
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amplifying Lewis acidity by oxidation: leveraging the redox-activity of bis(3,6-di-tert-butyl-catecholato)silane†
Thaddäus
Thorwart
,
Manuel
Schmitt
 and
Lutz
Greb
*
and
Lutz
Greb
*
Ruprecht-Karls-Universität Heidelberg, Anorganisch-Chemisches Institut, Im Neuenheimer Feld 270, Heidelberg 69120, Germany. E-mail: greb@uni-heidelberg.de
First published on 29th November 2024
Abstract
Bis(catecholato)silanes were showcased as strong Lewis acids, while their inherent redox activity remained unexplored in this context. In the present work, we study the oxidation of monomeric bis(3,6-di-tert-butyl-catecholato)silane (1), leading to the Lewis superacidic radicalic silylium ionradical 1˙+ (FIA 784 kJ mol−1). Oxidation of 1 with [N(p-C6H4Br)3][B(C6F5)4] yielded [1][B(C6F5)4], displaying strong catalytic activity in the Friedel–Crafts-dimerization, hydrodeoxygenation and carbonyl-olefin-metathesis. It demonstrates how Lewis acidity can be amplified through oxidation without needing an add-on redox-active substituent. Instead, it synergizes the constraining effect of catecholates with their inherent redox non-innocence to unlock enhanced catalytic performance.
Lewis acids are extensively used in all kinds of chemistry, ranging from materials science over organic chemistry and drug discovery to biological research.1–7 This broad applicability fostered a continuous development that led to numerous new p-block Lewis acids – an interest further propelled by the field of frustrated Lewis pairs.8 Frequently, the effect of a Lewis acid is related to its strength;9 a precept fueling the search for continuously stronger Lewis (super)acids.10 Typical strategies for increasing Lewis acidity include, among others, the installation of electron-withdrawing substituents, the introduction of cationic charge, or structural constraints. Other approaches aim to exploit a Lewis acidity enhancement on demand to overcome obstacles associated with the limited stability of the most potent Lewis acids. For instance, aryl- or alkyl-silylium ions are usually synthesized in situ by hydride abstraction through carbocations to mitigate side reactions, e.g., with the solvent or the counteranion.11–14 Another strategy relies on photochemical concepts to release Lewis acids upon irradiation.15–18 Moreover, redox approaches were shown to stimulate Lewis acidity. A change in the main group element oxidation states for increased reactivity was exemplified for pnictogen- and tin-compounds.19–22 As an alternative, the oxidation of redox-non-innocent ligands instead of the central elements was considered more recently (Fig. 1a). Paradies and coworkers investigated a row of ferrocenyl-substituted boranes and unlocked Lewis superacidity by oxidation (Fig. 1b).23 Caputo and coworkers presented a phenothiazine-substituted borane as transition metal free representative for this concept (Fig. 1c).24
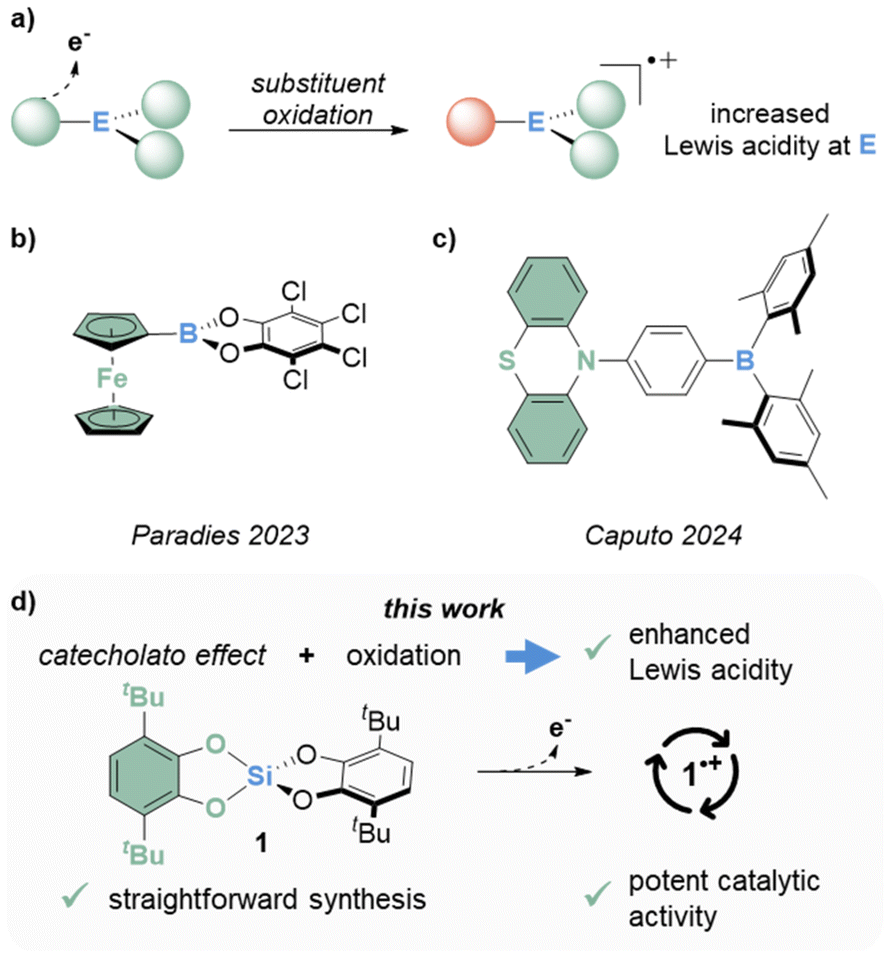 | ||
| Fig. 1 (a) Schematic representation of the strategy of Lewis acidity amplification by oxidation, (b/c) examples of previously reported boranes with redox-active substituents and (d) the redox-active silicon Lewis acid 1 presented in this work. | ||
In recent years, we pursued the search for novel main-group Lewis superacids based on the catecholato ligand.25–33 The bidentate ligand has been shown to have a multifold impact on the increase of Lewis acidity. First, it exhibits a bite angle that suits the geometry of hypervalent structures.34,35 Second, the pronounced electronegativity of the primary oxygen substituents stabilizes the non-bonding orbitals in the 3-center-4-electron (3c4e) bonds that supports hypercoordinate adduct formation. Lastly, the aromatic system allows for adjustment of the mesomeric π-acidity by introducing electron-withdrawing substituents in the ligand backbone. This synergistic impact on Lewis acidity was recently termed the catecholato effect.36 Interestingly, catecholates are mainly known as prototypical redox-active ligands, which can be oxidized to semiquinonate (sq) radicals or quinones. Even though this property was investigated holistically with transition metals,37 and also found application in the context of main group chemistry,38 it remained unconsidered in the design of catecholate Lewis acids. In contrast to previous approaches that require the aimed installation of a redox-active substituent (e.g., Fig. 1b and c), bis(catecholato)silanes are easily prepared and already exhibit considerable Lewis acidity through the catecholato effect.26 In this work, we identify bis(3,6-di-tert-butyl-catecholato)silane (1) as suitable model system that allows first insights on the oxidation chemistry of bis(catecholato)silanes (Fig. 1d). We then continue to investigate the chemical oxidation to 1˙+ and subsequently assess its increased Lewis acidity through computations and catalytic activity.
Common bis(catecholato)silanes readily undergo oligomerization via low-barrier dynamic covalent processes.28 Thus, to simplify the investigation for redox chemistry, we first screened for a bis(catecholato)silane representative that does not undergo oligomerization. Previous work suggested that the self-aggregation of bis(catecholato)silanes can be steered by suitable substitution of the catechol ligand.28 Whereas the parent compound bis(catecholato)silane (Si(catH)2) forms cyclic decamers as the favored configuration in the solid state (Fig. 2a), 3,5-substituted (tert-butyl or cumyl) catecholates are present as dimer, and related ortho-amidophenolates exhibit monomeric forms.28,39 These examples suggested that an increased steric demand around the silicon center will cause a decreased nuclearity of silicon atoms in the thermodynamic minimum. Thus, we proposed that a monomeric bis(catecholato)silane can be realized from a 3,6-substituted catechol ligand. In analogy, the use of 3,6-di-tert-butyl-catechol (H2cat3,6-tBu) was previously shown to prevent oligomerization of 3d metal complexes.40,41 Therefore, we synthesized H2cat3,6-tBu according to Ershov's procedure42 (Fig. 2b) and subsequently converted it with HSiCl3 in acetonitrile to give Si(cat3,6-tBu)2 (1) in 82% yield (Fig. 2c). While 1 was known from previous studies,43–45 a thorough characterization and unambiguous structure assignment has been lacking, while recent work even stated problems regarding its purification.45 We found that the present synthetic route yields analytically pure 1 as a colorless precipitate right from the reaction mixture.
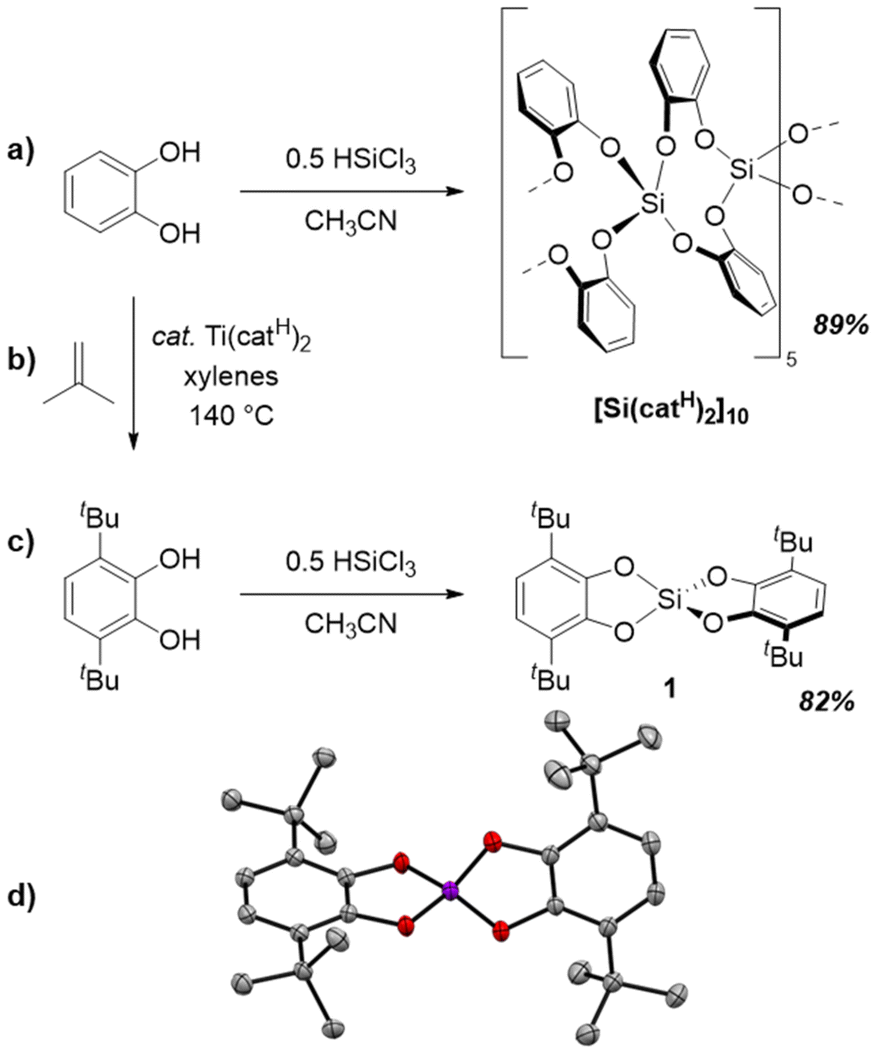 | ||
| Fig. 2 Illustration of the syntheses of (a) the decamer of bis(catecholato)silane, (b) 3,6-di-tert-buyl-catechol according to Ershov's procedure, and (c) the monomeric bis(3,6-di-tert-butyl-catecholato)silane (1) investigated in this work. (d) SCXRD derived molecular structure of 1 (hydrogen atoms omitted for clarity; ellipsoids shown at 50% probability). | ||
In contrast to previous representatives of bis(catecholato)silanes, 1 is exceptionally soluble in dichloromethane, which allowed us to undertake a more profound NMR spectroscopic investigation. Indeed, 1H, 13C, and 29Si NMR spectroscopy revealed sharp signals, indicating a monomeric form in solution (details see ESI†). SCXRD analysis of single crystals, grown by liquid diffusion of acetonitrile into a CH2Cl2 solution of 1, confirmed the monomeric, distorted tetrahedral entity in the solid state (Fig. 2d). Accompanying computations on the dimerization of 1 and related derivatives revealed a thermodynamic and not a kinetic inhibition of oligomerization (see ESI†). Of note, 1 showcases similar physicochemical properties to the previously reported bis(3,4,6-tri-iso-propyl-catecholato)silane (Si(cat3,4,6-iPr)2), for which the monomeric form could not be proven by scXRD.46
With this monomeric bis(catecholato)silane in hand, we proceeded with the strategy of Lewis acidity amplification by oxidation. Oxidation of 1 would result in Si(cat3,6-tBu)(sq3,6-tBu)˙+ (1˙+, Fig. 3a). The computed fluoride ion affinity (FIA, (DLPNO-CCSD(T)/def2-QZVPP//PBE0-D3(BJ)/def2-TZVP)) of 784 kJ mol−1 confirms a significant increase compared to 1 (404 kJ mol−1), surpassing the FIA of the prominent electrophilic phosphonium cation (EPC) [FP(C6F5)3]+ (ref. 47) and approaching even the strongest EPC known to date ([P(amFphF)2]+) (Fig. 3b).48 The same holds for the solvation-corrected FIAsolv, which have been found mandatory for comparing Lewis acids of varying charge states.49
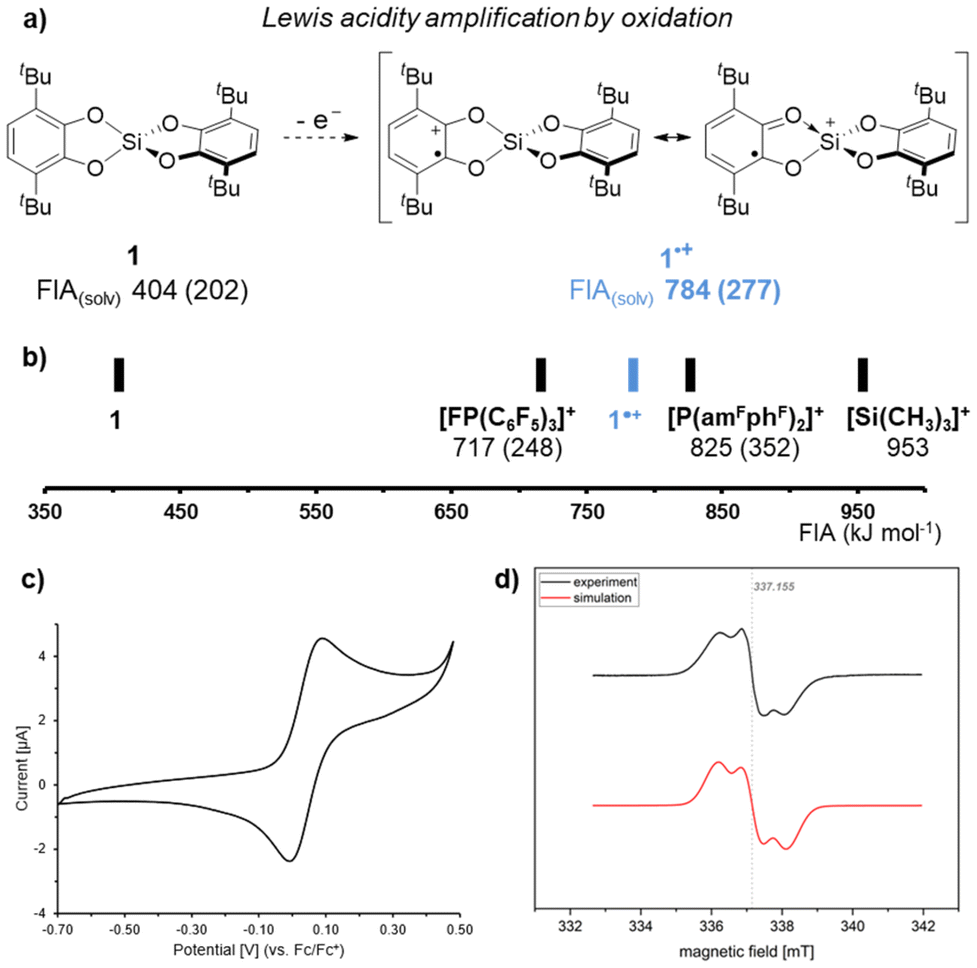 | ||
| Fig. 3 (a) Illustration for the strategy of enhancing the Lewis acidity of 1 upon oxidation; (b) comparison of the FIAs of 1 and 1˙+ with the electrophilic phosphonium cations [FP(C6F5)3]+ and [P(amFphF)2]+ as well as with the trimethylsilylium cation; FIAs refer to vacuum values, FIAs in CH2Cl2 are shown in parentheses; details on the FIA data are given in the ESI;† (c) cyclic voltammogram of 1 (details see ESI†). (d) Stacked EPR spectra from experiment ([1][B(C6F5)4] in o-C6H4F2) and simulation (1˙+). | ||
Thus, the oxidation of 1 was explored. Cyclovoltammetry revealed an oxidation potential of 0.04 V (vs. Fc/Fc+, details see ESI†) for 1 in dichloromethane (Fig. 3c). In accordance with the determined potential, the ferrocenium salt Fc[B(C6F5)]4 did not indicate any reactivity with 1 in dichloromethane. When using Ag[SbF6] instead, the immediate coloration of the reaction mixture was accompanied by a paramagnetic species as judged by EPR spectroscopy. However, various signals in the 19F NMR spectrum suggested an undefined reactivity, likely involving ligand scrambling after initial fluoride abstraction from SbF6− by 1˙+, supporting Lewis superacidic character. Thus, we tested silver salts with anions that were less prone to decomposition. When using AgOTf, AgNTf2, or Ag(o-C6H4F2)[Al(OtC4F9)4]50 such reactivity was indeed absent. However, in those cases, the oxidation processes were found slow, with full conversion not reached even after several days. In sight of the light-sensitivity of Ag(I) salts we aimed to reduce the reaction time by exploiting the increased oxidation capability of the Ag/I2 system.51 Indeed, the addition of half an equivalent of molecular iodine tremendously accelerated the conversion of 1 with the silver salts. Again, a deep coloration of the reaction mixture in conjunction with an EPR active sample strongly supported the initial oxidation. However, EPR spectra did not match the expected pattern for 1˙+ but indicated side reactivity under the harsh oxidation conditions.
Next, we considered [N(p-C6H4Br)3][B(C6F5)4] as a suitable oxidant with a preference for outer-sphere electron-transfer52 and a low inner-sphere reorganization.53 In an attempt to isolate the target radical cation, 1 was reacted with [N(p-C6H4Br)3][B(C6F5)4] in ortho-difluorobenzene (o-C6H4F2). While the oxidation process required several days, EPR monitoring suggested cleaner oxidation. After a complete reaction, an intensely colored oil was isolated, which solidified after trituration with n-hexane. Simulation of the room temperature recorded EPR spectrum revealed a resonance at giso = 2.00269 with a triplet hyperfine coupling constant of AisoH = 716 μT from two equivalent hydrogens (S = 1/2) (Fig. 3d). Comparison of the IR spectra of 1 and [1][B(C6F5)4] indicated altered C–O stretching modes (Fig. S13†), in line with typical catecholato and semiquinonato ligands.54 The compound was additionally characterized by mass spectrometry and elemental analysis, further supporting the proposed structural motif, but also indicating partial decomposition. This observation aligns with previously reported decomposition pathways for cat3,6-tBu and related tert-butyl-phenol derivatives under Lewis acidic55–58 or oxidative59,60 conditions. Unfortunately, the viscous nature of the compound prevented crystallization and analysis of a solid-state structure by scXRD. We also attempted oxidation in the presence of donors. However, scXRD analysis of proposed adducts with donors could not be achieved, as the resulting species suffered from the same obstacle. However, the isolated species allowed us to probe the initial hypothesis of whether 1˙+ displays an amplified Lewis acidity over 1. A Gutmann-Beckett classification was judged as unsuitable given the paramagnetic nature of the target species.61 Thus, we attempted to gauge the Lewis acidic potential by testing the catalytic activity of [1][B(C6F5)4]. Indeed, using 5 mol% [1][B(C6F5)4] enabled efficient and fast Friedel–Crafts dimerization of 1,1-diphenylethylene (Fig. 4a). Notably, [1][B(C6F5)4] was also found to be catalytically applicable in the reductive hydrodeoxygenation of benzophenone (Fig. 4b). To utilize the full potential of 1˙+ we next attempted the more challenging carbonyl-olefin-metathesis (COM) as its outcome is known to be critically dependent on the strength of the applied Lewis acid.62–68
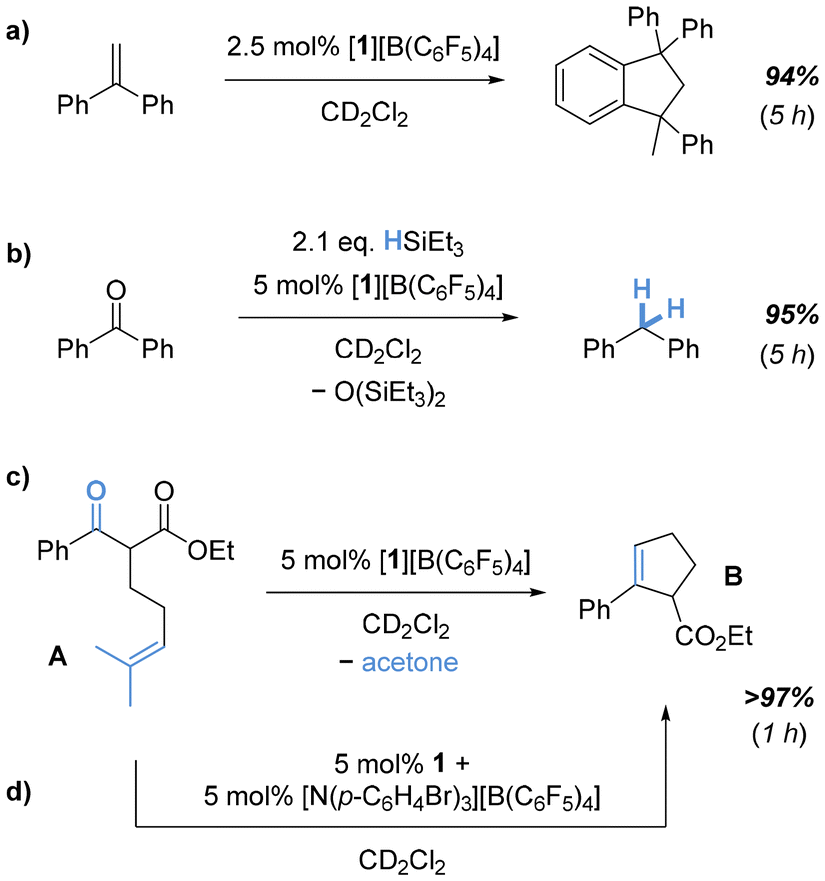 | ||
| Fig. 4 Representative (a) Friedel–Crafts dimerization, (b) hydrodeoxygenation, and (c) carbonyl-olefin-metathesis (COM) catalyzed by [1][B(C6F5)4]; (d) in situ generation of the catalytic species for the COM. | ||
Indeed, 5 mol% of [1][B(C6F5)4] enabled complete formation of the cyclization product B from the substrate A within one hour, thus ranging above the activity of the neutral bis(pertrifluoromethylcatecholato)silane30 but similar to the hitherto most active bis(catecholato)phosphonium ions29 (Fig. 4c). Strikingly, the catalytic activity of 1˙+ can be unlocked by in situ generation before substrate addition (Fig. 4d). Of note, the formation of B was not observed in the presence of 1 or the [N(p-C6H4Br)3]˙+ cation alone, supporting 1˙+ as catalytically active species.
Overall, the disclosed approach of combining two stable pre-catalysts for the generation of a Lewis superacid that takes profit from a combination of the catecholato effect and its redox activity states a valuable alternative to existing methods. The in situ oxidation approach allows the mitigation of long-term instability and represents a valuable extension of previous strategies for generating silylium ions.
Author contributions
T. T. and L. G. devised the project and designed the experiments. T. T. carried out the calculations, conducted experimental work and characterization, and analyzed the data. T. T. and L. G. wrote the manuscript. M. S. refined and finalized the scXRD structure.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data has been deposited under CCDC 2402241.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster) and GR5007/2-1. Open Access funding enabled and organized by Projekt DEAL. Dr R. Maskey is acknowledged for assistance with cyclic voltammetry measurement.References
- H. Yamamoto, Lewis acids in organic synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed.
- A. Corma and H. García, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS.
- H. Yamamoto, Lewis acid reagents: a practical approach, Knovel, Norwich, NY, 2007 Search PubMed.
- H. Yamamoto and K. Ishihara, Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Kobayashi and K. Manabe, Pure Appl. Chem., 2000, 72, 1373–1380 CrossRef CAS.
- S. J. Lippard and J. M. Berg, Principles of bioinorganic chemistry, University Science Books, Mill Valley, CA, 1994 Search PubMed.
- J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758–776 CAS.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- G. A. Olah and D. A. Klumpp, Superelectrophiles and their chemistry, Wiley-Interscience, Hoboken, 2008 Search PubMed.
- L. Greb, Chem. – Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- R. N. Perutz, Science, 2008, 321, 1168–1169 CrossRef CAS.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS.
- V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
- V. Lemieux, M. D. Spantulescu, K. K. Baldridge and N. R. Branda, Angew. Chem., Int. Ed., 2008, 47, 5034–5037 CrossRef CAS PubMed.
- A. Y. Khalimon, W. E. Piers, J. M. Blackwell, D. J. Michalak and M. Parvez, J. Am. Chem. Soc., 2012, 134, 9601–9604 CrossRef CAS PubMed.
- A. Y. Khalimon, B. K. Shaw, A. J. V. Marwitz, W. E. Piers, J. M. Blackwell and M. Parvez, Dalton Trans., 2015, 44, 18196–18206 RSC.
- R. Mizutsu, R. Asato, C. J. Martin, M. Yamada, Y. Nishikawa, S. Katao, M. Yamada, T. Nakashima and T. Kawai, J. Am. Chem. Soc., 2019, 141, 20043–20047 CrossRef CAS PubMed.
- M. Schorpp, R. Yadav, D. Roth and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e2022079 CrossRef.
- R. Prakash, J. Joseph, A. P. Andrews, B. Varghese and A. Venugopal, Inorg. Chem., 2023, 62, 14828–14832 CrossRef CAS.
- M. Yang, D. Tofan, C. H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., 2018, 130, 14064–14068 CrossRef.
- O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS.
- L. Köring, A. Stepen, B. Birenheide, S. Barth, M. Leskov, R. Schoch, F. Krämer, F. Breher and J. Paradies, Angew. Chem., Int. Ed., 2023, 62, e202216959 CrossRef PubMed.
- T. P. L. Cosby, A. Bhattacharjee, S. K. Henneberry, J. LeBlanc and C. B. Caputo, Chem. Commun., 2024, 60, 5391–5394 RSC.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS.
- D. Hartmann, M. Schädler and L. Greb, Chem. Sci., 2019, 10, 7379–7388 RSC.
- D. Roth, H. Wadepohl and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 20930–20934 CrossRef CAS.
- D. Hartmann, T. Thorwart, R. Müller, J. Thusek, J. Schwabedissen, A. Mix, J. H. Lamm, B. Neumann, N. W. Mitzel and L. Greb, J. Am. Chem. Soc., 2021, 143, 18784–18793 CrossRef CAS PubMed.
- D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845–15851 CrossRef CAS PubMed.
- T. Thorwart, D. Roth and L. Greb, Chem. – Eur. J., 2021, 27, 10422–10427 CrossRef CAS PubMed.
- N. Ansmann, D. Hartmann, S. Sailer, P. Erdmann, R. Maskey, M. Schorpp and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202203947 CrossRef CAS.
- N. Ansmann, T. Thorwart and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202210132 CrossRef CAS.
- Q. Luo and L. Greb, Eur. J. Inorg. Chem., 2023, 26, e202300186 CrossRef CAS.
- C. L. Frye, J. Am. Chem. Soc., 1964, 86, 3170–3171 CrossRef CAS.
- J. J. Harland, R. O. Day, J. F. Vollano, A. C. Sau and R. R. Holmes, J. Am. Chem. Soc., 1981, 103, 5269–5270 CrossRef CAS.
- L. Greb, Synlett, 2023, A–Q Search PubMed.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- R. Maskey and L. Greb, Catecholate Complexes of p-Block Elements: Redox Activity, Encycl. Inorg. Bioinorg. Chem., 2022, 1–15 Search PubMed.
- T. Thorwart, D. Hartmann and L. Greb, Chem. – Eur. J., 2022, 28, e202202273 CrossRef CAS.
- C.-M. Liu, E. Nordlander, D. Schmeh, R. Shoemaker and C. G. Pierpont, Inorg. Chem., 2004, 43, 2114–2124 CrossRef CAS.
- C. W. Lange, B. J. Conklin and C. G. Pierpont, Inorg. Chem., 1994, 33, 1276–1283 CrossRef CAS.
- I. Belostotskaya, N. Komissarova, E. Dzhuaryan and V. Ershov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1972, 21, 1535–1536 CrossRef.
- A. Prokof'ev, T. Prokofe'va, N. Bubnov, S. Solodovnikov, I. Belostotskaya, V. Ershov and M. Kabachnik, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1978, 27, 1726–1733 CrossRef.
- A. I. Prokof'ev, T. I. Prokof'eva, I. S. Belostotskaya, N. N. Bubnov, S. P. Solodovnikov, V. V. Ershov and M. I. Kabachnik, Tetrahedron, 1979, 35, 2471–2482 CrossRef.
- K. V. Arsenyeva, A. V. Klimashevskaya, M. V. Arsenyev, I. A. Yakushev, A. V. Cherkasov, P. V. Dorovatovskii, A. V. Maleeva, O. Y. Trofimova and A. V. Piskunov, Russ. Chem. Bull., 2024, 73, 117–130 CrossRef CAS.
- R. Maskey, T. Thorwart, S. F. Ebel, A. Jocic, D. Hartmann and L. Greb, Chem. – Eur. J., 2023, 29, e202300269 CrossRef CAS.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS.
- D. Roth, T. Thorwart, C. Douglas and L. Greb, Chem. – Eur. J., 2022, 29, e202203024 CrossRef.
- P. Erdmann, M. Schmitt, L. M. Sigmund, F. Krämer, F. Breher and L. Greb, Angew. Chem., Int. Ed., 2024, 63, e202403356 CrossRef CAS PubMed.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9262–9266 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- M. Quiroz-Guzman and S. N. Brown, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m171–m173 CrossRef CAS.
- A. Vlček Jr., Comments Inorg. Chem., 1994, 16, 207–228 CrossRef.
- V. B. Vol'eva, T. I. Prokof'eva, I. S. Belostotskaya, N. L. Komissarova, D. B. Gorbunov and L. N. Kurkovskaya, Russ. J. Org. Chem., 2011, 47, 1310 CrossRef.
- E. V. Ilyakina, O. G. Mishchenko, A. V. Piskunov, S. V. Maslennikov, I. V. Spirina, Y. A. Kurskii and A. V. Lado, Russ. J. Gen. Chem., 2009, 79, 724–727 CrossRef CAS.
- S. M. Kulikov, I. V. Kozhevnikov, M. N. Fomina and A. P. Krysin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 681–684 CrossRef.
- I. A. Novikova, V. B. Vol'eva, N. L. Komissarova, I. S. Belostotskaya and V. V. Ershov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1981, 30, 1732–1735 CrossRef.
- K. U. Ingold, Can. J. Chem., 1963, 41, 2807–2815 CrossRef CAS.
- J. J. Conradi and G. A. McLaren, J. Am. Chem. Soc., 1960, 82, 4745–4745 CrossRef CAS.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS.
- J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef CAS.
- A. Djurovic, M. Vayer, Z. Li, R. Guillot, J.-P. Baltaze, V. Gandon and C. Bour, Org. Lett., 2019, 21, 8132–8137 CrossRef CAS.
- H. Albright, H. L. Vonesh and C. S. Schindler, Org. Lett., 2020, 22, 3155–3160 CrossRef CAS PubMed.
- A. J. Davis, R. B. Watson, D. J. Nasrallah, J. L. Gomez-Lopez and C. S. Schindler, Nat. Catal., 2020, 3, 787–796 CrossRef CAS.
- R. Wang, Y. Chen, M. Shu, W. Zhao, M. Tao, C. Du, X. Fu, A. Li and Z. Lin, Chem. – Eur. J., 2020, 26, 1941–1946 CrossRef CAS PubMed.
- H. Albright, A. J. Davis, J. L. Gomez-Lopez, H. L. Vonesh, P. K. Quach, T. H. Lambert and C. S. Schindler, Chem. Rev., 2021, 121, 9359–9406 CrossRef CAS.
- S. R. Todtz, C. W. Schneider, T. Malakar, C. Anderson, H. Koska, P. M. Zimmerman and J. J. Devery III, J. Am. Chem. Soc., 2023, 145, 13069–13080 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2402241. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03176h |
| This journal is © The Royal Society of Chemistry 2025 |