
Selective hydroxylation of benzene to phenol via CuII(μ-O˙)CuII intermediate using a nonsymmetric dicopper catalyst†
Qin-Qin
Hu
a,
Qi-Fa
Chen
a,
Hong-Tao
Zhang
 *a,
Jia-Yi
Chen
*b,
Rong-Zhen
Liao
b and
Ming-Tian
Zhang
*a
*a,
Jia-Yi
Chen
*b,
Rong-Zhen
Liao
b and
Ming-Tian
Zhang
*a
aCenter of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: mtzhang@mail.tsinghua.edu.cn; zhanght18@tsinghua.org.cn
bKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, Hubei Key Laboratory of Materials Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: jiayi@hust.edu.cn
First published on 5th December 2024
Abstract
The one-step oxidation of benzene to phenol represents a significant and promising advancement in modern industries focused on the production of high-value-added chemical products. Nevertheless, challenges persist in achieving sufficient catalytic selectivity and preventing over-oxidation. Inspired by copper enzymes, we present a nonsymmetric dicopper complex ([CuII2(TPMAN)(μ-OH)(H2O)]3+, 1) for the selective oxidation of benzene to phenol. Utilizing H2O2 as the oxidant, complex 1 demonstrates remarkable catalytic activity (a TON of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 within 29 hours) and selectivity exceeding 97%, comparable to the finest homogeneous catalyst derived from first-row transition metals. It is noteworthy that the significant substituent effect, alongside a negligible kinetic isotope effect (KIE = 1.05), radical trapping experiments, and an inconsistent standard selectivity test of the ˙OH radicals, all contradict the conventional Fenton mechanism and rebound pathway. Theoretical investigations indicate that the active CuII(μ-O˙)CuII–OH species generated through the cleavage of the O–O bond in the CuII(μ-1,1-OOH)CuI intermediate facilitates the hydroxylation of benzene via an electrophilic attack mechanism. The nonsymmetric coordination geometry is crucial in activating H2O2 and in the process of O–O bond cleavage.
000 within 29 hours) and selectivity exceeding 97%, comparable to the finest homogeneous catalyst derived from first-row transition metals. It is noteworthy that the significant substituent effect, alongside a negligible kinetic isotope effect (KIE = 1.05), radical trapping experiments, and an inconsistent standard selectivity test of the ˙OH radicals, all contradict the conventional Fenton mechanism and rebound pathway. Theoretical investigations indicate that the active CuII(μ-O˙)CuII–OH species generated through the cleavage of the O–O bond in the CuII(μ-1,1-OOH)CuI intermediate facilitates the hydroxylation of benzene via an electrophilic attack mechanism. The nonsymmetric coordination geometry is crucial in activating H2O2 and in the process of O–O bond cleavage.
Introduction
Phenol is a vital chemical extensively utilized in the manufacturing of plastics, phenolic resins, pharmaceuticals, disinfectants, and various other products.1–3 Currently, the industrial synthesis of phenol predominantly relies on the three-step cumene process, which necessitates harsh reaction conditions and yields a mere 5% overall yield, accompanied by the equimolar production of acetone.4,5 Consequently, the one-step hydroxylation of benzene to phenol using inexpensive oxidants such as oxygen or hydrogen peroxide has emerged as a promising alternative approach, gathering substantial interest.6–10 However, this process faces two primary challenges: the activation of the phenylic C–H bond, which possesses a high bond dissociation energy of 113 kcal mol−1, and the prevention of over-oxidation of the resulting phenol.Significant advancements have been made in the development of heterogeneous materials11–19 and homogeneous molecular catalysts20–39 that facilitate this one-step phenol production. Nonetheless, these approaches remain distant from achieving large-scale industrial viability due to inadequate phenol yields and selectivity. To enhance the design of more efficient catalysts, extensive research has focused on elucidating the mechanism underlying aromatic hydroxylation, leading to the proposal of several reaction pathways. Notably, one mechanism suggests direct activation of the C–H bond by hydroxyl (˙OH) or hydroperoxyl (˙OOH) radicals produced via the Fenton-type reaction.40–44 Alternative pathways include an electrophilic aromatic substitution pathway and a rebound pathway mediated by highly energetic metal–oxygen species, such as FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species found in cytochrome-P450.45–50 Systematic investigations into molecular catalysts have revealed that the ligand denticity plays a crucial role in modulating the reaction activity and mechanism.51,52 For instance, mononuclear iron complexes, featuring tridentate N3 ligands, preferentially follow a hydroxyl radical pathway due to the instability of the iron–oxygen intermediate, thereby resulting in lower selectivity.53 In contrast, tetradentate N4 coordinated iron catalysts facilitate heterolytic cleavage of the O–O bond in the FeIII–OOH species, aided by a free coordination site, ultimately yielding more reactive FeVO intermediates that enhance both yield and selectivity.54 However, pentadentate N5 ligands in FeIII–OOH intermediates tend to favor homolytic cleavage of the O–O bond, leading to the generation of FeIVO and ˙OH, which activates benzene for hydroxylation.55
O species found in cytochrome-P450.45–50 Systematic investigations into molecular catalysts have revealed that the ligand denticity plays a crucial role in modulating the reaction activity and mechanism.51,52 For instance, mononuclear iron complexes, featuring tridentate N3 ligands, preferentially follow a hydroxyl radical pathway due to the instability of the iron–oxygen intermediate, thereby resulting in lower selectivity.53 In contrast, tetradentate N4 coordinated iron catalysts facilitate heterolytic cleavage of the O–O bond in the FeIII–OOH species, aided by a free coordination site, ultimately yielding more reactive FeVO intermediates that enhance both yield and selectivity.54 However, pentadentate N5 ligands in FeIII–OOH intermediates tend to favor homolytic cleavage of the O–O bond, leading to the generation of FeIVO and ˙OH, which activates benzene for hydroxylation.55
Inspired by copper-containing metalloenzymes, such as tyrosinase and catechol oxidase,56–61 which perform aromatic ring oxidation, copper-based catalysts have also attracted considerable attention due to their superior catalytic activity compared to other first-row transition metals, particularly in the cases of dicopper catalysts.21–25,31,32 Notably, the Cu2(6-hpa)(μ-OH) catalyst, with 6-hpa representing 1,2-bis[2-[bis(2-pyridylmethyl)aminomethyl]-6-pyridyl]ethane, exhibits a remarkable turnover number (TON) exceeding 12000 for phenol production,23 attributed to bimetallic cooperation catalysis via a μ–η1:η1-O2 type dicopper peroxo intermediate. Research in Cu–oxygen chemistry has shown that ligand denticity can significantly influence the coordination geometries of key Cu–O2 adducts and subsequently alter their reactivity, particularly regarding O–O bond cleavage.61–66 In recent years, a diverse range of dicopper–oxygen adducts, including Cu(μ–η1:η1-O2)Cu, Cu(μ–η2:η2-O2)Cu, and Cu(μ-oxo)2Cu intermediates, have been explored for the hydroxylation reaction of aromatic rings (Fig. 1a and b).57,67–70 Meanwhile, the Cu(μ–η1:η2-O2)Cu type peroxo intermediate remains largely uncharacterized in the context of selective benzene hydroxylation.
 | ||
| Fig. 1 (a) Hydroxylation of aromatics catalyzed by copper–oxygen intermediates; (b) different coordinated structures of dicopper–O2 intermediates have been well studied; and (c) the molecular structure of the nonsymmetric dicopper catalyst [CuII2(TPMAN)(μ-OH)(H2O)]3+ (1) in this work. | ||
Recently, we developed a dicopper water oxidation catalyst [CuII2(TPMAN)(μ-OH)(H2O)]3+(1, Fig. 1c), featuring a nonsymmetric ligand TPMAN (= 1-(7-((bis(pyridin-2-ylmethyl)amino)methyl)-1,8-naphthyridin-2-yl)-N-methyl-N-(pyridin-2-ylmethyl)methanamine), in which two CuII ions respectively possess different free coordination sites. This structure leads to the formation of a μ–η1:η2-O2 type dicopperperoxo intermediate, allowing for enhanced catalytic performance.65 In this study, we employ this structurally well-defined dicopper complex for the direct hydroxylation of benzene with H2O2 as the oxidant, demonstrating exceptional catalytic activity (a TON of 14000 within 29 hours) and selectivity (up to 97%) for phenol production. Mechanistic investigations reveal that the Cu(μ–η1:η2-O2)Cu intermediate can be effectively reduced by H2O2, directly generating the CuII(μ-1,1-OOH)CuI intermediate, which facilitates O–O bond cleavage. The resulting CuII(μ-O˙)CuII–OH species serves as the active entity for the electrophilic hydroxylation of benzene. Compared to previously reported dicopper catalysts,23,57,67–70 our uniquely coordinated dicopper site alters the activation pathway of H2O2, yielding a distinct copper-oxyl intermediate that enhances the selectivity of benzene hydroxylation.
Results and discussion
For the dicopper complex [CuII2(TPMAN)(μ-OH)(H2O)]3+ (1), the detailed synthetic procedure and characterization have been documented in our previous work.65 Subsequently, we conducted the hydroxylation of benzene utilizing complex 1 as the catalyst and 30% hydrogen peroxide (H2O2) as the oxidant. Under representative reaction conditions, 30 mmol benzene was dissolved in 20 mL of acetonitrile (CH3CN) and treated with 120 mmol H2O2 and 5 μmol triethylamine (Et3N) as the base, with 1 μmol catalyst 1 under heating at 45 °C for 6 hours. Following the reaction, the mixture was extracted with chloroform (CHCl3) and subsequently purified using a short neutral alumina column. After solvent removal, an analysis by 1H NMR confirmed the formation of phenol in the residue. Building on this preliminary finding, we optimized the reaction conditions, focusing on the catalyst, the ratio of oxidant/benzene, base concentrations and reaction temperature (refer to Tables 1 and S1–S3, Fig. S1†), and the products were detected and quantified by gas chromatography (Fig. S2†). Notably, the method of oxidant addition, whether one-pot or sequential, did not significantly influence the reaction outcome. Examining varying amounts of Et3N (Table S2†), we observed that when no Et3N (entry 1) was added or when excess Et3N was added (entry 3), the rate of phenol generation during the initial 7 hours was lower compared to when an optimal amount of Et3N was employed (entry 2). However, after 24 hours, the final conversion ratios and yields were comparable across all trials. Intriguingly, extending the reaction time to approximately 44 hours in the presence of high concentrations of Et3N resulted in catalytic selectivity reaching 100% (entry 3). We also evaluated various alternative bases (Table S3†), which exhibited performance analogous to that of Et3N. Additionally, the molar ratio of H2O2 to benzene significantly impacted the oxidation results (Table S1, entries 2, 4, and 5†). At a lower H2O2-to-benzene ratio, the reaction selectivity for phenol increased; however, this selectivity decreased to about 70% as the ratio increased. Conversely, the yield of phenol improved with higher ratios of hydrogen peroxide. Finally, we further optimized the catalytic activity of catalyst 1 by varying the reaction temperatures (Table 1). Our results indicate that moderate temperature enhances catalytic performance. At 65 °C after 20 h, the yield of phenol reached approximately 6.7%, based on the initial amount of benzene, with the selectivity reaching up to 92%. Notably, increasing the temperature beyond 70 °C did not yield any significant improvement in the reaction outcome.|
|
||||
|---|---|---|---|---|
| Entry | T (°C) | Conv.a (%) | Yieldb (%) | Selectivity for phenolc (%) |
| Conditions: 11 mmol of benzene, 44 mmol of 30% H2O2, 0.44 μmol of catalyst 1, and 5 μmol of Et3N in 6.3 mL of CH3CN.a Conversation% = n(consumed benzene)/n(starting benzene).b Yield% = n(phenol)/n(starting benzene).c Selectivity is calculated based on the products detected by GC. Selectivity% = n(phenol)/(n(phenol) + n(benzoquinone)). The reaction time is 20 h.d 24 h.e 12 h. | ||||
| 1 | 45 | 25 | 3.4 | 78 |
| 2 | 65 | 61 | 6.7 | 92 |
| 3 | 70 | 52 | 7.3d | 92 |
| 4 | 80 | 61 | 7.0e | 92 |
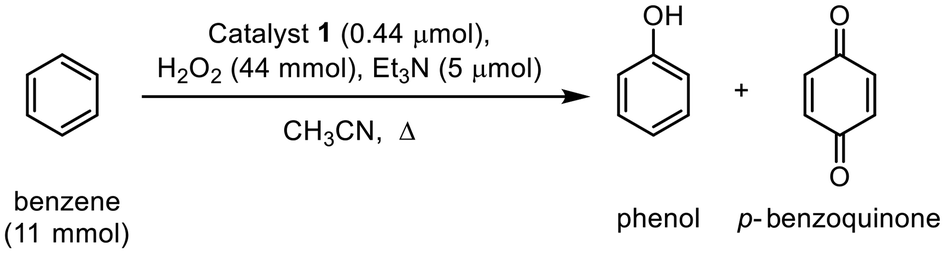
Subsequent investigations focused on the long-term catalytic performance of benzene hydroxylation using catalyst 1, with the time courses of phenol production illustrated in Fig. 2 and S5.† At a temperature of 65 °C, catalyst 1 exhibits a maximum turnover frequency (TOFmax) of approximately 859 h−1 and a maximum turnover number (TON) of 14000 after 29 hours, resulting in a phenol yield of 6.1% and a remarkable selectivity of 97%. This impressive catalytic performance is comparable to the highest TON for benzene hydroxylation using H2O2 in homogeneous catalysis (Table S4†).23 At a reduced reaction temperature of 45 °C, catalyst 1 still displayed a noteworthy TON of 8500 for phenol (Fig. S5†). It is important to note that p-benzoquinone was also generated during the early stages of the reaction alongside phenol, indicating that p-benzoquinone is a product of phenol over-oxidation.33 However, the yield of p-benzoquinone remained consistently low throughout the reaction period (Fig. S3 and S4†). These findings suggest that this nonsymmetric dicopper catalyst is capable of selectively promoting the hydroxylation of benzene to phenol, achieving the highest TON reported to date.
 | ||
| Fig. 2 Time profile formation of phenol in the hydroxylation of benzene using catalyst 1 (0.05 μmol) with 11 mmol of benzene, 44 mmol of 30% H2O2, and 5 μmol of Et3N in 6.3 mL of CH3CN at 65 °C. | ||
To further explore the reactivity of the active species, we examined various aromatic substrates, including phenol, toluene, and nitrobenzene, under identical reaction conditions (Table 2 and Fig. S6–S8†). For phenol, p-benzoquinone was the only product detected initially, while catechol was observed after 12 h, resulting in a total TON of 3200 after 23 hours. Using toluene as the substrate yielded o-cresol, p-cresol, and benzaldehyde as the major products, with a total TON of 1700 after 23 hours. The ratio of aromatic to aliphatic oxidations was approximately 2.7:1.0, and the distribution of cresols was about 1.6:1.0:0 (o:p:m), which is in contrast with the ratios typically associated with the Fenton mechanism.71 In the case of nitrobenzene, neither o-nitrophenol nor p-nitrophenol was detected. Consequently, the relative reactivity order based on the TON under the same reaction conditions was determined as follows: phenol > toluene > benzene ≫ nitrobenzene. This trend highlights the propensity for oxidation to occur preferentially on aromatic rings with electron-donating substituents, and the observed sensitivity to the electronic density of the aromatic ring suggests that the oxidation mechanism is likely metal-mediated through an electrophilic intermediate rather than involving radical pathways.
|
|
||||
|---|---|---|---|---|
| Entrya | Substrate | Products [%] (o/p/m) | TOFc (h−1) | TONd |
| a 0.44 μmol catalyst, 5 μmol base (Et3N), 44 mmol H2O2 (30%), 6.3 mL CH3CN, 45 °C, and 23 h. The substrate: 1 and 2: 11 mmol benzene; 3: 8.5 mmol phenol; 4: 8.2 mmol toluene; and 5: 8.2 mmol nitrobenzene. b 0.05 μmol catalyst, 5 μmol base (Et3N), 44 mmol H2O2 (30%), 6.3 mL CH3CN, 65 °C, and 23 h. c TOF of all products after 23 h. d TON of all products after 23 h. | ||||
| 1 | Benzene | Phenol [83] | 196 | 1200 |
| p-Benzoquinone [17] | ||||
| 2b | Benzene | Phenol [97] | 859 | 14000 |
| p-Benzoquinone [3] | ||||
| 3 | Phenol | Catechol [40] | 332 | 3200 |
| p-Benzoquinone [60] | ||||
| 4 | Toluene | Cresol [73] | 245 | 1700 |
| (60:40:trace) |
||||
| Benzaldehyde [27] | ||||
| 5 | Nitrobenzene | No product | — | — |
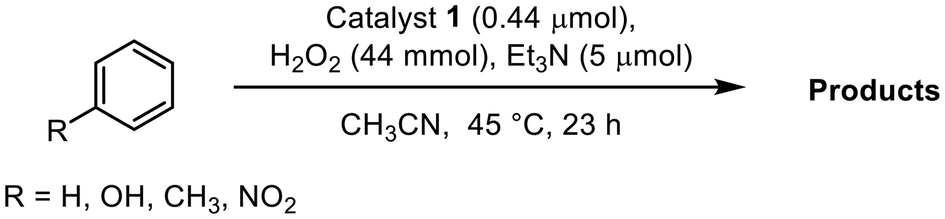
To investigate the reaction mechanism in depth, 5,5′-dimethyl-1-pyrroline N-oxide (DMPO) was employed as a radical scavenger to assess the formation of hydroxyl (˙OH) or hydroperoxyl (˙OOH) radicals, which are potential active species in the Fenton mechanism. As demonstrated in Fig. 3a, the rates of phenol production in the presence of either 0.5 mM or 5.0 mM DMPO were consistent with the rate observed in the absence of DMPO. Additionally, the introduction of carbon tetrachloride (CCl4) did not yield detectable chlorobenzene or biphenyl species at any point during the reaction. These results indicate that no obvious radical species, including hydroxyl (˙OH) or hydroperoxyl (˙OOH) radicals, exist in the reaction solution to catalyze hydroxylation of benzene through the radical-chain mechanism. Furthermore, the kinetic isotope effect (KIE) was investigated using hexadeuterio-benzene (d6-benzene) as a substrate, resulting in a KIE value of 1.05 determined from the initial rates of phenol production (Fig. 3b). This value suggests that the C–H bond cleavage of benzene is not part of the rate-determining step. This relatively low KIE value differs significantly from the KIE (approximately 1.7–1.8) reported for Fenton-type hydroxylation72 or those existing values greater than 4 for rebound mechanisms.42,73,74 Additionally, a standard selectivity test for the reaction involving the ˙OH radical was conducted using methylcyclohexane under standard conditions.75 The gas chromatography (GC) results reveal that the product ratio of tertiary alcohol to secondary alcohol is approximately 1:15 (Fig. S9†), which does not align with the expected selectivity ratio of 3:10 (ref. 75) for the ˙OH radical mechanism. Furthermore, the lack of hydroxylation reactivity observed with nitrobenzene is inconsistent with the Fenton-type mechanism.72 These findings seem to contradict the Fenton-type mechanism and the rebound process, while they are consistent with an electrophilic aromatic substitution mechanism.51,68,76
 | ||
| Fig. 3 (a) Time courses for phenol production in the reaction of benzene oxidation catalyzed by 1 in the absence (red) and presence of DMPO [0.5 mM (black) and 5 mM (blue)] during the first hour. (b) Time courses for phenol production in the oxidation of benzene (red) and d6-benzene (black) as the substrates catalyzed by 1 under the conditions described. | ||
Moreover, the reaction kinetics of benzene oxidation catalyzed by 1 was analyzed through the initial rates of phenol production. As depicted in Fig. S10–S12,† the concentrations of benzene, catalyst, and H2O2 each exhibit a first-order linear relationship with the rate of phenol production. Based on these dependencies, the kinetic equation governing the benzene oxidation reaction can be articulated as:
| d[Phenol]/dt = kcat[catalyst 1][benzene][H2O2], |
Based on the experimental results outlined above, computational studies were performed to elucidate the details of the catalytic mechanism. The initial [CuII2(TPMAN)(μ-OH)(H2O)] species (labeled as complex 1, Fig. 4a, for detailed structure, see Table S5†), featuring two penta-coordinated copper sites, can react with H2O2 to form the CuII(μ–η1:η1-OOH)CuII intermediate Int1 (Table S5†), accompanied by an energy release of 3.8 kcal mol−1. Following this, the deprotonation of Int1 by Et3N occurs, and the generated dicopperperoxo Int2 ([CuII(μ–η1:η2-O2)CuII], Table S6) is then reduced by 1/2 equivalent of H2O2, leading to the formation of Int3 ([CuII(μ-1,1-OOH)CuI], Table S6†), which has been confirmed to be a vital intermediate in our previous study,64,77 and 1/2 equivalent of O2. The deprotonation process is calculated to be exergonic by 10.0 kcal mol−1, while the reduction of Int2 to Int3 is endergonic by +1.8 kcal mol−1; thus, Int2 has been assigned as the starting point of the energy profile (Fig. 4b). Additionally, the possibility of a direct electrophilic attack of benzene on the peroxide moiety of Int2 can be ruled out, as the calculated energy barrier is +40.7 kcal mol−1 (Fig. S13, and Table S7†).
 | ||
| Fig. 4 (a) The generated process of Int3 begins with the nonsymmetric dicopper catalyst [CuII2(TPMAN)(μ-OH)(H2O)]3+(complex 1); the left superscript of the name of stationary points indicates the spin multiplicity. (b) Gibbs energy diagram for the formation of phenol catalyzed by Int3. The core structures of important intermediates are displayed, and the half arrow near the structure represents the spin direction of the unpaired electron. The AIMD simulation for the isomerization process is also shown. | ||
Subsequently, Int3 undergoes a homolytic O–OH bond cleavage process viaTS1, generating the diradical intermediate Int4′ (Table S8†) with a barrier of +7.6 kcal mol−1. The optimized structure of the transition state TS1 is depicted in Fig. 5. TS1 is a doublet and the Mulliken spin populations on Cu1, O1, Cu2, and O2 are 0.62, 0.42, 0.53, and −0.89, respectively. Frequency analysis of 2TS1 gives only one imaginary frequency of 295.5i cm−1, related to the cleavage of the O1–O2 bond. The length of the breaking O1–O2 bond and the forming hydrogen bond of O1–H1 measures 2.38 Å and 1.91 Å, respectively. The Ab Initio Molecular Dynamics (AIMD) calculations for Int4′ indicate that Int4′ can be regarded as a metastable intermediate since it can rapidly (within one ps, Fig. S14 and S15†) isomerize to the CuII(μ-O˙)CuII–OH intermediate Int4 without any applied potential. Int4 lies at −14.9 kcal mol−1 relative to Int3, with a ground state of the doublet. In 2Int4 (Fig. 5), the distance of the formed Cu2–O2 bond is 1.80 Å, consisting of a β-electron donated by the O2 moiety and an α-electron from Cu2. It should be pointed out that the decrease in Mulliken spin population on O1 (a Mulliken spin population of 0.30) and Cu2 (a Mulliken spin population of −0.11) atoms is due to the interaction of the remaining β-electron on Cu2 with the O1 radical, which possesses an α-electron. Additionally, the formation of the CuII(μ-OH)CuII–O˙ intermediate Int4′′ (Table S9) has also been considered, with a barrier of +23.5 kcal mol−1 (TS1′, Fig. 4b, Table S9†) relative to Int3 and +15.9 kcal mol−1 higher than that of TS1. Compared to Int4′′, the formation of Int4 is kinetically more favorable; therefore, the pathway initiating from Int4 is described in detail here, and other pathways will be briefly discussed when necessary.
 | ||
| Fig. 5 Optimized structures of TS1, Int4, TS2, and TS3. Distances are shown in Å in black, Mulliken spin populations on selected atoms are shown in red, and the imaginary frequencies (in cm−1) for transition states are also shown. For clarity, unimportant hydrogen atoms are not shown. | ||
The O1 radical in Int4 then electrophilically attacks the benzene substrate directly, forming the essential O1–C1 bond through a quartet transition state TS2, with a significant Mulliken spin population distributed at the O1 (a Mulliken spin population of 0.69) and benzene (a Mulliken spin population of 0.53). The barrier of TS2 is calculated to be +14.0 kcal mol−1 relative to Int4 plus benzene, and the length of the O1–C1 bond in TS2 is 1.92 Å. The generated 4Int5 (Table S10†) is +5.2 kcal mol−1 higher in energy than 2Int4, which contains a benzene radical (a Mulliken spin population of 0.92) and two CuII ion centers. Notably, when nitrobenzene acts as a substrate, the O1–C1 bond formation via the electrophilic attack of Int4 needs to overcome higher barriers of about +15.4–16.7 kcal mol−1 (Tables S11 and S12†), consistent with the above-mentioned experimental findings. If CH3CN as the substrate is activated by Int4 abstracting the hydrogen atom, the energy barrier is also obviously higher than that of the benzene activation (Tables S11 and S12†). Thus, the CH3CN solvent would not interfere with the benzene hydroxylation reaction, and no CH3CN-related products are observed in the gas chromatograph (Fig. S8†). These results further confirm that Int4 activates the benzene via electrophilic attack rather than the abstraction of hydrogen atoms.
The ground state of transition state TS3 (Fig. 5) is a doublet, showing that the electron on the benzene radical is flipped (a Mulliken spin population of −0.43) and coupled with Cu1II (a Mulliken spin population of 0.32) antiferromagnetically. TS3 involves proton transfer from C1 to the OH moiety and electron transfer from the benzene radical to the Cu1II ion. The C1–H2, H2–O2, and Cu2–O1 bond distances in 2TS3 are 1.16, 1.79, and 1.98 Å, respectively; the barrier of TS3 is +6.3 kcal mol−1, and the imaginary frequency is 198.8i cm−1. Downhill from TS3, Int6 (Table S10†) is formed, and it is associated with a large energy release of 49.7 kcal mol−1 relative to Int5. Based on the Mulliken spin population analysis of Int6, Cu1 accepts the β-electron transferred from the benzene radical, resulting in its oxidation state becoming +1; Cu2 remains at +2 and coordinated with the generated H2O. Finally, the oxidant H2O2 reacts with Int6, yielding H2O molecules and the target product phenol and regenerating species Int3 to catalyze the next cycle.
According to the Gibbs free energy diagram derived from the DFT calculations, the electrophilic attack of the catalytically active species Int4 on the benzene (TS2) is the rate-determining step, with a calculated barrier of +14.0 kcal mol−1. This finding aligns with the experimental evidence indicating that the C–H bond cleavage of benzene was not involved in the rate-determining step. The additive base (Et3N) is solely responsible for the deprotonation of Int1 to form Int2, initiating the catalytic process via the generated active intermediate Int3. Moreover, the generation of catalytically active species Int4 (CuII(μ-O˙)CuII–OH) benefits from the nonsymmetric coordination geometry of Int3, which provides vacant coordination sites to receive the OH group.
Conclusions
In this study, we successfully demonstrated the efficacy of a nonsymmetric dicopper water oxidation catalyst ([CuII2(TPMAN)(μ-OH)(H2O)]3+, 1) for the direct oxidation of benzene to phenol using the environmentally benign oxidant H2O2. Its catalytic performance was found to be comparable to the most effective copper-based molecular catalyst reported to date. Investigations into the substituent effects on the benzene ring revealed that electron-donating groups enhanced the oxidation reaction, while electron-withdrawing groups exhibited the opposite effect, thereby suggesting the involvement of an electrophilically active intermediate in the reaction mechanism. Notably, experimental results indicating no interaction with the radical scavenger DMPO, coupled with a kinetic isotope effect (KIE) of 1.05 and an inconsistent standard selectivity test of the ˙OH radical, contradict the traditional Fenton and rebound mechanisms as pathways for this reaction. Additionally, density functional theory (DFT) calculations elucidated how the unique geometrical arrangement of the dicopper core modulates the activation pathway of H2O2, leading to the formation of an electrophilic CuII(μ-O˙)CuII–OH species. This species is pivotal in facilitating the selective hydroxylation of benzene to phenol in a controllable manner. Overall, this work advances the understanding of the mechanism underlying the one-step oxidation of benzene and provides valuable insights for the design of efficient catalysts for benzene hydroxylation.Experimental section
Materials and instruments
Chemicals were obtained commercially with the highest purity and were used without further purification unless otherwise specified. The ligand TPMAN was synthesized following our previous procedure.65 Gas chromatography (GC) data were collected using a Shimadzu 2010 Plus GC system with an Rxi®-5 ms capillary column with nitrobenzene utilized as a standard. NMR spectra were recorded on a 400 MHz Bruker BioSpin Advance III NMR spectrometer.Synthesis of [CuII2(TPMAN)(μ-OH)(H2O)](CF3SO3)3 (1(OTf)3)
The synthesis procedure of 1 has been reported in our previous work.65 A solution of the TPMAN ligand (90 mg, 0.19 mmol) in acetonitrile (10 mL), was added dropwise into a stirred solution of Cu(CF3SO3)2 (138.80 mg, 0.38 mmol) in 8 mL of MeCN:H2O (v:v = 3:1). The solution was rapidly converted to deep blue-green and allowed to stir overnight. The resulting solution was concentrated under reduced pressure and an oil was obtained. Ether (20 mL) was added, and a blue crude product was isolated after ultrasonic washing of the resulting mixture for several minutes, followed by copious washing with ethyl ether. The syrup product was recrystallized from acetonitrile and ether. The product was obtained as a blue powder (80 mg, yield 35%). HRMS (ESI+): m/z [M − H2O-3(CF3SO3)]3+, exp.: 206.0361, cal.: 206.0368; [M − H2O-2(CF3SO3)]2+, exp.: 383.5371, cal.: 383.5312. Elemental analysis for C32H32Cu2F9N7O11S3, calcd: C, 35.43; H, 2.97; N, 9.04; found: C, 35.19; H, 3.57; N, 8.76.
Catalytic reaction conditions for the hydroxylation of aromatic compounds
Under an argon atmosphere, a solution containing compound 1 (0.48 mg, 0.44 μmol), substrates (benzene (860 mg, 11 mmol), toluene (780 mg, 8.5 mmol), phenol (770 mg, 8.2 mmol), and nitrobenzene (1010 mg, 8.2 mmol)), and triethylamine (Et3N) (0.5 mg, 5.0 μmol) was prepared in acetonitrile (6.3 mL) within a Schlenk flask and heated to 65 °C while stirring. Subsequently, 4.5 mL of 30% aqueous hydrogen peroxide (H2O2, 44 mmol) was introduced under an argon atmosphere. A portion of the reaction solution was extracted using a syringe, dried with anhydrous sodium sulfate (Na2SO4), and then analyzed by gas chromatography (GC), using nitrobenzene as a standard.Calibration solution preparation
Calibration solutions were prepared to ascertain the correction factors (fi). The calibration curves were established by plotting the linear relationship between the peak area ratio (Si/S0) and the mass ratio (mi/m0). Here, Si and S0 denote the peak areas of the analyte and the internal standard, respectively, while mi and m0 refer to the masses of the analyte and the internal standard.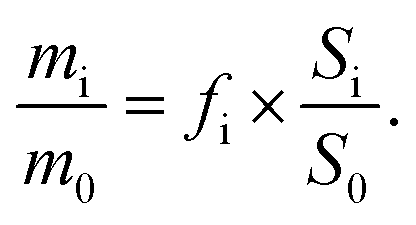 | (1) |
Quantification of the hydroxylation products
Correction factors were used to quantify the mass of the hydroxylation products (ms) using eqn (2). Ss and S0 are the peak areas of the analyte and internal standard, while ms and m0 are the masses of the analyte and internal standard, respectively. | (2) |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC Grant No. 219330007 and 22193011).References
- R. A. Sheldon and J. K. Kochi, in Metal-Catalyzed Oxidations of Organic Compounds, ed. R. A. Sheldon and J. K. Kochi, AcademicPress, New York, 1981, pp. 315–339 Search PubMed.
- M. Weber and M. Kleine-Boymann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- K. Weissermel and H. J. Arpe, Industrial Organic Chemistry, John Wiley & Sons, 3rd edn, 2008 Search PubMed.
- R. Molinari and T. Poerio, Asia-Pac. J. Chem. Eng., 2009, 5, 191–206 CrossRef.
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89–103 CrossRef CAS.
- D. P. Ivanov, L. V. Pirutko and G. I. Panov, J. Catal., 2014, 311, 424–432 CrossRef CAS.
- R. Bal, M. Tada, T. Sasaki and Y. Iwasawa, Angew. Chem., Int. Ed., 2006, 45, 448–452 CrossRef CAS PubMed.
- J. W. Han, J. Jung, Y. M. Lee, W. Nam and S. Fukuzumi, Chem. Sci., 2017, 8, 7119–7125 RSC.
- S. S. Acharyya, S. Ghosh, R. Tiwari, C. Pendem, T. Sasaki and R. Bal, ACS Catal., 2015, 5, 2850–2858 CrossRef CAS.
- Y. Zhu, W. Sun, J. Luo, W. Chen, T. Cao, L. Zheng, J. Dong, J. Zhang, M. Zhang, Y. Han, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 3861 CrossRef PubMed.
- S. Bhandari, R. Khatun, T. S. Khan, D. Khurana, M. K. Poddar, A. Shukla, V. V. D. N. Prasad and R. Bal, Green Chem., 2022, 24, 9303 RSC.
- J. Xie, X. Li, J. Guo, L. Luo, J. J. Delgado, N. Martsinovich and J. Tang, Nat. Commun., 2023, 14, 4431 CrossRef CAS.
- Y.-J. Lyu, T. Qi, H.-Q. Yang and C.-W. Hu, Catal. Sci. Technol., 2018, 8, 176–186 RSC.
- G. Wen, S. Wu, B. Li, C. Dai and D. S. Su, Angew. Chem., Int. Ed., 2015, 54, 4105–4109 CrossRef CAS PubMed.
- S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, ACS Sustainable Chem. Eng., 2017, 5, 3637–3640 CrossRef CAS.
- L. Meng, X. Zhu and E. J. M. Hensen, ACS Catal., 2017, 7, 2709–2719 CrossRef CAS PubMed.
- W. Laufer, J. P. M. Niederer and W. F. Hoelderich, Adv. Synth. Catal., 2002, 344, 1084–1089 CrossRef CAS.
- Y. Zhao, H. Cao, L. Tao, Z. Qiao and C. Ding, Dalton Trans., 2023, 52, 5399–5417 RSC.
- T. Shen, Z. Song, J. Li, S. Bai, G. Liu, X. Sun, S. Li, W. Chen, L. Zheng and Y. F. Song, Small, 2023, 19, e2303420 CrossRef.
- G. Capocasa, G. Olivo, A. Barbieri, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2017, 7, 5677–5686 RSC.
- L. Vilella, A. Conde, D. Balcells, M. M. Díaz-Requejo, A. Lledós and P. J. Pérez, Chem. Sci., 2017, 8, 8373–8383 RSC.
- A. Conde, M. Mar Díaz-Requejo and P. J. Pérez, Chem. Commun., 2011, 47, 8154–8156 RSC.
- T. Tsuji, A. A. Zaoputra, Y. Hitomi, K. Mieda, T. Ogura, Y. Shiota, K. Yoshizawa, H. Sato and M. Kodera, Angew. Chem., Int. Ed., 2017, 56, 7779–7782 CrossRef CAS PubMed.
- M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856–2863 RSC.
- L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Dalton Trans., 2015, 44, 8013–8020 RSC.
- O. Shoji, S. Yanagisawa, J. K. Stanfield, K. Suzuki, Z. Cong, H. Sugimoto, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2017, 56, 10324–10329 CrossRef CAS PubMed.
- K. Ikeda, K. Yoshizawa and Y. Shiota, Inorg. Chem., 2021, 61, 10–14 CrossRef PubMed.
- H. Qi, D. Xu, J. Lin and W. Sun, Mol. Catal., 2022, 528, 112441 CrossRef CAS.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed.
- S. Muthuramalingam, K. Anandababu, M. Velusamy and R. Mayilmurugan, Catal. Sci. Technol., 2019, 9, 5991–6001 RSC.
- E. Borrego, L. Tiessler-Sala, J. J. Lázaro, A. Caballero, P. J. Pérez and A. Lledós, Organometallics, 2022, 41, 1892–1904 CrossRef CAS PubMed.
- S. Kumari, S. Muthuramalingam, A. K. Dhara, U. P. Singh, R. Mayilmurugan and K. Ghosh, Dalton Trans., 2020, 49, 13829–13839 RSC.
- J. Kumari, S. M. Mobin, S. Mukhopadhyay and K. M. Vyas, Inorg. Chem. Commun., 2019, 105, 217–220 CrossRef CAS.
- L. Carneiro and A. R. Silva, Catal. Sci. Technol., 2016, 6, 8166–8176 RSC.
- O. Y. Lyakin, A. M. Zima, N. V. Tkachenko, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2018, 8, 5255–5260 CrossRef CAS.
- N. V. Tkachenko, R. V. Ottenbacher, O. Y. Lyakin, A. M. Zima, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, ChemCatChem, 2018, 10, 4052–4057 CrossRef CAS.
- N. V. Tkachenko, O. Y. Lyakin, A. M. Zima, E. P. Talsi and K. P. Bryliakov, J. Organomet. Chem., 2018, 871, 130–134 CrossRef CAS.
- A. M. Zima, O. Y. Lyakin, D. P. Lubov, K. P. Bryliakov and E. P. Talsi, Mol. Catal., 2020, 483, 110708 CrossRef CAS.
- E. Masferrer-Rius, M. Borrell, M. Lutz, M. Costas and R. J. M. K. Gebbink, Adv. Synth. Catal., 2021, 363, 3783–3795 CrossRef CAS.
- S. R. Kalahrudi, A. Shakeri, A. Ghadimi and H. Mahdavi, J. Membr. Sci., 2020, 611, 118230 CrossRef.
- C. Walling and R. A. Johnson, J. Am. Chem. Soc., 1975, 97, 363–367 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- S. Ito, A. Mitarai, K. Hikino, M. Hirama and K. Sasaki, J. Org. Chem., 1992, 57, 6937–6941 CrossRef CAS.
- X. Jia, C. Liu, X. Xu, F. Wang, W. Li, L. Zhang, S. Jiao, G. Zhu and X. Wang, RSC Adv., 2023, 13, 19140–19148 RSC.
- L. T. Burka, T. M. Plucinski and T. L. Macdonald, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6680–6684 CrossRef CAS PubMed.
- S. P. de Visser and S. Shaik, J. Am. Chem. Soc., 2003, 125, 7413–7424 CrossRef CAS PubMed.
- C. M. Bathelt, L. Ridder, A. J. Mulholland and J. N. Harvey, J. Am. Chem. Soc., 2003, 125, 15004–15005 CrossRef CAS PubMed.
- M.-J. Kang, W. J. Song, A.-R. Han, Y. S. Choi, H. G. Jang and W. Nam, J. Org. Chem., 2007, 72, 6301–6304 CrossRef CAS PubMed.
- M. Asaka and H. Fujii, J. Am. Chem. Soc., 2016, 138, 8048–8051 CrossRef CAS PubMed.
- S. Shaik, P. Milko, P. Schyman, D. Usharani and H. Chen, J. Chem. Theory Comput., 2011, 7, 327–339 CrossRef CAS PubMed.
- S. Muthuramalingam, K. Anandababu, M. Velusamy and R. Mayilmurugan, Inorg. Chem., 2020, 59, 5918–5928 CrossRef CAS PubMed.
- A. Rajeev, M. Balamurugan and M. Sankaralingam, ACS Catal., 2022, 12, 9953–9982 CrossRef CAS.
- B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 15337–15345 RSC.
- J. N. Rebilly, W. Zhang, C. Herrero, H. Dridi, K. Senechal-David, R. Guillot and F. Banse, Chem. – Eur. J., 2020, 26, 659–668 CrossRef CAS PubMed.
- S. Xu, A. Draksharapu, W. Rasheed and L. Que, Jr., J. Am. Chem. Soc., 2019, 141, 16093–16107 CrossRef CAS PubMed.
- L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. P. Stack, Science, 2005, 308, 1890–1892 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- L. Chiang, W. Keown, C. Citek, E. C. Wasinger and T. D. Stack, Angew. Chem., Int. Ed., 2016, 55, 10453–10457 CrossRef CAS PubMed.
- A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rubhausen, O. Troeppner, I. Ivanovic-Burmazovic, E. C. Wasinger, T. D. Stack and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398–5401 CrossRef CAS PubMed.
- M. F. Qayyum, R. Sarangi, K. Fujisawa, T. D. P. Stack, K. D. Karlin, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 17417–17431 CrossRef CAS PubMed.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS PubMed.
- H.-T. Zhang, F. Xie, Y.-H. Guo, Y. Xiao and M.-T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202310775 CrossRef CAS PubMed.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- Q.-Q. Hu, X.-J. Su and M.-T. Zhang, Inorg. Chem., 2018, 57, 10481–10484 CrossRef CAS PubMed.
- X.-J. Su, C. Zheng, Q.-Q. Hu, H.-Y. Du, R.-Z. Liao and M.-T. Zhang, Dalton Trans., 2018, 47, 8670–8675 RSC.
- A. Kunishita, J. D. Scanlon, H. Ishimaru, K. Honda, T. Ogura, M. Suzuki, C. J. Cramer and S. Itoh, Inorg. Chem., 2008, 47, 8222–8232 CrossRef CAS PubMed.
- P. L. Holland, K. R. Rodgers and W. B. Tolman, Angew. Chem., Int. Ed., 1999, 38, 1139–1142 CrossRef CAS PubMed.
- G. Battaini, E. Monzani, A. Perotti, C. Para, L. Casella, L. Santagostini, M. Gullotti, R. Dillinger, C. Näther and F. Tuczek, J. Am. Chem. Soc., 2003, 125, 4185–4198 CrossRef CAS PubMed.
- T. Abe, Y. Kametani, K. Yoshizawa and Y. Shiota, Inorg. Chem., 2021, 60, 4599–4609 CrossRef CAS PubMed.
- H. Marusawa, K. Ichikawa, N. Narita, H. Murakami, K. Ito and T. Tezuka, Bioorg. Med. Chem., 2002, 10, 2283–2290 CrossRef CAS PubMed.
- R. Augusti, A. O. Dias, L. L. Rocha and R. M. Lago, J. Phys. Chem. A, 1998, 102, 10723–10727 CrossRef CAS.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- J. C. Schöneboom, S. Cohen, H. Lin, S. Shaik and W. Thiel, J. Am. Chem. Soc., 2004, 126, 4017–4034 CrossRef PubMed.
- S. N. Sharma, H. R. Sonawane and S. Dev, Tetrahedron, 1985, 41, 2483–2491 CrossRef CAS.
- K. Honda, J. Cho, T. Matsumoto, J. Roh, H. Furutachi, T. Tosha, M. Kubo, S. Fujinami, T. Ogura, T. Kitagawa and M. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 3304–3307 CrossRef CAS PubMed.
- Q.-F. Chen, K.-L. Xian, H.-T. Zhang, X.-J. Su, R.-Z. Liao and M.-T. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202317514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02872d |
| This journal is © The Royal Society of Chemistry 2025 |