
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced energy and electron transfer processes in a supramolecular system combining a tetrapyrenylporphyrin derivative and arene–ruthenium metalla-prisms†
Santiago Luis
Pons Alles
 a,
Daniele
Veclani
b,
Andrea
Barbieri
b,
Bruno
Therrien
*a and
Barbara
Ventura
*b
a,
Daniele
Veclani
b,
Andrea
Barbieri
b,
Bruno
Therrien
*a and
Barbara
Ventura
*b
aInstitute of Chemistry, University of Neuchâtel, Avenue de Bellevaux 51, CH-2000 Neuchatel, Switzerland. E-mail: Bruno.Therrien@unine.ch
bInstitute for Organic Synthesis and Photoreactivity (ISOF) – National Research Council (CNR), Via P. Gobetti 101, 40129 Bologna, Italy. E-mail: barbara.ventura@isof.cnr.it
First published on 20th January 2025
Abstract
A supramolecular system, consisting of a tetrapyrenylporphyrinic core surrounded by arene–ruthenium prisms, has been assembled and characterized by means of electrochemical and photophysical techniques. The photophysical study shows that quantitative energy transfer from the peripheral pyrenyl units towards the central porphyrin core is operative in the tetrapyrenylporphyrinic system. Interestingly, encapsulation of the pyrenyl units into the ruthenium cages affects the photophysics of the central porphyrin component, since its emission quantum yield is reduced in the supramolecular array. Femtosecond transient absorption analysis evidenced a complex interplay of deactivation pathways, including energy and electron transfer processes from the porphyrin to the metalla-prisms, associated with different conformations of the system allowed by the flexibility of the linkers. Moreover, the non-emissive arene–ruthenium cages present a peculiar excited-state dynamics, here disentangled for the first time by means of transient absorption investigations.
Introduction
Ruthenium-based complexes have been used in many fields, such as catalysis,1,2 solar energy conversion,3 imaging and sensing,4,5 as well as in medicinal chemistry.6–9 Arene–ruthenium complexes are an important family of coordination compounds, in which the metal is stabilized in a +2 oxidation state by an aromatic ligand (arene) which occupies three of the six coordination sites, limiting the introduction of other ligands. However, these additional ligands can modify the properties of the complexes,10 and accordingly, their biological and photophysical properties. The three remaining sites are positioned at 90° from each other, allowing for the formation of shape-controlled supramolecular assemblies.11 Accordingly, depending on the ligand used, different structures can be obtained through self-assembly of the different building blocks, including 3D structures such as rectangular, prismatic or cubic assemblies.12–14 Some of these 3D architectures possess an internal hydrophobic cavity of tuneable size that can accomodate planar aromatic molecules,15–17 which are primarily interacting with the host by π–π interactions.The combination of coordination complexes with fluorophores has raised interest in recent years.18 Indeed, self-assembled supramolecular structures offer an efficient strategy for the construction of energy transfer systems,19–23 including systems based on fluorescence resonance energy transfer (FRET). FRET is a nonradiative energy transfer process taking place between two fluorophores, in which the excitation energy can be transferred from an excited donor to a nearby ground-state acceptor, through nonradiative dipole–dipole coupling.18,24,25 This process is highly dependent on the donor–acceptor distance, which makes FRET interesting for applications in many fields such as light harvesting,26 fluorescence sensing/imaging,27,28 or determination of intermolecular interactions in biological systems.29,30
Multi-step FRET systems have gained interest as they possess advantages over one-step FRET systems, such as the possibility of monitoring longer range (>10 nm) interactions and higher efficiency of long-range energy transfer.18,25 Arene–ruthenium metalla-assemblies represent an interesting scaffold for the construction of multi-step FRET systems, as they offer many options in terms of number, location and nature of fluorophores. Host–guest interactions are particularly relevant in this context, as organic fluorophores can be encapsulated in hydrophobic cavities,18,31 thus enhancing their solubility, while shortening the donor–acceptor distance, and ultimately promoting higher FRET efficiencies.
In the present study, a novel supramolecular structure has been designed, based on the combination of a porphyrinic core with four diethyleneglycol (DEG) spacers terminated with pyrenyl units (P-Pyr4). The pyrenyl entities can be encapsulated into prismatic arene–ruthenium metalla-prisms (Rucage), in view to increase the water solubility of the pyrenyl-functionalized porphyrin and to avoid aggregation, thus forming a P-Pyr4@(Rucage)4 hybrid host–guest system (Fig. 1). Electrochemical and photophysical characterization of this system has been performed, with special emphasis on the elucidation of the excited state dynamics using transient absorption spectroscopy. The analysis has been supported by a detailed theoretical investigation that allowed to characterize the interaction among the appended pyrenyl units and the metalla-cages, and the different conformations that the P-Pyr4@(Rucage)4 system can adopt in solution. The results suggest that in solution the supramolecular structure is retained, and that the metalla-prisms not only increase the solubility and prevent aggregation, but also modify the photophysical behaviour of the pyrenyl-functionalized porphyrin derivative. This study provides new perspectives for supramolecular arene–ruthenium hybrid systems, like energy collection and charge separation applications.
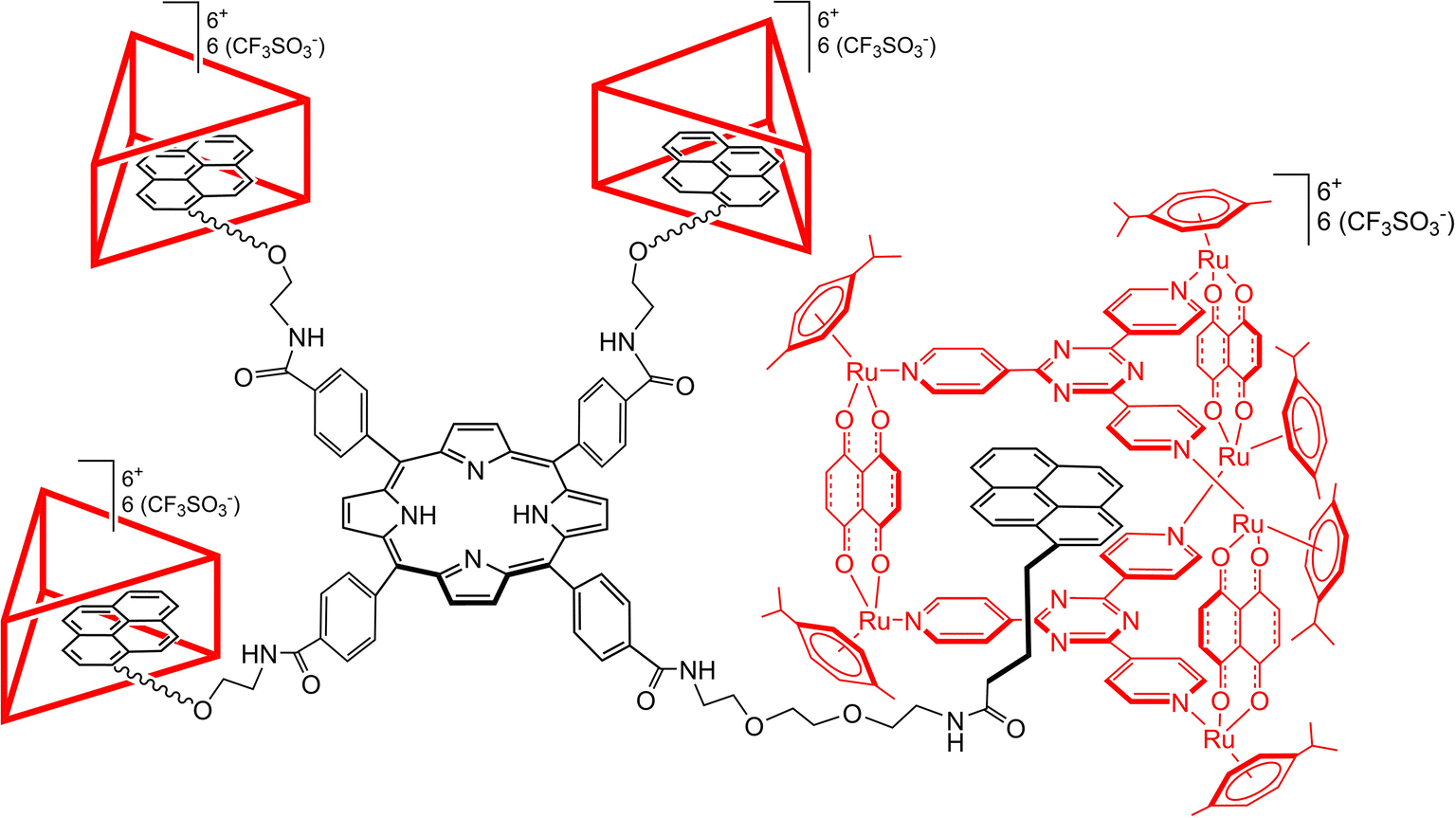 | ||
| Fig. 1 Schematic representation of the P-Pyr4@(Rucage)4 hybrid host–guest system. | ||
Experimental
General
NMR spectra were recorded on a Bruker Avance Neo Ascend 600 MHz spectrometer. Mass spectra were performed by the ISIC's Mass spectrometry service by nanochip-ESI/LTQ-Orbitrap in positive ion mode.Synthetic procedures
Panel,32Clip,33Rucage,9 and P-Pyr4 precursor34 (see Fig. 2) were prepared according to published methods. Tetraphenylporphyrin (TPP), pyrene (Pyr), 1-pyrenebutyric acid, silver triflate, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCl), 4-dimethylaminopyridine (DMAP) and all other reagents were commercially available and used as received.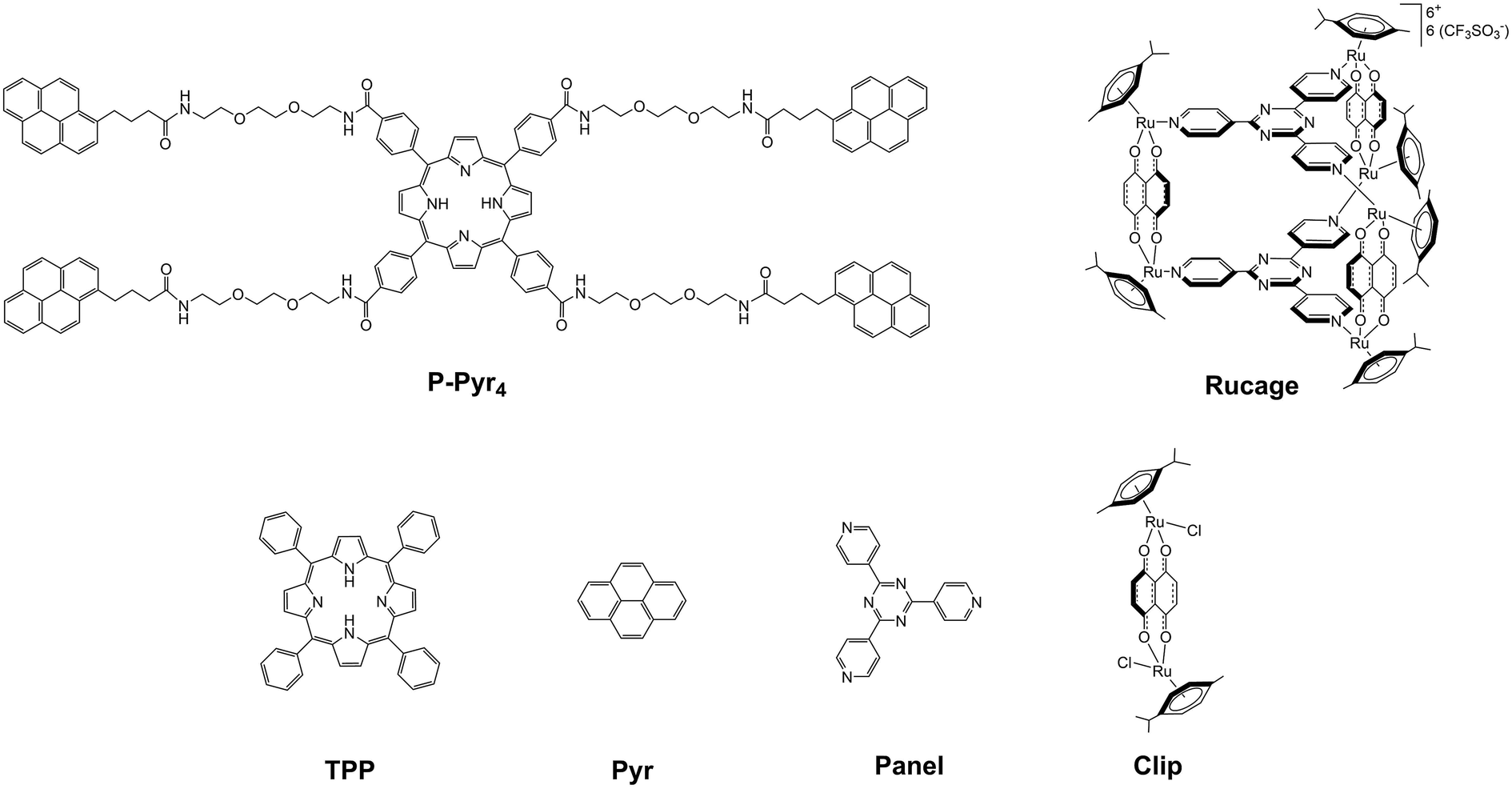 | ||
| Fig. 2 Molecular structures and codenames of the considered models and building blocks. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH gradient up to 9:1 (yield 255 mg, 107 μmol, 41%).
:MeOH gradient up to 9:1 (yield 255 mg, 107 μmol, 41%).
1H NMR (600 MHz, CDCl3) δ 8.71 (s, 8H, Hβpyrrole), 8.16 (d, J = 8.3 Hz, 8H, Hph), 8.10 (m, 10H, Hph and Hpyr), 8.09 (s, 2H, Hpyr), 7.95 (dd, J = 7.6, 1.1 Hz, 4H, Hpyr), 7.87 (dd, J = 7.7, 1.2 Hz, 4H, Hpyr), 7.84 (d, J = 7.8 Hz, 4H, Hpyr), 7.82 (s, 2H, Hpyr), 7.78 (m, 14H, Hpyr), 7.65 (d, J = 7.7 Hz, 4H, Hpyr), 6.99 (t, J = 5.3 Hz, 4H, NHCOporph), 6.02 (t, J = 5.5 Hz, 4H, NHCO), 3.74 (dt, J = 21.6, 5.1 Hz, 16H, HDEG), 3.65 (m, 16H, HDEG), 3.57 (t, J = 5.2 Hz, 8H, HDEG), 3.48 (m, 8H, HDEG), 3.23 (t, J = 7.7 Hz, 8H, Hbutyl), 2.25 (t, J = 7.3 Hz, 8H, Hbutyl), 2.11 (p, J = 7.5 Hz, 8H, Hbutyl). 13C NMR (151 MHz, CDCl3) δ 172.91 (COCH2), 167.67 (COCph), 145.18 (Cph, Cporph), 135.73 (Cporph), 134.63 (Cporph), 133.98 (CphCO), 131.15 (Cpyr), 130.63 (Cpyr), 129.70 (Cpyr), 128.56 (Cpyr), 127.28 (CHph), 127.18 (CHph), 126.52 (CHpyr), 125.68 (CHpyr), 125.56 (CHpyr), 124.83 (CHpyr), 124.75 (CHpyr), 124.70 (CHpyr), 124.63 (CHpyr), 124.58 (CHpyr), 123.18 (CHpyr), 119.25 (CHβpyrrole), 70.33 (CH2DEG), 70.31 (CH2DEG), 69.96 (CH2DEG), 69.92 (CH2DEG), 53.55 (DCM), 40.03 (CH2NH), 39.26 (CH2NH), 35.99 (CH2CO), 32.68 (CH2Cpyr), 27.39 (CH2CH2CH2). 1H and 13C NMR spectra are reported in Fig. S1 and S2.† UV/Vis (DCM): λmax (ε [M−1 cm−1]) = 236 (213000), 244 (288000), 267 (121000), 278 (197000), 315 (64000), 329 (126000), 345 (178000), 421 (427000), 517 (20000), 553 (9000), 592 (6000), 646 (4000). HRMS-ESI m/z: [P-Pyr4 + 2Na]2+ calcd for C152H142N12Na2O16 1218.5226; found 1218.5240. [P-Pyr4 + H + Na]2+ calcd for C152H143N12NaO16 1207.5316; found 1207.5330 (see Fig. S5†).
Electrochemistry
Cyclic and square wave voltammograms were acquired using a Metrohm Autolab PG-STAT 302 electrochemical workstation controlled by the NOVA 2.0 software. The experiments were conducted in dry DCM (VWR, Uvasol®) solutions containing 0.1 M tetra-n-butylammonium hexafluorophosphate (Sigma-Aldrich, electrochemical grade) and approximately 2 × 10−4 M analyte, under a dry nitrogen atmosphere, at a scan rate of ν = 100 mV s−1. The experimental setup involved a single-compartment three-electrode cell fitted with platinum wire counter and pseudo-reference electrodes, along with a glassy carbon working electrode (BioLogic). All recorded redox potentials are referenced to the ferrocene/ferrocenium (Fc/Fc+) redox couple, which was added at the conclusion of each experiment, serving as an internal reference system.Absorption and emission spectroscopy, photophysics
Spectroscopic grade DCM (Merck, Uvasol®) was passed through an alumina column to remove traces of acid. TPP was purchased from Porphychem, α-NPO (2-(1-naphthyl)-5-phenyloxazole) from Lambdaphysik and Pyr from Acros.Absorption spectra were recorded with a Perkin–Elmer Lambda 650 UV-Vis or a Lambda 950 UV-Vis-NIR spectrophotometer in 1 cm quartz cuvettes. Emission spectra were collected with an Edinburgh FLS920 spectrofluorometer, equipped with a Peltier-cooled Hamamatsu R928 PMT (280–850 nm). The spectra have been corrected for the wavelength-dependent phototube response. Measurements in the NIR region were performed with a FLS920 fluorimeter (Edinburgh) equipped with a Hamamatsu R5509-72 InP/InGaAs phototube multiplier supercooled at 193 K in a liquid nitrogen cooled housing, and a TM300 emission monochromator with a NIR grating blazed at 1000 nm (sensitivity range: 400–1700 nm).
Fluorescence quantum yields were evaluated with the comparative method developed by Demas and Crosby,35 by correcting the spectra for the wavelength-dependent photomultiplier response. The standards used were α-NPO in aerated cyclohexane (ϕfl = 0.94)36 for the pyrene unit and TPP in toluene (ϕfl = 0.11)37 for the porphyrin component. Measurements at 77 K were performed using Pyrex tubes dipped in liquid nitrogen in a quartz Dewar. Excitation spectra were corrected for the wavelength-dependent lamp intensity.
Excited state lifetimes in the nanosecond range were determined by using an IBH time-correlated single-photon counting apparatus with nanoLED excitation sources at 331 nm and 465 nm.
Pump–probe transient absorption measurements were performed with an Ultrafast Systems HELIOS (HE-VIS-NIR) femtosecond transient absorption spectrometer by using, as excitation source, a Newport Spectra Physics Solstice-F-1K-230 V laser system, combined with a TOPAS Prime (TPR-TOPAS-F) optical parametric amplifier (pulse width: 100 fs, 1 kHz repetition rate) tuned at 420 nm and 700 nm. The pump energy on the sample was 1 μJ per pulse or 10 μJ per pulse, respectively. Excitation at 420 nm required a 10 times reduction of the laser power due to the photoinstability of the porphyrin components upon excitation with high energy photons, leading to weak transient signals. The overall time resolution of the system is 300 fs. Air-equilibrated solutions in 0.2 cm optical path cells were analysed under continuous stirring. The Surface Xplorer V4 software from Ultrafast Systems was used for data acquisition and analysis. The 3D data surfaces were corrected for the chirp of the probe pulse prior to analysis.
Estimated errors are 10% on transient absorbance lifetimes, 10% for luminescence lifetimes, 10% for molar absorption coefficients and 10% on quantum yields.
Modelling
Different computational approaches have been employed to analyse a series of molecular P-Pyr4@(Rucage)4 models in order to aid the interpretation of the experimental results. The simplest models, which consist of Pyr and modified Clip (Clip-2, where Cl is replaced with pyridine moieties, see Fig. S6†), were subjected to density functional theory calculations (DFT). Geometry optimizations were performed using range-separated hybrid functionals (LC-PBE functional),38 with a Def2-SVP basis set39,40 for all atoms, with relative effective core potentials for Ru atoms. Recognizing the significant influence of solvation parameters, environmental effects (i.e., DCM) were incorporated using a conductor-like polarizable continuum model (CPCM).41 The directions and intensities of the dipole moments were determined for Pyr, while the transition electric dipole moments for modified Clip were obtained through time-dependent DFT calculations (TD-DFT),42,43 which were conducted at the same level of theory as used for geometry optimizations. All DFT and TD-DFT calculations were performed using ORCA 5.0.4 software.44Complex models were analysed using the advanced GFN2-xTB method,45 implemented into the xTB software,45–47 to overcome the limitations associated with DFT calculations. This method is primarily designed for the rapid, robust, and reasonably accurate computation of large molecules, comprising more than 1000 atoms, while maintaining a good accuracy performance and computational cost for the target properties.48 Geometry optimization was conducted with very tight convergence criteria for energies and gradients (Econv = 1 × 10−7 Eh; Gconv = 2 × 10−4 Eh α−1). Implicit solvent effects (i.e., DCM) were incorporated by employing the Analytical Linearized Poisson–Boltzmann (ALPB) method.49
For P-Pyr4@(Rucage)4, six different conformers were generated: the first two exhibit linear conformation of the branches alternatively below and above the porphyrin plane (P-Pyr4@(Rucage)4_I) or along the plane (P-Pyr4@(Rucage)4_II). In the remaining models, the conformations of the branches were progressively modified (from 1 to 4 branches, labelled P-Pyr4@(Rucage)4_III, P-Pyr4@(Rucage)4_IV, P-Pyr4@(Rucage)4_V, P-Pyr4@(Rucage)4_VI) to enable interactions between the porphyrin core and the cages. A simplified mono-functionalised system, labelled P-Pyr@(Rucage) was used to study the inclusion of Pyr inside the Rucage, using relaxed scan calculations (Fig. S19†). Triflate anions were kept as the counterions and placed in proximity of the Rucage to neutralize its +6 positive charge; indeed, preliminary calculations suggested that the positively charged models were unstable without proper neutralization.
The dissociation energy (De), the interaction energy (IE) and the deformation energy (Def)50,51 were obtained as reported in eqn (1).
| De = |IE| − Def | (1) |
The De is defined in eqn (2):
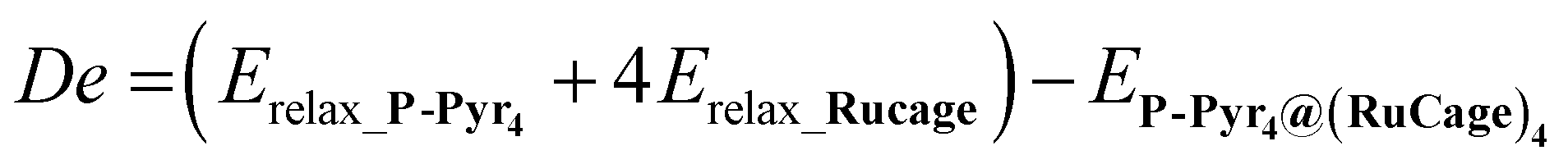 | (2) |
| IE = EP-Pyr4@(RuCage)4 − (EP-Pyr4 + 4ERucage) | (3) |
Def evaluates the energy cost to bring each fragment from its optimized structure as an isolated species to that in the complex (eqn (4)) and quantitatively describes the structural deformations of the molecules.
| DefP-Pyr4 = Erelax\_P-Pyr4 − EP-Pyr4 | (4) |
Energy transfer
In the context of a general energy transfer process, one can derive the intramolecular rate constant from either eqn (5) or (6):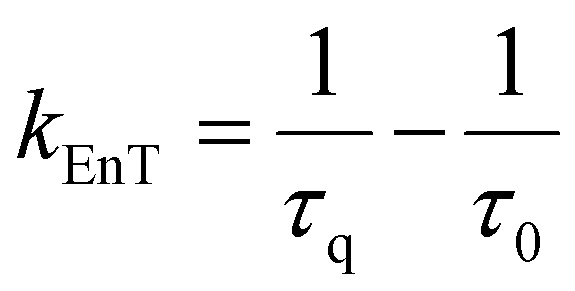 | (5) |
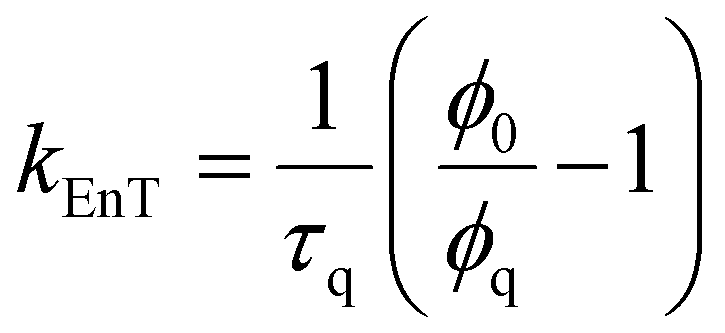 | (6) |
The overall efficiency of the intramolecular energy transfer process can be assessed using eqn (7):
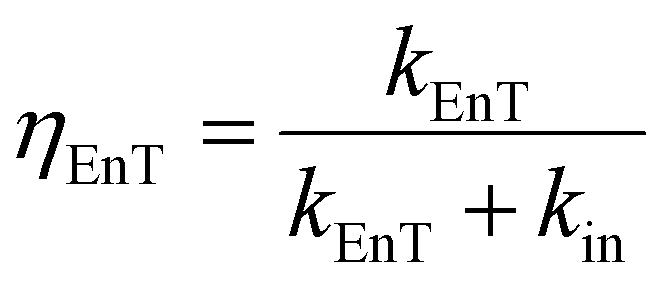 | (7) |
The calculation of the interchromophoric distance-dependent energy transfer rates, grounded in the Förster theory, involves the following eqn (8)–(10):
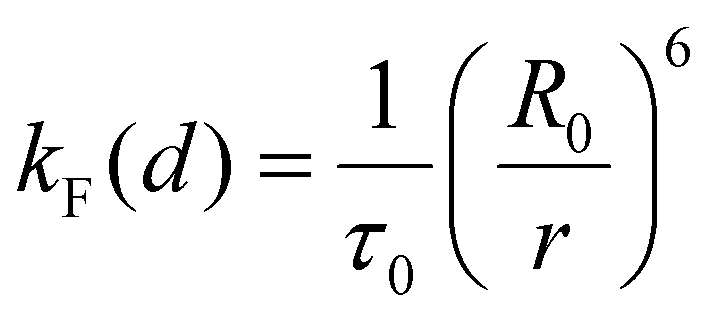 | (8) |
 | (9) |
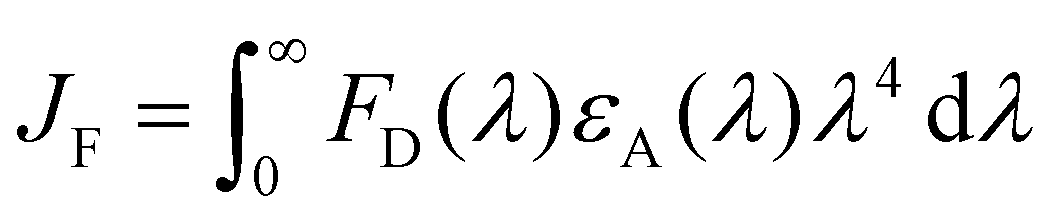 | (10) |
Results and discussion
Synthetic procedures
The tetrapyrenylporphyrin derivative (P-Pyr4) was obtained in good yield from 1-pyrenebutyric acid and the corresponding tetrapegylated amido-porphyrin (Scheme 1). Upon formation of P-Pyr4, new signals associated to the pyrenyl protons were observed in the 1H NMR spectrum, especially those of the pyrenyl aromatic protons between 7.6 and 8.1 ppm (Fig. S1†). Under electrospray condition P-Pyr4 shows in the mass spectrum a dicationic peak at m/z = 1196.5420, in agreement with a di-protonated compound [M + 2 H+]2+ (Fig. S5†).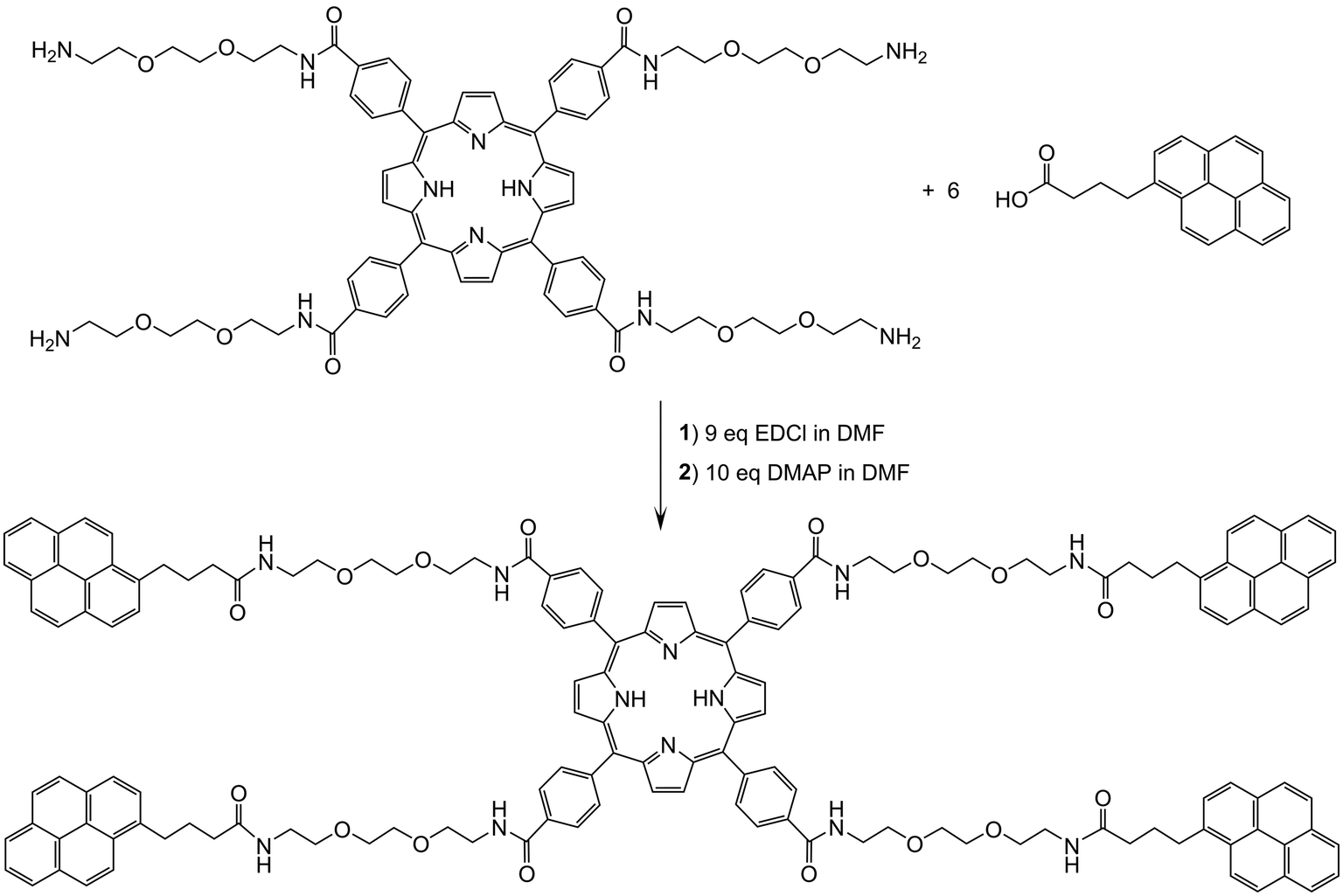 | ||
| Scheme 1 Synthesis of P-Pyr4 from 1-pyrenebutyric acid and the DEG-porphyrin precursor. | ||
Then, P-Pyr4 was added during the synthesis of the arene ruthenium metalla-prisms, thus allowing the formation of the tetra-encapsulated system P-Pyr4@(Rucage)4 to take place. This final assembly was performed in methanol, to force the pyrenyl units to hide inside the hydrophobic cavity of the metalla-prism, [(η6-p-cymene)6Ru6(μ-dioxidonaphthoquinonato)3(tpt)2]6+ (Rucage). Therefore, P-Pyr4@(Rucage)4 is cationic with an overall charge of 24+, and the product isolated as a salt, with triflate counter ions, is [P-Pyr4@(Rucage)4](CF3SO3)24. In solution, dynamic exchange between the host and the guest cannot be totally ruled out, as the binding constant for such 1:1 pyrenyl-functionalized derivatives and arene ruthenium metalla-prisms has been estimated to be around 7.5 × 104 M−1 in acetonitrile.53 However, in the range of concentrations used, we can assume that an intact P-Pyr4@(Rucage)4 system is predominant, as suggested by the 1H NMR spectrum of P-Pyr4@(Rucage)4 (Fig. S3†), showing an intense broadening of the pyrenyl signals.
Electrochemistry
In order to investigate the possibility of redox reactions within the units of the studied systems, the electrochemical characterization of Rucage and its models Clip and Panel has been performed in DCM with tetrabutylammonium hexafluorophosphate supporting electrolyte (0.1 M). Ferrocene was used as internal reference.The cyclic voltammogram and square-wave voltammogram of Rucage are shown in Fig. 3, while those of Panel and Clip are displayed in Fig. S7 and S8.† The electrochemical data are collected in Table 1. Panel shows three reversible reduction waves (Fig. S7†), that can be attributed to the reduction of the three pyridine rings of the molecule.54 Conversely, Clip presents two reduction and two oxidation waves (Fig. S8†), all the processes being quasi-reversible. These waves correspond to oxidation of the Ru centres and reduction of the organic naphthoquinonato ligands, respectively. With regard to Rucage, a complex behaviour is observed (Fig. 3): four reduction and two quasi-reversible oxidation waves are detected. By comparison with the data of the models, two of the four reduction processes, i.e. E3–4red,Rucage (see Table 1), can be ascribed to the reduction of the panels, with the potentials slightly negatively shifted with respect to the model (Table 1). The reduction wave E1red,Rucage at −0.91 V likely corresponds to the first reduction of the organic ligand of a Ru clip, largely positively shifted with respect to the model (E1red,Clip = −1.32 V, Table 1). The reduction process E2red,Rucage at −1.44 V can reasonably originate from the merging of two independent redox processes, namely, the second reduction of the Clip positively shifted (Table 1), and the first reduction of the Panel. Eventually, the two oxidation processes, E1–2ox,Rucage, originate from the oxidation of the metal centres of the clips. Also in this case, the potentials are positively shifted with respect to Clip alone, with the first oxidation peak particularly affected. These positive shifts are likely due to the increase of the residual positive charge on the Ru atoms induced by the coordination of the pyridine moieties of the Panel, as indicated by the calculation of the Mulliken charges. Indeed, in the Clip the two Ru atoms show similar negative Mulliken charge values (≈−0.21), while in the Clip-2 model (where pyridine moieties replace the Cl atoms) an increase in the Mulliken charge values up to −0.08 is observed.
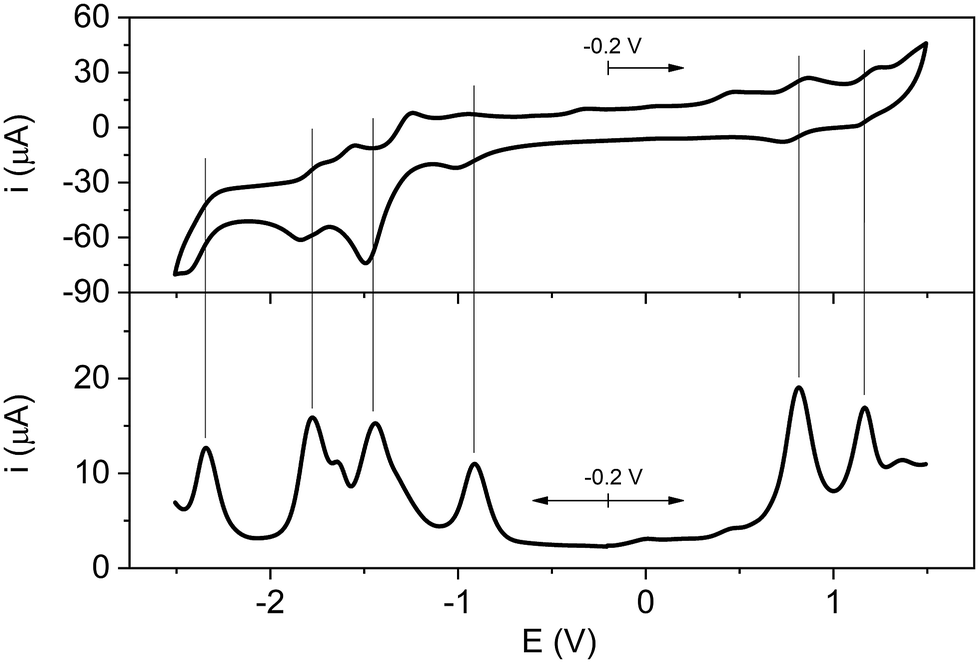 | ||
| Fig. 3 Cyclic (top panel) and square wave (bottom panel) voltammetry scans of Rucage in DCM solution. CV anodic scan starting from −0.2 V; SWV anodic and cathodic scans from −0.2 V (the sign of the cathodic current is reversed). | ||
| E 4red | E 3red | E 2red | E 1red | E 1ox | E 2ox | |
|---|---|---|---|---|---|---|
| a Potentials (in Volts) reported vs. Fc/Fc+, determined by the peaks of the square wave voltammograms. | ||||||
| Panel | — | −2.24 | −1.76 | −1.34 | — | — |
| Clip | — | — | −1.60 | −1.32 | 0.42 | 0.90 |
| Rucage | −2.34 | −1.78 | −1.44 | −0.91 | 0.81 | 1.17 |
Photophysical characterization of P-Pyr4@(Rucage)4
The photophysical characterization of the supramolecular system P-Pyr4@(Rucage)4 and all models has been performed in DCM. Due to the complexity of the system, two sets of models have been considered: (i) P-Pyr4 and Rucage as first level components and (ii) their further constituents, i.e., tetra-phenyl porphyrin (TPP) and pyrene (Pyr) as models for P-Pyr4, and Panel and Clip as models for Rucage (Fig. 2).Comparison of the absorption spectra of P-Pyr4 and Rucage with those of their respective models and their weighted sum is reported in Fig. S9 and S10.† In case of P-Pyr4, the spectrum of the array well matches the porphyrin features in the region 350–700 nm, where only a slight decrease and broadening of the Soret band are observed, while the bands of the pyrenyl units appear red-shifted by 5–8 nm (Fig. S9†). These results indicate the presence of weak electronic interactions between the different units in the array. Conversely, comparison of the spectrum of Rucage with the sum of the spectra of the models shows large differences (Fig. S10†), indicating strong electronic interactions among the units in the cage. It can be noticed that the cage, as well as the Clip component, presents absorption features up to 800 nm.
The absorption spectrum of P-Pyr4@(Rucage)4 is shown in Fig. 4, together with those of its components, P-Pyr4 and Rucage, and their weighted sum. The spectrum reasonably matches the sum of the spectra of the components, with a clear decrease only in the Soret region and in the pyrene peaks. These data indicate the presence of ground state interactions among the cages and the other units of the supramolecular system.
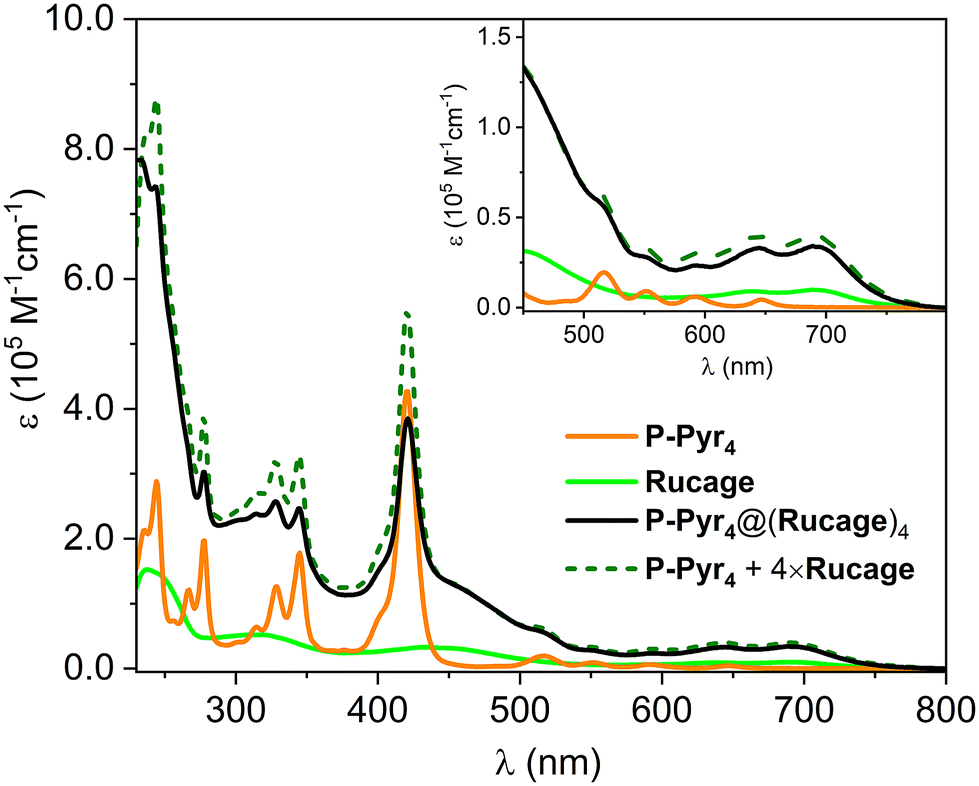 | ||
| Fig. 4 Absorption spectra of P-Pyr4@(Rucage)4 and its respective models P-Pyr4 and Rucage in DCM. The weighted sum of the spectra of the models is also reported. Inset: magnification of the 450–800 nm region. | ||
Rucage and its components were found to be non-emissive both at room temperature and at 77 K, even probing the NIR spectral range up to 1400 nm. Also, selective excitation of the Ru component at 700 nm in P-Pyr4@(Rucage)4 resulted in no appreciable emission at both temperatures.
On the other hand, the porphyrin component in P-Pyr4 displays typical porphyrin fluorescence, when selectively excited at 515 nm, with a quantum yield of 0.10 and a lifetime of 9.2 ns (Table 2 and Fig. S11†). Interestingly, the fluorescence of the pyrenyl units in P-Pyr4 is almost completely quenched, as observed upon their excitation at 345 nm (Fig. 5). It is evident that this quenching is accompanied by the full sensitization of the porphyrin emission, that appears comparable to that of the TPP model (Fig. 5). These results point to a very efficient energy transfer from the pyrene moieties to the central porphyrin core in P-Pyr4 (vide infra).
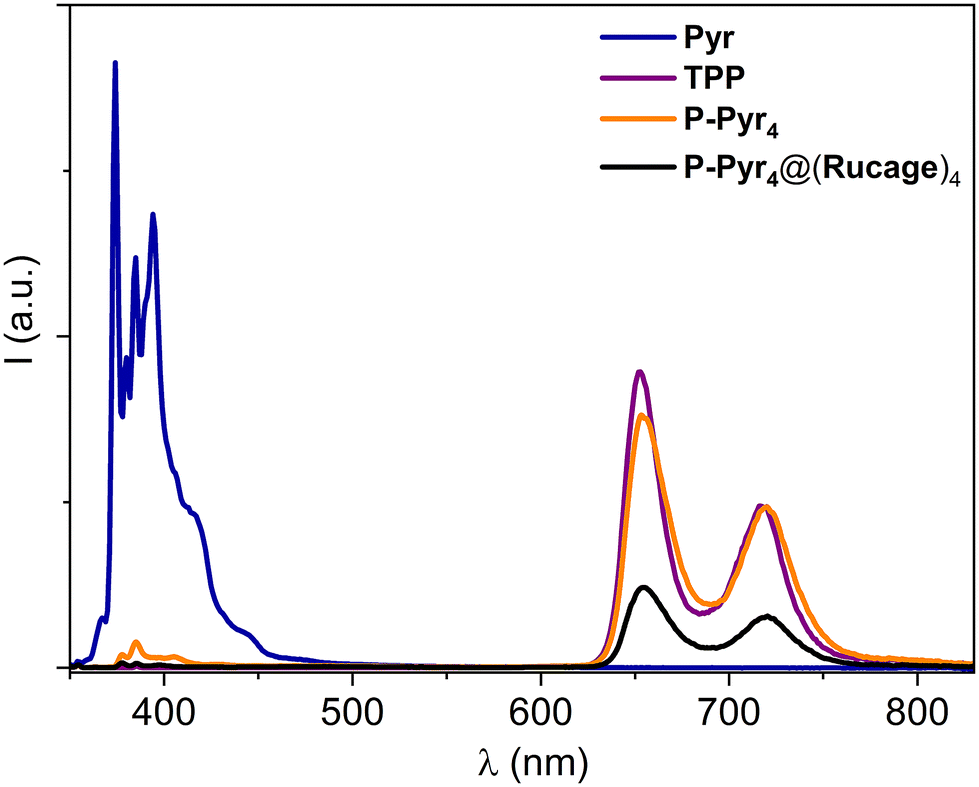 | ||
| Fig. 5 Corrected emission spectra of isoabsorbing solutions of P-Pyr4, P-Pyr4@(Rucage)4 and models Pyr and TPP in DCM; excitation at 345 nm (A345 = 0.11); at this wavelength the pyrenyl units are almost exclusively excited (90% of the absorbed photons). | ||
| λ max (nm) | ϕ fl | τ (ns) | ||
|---|---|---|---|---|
| a Emission maxima from corrected spectra. b Fluorescence quantum yields. c Measured with reference to α-NPO in aerated cyclohexane (ϕfl = 0.94),36 excitation at 321 nm. d Porphyrin emission quantum yield, measured with reference to TPP in aerated toluene (ϕfl = 0.11),37 excitation at 515 nm. e Excited state lifetimes measured with the single photon counting technique, excitation at 465 nm for the porphyrin component and 331 nm for pyrene (in brackets: percentages of pre-exponential values). f Data in line with those reported in ref. 10. g Lifetimes measured with single photon counting technique. An ultrafast component is also identified in P-Pyr4@(Rucage)4 by means of transient absorption analysis (see Discussion). | ||||
| Pyr | 374, 385, 394 | 0.063c | 26.3 | |
| TPP | 652, 718 | 0.091d | 8.1 | |
| P-Pyr4 | 1Pyr | 378, 385, 405 | ca. 1.0 × 10−3 | 0.37 |
| 1Porph | 654, 720 | 0.10d | 9.2 | |
| P-Pyr4@(Rucage)4 | 1Pyr | 378, 385, 398 | ca. 1.0 × 10−3 | 0.36 |
| 1Porph | 654, 720 | 0.030d | 0.40 (70%); 9.1 (30%)g | |
Selective excitation of the pyrene components in P-Pyr4@(Rucage)4 leads again to the complete quenching of their fluorescence in the array, but, in this case, the quenching is accompanied by only a 30% recovery of the porphyrin emission (Fig. 5). Indeed, the yield estimated for the porphyrin fluorescence in P-Pyr4@(Rucage)4 upon excitation at 515 nm is one third of that of TPP (Table 2 and Fig. S11†). Quenching of the porphyrin fluorescence in P-Pyr4@(Rucage)4 is attested also by lifetime measurements, which reveal the presence of a short component of 400 ps in addition to the value of ca. 9 ns of the unquenched porphyrin component (Table 2). It can be thus inferred that, in P-Pyr4@(Rucage)4, an efficient energy transfer from the pyrene to the porphyrin units is still operative, but the sensitized porphyrin singlet is partially depopulated by processes involving the cage components.
Further confirmations that an effective Pyr →Porph energy transfer occur in both arrays comes from different experiments. The excitation spectra of P-Pyr4 and P-Pyr4@(Rucage)4, collected at 720 nm, where only emission from the porphyrin is present, well match the absorption spectrum of P-Pyr4 in both porphyrin and pyrenyl domains (Fig. S12†). Also, upon excitation of the pyrene component at 331 nm, a lifetime of ca. 370 ps was measured at 380 nm in both P-Pyr4 and P-Pyr4@(Rucage)4 (Fig. S13† and Table 2), corresponding to the quenched pyrene lifetime, accompanied by a rise of porphyrin emission in the 650–750 nm range with the same time constant (Fig. S13†), attesting the sensitization of the porphyrin singlet. A detailed analysis of the Pyr →Porph energy transfer process is discussed later on in the Energy transfer section.
In order to shed light on the photoinduced processes at the basis of the observed porphyrin quenching in P-Pyr4@(Rucage)4, transient absorption measurements with femtosecond resolution have been performed. Two excitation wavelengths have been selected: 700 nm, where only the cage components absorb, and 420 nm, where the porphyrin components are prevalently excited (see Fig. 4).
Excitation at 700 nm of P-Pyr4@(Rucage)4 leads to the observation of an intense absorption band at 570 nm, with a broad tail extending in the NIR region and ground state bleaching features below 470 nm (Fig. 6). The kinetics is represented by a bi-exponential function all over the entire spectral range: a short decay of ca. 10 ps dominates the evolution in the visible region, paralleled by a rise of a band in the 900–1200 nm range, while the second process is represented by a decay of 2.5 ns. The observed behaviour can be tentatively interpreted as follows: (i) an initial fast process (10 ps) leads to the population of the lowest MLCT triplet state of the Ru cage component, whose spectrum is characterized by bands at ca. 540 nm and at ca. 1020 nm. This process can be ascribed to solvent induced vibrational relaxation of the triplet state, being intersystem crossing an ultrafast process; (ii) the formed triplet then decays on a longer time scale (2.5 ns). These features are very similar to those observed upon excitation of Rucage at 700 nm (Fig. S14†), indicating that the photophysics of the cage is not affected by the presence of the pyrenyl unit inside its cavity. By looking at the behaviour of model Clip, upon excitation at the same wavelength, a quite similar scenario is observed (Fig. S15†), with the difference of red-shifted spectra (maxima at 620 nm for the end-of-pulse spectrum and 640 and 1080 nm for the triplet state) and faster kinetics (ca. 5 ps and 400 ps) with respect to the cage system. The slower kinetics observed in the latter can be attributed to the presence of the panel ligands, whose orbitals might be involved in the charge-transfer transitions.
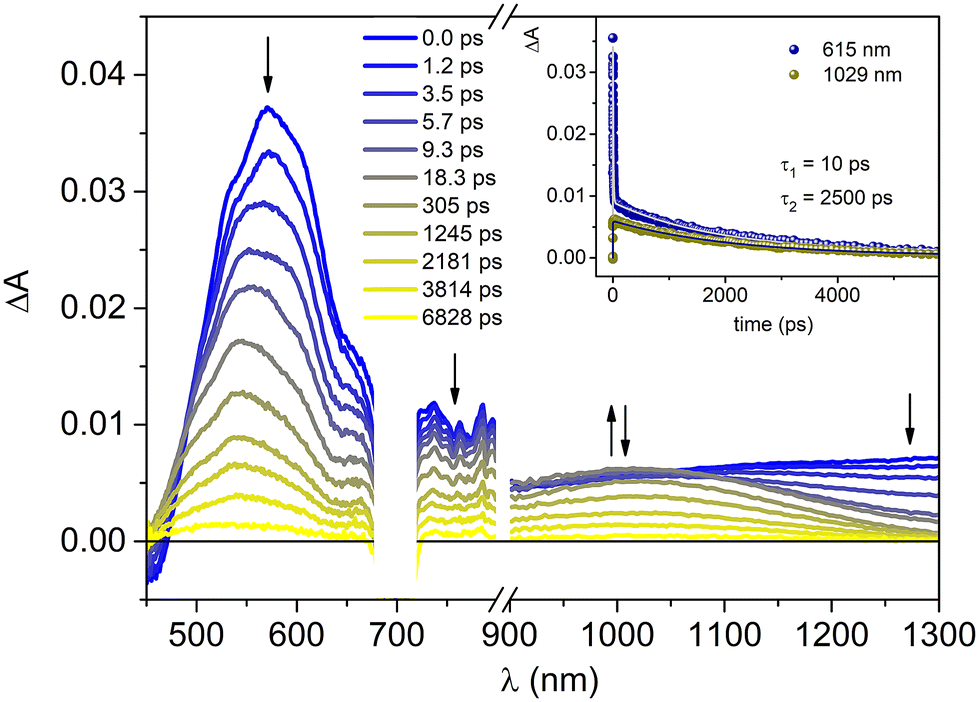 | ||
| Fig. 6 Transient absorption spectra of P-Pyr4@(Rucage)4 in DCM at different delays. Excitation at 700 nm (A700 = 0.34, 0.2 cm optical path, 10 μJ per pulse). Inset: ΔA time evolutions (dots) and fittings (lines) at the indicated wavelengths. | ||
Upon excitation at 420 nm, the end-of-pulse spectrum observed for the array P-Pyr4@(Rucage)4 (Fig. 7a) is different from that of P-Pyr4 (Fig. 7b), the latter presenting usual features of porphyrinic systems with positive absorption in the visible region overlaid with ground state bleaching of the Q-bands, stimulated emission at 654 and 720 nm and a positive band in the NIR region at 1080 nm. In P-Pyr4@(Rucage)4, indeed, the spectrum shows a more pronounced absorption in the 550–650 nm region. However, the most important difference resides in the kinetics: while for P-Pyr4 a slow decay of the signal is observed (Fig. 7b, inset), compatible with a lifetime of 9.2 ns as measured from fluorescence analysis (which is outside the maximum timescale of the transient instrumentation), in P-Pyr4@(Rucage)4 a more complex scenario is detected, with multi-exponential decays that include short components (Fig. 7a, inset). To get further insights in the comprehension of the transient data, a global fit analysis has been performed on the visible region of the matrix. Fig. S16† shows the spectral distribution of the amplitudes of the calculated lifetimes.55 A lifetime of 15 ps is associated to a spectrum with bands between 550 and 650 nm, indicative of a free-base porphyrin cation,56,57 while a component of 400 ps (fixed in the analysis) is associated to a typical spectrum of a porphyrin singlet. The spectrum corresponding to a lifetime of ca. 5 ns is similar to that of Rucage. As a confirmation, the transient absorption of Rucage excited at the same wavelength is reported in Fig. S17:† it can be observed that it reproduces well what observed upon excitation at 700 nm (Fig. 6), both in spectral features and decay lifetimes. The decay of 5 ns detected in the complex can thus be ascribed to the Rucage triplet (the longer lifetime with respect to that of the empty cage can derive from a rigidification of the system once hosting the pyrene moiety). Finally, a spectrum with an “infinite” lifetime on the timescale of the instrument matches the typical spectrum of a porphyrin triplet.58
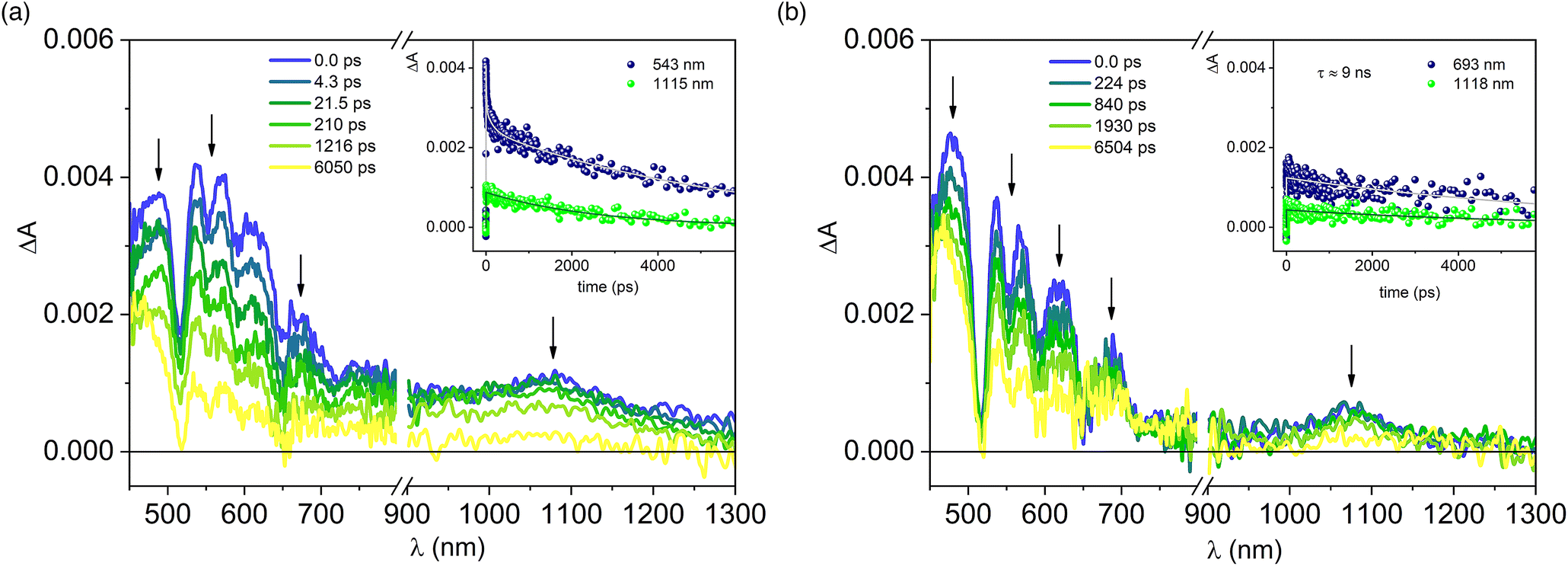 | ||
| Fig. 7 Transient absorption spectra of P-Pyr4@(Rucage)4 (a) and P-Pyr4 (b) in DCM at different delays. Excitation at 420 nm (A420 = 0.20, 0.2 cm optical path, 1 μJ per pulse). Inset: ΔA time evolutions (dots) and fittings (lines) at the indicated wavelengths. | ||
On the basis of these indications a plausible description of the processes occurring in P-Pyr4@(Rucage)4 can be drawn. Different conformations of the array can exist in solution due to the flexibility of the pegylated branches, as supported by GFN2-xTB calculations (see the Modelling section below). This leads to different distances between the porphyrin core and the metalla-prisms that can affect the photophysics of the system.
Conformations where one to four branches are folded towards the porphyrin core (P-Pyr4@(Rucage)4_III–VI) are more stable in solution than those where the branches are open in an extended “star” shape (P-Pyr4@(Rucage)4_I–II). Distances in the order of 5–6 Å between the porphyrin and the metalla-cages in these conformations are found, considering the minimum distances among the centre of the porphyrin and the centre of the naphtoquinonato moiety of the Clip (Table S5†) or among the centre of the porphyrin and the Ru atoms (Table S6†). These short distances allow for an ultrafast deactivation of the porphyrin singlet by electron transfer towards the cage. The energy of the charge separated (CS) state Porph+–Rucage− can be estimated as 1.43 eV, by considering the measured reduction potential of the Rucage (−0.91 V vs. Fc+/Fc, Table 1) and the oxidation potential of TPP (+0.52 V in DCM, vs. Fc+/Fc).59 Population of the CS state from the porphyrin singlet is thus thermodynamically allowed (Scheme 2). On these arguments, the detection, at the end-of pulse, of the formed Porph+ species can be explained (the features of the reduced Rucage are not clearly observable). The lifetime of this CS state is thus in the order of 15 ps.
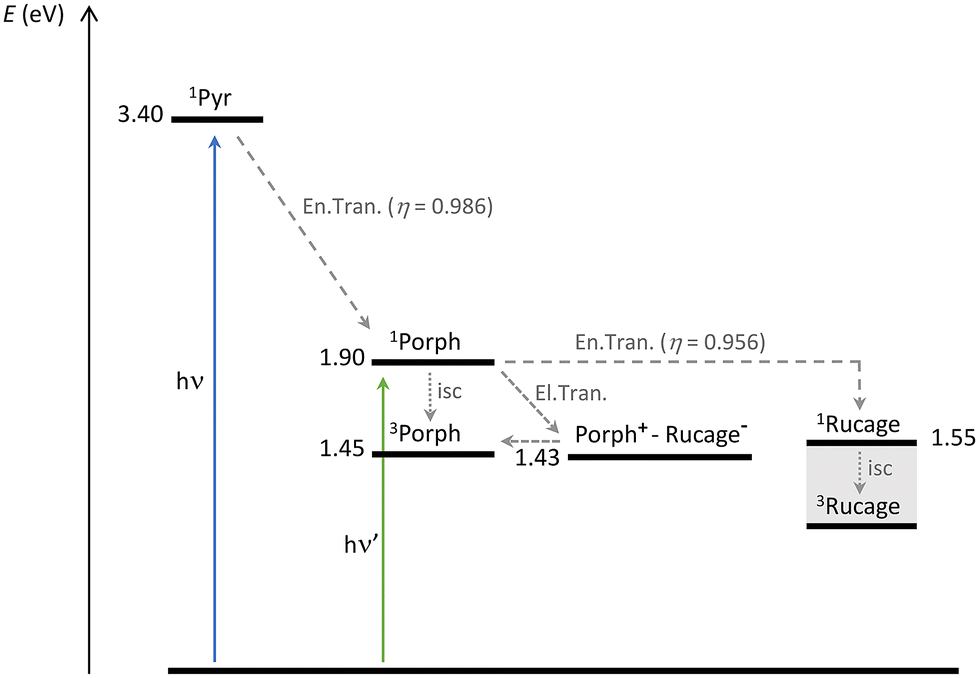 | ||
| Scheme 2 Energy level diagram and photoinduced processes for the system P-Pyr4@(Rucage)4. Singlet and triplet energy levels for the pyrene and porphyrin components are derived from data of the present paper and from the literature.61 The singlet energy level of Rucage has been estimated as onset of the absorption spectrum, while the triplet level could not be estimated and its position in the scheme is qualitative. The energy of the CS state Porph+–Rucage− has been calculated as described in the text. | ||
Accordingly, in conformations where the distance between the porphyrin and the metalla-prisms is higher, in the order of 13–16 Å (see below), the decay of the porphyrin singlet occurs in the order of hundreds of ps, corresponding to the measured 400 ps in the fluorescence decay (Table 2).60 This deactivation can be ascribed to energy transfer towards the cage components (as discussed in the Energy transfer section); the Rucage triplet that forms then decays in 5 ns.
Finally, the last species that can be identified from the transient analysis on long time scales is the porphyrin triplet. Its population can derive both by charge recombination (the CS state and the porphyrin triplet are almost isoenergetic, Scheme 2) or by intersystem crossing in the fraction of the porphyrin population that is not quenched by photoinduced reactions. The latter condition is observed in conformations where the porphyrin and the cage are too far from each other to enable any photoinduced process among them, as for P-Pyr4@(Rucage)4_I–II (average distance of the order of 24–25 Å, see below), and is confirmed by the lifetime of 9.1 ns detected with fluorescence analysis (Table 2).
In order to get further information on the energy transfer dynamics between the triplet states of the porphyrin and the cage components in Pyr4@(Rucage)4, the production of singlet oxygen from optically matched solutions of TPP, P-Pyr4, Rucage and P-Pyr4@(Rucage)4 has been measured. Excitation has been performed at 400 nm, where the porphyrin unit is selectively excited in P-Pyr4 and is absorbing 50% of the light in P-Pyr4@(Rucage)4. With respect to TPP, taken as standard (ϕΔ = 0.60 in DCM),62 the yield is only slightly lower for P-Pyr4 (ϕΔ = 0.55) while it is reduced to ϕΔ = 0.08 in Pyr4@(Rucage)4 (Fig. S18†). The latter corresponds to the yield of formation of the porphyrin triplet in the array, by considering a 50% absorption of the porphyrin in the array and a 70% quenching of the porphyrin singlet. This result rules out the occurrence of an energy transfer from the porphyrin triplet to the cage triplet, while a singlet–singlet energy transfer is possible (a detailed analysis is reported in the Energy transfer section). It can be also noticed that for Rucage the singlet oxygen signal is not detected.63 This results can be attributed either to a non-effective encounter of the cage with molecular oxygen, due to the short triplet lifetime, or to a lower energy of the cage triplet with respect to that of singlet oxygen (0.98 eV).
Modelling
Computational calculations were employed to support the interpretation of experimental findings. Initially, DFT and TD-DFT calculations were applied to Pyr and modified Clip-2 models to obtain electric dipole and transient electric dipole moments, which were further used for the energy transfer calculations. The ground state optimized structure of Pyr shows an electric dipole moment of 3 × 10−4 D, oriented on the plane of the carbon atoms, as reported in Fig. S20a.† In the modified Clip-2 the electric transition dipole moment of 6.6 D, calculated between the ground state and the first excited S1 state, is located in the middle of the aromatic rings of the dihydroxynaphthoquinonato moiety, and is parallel to it (Fig. S20b, c†).The GFN2-xTB method has been initially used to investigate the encapsulation of Pyr in Rucage by relaxed scan calculations. The obtained energy profile (ΔE) is reported in Fig. S21.† At the starting point, P-Pyr is placed at 45.5 Å from the Rucage and no interaction is observed from the zero value of ΔE. A decrease in ΔE is detected approximately at 23 Å, with a value of −8.6 kcal mol−1, indicating that the first interaction between the P-Pyr pegylated branch and the Rucage is taking place, while the Pyr is involved in an intramolecular interaction. Then, a stronger decrease in the ΔE value is observed (from −8.6 to −31.0 kcal mol−1) in the range 15.0–6.5 Å, due to the progressive inclusion of the Pyr moiety inside the Rucage (Fig. S21†). In the last part of the curve, negligible decrease in ΔE is reported with an energy minimum of −34.1 kcal mol−1 when the two entities are ca. 3.0 Å apart. The quantitative study of the Porph/Pyr/Rucage interaction has been completed with a qualitative analysis performed by Independent Gradient Model (IGM) method.64,65 IGM analysis (Fig. S22†) demonstrates the presence of π–π stacking, CH–π and CH–O interactions, all contributing to the stabilisation of the supramolecular system.
To investigate the stability of various P-Pyr4@(Rucage)4 conformations, six conformers were examined (Fig. 8), and the results are reported in Table 3 (refer also to Tables S1 and S2†). All investigated models exhibit highly positive De values, indicating the establishment of stable interactions between P-Pyr4 and four Rucage assemblies. As depicted in Table 3, De values increase with the increasing number of Rucage molecules interacting with the P-Pyr4 moiety. A similar trend is observed in |IE| and Def values and their individual components (Tables S1 and S2†), corresponding to an increasing number of interactions between Rucage and P-Pyr4, with a consequent increase in the deformation of the systems (Fig. S23†).
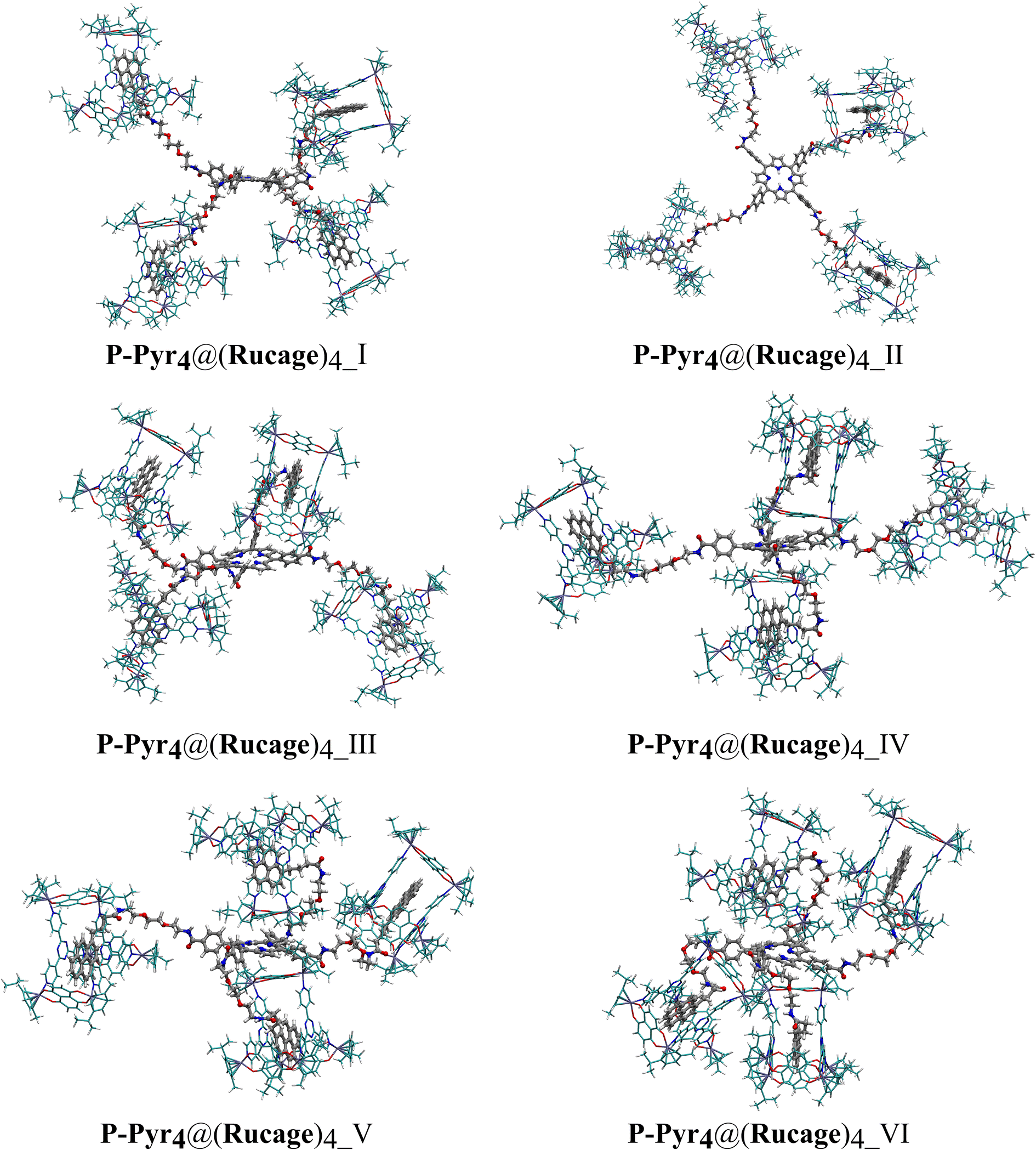 | ||
| Fig. 8 Optimized structures of P-Pyr4@(Rucage)4 conformations. To facilitate the visualization of the units, triflate anions are removed and the C atoms of P-Pyr4 are coloured in silver, while those of Rucage are reported in cyan. | ||
| Conformer | De | |IE| | Def |
|---|---|---|---|
| P-Pyr4@(Rucage)4_I | 175.8 | 190.3 | 14.4 |
| P-Pyr4@(Rucage)4_II | 193.2 | 210.3 | 17.1 |
| P-Pyr4@(Rucage)4_III | 216.4 | 243.8 | 27.4 |
| P-Pyr4@(Rucage)4_IV | 223.8 | 252.5 | 28.7 |
| P-Pyr4@(Rucage)4_V | 241.1 | 273.7 | 32.6 |
| P-Pyr4@(Rucage)4_VI | 293.4 | 344.6 | 51.2 |
The first two models exhibit a linear conformation of the branches. P-Pyr4@(Rucage)4_I, with branches oriented below and above the porphyrin plane, is found to be less stable compared to P-Pyr4@(Rucage)4_II (Table 3), where branches are positioned parallel to the porphyrin plane. These differences in stability can be attributed to the interactions between two Rucage units and the interspersed triflate anions in P-Pyr4@(Rucage)4_II (Fig. S24†). The |IE| decomposition, as reported in Table S1,† reveals similar values for both systems, ranging between 41.6 and 56.4 kcal mol−1, indicating different interactions between Rucage and the P-Pyr4 branches (Fig. 8), with a centre of porphyrin – centre of naphtoquinonato pillar average distance (dPorph–Rucage, Table S5†) of 24.7 Å for P-Pyr4@(Rucage)4_I and 24.1 Å for P-Pyr4@(Rucage)4_II.
In P-Pyr4@(Rucage)4_III one of the branches undergoes self-folding, facilitated by the interaction between Rucage and the porphyrin core (Fig. 8), resulting in a minimum dPorph–Rucage distance of 5.7 Å (Porph-Ruc1 in Table S5†). This interaction leads to a De stabilization of approximately 23 kcal mol−1 compared to P-Pyr4@(Rucage)4_II (Table 3). Indeed, the decomposition of |IE| reveals a significant increase, rising from 56.4 kcal mol−1 in P-Pyr4@(Rucage)4_II to 74.6 kcal mol−1 in P-Pyr4@(Rucage)4_III (Table S1†) for the Rucage involved in the folding process (Rucage-3).
In a similar way, P-Pyr4@(Rucage)4_IV exhibits two self-folding branches, resulting in a slight increase in stability by 7.4 kcal mol−1 (Table 3), and similar minimum dPorph–Rucage distances of ca. 5.0 Å for the first folded Rucage and ca. 5.5 Å for the second one (Table S5†). Once again, both cages involved in the interaction with P-Pyr4 exhibit higher |IE| values (Table S1†).
In P-Pyr4@(Rucage)4_V and P-Pyr4@(Rucage)4_VI, a folding of the third and fourth branches is observed, resulting in an increase in De of 17.3 and 69.6 kcal mol−1, respectively, compared to P-Pyr4@(Rucage)4_IV (Table 3). However, in these systems, the third and fourth Rucage units are unable to fully interact with the porphyrin core, as evidenced by the |IE| values in Table S1† and by the dPorph–Rucage distances of 13.1 Å (third branch, Table S5†) in P-Pyr4@(Rucage)4_V and 12.7 and 16.2 Å (third and fourth branches, Table S5†) in P-Pyr4@(Rucage)4_VI due to the steric hindrance of the other two Rucage units.
Overall, the scan calculations highlight that Pyr encapsulation is a diffusive process, given the absence of energy barriers, through the formation of non-covalent interactions detected by the IGM method. The analysis of the stability of the six P-Pyr4@(Rucage)4 conformers has revealed that the folding of the branches results in an increased stability, identifying the P-Pyr4@(Rucage)4_VI model as the most stable. However, other conformers with fewer folded branches can coexist, albeit in lower proportions.
Energy transfer
| k EnT (s−1) | η EnT | J F (cm3 M−1)c | κ 2 | R 0 (Å) | r (Å) | d (Å) | |
|---|---|---|---|---|---|---|---|
| a Experimental energy transfer rate constant calculated from eqn (5) and (6). b Experimental efficiency of the energy transfer process calculated from eqn (7). c Overlap integral calculated from eqn (10). d Förster critical distance calculated from eqn (8). e Interchromophoric distance calculated for kF = kEnT. f Average donor–to–acceptor interchomophoric distance calculated from molecular modelling. | |||||||
| Pyr → Porph | 2.66 × 109 | 0.986 | 2.18 × 10−13 | 0.667 | 35.7 | 17.6 | 20.4 |
| Pyr → Rucage | 7.51 × 107 | 0.027 | 0.68 × 10−13 | 6 × 10−6 | 4.2 | 7.7 | 7.7 |
| Porph → Rucage | 2.39 × 109 | 0.956 | 1.57 × 10−13 | 0.667 | 29.9 | 17.9 | 20.3 |
Conclusions
A novel supramolecular system has been assembled by combining a tetrapyrenylporphyrinic core with arene–ruthenium metalla-prisms, the latter being able to encapsulate the pyrenyl residues, thus leading to a large complex which is stable and does not aggregate in DCM solution. Detailed electrochemical and photophysical investigations elucidated the complex excited state dynamics of the supramolecular array. Femtosecond transient absorption analysis, in particular, evidenced fast excited states deactivation processes in the arene–ruthenium complexes, which were found to be non-emissive both at room temperature and at 77 K, and energy and electron transfer processes in the complete array. Different kinetics have been observed for the photoinduced reactions occurring between the central porphyrin core and the peripheral metalla-cages, indicating the presence in solution of various conformations, where the components are placed at different distances. Detailed computational analysis elucidated the stability of the different conformations and the encapsulation interactions between the pyrenyl groups and the metalla-cages. The results show novel photophysical insights on pyrenyl-functionalized porphyrins/metalla arene–ruthenium structures, that can lead to promising applications in biological and energy conversion contexts.Author contributions
Conceptualization, verification and funding acquisition: B. T., B. V.; investigation: S. L. P. A., B. V., A. B.; formal analysis: D. V.; data curation, writing – original draft and writing – review and editing: all authors.Data availability
Data supporting this article have been presented in the ESI.†Conflicts of interest
There are no conflicts to declare.Note after first publication
This article replaces the version published on 20th January 2025, which contained an incorrect version of eqn (2) due to a production error.Acknowledgements
We thank the European Union for financial support for the H2020-MSCA-ITN-2017-765297 project “NOAH”. Italian CNR (Project “Light-Induced Processes (LIP)”) is also acknowledged.References
- M. D. Karkas, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- A. Carella, F. Borbone and R. Centore, Front. Chem., 2018, 6, 481 CrossRef CAS PubMed.
- C. E. Elgar, N. A. Yusoh, P. R. Tiley, N. Kolozsvari, L. G. Bennett, A. Gamble, E. V. Pean, M. L. Davies, C. J. Staples, H. Ahmad and M. R. Gill, J. Am. Chem. Soc., 2023, 145, 1236–1246 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- N. P. Barry, O. Zava, J. Furrer, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 5272–5277 RSC.
- J. Mattsson, O. Zava, A. K. Renfrew, Y. Sei, K. Yamaguchi, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 8248–8255 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- J. W. Yi, N. P. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461–471 CrossRef CAS PubMed.
- J. Shirdel, A. Penzkofer, R. Procházka, Z. Shen, J. Strauss and J. Daub, Chem. Phys., 2007, 331, 427–437 CrossRef CAS.
- B. Therrien, CrystEngComm, 2015, 17, 484–491 RSC.
- E. Orhan, A. Garci and B. Therrien, Inorg. Chim. Acta, 2017, 461, 78–83 CrossRef CAS.
- F. Schmitt, N. P. Barry, L. Juillerat-Jeanneret and B. Therrien, Bioorg. Med. Chem. Lett., 2012, 22, 178–180 CrossRef CAS PubMed.
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 2009, 4695–4700 CrossRef.
- J. Freudenreich, N. P. E. Barry, G. Süss-Fink and B. Therrien, Eur. J. Inorg. Chem., 2010, 2010, 2400–2405 CrossRef.
- J. Mattsson, P. Govindaswamy, J. Furrer, Y. Sei, K. Yamaguchi, G. Süss-Fink and B. Therrien, Organometallics, 2008, 27, 4346–4356 CrossRef CAS.
- D. Chen, T. Xiao, E. Monflier and L. Wang, Commun. Chem., 2024, 7, 88 CrossRef CAS PubMed.
- K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed.
- D. Li, X. Liu, L. Yang, H. Li, G. Guo, X. Li and C. He, Chem. Sci., 2023, 14, 2237–2244 RSC.
- Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J. H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed.
- A. Sautter, B. K. Kaletas, D. G. Schmid, R. Dobrawa, M. Zimine, G. Jung, I. H. van Stokkum, L. De Cola, R. M. Williams and F. Wurthner, J. Am. Chem. Soc., 2005, 127, 6719–6729 CrossRef CAS PubMed.
- Z. Zhang, Z. Zhao, Y. Hou, H. Wang, X. Li, G. He and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 8862–8866 CrossRef CAS PubMed.
- P. P. Jia, L. Xu, Y. X. Hu, W. J. Li, X. Q. Wang, Q. H. Ling, X. Shi, G. Q. Yin, X. Li, H. Sun, Y. Jiang and H. B. Yang, J. Am. Chem. Soc., 2021, 143, 399–408 CrossRef CAS PubMed.
- T. T. Kim Cuc, P. Q. Nhien, T. M. Khang, H.-Y. Chen, C.-H. Wu, B.-T. B. Hue, Y.-K. Li, J. I. Wu and H.-C. Lin, ACS Appl. Mater. Interfaces, 2021, 13, 20662–20680 CrossRef CAS PubMed.
- J. Otsuki, J. Mater. Chem. A, 2018, 6, 6710–6753 RSC.
- G. Chen, F. Song, X. Xiong and X. Peng, Ind. Eng. Chem. Res., 2013, 52, 11228–11245 CrossRef CAS.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- T. Chen, B. He, J. Tao, Y. He, H. Deng, X. Wang and Y. Zheng, Adv. Drug Delivery Rev., 2019, 143, 177–205 CrossRef CAS PubMed.
- M. Dimura, T. O. Peulen, C. A. Hanke, A. Prakash, H. Gohlke and C. A. Seidel, Curr. Opin. Struct. Biol., 2016, 40, 163–185 CrossRef CAS PubMed.
- F. C. Ho, Y. J. Huang, C. C. Weng, C. H. Wu, Y. K. Li, J. I. Wu and H. C. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 53257–53273 CrossRef CAS PubMed.
- H. L. Anderson, S. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2231–2245 RSC.
- N. P. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172–7180 RSC.
- T. Gianferrara, A. Bergamo, I. Bratsos, B. Milani, C. Spagnul, G. Sava and E. Alessio, J. Med. Chem., 2010, 53, 4678–4690 CrossRef CAS PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- I. B. Berlman, in Handbook of Fluorescence Spectra of Aromatic Molecules, ed. A. Press, New York and London, 2nd edn, 1971, p. 296 Search PubMed.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1–13 CrossRef CAS.
- K. A. Nguyen, P. N. Day and R. Pachter, J. Chem. Phys., 2011, 135, 074109 CrossRef PubMed.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- F. Neese, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- P. Pracht, E. Caldeweyher, S. Ehlert and S. Grimme, ChemRxiv, 2019, preprint, DOI:10.26434/chemrxiv.8326202.v1.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e01493 Search PubMed.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- S. Halbert and H. Gérard, New J. Chem., 2015, 39, 5410–5419 RSC.
- E. Pancani, D. Veclani, M. Agnes, A. Mazza, A. Venturini, M. Malanga and I. Manet, RSC Adv., 2023, 13, 10923–10939 RSC.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, pp. 443–475 Search PubMed.
- M. A. Furrer, F. Schmitt, M. Wiederkehr, L. Juillerat-Jeanneret and B. Therrien, Dalton Trans., 2012, 41, 7201–7211 RSC.
- A completely irreversible oxidation process, located at ca. Epeak = +0.80 V, probably attributable to the generation of a reduced species during the cathodic scan, is also observed.
- A lifetime of 400 ps has been fixed in the analysis, in order to match the 400 ps component measured with single photon counting analysis of the luminescence decay.
- E. A. Alemán, J. Manríquez Rocha, W. Wongwitwichote, L. A. Godínez Mora-Tovar and D. A. Modarelli, J. Phys. Chem. A, 2011, 115, 6456–6471 CrossRef PubMed.
- J. Fajer, D. C. Borg, A. Forman, D. Dolphin and R. H. Felton, J. Am. Chem. Soc., 1970, 92, 3451–3459 CrossRef CAS PubMed.
- S. Durot, L. Flamigni, J. Taesch, T. T. Dang, V. Heitz and B. Ventura, Chem. – Eur. J., 2014, 20, 9979–9990 CrossRef CAS PubMed.
- T. T. Dang, S. Durot, L. Monnereau, V. Heitz, A. Barbieri and B. Ventura, Dalton Trans., 2017, 46, 9375–9381 RSC.
- Single wavelength decay analysis of the transient matrix of P-Pyr4@(Rucage)4 leads to identify a component of ca. 400 ps, which can be different in the global analysis due to the complex overlay of signals.
- M. Gouterman and G.-E. Khalil, J. Mol. Spectrosc., 1974, 53, 88–100 CrossRef CAS.
- J. A. S. Cavaleiro, H. Görner, P. S. S. Lacerda, J. G. MacDonald, G. Mark, M. G. P. M. S. Neves, R. S. Nohr, H.-P. Schuchmann, C. von Sonntag and A. C. Tomé, J. Photochem. Photobiol. A: Chem., 2001, 144, 131–140 CrossRef CAS.
- The same result has been obtained upon excitation of Clip-2, Rucage and P-Pyr4@(Rucage)4 at 700 nm.
- C. Lefebvre, H. Khartabil, J. C. Boisson, J. Contreras-Garcia, J. P. Piquemal and E. Henon, ChemPhysChem, 2018, 19, 724–735 CrossRef CAS PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-Garcia and E. Henon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra, electrochemical, photophysical and theoretical data. See DOI: https://doi.org/10.1039/d4dt03154g |
| This journal is © The Royal Society of Chemistry 2025 |