
A multi-centre metal-free COF@g-C3N4 catalyst assembled with covalent bonds for photocatalytic CO2 reduction†
Xiang
Wang
a,
Zhenping
Wang
a,
Yanling
Liu
*a,
Wanzhen
Peng
a,
Xiangyi
Fu
a,
Jie
Zhou
 *a,
Lizhi
Han
a,
Yingjie
Hua
a and
Zi-Yan
Zhou
*bc
*a,
Lizhi
Han
a,
Yingjie
Hua
a and
Zi-Yan
Zhou
*bc
aKey Laboratory of Electrochemical Energy Storage and Energy Conversion of Hainan Province, School of Chemistry and Chemical Engineering, Hainan Normal University, Haikou, Hainan 571158, China. E-mail: Liuyl7809@163.com; 920486@hainnu.edu.cn
bShandong Engineering Research Center of Green and High-value Marine Fine Chemicals, Weifang University of Science and Technology, Shouguang, Shandong 262700, China. E-mail: zyzhou@sdut.edu.cn
cCollege of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, 255000 Shandong, China
First published on 5th December 2024
Abstract
Photocatalytic carbon dioxide (CO2) reduction technology using solar energy can convert carbon dioxide into fuels and chemicals, and is one of the most effective strategies to mitigate the energy crisis and greenhouse effect. In recent years, covalent organic frameworks (COFs) have flourished due to their unique advantages and have received extensive attention in the field of photocatalytic reduction of CO2. Here, we use the pre-designability of COFs to preserve the aldehyde group at the end of the COF skeleton by the motif strategy, while ensuring its excellent photosensitivity. This facilitates further assembly with amino-terminated g-C3N4 through covalent bonding, resulting in composite catalysts (COF@g-C3N4). This COF@g-C3N4 material can take g-C3N4 as the active center to undertake the main catalytic reaction function, COF as the photosensitive center to absorb light energy and generate photogenerated carriers, and covalent bonds as electron transport bridges, effectively facilitating the transfer of electrons. These three components operate independently yet synergistically to accomplish the photocatalytic CO2 reduction reaction. In addition, by integrating theoretical calculations with experimental results, the electron transfer and reaction mechanism in the photocatalytic process of COF@g-C3N4 were thoroughly explored, and a rational photocatalytic process was proposed. This multi-center metal-free catalyst, COF@g-C3N4, not only exhibits good photocatalytic performance but also is more economical and environmentally friendly, which is worthy of attention.
Introduction
Photocatalytic CO2 reduction technology is considered to be one of the most promising new energy technologies in the 21st century, and it is a recognized technology that can achieve China's dual-carbon goal, which is now booming.1 So far, more and more catalytic materials have been used for photocatalytic CO2 reduction, such as nonmetallic oxides,2 metal oxides,3 metal sulfides4 and their heterojunction materials.5 Although encouraging results have been achieved, inorganic photocatalysts still have undesirable defects; for example, inorganic semiconductor photocatalysts usually have weak visible light response and CO2 adsorption capacities and too fast photogenerated electron–hole pair self-complexation, the tunability of the composition and photochemical properties is very limited, and it is very difficult to modify and change their properties.6 In addition, problems such as the complicated synthesis steps, complex structure, unclear catalytic sites, and lack of clear structural information to identify catalytic sites of inorganic catalysts are also major factors that are not conducive to in-depth research on the mechanism of photocatalytic reduction of CO2.7 Therefore, it is of great significance to develop organic crystalline photocatalyst materials with well-defined structures and high efficiency and stability for artificial photosynthesis.Covalent organic frameworks (COFs), as an emerging class of organic crystalline porous materials, have shown great potential for application in the field of photocatalytic reduction of CO2 due to their advantages of structural designability, large surface area, stability, high porosity, and large light absorption range.8 However, the photocatalytic CO2 reduction efficiency is still relatively low because metal-free COFs usually lack efficient catalytic active sites.9 Currently, in order to solve the activity problem of COF materials, metal ions or metal complexes are usually anchored as catalytic active centers through restriction effects or coordination.10 Nevertheless, due to the relatively weak ligand bonds, the composite catalysts obtained from this post-modification are usually limited by poor chemical and thermal stability; in particular, under harsh reaction conditions, the metal ions or metal complexes are prone to decomposition and separation from COFs, and cannot be reused.11 In summary, if more robust and stable covalent bonds can be utilized to link COFs with crystalline polymeric materials, which exhibit photocatalytic activity (such as functionalized polymers and organic molecular polymers), the chemical stability and thermal stability of the composite material can be significantly improved, and because of the strong electron-conducting ability of covalent bonding, it can greatly enhance the photogenerated charge carrier separation and transfer ability of COF-based composites, which in turn improves their photocatalytic reduction of CO2 performance.
Here, we carefully prepared COF-TpBb12 with excellent photosensitivity by using Tp-CHO and Bb-NH2, and employed a COF-motif strategy to retain –CHO groups at the termini of the COF. Then, g-C3N4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 with catalytic activity and terminal –NH2 is assembled together by the solvothermal method to prepare the COF@g-C3N4 composite catalyst. In this composite catalyst, each component plays its unique role while working together as a whole. Specifically, COF-TpBb is used as the photosensitive center of the system, g-C3N4 is used as the active center, and covalent bonds are used as the electron transmission bridge to jointly complete the photocatalytic CO2 reduction reaction (CO2RR). Under appropriate conditions, the main product of the photocatalytic CO2 reduction by the composite catalyst is CO, and the yield can reach 195 μmol g−1 h−1 with a selectivity of 92%. A series of controlled experiments also investigated the effects of conditions such as loading amount, sacrificial agent, and water content on the yield and proportion of reduction products. Finally, density functional theory (DFT) calculations were utilized to delve into the mechanism of photocatalytic CO2 reduction, and based on both theoretical and experimental results, reasonable hypotheses were made about possible electron transfer pathways and the photocatalytic reaction mechanism.
13 with catalytic activity and terminal –NH2 is assembled together by the solvothermal method to prepare the COF@g-C3N4 composite catalyst. In this composite catalyst, each component plays its unique role while working together as a whole. Specifically, COF-TpBb is used as the photosensitive center of the system, g-C3N4 is used as the active center, and covalent bonds are used as the electron transmission bridge to jointly complete the photocatalytic CO2 reduction reaction (CO2RR). Under appropriate conditions, the main product of the photocatalytic CO2 reduction by the composite catalyst is CO, and the yield can reach 195 μmol g−1 h−1 with a selectivity of 92%. A series of controlled experiments also investigated the effects of conditions such as loading amount, sacrificial agent, and water content on the yield and proportion of reduction products. Finally, density functional theory (DFT) calculations were utilized to delve into the mechanism of photocatalytic CO2 reduction, and based on both theoretical and experimental results, reasonable hypotheses were made about possible electron transfer pathways and the photocatalytic reaction mechanism.
Results and discussion
Here, we carefully prepared COF-TpBb12,14 as the photosensitive center of the system as reported and utilized g-C3N4 as the active center, and further assembled COF-TpBb with g-C3N4 by a solvothermal method, which successfully yielded the reddish COF@g-C3N4 powder, and this composite was successfully used as a photocatalyst for the CO2RR.COF-TpBb was synthesized as shown in Fig. 1a by condensation of the two precursors Tp-CHO and Bb-NH2 under vacuum solvothermal conditions. As can be seen in the Fourier transform infrared (FT-IR) spectrum in Fig. 1b, the –CHO vibrational peaks of the Tp-CHO precursor at 1660 cm−1 and the –NH2 vibrational peaks of Bb-NH2 at 3000–3500 cm−1 are very obvious, whereas these vibrational peaks disappeared obviously in the IR spectrum of COF-TpBb, which proves that the two kinds of raw materials have undergone condensation reaction and COF-TpBb has been successfully synthesized. The powder X-ray diffraction (PXRD) pattern (Fig. 1c) shows that the prepared COF-TpBb possesses fairly excellent crystallinity, with the main characteristic diffraction peak at 3.5°, which corresponds to the (100) crystal plane. In order to elucidate the crystal structure, a crystal model for the parallel stacking of COF-TpBb (Fig. S1†) was established using the Material Studio software by selecting the P6/MMM space group. The simulated PXRD pattern of COF-TpBb agrees well with the experimental results, and further refinement of the PXRD pattern by Pawley also agrees well with the experimental data and residual factors of Rp = 2.52% and Rwp = 3.56%, proving the reliability of the computational model. Its optimized best cell length is a = b = 24.907 Å, c = 3.438 Å, α = β = 90°, and γ = 120°.
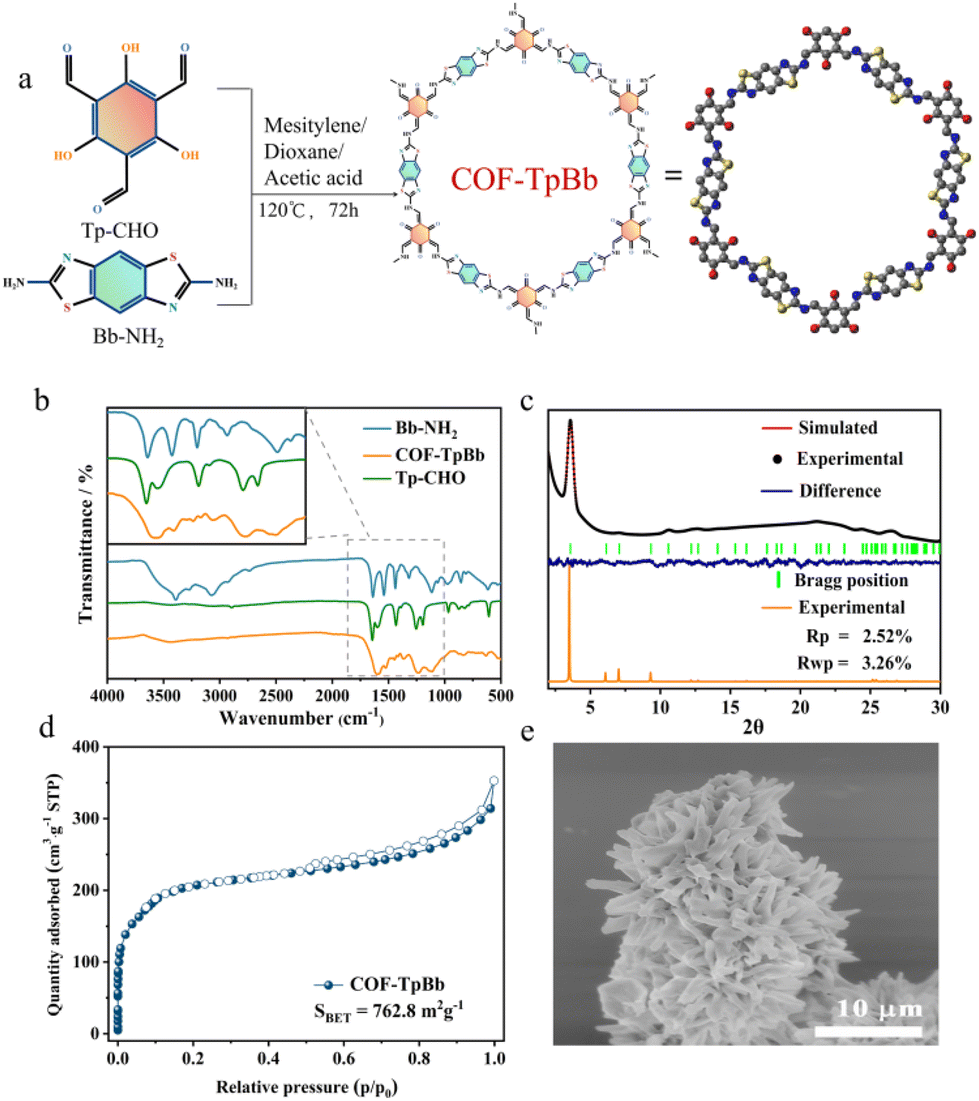 | ||
| Fig. 1 Preparation and characterization. (a) Schematic of the synthesis of COF-TpBb via the condensation of Tp-CHO and Bb-NH2; (b) IR spectra of COF-TpBb, Tp-CHO and Bb-NH2; (c) experimental and simulated PXRD patterns of COF-TpBb; (d) N2 adsorption curve of COF-TpBb at 77 K; and (e) SEM image of COF-TpBb. | ||
The specific surface area of COF-TpBb was investigated by N2 adsorption–desorption measurements at 77 K. As shown in Fig. 1d, the Brunauer–Emmett–Teller (BET) surface area of COF-TpBb was calculated to be 762.8 m2 g−1, and the maximum adsorption capacity could reach 350 cm3 g−1. The thermogravimetric diagram (Fig. S2†) shows that COF-TpBb is thermally well stabilized, and there is no significant weight loss until 280 °C.
The morphology of COF-TpBb was characterized using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). From the SEM image of Fig. 1e, it can be clearly observed that COF-TpBb is a sea urchin sphere structure made up of small nanosheets of about 1 μm stacked together, which is distinctly different from the block structure made up of fine particles stacked together for the two precursors Tp-CHO (Fig. S3†) and Bb-NH2 (Fig. S4†), and it can further illustrate the successful synthesis of COF-TpBb. The TEM image (Fig. S5†) of COF-TpBb also similarly demonstrated its lamellar structure.
As shown in Fig. 2a, to complete the photocatalytic CO2RR, we further assembled COF-TpBb with g-C3N4 using a solvothermal method to obtain the COF@g-C3N4 composite material. As shown in Fig. 2b, the IR curve of COF@g-C3N4-0.2 is closer to the IR curve of g-C3N4, probably because of the higher content of g-C3N4. As illustrated in Fig. 2c, the XRD pattern of COF@g-C3N4-0.2 contains the main characteristic diffraction peak of the COF at 3.5° and the main characteristic diffraction peak of g-C3N4 at 27°, proving that the two in COF@g-C3N4-0.2 maintain their respective crystalline states.
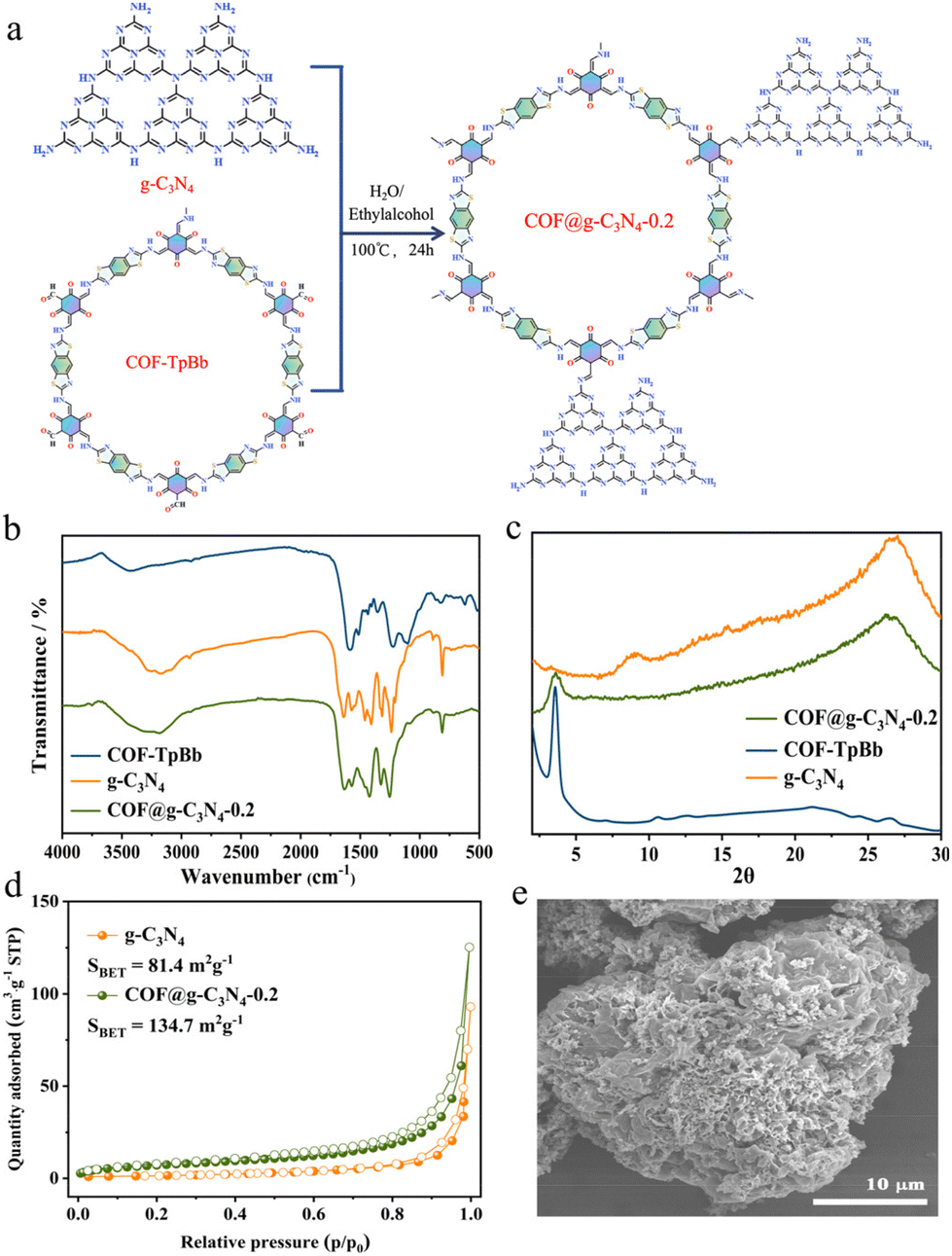 | ||
| Fig. 2 Preparation and characterization. (a) Schematic of the synthesis of COF@g-C3N4-0.2 via the condensation of COF-TpBb and g-C3N4; (b) IR spectra, (c) XRD patterns and (d) N2 adsorption curve at 77 K of COF@g-C3N4-0.2, COF-TpBb and g-C3N4; and (e) SEM image of COF@g-C3N4-0.2. | ||
The specific surface area determined by the N2 adsorption isotherm (Fig. 2d) shows that the BET of g-C3N4 itself is relatively small, only 81.4 m2 g−1, which is much smaller than that of COF-TpBb. Therefore, compared with g-C3N4, after its assembly, the BET of the obtained COF@g-C3N4-0.2 is increased to 134.7 m2 g−1, and the increased surface area can provide more catalytic sites. As can be seen from the thermogravimetric diagram in Fig. S6 and S7,† COF@g-C3N4-0.2 inherits the high thermal stability of g-C3N4 and there is no obvious weight loss before 500 °C.
It can be seen from the SEM image in Fig. 2e that the morphology of COF@g-C3N4-0.2 is more similar to that of pure g-C3N4, which is a lamellar structure with a large number of voids, except that the voids are smaller and the lamellar structure is larger compared to that of g-C3N4. Similarly, as can be seen from the TEM images in Fig. S8 and S9,† g-C3N4 has an obvious pore structure, while the pores of COF@g-C3N4-0.2 are smaller and more lamellar structures are observed.
The structure of COF@g-C3N4-0.2 was further studied by X-ray photoelectron spectroscopy (XPS) measurements. The peak positions of all XPS spectra were corrected according to the C–C peak at 284.6 eV, and the XPS fitting of all elements is provided in the ESI (Fig. S10–12†). The S 2p3/2 at 164.6 eV and S 2p1/2 at 168.0 eV observed from the XPS spectra in Fig. S13† can be attributed to S in the thiazole ring in the Bb-NH2 ligand.4c,15 In the XPS spectrum of the S of COF@g-C3N4-0.2 in Fig. S14,† only a single peak of S 2p3/2 is observed. This may be due to the low content of COF-TpBb in the composites, resulting in the 2p1/2 peak of the S being not obvious. However, since there is no S in g-C3N4, the XPS peak of S comes from COF-TpBb, which proves the successful loading of COF-TpBb and g-C3N4. Similarly, as shown in Fig. S15 and S16,† the O 1s of COF-TpBb has three characteristic peaks, which are attributed to the enone conversion (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH) of the Tp-CHO monomer and adsorbed water (H2O). In COF@g-C3N4-0.2, also due to the low content of COF-TpBb, it only shows a single characteristic peak of adsorbed water (H2O).
O, C–OH) of the Tp-CHO monomer and adsorbed water (H2O). In COF@g-C3N4-0.2, also due to the low content of COF-TpBb, it only shows a single characteristic peak of adsorbed water (H2O).
In addition, the positions of the three main characteristic peaks in the high-resolution XPS spectra of the C 1s of COF@g-C3N4-0.2, COF-TpBb and g-C3N4 are not significantly different (Fig. S17–S19†), except that COF-TpBb is more strongly conjugated due to the presence of benzothiazole, and its CC peak at position 286.6 eV exhibits stronger intensity. As for the N 1s (Fig. S20–S22†), all three samples contain tertiary and aromatic N peaks at 398.3 eV and 400.2 eV, except that g-C3N4 does not have the characteristic peak near 405 eV, which is attributed to the N in the thiazole ring of monomer Bb-NH2.
Since light absorption and charge separation are key factors in determining the activity of photocatalytic reactions, we first conducted the solid-state UV-vis spectroscopy (UV) of the photocatalyst. As shown in Fig. 3a, the absorption spectrum of g-C3N4 only reaches ∼400 nm, while the UV spectrum of COF-TpBb can reach 550 nm and tails to 800 nm. COF@g-C3N4-0.2 has a new and obvious absorption peak at 500 nm, and the photosensitivity in the entire visible light range is enhanced. This is due to the loading of COF-TpBb with excellent photosensitivity, providing a prerequisite for effective photocatalysis. According to the calculation results of the Tauc plot (Fig. S23 and S24†), the corresponding band gaps of g-C3N4 and COF-TpBb are 2.92 eV and 1.77 eV (vs. NHE), respectively. The electronic band positions of the prepared materials are calculated by Mott–Schottky testing to clarify the semiconductor characteristics of the samples.16 As shown in Fig. S25 and S26,† the flat-band potentials of g-C3N4 and COF-TpBb are −1.30 and −0.98 V vs. Ag/AgCl (i.e., −1.10 and −0.78 V vs. NHE), respectively, which are equal to the potentials of their conduction bands (CB). According to the formula Eg = EVB − ECB, the potentials of their valence bands (VB) are calculated to be 1.82 V and 0.99 V vs. NHE, respectively. Due to their suitable band structures, they can theoretically reduce CO2 to CO.
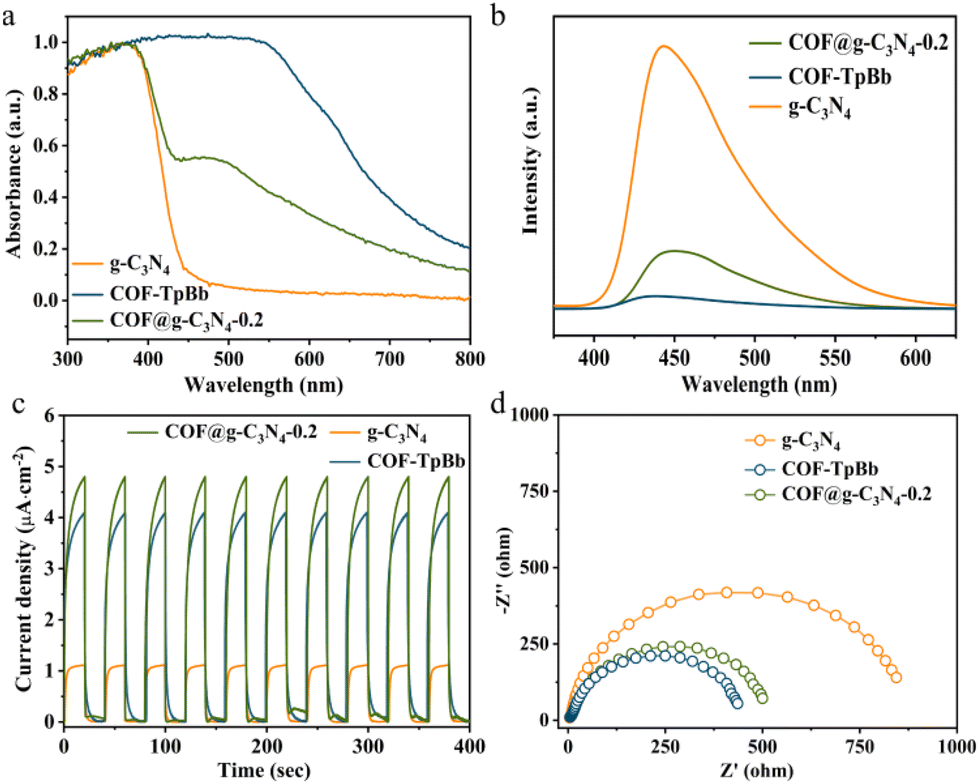 | ||
| Fig. 3 Photophysical and electrochemical properties. (a) Solid-state UV-vis diffuse reflectance spectra, (b) photoluminescence spectra, (c) transient photocurrent responses and (d) electrochemical impedance spectroscopy of COF@g-C3N4-0.2, COF-TpBb and g-C3N4. | ||
The photogenerated electron transfer properties of the samples were investigated using a combination of steady-state fluorescence (PL) and transient fluorescence spectroscopy, transient photocurrent testing and electrochemical impedance spectroscopy (EIS). As shown in Fig. 3b, compared with the strong peak of g-C3N4 at about 445 nm, the PL intensity of COF-TpBb is negligible, while the PL intensity of COF@g-C3N4-0.2 is significantly reduced, proving that the recombination rate of photogenerated electron–hole pairs in the composite material is lower. At the same time, the time-resolved fluorescence decay spectrum was measured. As shown in Fig. S27–29,† the average lifetime of photogenerated carriers in COF@g-C3N4-0.2 is 6.53 ns, which is longer than the 1.56 ns of COF-TpBb and 4.94 ns of g-C3N4. This indicates that there is indeed a favorable transfer of electrons between COF-TpBb and g-C3N4 in COF@g-C3N4-0.2, and the photogenerated carriers have a longer survival time on the surface of COF@g-C3N4-0.2.
As shown in Fig. 3c, under visible light irradiation, the photocurrent intensities of all three materials show an obvious tendency to rise and fall with switching characteristics and can be repeated for many cycles, proving that all three materials are photostable. Among them, COF@g-C3N4-0.2 has the largest photocurrent response intensity, slightly higher than that of COF-TpBb, and both are much greater than that of g-C3N4. In EIS (Fig. 3d), the Nyquist semicircle radius of COF@g-C3N4-0.2 in the high-frequency region is similar to that of COF-TpBb and much smaller than that of g-C3N4, proving that COF@g-C3N4-0.2 has better conductivity. All the above photochemistry and electrochemistry characterization studies prove that COF@g-C3N4-0.2 has better separation and transfer efficiency of carriers.
In a mixed solvent of MeCN and H2O (v/v = 9:1), using TEOA as a sacrificial agent, the photocatalytic performance of COF@g-C3N4-0.2 was studied under visible light irradiation (λ ≥ 400 nm). As a control, the photocatalytic performances of pure COF-TpBb and g-C3N4 were also studied. As shown in Fig. 4a, the main product of photocatalytic CO2 reduction by COF@g-C3N4-0.2 is CO (∼195 μmol g−1 h−1), accompanied by a small amount of H2 production (∼17 μmol g−1 h−1). No other liquid and gas products such as HCOOH and CH4 are observed to be generated, and the selectivity for CO is as high as 92%. The yield and selectivity of this reduction product are significantly higher than the yields of CO and H2 obtained when g-C3N4 or COF-TpBb is used as a catalyst (∼45 μmol g−1 h−1, ∼12 μmol g−1 h−1 or n.d., n.d.). This photocatalytic result is in line with our assumption of constructing a composite photocatalyst with COF-TpBb as the photosensitive center of the system and g-C3N4 as the active center.
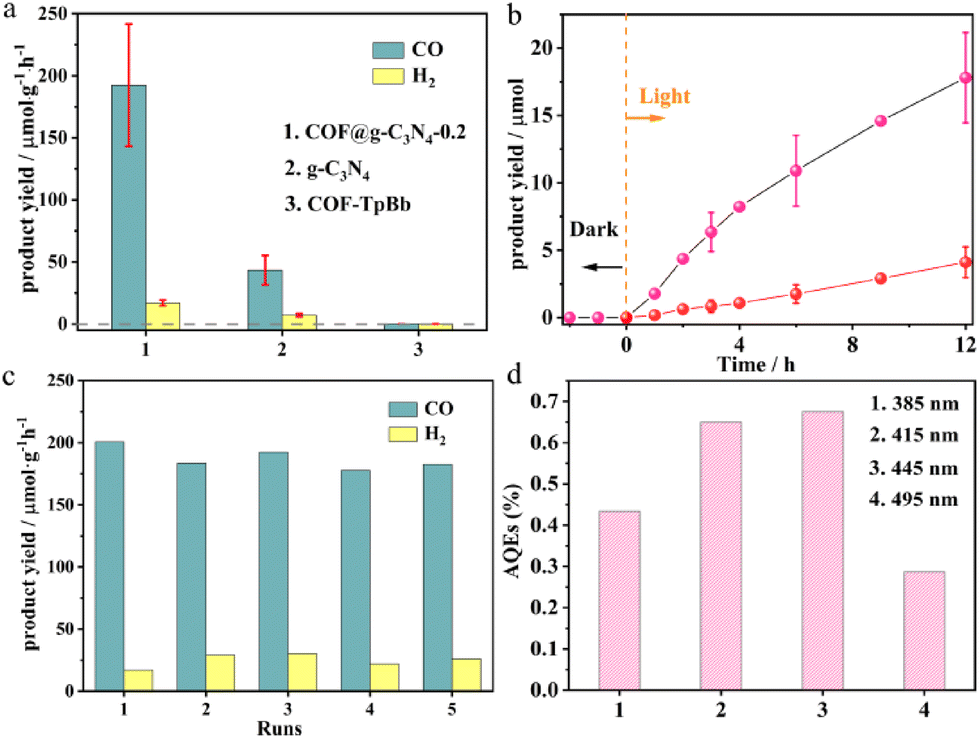 | ||
| Fig. 4 Photocatalytic performances. (a) Photocatalytic properties of COF@g-C3N4-0.2, COF-TpBb and g-C3N4; (b) time-dependent yields of CO2 reduction products and (c) recycling experiments using COF@g-C3N4-0.2 as the catalyst; and (d) apparent quantum efficiency of COF@g-C3N4-0.2 for photocatalytic processes at different wavelengths. | ||
In addition, in order to evaluate the catalytic stability and reusability of COF@g-C3N4-0.2, we have carried out long-term testing and cyclic testing. During the 12 h photocatalytic reaction (Fig. 4b), the CO yield continued to increase with time, and the final CO yield could reach 17.5 μmol at the 12th hour, while maintaining a high selectivity. The cycling experiments were divided into the same-batch catalyst recycling retesting experiments (Fig. 4c) and the same-condition tests with different batches of catalysts (Fig. S30†), and they both demonstrated that the photocatalytic process maintained the original photocatalytic efficiency during the five rounds of cycling, with no significant decrease in the yield and selectivity of CO. Then the apparent quantum efficiency of COF@g-C3N4-0.2 in the photocatalytic process at different wavelengths was evaluated. As shown in Fig. 4d, the difference in quantum efficiency at 415 nm and 445 nm is not significant, being 0.65% and 0.67%, respectively, which are higher than the 0.43% at 385 nm and 0.29% at 495 nm.
The effects of different COF-TpBb loadings in COF@g-C3N4 on the photocatalytic performance were tested under the same photocatalytic reaction conditions. Depending on the different masses of COF-TpBb loaded in the COF@g-C3N4 composites, four composite materials, COF@g-C3N4-0.05, COF@g-C3N4-0.1, COF@g-C3N4-0.2, and COF@g-C3N4-0.3, were prepared, respectively (the synthesis method is detailed in the ESI†). As shown in Fig. 5a, when the content of loaded COF-TpBb is 5%, compared with the production of CO (∼45 μmol g−1 h−1) and H2 (∼12 μmol g−1 h−1) of g-C3N4, the CO production of COF@g-C3N4-0.05 is significantly increased, while the H2 production is only slightly increased, which greatly enhances the system's CO selectivity of the photocatalytic system. When the content of loaded COF-TpBb was gradually increased to 20%, the production of CO also synchronously increased to four times that of g-C3N4 for a catalyst. In contrast, H2 only increases a little bit, obtaining the best production and selectivity. However, when the loading ratio of COF-TpBb further increases, its yield and selectivity decrease. This may be due to the existence of COF-TpBb as a photosensitive center; if its content is excessive, it hinders the contact between CO2 and the active center g-C3N4, thereby reducing the reactivity of the reaction. These results indicate that the assembly of COF-TpBb and g-C3N4 leads to excellent CO yield and selectivity for CO.
 | ||
| Fig. 5 Photocatalytic performances. (a) The yields of CO2 reduction products with different loading of catalysts; (b) changing sacrificial reaction conditions; (c) changing other reaction conditions; and (d) changing the ratio of solvents. | ||
To further investigate the effects of different reaction conditions on the photocatalytic system, a series of controlled photocatalytic CO2 reduction experiments were carried out using COF@g-C3N4-0.2 as the catalyst. Firstly, the sacrificial reagents were selected; as can be seen in Fig. 5b, the photocatalytic efficiency was significantly reduced when triethylamine (TEA), ethylenediaminetetraacetic acid (EDTA), ascorbic acid (AA), and methanol (MeOH) were used as sacrificial electron donors, which suggests that the selection of the sacrificial electron donors has a great influence on the photocatalytic activity. As shown in Fig. 5c, when no bpy, Co2+, or sacrificial agent was added to the photocatalytic system, negligible reduction products were generated, demonstrating that either electron mediators or electron donors are essential and important components of the system. In addition, under the conditions of no illumination, no catalyst, or using Ar instead of CO2, the generation of CO is also not detected, indicating that the source of CO is not the decomposition of the organic matter in the photocatalytic system (it is indeed from CO2). It is noteworthy that the photocatalytic products of mechanically mixed COF-TpBb and g-C3N4 are also almost undetectable. This may be due to the fact that light is absorbed by COF-TpBb but cannot transfer photogenerated electrons to g-C3N4. The above experimental results proved that all the above reaction conditions played a key role in CO2 reduction in this photocatalytic system.
Finally, we adjusted the ratio of solvents to explore the effect of water on the reaction system (Fig. 5d). When there is no water in the system, the selectivity of the CO2 reduction system for CO reaches 99%, but the yield is only about 100 μmol g−1 h−1. When the ratio of MeCN:H2O in the system is 8:1, the yield nearly doubles, but only the selectivity drops to 92%. When the water content in the system continues to increase, the H2 content in its photocatalytic system also increases. When the ratio of MeCN:H2O is 2:1, the production of CO and H2 is almost equal, and when the ratio of MeCN:H2O is 1:2, there is almost no production of CO.
To further deeply study the mechanism of photocatalytic reduction of CO2 by COF@g-C3N4, theoretical calculations were carried out using Gaussian 16. As shown in Fig. S31,† the calculation model ([Cat.]) is constructed from the finite cluster structure of COF@g-C3N4, and the long-range interactions of each local center are omitted. The geometric optimization of the ground state is carried out at the 6-31G* level by the B3LYP method of DFT calculation.17 Time-dependent density functional theory (TDDFT) calculations and orbital composition analysis are also carried out at the same level.
The calculation results show that [Cat.] has multiple excitation modes in the visible light region (Fig. 6a and Fig. S32†). The single excitation mode corresponding to visible light with a wavelength of 675 nm is HOMO → LUMO. Visible light with a wavelength of 513 nm corresponds to multiple excitation modes of HOMO−4 → LUMO, HOMO−1 → LUMO+1, and HOMO → LUMO+4. In contrast, the visible light with wavelengths of 468 nm and 459 nm both correspond to the double excitation mode of HOMO−4 → LUMO+1 and HOMO−2 → LUMO+4. According to the orbital composition analysis, the electron cloud of the highest occupied molecular orbital (HOMO) is concentrated on COF-TpBb. The electron cloud of the lowest unoccupied molecular orbital (LUMO) is concentrated on the junction of COF-TpBb and g-C3N4. So, after excitation, the electron cloud moves to g-C3N4. In addition, as shown in the electrostatic potential pattern (Fig. 6b), the red region is an electron-rich region with electrophilic activity, and the blue region is an electron-deficient region with nucleophilic activity, which also indicates that g-C3N4 is the active center for photocatalytic reduction of CO2.
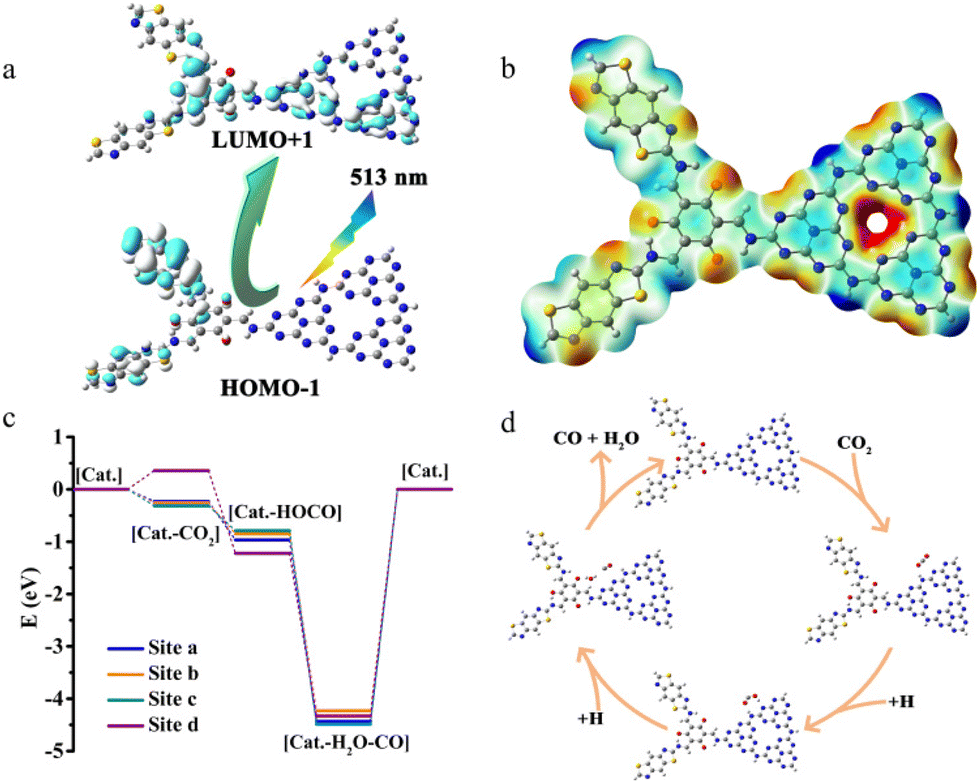 | ||
| Fig. 6 Mechanism and DFT calculations. (a) Schematic illustration of the transition of electron cloud distribution under light irradiation; (b_) electrostatic potential pattern of [Cat.]; (c) energy profile for the CO2 reduction pathway; and (d) a proposed reaction pathway for the photocatalytic CO2 reduction over COF@g-C3N4. | ||
Four sites, site a (N on the periphery of the triazine ring of g-C3N4), site b (N on the thiazole of Bb-NH2), site c (O on the Tp-CHO), and site d (N inside the triazine ring of g-C3N4), were chosen to perform energetic simulations of CO2 adsorption and the generation of the reaction intermediates in order to further investigate the photocatalytic reaction mechanism. The ground-state models [Cat.], [Cat.-CO2]-a (-b, -c, -d), [Cat.-OCOH]-a (-b, -c, -d), and [Cat.-CO-H2O]-a (-b, -c, -d) are geometrically optimized in the solvent-free state (Fig. S33–35†). The vibrational frequencies calculated by the above models at the same level are all positive, proving that they can all be stabilized, and the energy (ΔE) of [Cat.] is set to 0.000 eV for the next calculation. Calculations showed that the CO2 adsorption energies at the four sites were −0.230 eV, −0.258 eV, −0.314 eV and 0.358 eV, respectively. As shown in Fig. 6c, although site d has the lowest energy during the first hydrogenation process, its energy during the first step of CO2 adsorption is positive and CO2 adsorption is unstable and less likely to occur. Whereas the energy at which the hydrogenation process occurs at the latter four sites is reduced, the most stable site is site a, i.e., on the N on the periphery of the triazine ring of g-C3N4.
According to the results of experiments and theoretical calculations, we made a reasonable speculation on the photocatalytic process of the COF@g-C3N4 catalyst (Fig. 6d). Under visible light irradiation, COF-TpBb acts as the photosensitive center of the system and first generates photogenerated electrons. Subsequently, the photogenerated electrons are transferred to the catalytically active N sites on the periphery of the triazine ring on g-C3N4.
As shown in the figure, the photocatalyst COF@g-C3N4 first adsorbs CO2 and then completes the photoreduction process through the classic two-step hydrogenation process. In the first hydrogenation, CO2 is converted into the intermediate HOCO, and then further hydrogenation reaction yields CO and H2O.18 In addition, the photogenerated holes in the reaction process are consumed by TEOA to maintain the charge balance of the entire photocatalytic system.
Conclusions
In conclusion, we successfully prepared COF-TpBb with aldehyde groups retained at the terminal positions and good photosensitivity by using the COF-motif strategy. Furthermore, we assembled it with g-C3N4 with amino groups at the terminal positions by using the solvothermal method and obtained the covalently bonded composite catalyst COF@g-C3N4. Under suitable conditions, this catalyst can achieve photocatalytic reduction of CO2 to CO, and the yield can reach 195 μmol g−1 h−1 with a selectivity of 92%, accompanied by a small amount of H2. It was learned from the controlled experiments that the loading amount of COF-TpBb during synthesis, the choice of sacrificial agent in the system, and the water content in the catalytic process are all necessary factors that affect the final photocatalytic effect. Furthermore, we discussed the mechanism of the photocatalytic process by combining DFT calculations and experimental results, and made reasonable speculations on possible electron transfer paths and photocatalytic reaction mechanisms. Finally, our synthesized composite catalysts with COF-TpBb as the photosensitive center, g-C3N4 as the active center, and covalent bond as the electron transport bridge realized each of their respective roles and overall synergistic co-completion of the photocatalytic reaction, which is in line with our pre-designed ideas.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was financially supported by the Guangdong Basic and Applied Basic Research Foundation (no. 2022A1515110934), the Talent Research Initiation Fund Project of Hainan Normal University (no. HSZK-KYQD-202412, HSZK-KYQD-202411), the Weifang Institute of Science and Technology high-level talents research start-up fund project (no. KJRC2023010, 2023KJ08), the Natural Sciences Cross-Cutting Projects (no. RH2300003804), and the Grants for the Innovation Center of Academician Sun Shigang's Team in Hainan Province.References
- (a) M. Lu, M. Zhang, J. Liu, Y. Chen, J. P. Liao, M. Y. Yang, Y. P. Cai, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202200003 CrossRef CAS; (b) E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC; (c) A. Verma and Y.-P. Fu, Dalton Trans., 2024, 53, 4890–4899 RSC; (d) M. A. Khan, S. Mutahir, I. Shaheen, Y. Qunhui, M. Bououdina and M. Humayun, Coord. Chem. Rev., 2025, 522, 216227 CrossRef CAS; (e) Y.-S. Xia, L. Zhang, J.-N. Lu, X.-H. Zhao, L.-Z. Dong, J. Liu and Y.-Q. Lan, Nat. Synth., 2024, 3, 406–418 CrossRef CAS.
- (a) J. Yang, L. Jing, X. Zhu, W. Zhang, J. Deng, Y. She, K. Nie, Y. Wei, H. Li and H. Xu, Appl. Catal., B, 2023, 320, 122005 CrossRef CAS; (b) S. Barman, A. Singh, F. A. Rahimi and T. K. Maji, J. Am. Chem. Soc., 2021, 143, 16284–16292 CrossRef CAS.
- (a) D. Mao, Y. Hu, S. Yang, J. Liang, H. He, S. Yang, Z. Xu, C. Sun, S. Zheng, Z. Jiang, X. Qu and C.-S. Lee, Chem. Eng. J., 2023, 452, 139304 CrossRef CAS; (b) X. Liu, L. Yang, M. Huang, Q. Li, L. Zhao, Y. Sang, X. Zhang, Z. Zhao, H. Liu and W. Zhou, Appl. Catal., B, 2022, 319, 121887 CrossRef CAS; (c) X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- (a) C. Choi, F. Zhao, J. L. Hart, Y. Gao, F. Menges, C. L. Rooney, N. J. Harmon, B. Shang, Z. Xu, S. Suo, Q. Sam, J. J. Cha, T. Lian and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302152 CrossRef CAS; (b) B. Xiao, T. Lv, J. Zhao, Q. Rong, H. Zhang, H. Wei, J. He, J. Zhang, Y. Zhang, Y. Peng and Q. Liu, ACS Catal., 2021, 11, 13255–13265 CrossRef CAS; (c) W. J. Dong, I. A. Navid, Y. Xiao, J. W. Lim, J. L. Lee and Z. Mi, J. Am. Chem. Soc., 2021, 143, 10099–10107 CrossRef CAS PubMed.
- (a) C. Xia, R.-T. Guo, Z.-X. Bi, Z.-R. Zhang, C.-F. Li and W.-G. Pan, Dalton Trans., 2023, 52, 12742–12754 RSC; (b) J. Jing, J. Yang, W. Li, Z. Wu and Y. Zhu, Adv. Mater., 2021, 34, e2106807 CrossRef PubMed; (c) N. Sun, M. Zhou, X. Ma, Z. Cheng, J. Wu, Y. Qi, Y. Sun, F. Zhou, Y. Shen and S. Lu, J. CO2 Util., 2022, 65, 102220 CrossRef CAS; (d) L. Zhao, J. Bian, X. Zhang, L. Bai, L. Xu, Y. Qu, Z. Li, Y. Li and L. Jing, Adv. Mater., 2022, 34, e2205303 CrossRef PubMed.
- (a) J. Xu, Z. Ju, W. Zhang, Y. Pan, J. Zhu, J. Mao, X. Zheng, H. Fu, M. Yuan, H. Chen and R. Li, Angew. Chem., Int. Ed., 2021, 60, 8705–8709 CrossRef CAS; (b) S. Gong, G. Zhu, R. Wang, F. Rao, X. Shi, J. Gao, Y. Huang, C. He and M. Hojamberdiev, Appl. Catal., B, 2021, 297, 120413 CrossRef CAS; (c) L. Gu and A. D. Chowdhury, Dalton Trans., 2023, 52, 15958–15967 RSC.
- (a) L. Zhang, R.-H. Li, X.-X. Li, J. Liu, W. Guan, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210550119 CrossRef CAS; (b) Y. He, L. Yin, N. Yuan and G. Zhang, Chem. Eng. J., 2024, 481, 148754 CrossRef CAS; (c) Z. Chen, H. Zhang, P. Guo, J. Zhang, G. Tira, Y. J. Kim, Y. A. Wu, Y. Liu, J. Wen, T. Rajh, J. Niklas, O. G. Poluektov, P. D. Laible and E. A. Rozhkova, J. Am. Chem. Soc., 2019, 141, 11811–11815 CrossRef CAS PubMed.
- (a) R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC; (b) W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X. W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y. B. Zhang, H. Tian, W. H. Zhu and A. I. Cooper, Nature, 2022, 604, 72–79 CrossRef CAS PubMed; (c) Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS; (d) X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- (a) Y. Yusran, X. Guan, H. Li, Q. Fang and S. Qiu, Natl. Sci. Rev., 2020, 7, 170–190 CrossRef CAS PubMed; (b) J. F. Kurisingal, H. Kim, J. Hyeak Choe and C. Seop Hong, Coord. Chem. Rev., 2022, 473, 214835 CrossRef; (c) H. Zhong, R. Sa, H. Lv, S. Yang, D. Yuan, X. Wang and R. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS.
- (a) M. Lu, M. Zhang, J. Liu, T.-Y. Yu, J.-N. Chang, L.-J. Shang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed; (b) Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS; (c) J.-X. Cui, Y.-M. Fu, B. Meng, J. Zhou, Z.-Y. Zhou, S.-M. Liu and Z.-M. Su, J. Mater. Chem. A, 2022, 10, 13418–13427 RSC; (d) M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei, H. Xie, P. K. Wong and L. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- (a) J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed; (b) H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS.
- J.-X. Cui, L.-J. Wang, L. Feng, B. Meng, Z.-Y. Zhou, Z.-M. Su, K. Wang and S. Liu, J. Mater. Chem. A, 2021, 9, 24895–24902 RSC.
- (a) K. Eid, M. H. Sliem, M. Al-Ejji, A. M. Abdullah, M. Harfouche and R. S. Varma, ACS Appl. Mater. Interfaces, 2022, 14, 40749–40760 CrossRef CAS; (b) Q. Lu, A. Abdelgawad, J. Li and K. Eid, Int. J. Mol. Sci., 2022, 23, 15129 CrossRef CAS PubMed; (c) Q. Lu, K. Eid and W. Li, Nanomaterials, 2022, 12, 2379 CrossRef CAS PubMed; (d) Q. Lu, K. Eid, W. Li, A. M. Abdullah, G. Xu and R. S. Varma, Green Chem., 2021, 23, 5394–5428 RSC; (e) B. Salah, A. Abdelgawad, Q. Lu, A. K. Ipadeola, R. Luque and K. Eid, Green Chem., 2023, 25, 6032–6040 RSC.
- H. Zhong, R. J. Sa, H. W. Lv, S. L. Yang, D. Q. Yuan, X. C. Wang and R. H. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS.
- (a) J. N. Chang, Q. Li, Y. Yan, J. W. Shi, J. Zhou, M. Lu, M. Zhang, H. M. Ding, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202209289 CrossRef CAS; (b) X. Yan, B. Wang, J. Zhao, G. Liu, M. Ji, X. Zhang, P. K. Chu, H. Li and J. Xia, Chem. Eng. J., 2023, 452, 139271 CrossRef CAS.
- F.-M. Zhang, J.-L. Sheng, Z.-D. Yang, X.-J. Sun, H.-L. Tang, M. Lu, H. Dong, F.-C. Shen, J. Liu and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106–12110 CrossRef CAS PubMed.
- (a) S. Gao, Q. Zhang, X. Su, X. Wu, X.-G. Zhang, Y. Guo, Z. Li, J. Wei, H. Wang, S. Zhang and J. Wang, J. Am. Chem. Soc., 2023, 145, 9520–9529 CrossRef CAS; (b) J. Cui, C.-X. Li, J. Zhou, Y. Hua, Z.-Y. Zhou and Z.-M. Su, Sep. Purif. Technol., 2024, 346, 127466 CrossRef CAS; (c) X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- (a) A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS; (b) R. Chen, F. Fan and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202117567 CrossRef CAS PubMed; (c) M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02996h |
| This journal is © The Royal Society of Chemistry 2025 |