
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalysed C–H bond functionalisation route to 1,2-dihydroferroceno[c]isoquinoline and its annellated derivatives and the reactivity of these compounds†
Věra
Varmužová
a,
Ivana
Císařová
a,
Jiří
Schulz
a,
Květa
Kalíková
b and
Petr
Štěpnička
 *a
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 00 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
bDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 00 Prague, Czech Republic
First published on 3rd December 2024
Abstract
C–H bond functionalisation has developed into a powerful synthetic methodology that is applicable to a wide array of substrates, including organometallic compounds. In this study, racemic, planar-chiral 1,2-dihydroferroceno[c]isoquinoline and analogous helical compounds with one or two additional ortho-fused benzene rings were synthesised by palladium-catalysed C–H bond activation/cyclisation of N-[(bromoaryl)methyl]-N-(methylsulfonyl)aminoferrocenes. These starting materials are readily accessible from FcNHSO2Me (Fc = ferrocenyl) and appropriate vicinal bromo-(bromomethyl)arenes. The racemic products were successfully enantioseparated using chiral chromatography and the representative compound, 1,2-dihydro-2-(methylsulfonyl)ferroceno[c]isoquinoline, was converted to the unstable ferroceno[c]isoquinoline and further used to prepare a heterobimetallic, Fe/Ru bis-metallocene complex via a reaction with [(η5-C5Me5)Ru(MeCN)3][PF6]. All compounds were characterised by spectroscopic methods (NMR, FTIR, and UV-vis) and mass spectrometry and, in most cases, the structures were determined by single-crystal X-ray diffraction analysis. In addition, the representative compounds were examined by cyclic voltammetry, and the results were rationalised with the aid of DFT calculations.
Introduction
Transition metal-catalysed C–H bond functionalisation enables the synthesis of more complex molecules from accessible C–H substrates through the formation of new C–C and C–heteroatom bonds. Compared with conventional methods, this approach is more atom-economic but also more challenging due to the generally low reactivity of the C–H bonds. Hence, harsh reaction conditions or carefully designed catalysts and various directing groups are typically needed to achieve satisfactory results.1In ferrocene chemistry,2 reactions involving directed C–H functionalisation provide alternative access to planar chiral compounds.3 In this respect, ortho-condensed systems in which the ferrocene cyclopentadienyl rings are annellated with a (hetero)aromatic moiety are particularly interesting. Although the first compounds of this type, bis(η5-indenyl)iron(II) (A),4 ferroceno[2,3]indenone (B)5 and the corresponding indene C6 (Scheme 1, top), were prepared shortly after the discovery of ferrocene itself,2a interest in these compounds was renewed with the advent of synthetic methods based on the C–H bond activation. Thus, compound B was obtained using Pd-catalysed cyclisation of (2-halobenzoyl)ferrocenes,7 and a similar method was utilised to synthesise similar compounds with a terminal pyridine ring.8,9 More recently, routes based on asymmetric, Pd-mediated C–H bond functionalisation led to ferrocenopyridines (D),10 quinoline derivatives (E and F),11 and (iso)quinolinones (G)12 (Scheme 1).
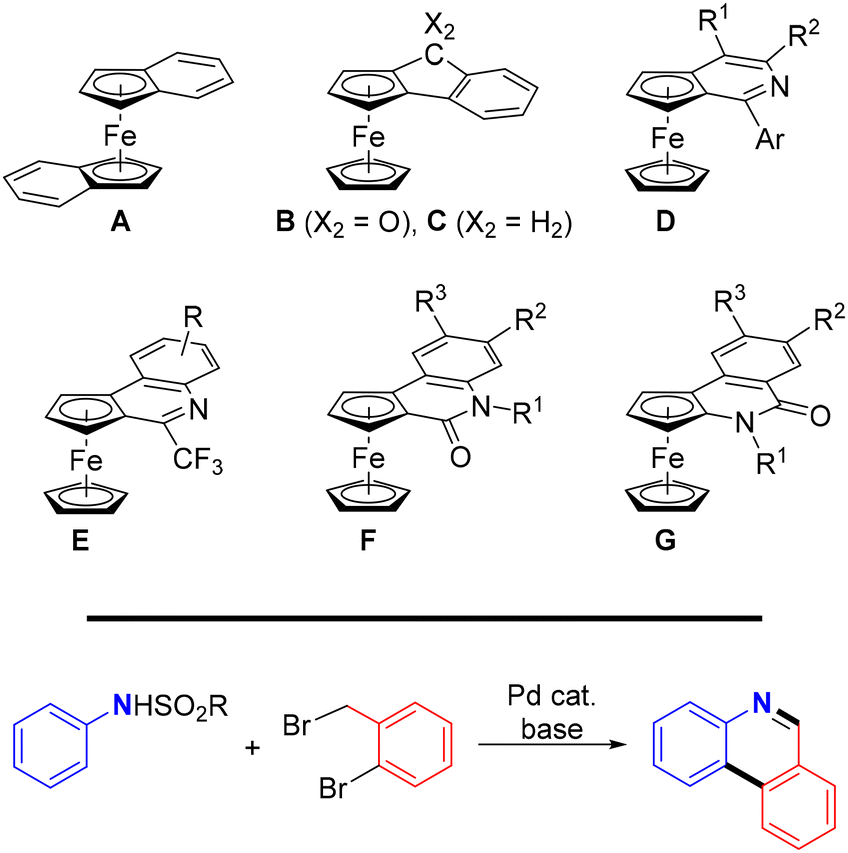 | ||
| Scheme 1 (top) Examples of annellated ferrocenes relevant to this study and (bottom) Pd-catalysed cascade reaction producing phenanthridines from N-sulfonyl anilines. | ||
In our research, we were inspired by the recently disclosed13 one-pot cascade reaction involving nucleophilic alkylation, C–H bond activation, and aromatisation that converted N-sulfonyl aryl amines into phenanthridines (Scheme 1, bottom), and decided to apply this approach to the synthesis of analogous ferrocene-based compounds. The results from our study are reported here.
Results and discussion
Synthesis of 1,2-dihydro-2-(methylsulfonyl)ferroceno[c]isoquinoline and analogues with an extended π-system
The starting material, viz. [(methylsufonyl)amino]ferrocene (1), was prepared by the reaction of aminoferrocene (FcNH2, Fc = ferrocenyl) with methanesulfonyl chloride in tetrahydrofuran (THF) in the presence of pyridine.14 It was isolated as an air-stable, orange crystalline solid in 83% yield after column chromatography. Unfortunately, the direct reaction of 1 with 2-bromobenzyl bromide (2a) (Scheme 2) did not proceed under the conditions reported for the aforementioned direct synthesis of phenanthridines13 despite numerous attempts.15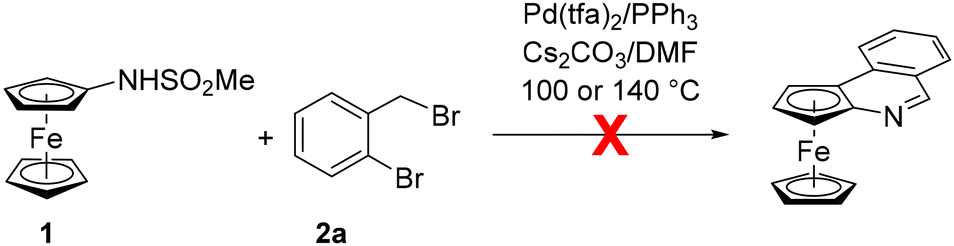 | ||
| Scheme 2 Attempted direct annulation of 1 with 2a. Conditions: 1 (0.36 mmol), 2a (0.36 mol), Pd(tfa)2 (5 mol%), PPh3 (20 mol%), and Cs2CO3 (4 equiv. vs. 1) were reacted in anhydrous N,N-dimethylformamide (4 mL) at 100 or 140 °C for 1–2 days; Pd(tfa)2 = palladium(II) trifluoromethanesulfonate. | ||
To achieve our synthetic goal, we then resorted to a stepwise approach and employed a “preassembled” intermediate. In the first step, compound 1 was alkylated with 2a in the presence of Cs2CO3 in acetonitrile at 70 °C to produce N-(2-bromobenzyl)sulfonamide 3a in a 94% isolated yield (Scheme 3; N.B. a similar reaction in N,N-dimethylformamide gave 3a in only 45% yield). Compound 3a already reacted under the conditions provided in Scheme 2 to produce a mixture of the targeted compound 4a and the corresponding dehalogenation product 5a (see Scheme 3). However, as the majority of the starting material 3a (71%) remained unconsumed, we pursued a more efficient catalyst. Indeed, upon replacing Pd(tfa)2 with Generation 3 Buchwald's Pd precatalyst BG316 (Scheme 4), increasing the catalyst amount to 10 mol% Pd and extending the reaction time, 3a was fully converted into a 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 mixture of 4a and 5a (Table 1). With the following optimisation experiments, we tried to increase the reaction efficiency (i.e., the conversion and selectivity towards the cyclisation product 4a) at a lower catalyst amount (1 mol% Pd).
:6 mixture of 4a and 5a (Table 1). With the following optimisation experiments, we tried to increase the reaction efficiency (i.e., the conversion and selectivity towards the cyclisation product 4a) at a lower catalyst amount (1 mol% Pd).
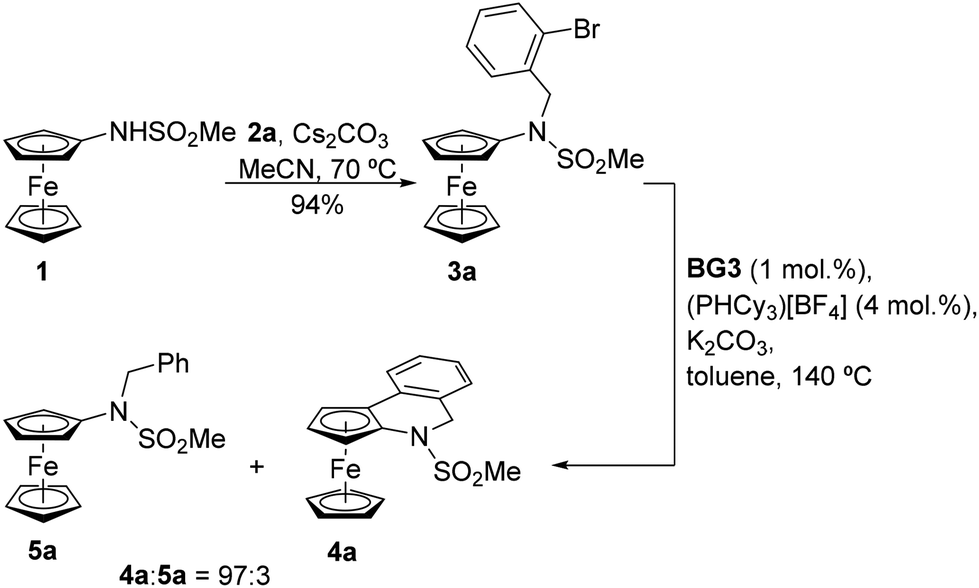 | ||
| Scheme 3 Synthesis and Pd-catalysed cyclisation of 3a. | ||
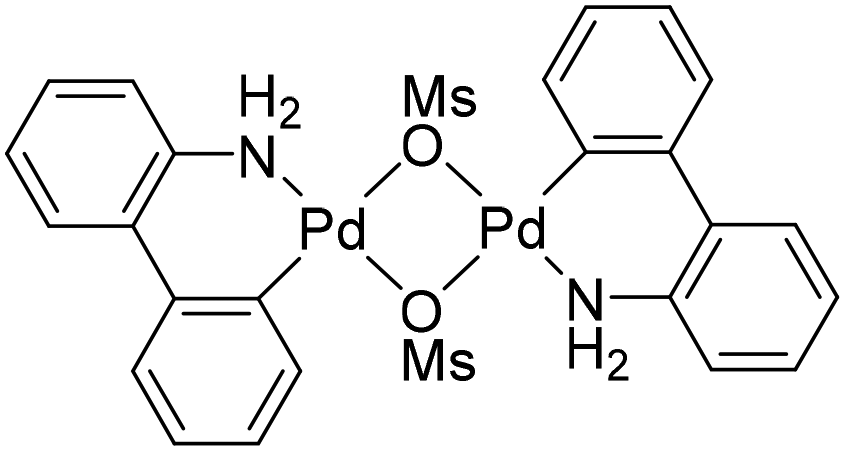 | ||
| Scheme 4 Precatalyst BG3. | ||
| Pd source [mol%] | Ligand [equiv.]b | Base | Solvent | Time [h] |
3a:4a:5a |
|---|---|---|---|---|---|
| a All reactions were performed at 140 °C. DMF = N,N-dimethylformamide, DCE = 1,2-dichloroethane, Cy = cyclohexyl; the structure of BG3 is shown in Scheme 4. b Equivalents of the phosphine ligand per palladium. | |||||
| Pd(tfa)2 [5] | PPh3 [4] | Cs2CO3 | DMF | 13 | 71:21:8 |
| BG3 [10] | PPh3 [4] | AcOK | Toluene | 144 | 0:94:6 |
| Pd(OAc)2 [1] | PPh3 [2] | AcOK | Toluene | 24 | 68:30:2 |
| Pd(OAc)2 [1] | PPh3 [4] | AcOK | Toluene | 24 | 60:38:2 |
| BG3 [1] | PPh3 [2] | AcOK | Toluene | 24 | 15:83:2 |
| BG3 [1] | PPh3 [4] | AcOK | Toluene | 24 | 2:94:4 |
| BG3 [1] | PPh3 [4] | AcONa | Toluene | 24 | 76:20:4 |
| BG3 [1] | PPh3 [4] | AcOCs | Toluene | 24 | 42:55:3 |
| BG3 [1] | PPh3 [4] | AcOK | DCE | 24 | 79:20:1 |
| BG3 [1] | PPh3 [4] | K2CO3 | Toluene | 24 | 66:29:5 |
| BG3 [1] | PPh3 [4] | K3PO4 | Toluene | 24 | 58:38:4 |
| BG3 [1] | PCy3·H[BF4] [4] | AcOK | Toluene | 24 | 0:97:3 |
| BG3 [1] | PCy3·H[BF4] [4] | K2CO3 | Toluene | 24 | 0:97:3 |
The results shown in Table 1 confirm the generally better efficiency of the catalysts generated from BG3 compared to those resulting from palladium carboxylates. A further improvement was noted upon changing triphenylphosphine to the more basic tricyclohexylphosphine (PCy3; the phosphine was generated in situ from the easier-to-handle, air-stable phosphonium salt and the base, which was used in excess),17 whereas the amount of auxiliary phosphine (2 vs. 4 equiv. per Pd) had a lower impact on the reaction course. Potassium acetate or carbonate were identified as suitable bases, and the reactions performed in toluene proceeded better than those in N,N-dimethylformamide and 1,2-dichloroethane. Eventually, the combination of a BG3/PCy3 catalyst (1 mol% Pd, Pd:P = 1:4) and K2CO3 in toluene enabled the cyclisation with full conversion of the starting material and 97% selectivity for 4a when the reaction was performed in a pressure tube at 140 °C. Compounds 4a and 5a could not be efficiently separated by column chromatography; a pure sample of 4a was obtained by crystallisation from THF/hexane.
Having established a reliable route to 4a, we focused on the synthesis of congeners with a larger conjugated π-system. Extension of the aromatic system by one fused benzene ring was easily achieved. The required starting material 3b was obtained in good yield (>90%) by alkylation of 1 with 1-bromo-2-(bromomethyl)naphthalene (2b) under conditions used to prepare 3a (Scheme 5). Amide 3b was subsequently cyclised under the developed conditions (BG3/PCy3·H[BF4], K2CO3, toluene, 140 °C/24 h) to produce the corresponding annellated compound 4b contaminated with a minor amount of the dehalogenation product 5b (11%; Scheme 5). Even in this case, the side product could not be efficiently separated by column chromatography; however, the crystallisation of the product mixture from hot heptane produced two types of crystals, which were manually separated.
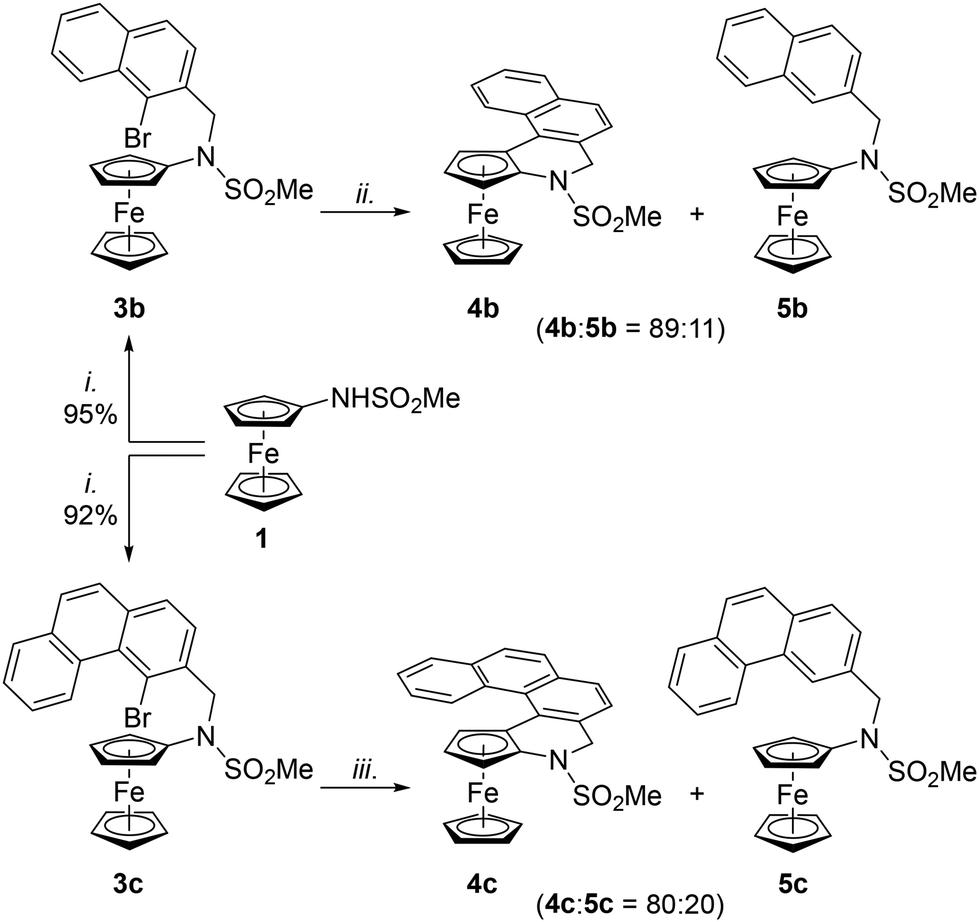 | ||
| Scheme 5 Synthesis and cyclisation of 3b and 3c. Conditions: i. 2b or 2c, Cs2CO3, MeCN, 70 °C/3 h; ii. BG3 (1 mol%), (PHCy3)[BF4] (Pd:P = 1:4), toluene, 140 °C/24 h; iii. same as ii. except that 1,4-dioxane was used as the solvent. | ||
The alkylating agent needed for the synthesis of the compound possessing three fused benzene rings, 4-bromo-3-(bromomethyl)phenanthrene (2c), was synthesised from the commercially available 2,3-dihydrophenanthren-4(1H)-one (8). This compound was converted to 4-bromophenanthrene-3-carbaldehyde (10) in two steps using a literature procedure.18 Next, the aldehyde was reduced to the corresponding alcohol 11 using Na[BH4] in THF/methanol, and the alcohol was brominated with PBr3 to produce the targeted dibromide 2c (Scheme 6). The yield over the last two synthetic steps was 56%.
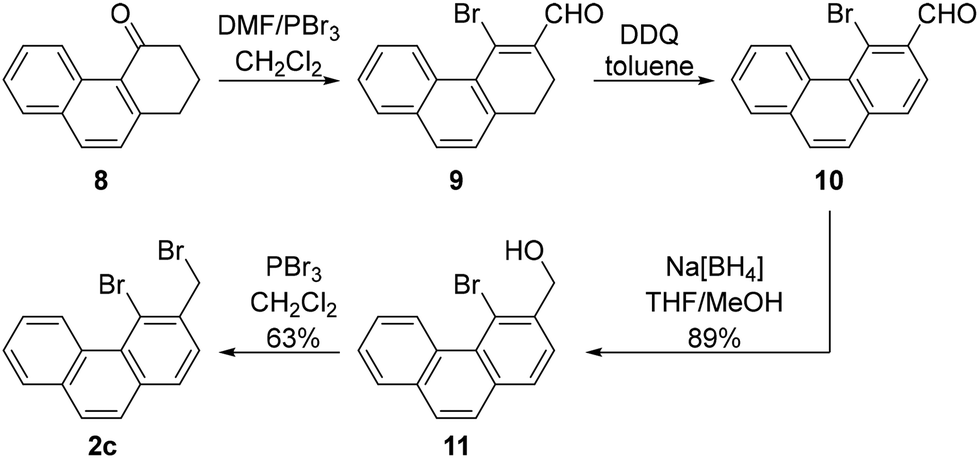 | ||
| Scheme 6 Synthesis of 4-bromo-3-(bromomethyl)phenanthrene (2c). | ||
Alkylation of 1 with 2c smoothly proceeded to produce starting material 3c. However, the following cyclisation step required additional optimisation to proceed satisfactorily. In particular, the reaction had to be performed in anhydrous 1,4-dioxane, in which it proceeded with full conversion and afforded an 80:20 mixture of 4c and 5c (Scheme 5; N.B. the amount of the dehalogenated side product 5c was considerably greater when non-strictly dried and deoxygenated solvent was employed).
With compounds 4a–c in hand, we attempted to remove the sulfonate group and to synthesise the corresponding, fully aromatic compounds, ferrocenoisoquinolines. Following a literature procedure for NH group deprotection,19,20 a mixture of 4a and 5a (97:3) was treated with sodium bis(2-methoxyethoxy)aluminium hydride (Red-Al) in toluene. However, instead of the expected sulfonate group removal, this reaction directly produced ferrocenoisoquinoline 6a in a 30–35% yield (Scheme 7).
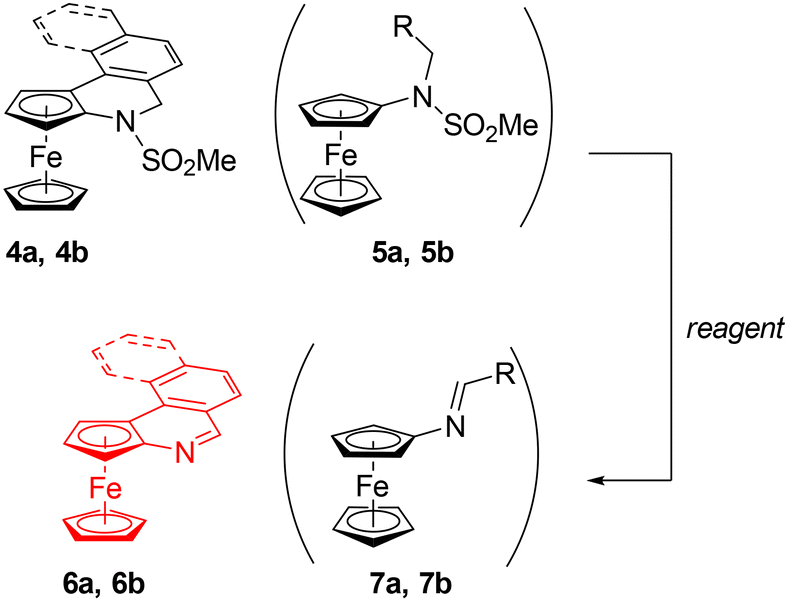 | ||
| Scheme 7 Synthesis of ferrocenoisoquinolines 6 (R = Ph (a), 2-naphthyl (b); see text for details). | ||
Next, we tried to “deprotect” the 4a/5a mixture using a base. No reaction was observed when the mixture was refluxed with 5% KOH in methanol or heated with Cs2CO3 in N,N-dimethylformamide at 140 °C. Eventually, the deprotection, again with concomitant aromatisation, was achieved upon treating the 4a/5a mixture with potassium tert-butoxide (5 equiv.) in toluene at 100 °C for 2 h. The conversion of 4a into 6a was indicated by a pronounced colour change from orange to deep red. Compound 6a was isolated by chromatography as a red solid with a 71% yield over the two steps (i.e., from 3a). A small amount of impure Schiff base 7a was also obtained and adequately identified.
Unfortunately, compound 6a was unstable and readily converted into an insoluble black solid. The decomposition was faster in a solution (especially in halogenated solvents), which precluded the crystallisation of this compound. Solid samples could be stored at −18 °C but still had to be freshly purified by chromatography before use. Efforts to prepare a more stable derivative by the methylation of 6a with MeI or [Me3O][BF4] and by the reaction with picric acid were unsuccessful; these reactions uniformly led to dark intractable solids.
The mixture of 4b and 5b also reacted with t-BuOK to give a mixture of the respective aromatised compounds 6a and 7b (Scheme 7). These compounds were separated by column chromatography; the yield of 6b was 67%. However, compound 6b was even less stable than 6a and, hence, no further experiments focused on the properties and reactivity of these aromatic derivatives were made.
Notably, our attempts to conduct the cyclisation reaction asymmetrically were unsuccessful. The cyclisation reactions were performed similarly to the synthesis of the racemic compounds but with catalysts based on BG3 and common chiral phosphine ligands (see ESI, Scheme S1†) that were successfully applied in asymmetric, Pd-catalysed reactions leading to chiral helicenes.21 In the present case, however, these conditions either entirely failed (for (S)-BINAP and (R)-H8BINAP; yields <5%) or led to racemic cyclisation product 4a (for (S)-SEGPHOS and (R)-2,2′-bis[bis(3,5-dimethylphenyl)phosphino]-6,6′-dimethoxybiphenyl).
Therefore, the separation of the enantiomers was examined using supercritical fluid chromatography (SFC)22 on a modified amylose column with a CO2–methanol mixture as the eluent (for details, see the ESI†). Under these conditions, the enantiomers of 4a–4c were baseline resolved, with increasing amounts of CO2 in the mobile phase (70:30 → 90:10) improving peak resolution. The retention time increased with increasing molecular weight, as expected. Thus, all six species present in a model mixture of the three compounds 4a–4c could be separated (Fig. 1). Efficient separation of the enantiomers of 4a was also achieved by conventional HPLC analysis on the same column using n-heptane/2-propanol (80/20) as the eluent.
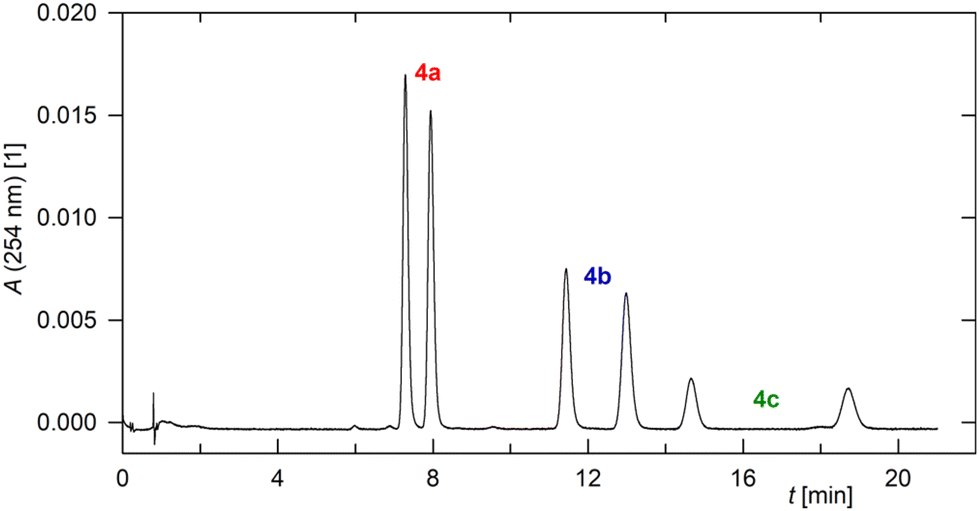 | ||
| Fig. 1 SFC chromatogram of an equimolar 4a–4c mixture (Lux® i-Amylose-3 column, 150 × 4.6 mm, particle size 3 μm, CO2–methanol 80:20, 2 mL min−1, 40 °C, back pressure regulator 2000 psi, UV detection at 254 nm). | ||
Structural characterisation of the prepared compounds
All compounds were characterised using a combination of electrospray ionisation mass spectrometry, elemental analysis (conventional or from high-resolution MS), and UV-vis, FTIR and NMR spectroscopies. In addition, the solid-state structures of compounds 1, 3a–c, and 4a–c were determined by single-crystal X-ray diffraction analysis. Only the structures of the annellated compounds 4a–c are presented here; other structures are discussed in the ESI.†The identities of the prepared compounds were initially verified by the mass spectra, which revealed ions due to the molecular ions (M+) or their simple adducts ([M + H]+ or [M + Na]+), whereas the individual structures were corroborated by the NMR spectra. In particular, the 1H NMR spectra displayed characteristic sets of signals due to the ferrocene moiety. Besides a strong singlet of the C5H5 ring, two virtual triplets arising from the AA′BB′ spin system were observed for 1, 3a–c and 5a–c, whereas an AMX pattern due to the C5H3 ring was detected in the spectra of the annellated compounds 4a–c, 6a and 6b. The methylene protons in 3a–c gave rise to singlets at δH 4.9–5.2, while the planar chiral compounds 4a–c, where these protons become diastereotopic, displayed AB doublets at δH 4.9–5.3 (only compound 4b showed a broad singlet). The corresponding 13C NMR methylene signals were detected at δC 56–58 and δC 51–53, respectively. The 13C NMR spectra revealed all other expected signals, including the low-field resonance due to ferrocene Cipso–N (3a–c: δC ≈ 101; 4a–c: δC ≈ 97; cf. δC 105.6 for aminoferrocene in CDCl323).
The NMR spectra also showed signals at approximately δH 2.4–3.0 and δC ≈ 37, assigned to the sulfonamide moiety. Its presence was further confirmed by intense absorption bands at approximately 1340 (νasSO2) and 1160 (νsSO2) cm−1 in the FTIR spectra.24 For the parent compound 1, the sulfonate bands were shifted to lower energies (maxima at 1307 and 1148 cm−1), and the spectrum displayed a strong νNH band at 3245 cm−1.24
The NMR spectra of isoquinolines 6a and 6b revealed diagnostic imine resonances at δH ≈ 8.9–9.1/δC ≈ 157 and the expected signals from the aromatic fragments. Furthermore, the colour change associated with the conversion of 4a to 6a, reflecting the different extents of conjugation in these molecules, was nicely manifested in the UV-vis spectra (Fig. 2). The spectrum of 4a displayed a band at 452 nm, which was ascribed to the forbidden d–d transition at the ferrocene unit25 (N.B. ferrocene itself displays this band at 440 nm in an EtOH solution as well as in the gas phase).26 A shoulder at a strong band extending from the UV region was also detected. For isoquinoline 6a, the low-energy band was bathochromically shifted to 505 nm (albeit with a practically unchanged intensity), whereas the shoulder developed into a separate band at 398 nm.
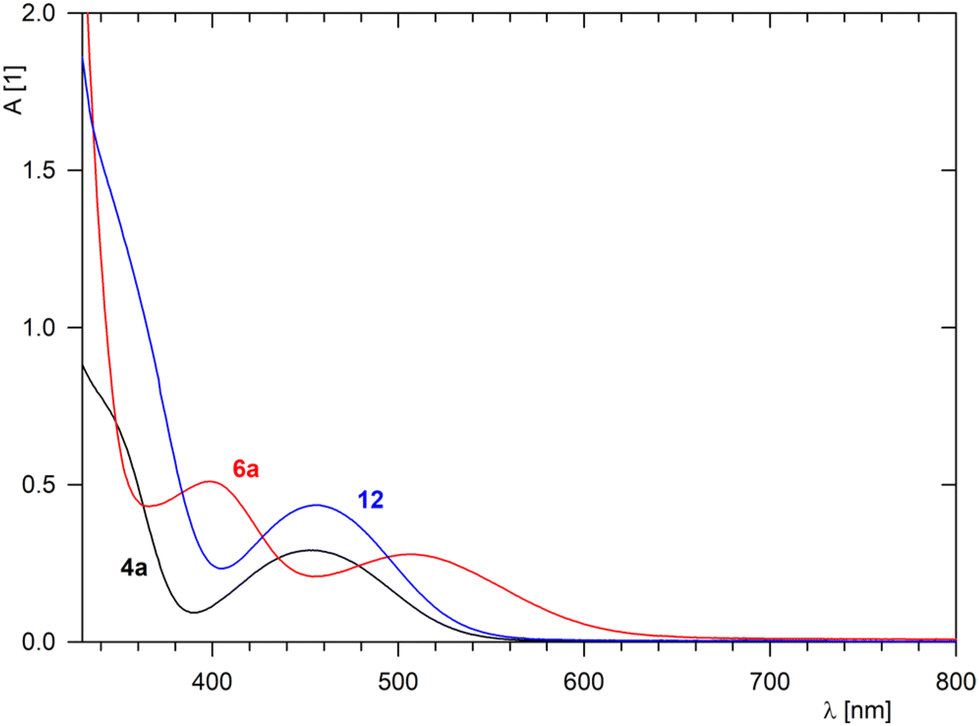 | ||
| Fig. 2 UV-vis spectra of 4a, 6a, and complex 12 (c = 0.75 mM in dichloromethane, optical path 1 cm). | ||
The structures of 4a·0.25THF, 4b, and 4c determined by X-ray diffraction analysis are shown in Fig. 3, and the selected geometric parameters are listed in Table 2. The compounds were prepared and isolated as racemic mixtures and, correspondingly, crystallised with the symmetry of centric space groups (P21/n or P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The asymmetric units of 4a·0.25THF and 4b contained two practically identical molecules (see the ESI†).
). The asymmetric units of 4a·0.25THF and 4b contained two practically identical molecules (see the ESI†).
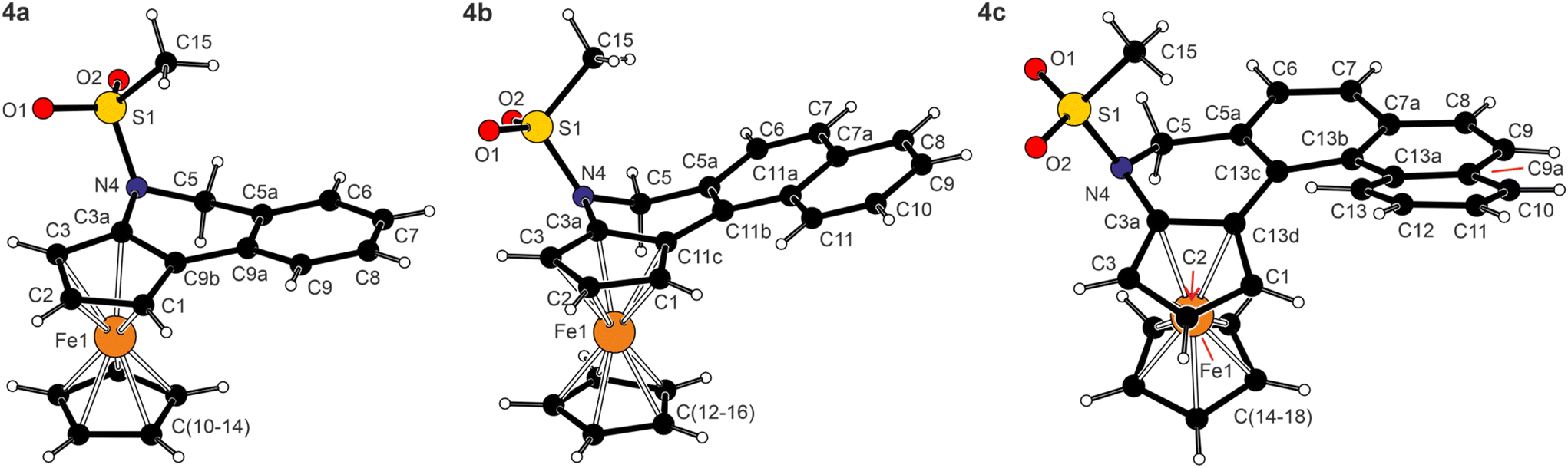 | ||
| Fig. 3 Molecular structures of 4a (molecule 1), 4b (molecule 1) and 4c (for displacement ellipsoid plots and additional structure diagrams, see the ESI†). | ||
| Parametera | 4a (molecule 1) | 4a (molecule 2) | 4b (molecule 1) | 4b (molecule 2) | 4c |
|---|---|---|---|---|---|
| a Definitions: ϕ is the dihedral angle between the substituted cyclopentadienyl ring and the plane of the terminal C6H4 ring; τ1 is the torsion angle C1–C9b–C9a–C9 for 4a, C1–C11c–C11b–C11a for 4b, and C1–C13d–C13c–C13b for 4c; τ2 is the torsion angle C11c–C11b–C11a–C11 for 4b, and C13d–C13c–C13b–C13a for 4c; τ3 is the torsion angle C13c–C13b–C13a–C13 in 4c; tilt is the dihedral angle of the cyclopentadienyl least squares; and n.a. = not applicable. | |||||
| Fe–C (range) | 2.027(2)–2.052(2) | 2.041(1)–2.056(1) | 2.022(8)–2.050(8) | 2.01(1)–2.104(8) | 2.026(1)–2.068(1) |
| Tilt | 4.4(1) | 0.44(9) | 1.5(5) | 6.2(6) | 2.46(8) |
| N4–S1 | 1.646(1) | 1.648(1) | 1.638(6) | 1.603(8) | 1.646(1) |
| S1–O1/O2 | 1.433(1)/1.434(1) | 1.434(1)/1.436(1) | 1.430(6)/1.441(6) | 1.421(6)/1.449(7) | 1.434(1)/1.427(1) |
| O1–S1–O2 | 118.97(8) | 119.21(7) | 119.8(3) | 118.8(4) | 119.25(6) |
| N4–S1–C(Me) | 107.34(8) | 107.16(8) | 106.7(4) | 107.5(4) | 106.87(7) |
| ϕ | 12.79(9) | 13.49(8) | 20.7(4) | 25.4(5) | 42.44(7) |
| τ 1 | −13.7(3) | −10.2(3) | 17(1) | −22(2) | 19.0(2) |
| τ 2 | n.a. | n.a. | −9(1) | −8(1) | 21.9(2) |
| τ 3 | n.a. | n.a. | n.a. | n.a. | 18.6(2) |
The combination of three types of rings (cyclopentadienyl, 1,2-dihydropyridine and benzene) made the structures of 4a–c rather irregular compared with archetypal helicenes built exclusively from ortho-fused benzene rings.27 In particular, the C5 atom and, to a lesser extent, the adjacent carbon C5a of the dihydropyridine moiety were displaced from the pivotal cyclopentadienyl plane (see the torsion angles in the ESI, Fig. S10†). This also resulted in tilting of the adjacent benzene ring [(C5a, C6–9, C9a) for 4a and analogously in other compounds] by approximately 13° in 4a and by 21–22° in 4b and 4c. However, this twisting alleviated steric collisions within the condensed ring system, thereby reducing the tendency towards a helical arrangement in smaller molecules of 4a and 4b. Most likely, the limit was exceeded upon the addition of yet another ortho-fused benzene ring, such as in 4c; here, the rings were already arranged in a screw-like manner (see τ angles in Table 2). For example, the benzene rings in the phenanthrene fragment of 4c were mutually tilted by 11.12(6)° (C5a–C13c vs. C7a–C13b) and 10.91(6)° (C7a–C13b vs. C9a–C13a), and the terminal C6H4 ring was twisted by 42.44(7)° from the C5H3 plane. In contrast, the carbon atoms in the entire naphthalene fragment of 4b were coplanar within less than 0.1 Å. For all molecules, the sulfonate arm was directed above the ferrocene unit, and its oxygen atoms were oriented away from the ring system. The geometry of the N-SO2Me fragment in 4a–c remained similar to that in precursors 1 and 3a–c (see the ESI†).
Synthesis of the Fe/Ru complex 12 and electrochemistry
Compound 4a was further used to prepare heterobimetallic Fe/Ru complex 12via a reaction with [(η5-C5Me5)Ru(MeCN)3] [PF6]28 in 1,2-dichloroethane at 40 °C (Scheme 8). Evaporation followed by chromatography over an alumina column produced compound 12 as an orange solid in a 75% yield. A similar reaction with 6a (in 1,2-dichloroethane or THF) led to a complete decomposition of the ferrocene derivative.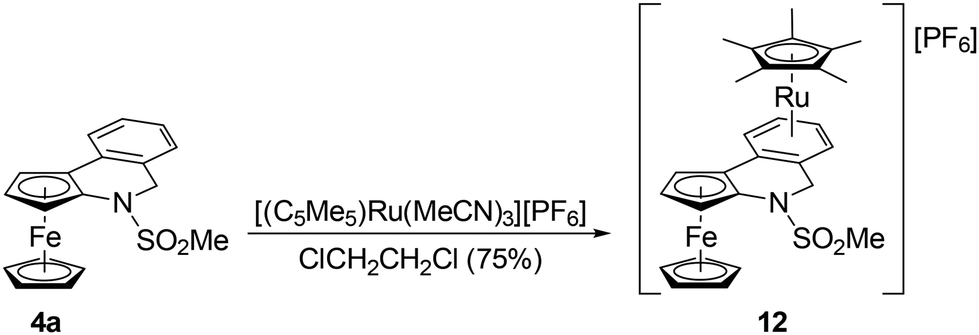 | ||
| Scheme 8 Synthesis of complex 12. | ||
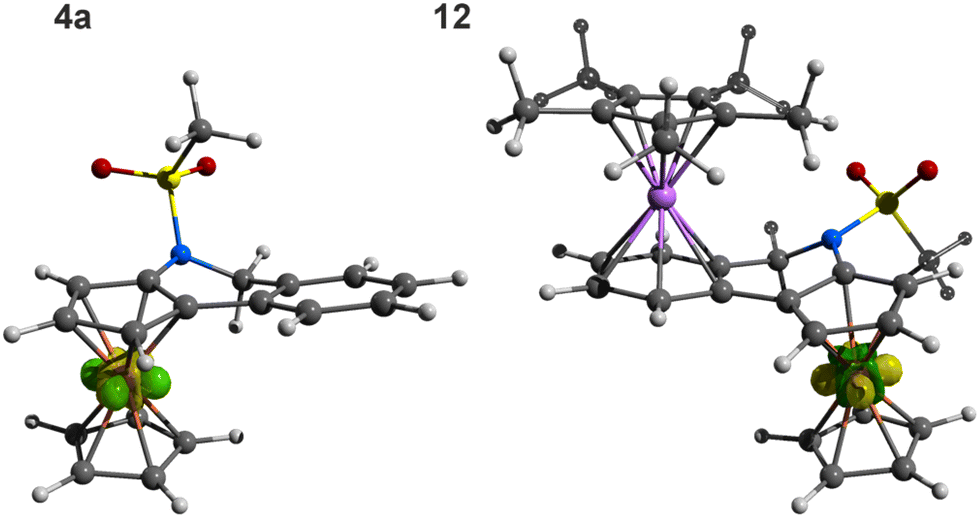
Complex 12 was spectroscopically characterised, and the solid-state structure of solvate 12·2CH2Cl2 was established by X-ray diffraction analysis. The formation of 12 was indicated by the NMR spectra showing signals due to the introduced (η5-C5Me5)Ru moiety (δH 1.83, δC 10.30 and 97.55 in acetone-d6) and resonances expected for π-coordinated 4a, especially the high-field signals due to the C6H4 ring (δH 6.06–6.31; δC 82.88–88.08 for the four CH groups, and 97.05/97.96 for Cipso). The presence of the counterion was corroborated by a characteristic septet in the 31P{1H} NMR spectrum (δP −138.2, 1JPF = 708 Hz) and a broad intense ν3(PF6−) band centred at approximately 845 cm−1 in the FTIR spectrum.29 Notably, the coordination of (η5-C5Me5)Ru+ to 4a did not change the UV-vis spectrum except for an increase in the intensity of the band at approximately 455 nm (see Fig. 2).
The structure of 12 (Fig. 4) consisted of two isoelectronic (cyclopentadienyl)metal fragments coordinated in an anti-fashion with respect to the bridging, non-conjugated bis-six electron donor ligand 4a. The two metallocene moieties adopted their usual geometry (cf. the structure of [(η5-C5Me5)Ru(η6-C6H6)][BPh4]30). While the π-bound rings in the ferrocene unit were tilted by 4.95(8)°, a smaller deformation was found for the (η5-C5Me5)Ru(η6-C6H4) fragment (0.86(7)°). The planes of the C5H3 and C6H4 rings were mutually tilted by 12.36(7)°; this value was similar to that in uncoordinated 4a (in fact, the “molecule” of 4a retained many of its structural features, including an overall conformation, upon coordination). Consistent with the Chatt–Dewar–Duncanson bonding model,31 the C–C bonds in the C6H4 ring were elongated from 1.388(3)–1.405(2) Å in 4a to 1.414(2)–1.427(2) Å in 12.
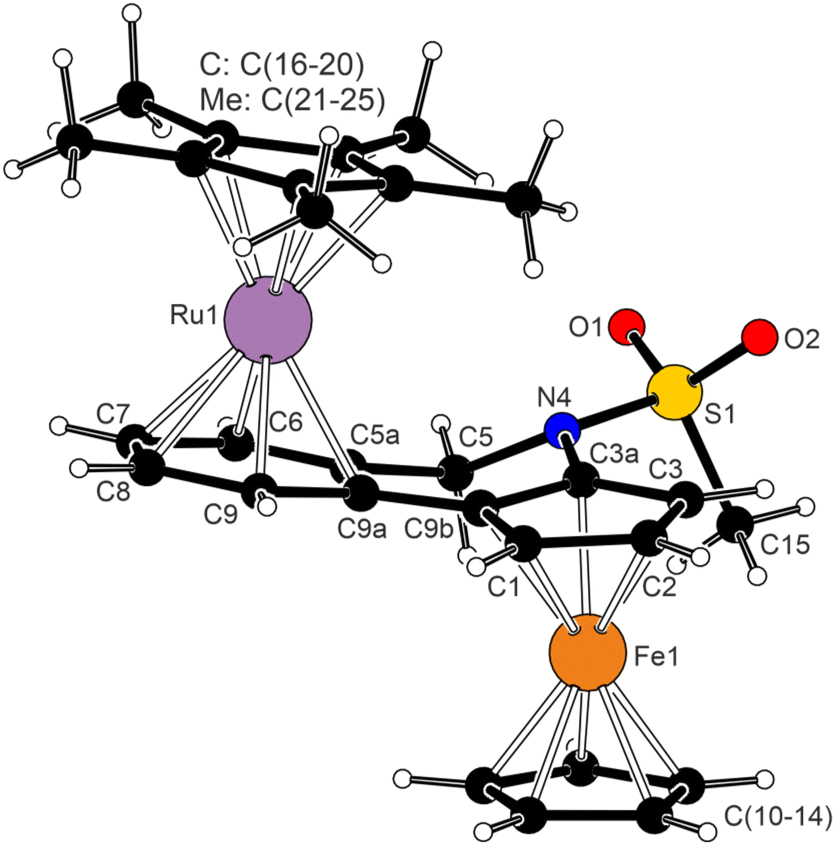 | ||
| Fig. 4 View of the complex cation in the structure of 12·2CH2Cl2. Selected distances and angles (in Å and °): Ru1–C5A 2.220(1), Ru1–C6 2.219(1), Ru1–C7 2.222(1), Ru1–C8 2.224(1), Ru1–C9 2.223(1), Ru1–C9A 2.237(1), Ru1–C(16–20) 2.182(1)–2.194(1), Fe–C 2.038(1)–2.063(2), S1–N4 1.646(1), S1–O1/2 1.430(1)/1.431(1), N4–S1–C15 106.37(7), O1–S1–O2 119.23(7). | ||
In addition to spectroscopic and structural characterisation, representative compounds 1, 3a, 4a, 6a, and 12 were studied by cyclic voltammetry at a glassy carbon disc electrode in dichloromethane containing 0.1 M Bu4N[PF6] as the supporting electrolyte. In the accessible potential range, the compounds underwent several redox transitions. Typically, the “primary” oxidations (Fig. 5) were followed by additional, ill-defined oxidation step(s) that were affected by adsorption phenomena and resulted in blocking of the electrode surface. Therefore, further attention was given only to the primary oxidations.
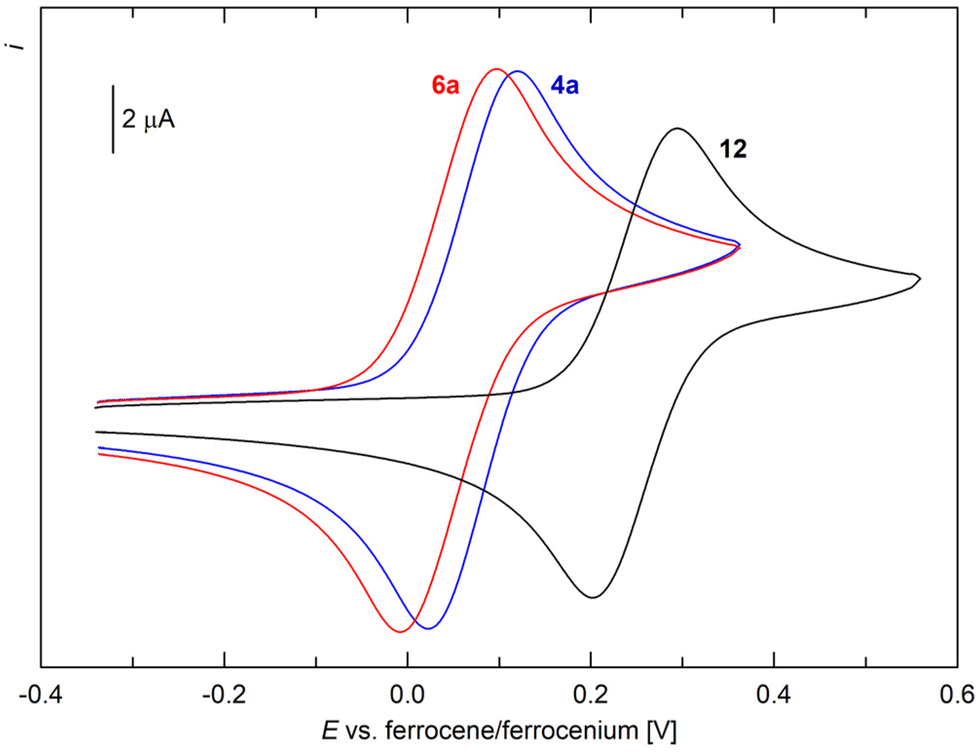 | ||
| Fig. 5 Cyclic voltammograms of 4a, 6a, and 10 as recorded on a glassy carbon disc electrode in CH2Cl2 (0.1 M Bu4N[PF6], scan rate 100 mV s−1). | ||
For 1 and 3a, this oxidation was observed as a standard, one-electron reversible transition at E°′ = 0.00 and 0.06 V vs. the ferrocene–ferrocenium reference,32 respectively (see the ESI†). These values, close to zero, indicated that the electronic properties of the ferrocene core were only marginally perturbed by the appended substituents; the former value also corresponded with the Hammett σp constant for the HNSO2Me group, close to zero (0.03).33
The oxidation of 4a occurred as a diffusion-controlled, reversible redox transition at 0.08 V, whereas the oxidation of freshly prepared 6a was detected at 0.05 V (Fig. 5). Conversely, the primary oxidation of complex 12 was anodically shifted to 0.25 V, although the characteristics of a one-electron reversible redox transition were maintained. These results were consistent with the cationic nature of the complex, which inherently renders electron removal more difficult. No oxidation corresponding to a possible RuII-to-RuIII transition was observed within the accessible potential range.34
Considering their nature, the discussed redox processes were tentatively attributed to ferrocene/ferrocenium redox transitions and this assignment was corroborated by the DFT calculations of 1, 3a, and 12. The calculations showed that the highest occupied molecular orbitals (HOMOs) of these molecules were predominantly localised on the ferrocene unit (see Fig. 6 and ESI†) and that the change in electron density associated with electron removal35 exclusively occurred at the ferrocene unit.
 | ||
| Fig. 6 Electron density changes associated with electron removal, ρ(M+) − ρ(M), mapped at the geometry of the parent species M (M = 4a and the cation of 12); isosurfaces at ±0.02 a.u. are shown. For details, see the ESI.† | ||
Conclusion
In summary, we have developed a protocol for Pd-catalysed intramolecular C–H bond activation/C–C bond formation suitable for converting readily accessible N-[(bromoaryl)methyl]-N-(methylsulfonyl)aminoferrocenes into 1,2-dihydro-2-(methylsulfonyl)ferroceno[c]isoquinoline and the homologous compounds with one or two additional benzene rings ortho-fused onto the dihydroisoquinoline moiety. This method expands not only the palette of synthetic methods leading to planar-chiral ferrocenes annellated with N-heterocyclic moieties9,36 but also the family of rare helical molecules containing a pivotal ferrocene moiety.37 Attempts to perform the cyclisation reaction in an asymmetric manner using chiral phosphines as auxiliary ligands failed, but the compounds could be efficiently resolved into enantiomers using chromatography with chiral stationary phases. In addition, we demonstrated that the terminal ring in the model compound N-(methylsulfonyl)-1,2-dihydro-2-(methylsulfonyl)ferroceno[c]isoquinoline could be coordinated to the (η5-C5Me5)Ru+ fragment and that the ferrocenodihydroisoquinolines could be transformed into the corresponding isoquinolines. Unfortunately, the latter compounds readily degraded, which made further reactivity studies impossible.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
This work was supported by the Charles University Research Centre Program (project no. UNCE/24/SCI/010). Computational resources were provided through the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.References
- Representative reviews: (a) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275 CrossRef CAS; (f) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (g) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692 CrossRef CAS PubMed.
- (a) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC; (b) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008 Search PubMed.
- Selected reviews: (a) L. A. López and E. López, Dalton Trans., 2015, 44, 10128 RSC; (b) D.-Y. Zhu, P. Chen and J.-B. Xia, ChemCatChem, 2016, 8, 68 CrossRef CAS; (c) D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351 CrossRef CAS PubMed; (d) C.-X. Liu, Q. Gu and S.-L. You, Trends Chem., 2020, 2, 737 CrossRef CAS; (e) Z.-Z. Zhang, D.-Y. Huang and B.-F. Shi, Org. Biomol. Chem., 2022, 20, 4061 RSC; (f) T. Čarný and R. Šebesta, Synlett, 2024, 165–182 Search PubMed.
- E. O. Fischer and D. Seus, Z. Naturforsch., 1953, 8b, 694 CrossRef CAS.
- D. E. Bublitz, W. E. McEwen and J. Kleinberg, J. Am. Chem. Soc., 1962, 84, 1845 CrossRef CAS.
- M. Cais, A. Modiano and A. Raveh, J. Am. Chem. Soc., 1965, 87, 5607 CrossRef CAS.
- (a) D.-W. Gao, Q. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2014, 136, 4841 CrossRef CAS; (b) R. Deng, Y. Huang, X. Ma, G. Li, R. Zhu, B. Wang, Y.-B. Kang and Z. Gu, J. Am. Chem. Soc., 2014, 136, 4472 CrossRef CAS.
- D.-W. Gao, C. Zheng, Q. Gu and S.-L. You, Organometallics, 2015, 34, 4618 CrossRef CAS.
- For a review of ferrocene-fused nitrogen heterocycles, see: O. Bernardo, S. González-Pelayo and L. A. López, Eur. J. Inorg. Chem., 2022, e202100911 CrossRef CAS.
- S. Luo, Z. Xiong, Y. Lu and Q. Zhu, Org. Lett., 2018, 20, 1837 CrossRef CAS PubMed.
- A.-A. Zhang, C. Chen, Y. Gao, M. Mo, R.-Z. Shen, Y.-H. Zhang, N. Ishida, M. Murakami and L. Liu, Green Synth. Catal., 2021, 2, 311 CrossRef.
- (a) X. Ma and Z. Gu, RSC Adv., 2014, 4, 36241 RSC; (b) L. Liu, A.-A. Zhang, R.-J. Zhao, F. Li, T.-J. Meng, N. Ishida, M. Murakami and W.-X. Zhao, Org. Lett., 2014, 16, 5336 CrossRef CAS PubMed; (c) L. Liu, H. Liu, Z. Zuo, A.-A. Zhang, Z. Li, T. Meng, W. Wu, Y. Hua and G. Mao, Chin. Chem. Lett., 2021, 32, 239 CrossRef CAS.
- W. Han, X. Zhou, S. Yang, G. Xiang, B. Cui and Y. Chen, J. Org. Chem., 2015, 80, 11580 CrossRef CAS PubMed.
- For the preparation of an analogous compound bearing a phosphine substituent at the ferrocene unit, see: V. Varmužová, F. Horký and P. Štěpnička, New J. Chem., 2021, 45, 3319 RSC.
- Only minor amounts of 4a were detected in the crude reaction mixtures obtained at 100 or 140 °C. Signals due to 6a were detected in neither case.
- (a) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC; (b) N. C. Bruno, N. Niljianskul and S. L. Buchwald, J. Org. Chem., 2014, 79, 4161 CrossRef CAS.
- S. Ricard and A. Gagnon, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2016, pp. 1–14; accessible through DOI:10.1002/047084289X.rn01905.
- I. Caivano, S. Bingel, I. Císařová, D. Nečas and M. Kotora, Catal. Today, 2022, 390–391, 48 CrossRef CAS.
- (a) S. Wu, C. Liu, G. Luo, Z. Jin, P. Zheng and Y. R. Chi, Angew. Chem., Int. Ed., 2019, 58, 18410 CrossRef CAS PubMed; (b) Y.-Y. Li, S. Li, T. Fan, Z.-J. Zhang, J. Song and L.-Z. Gong, ACS Catal., 2021, 11, 14388 CrossRef CAS.
- 10 equiv. of Red-Al were used and the reaction was conducted in toluene at 0 °C/12 h and at room temperature/24 h. The reaction mixture contained only 6a (30 and 35%, respectively) and the unreacted starting material.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007 RSC.
- (a) L. T. Taylor, J. Supercrit. Fluids, 2009, 47, 566 CrossRef CAS; (b) K. De Klerck, De. Mangelings and Y. Vander Heyden, J. Pharm. Biomed. Anal., 2012, 69, 77 CrossRef CAS PubMed; (c) C. West, TrAC, Trends Anal. Chem., 2019, 120, 115648 CrossRef CAS; (d) D. Speybrouck, M. Howsam and E. Lipka, TrAC, Trends Anal. Chem., 2020, 133, 116090 CrossRef CAS.
- D. C. D. Butler and C. J. Richards, Organometallics, 2002, 21, 5433 CrossRef CAS.
- (a) J. N. Baxter, J. Cymerman-Craig and J. B. Willis, J. Chem. Soc., 1955, 669 RSC; (b) A. R. Katritzky and R. A. Jones, J. Chem. Soc., 1960, 4497 RSC.
- U. Salzner, J. Chem. Theory Comput., 2013, 9, 4064 CrossRef CAS.
- (a) D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 516 CrossRef CAS; (b) A. T. Armstrong, F. Smith, E. Elder and S. P. McGlynn, J. Chem. Phys., 1967, 46, 4321 CrossRef CAS.
- J. B. M. Somers, J. H. Borkent and W. H. Laarhoven, Recl. Trav. Chim. Pays-Bas, 1981, 100, 110 CrossRef CAS.
- C. Slugovc, E. Rüba, R. Schmid, K. Kirchner and K. Mereiter, Monatsh. Chem., 2000, 131, 1241 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistry, 6th edition, Wiley, Hoboken, 2009, ch. 2.8, pp. 221–237 Search PubMed.
- G. Albertin, S. Antoniutti, J. Castro and G. Gasparetto, New J. Chem., 2019, 43, 2676 RSC.
- (a) M. J. S. Dewar, Bull. Soc. Chem. Fr., 1951, 18, C71 Search PubMed; (b) J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- (a) G. Gritzner and J. Kůta, Pure Appl. Chem., 1984, 56, 461 CrossRef; (b) R. R. Gagné, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- Electrochemical oxidation of [(η5-C5Me5)Ru(η6-C6H6)]+ in MeCN/0.1 M Bu4N[BF4] was detected at +2.11 V vs. SCE: O. V. Gusev, M. A. Ievlev, M. G. Peterleitner, S. M. Peregudova, L. I. Denisovich, P. V. Petrovskii and N. A. Ustynyuk, J. Organomet. Chem., 1997, 534, 57 CrossRef CAS.
- (a) J. Schulz, F. Uhlík, J. M. Speck, I. Císařová, H. Lang and P. Štěpnička, Organometallics, 2014, 33, 5020 CrossRef CAS; (b) K. Škoch, I. Císařová, F. Uhlík and P. Štěpnička, Dalton Trans., 2018, 47, 16082 RSC; (c) K. Škoch, J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 3060 CrossRef; (d) P. Vosáhlo, J. Schulz, I. Císařová and P. Štěpnička, Dalton Trans., 2021, 50, 6232 RSC; (e) M. Franc, J. Schulz and P. Štěpnička, Dalton Trans., 2024, 53, 11445 RSC.
- Relevant recent examples: (a) V. Mamane and Y. Fort, J. Org. Chem., 2005, 70, 8220 CrossRef CAS PubMed; (b) J. Wang, Z.-H. Zhu, M.-W. Chen, Q.-A. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2019, 58, 1813 CrossRef CAS; (c) Z.-B. Zhao, X. Li, M.-W. Chen, Z. K. Zhao and Y.-G. Zhou, Chem. Commun., 2020, 56, 7309 RSC; (d) M. Ito, M. Okamura, K. S. Kanyiva and T. Shibata, Organometallics, 2019, 38, 4029 CrossRef CAS; (e) K. Yoshida, Q. Liu, R. Yasue, S. Wada, R. Kimura, T. Konishi and M. Ogasawara, ACS Catal., 2020, 10, 292 CrossRef CAS; (f) Y. An, X.-Y. Zhang, Y.-N. Ding, Y. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2022, 24, 7294 CrossRef CAS PubMed; (g) N. Lin, B. Hong, J. Zhao and Z. Gu, Angew. Chem., Int. Ed., 2023, 62, e202215530 CrossRef.
- Selected examples: (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680 RSC (review); (b) A. Urbano, G. Hernández-Torres, A. M. del Hoyo, A. Martínez-Carrión and M. C. Carreño, Chem. Commun., 2016, 52, 6419 RSC; (c) T. Shibata, N. Uno, T. Sasaki and K. S. Kanyiva, J. Org. Chem., 2016, 81, 6266 CrossRef CAS; (d) M. Akiyama and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 2040 CrossRef CAS; (e) A. Urbano, A. M. del Hoyo, A. Martínez-Carrión and M. C. Carreño, Org. Lett., 2019, 21, 4623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, including all synthetic procedures and characterisation data, details on electrochemical measurements, structure determination and DFT calculations, Cartesian coordinates of DFT-optimised structures and copies of the NMR spectra. CCDC 2389173–2389180. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03063j |
| This journal is © The Royal Society of Chemistry 2025 |