
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-infrared phosphorescence in a ruthenium(II) complex equipped with a pyridyl-1,2-azaborine ligand†
Matteo
Pompei
a,
Filippo
Monti
 *b,
Letizia
Sambri
a,
Nicola
Armaroli
b and
Andrea
Baschieri
*b
*b,
Letizia
Sambri
a,
Nicola
Armaroli
b and
Andrea
Baschieri
*b
aDepartment of Industrial Chemistry “Toso Montanari”, University of Bologna, Via Piero Gobetti 85, 40129 Bologna, Italy
bInstitute for Organic Synthesis and Photoreactivity (ISOF), National Research Council of Italy (CNR), Via Piero Gobetti 101, 40129 Bologna, Italy. E-mail: filippo.monti@isof.cnr.it; andrea.baschieri@isof.cnr.it
First published on 28th November 2024
Abstract
The 4-methyl-2-(pyridin-2-yl)-2,1-borazaronaphthalene molecule Hazab-py has been successfully used, for the first time, as a ligand in a ruthenium(II) polypyridine complex A (with the formula [Ru(dtbbpy)2(azab-py)]+, where dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine). This compound was characterized by NMR spectroscopy and high-resolution mass spectrometry (MS), and its electrochemical and photophysical properties were fully investigated and compared to those of its homoleptic analogue [Ru(dtbbpy)3]2+ (B), an archetypical mono-cationic cyclometalated complex C (with the formula [Ru(dtbbpy)2(ppy)]+, where Hppy = 2-phenylpyridine), and the more structurally similar analogue [Ru(dtbbpy)2(naft-py)]+ (D), where the B–N unit of the azaborine ligand is replaced by a standard C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C one, resulting in the 2-(naphthalen-2-yl)pyridine ligand (Hnaft-py). The presence of the novel 1,2-azaborine ligand induces a 0.51 V decrease in the redox gap of complex A, compared to that of B, leading to electrochemical and photophysical properties that resemble those of C and D. Accordingly, the azaborine complex displays an emission band extending up to the near infrared region of the spectrum (with the maximum at 765 nm in room-temperature acetonitrile solution), arising from a triplet metal-to-ligand charge-transfer (3MLCT) state. As in the case of other mono-cationic cyclometalated ruthenium(II) complexes, A shows modest photoluminescence quantum yields (PLQYs), but higher PLQYs when compared to those of its direct CC analogue D (e.g., PLQY = 0.6 vs. 0.1% in a PMMA matrix at 298 K). Density functional theory (DFT) calculations were used to provide complete rationalization of the electronic properties of all the complexes and to identify lower-lying metal-centred triplets (3MC), responsible for the low PLQYs of such an azaborine-based ruthenium(II) complex.
C one, resulting in the 2-(naphthalen-2-yl)pyridine ligand (Hnaft-py). The presence of the novel 1,2-azaborine ligand induces a 0.51 V decrease in the redox gap of complex A, compared to that of B, leading to electrochemical and photophysical properties that resemble those of C and D. Accordingly, the azaborine complex displays an emission band extending up to the near infrared region of the spectrum (with the maximum at 765 nm in room-temperature acetonitrile solution), arising from a triplet metal-to-ligand charge-transfer (3MLCT) state. As in the case of other mono-cationic cyclometalated ruthenium(II) complexes, A shows modest photoluminescence quantum yields (PLQYs), but higher PLQYs when compared to those of its direct CC analogue D (e.g., PLQY = 0.6 vs. 0.1% in a PMMA matrix at 298 K). Density functional theory (DFT) calculations were used to provide complete rationalization of the electronic properties of all the complexes and to identify lower-lying metal-centred triplets (3MC), responsible for the low PLQYs of such an azaborine-based ruthenium(II) complex.
Introduction
In the last few years, azaborine chemistry has seen a significant expansion.1,2 The introduction of boron–nitrogen units into aromatic benzenoid moieties has been intensively explored to tune the properties of the related organic molecules. The replacement of two aromatic sp2 carbon atoms in a benzene ring with one boron and one nitrogen atoms can lead to three structural isomers: (i) 1,2-azaborine,3 (ii) 1,3-azaborine4 or (iii) 1,4-azaborine.5The BN/CC isosterism can be a powerful tool to modify the molecular properties and stimulate research in materials science,6,7 and for biomedical applications.8–10 Moreover, azaborine-containing ligands have been demonstrated to promote different reactivities and selectivities in transition-metal complexes for catalysis.11,12 Azaborines also find applications in optoelectronic devices, such as organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), organic photovoltaics (OPVs), and sensors.13 In 2023, 1,4-azaborines were used by Huang and coworkers as donor units in organic solar cells,14 while Zysman-Colman and coworkers synthesized a 1,4-azaborine-triazine derivative displaying thermally activated delayed fluorescence (TADF), tested as an active material in OLEDs.15
In coordination chemistry, five- or six-membered 1,2-azaborine rings have often been used as cyclopentadienyl alternatives in the formation of organometallic compounds.16 Indeed, several 1,2-azaborine compounds based on zirconium(IV),17–20 titanium(IV),21 ytterbium(III),22 chromium(III),23–27 molybdenum(III),28 palladium(II),12 iron(II),29,30 and ruthenium(II)31–34 have been synthesized and deeply investigated. However, all the above-cited organometallic complexes have been studied for other purposes rather than luminescence, since they are not emissive.
The photophysical properties of azaborines (considered as single organic molecules) have already been investigated in recent years.35–42 However, only in a few examples, the photophysical properties of such B–N compounds have been directly compared with those of their isoelectronic CC counterparts.43,44 In such literature examples, interesting differences in the absorption and emission properties have been reported between azaborines and their CC analogues.
Recently, our research group published a paper on a series of iridium(III) complexes having an 1,2-azaborine derivative as an ancillary ligand.45 In that study, we decided to functionalize the boron atom of a 2,1-borazarolenaphthane core with a pyridyl moiety, in order to obtain a suitable chelating ligand for the iridium(III) centre. The obtained 4-methyl-2-(pyridin-2-yl)-2,1-borazaronaphthalene ligand (Hazab-py, Scheme 1) coordinates the metal centre through its two nitrogen atoms (i.e., one from the azaborine unit and the other from the pyridyl moiety), forming a typical five-membered metal-containing ring. To directly evaluate the effects of this new azaborine ligand on the properties of the related complexes, we also synthesized and characterized the isoelectronic CC equivalent (Hnaft-py, Scheme 2) and its isosteric iridium(III) complexes. Remarkably, a blue shift in the emission spectra of the azaborine-based complex is observed when compared to that of its isosteric CC counterpart.45
 | ||
| Scheme 1 Synthesis of the azaborine complex A. Complex B was used for comparison in the characterization. | ||
 | ||
| Scheme 2 Synthesis of the isoelectronic CC complexes C and D. | ||
Encouraged by the above results, and since only a few examples of azaborine-based ruthenium(II) complexes are reported in the literature (all of which are non-emissive, see above for references), we wanted to explore the effect of our azaborine ligand on the electrochemical and photophysical properties of a prototypical ruthenium(II) complex.
Herein, we report the synthesis and characterization of a new near-infrared phosphorescent ruthenium(II) complex with the formula [Ru(dtbbpy)2(azab-py)][BF4] (A, Scheme 1) equipped with two neutral 4,4′-di-tert-butyl-2,2′-bipyridine ligands (dtbbpy) and one anionic pyridyl-1,2-azaborine chelator (azab-py−). The properties of complex A are compared with those of the prototypical and well-known [Ru(dtbbpy)3][BF4]2 complex B,46,47 which could be readily obtained as a reaction by-product (Scheme 1). For a more rigorous comparison, we also synthesised another reference complex, having the same charge as A, since it is equipped with an anionic cyclometalating 2-phenylpyridine ligand (i.e., [Ru(dtbbpy)2(ppy)][BF4], C, Scheme 2).48 Lastly, as already done for the iridium-based series,45 we used the CC isoelectronic counterpart of our azaborine ligand (Hnaft-pyvs.Hazab-py) to obtain the corresponding ruthenium-based complex [Ru(dtbbpy)2(naft-py)][BF4] (D, Scheme 2). It is worth noting that, in the case of ruthenium, the cyclometalation only occurs at the C-3 position of the naphthyl core, and any attempt to achieve C-1 cyclometalation was not successful.
Results and discussion
Synthesis and structural characterization
To set up an effective synthetic protocol to get complex A, the [Ru(dtbbpy)2Cl2] precursor was prepared following a standard procedure.49 Accordingly, RuCl3 was accurately reacted with 2 equivalents of 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) dissolved in degassed ethanol at reflux, under an inert atmosphere and protected from light. Once the formation of [Ru(dtbbpy)2Cl2] was complete, ethanol solvent was replaced with dichloromethane, and the precursor was treated in situ with AgBF4 to remove the chloride ions from the ruthenium(II) coordination sphere, increasing the metal reactivity. The as-obtained solvato complex (1) was reacted at room temperature with an excess of the azaborine ligand (Hazab-py), previously deprotonated with NaH in dry dichloromethane. As mentioned in the introduction, the 4-methyl-2-(pyridin-2-yl)-1,2-dihydrobenzo[e][1,2]azaborine ligand (Hazab-py) was synthesized following a procedure previously reported by us.45The reaction mixture was stirred for 24 hours under an inert nitrogen atmosphere. After a standard workup, followed by a purification by column chromatography on basic alumina, complex A was obtained in good yield (40%, Scheme 1). Despite the amount of dtbbpy ligand being limited, the homoleptic complex [Ru(dtbbpy)3][BF4]2 (B) was also found in the reaction crude product as a minor by-product (13% yield). While this complex is well-known in the literature,46,50–52 it was nonetheless investigated and directly compared to A.
Complexes A and B were characterized by NMR spectroscopy and mass spectrometry (Fig. S1–S10†). The 1H NMR spectrum of complex A shows 21 signals in the aromatic region and 5 singlets in the aliphatic region: this number fits with the expected signals. In contrast, the 1H NMR spectrum of B shows the presence of just 3 peaks in the aromatic region and 1 singlet in the aliphatic zone, due to the D3 symmetry of the complex.
Finally, to better compare the properties of complex A, two additional reference complexes C and D were also synthesized (Scheme 2). For this scope, the [Ru(dtbbpy)2Cl2] precursor was synthesized with a different reaction, using a large excess of LiCl in DMF at reflux to avoid the formation of B and, unlike the previous case reported in Scheme 1, it was isolated and characterized by 1H-NMR to confirm its structure (Fig. S11†).
To obtain complex C, the ruthenium(II) precursor was reacted with AgBF4 to remove the chlorine atoms and subsequently reacted with 2-phenylpyridine (Hppy), used as a cyclometallating ligand. It is worth noting that, unlike B, the reference complex C is mono-cationic and heteroleptic as A.
To get an even more similar reference complex, we also synthesized the Hnaft-py ligand (i.e., the CC equivalent of Hazab-py) following the procedure recently reported by us.45 As we already demonstrated in the case of iridium(III) complexes, the ligand Hnaft-py can undergo cyclometallation at two different positions (i.e., C-1 and C-3). Therefore, we expected to obtain an isomeric mixture of two ruthenium(II) complexes; however, in this specific case, we obtained only one product (D), which is the less sterically hindered complex and probably the most thermodynamically stable.45 Apart from the isomerism, this complex has a structure similar to that of A, with the notable substitution of the B–N fragment by the isoelectronic CC unit. Even in these cases, all complexes were fully characterized by NMR spectroscopy, mass spectrometry and elemental analysis (Fig. S12–S24†).
Complexes B–D were used as reference compounds for all the subsequent structural, photophysical and electrochemical characterization studies, to properly identify the effects of the new azaborine ligand on ruthenium(II) complexes.
DFT calculations: ground-state properties
Initially, the structural and electronic properties of [Ru(dtbbpy)2(azab-py)]+ (A) were explored by DFT methods, adopting the same level of theory successfully used for previously reported azaborine-based iridium(III) complexes (see the Experimental section for further details).45 The reference complexes B–D were compared to A to explore the effect of replacing the anionic azaborine ligand with (i) a neutral N^N analogue like dtbbpy (as in B), (ii) an archetypical anionic C^N counterpart as the ppy− ligand (in C) or (iii) a more similar CC analogue naft-py− (as in D). Since it was synthetically impossible to obtain the same cyclometallation mode observed in the azab-py− ligand also for the naft-py− counterpart (see above), we have also theoretically investigated complex D′, which is an analogue of A in which the B–N fragment is merely replaced with the isoelectronic CC part.
The energy diagrams and the frontier molecular orbitals of complexes A–D and D′ are depicted in Fig. 1. Notably, due to the D3 point-group symmetry of complex B, some of its molecular orbitals are degenerate.
 | ||
| Fig. 1 Energy diagram showing the frontier molecular orbitals of complexes A–D, calculated in acetonitrile. For some relevant orbitals, the corresponding isosurface is also displayed (isovalues = 0.04 e1/2 bohr−3/2). Along the series, relevant orbitals with similar topologies are plotted with the same colour for an easier comparison (see the legend). The CC theoretical analogue of complex A is displayed as D′, for the sake of comparison. | ||
In all complexes, the three highest occupied molecular orbitals are dominated by the contribution of the pseudo-t*2g (dπ) orbitals of ruthenium(II), despite minor contributions from the π orbitals of the azaborine (or C^N ligands) being present in the HOMO of complex A (or C, D and D′).
Complex B displays the lowest-energy HOMO of the whole series, basically due to its bis-cationic nature compared to all the other complexes (i.e., 2+ vs. 1+). Indeed, by replacing one neutral dtbbpy ligand with the anionic azaborine-based one, a HOMO destabilization of 0.74 eV is observed (compare B and A in Fig. 1). Similarly, if the azaborine ligand is replaced by a regular C^N cyclometalating ligand, like ppy− (as in C) or naft-py− (as in D or D′), a further destabilization is observed, but of just about 0.12 eV (Fig. 1). For a deeper understanding of this more subtle phenomenon, the readers could have a look at the orbital-interaction diagrams in Fig. S25–S28,† showing how the π and σ orbitals of the anionic ligands interact with the set of ruthenium(II) pseudo-t*2g orbitals, lifting up their energy.
In contrast, the lowest unoccupied orbitals of all the investigated complexes are less affected by the replacement of their third ligand. Indeed, both the LUMO and LUMO+1 are always centered on the π* orbitals of the two dtbbpy ligands that all the complexes have in common. Consequently, the LUMO levels in the mono-cationic complexes differ by less than 0.05 eV, and a LUMO stabilization of about 0.3 eV is observed for the bis-cationic complex B (Fig. 1).
Accordingly, the homoleptic complex B presents the widest HOMO–LUMO gap of the series (i.e., 3.61 eV), mainly due to HOMO stabilization. For the heteroleptic (and mono-cationic) ones, the gap decreases in the order: A > C > D > D′. It is worth noting that, if the azaborine-based complex A is compared to its theoretical direct analogue D′, a 0.10 eV reduction in the HOMO–LUMO gap is observed (i.e., 3.18 vs. 3.08 eV, see Fig. 1).
Electrochemistry
To explore the effect of the anionic azaborine ligand on the electronic properties of its corresponding ruthenium(II) complex, cyclic and square-wave voltammetry experiments were carried out in room-temperature acetonitrile solutions of complex A and all the reference complexes B–D; the well-known archetypical complex [Ru(bpy)3][ClO4]2 (E) was also investigated for the sake of completeness. All cyclic voltammograms are reported in Fig. 2, square-wave voltammograms in Fig. S29,† and the recorded redox potentials in Table 1 and S1,† relative to the ferrocene/ferrocenium (Fc/Fc+) couple.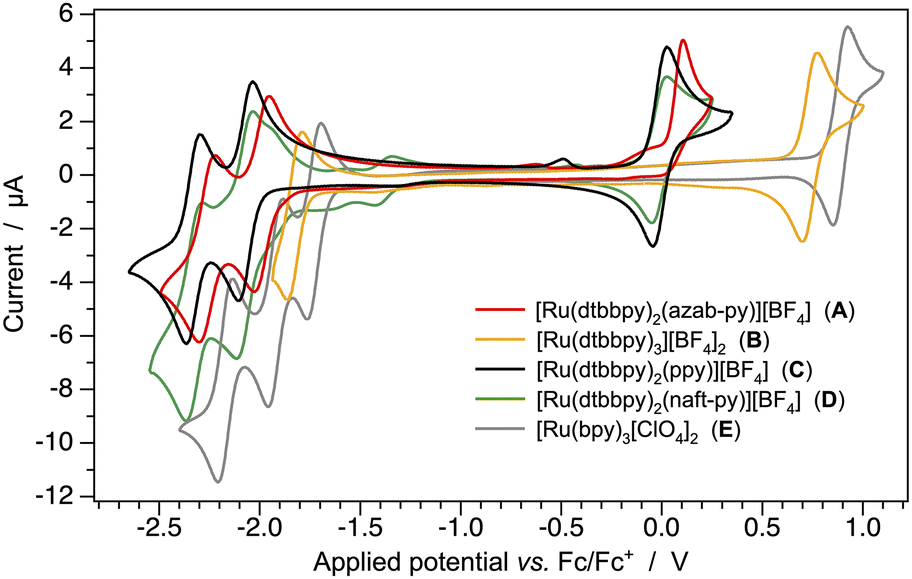 | ||
| Fig. 2 Cyclic voltammograms of complexes A–D (1.0 mM) in acetonitrile solution at 298 K, together with that of the archetypical complex [Ru(bpy)3][ClO4]2 (E), used as further reference. | ||
| E ox (ΔEp)a [V (mV)] | E red (ΔEp)a [V (mV)] | ΔEredoxb [V] | |
|---|---|---|---|
| a The reported potential values are obtained by cyclic voltammetry and reported vs. the ferrocene/ferrocenium couple, used as an internal reference. The values in parentheses are the peak-to-peak separation (ΔEp); redox processes are reversible, unless otherwise stated (irr.). b ΔEredox = Eox − Ered. | |||
| A | +0.06 (irr.) | −1.991 (70), −2.262 (73) | 2.05 |
| B | +0.735 (72) | −1.825 (69) | 2.560 |
| C | −0.009 (68) | −2.071 (70), −2.333 (67) | 2.062 |
| D | −0.013 (73) | −2.075 (77), −2.328 (74) | 2.088 |
| E | +0.889 (73) | −1.730 (70), −1.923 (73), −2.172 (72) | 2.619 |
As already well documented in the literature,51 the presence of the electron-donating tert-butyl groups on the 2,2′-bipyridine ligands are able to cathodically shift the redox processes of the corresponding homoleptic ruthenium(II) complex by about 0.1 V (compare Bvs.E, Fig. 2 and Table 1). This effect is strongly amplified if one neutral dtbbpy ligand is replaced by an anionic one (as in A, C or D), a phenomenon that is basically due to electrostatic reasons.
Indeed, the first oxidation process in the azaborine-based complex A occurs at much lower potentials when compared to that of B (i.e., +0.06 vs. +0.735 eV, Table 1). Notably, such a process is further shifted to lower potentials by approx. 0.07 V, if a standard anionic C^N cyclometalating ligand is used instead of the azab-py− azaborine derivative (compare Avs.C or D, Fig. 2 and Table 1). However, it should be noted that, in all the investigated complexes, the first oxidation always involves the formal oxidation of Ru(II) to Ru(III), as demonstrated by spin-unrestricted calculations preformed on the corresponding oxidized radicals (Fig. S30†). It is worth noting that, for the azaborine complex A, such an oxidation process is found to be irreversible at any scan rate from 100 to 1000 mV s−1 (Fig. S31†), while good first-oxidation reversibility is observed for all other complexes.
A similar trend is also observed in the first reduction processes, though less pronounced, as the replacement of one dtbbpy ligand (as in B) with the anionic azaborine (in A) or C^N cyclometallating alternatives (in C and D) has an indirect effect. Indeed, in all the complexes, the first reduction process consistently involves the formal reduction of one of the dtbbpy ligands (Fig. S30†). For instance, the first reduction potential of A is more negative by 0.166 V when compared to that of the homoleptic complex B, and a further 0.08 V cathodic shift is observed in the case of C and D.
As suggested by DFT calculations (Fig. 1), the redox gap of the azaborine-based ruthenium(II) complex (A) is consequently about 0.51 V narrower than that of the archetypical [Ru(dtbbpy)3][BF4]2 complex (B), and it is substantially comparable to that of the cyclometalated complexes C and D (Table 1), indicating that, in this case, the BN/CC isosterism seems not to affect dramatically the electronic properties of the complexes.
Photophysical properties and excited-state calculations
The UV-Vis absorption spectra of all the ruthenium(II) complexes were recorded in both acetonitrile and dichloromethane solutions at 298 K (Fig. 3 and S32†). The absorption profiles of all the samples are virtually not affected by solvent polarity, and only a minor red shift is observed on passing from acetonitrile to dichloromethane (Fig. S32†). It should be also noted that the spectrum of the well-known [Ru(dtbbpy)3]2+ complex (B) and that of the cyclometalated [Ru(dtbbpy)2(ppy)]+ equivalent (C) are in line with literature data.51,53 | ||
| Fig. 3 Absorption spectra of complexes A–D in acetonitrile solution at 298 K. The lowest-energy transitions are magnified in the inset. | ||
As commonly observed in ruthenium(II) polypyridine complexes,54 the intense absorption peaks at 275–300 nm can be assigned to spin-allowed ligand-centered (1LC) π → π* transitions. The bands at longer wavelengths, magnified in the inset of Fig. 3, are attributed to spin-allowed metal-to-ligand charge-transfer (1MLCT) processes, while direct singlet-to-triplet 3MLCT transitions can be detected as a shoulder at even lower energy. Notably, in line with DFT and electrochemical findings, the lowest-energy MLCT band of the homoleptic (and bis-cationic) complex B is considerably blue-shifted compared to that of the azaborine-based complex A, and a further red shift is detected when C^N cyclometalating ligands are used, as in C and D (Fig. 3, inset).
To gain a more rigorous understanding of the excited-state scenario in complexes A–D, the lowest triplet states were investigated using the TD-DFT approach. Tables S2–S6† show the triplet excitation below 2.5 eV, using the Natural Transition Orbital (NTO) formalism,55 and Fig. 4 summarizes the lowest triplet vertical excitation computed from the ground-state minimum of all the complexes examined (see the Experimental section for further details).
 | ||
| Fig. 4 Energy diagram of the lowest-lying triplet states for complexes A–D, computed in acetonitrile as vertical excitation from the respective ground-state minimum-energy geometries. | ||
TD-DFT results clearly indicate that, for all complexes, the lowest triplet states are MLCT in nature, resulting in a charge-transfer from the ruthenium(II) d orbitals to the π* orbitals on the dtbbpy ligands (Fig. 4). In particular, the energy of the lowest triplet state (T1) increases in the order: D ≈ C < A ≪ B. Such a trend is in agreement with the onset of the experimental absorption profiles (Fig. 3, inset).
Notably, for complex A, equipped with the novel azab-py− ligand, the lowest triplet state involving such an azaborine unit is T7, which is found to be 0.52 eV above T1 (or 2.42 eV above S0, see also Table S2†); accordingly, a direct contribution of such a ligand in the emitting state of A can be safely ruled out. A similar scenario is also observed for C, with T7 being a mixed 3LC/3MLCT involving the ppy− cyclometalating ligand and located 2.39 eV above the ground state. When the π-extended CC analogue of the azaborine ligand is used in D, both T7 and T8 are found to involve such a cyclometalating ligand, but such states are again found well above T1 (Fig. 4 and Table S5†). It is important to mention that the different cyclometalation mode of the naft-py− ligand (which can form the Ru–C bond at C-3 or C-1 positions, as in D or D′) has virtually no impact on the nature and energy of the lowest triplet vertical excitation of the related ruthenium(II) complexes (compare Tables S5 and S6†).
Fig. 5 shows the normalized emission spectra of all the complexes, recorded in both acetonitrile and dichloromethane solutions at 298 K, and in butyronitrile glass at 77 K; all the related photophysical data are summarized in Table 2.
| Oxygen-free acetonitrile solution, 298 K | Oxygen-free dichloromethane solution, 298 K | Butyronitrile, 77 K | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ em [nm] | PLQYa [%] | τ [ns] | k r [105 s−1] | k nr [106 s−1] | λ em [nm] | PLQYa [%] | τ [ns] | k r [105 s−1] | k nr [106 s−1] | λ em [nm] | τ [μs] | |
| a λ exc = 440 nm. b λ exc = 465 nm. c Radiative constant: kr = PLQY/τ. d Non-radiative constant: knr = 1/τ − kr. shShoulder. | ||||||||||||
| A | 765 | 0.073 | 15 | 0.49 | 67 | 700 | 0.19 | 26 | 0.74 | 38 | 644sh, 690 | 0.434 |
| B | 630 | 14.3 | 832 | 1.72 | 1.03 | 617 | 15.4 | 910 | 1.69 | 0.930 | 578, 625, 675sh | 5.36 |
| C | 814 | 0.037 | 17 | 0.22 | 59 | 792 | 0.12 | 33 | 0.38 | 30 | 722, 785sh | 0.423 |
| D | 800 | 0.041 | 39 | 0.11 | 26 | 784 | 0.11 | 53 | 0.21 | 19 | 635sh, 714, 785sh | 0.454 |
 | ||
| Fig. 5 (Top) Normalized emission spectra of complexes A–D in acetonitrile (solid) and dichloromethane (dotted) at 298 K and (bottom) in butyronitrile glass at 77 K (dashed). Sample concentration: ≈15 μM. | ||
As is typically expected for 3MLCT transition, the emission spectra of all the investigated compounds display (i) a broad and unstructured emission profile at 298 K; (ii) a red-shifted band in the more polar acetonitrile solvent vs. dichloromethane; (iii) a strong blue shift in the butyronitrile rigid matrix at 77 K, with associated spectra displaying vibronic progressions. Moreover, in line with DFT and electrochemical findings, the emission maximum of the homoleptic complex B is found at much higher energy, compared to the other compounds (Fig. 5 and Table 2). Indeed, the emission spectrum of the azaborine complex A is very similar to those of the cyclometalated complexes C and D, displaying only a minor blue-shift compared to them, but still extending in the near-infrared region of the electromagnetic spectrum (Table 2 and Fig. 5).
To rigorously attribute the 3MLCT nature of the emitting states, the lowest triplet states of all the complexes were fully optimized by a spin-unrestricted DFT approach (Fig. S33†). Such calculations further confirm that, in all cases, the emitting state has the excited electron located on the lowest π* orbitals of a dtbbpy ligand and the hole on the highest ruthenium pseudo-t*2g (dπ) orbital. Notably, two virtually isoenergetic triplets are found in the case of the heteroleptic complexes (A, C and D) due to the presence of two non-equivalent dtbbpy ligands (i.e., T1 and T2 in Fig. S33†). In addition, the adiabatic energy difference between S0 and T1 is calculated to be 1.61, 2.04, 1.52 and 1.59 eV for A, B, C and D, respectively; such theoretical findings are in excellent agreement with the experimental emission maxima recorded in acetonitrile solution at 298 K (i.e., 1.62, 1.97, 1.52 and 1.55 eV, Table 2).
Notably, the photoluminescence quantum yield (PLQY) of the azaborine complex A is extremely low and dependent on the solvent (i.e., 0.07% vs. 0.19% in acetonitrile vs. dichloromethane solution at 298 K, Table 2), while that of B is rather solvent-independent and around 15%. The much lower PLQY of A, compared to that of B, is mainly due to a 50-times higher non-radiative rate constant (knr) of its emitting state, rather than a significant change in the radiative constant (kr, Table 2). Such findings further confirm the similar nature of the emitting state in both compounds, but suggest the presence of effective non-radiative pathways in the azaborine-based complex.
Nonetheless, when the photophysical parameters of A are directly compared to those of its CC analogue D, a much similar scenario is observed. However, it must be emphasized that the azaborine complex A exhibits nearly double the PLQY of D (Table 2), primarily due to a significantly higher kr of the emitting state. Despite the overall modest performances of both A and D, these novel complexes display higher emission efficiencies than the archetypical (and previously reported) cyclometalated complex C.53
As already reported in the literature,54 non-emissive metal-centered states of triplet multiplicity (3MC) can be responsible for the poor PLQYs observed in ruthenium(II) complexes, providing fast radiationless channels to the ground state. The population of such 3MC levels formally results from the excitation of an electron from the occupied pseudo-t*2g (dπ) orbitals of the ruthenium(II) ion to the unoccupied pseudo-e*g (dσ*) orbitals of the metal itself, which are usually found at high energy and, as a consequence, not readily populated.54 Commonly, the relaxed geometry of a 3MC state in a polypyridine ruthenium(II) complex involves the elongation of selected Ru–N bonds,56 possibly resulting in the decoordination of one or more pyridine moieties from the metal coordination sphere.57,58
Accordingly, unrestricted DFT calculations were carried out to explore the presence of accessible 3MC states in the novel azaborine-based ruthenium(II) complex A, for appropriate rationalization of its low PLQYs (Table 2). Due to the asymmetric nature of the azaborine ligand, there are three possible axial deformations leading to the population of different 3MC states, through the elongation of trans-Ru–N bonds. As reported in Fig. 6, all the three 3MC states were properly optimized and two of them are found at lower energy, compared to the 3MLCT states (i.e., T1 and T2). Nevertheless, energy barriers are likely expected between such 3MC states and the emissive 3MLCT ones; thus these theoretical findings may provide a reasonable explanation for the modest (but still appreciable) emission performances of A, the first known ruthenium(II) complex equipped with an azaborine ligand.
 | ||
| Fig. 6 Schematic energy diagram reporting the ground state (S0), the emitting triplets (T1 and T2, see Fig. S33† for further details), and the three metal-centered (3MC) states calculated for A in acetonitrile. The fully relaxed minimum-energy geometries are also reported, together with the associated spin-density distribution (isovalues: 0.002 e bohr−3). For 3MC states, double-ended arrows indicate the axis of the elongated N–Ru–N bonds. Since minima are fully relaxed, the x axis of the graph is arbitrary, spanning all the possible degrees of freedom of A. | ||
It must be also stressed that complexes A, C and D are red-to-NIR (near-infrared) emitters, so that the energy gap between the emissive and the ground states is very close in energy. Indeed, such a small energy difference can enable fast non-radiative pathways from the emissive triplet to S0 due to vibrational overlap between such electronic states (i.e., energy-gap law).
The emission properties of complexes A–D were also investigated in the solid state by dispersing the emitters in a poly(methyl methacrylate) (PMMA) matrix at a concentration of 1% by weight; related spectra and photophysical parameters are reported in Fig. S34† and Table 3.
The emission profiles of all complexes mostly preserve the same characteristic already discussed for the spectra in solution, with two notable differences: (i) a blue shift is observed in the PMMA matrix, due to the lack of solvent stabilization of the emitting states and (ii) a much higher PLQY is obtained for A compared to the solution data, which can be attributed to the combined effect of its fast kr (relative to its CC analogue D) and the solid-state matrix limiting the excited-state distortions needed to populate the above-mentioned 3MC states. Indeed, for all the complexes, the knr values in the PMMA matrix are considerably lower than those in solution, while the kr values are comparable (see Tables 2 and 3).
Conclusions
A near-infrared emissive ruthenium(II) polypyridine complex equipped with a 1,2-azaborine ligand has been reported for the first time. Such [Ru(dtbbpy)2(azab-py)]+ complex (A) has been obtained with satisfactory yields (40%), together with the corresponding homoleptic by-product [Ru(dtbbpy)3]2+ (B), and its electrochemical and photophysical performances are compared to those of similar complexes having more standard anionic cyclometallating ligands (C and D).Notably, complex A displays a phosphorescence emission from a 3MLCT state, peaking at 765 nm and extending up to 1100 nm in room-temperature acetonitrile solution. However, the quantum yield of its near-infrared photoluminescence is low, particularly in solution at 298 K (i.e., below 0.2%). Nevertheless, the emission performances of the azaborine complex A are remarkably superior when compared to those of its direct CC analogue (D). To provide a full picture of the excited states of the novel complex A, DFT calculations were used to identify and characterize accessible 3MC states, which are known to quench the 3MLCT emissive states in transition-metal complexes by providing fast non-radiative deactivation pathways to the ground state.
This work opens the way to a novel class of luminescent ruthenium(II) complexes, which might be expanded by suitably designed 1,2-azaborines to optimize their luminescence performances, allowing their exploration in sensing or optoelectronic applications.
Experimental section
General information
4-Methyl-2-(pyridin-2-yl)-2,1-borazaronaphthalene (Hazab-py) and 2-(naphthalen-2-yl)pyridine (Hnaft-py) were prepared according to reported methods.45 Analytical grade solvents and commercially available reagents were used as received, unless otherwise stated. Chromatographic purifications were performed using aluminium oxide 90 active neutral (70–230 mesh). 1H, 19F, 11B and 13C NMR spectra were recorded on Varian Mercury (400 MHz for 1H) and Agilent (500 MHz for 1H) spectrometers. Chemical shifts (δ) are reported in ppm relative to residual solvent signals for 1H and 13C NMR (1H NMR: 7.26 ppm for CDCl3 and 5.33 ppm for CD2Cl2; 13C NMR: 77.0 ppm for CDCl3 and 53.84 ppm for CD2Cl2) or relative to an internal standard as a chemical shift reference for 19F and 11B NMR (19F NMR: −163 ppm for C6F6; 11B NMR: 0 ppm for BF3·Et2O). 13C{1H} NMR spectra were acquired with the 1H broadband decoupled mode. Coupling constants are given in Hz. The abbreviations used to indicate the multiplicity of signals are: s, singlet; d, doublet; t, triplet; q, quartet; dd, double doublet; and m, multiplet. The high-resolution mass spectra (HRMS) were obtained with an ESI-QTOF (Agilent Technologies, model G6520A) instrument, and the m/z values are referred to the monoisotopic mass. ESI-MS analyses were performed by direct injection of acetonitrile solutions of the compounds using a WATERS ZQ 4000 mass spectrometer.General procedure for the synthesis of complexes A and B
In a 50 mL round bottom flask, RuCl3·3H2O (37 mg, 0.14 mmol, 1.0 equiv.) and 4,4′-di-tert-butyl-2,2′-bipyridine (75 mg, 0.28 mmol, 2.0 equiv.) were dissolved in deoxygenated ethanol (6 mL) and the reaction vessel was shielded from light. The resulting solution was refluxed under a nitrogen atmosphere for 1 h. Afterward, the solution was cooled to room temperature and anhydrous CH2Cl2 (15 mL) was added. AgBF4 (82 mg, 0.42 mmol, 3.0 equiv.) was added in the absence of light. The mixture was kept in the dark and stirred under nitrogen at reflux for further 6 hours. The solution was evaporated to dryness, and the resulting solid was used in the next step of the synthesis without further purification.A solution of the Hazab-py compound (35 mg, 0.16 mmol, 1.2 equiv.) and NaH 60% dispersion in mineral oil (16 mg, 0.4 mmol, 2.9 equiv.) in anhydrous CH2Cl2 (8 mL) was stirred for 2 h at room temperature and then added dropwise to the previous solid. The mixture was stirred for further 24 h in the dark, under a nitrogen atmosphere, at room temperature. After this time, the solvent was evaporated, and the crude product was purified by flash chromatography on neutral Al2O3 using a mixture of dichloromethane/methanol from 99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 to 98:2.
:0.5 to 98:2.
General procedure for the synthesis of complexes C and D
In a 50 mL round bottom flask, RuCl3·3H2O (360 mg, 1.38 mmol, 1.0 equiv.), 4,4′-di-tert-butyl-2,2′-bipyridine (741 mg, 2.77 mmol, 2.0 equiv.) and LiCl (585 mg, 13.8 mmol, 10 equiv.) were dissolved in deoxygenated N,N-dimethylformamide (5 mL). The resulting solution was refluxed under a nitrogen atmosphere overnight. Afterward, the solution was cooled to room temperature and ice-cold acetone (50 mL) was added dropwise. The solution was filtered under vacuum and the precipitate was washed with 4 °C pre-cooled water and acetone. The solid was discarded and the solution was evaporated to dryness. The resulting crude product was purified by flash chromatography on neutral Al2O3 using dichloromethane as the eluent. Finally, the resulting solid was washed with ethyl ether. [Ru(dtbpy)2Cl2] was obtained as a dark red-violet solid in 31.2% yield (305 mg, 0.43 mmol). 1H NMR (400 MHz, CDCl3) δ 10.18 (s, 1H), 8.07 (s, 1H), 7.93 (s, 1H), 7.56 (d, J = 4.0 Hz, 2H), 6.93 (d, J = 4.4 Hz, 1H), 1.49 (s, 9H), 1.29 (s, 9H).:0 to 98:2. This solid was collected and washed with hexane (15 mL) to afford complex C as a purple solid in 84.2% yield (148 mg, 0.168 mmol). The 1H NMR spectrum was consistent with that previously reported in the literature.531H NMR (500 MHz, CD2Cl2) δ 8.30 (d, J = 1.9 Hz, 1H), 8.22 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 5.3, 2.0 Hz, 2H), 7.96 (d, J = 6.3 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.81 (d, J = 6.4 Hz, 1H), 7.72 (d, J = 5.8 Hz, 1H), 7.67–7.62 (m, 2H), 7.57 (d, J = 6.1 Hz, 1H), 7.53 (d, J = 5.3 Hz, 1H), 7.42 (dd, J = 6.0, 1.9 Hz, 1H), 7.22–7.18 (m, 2H), 7.16 (dd, J = 6.1, 2.1 Hz, 1H), 6.93–6.89 (m, 2H), 6.86 (t, J = 7.9 Hz, 1H), 6.50 (d, J = 7.2 Hz, 1H), 1.44 (s, 9H), 1.41 (s, 9H), 1.40 (s, 9H), 1.39 (s, 9H). ESI-MS: 792 [M]+.
:0.5). This solid was collected and washed with exane (15 mL) to afford complex D as a purple solid in 12.1% yield (21 mg, 0.023 mmol). 1H NMR (500 MHz, CD2Cl2) δ 8.32 (d, J = 2.1 Hz, 1H), 8.28 (s, 1H), 8.24 (d, J = 2.1 Hz, 1H), 8.19–8.14 (m, 2H), 8.13 (d, J = 2.3 Hz, 1H), 8.06 (d, J = 6.1 Hz, 1H), 7.81–7.71 (m, 4H), 7.63–7.59 (m, 2H),7.44 (dd, J = 5.7, 2.0 Hz, 1H), 7.29–7.15 (m, 5H), 7.08 (dd, J = 6.4, 2.1 Hz, 1H), 6.99 (t, J = 7.1 Hz, 1H), 6.72 (s, 1H), 1.45 (s, 18H), 1.37 (s, 9H), 1.36 (s, 9H); 13C NMR (126 MHz, CD2Cl2) δ 187.2 (C), 167.2 (C), 160.5 (C), 159.1 (C), 158.0 (C), 157.7 (C), 157.4 (C), 156.7 (C), 156.4 (C), 155.2 (C), 153.6 (CH), 150.2 (CH), 149.4 (CH), 149.1 (CH), 148.5 (CH), 145.3 (C), 135.2 (CH), 134.6 (C), 132.8 (CH), 129.9 (C), 128.7 (CH), 126.2 (CH), 124.9 (CH), 124.3 (CH), 123.6 (CH), 123.5 (CH), 123.4 (CH), 122.8 (CH), 122.7 (CH), 122.4 (CH), 119.6 (CH), 119.5 (CH), 119.43 (CH), 119.38 (CH), 119.2 (CH), 35.3 (C), 35.1 (C), 35.0 (C), 34.98 (C), 30.2 (2 CH3), 30.1 (CH3), 30.06 (CH3); 19F NMR (470 MHz, CD2Cl2) δ −153.15, −153.21. ESI-MS: 842 [M]+. HRMS (ESI-QTOF) for C51H58BN5F4Ru: ([M]+): m/z calcd: 922.3829; found: 922.3814. Anal. calc. for D = C51H58BN5F4Ru: C 65.94% H 6.29% N 7.54%; found: C 66.08% H 8.62% N 5.92%.
Electrochemical characterization
Voltammetric experiments were performed using a Metrohm d AutoLab PGSTAT 302N electrochemical work-station in combination with the NOVA 2.1.6 software package. All the measurements were carried out at room temperature in acetonitrile solutions with a sample concentration of approximately 1.0 mM and using 0.1 M tetrabutylammonium hexafluorophosphate (electrochemical grade, TBAPF6) as the supporting electrolyte. Oxygen was removed from the solutions by bubbling nitrogen. All the experiments were carried out using a three-electrode setup (BioLogic VC-4 cell, volume range: 1–3 mL) with a glassy carbon working electrode (having an active surface disk of 1.6 mm diameter), a Ag/AgNO3 redox couple (0.01 M in acetonitrile, with 0.1 M TBAClO4 supporting electrolyte) as the reference electrode, and a platinum wire as the counter electrode. At the end of each measurement, ferrocene was added as the internal reference. Cyclic voltammograms (CV) were typically recorded at a scan rate of 100 mV s−1. Osteryoung square-wave voltammograms (OSWV) were recorded with a scan rate of 25 mV s−1, a SW amplitude of ±20 mV, and a frequency of 25 Hz.Photophysics
The spectroscopic investigations were carried out in spectrofluorimetric grade acetonitrile and dichloromethane. The absorption spectra were recorded with a PerkinElmer Lambda 950 spectrophotometer. For the photoluminescence experiments, the sample solutions were placed in fluorimetric Suprasil quartz cuvettes (10.00 mm) and dissolved oxygen was removed by bubbling argon for 30 min. The uncorrected emission spectra were obtained with an Edinburgh Instruments FLS920 spectrometer equipped with a Peltier-cooled Hamamatsu R928 photomultiplier tube (PMT, spectral window: 185–850 nm) and an R5509-72 InP/InGaAs PMT supercooled at 193 K in a liquid nitrogen cooled housing and a TM300 emission monochromator with a NIR grating blazed at 1000 nm. An Osram XBO xenon arc lamp (450 W) was used as the excitation light source. The corrected spectra were acquired by means of a calibration curve, obtained by using an Ocean Optics deuterium–halogen calibrated lamp (DH-3plus-CAL-EXT). The photoluminescence quantum yields (PLQYs) in solution were obtained from the corrected spectra on a wavelength scale (nm) and measured according to the approach described by Demas and Crosby,59 using an air-equilibrated water solution of tris(2,2′-bipyridyl)ruthenium(II) dichloride as a reference (PLQY = 0.040).60 The emission lifetimes (τ) were measured through the time-correlated single photon counting (TCSPC) technique using an HORIBA Jobin Yvon IBH FluoroHub controlling a spectrometer equipped with a pulsed NanoLED as the excitation source and a red-sensitive Hamamatsu R-3237-01 PMT (185–850 nm) as the detector. The analysis of the luminescence decay profiles was accomplished with the DAS6 Decay Analysis software provided by the manufacturer, and the quality of the fit was assessed with the χ2 value close to unity and with the residuals regularly distributed along the time axis. To record the 77 K luminescence spectra, samples were put in quartz tubes (2 mm inner diameter) and inserted into a special quartz Dewar flask filled with liquid nitrogen. The poly(methyl methacrylate) (PMMA) films containing 1% (w/w) of the complexes were obtained by dropcasting and the thickness of the films was not controlled. Solid-state PLQY values were calculated with corrected emission spectra obtained from an Edinburgh FLS920 spectrometer equipped with an integrating sphere coated with barium sulphate, following the procedure described by Würth et al.61 Experimental uncertainties are estimated to be ±8% for τ determinations, ±10% for PLQYs, and ±2 nm and ±5 nm for absorption and emission peaks, respectively.Computational details
Density functional theory (DFT) calculations were performed using the B.01 revision of the Gaussian 16 program package,62 in combination with the M06 global-hybrid meta-GGA exchange–correlation functional.63,64 The fully relativistic Stuttgart/Cologne energy-consistent pseudopotential with multielectron fit was used to replace the first 28 inner-core electrons of the ruthenium metal centre (i.e., ECP28MDF) and was combined with the associated triple-ζ basis set (i.e., cc-pVTZ-PP basis);65 on the other hand, the Pople 6-31G(d,p) basis was adopted for all other atoms.66,67 The efficacy of the adopted computational protocol has already been validated on similar studies as reported in the literature.45 All the reported complexes were fully optimized, using a time-independent DFT approach, in their ground state (S0) and lowest triplet states; all the optimization procedures were performed using the polarizable continuum model (PCM) to simulate acetonitrile solvation effects.68–70 Frequency calculations were always used to confirm that every stationary point found by geometry optimizations was actually a minimum on the corresponding potential-energy surface (no imaginary frequencies). To investigate the nature of the emitting states, geometry optimizations and frequency calculations were performed at the spin-unrestricted UM06 level of theory (imposing a spin multiplicity of 3), using the S0 minimum-energy geometry as an initial guess. To locate and fully relax metal-centered triplet excited states (3MC), an analogous procedure was adopted, but using properly designed initial geometries as guesses (i.e., by selectively elongating the axial Ru–N bonds to approx. 2.5 Å).56 Time-dependent DFT calculations (TD-DFT),71,72 carried out at the same level of theory used for geometry optimizations, were used to calculate the first 16 triplet excitation from S0 minima and their nature was assessed with the support of Natural Transition Orbital (NTO) analysis.55 Charge decomposition analysis was performed and orbital-interaction diagrams were obtained using Multiwfn 3.8 – a Multifunctional Wavefunction Analyzer.73 All the pictures showing molecular geometries, orbitals and spin-density surfaces were created using GaussView 6.74Author contributions
A. B., F. M. and L. S. supervised and coordinated the project. L. S. and N. A. were responsible for funding acquisition. M. P. synthesized the compounds and, together with A. B., was responsible for their structural characterization. The photophysical and electrochemical studies were carried out by F. M. and M. P., and discussed with N.A.; F. M. was also responsible for the DFT calculations. The original draft was prepared by A. B. and F. M.; all the authors reviewed, edited, and approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender No. 104 published on 2.2.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU – Project 2022HX5CHP “HEPIrCOS”; Investment 1.5 (project ECOSISTER – Ecosystem for Sustainable Transition in Emilia-Romagna, grant number ECS00000033, Concession Decree No. 1052 of 23/06/2022, within Spoke 1 – Materials for sustainability and ecological transition and Spoke 2 – Clean energy production, storage and saving). Funding from the University of Bologna is also gratefully acknowledged.References
- Z. X. Giustra and S. Y. Liu, The state of the art in azaborine chemistry: New synthetic methods and applications, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed.
- C. Chen, C. Z. Du and X. Y. Wang, The rise of 1,4-BN-heteroarenes: Synthesis, properties, and applications, Adv. Sci., 2022, 9, e2200707 CrossRef.
- G. E. Rudebusch, L. N. Zakharov and S. Y. Liu, Rhodium-catalyzed boron arylation of 1,2-azaborines, Angew. Chem., Int. Ed., 2013, 52, 9316–9319 CrossRef CAS PubMed.
- S. Xu, L. N. Zakharov and S. Y. Liu, A 1,3-dihydro-1,3-azaborine debuts, J. Am. Chem. Soc., 2011, 133, 20152–20155 CrossRef CAS PubMed.
- H. Braunschweig, A. Damme, J. O. Jimenez-Halla, B. Pfaffinger, K. Radacki and J. Wolf, Metal-mediated synthesis of 1,4-di-tert-butyl-1,4-azaborine, Angew. Chem., Int. Ed., 2012, 51, 10034–10037 CrossRef CAS.
- W. Luo, P. G. Campbell, L. N. Zakharov and S. Y. Liu, A single-component liquid-phase hydrogen storage material, J. Am. Chem. Soc., 2011, 133, 19326–19329 CrossRef CAS PubMed.
- P. G. Campbell, L. N. Zakharov, D. J. Grant, D. A. Dixon and S. Y. Liu, Hydrogen storage by boron-nitrogen heterocycles: A simple route for spent fuel regeneration, J. Am. Chem. Soc., 2010, 132, 3289–3291 CrossRef CAS PubMed.
- P. Zhao, D. O. Nettleton, R. G. Karki, F. J. Zecri and S. Y. Liu, Medicinal chemistry profiling of monocyclic 1,2-azaborines, ChemMedChem, 2017, 12, 358–361 CrossRef CAS.
- B. A. Haney, C. L. Schrank and W. M. Wuest, Synthesis and biological evaluation of an antibacterial azaborine retinoid isostere, Tetrahedron Lett., 2021, 62, 152667 CrossRef CAS PubMed.
- D. H. Knack, J. L. Marshall, G. P. Harlow, A. Dudzik, M. Szaleniec, S. Y. Liu and J. Heider, BN/CC isosteric compounds as enzyme inhibitors: N- and B-ethyl-1,2-azaborine inhibit ethylbenzene hydroxylation as nonconvertible substrate analogues, Angew. Chem., Int. Ed., 2013, 52, 2599–2601 CrossRef CAS.
- S. Xu, F. Haeffner, B. Li, L. N. Zakharov and S. Y. Liu, Monobenzofused 1,4-azaborines: Synthesis, characterization, and discovery of a unique coordination mode, Angew. Chem., Int. Ed., 2014, 53, 6795–6799 CrossRef CAS.
- S. Xu, Y. Zhang, B. Li and S. Y. Liu, Site-selective and stereoselective trans-hydroboration of 1,3-enynes catalyzed by 1,4-azaborine-based phosphine-pd complex, J. Am. Chem. Soc., 2016, 138, 14566–14569 CrossRef CAS.
- J.-Y. Wang and J. Pei, BN-embedded aromatics for optoelectronic applications, Chin. Chem. Lett., 2016, 27, 1139–1146 CrossRef CAS.
- S. Chen, M. Dong, Y. Bai, Y. Chen, Y. Fu, L. Shao, X. Lu, C. Liu, K. Zhang, H. Wu and F. Huang, 1,4-azaborine based unfused non-fullerene acceptors for organic solar cells, J. Mater. Chem. A, 2023, 11, 3653–3662 RSC.
- P. Sudhakar, S. Kuila, K. Stavrou, A. Danos, A. M. Z. Slawin, A. Monkman and E. Zysman-Colman, Azaborine as a versatile weak donor for thermally activated delayed fluorescence, ACS Appl. Mater. Interfaces, 2023, 15, 25806–25818 CrossRef CAS.
- A. J. Ashe, Aromatic borataheterocycles: Surrogates for cyclopentadienyl in transition-metal complexes, Organometallics, 2009, 28, 4236–4248 CrossRef CAS.
- H. Yang, X. Fang, J. W. Kampf and A. J. Ashe, Bridged 1,2-azaborolyl zirconium complexes: Heterocyclic analogs of the ansa-zirconocene olefin polymerization catalysts, Polyhedron, 2005, 24, 1280–1288 CrossRef CAS.
- A. J. Ashe, H. Yang, X. Fang and J. W. Kampf, Synthesis and coordination chemistry of 3a,7a-azaborindenyl, a new isoelectronic analogue of the indenyl ligand, Organometallics, 2002, 21, 4578–4580 CrossRef CAS.
- J. Pan, J. W. Kampf and A. J. Ashe III, The preparation and crystal structures of η1-derivatives of 2-phenyl-1,2-azaboratabenzene, Organometallics, 2008, 27, 1345–1347 CrossRef CAS.
- X. Fang, H. Yang, J. W. Kampf, M. M. Banaszak Holl and A. J. Ashe, Syntheses of ring-fused B−N heteroaromatic compounds, Organometallics, 2005, 25, 513–518 CrossRef.
- Y. Chen, X. Fang and W. Dan, 1,2-azaborolyl-ligated titanium(IV) half-metallocenes: Synthesis, characterization, and catalytic activity in ethylene polymerization, Organometallics, 2015, 35, 15–19 CrossRef.
- X. Fang, Y. Deng, Q. Xie and F. Moingeon, A bis(1,2-azaborolyl)yttrium alkyl complex: Synthesis, structure, and polymerization study, Organometallics, 2008, 27, 2892–2895 CrossRef CAS.
- J. Pan, J. W. Kampf and A. J. Ashe, Tricarbonylchromium complexes of 1,2-dihydro-1,2-benzazaborines, Organometallics, 2008, 28, 506–511 CrossRef.
- J. Pan, J. W. Kampf and A. J. Ashe, Switchable haptotropic migrations of tricarbonylchromium complexes of 1,2-dihydro-2-phenyl-1,2-azaborine, Organometallics, 2005, 25, 197–202 CrossRef.
- A. J. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S. Y. Liu, A hybrid organic/inorganic benzene, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS PubMed.
- J. Pan, J. Wang, M. M. Banaszak Holl, J. W. Kampf and A. J. Ashe, Haptotropic migration from the six- to the five-membered ring of (3a,7a-azaborindenyl)tricarbonylchromium anion, Organometallics, 2006, 25, 3463–3467 CrossRef CAS.
- A. D. Rohr, J. W. Kampf and A. J. Ashe, Syntheses of [6,6]-fused-ring 1,2-azaborines, Organometallics, 2014, 33, 1318–1321 CrossRef CAS.
- A. J. Ashe, X. Fang, X. Fang and J. W. Kampf, Synthesis of 1,2-dihydro-1,2-azaborines and their conversion to tricarbonyl chromium and molybdenum complexes, Organometallics, 2001, 20, 5413–5418 CrossRef CAS.
- S. Y. Liu, I. D. Hills and G. C. Fu, Synthesis, resolution, and aldol reactions of a planar-chiral lewis acid complex, J. Am. Chem. Soc., 2005, 127, 15352–15353 CrossRef CAS PubMed.
- S.-Y. Liu, M. M. C. Lo and G. C. Fu, The synthesis of an enantiopure planar-chiral lewis acid complex via kinetic resolution and its application in stereoselective additions to imines, Tetrahedron, 2006, 62, 11343–11349 CrossRef CAS.
- A. J. Ashe 3rd and X. Fang, A synthesis of aromatic five- and six-membered B-N heterocycles via ring closing metathesis, Org. Lett., 2000, 2, 2089–2091 CrossRef.
- J. Pan, J. W. Kampf and A. J. Ashe, 1,2-azaboratabenzene: A heterocyclic π-ligand with an adjustable basicity at nitrogen, Organometallics, 2004, 23, 5626–5629 CrossRef CAS.
- J. Pan, J. W. Kampf and A. J. Ashe, The ligand properties of 2-vinyl-1,2-azaboratabenzene, J. Organomet. Chem., 2009, 694, 1036–1040 CrossRef CAS.
- Z. Liu, J. Xu, W. Ruan, C. Fu, H. J. Zhang and T. B. Wen, A half-sandwich 1,2-azaborolyl ruthenium complex: Synthesis, characterization, and evaluation of its catalytic activities, Dalton Trans., 2013, 42, 11976–11980 RSC.
- A. J. Marwitz, J. T. Jenkins, L. N. Zakharov and S. Y. Liu, 1,2-azaborine cations, Angew. Chem., Int. Ed., 2010, 49, 7444–7447 CrossRef CAS PubMed.
- C. J. Saint-Louis, R. N. Shavnore, C. D. C. McClinton, J. A. Wilson, L. L. Magill, B. M. Brown, R. W. Lamb, C. E. Webster, A. K. Schrock and M. T. Huggins, Synthesis, computational, and spectroscopic analysis of tunable highly fluorescent BN-1,2-azaborine derivatives containing the N-BOH moiety, Org. Biomol. Chem., 2017, 15, 10172–10183 RSC.
- N. N. T. Nguyen, H. Mubarok, T. Lee, T. Q. Tran, J. Jung and M. H. Lee, Highly emissive planarized B,N-diarylated benzonaphthoazaborine compounds for narrowband blue fluorescence, RSC Adv., 2022, 12, 29892–29899 RSC.
- N. Yan, F. Wang, J. Wei, J. Song, L. Yan, J. Luo, Z. Fang, Z. Wang, W. Zhang and G. He, Highly emissive B←N unit containing four-coordinate C,N-chelated organoboron compound for the detection of fluoride ions, Dyes Pigm., 2019, 166, 410–415 CrossRef CAS.
- R. W. Lamb, A. K. Schrock, M. T. Huggins and C. E. Webster, Predicting absorption and emission maxima of polycyclic aromatic azaborines: Reliable transition energies and character, J. Phys. Chem. A, 2021, 125, 3–12 CrossRef CAS PubMed.
- C. J. Saint-Louis, L. L. Magill, J. A. Wilson, A. R. Schroeder, S. E. Harrell, N. S. Jackson, J. A. Trindell, S. Kim, A. R. Fisch, L. Munro, V. J. Catalano, C. E. Webster, P. P. Vaughan, K. S. Molek, A. K. Schrock and M. T. Huggins, The synthesis and characterization of highly fluorescent polycyclic azaborine chromophores, J. Org. Chem., 2016, 81, 10955–10963 CrossRef CAS PubMed.
- K. Mitsudo, K. Shigemori, H. Mandai, A. Wakamiya and S. Suga, Synthesis and properties of dithieno-fused 1,4-azaborine derivatives, Org. Lett., 2018, 20, 7336–7340 CrossRef CAS.
- A. D. Campbell, K. Ellis, L. K. Gordon, J. E. Riley, V. Le, K. K. Hollister, S. O. Ajagbe, S. Gozem, R. B. Hughley, A. M. Boswell, O. Adjei-sah, P. D. Baruah, R. N. Malone, L. M. Whitt, R. J. Gilliard and C. J. Saint-Louis, Solvatochromic and aggregation-induced emission active nitrophenyl-substituted pyrrolidinone-fused-1,2-azaborine with a pre-twisted molecular geometry, J. Mater. Chem. C, 2023, 11, 13740–13751 RSC.
- A. W. Baggett, M. Vasiliu, B. Li, D. A. Dixon and S. Y. Liu, Late-stage functionalization of 1,2-dihydro-1,2-azaborines via regioselective iridium-catalyzed C-H borylation: The development of a new N,N-bidentate ligand scaffold, J. Am. Chem. Soc., 2015, 137, 5536–5541 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, B. Li, L. N. Zakharov, M. Vasiliu, D. A. Dixon and S. Y. Liu, A modular synthetic approach to monocyclic 1,4-azaborines, Angew. Chem., Int. Ed., 2016, 55, 8333–8337 CrossRef CAS PubMed.
- A. Baschieri, F. Aleotti, E. Matteucci, L. Sambri, M. Mancinelli, A. Mazzanti, E. Leoni, N. Armaroli and F. Monti, A pyridyl-1,2-azaborine ligand for phosphorescent neutral iridium(III) complexes, Inorg. Chem., 2023, 62, 2456–2469 CrossRef CAS PubMed.
- H. Rudmann, S. Shimada and M. F. Rubner, Solid-state light-emitting devices based on the tris-chelated ruthenium(II) complex. 4. High-efficiency light-emitting devices based on derivatives of the tris(2,2′-bipyridyl) ruthenium(II) complex, J. Am. Chem. Soc., 2002, 124, 4918–4921 CrossRef CAS PubMed.
- D. van der Westhuizen, K. G. von Eschwege and J. Conradie, Electrochemistry and spectroscopy of substituted [Ru(phen)3]2+ and [Ru(bpy)3]2+ complexes, Electrochim. Acta, 2019, 320, 134540 CrossRef CAS.
- P. G. Bomben, K. C. Robson, P. A. Sedach and C. P. Berlinguette, On the viability of cyclometalated Ru(II) complexes for light-harvesting applications, Inorg. Chem., 2009, 48, 9631–9643 CrossRef CAS.
- S. Vierucci, S. Muzzioli, P. Righi, V. Borzatta, G. Gorni and I. Zama, A greener procedure for the synthesis of [Bu4N]2-cis-[Ru(4-carboxy-4′-carboxylate-2,2′-bipyridine)2(NCS)2] (N719), a benchmark dye for DSSC applications, RSC Adv., 2016, 6, 55768–55777 RSC.
- T. B. Hadda and H. Le Bozec, Preparation and characterization of ruthenium complexes with the new 4,4′,4′′-tri-tert-butylterpyridine ligand and with 4,4′-di-tert-butylbipyridine, Polyhedron, 1988, 7, 575–577 CrossRef.
- S. Bernhard, J. A. Barron, P. L. Houston, H. D. Abruna, J. L. Ruglovksy, X. Gao and G. G. Malliaras, Electroluminescence in ruthenium(II) complexes, J. Am. Chem. Soc., 2002, 124, 13624–13628 CrossRef CAS PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal Jr. and S. Bernhard, Accelerated luminophore discovery through combinatorial synthesis, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- T. C. Motley, L. Troian-Gautier, M. K. Brennaman and G. J. Meyer, Excited-state decay pathways of tris(bidentate) cyclometalated ruthenium(II) compounds, Inorg. Chem., 2017, 56, 13579–13592 CrossRef CAS PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Photochemistry and photophysics of coordination compounds: Ruthenium”, in Photochemistry and photophysics of coordination compounds, I, 2007, 117–214 CAS.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- A. Soupart, I. M. Dixon, F. Alary and J.-L. Heully, DFT rationalization of the room-temperature luminescence properties of Ru(bpy)32+ and Ru(tpy)22+ : 3MLCT–3MC minimum energy path from NEB calculations and emission spectra from VRES calculations, Theor. Chem. Acc., 2018, 137, 37 Search PubMed.
- K. Nisbett, Y. J. Tu, C. Turro, J. J. Kodanko and H. B. Schlegel, DFT investigation of ligand photodissociation in [RuII(tpy)(bpy)(py)]2+ and [RuII(tpy)(Me2bpy)(py)]2+ complexes, Inorg. Chem., 2018, 57, 231–240 CrossRef CAS PubMed.
- M. Kayanuma, Photosubstitution reaction of a bidentate ligand in a Ru(II) complex in aqueous solution, Comput. Theor. Chem., 2022, 1213, 113745 CrossRef CAS.
- G. A. Crosby and J. N. Demas, Measurement of photoluminescence quantum yields. Review, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Relative and absolute determination of fluorescence quantum yields of transparent samples, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Revision B.01, Gaussian Inc., Wallingford, CT, USA, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y-Pd, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, A complete basis set model chemistry. I. The total energies of closed–shell atoms and hydrides of the first–row elements, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, A complete basis set model chemistry. II. Open–shell systems and the total energies of the first–row atoms, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- J. Tomasi and M. Persico, Molecular-interactions in solution - An overview of methods based on continuous distributions of the solvent, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, “Continuum solvation models”, in Solvent effects and chemical reactivity, ed. O. Tapia and J. Bertrán, Springer Netherlands, 2002, vol. 17, pp. 1–80 Search PubMed.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with time-dependent density functional theory, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- A. D. Laurent, C. Adamo and D. Jacquemin, Dye chemistry with time-dependent density functional theory, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, Gaussview, version 6, Semichem Inc., Shawnee Mission, KS, USA, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03115f |
| This journal is © The Royal Society of Chemistry 2025 |