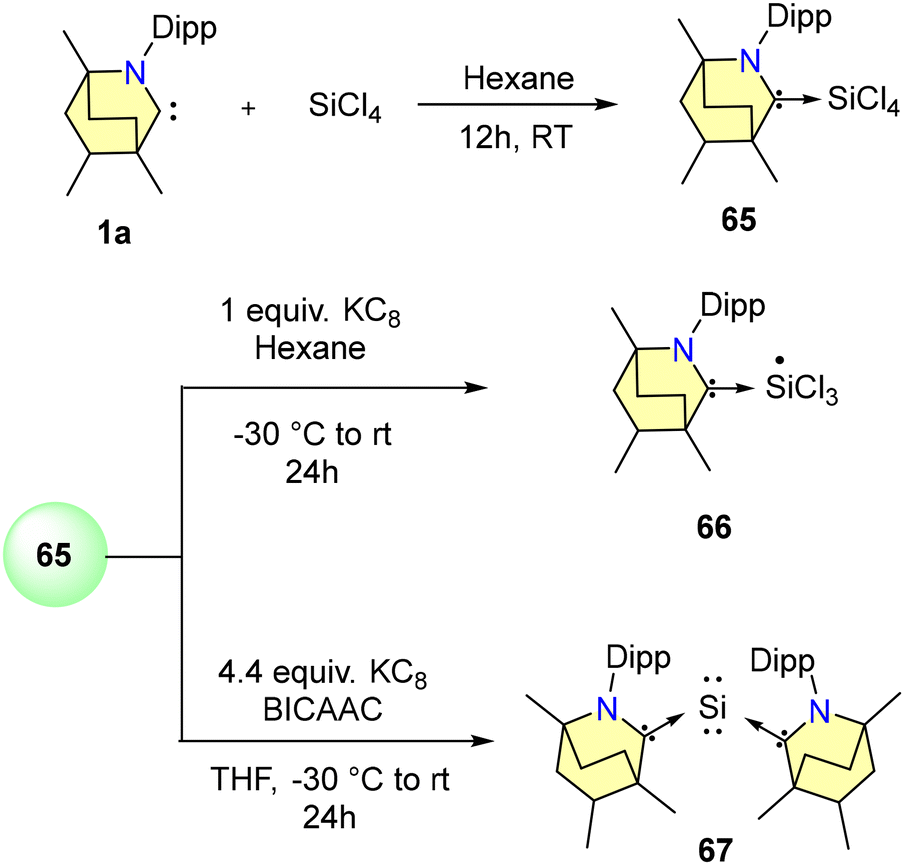 | ||
| Scheme 42 Synthesis of BICAAC stabilised low-valent silicon compounds. | ||
 | ||
| Scheme 43 Synthesis of a BICAAC-Si(III) radical. | ||
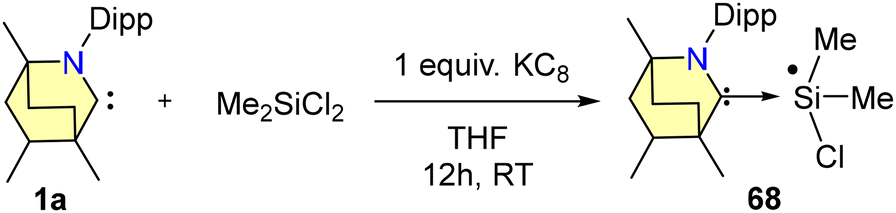
The reduction of 65 with either one equivalent or an excess of KC8 produced 66 and 67, respectively (Scheme 42). Compound 66 is a mono-radical and the formal oxidation state of silicon is +3. 67 is a bent, two-coordinated Si(0) compound. Furthermore, the singlet ground state and silylone nature of complex 67 were supported by DFT calculations. From SCXRD techniques, it was revealed that the Si(0) adopts a bent geometry and is coordinated with two BICAAC units in the monoclinic system with a C2/c space group. Additionally, Si(III) radical 68 was also formed via a single-pot reaction of Me2SiCl2 with a BICAAC and KC8 in THF (Scheme 43). It was isolated with a yield of 73% as an orange-red solid.
Bertrand et al., in their 1st report of isolating BICAACs, synthesized selenium and phosphinidene adducts [(MeBICAAC)Se (69) and (MeBICAAC)PPh (70)] of MeBICAAC (1a) to study the electronic properties of BICAACs (Scheme 39).9 The 31P{1H} NMR signal displayed a remarkable shift to +90 ppm for compound 70, diverging from the +56 to +69 ppm range seen in CAACs.73 Similarly, the 77Se NMR spectrum of compound 69 revealed a distinctively downfield signal at +645 ppm, in stark contrast to +492 ppm and +87 ppm exhibited by (Me2CAAC)Se and (IPr)Se, respectively.74,75 This pronounced shift highlighted the better π-accepting and σ-donating capabilities inherent to BICAACs compared to their CAAC counterparts. Furthermore, treating 1a with dibromine forms bromide salt 71 (Scheme 44).
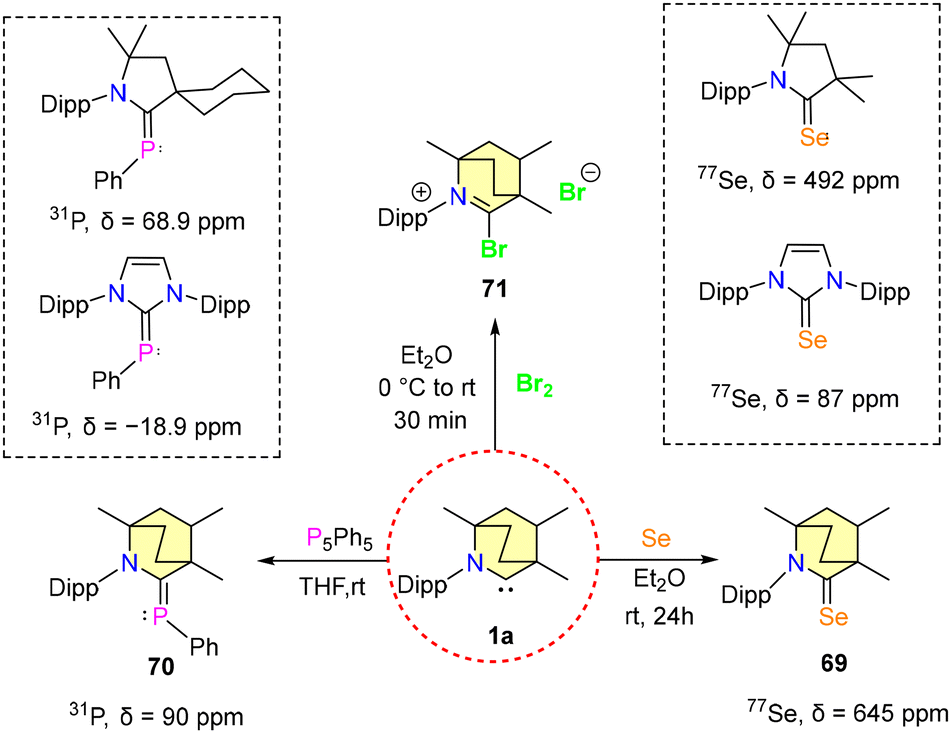 | ||
| Scheme 44 Reactivity of 1a towards Se, P5Ph5 and Br2. | ||
In 2023, Singh et al. reported the synthesis of (BICAAC)ECl3 adducts (E = P, 72 and Sb, 73) and their three-electron reduction to form (BICAAC)2E2 (E = P, 74 and Sb, 75) complexes (Schemes 45 and 46).76 The single crystal X-ray analysis revealed that P and Sb have seesaw geometry in compounds 72–73. The 31P{1H} NMR of (BICAAC)PCl3 (72) showed a single peak at 33.0 ppm, whereas the compound (BICAAC)2P2 (74) showed two closely spaced singlets of equal intensity at 65.5 and 65.3 pm (due to two diastereomers). The 31P{1H} signal of 74 stands relatively deshielded compared to its corresponding NHC analogue, specifically the (IPr)2P2 compound (δ = −52.4 ppm) (Scheme 41).77 The corresponding (cyCAAC)2P2 shows the 31P{1H} signal at 59.4 ppm.78 This distinctive shift is attributed to the increased electrophilicity of the BICAAC carbene. The Ccarbene–P bond distance in 74 [1.734(14) Å] was found to be shorter when compared with the Ccarbene–P bond distance in (IPr)2P2 [1.7504(17) Å] and comparable with the Ccarbene–P bond length of (cyCAAC)2P2 [1.719(7) Å] (Scheme 41). Meanwhile, the P–P bond distance in 74 [2.203(6) Å] is comparable with the P–P single bond distance of molecular tetrahedral P4 (2.21 Å) and also comparable with that of (IPr)2P2 [2.2052(10) Å] and (cyCAAC)2P2 [2.184(3) Å] (Scheme 41).77,78 The experimental data indicate that the bis(phosphinidene) (74) is better represented as bis(phosphaalkene), whereas the antimony analogue (75) is described as a BICAAC stabilized Sb2 core.
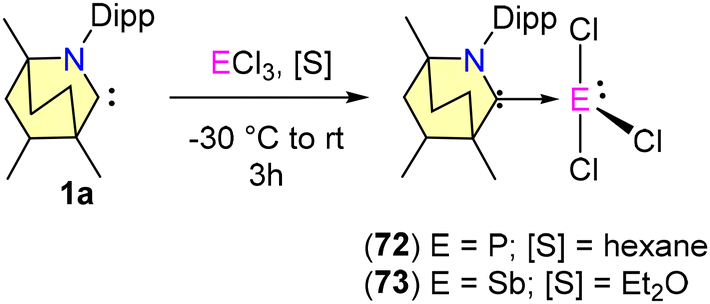 | ||
| Scheme 45 Synthesis of (BICAAC)ECl3 adducts (E = P and Sb). | ||
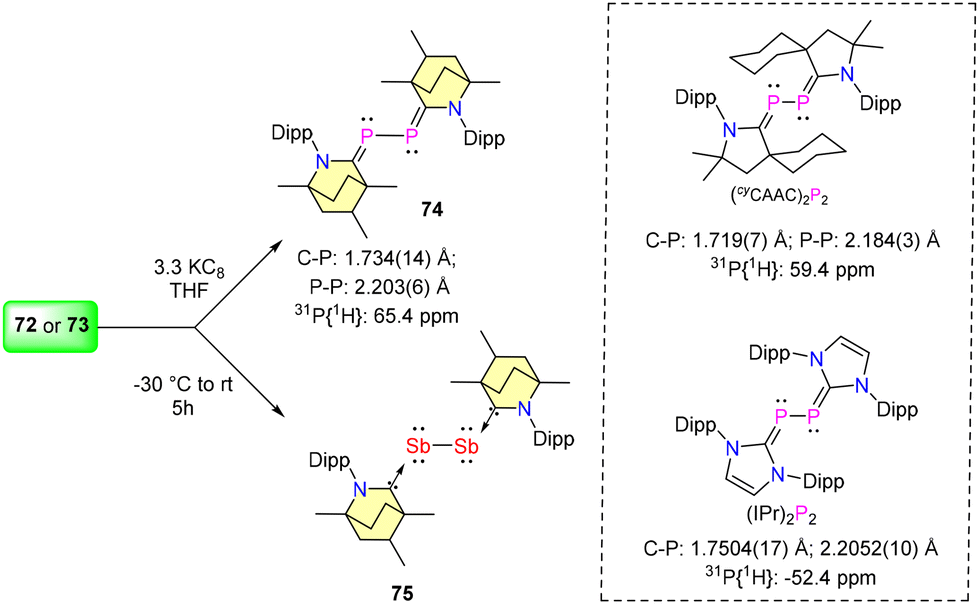 | ||
| Scheme 46 Synthesis and comparison of (BICAAC)2E2 complexes. | ||
In 2023, our group reported the synthesis of BICAAC stabilised halophosphaalkenes (76–78), which were obtained by reacting free BICAACs with PX3 (X, X = Cl, Br, and I) in THF, as shown in Scheme 47.79 Compounds 76–78 were characterized by single crystal XRD techniques and heteronuclear NMR spectroscopy. The 31P{1H} studies revealed that compound 76 shows a downfield shift at 115.35 ppm compared to 77 (103.12 ppm) and 78 (56.13 ppm), and this is mainly due to the electronegativity differences between halogens. Furthermore, the reduction of compound 77 with 1 equiv. of KC8 resulted in the formation of 74. Note that compound 74 can also be synthesized from the direct reduction of (BICAAC)PCl3 as discussed (vide supra).76 There have been several reports where NHC and CAACs have been used as supporting backbone ligands for the isolation of phosphenium cations, but none have been found to show air and moisture stability.80–83 Recently, our group employed a BICAAC for the isolation of an air- and water-stable phosphenium cation 78.84 [(BICAAC)PPh2]OTf was obtained in good yields from the reaction of the BICAAC with Ph2PCl in THF, followed by ion exchange with LiOTf. The increased π-accepting properties of the BICAAC have been reflected by 31P{1H} NMR spectroscopic data, as compound 78 exhibits a singlet peak at +13.73 ppm, which is downfield shifted in comparison with its NHC85,86 [(Me2Im)P (Ph)2][PF6] (31P{1H} = −25.89 ppm) and CAAC82,83 [(Me2CAAC)P(Ph)2][SbF6] (31P{1H} = 0 ppm) analogues. Furthermore, the cyclic voltammogram of 78 showed one electron reversible redox process. The reduction of 78 with 1 equiv. of KC8 resulted in the formation of radical phosphine 79. Moreover, the oxidation of 78 using mCPBA yielded 80, which proved the oxophilic nature of phosphorus (Scheme 48). Remarkably, complex 78 showed selective binding to fluoride ions and no interaction with other halides (Cl− and Br−) was observed.
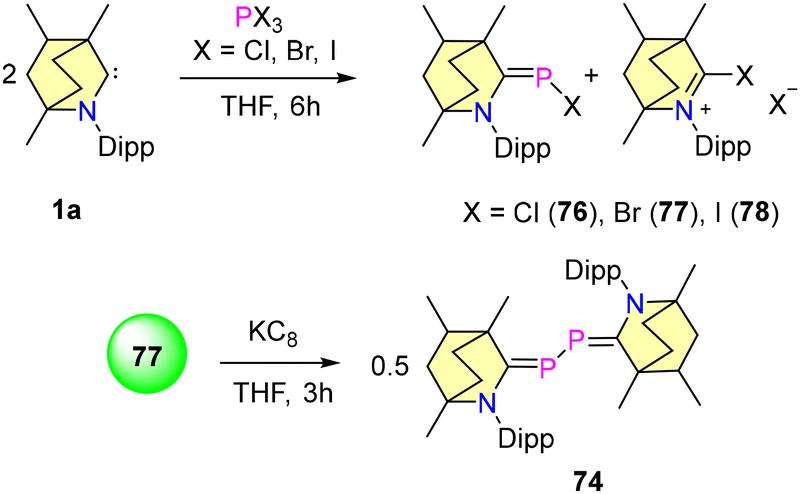 | ||
| Scheme 47 Synthesis of BICAAC stabilized halophosphaalkenes and bisphosphinidenes. | ||
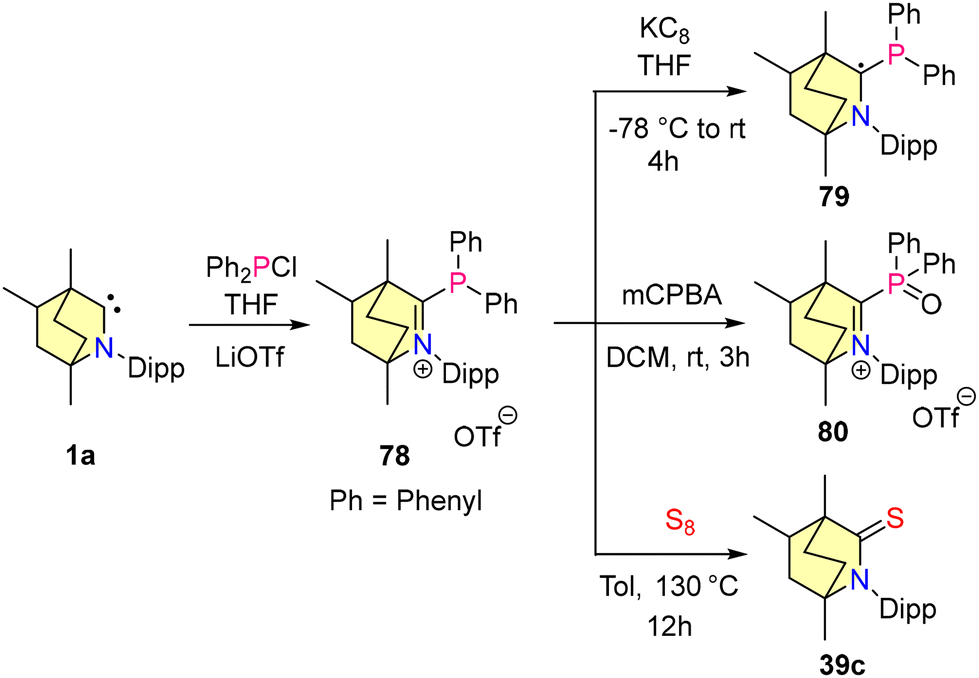 | ||
| Scheme 48 Synthesis of a BICAAC stabilized phosphenium cation (78) and its reactivity. | ||
7. Conclusions
This review focuses on the synthesis and properties of BICAACs, their usefulness in the synthesis of metal complexes, their applications as metal-free catalysts, and their role in the isolation of low-valent main-group compounds. The decreased HOMO–LUMO gap of BICAACs, combined with their exceptional nucleophilic and electrophilic nature and a unique steric environment due to the bicyclic backbone, makes them distinctive ligands. This is exemplified by the isolation of air- and moisture-stable radicals and phosphenium cations using BICAACs. Additionally, BICAACs have shown promise in the field of metal-free catalysis. BICAAC-coinage metal amides exhibit superior photoluminescence properties and quantum yields compared to CAACs and NHCs, likely due to the system's rigidity, which reduces various vibrational relaxation processes of the excited photoelectrons.Compared to NHCs and five-membered CAACs, the chemistry of BICAACs has been less explored, possibly due to their relatively recent introduction to carbene chemistry. Therefore, there is significant potential for further investigation. For instance, the chemistry of s-block elements with BICAACs remains largely unexplored. Similarly, there are few examples of stable organic radicals and main-group radicals with BICAACs, and no reports of BICAAC-supported Al, Sn, or Bi complexes. Furthermore, the functionalization of BICAACs to create chelating ligands has not been attempted, although the related chemistry of NHCs is well established. The metal-mimicking metal-free catalysis of BICAACs is also in its infancy but holds great promise for further exploration. Due to its chiral structure, BICAACs can be envisioned for applications relevant to chirality (akin to CAACs).87 Moreover, theoretical advances in this area could encourage experimental chemists to explore their chemistry. In conclusion, the field of carbene chemistry is undergoing rapid expansion, with the isolation of numerous exotic and previously unattainable species, and we believe that BICAACs will make significant contributions to this domain.
Author contributions
All the authors contributed to writing the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by SERB, New Delhi, India (CRG/2023/002353), CSIR, India (01/3041/21/EMR-II) and ASHOKA-IITD MFIRP. Ankita thanks PMRF for the financial support and fellowship. We thank CRF, IIT Delhi for the synthesis lab facility.References
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- (a) P. R. Sultane, G. Ahumada, D. Janssen-Müller and C. W. Bielawski, Angew. Chem., Int. Ed., 2019, 58, 16320–16325 CrossRef CAS; (b) S. Appel, P. Brüggemann and C. Ganter, Chem. Commun., 2020, 56, 9020–9023 RSC; (c) H. Kim, M. Kim, H. Song and E. Lee, Chem. – Eur. J., 2021, 27, 3849–3854 CrossRef CAS; (d) M. Kim, H. Kim, S. Kim, S. Hong and E. Lee, Organometallics, 2022, 41, 1905–1910 CrossRef CAS; (e) J. Volk, M. Heinz, M. Leibold, C. Bruhn, T. Bens, B. Sarkar, M. C. Holthausen and U. Siemeling, Chem. Commun., 2022, 58, 10396–10399 RSC; (f) S. Solé, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2001, 292, 1901–1903 CrossRef; (g) D. Magis, J. J. Cabrera-Trujillo, J. Vignolle, J.-M. Sotiropoulos, D. Taton, K. Miqueu and Y. Landais, J. Am. Chem. Soc., 2024, 146, 16802–16813 CrossRef CAS PubMed; (h) Y. K. Loh, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2023, 145, 2064–2069 CrossRef CAS.
- (a) S. P. Nolan, N-Heterocyclic Carbenes, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 CrossRef; (b) H. V. Huynh, The Organometallic Chemistry of N-Heterocyclic Carbenes, John Wiley & Sons, Ltd, Chichester, UK, 2017 CrossRef; (c) N. I. Korotkikh, G. F. Rayenko, O. P. Shvaika, T. M. Pekhtereva, A. H. Cowley, J. N. Jones and C. L. B. Macdonald, J. Org. Chem., 2003, 68, 5762–5765 CrossRef CAS; (d) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (e) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS; (f) P. W. Antoni, C. Golz, J. J. Holstein, D. A. Pantazis and M. M. Hansmann, Nat. Chem., 2021, 13, 587–593 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- (a) B. Rao, H. Tang, X. Zeng, L. L. Liu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 14915–14919 CrossRef CAS; (b) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS.
- E. Tomas-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef CAS PubMed.
- C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS.
- K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016, 108, 8–14 CrossRef CAS.
- K. Denk, P. Sirsch and W. A. Herrmann, J. Organomet. Chem., 2002, 649, 219–224 CrossRef CAS.
- K. C. Mondal, B. Dittrich, B. Maity, D. Koley and H. W. Roesky, J. Am. Chem. Soc., 2014, 136, 9568–9571 CrossRef CAS PubMed.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123 CrossRef CAS.
- F. Vermersch, S. Yazdani, G. P. Junor, D. B. Grotjahn, R. Jazzar and G. Bertrand, Angew. Chem., Int. Ed., 2021, 60, 27253–27257 CrossRef CAS.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2015, 2416–2425 CrossRef CAS.
- K. C. Mondal, S. Roy, B. Maity, D. Koley and H. W. Roesky, Inorg. Chem., 2016, 55, 163–169 CrossRef CAS.
- (a) E. A. LaPierre, L. K. Watanabe, B. O. Patrick, J. M. Rawson, H. M. Tuononen and I. Manners, J. Am. Chem. Soc., 2023, 145, 9223–9232 CrossRef CAS PubMed; (b) B. Li, S. Kundu, A. C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2016, 56, 397–400 CrossRef; (c) B. Das, A. Makol and S. Kundu, Dalton Trans., 2022, 51, 12404–12426 RSC; (d) J. E. Walley and R. J. Gilliard Jr, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, pp. 1–14; (e) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed; (f) M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef; (g) M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed; (h) A. Maiti, J. Stubbe, N. I. Neuman, P. Kalita, P. Duari, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Angew. Chem., Int. Ed., 2020, 59, 6729–6734 CrossRef CAS; (i) S. A. Gonsales, Z. C. Mueller, F. Zhao, P. H. S. Paioti, L. Karmazin, J. Wan, F. Liu, K. N. Houk and A. H. Hoveyda, J. Am. Chem. Soc., 2021, 143, 20640–20644 CrossRef CAS PubMed; (j) V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2014, 54, 1919–1923 CrossRef PubMed; (k) R. K. Singh, T. K. Khan, S. Misra and A. K. Singh, J. Organomet. Chem., 2021, 956, 122133 CrossRef CAS.
- S. Kang, S. E. Byeon and H. J. Yoon, Bull. Korean Chem. Soc., 2021, 42, 712–723 CrossRef CAS.
- H. Song, E. Pietrasiak and E. Lee, Acc. Chem. Res., 2022, 55, 2213–2223 CrossRef CAS PubMed.
- S. K. Kushvaha, A. Mishra, H. W. Roesky and K. C. Mondal, Chem. Asian J., 2022, 17(7), e202101301 CrossRef.
- A. Tyagi, S. Mondal, Anmol, V. Tiwari, T. Karmakar and S. Kundu, Chem. Commun., 2023, 59, 110–113 RSC.
- (a) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (b) M.-M. Li, X. Chen, Y. Deng and J. Lu, RSC Adv., 2021, 11, 38060–38078 RSC; (c) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC.
- (a) Y. Pan, X. Jiang, Y.-M. So, C. T. To and G. He, Catalysts, 2020, 10, 71 CrossRef CAS; (b) E. Peris, Chem. Rev., 2017, 118, 9988–10031 CrossRef PubMed.
- R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed.
- N. V. Tzouras, E. A. Martynova, X. Ma, T. Scattolin, B. Hupp, H. Busen, M. Saab, Z. Zhang, L. Falivene, G. Pisanò, K. Van Hecke, L. Cavallo, C. S. J. Cazin, A. Steffen and S. P. Nolan, Chem. – Eur. J., 2021, 27, 11904–11911 CrossRef CAS.
- S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC.
- (a) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS; (b) R. Yadav, S. Sinhababu, R. Yadav and S. Kundu, Dalton Trans., 2022, 51, 2170–2202 RSC.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS; (b) S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- D. Magis, J. J. Cabrera-Trujillo, J. Vignolle, J.-M. Sotiropoulos, D. Taton, K. Miqueu and Y. Landais, J. Am. Chem. Soc., 2024, 146, 16802–16813 CrossRef CAS.
- (a) S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390 CrossRef CAS PubMed; (b) M. Marinelli, C. Santini and M. Pellei, Curr. Top. Med. Chem., 2016, 16, 2995–3017 CrossRef CAS; (c) M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC; (d) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (e) J. Hossain, R. Akhtar and S. Khan, Polyhedron, 2021, 201, 115151 CrossRef CAS.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS.
- D. Pichon, M. Soleilhavoup, J. Morvan, G. P. Junor, T. Vives, C. Crévisy, V. Lavallo, J.-M. Campagne, M. Mauduit, R. Jazzar and G. Bertrand, Chem. Sci., 2019, 10, 7807–7811 RSC.
- J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS.
- (a) K. K. Manar, S. Chakrabortty, V. K. Porwal, D. Prakash, S. K. Thakur, A. R. Choudhury and S. Singh, ChemistrySelect, 2020, 5, 9900–9907 CrossRef CAS; (b) G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2008, 693, 1674–1682 CrossRef CAS PubMed.
- E. A. Romero, T. Zhao, R. Nakano, X. Hu, Y. Wu, R. Jazzar and G. Bertrand, Nat. Catal., 2018, 1, 743–747 CrossRef CAS.
- (a) F. Chotard, V. Sivchik, M. Linnolahti, M. Bochmann and A. S. Romanov, Chem. Mater., 2020, 32, 6114–6122 CrossRef CAS; (b) A. S. Romanov, S. T. E. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington and M. Bochmann, Adv. Opt. Mater., 2018, 6, 1801347 CrossRef.
- X.-F. Song, Z.-W. Li, W.-K. Chen, Y.-J. Gao and G. Cui, Inorg. Chem., 2022, 61, 7673–7681 CrossRef CAS.
- Y. Gao, S. Yazdani, A. Kendrick, G. P. Junor, T. Kang, D. B. Grotjahn, G. Bertrand, R. Jazzar and K. M. Engle, Angew. Chem., Int. Ed., 2021, 60, 19871–19878 CrossRef CAS PubMed.
- Y. Gao, N. Kim, S. D. Mendoza, S. Yazdani, A. F. Vieira, M. Liu, A. Kendrick, D. B. Grotjahn, G. Bertrand, R. Jazzar and K. M. Engle, ACS Catal., 2022, 12, 7243–7247 CrossRef CAS PubMed.
- S. Yazdani, G. P. Junor, J. L. Peltier, M. Gembicky, R. Jazzar, D. B. Grotjahn and G. Bertrand, ACS Catal., 2020, 10, 5190–5201 CrossRef CAS.
- S. K. Thakur, M. Kaur, K. K. Manar, M. Adhikari, A. R. Choudhury and S. Singh, Chem. – Eur. J., 2022, 28, e202202237 CrossRef CAS PubMed.
- S. Pelties and R. Wolf, Z. Anorg. Allg. Chem., 2013, 639, 2581–2585 CrossRef CAS.
- (a) N. M. Rajendran, N. Gautam, P. Sarkar, J. Ahmed, A. Das, S. Das, S. K. Pati and S. K. Mandal, Chem. Commun., 2021, 57, 5282–5285 RSC; (b) A. P. Singh, P. P. Samuel, H. W. Roesky, M. C. Schwarzer, G. Frenking, N. S. Sidhu and B. Dittrich, J. Am. Chem. Soc., 2013, 135, 7324–7329 CrossRef CAS PubMed.
- M. Nagyházi, Á. Lukács, G. Turczel, J. Hancsók, J. Valyon, A. Bényei, S. Kéki and R. Tuba, Angew. Chem., Int. Ed., 2022, 61, e202204413 CrossRef PubMed.
- J. Deme, M. Nagyházi, Z. May, J. Hancsók, J. Valyon, S. Kéki, R. Tuba and G. Turczel, React. Kinet., Mech. Catal., 2022, 135, 2519–2531 CrossRef CAS.
- S. Chakrabortty, M. Kaur, M. Adhikari, K. K. Manar and S. Singh, Inorg. Chem., 2021, 60, 6209–6217 CrossRef CAS.
- H. Lebel, M. K. Janes, A. B. Charette and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5046–5047 CrossRef CAS PubMed.
- C. Metallinos, F. B. Barrett, J. L. Chaytor and M. E. A. Heska, Org. Lett., 2004, 6, 3641–3644 CrossRef CAS PubMed.
- M. Nagyházi, B. Almási, Á. Lukács, A. Bényei, T. Nagy, S. Kéki and R. Tuba, J. Mol. Struct., 2022, 1256, 132483 CrossRef.
- M. Kaur, M. Adhikari, K. K. Manar, Y. Yogesh, D. Prakash and S. Singh, Inorg. Chem., 2024, 63, 1513–1523 CrossRef CAS.
- P. Bharadwaz, R. D. Dewhurst and A. K. Phukan, Adv. Synth. Catal., 2018, 360, 4543–4561 CrossRef CAS.
- R. Logdi, A. Bag and A. K. Tiwari, J. Mol. Graphics Modell., 2019, 93, 107437 CrossRef CAS.
- S. Dutta, S. De, S. Bose, E. Mahal and D. Koley, Eur. J. Inorg. Chem., 2020, 2020, 638–655 CrossRef CAS.
- K. K. Manar, V. K. Porwal, R. S. Kamte, M. Adhikari, S. K. Thakur, D. Bawari, A. R. Choudhury and S. Singh, Dalton Trans., 2019, 48, 17472–17478 RSC.
- N. Kuhn, G. Henkel, T. Kratz, J. Kreutzberg, R. Boese and A. H. Maulitz, Chem. Ber., 1993, 126, 2041–2045 CrossRef CAS.
- T. Böttcher, S. Steinhauer, L. C. Lewis-Alleyne, B. Neumann, H.-G. Stammler, B. S. Bassil, G.-V. Röschenthaler and B. Hoge, Chem. – Eur. J., 2015, 21, 893–899 CrossRef PubMed.
- J. Monot, L. Fensterbank, M. Malacria, E. Lacôte, S. J. Geib and D. P. Curran, Beilstein J. Org. Chem., 2010, 6, 709–712 CrossRef PubMed.
- P. K. Hota, S. Maji, J. Ahmed, N. M. Rajendran and S. K. Mandal, Chem. Commun., 2020, 56, 575–578 RSC.
- J. L. Peltier, E. Tomás-Mendivil, D. R. Tolentino, M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2020, 142, 18336–18340 CrossRef CAS.
- N. Gautam, R. Logdi, P. Sreejyothi, N. M. Rajendran, A. K. Tiwari and S. K. Mandal, Chem. Commun., 2022, 58, 3047–3050 RSC.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS.
- G. Kundu, R. Dixit, S. Tothadi, K. Vanka and S. S. Sen, Dalton Trans., 2022, 51, 14452–14457 RSC.
- N. Gautam, R. Logdi, S. P. A. Roy, A. K. Tiwari and S. K. Mandal, Chem. Sci., 2023, 14, 5079–5086 RSC.
- S. Bansal, A. Biswas, A. Kundu, M. Adhikari, S. Singh and D. Adhikari, ACS Catal., 2024, 14, 8087–8095 CrossRef CAS.
- (a) B. Das, R. Yadav, A. Sharma, T. Karmakar and S. Kundu, Chem. Commun., 2023, 59, 5285–5288 RSC; (b) Y. Kim, J. E. Byeon, G. Y. Jeong, S. S. Kim, H. Song and E. Lee, J. Am. Chem. Soc., 2021, 143, 8527–8532 CrossRef CAS.
- A. Das, S. Saha, S. Maji, P. Sarkar, A. Jose, M. M. Bhatt, A. Bhunia, A. Dutta, S. K. Pati and S. K. Mandal, Chem. – Eur. J., 2024, 30, e202303411 CrossRef CAS PubMed.
- (a) M. Adhikari, S. De, K. K. Manar, S. K. Thakur, R. S. Kamte, D. Koley and S. Singh, Eur. J. Inorg. Chem., 2024, e202400129 CrossRef CAS; (b) Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- S. K. Thakur, S. De, M. Adhikari, K. K. Manar, D. Koley and S. Singh, Chem. – Asian J., 2024, e202400730 CrossRef.
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- M. Tretiakov, Y. G. Shermolovich, A. P. Singh, P. P. Samuel, H. W. Roesky, B. Niepötter, A. Visscher and D. Stalke, Dalton Trans., 2013, 42, 12940–12946 RSC.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- M. Adhikari, S. K. Thakur and S. Singh, Eur. J. Inorg. Chem., 2023, e202300379 CrossRef CAS.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. SchaeferIII, P. v R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 CrossRef CAS PubMed.
- R. Yadav, B. Das, A. Singh, Anmol, A. Sharma, C. Majumder and S. Kundu, Dalton Trans., 2023, 52, 16680–16687 RSC.
- L. L. Liu, D. A. Ruiz, F. Dahcheh and G. Bertrand, Chem. Commun., 2015, 51, 12732–12735 RSC.
- Y. Wang, H. P. Hickox, Y. Xie, P. Wei, D. Cui, M. R. Walter, H. F. Schaefer III and G. H. Robinson, Chem. Commun., 2016, 52, 5746–5748 RSC.
- L. Gu, Y. Zheng, E. Haldón, R. Goddard, E. Bill, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 8790–8794 CrossRef CAS PubMed.
- Y. Livshits-Kritsman, B. Tumanskii, G. Ménard and R. Dobrovetsky, Chem. Commun., 2020, 56, 1341–1344 RSC.
- R. Yadav, A. Sharma, B. Das, C. Majumder, A. Das, S. Sen and S. Kundu, Chem. – Eur. J., 2024, e202401730 CrossRef CAS.
- S. Gaillard and J.-L. Renaud, Dalton Trans., 2013, 42, 7255 RSC.
- B. E. Maryanoff and R. O. Hutchins, J. Org. Chem., 1972, 37, 3475–3480 CrossRef CAS.
- (a) D. Pichon, M. Soleilhavoup, J. Morvan, G. P. Junor, T. Vives, C. Crévisy, V. Lavallo, J.-M. Campagne, M. Mauduit, R. Jazzar and G. Bertrand, Chem. Sci., 2019, 10, 7807–7811 RSC; (b) J. Lorkowski, D. Bouetard, P. Yorkgitis, M. Gembicky, T. Roisnel, N. Vanthuyne, D. Munz, L. Favereau, G. Bertrand, M. Mauduit and R. Jazzar, Angew. Chem., 2023, 135, e202305404 CrossRef; (c) M. Deng, N. F. M. Mukthar, N. D. Schley and G. Ung, Angew. Chem., 2020, 132, 1244–1247 CrossRef; (d) A. M. T. Muthig, J. Wieland, C. Lenczyk, S. Koop, J. Tessarolo, G. H. Clever, B. Hupp and A. Steffen, Chem. – Eur. J., 2023, 29, e202300946 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |