
Reactions of chalcogens and borane with phosphazane macrocycles assembled from diethanolamine and P2N2 building blocks†
Manu
Goyal
 ,
Chandrakala
Negi
,
Nitish Kumar
Garg
,
Shalender
Jain
and
Sanjay
Singh
*
,
Chandrakala
Negi
,
Nitish Kumar
Garg
,
Shalender
Jain
and
Sanjay
Singh
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Mohali, Knowledge City, Sector 81, SAS Nagar, Mohali 140306, Punjab, India. E-mail: sanjaysingh@iisermohali.ac.in
First published on 24th January 2025
Abstract
Phosphazanes of the type [ClP(μ-NR)]2 are excellent building blocks for the formation of a range of macrocycles. The condensation reaction of the bifunctional linkers, N-substituted diethanolamine with cyclodiphosphazane, [ClP(μ-NtBu)]2 leads to the formation of dimeric macrocycles, [{P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2; (R = Me (1), Ph (2)). Furthermore, the PIII centres of 1 and 2 were oxidized with chalcogens (O, S, and Se) to afford the corresponding PV macrocycles – [{(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (3), Ph (4)); [{(S)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (5), Ph (6)) and [{(Se)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (7), Ph (8)). An investigation of the Lewis basic behavior of the dimeric macrocycles 1 and 2 was performed by treating them with BH3·SMe2, which resulted in Lewis adduct formation, incorporating a total of six BH3 molecules in the macrocyclic skeleton of 1, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) and four BH3 molecules in the macrocyclic skeleton of 2, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10). Compounds 9 and 10 constitute the first examples of the Lewis adduct of a P2N2 macrocycle with a main group Lewis acid. All the new compounds have been fully characterized using multinuclear NMR, HRMS, and single-crystal X-ray diffraction (compounds 1–3, 8 and 9).
)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (3), Ph (4)); [{(S)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (5), Ph (6)) and [{(Se)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (7), Ph (8)). An investigation of the Lewis basic behavior of the dimeric macrocycles 1 and 2 was performed by treating them with BH3·SMe2, which resulted in Lewis adduct formation, incorporating a total of six BH3 molecules in the macrocyclic skeleton of 1, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) and four BH3 molecules in the macrocyclic skeleton of 2, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10). Compounds 9 and 10 constitute the first examples of the Lewis adduct of a P2N2 macrocycle with a main group Lewis acid. All the new compounds have been fully characterized using multinuclear NMR, HRMS, and single-crystal X-ray diffraction (compounds 1–3, 8 and 9).
Introduction
Over the past century, Lewis acid–base adducts have garnered attention for their potential applications as reagents, hydrogen storage materials, polymer precursors, and in the field of medicine.1,2 While the Lewis base characteristics of smaller molecules, such as amines and phosphines, have been extensively studied,3,4 larger molecules containing phosphorus and nitrogen, particularly those in macrocyclic systems, remain less explored.5 This lack of investigation primarily stems from the challenges associated with developing general synthetic methods for macrocyclization, complicated by the low bond energy of carbon–element covalent bonds. These challenges result in increased kinetic liability, bond polarity, and variable oxidation states, leading to the formation of multiple products.6–11 As a result, these limitations impact the synthesis, reactivity, yield, and purification of macrocyclic systems that involve main-group elements.12In spite of myriad challenges, cyclodiphosphazanes, [R′P(μ-NR)]2, have garnered attention and emerged as a reactive and highly symmetrical synthon with a rigid framework, pre-organized to form stable macrocyclic frameworks [{P(μ-NR)}2(μ-LL′)]n, where LL′ represents an organic bifunctional linker.13–15 Their ability to encapsulate small molecules or ions within their cavities, coupled with the relatively high bond energy of their saturated P–N bonds (comparable to the energy of C–C bonds), positions them favourably (ca. 290 vs. 348 kJ mol−1, respectively).16 Various approaches for ion binding, such as recognition via hydrogen bonding, organometallic ligands, Lewis acid–base interactions17–19 (Fig. 1), and diprotic receptors, have been explored.20,21
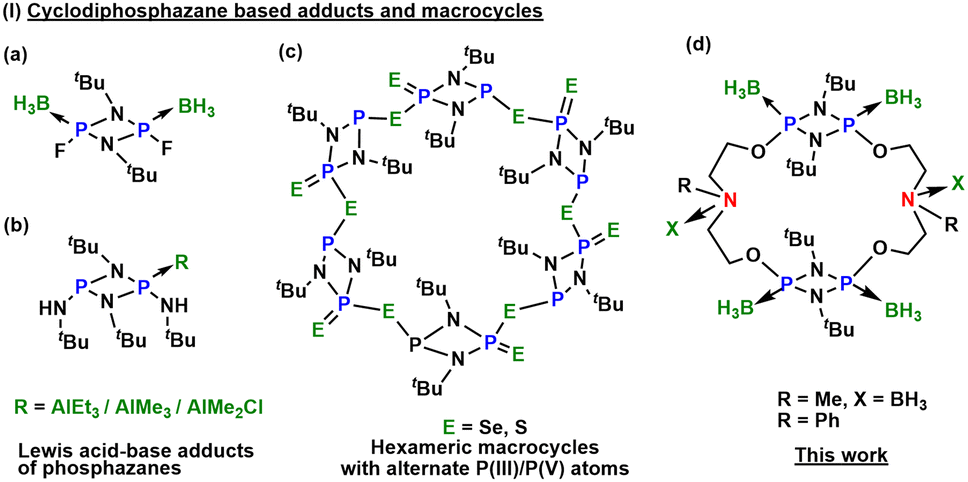 | ||
| Fig. 1 (a and b) Selected examples of cyclodiphosphazane [P(μ-NtBu)]2-based adducts with main group Lewis acids; (c) an example of the heteroleptic PV/PIII macrocycle; and (d) the newly synthesized first main group Lewis acid base adduct with phosphazane macrocycles. | ||
Over the past two decades, Wright and co-workers reported inorganic and hybrid organic–inorganic macrocyclic systems using cyclodiphosphazane.22,23 Additionally, the research groups of Chivers and Garcia have also published [P(μ-NtBu)]2-based PIII/PV macrocycles of different ring sizes. Depending on the steric and electronic demands of the molecules, some of these serve as hosts for anionic and neutral guest molecules.24,25 The research studies of Kumaraswamy and Balakrishna have harnessed the donor properties of phosphorus atoms to isolate various metal-containing macrocycles, homo- or hetero-polynuclear complexes, and coordination polymers.26–28 Our interest in this area stems from the extension of our previous studies, wherein the dimer [ClP(μ-NtBu)]2 was transformed into a PV derivative, serving as a building block to synthesize hexameric macrocyclic frameworks.11,29 In this work, we detail the syntheses of inorganic–organic hybrid dimeric macrocycles, [{P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (1), Ph (2)), by employing dichlorocyclodiphosphazane, [ClP(μ-NtBu)]2, as the inorganic building block with bifunctional organic linkers N-methyldiethanolamine and N-phenyldiethanolamine. The dimeric macrocycles 1 and 2 were found to be air- and moisture-sensitive owing to the presence of reactive PIII centres. It encouraged us to explore the oxidation of these macrocycles with different chalcogens to afford relatively robust PV derivatives 3–8, [{(E)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me, Ph and E = O, S, Se). Compounds 1 and 2 have an electron-rich framework featuring four O, two N, and four P centres possessing lone pairs in the primary macrocyclic backbone. This encouraged us to investigate the Lewis basic characteristics of macrocycles 1 and 2 with a non-metal Lewis acid, BH3·SMe2, affording the first examples of borane coordinated phosphazane macrocycles, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) and [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10).
Results and discussion
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of [ClP(μ-NtBu)]2 with the difunctional linkers N-methyldiethanolamine and N-phenyldiethanolamine in the presence of Et3N afforded compounds 1 and 2 in 83 and 71% yields, respectively (Scheme 1). The dimeric nature of both the macrocycles was indicated by their HRMS spectra in which 1 exhibited a signal at m/z = 643.3548 (calculated 643.3517 [M + H]+) for C26H59N6O4P4, while 2 showed a signal at m/z = 767.3875 (calculated 767.3861 [M + H]+) for C36H63N6O4P4. The 31P{1H} NMR spectra of 1 and 2 showed a signal at δ = 134.4 and 136.6 ppm, respectively, indicating the presence of a symmetrical PIII environment in these compounds (see ESI Fig. S2 and S6†). The 1H NMR spectrum of 1 revealed resonances consistent with the anticipated chemical shifts, including a signal at 2.38 ppm for six protons of two N-Me groups of the linker, while the 1H NMR spectrum of 2 showed signals for ten protons of two N-Ph groups of the linker in between 6.73 and 7.27 ppm. Additionally, two poorly resolved triplets at 2.69 (8H) and 3.98 ppm (8H) ppm in 1 and two triplets at 3.62 (8H) and 4.05 (8H) ppm in 2 were observed corresponding to the –NCH2CH2O– groups of the linkers (see ESI Fig. S1, S3, S5 and S7†). Furthermore, the dimeric nature of the molecules, as anticipated by HRMS observations (see ESI Fig. S4 and S8†), was confirmed by single-crystal X-ray structure analysis vide infra (Fig. 2). Compounds 1 and 2 showed signs of hydrolysis in an open atmosphere. To enhance the stability of 1 and 2 towards air and moisture, we oxidized the PIII centres to PV by using mCPBA (meta-chloroperbenzoic acid) as an oxidizing agent. The 1:4 reaction of macrocycles 1 and 2 with mCPBA at room temperature afforded the corresponding P(O) macrocycles 3 and 4 (Scheme 2). In the 31P{1H} NMR spectra of 3 and 4, the signal at −4.17 ppm for 3 and at −2.72 ppm for 4 (expected range for P(O) moieties) demonstrated the oxidation of all the four P centres of the precursors (see ESI Fig. S10 and S14†). The HRMS spectra also supported oxidation of all four PIII centres and showed a signal at m/z = 707.3318 (calculated 707.3345 [M + H]+) for C26H59N6P4O8 (3) and m/z = 831.3649 (calculated 831.3658 [M + H]+) for C36H63N6P4O8 (4) (see ESI Fig. S12 and S16†). In the 1H NMR spectrum of 3, two equivalents each of THF and the side product, m-chlorobenzoic acid (signals at 7.38, 7.52, 7.93, and 8.04 ppm), were found per equivalent of the macrocycle (see ESI Fig. S9†). The presence of N⋯H–O hydrogen bonds involving m-chlorobenzoic acid and the macrocycle 3 was later confirmed from single crystal X-ray diffraction (Fig. 2, vide infra). Likewise, in the 1H NMR spectrum of 4, the signals observed were in accordance with the expected structure along with the presence of four equivalents of m-chlorobenzoic acid per equivalent of the macrocycle 4 (see ESI Fig. S13†). Oxidation of macrocycles 1 and 2 with N-methyl morpholine N-oxide (NMMO) and triethylamine N-oxide did not proceed.
:1 reaction of [ClP(μ-NtBu)]2 with the difunctional linkers N-methyldiethanolamine and N-phenyldiethanolamine in the presence of Et3N afforded compounds 1 and 2 in 83 and 71% yields, respectively (Scheme 1). The dimeric nature of both the macrocycles was indicated by their HRMS spectra in which 1 exhibited a signal at m/z = 643.3548 (calculated 643.3517 [M + H]+) for C26H59N6O4P4, while 2 showed a signal at m/z = 767.3875 (calculated 767.3861 [M + H]+) for C36H63N6O4P4. The 31P{1H} NMR spectra of 1 and 2 showed a signal at δ = 134.4 and 136.6 ppm, respectively, indicating the presence of a symmetrical PIII environment in these compounds (see ESI Fig. S2 and S6†). The 1H NMR spectrum of 1 revealed resonances consistent with the anticipated chemical shifts, including a signal at 2.38 ppm for six protons of two N-Me groups of the linker, while the 1H NMR spectrum of 2 showed signals for ten protons of two N-Ph groups of the linker in between 6.73 and 7.27 ppm. Additionally, two poorly resolved triplets at 2.69 (8H) and 3.98 ppm (8H) ppm in 1 and two triplets at 3.62 (8H) and 4.05 (8H) ppm in 2 were observed corresponding to the –NCH2CH2O– groups of the linkers (see ESI Fig. S1, S3, S5 and S7†). Furthermore, the dimeric nature of the molecules, as anticipated by HRMS observations (see ESI Fig. S4 and S8†), was confirmed by single-crystal X-ray structure analysis vide infra (Fig. 2). Compounds 1 and 2 showed signs of hydrolysis in an open atmosphere. To enhance the stability of 1 and 2 towards air and moisture, we oxidized the PIII centres to PV by using mCPBA (meta-chloroperbenzoic acid) as an oxidizing agent. The 1:4 reaction of macrocycles 1 and 2 with mCPBA at room temperature afforded the corresponding P(O) macrocycles 3 and 4 (Scheme 2). In the 31P{1H} NMR spectra of 3 and 4, the signal at −4.17 ppm for 3 and at −2.72 ppm for 4 (expected range for P(O) moieties) demonstrated the oxidation of all the four P centres of the precursors (see ESI Fig. S10 and S14†). The HRMS spectra also supported oxidation of all four PIII centres and showed a signal at m/z = 707.3318 (calculated 707.3345 [M + H]+) for C26H59N6P4O8 (3) and m/z = 831.3649 (calculated 831.3658 [M + H]+) for C36H63N6P4O8 (4) (see ESI Fig. S12 and S16†). In the 1H NMR spectrum of 3, two equivalents each of THF and the side product, m-chlorobenzoic acid (signals at 7.38, 7.52, 7.93, and 8.04 ppm), were found per equivalent of the macrocycle (see ESI Fig. S9†). The presence of N⋯H–O hydrogen bonds involving m-chlorobenzoic acid and the macrocycle 3 was later confirmed from single crystal X-ray diffraction (Fig. 2, vide infra). Likewise, in the 1H NMR spectrum of 4, the signals observed were in accordance with the expected structure along with the presence of four equivalents of m-chlorobenzoic acid per equivalent of the macrocycle 4 (see ESI Fig. S13†). Oxidation of macrocycles 1 and 2 with N-methyl morpholine N-oxide (NMMO) and triethylamine N-oxide did not proceed.
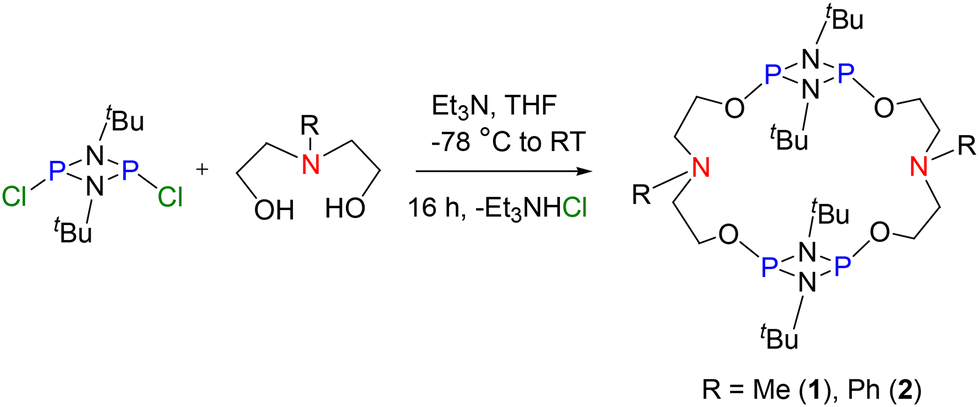 | ||
| Scheme 1 Synthesis of macrocycles – [{P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (1) and [{P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (2). | ||
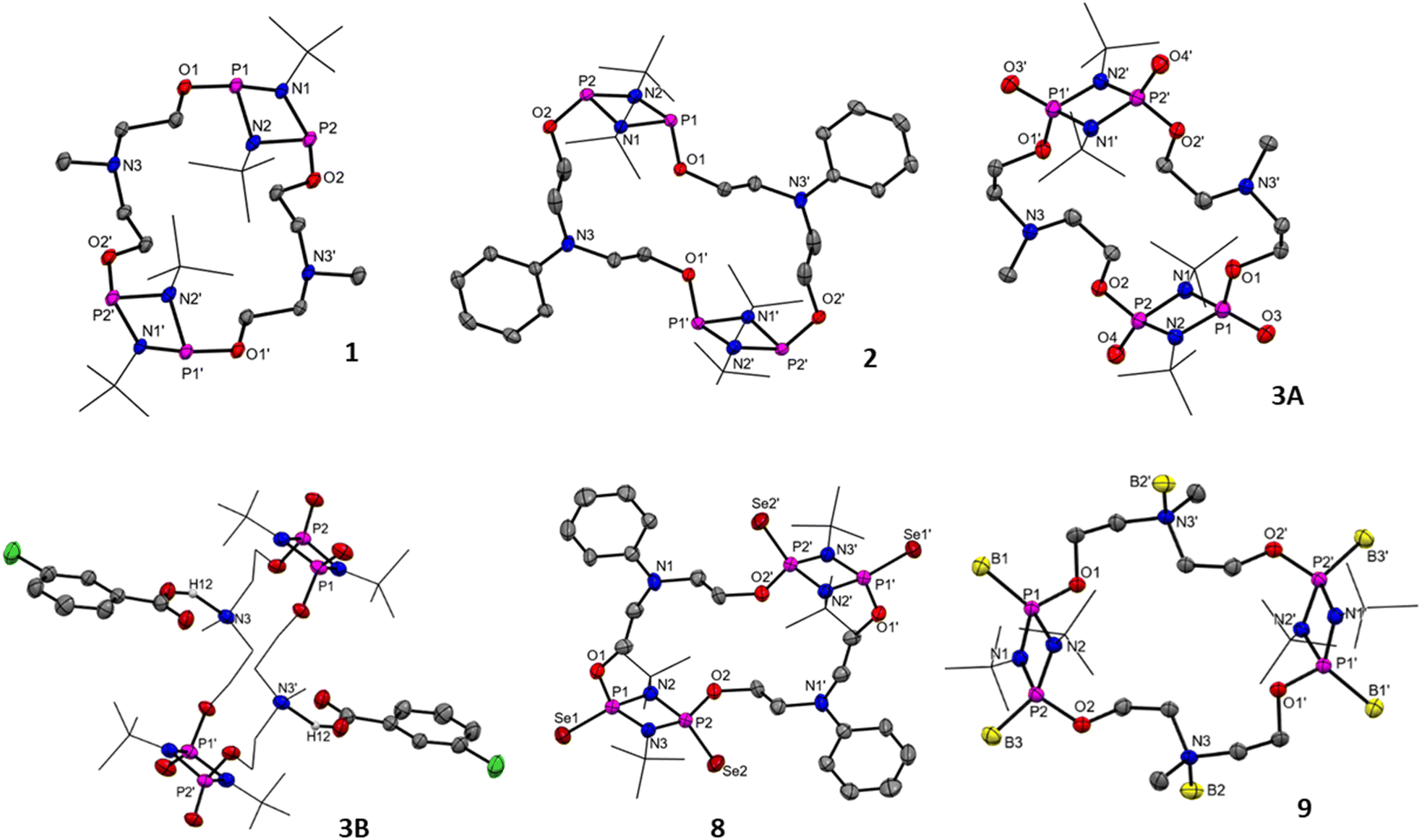 | ||
| Fig. 2 Single crystal X-ray structure of macrocycles 1, 2, 3·(THF)2(m-ClC7H5O2)2, 8·(C6D6)2, and 9·(DCM)2. Thermal ellipsoids are shown at a 50% probability level. All hydrogen atoms, co-crystallized solvents (for 3, 8 and 9) and m-chlorobenzoic acid (in 3A) have been omitted for clarity. Selected bond lengths [Å] and bond angles [°] for 1: P1–O1 1.631(3), P2–O2 1.624(3), P1–N1 1.733(4), P1–N2 1.6724(3), P2–N1 1.730(3), P2–N2 1.731(4); P2–N1–P1 96.29(18), P1–N2–P2 96.59(18), O1–P1–P2 125.20(11), O1–P1–N1 107.13(16), O1–P1–N2 107.29(16); 2: P1–O1 1.646(3), P2–O2 1.618(3), P1–N1 1.705(4), P1–N2 1.701(4), P2–N2 1.717(4); O1–P1–P2 114.48(11), O1–P1–N1 104.23(17), O1–P1–N2 103.34(18), N2–P1–N1 81.20(18); 3: P1–O3 1.466(3), P1–O1 1.587(3), P1–N1 1.674(3), P2–O2 1.581(3), P2–O4 1.464(3); O3–P1–O1 112.60(15), O3–P1–N1 119.86(16), O1–P1–N1 108.56(15); 8: Se2–P2 2.0746(13), Se1–P1 2.0707(13), P2–O2 1.589(3), P2–N3 1.682(4), P1–O1 1.593(3); Se2–P2–O2 113.8(13), Se1–P1–O1 108.66(13), O2–P2–N3 107.36(18), N3–P2–Se2 120.42(14); and 9: P2–O2 1.596(3), P2–N2 1.675(3), P2–N1 1.677(3), P2–B3 1.896(5), P1–B1 1.881(5), P1–O1 1.584(3); O2–P2–N2 106.64(15), O2–P2–N1 107.07(16), O2–P2–B3 113.6(2), O1–P1–B1 107.4(2), N2–P2–B3 121.2(2), N2–P2–N1 84.14(17). | ||
 | ||
| Scheme 2 Oxidation of PIII centres to PV centres and syntheses of macrocycles – [{(E)P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me; E = O (3), S (5), Se (7)) and (R = Ph; E = O (4), S (6), Se (8)). | ||
Subsequently, we also attempted to oxidize the macrocycles with heavier chalcogens (elemental S and Se). The 1:4, neat reaction of compounds 1 and 2 with S and Se under heating or in toluene under reflux conditions afforded the expected oxidation of the dimeric macrocycle with the P(S) and P(Se) groups, respectively. The HRMS spectrum at m/z = 770.2319 (calculated 770.2353 [M]+) for C26H58N6O4P4S4 and 895.2734 (calculated 895.2744 [M + H]+) for C36H63N6O4P4S4 showed the formation of oxidized products [{(S)P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (5) and [{(S)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (6), respectively, while at m/z = 961.0259 (calculated 961.0230 [M + H]+) for C26H59N6O4P4Se4 and m/z = 1085.0408 (calculated 1085.0547 [M + H]+) for C36H63N6O4P4Se4 also supported the formation of products [{(Se)P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (7) and [{(Se)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (8), respectively (see ESI Fig. S20, S24, S28 and S32†). In the 31P{1H} NMR spectrum, the absence of a signal in the PIII region and the presence of a new signal at 17.5 ppm for 5 and 16.6 ppm for 6 also indicated the oxidation of all four PIII centres to (S)PV (see ESI Fig. S18 and S22†). Furthermore, the changes in the chemical shift values in the signals in the 1H NMR spectra of 5 and 6 compared to the parent molecules were attributed to the formation of the expected products (see ESI Fig. S17 and S21†). The NMR characterization of Se-containing macrocycles 7 and 8 also showed a shift in the position of signals, indicating the formation of the expected products. The 31P{1H} NMR spectrum of 7 showed a signal at 49.3 ppm containing the characteristic 77Se satellites (1J31P,77Se = 975 Hz); similarly, 8 showed a signal at 46.5 ppm (77Se satellites with 1J31P,77Se = 954 Hz) and confirmed the oxidation of all four PIII centres to PV by Se (see ESI Fig. S26 and S30†). The 1H and 13C{1H} NMR spectra of 7 and 8 confirmed the macrocycle structure of the product to be retained upon oxidation of PIII centres. Further, the oxidation of all PIII centres with Se was confirmed with the single crystal X-ray structure of [{SeP(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (8) (Fig. 2 and ESI Table S1†). Multiple attempts to crystalize compounds 4–7 were unsuccessful.
Towards exploring the reaction chemistry or Lewis basic behavior of P and N centres in macrocycles 1 and 2, we performed reaction with BH3·SMe2. Interestingly, the reaction of macrocycles 1 and 2 with six equivalents of BH3·SMe2 resulted in the coordination of six and four BH3 units in the respective products 9 and 10 (Scheme 3). The HRMS spectrum showed a signal at m/z = 712.65217 (calcd 712.5233 [M − BH2]+) for C26H74N6O4P4B5 corresponding to the macrocycle with six BH3 units, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) (see ESI Fig. S37†). Complex 9 was stable in an open atmosphere for a few hours but sensitive to moisture. In the 31P{1H} NMR spectrum, the presence of a sole signal at 110.2 ppm confirmed a symmetrical structure and BH3 coordination to all the four P centres (see ESI Fig. S34†). Similarly, the reaction of BH3·SMe2 with macrocycle 2 afforded a white compound characterized as [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10), which showed a broad signal in the 31P{1H} NMR spectrum at 109.2 ppm (see ESI Fig. S39†). The 11B NMR spectrum of 9 showed two broad signals at −11.4 ppm (for N-BH3) and −38.6 ppm (for P-BH3) (see ESI Fig. S35†). In contrast, the 11B NMR spectrum of 10 showed only one signal at −37.7 ppm (for P-BH3 units). The BH3 coordination to the N-Ph centres was not observed (see ESI Fig. S40†). The HRMS spectrum of 10 showed a signal at m/z = 823.5162 (calcd 823.5195 [M + H]+) for C36H75N6O4P4B4, also confirming the coordination of only four BH3 units. The unambiguous structure of 9 was elucidated by the single-crystal X-ray diffraction technique (Fig. 2).
 | ||
| Scheme 3 Synthesis of [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) and [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10). | ||
Solid state structure of macrocycles 1–3, 8 and 9
The solid-state structures of compounds 1–3, 8, and 9 were elucidated by the single crystal X-ray diffraction method (Fig. 2 and Table S1, ESI†). Single crystals of 1 suitable for X-ray diffraction were obtained from the C6D6 solution as colorless needles and of 2 from the THF solution as colorless blocks. Complex 1 crystallized in the monoclinic crystal system with a P21/c space group and 2 crystallized in the triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The solid-state structure of [{P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (1) confirmed the dimeric nature of the macrocycle containing 20 atoms and C8P4N4O4 in the inner core arranged in a centrosymmetric manner. The separation between transannular phosphorus atoms P1⋯P1′ in 1 was 9.86 Å, while the same in 2 was found to be 10.02 Å, indicating large macrocyclic cavities in 1 and 2. These P1⋯P1′ separations along with the separation between the N-atom of two diethanolamine moieties N3⋯N3′ (6.17 Å in 1 and 6.96 Å in 2) make these cavities somewhat elliptical in shape. Compound 3 crystallized in the triclinic system with the P space group. The single crystal X-ray structure of 3 confirmed the macrocyclic structure with an elliptical cavity of size 8.730 Å × 6.257 Å measured across the transannular P1⋯P1′ and N3⋯N3′, respectively (Fig. 2, 3A). It is interesting to note that oxidation of 1 into 3 led to a decrease in the cavity size. The co-crystallization of two molecules – each of THF and m-chlorobenzoic acid per molecule of 3 (also observed in the 1H and 13C{1H} NMR spectra of 3) – was confirmed by SCXRD analysis for 3. The N-center of the N-methyldiethanolamine linker was involved in N⋯H–O hydrogen bonds with m-chlorobenzoic acid with the N⋯H distance of 1.817 Å (Fig. 2, 3B). Compound 8 crystallized as colorless block-shaped crystals from a solution in C6D6. Compound 8 crystallized in the monoclinic system with a P21/c space group. All four P-centres of 8 were oxidized with Se, attending the pentavalent state. Interestingly, the cavity size of 10.02 Å × 6.96 Å for 8 measured across the transannular P1⋯P1′ and N1⋯N1′ centres remained unchanged compared to its precursor 2. The borane coordinated macrocycle 9 crystallized in the monoclinic system with a P21/n space group. In the solid-state structure of 9, all four P-centres, as well as both N-centres of the linker, were coordinated with the BH3 molecules with the P–B bond length as 1.895(1) Å and the N–B distance as 1.637(1) Å. Due to this Lewis acid–base coordination, the P1⋯P1′ distance (8.702 Å) was shortened, and the N3⋯N3′ distance (7.103 Å) was elongated compared to the parent macrocycle 1, vide supra.
space group. The solid-state structure of [{P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (1) confirmed the dimeric nature of the macrocycle containing 20 atoms and C8P4N4O4 in the inner core arranged in a centrosymmetric manner. The separation between transannular phosphorus atoms P1⋯P1′ in 1 was 9.86 Å, while the same in 2 was found to be 10.02 Å, indicating large macrocyclic cavities in 1 and 2. These P1⋯P1′ separations along with the separation between the N-atom of two diethanolamine moieties N3⋯N3′ (6.17 Å in 1 and 6.96 Å in 2) make these cavities somewhat elliptical in shape. Compound 3 crystallized in the triclinic system with the P space group. The single crystal X-ray structure of 3 confirmed the macrocyclic structure with an elliptical cavity of size 8.730 Å × 6.257 Å measured across the transannular P1⋯P1′ and N3⋯N3′, respectively (Fig. 2, 3A). It is interesting to note that oxidation of 1 into 3 led to a decrease in the cavity size. The co-crystallization of two molecules – each of THF and m-chlorobenzoic acid per molecule of 3 (also observed in the 1H and 13C{1H} NMR spectra of 3) – was confirmed by SCXRD analysis for 3. The N-center of the N-methyldiethanolamine linker was involved in N⋯H–O hydrogen bonds with m-chlorobenzoic acid with the N⋯H distance of 1.817 Å (Fig. 2, 3B). Compound 8 crystallized as colorless block-shaped crystals from a solution in C6D6. Compound 8 crystallized in the monoclinic system with a P21/c space group. All four P-centres of 8 were oxidized with Se, attending the pentavalent state. Interestingly, the cavity size of 10.02 Å × 6.96 Å for 8 measured across the transannular P1⋯P1′ and N1⋯N1′ centres remained unchanged compared to its precursor 2. The borane coordinated macrocycle 9 crystallized in the monoclinic system with a P21/n space group. In the solid-state structure of 9, all four P-centres, as well as both N-centres of the linker, were coordinated with the BH3 molecules with the P–B bond length as 1.895(1) Å and the N–B distance as 1.637(1) Å. Due to this Lewis acid–base coordination, the P1⋯P1′ distance (8.702 Å) was shortened, and the N3⋯N3′ distance (7.103 Å) was elongated compared to the parent macrocycle 1, vide supra.
Conclusions
In conclusion, we have reported the first examples of the Lewis base interaction of phosphazane-based macrocycles with a main-group Lewis acid. The dimeric macrocycles, [{P(μ-NtBu)}2{O(CH2)2N(R)(CH2)2O}]2 (R = Me (1) and Ph (2)), having PIII centres were assembled by condensation with the elimination of HCl. Furthermore, the PIII centres in 1 and 2 were oxidized to a comparatively more stable PV with chalcogens (O, S, and Se) and afforded macrocycles 3–8. The Lewis basic behavior of the dimeric macrocycle has been studied by treating it with BH3·SMe2, which resulted in the formation of Lewis adducts, [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9) and [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10).Experimental
General
All manipulations were performed under a nitrogen/argon atmosphere using a Schlenk line or glove box techniques. All chemicals were purchased from Sigma-Aldrich and used without further purification. [ClP(μ-NtBu)]2 was prepared as per the reported procedure.23 The 1H, 13C{1H}, and 31P{1H} NMR spectra were recorded by using a Bruker 400 MHz spectrometer; chemical shift values are reported in ppm. High-resolution mass spectrometry was performed using a Waters SYNAPT G2-S. Melting points were obtained in sealed capillaries using a Büchi B-540 melting point instrument.Synthesis of [{P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (1)
A solution of [ClP(μ-NtBu)]2 (1.0 g, 3.6 mmol) in THF (30 mL) was added dropwise at −78 °C to a stirred solution of N-methyldiethanolamine (0.43 g, 3.6 mmol) and Et3N (2.0 mL, excess) in THF (30 mL) over 30 min. The reaction mixture was allowed to warm to room temperature and further stirred for 12 h. The volatiles were removed under vacuum, and the residue was extracted in hexane (60 mL). The volume of the resulting colorless solution was reduced under vacuum (∼20 mL) and kept at −10 °C, which afforded colorless crystals suitable for single-crystal X-ray diffraction. Yield: 0.96 g (83%). Mp: 127 °C. 1H NMR (400 MHz, CDCl3): δ = 1.28 (s, 36H, tBu), 2.38 (s, 6H, –NCH3), 2.69 (t, 3JH–H = 8 Hz, 8H, NCH2), 3.98 (broad t, 8H, OCH2) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 134.3 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 31.1, 44.2, 51.1, 57.9, 59.9 ppm. HRMS (AP+): m/z calculated for C26H59N6O4P4: (643.3548) [M + H]+; found: 643.3517.Synthesis of [{P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (2)
The procedure described above for 1 was followed to prepare 2 by taking [ClP(μ-NtBu)]2 (1.0 g, 3.6 mmol) in THF (30 mL) and N-phenyldiethanolamine (0.66 g, 3.6 mmol) and Et3N (2.0 mL, excess) in THF (30 mL). Crystals suitable for single-crystal X-ray diffraction of 2 were grown from a THF solution at −20 °C. Yield: 1.0 g (71%). Mp: 135 °C. 1H NMR (400 MHz, CDCl3): δ = 1.36 (s, 36H, tBu), 3.64 (t, 3JH–H = 8 Hz, 8H, NCH2), 4.05 (t, 8H, OCH2), 6.73–6.80 (closely placed d and t, 6H, –PhN), 7.27 (t, 4H, –PhN). 31P{1H} NMR (162 MHz, CDCl3): δ = 136.6 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 25.6, 31.1, 51.1, 58.4, 111.3, 116.3, 129.5 and 147.3 ppm. 19.5 ppm. HRMS (ES+): m/z calculated for C36H63N6O4P4: (767.3861) [M + H]+; found: 767.3875.Synthesis of [{(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) )P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (3)
)P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (3)
A solution of 1 (0.64 g, 1.0 mmol) in THF (20 mL) was added to a solution of mCPBA (0.70 g, 4.0 mmol) in 20 mL of THF at −78 °C, and then stirred at room temperature for 10 h. The solution was concentrated and kept for crystallization at 4 °C, which afforded colorless crystals of 3 suitable for single-crystal X-ray diffraction. Compound 3 was found to be a white solid. Yield: 1.29 g (96%; including m-chlorobenzoic acid). Mp: 92–94 °C. 1H NMR (400 MHz, CDCl3): δ = 1.43 (s, 36H, tBu), 2.44 (s, 6H, –NCH3), 2.87 (broad t, 8H, NCH2), 4.25 (broad t, 8H, OCH2) (residual THF signals were found at δ = 1.8 & 3.7 ppm and m-chlorobenzoic acid (two molecules) at δ = 7.38 (t, 3JH–H = 8 Hz, 2H), 7.52 (d, 2H, 3JH–H = 8 Hz), 7.93 (d, 3JH–H = 8 Hz, 2H), 8.04 (s, 2H) ppm). 31P{1H} NMR (162 MHz, CDCl3): δ = −4.2 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 30.2, 43.3, 55.1, 57.1 and 66.0 ppm (THF signals at δ = 25.6, 68.0 ppm and m-chlorobenzoic acid signals at δ = 128.0, 129.7, 130.0, 132.2, 133.1, 134.4, 168.7 ppm). HRMS (ES+): m/z calculated for C26H59N6O8P4: (707.3345) [M + H]+; found: 707.3318.
Synthesis of [{(O)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (4)
The procedure described above for 3 was followed to synthesize 4 by using a solution of 2 (0.77 g, 1.0 mmol) in THF (20 mL) and a solution of mCPBA (0.70 g, 4.0 mmol) in THF (20 mL). Yield: 1.41 g (95%) as a white solid including m-chlorobenzoic acid. Mp: 104 °C. 1H NMR (400 MHz, CDCl3): 1.44 (s, 36H, tBu), 3.80 (t, 3JH–H = 8 Hz, 8H, NCH2), 4.33 (t, 8H, OCH2), 6.79–6.81 (overlapped d and t, 6H, –PhN), 7.28 (t, 4H, –PhN) [m-chlorobenzoic acid (four molecules) at δ = 7.43 (t, 3JH–H = 8 Hz, 4H), 7.57 (d, 3JH–H = 8 Hz, 2H), 7.99 (d, 3JH–H = 8 Hz, 2H), 8.09 (s, 4H)]. 31P{1H} NMR (162 MHz, CDCl3): δ = −2.7 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 30.3, 52.4, 55.1, 65.3, 112.0, 117.8, 129.7 and 146.5 ppm. (m-chlorobenzoic acid signals at δ = 128.2, 130.1, 131.5, 133.5, 134.5, and 168.7 ppm.) HRMS (ES+): m/z calculated for C36H63N6O8P4: (831.3658) [M + H]+; found: 831.3649.
Synthesis of [{(S)P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (5)
A solution of 1 (0.64 g, 1.0 mmol) and sulfur (0.13 g, 4.2 mmol) in toluene was refluxed for 24 h and the reaction progress was monitored by in situ31P{1H} NMR spectroscopy. After completion of the reaction (∼24 h), all volatiles were removed under vacuum, and the product was washed with hexane. Yield: 0.74 g (95%). Mp: 186 °C. 1H NMR (400 MHz, C6D6): δ = 1.52 (s, 36H, tBu), 2.36 (s, 6H, –NCH3), 2.82 (8H, NCH2), 3.05 (8H, OCH2) ppm. 31P{1H} NMR (162 MHz, C6D6): δ = 17.5 ppm. 13C{1H} NMR (100 MHz, C6D6) δ = 21.4, 30.7, 41.3, 57.0 and 57.3 ppm. HRMS (AP+): m/z calculated for C26H58N6O4P4S4: (770.2353) [M]+; found: 770.2319.
Synthesis of [{(S)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (6)
The procedure described above for 5 was followed to synthesize 6, by using a solution of 2 (0.77 g, 1.0 mmol) and sulfur (0.13 g, 4.2 mmol) in toluene. Yield: 0.82 g (91%). Mp: 192 °C. 1H NMR (400 MHz, CDCl3): δ = 1.51 (s, 36H, tBu), 3.10 (t, 3JH–H = 8 Hz, 8H, NCH2), 3.73 (t, 8H, OCH2), 6.77–6.87 (closely placed d and t, 6H, –PhN), 7.29 (t, 4H, –PhN) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 16.6 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 30.5, 51.7, 57.2, 112.8, 117.9, 129.7 and 146.0 ppm. HRMS (ES+): m/z calculated for C36H63N6O4P4S4: (895.2744) [M + H]+; found: 895.2733.
Synthesis of [{(Se)P(μ-NtBu)}2{O(CH2)2N(Me)(CH2)2O}]2 (7)
A mixture of 1 (0.64 g, 1.0 mmol) and selenium (0.33 g, 4.2 mmol) in toluene was refluxed for 24 h and the reaction progress was monitored by in situ31P{1H} NMR spectroscopy. After completion of the reaction (∼24 h), all volatiles were removed under vacuum and the product was washed with hexane. Yield: 0.93 g (97%). Mp: 191 °C. 1H NMR (400 MHz, C6D6): δ = 1.51 (s, 36H, tBu), 2.04 (s, 6H, –NCH3), 2.38 (t, 3JH–H = 8 Hz, 8H, –NCH2), 4.01 (broad t, 8H, OCH2). 31P{1H} NMR: (162 MHz, C6D6): δ = 49.3 ppm (s with a satellite doublet due to [(77Se)P(μ-NtBu)2P(Se)], 1J31P,77Se = 975 Hz). 13C{1H} NMR (100 MHz, C6D6): δ = 29.4, 42.6, 55.9, 57.1 and 66.7 ppm. HRMS (ES+): m/z calculated for C26H59N6O4P4Se4: (961.0230) [M + H]+; found: 961.0259.
Synthesis of [{(Se)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (8)
The procedure described above for 7 was followed to synthesize 8, by using a solution of 2 (0.77 g, 1.0 mmol) and selenium (0.33 g, 4.2 mmol) in toluene. The crystals suitable for single-crystal X-ray diffraction analysis were obtained from a deuterated benzene solution of 8 at room temperature. Yield: 1.04 g (96%). Mp: 198 °C. 1H NMR (400 MHz, CDCl3): δ = 1.53 (s, 36H, tBu), 3.37 (t, 3JH–H = 8 Hz, 8H, NCH2), 4.20 (t, 8H, OCH2), 6.76–6.80 (overlapped d and t, 6H, –PhN), 7.26 (t, 4H, –PhN) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 46.5 ppm (s with a satellite doublet due to [(77Se)P(μ-NtBu)2P(Se)], 1J31P,77Se = 954 Hz). 13C{1H} NMR (100 MHz, CDCl3): δ = 30.0, 51.2, 57.1, 66.5, 112.3, 117.8, 129.8 and 146.6 ppm. HRMS (ES+): m/z calculated for C36H63N6O4P4Se4: (1085.0547) [M + H]+; found: 1085.0408.
Synthesis of [{(BH3)P(μ-NtBu)}2{O(CH2)2N(BH3)(Me)(CH2)2O}]2 (9)
To a solution of 1 (0.65 g, 1.0 mmol) in toluene, BH3·SMe2 (0.6 mL, 6.2 mmol) was added at −70 °C, followed by gradually increasing the temperature, and formation of a white precipitate was observed. The reaction mixture was stirred at room temperature for 2 h. All volatiles were removed under vacuum and the residue was washed with hexane to afford 9. Suitable crystals of 9 for single-crystal X-ray diffraction analysis were obtained from a solution of DCM and few drops of THF; the crystallized compound contained an equimolar amount of DCM. Yield: 0.71 g (93%). Mp: 187 °C. 1H NMR (400 MHz, CDCl3): δ = 1.49 (s, broad, 54H, tBu and BH3), 2.77 (s, 6H, –NCH3), 2.97 (4H, –NCH2), 3.25 (4H, –NCH2), 4.53 (8H, OCH2) ppm and at 5.32 ppm (s, one molecule of DCM). 31P{1H} NMR (162 MHz, CDCl3): δ = 110.2 ppm. 11B NMR (128.4 MHz, CDCl3): δ = −11.4 ppm and −38.6 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 30.4, 53.2, 55.7, 62.3 and 63.1 ppm. HRMS (ES+): m/z calculated for C26H74N6O4P4B5: (712.5233) [M − BH2]+; found: 712.6521.Synthesis of [{(BH3)P(μ-NtBu)}2{O(CH2)2N(Ph)(CH2)2O}]2 (10)
The procedure described above for 9 was followed to synthesize 10, by using a solution of 2 (0.77 g, 1.0 mmol) in toluene and BH3·SMe2 (0.4 mL, 4.2 mmol). Yield: 0.77 g (94%). Mp: 163 °C. 1H NMR (400 MHz, CDCl3): δ = 1.38 (s, broad, 48H, tBu and BH3), 3.57 (t, 3JH–H = 8 Hz, 8H, NCH2), 4.08 (t, 8H, OCH2), 6.64–6.74 (closely placed d and t, 6H, –PhN), 7.19 (t, 4H, –PhN) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 109.2 ppm. 11B NMR (128.4 MHz, CDCl3): δ = −37.7 ppm. 13C{1H} NMR (100 MHz, CDCl3): δ = 30.0, 51.6, 55.5, 63.3, 112.48, 118.4, 129.7 and 146.4 ppm. HRMS (ES+): m/z calculated for C36H75N6O4P4B4: (823.5195) [M + H]+; found: 823.5162.Data availability
All data included and leading to conclusions presented in this manuscript are included within the manuscript and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the Department of Science and Technology (DST), New Delhi [DST/SERB (EMR/2016/003911)]. We are grateful to IISER Mohali for central NMR and HRMS facilities and the Department of Chemical Sciences for the X-ray diffraction facility. Useful discussions and help rendered by Dr. Angshuman Roy Choudhury are gratefully acknowledged for the X-ray structure of compounds reported in the paper. The DST-FIST 400 MHz NMR facility, SR/FST/CS-II/2019/94 (C) (TPN No. 32545), is also gratefully acknowledged.References
- K. Takrouri, V. M. Dembistky and M. Srebnik, Studies Inorg. Chem., 2005, 22, 495–549 CrossRef CAS
.
- A. Jabbour, R. Smoum, K. Takrouri, E. Shalom, B. Zaks, D. Steinberg, A. Rubinstein, I. Goldberg, J. Katzhendler and M. Srebnik, Pure Appl. Chem., 2006, 78, 1425–1453 CrossRef CAS
- B. Carboni and L. Monnier, Tetrahedron, 1999, 55, 1197–1248 CrossRef CAS
- A. Staubitz, A. P. M. Robertson, M. E. Solan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed
- K. Worm, F. P. Schmidtchen, A. Schier, A. Schafer and M. Hesse, Angew. Chem., Int. Ed. Engl., 1994, 33, 327–329 CrossRef
- J. S. Ritch and T. Chivers, Angew. Chem., Int. Ed., 2007, 46, 4610–4613 CrossRef CAS PubMed
- R. E. Mulvey, Chem. Commun., 2001, 1049–1056 RSC
- W. Clegg, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and D. M. Tooke, Angew. Chem., Int. Ed., 2001, 40, 3902–3905 CrossRef CAS PubMed
- S. Gonzàlez-Calera, D. J. Eisler, J. M. Goodman, M. McPartlin, S. Singh and D. S. Wright, Dalton Trans., 2009, 1293–1296 RSC
- S. Gonzàlez-Calera, D. J. Eisler, J. V. Morey, M. McPartlin, S. Singh and D. S. Wright, Angew. Chem. Int. Ed., 2008, 47, 1111–1114 CrossRef PubMed
- D. Bawari, B. Prashanth, S. Ravi, K. R. Shamasundar, S. Singh and D. S. Wright, Chem. – Eur. J., 2016, 22, 12027–12033 CrossRef CAS PubMed
-
(a) A. J. Plajer, R. Garcia-Rodriguez, C. G. M. Benson, P. D. Matthews, A. D. Bond, S. Singh, L. H. Gade and D. S. Wright, Angew. Chem. Int. Ed., 2017, 56, 9087–9090 CrossRef CAS PubMed
-
(a) F. Dodds, F. Garcia, R. A. Kowenicki, M. McPartlin, A. Steiner and D. S. Wright, Chem. Commun., 2005, 3733–3735 RSC
-
(a) E. L. Doyle, F. Garcia, S. M. Humphrey, R. A. Kowenicki, L. Riera, A. D. Woods and D. S. Wright, Dalton Trans., 2004, 4, 807–812 RSC
-
(a) V. S. Kashid, J. T. Mague and M. S. Balakrishna, J. Chem. Sci., 2017, 129, 1531–1537 CrossRef CAS
- H. C. Niu, A. J. Plajer, R. Garcia-Rodriguez, S. Singh and D. S. Wright, Chem. – Eur. J., 2018, 24, 3073–3082 CrossRef CAS PubMed
- M. Rastatter and P. W. Roesky, Eur. J. Inorg. Chem., 2008, 5287–5291 CrossRef
- M. Rastatter, P. W. Roesky, D. Gudat, G. B. Deacon and P. C. Junk, Chem. – Eur. J., 2007, 13, 7410–7415 CrossRef PubMed
- J. S. Jessup, R. T. Paine and C. F. Campana, Phosphorus Sulfur Relat. Elem., 1981, 9, 279–284 CrossRef CAS
- M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650–664 RSC
- A. Bashall, A. D. Bond, E. L. Doyle, F. Garcia, S. Kidd, G. T. Lawson, M. C. Parry, M. McPartlin, A. D. Woods and D. S. Wright, Chem. – Eur. J., 2002, 8, 3377–3385 CrossRef CAS PubMed
- A. J. Plajer, H. C. Niu, F. J. Rizzuto and D. S. Wright, Dalton Trans., 2018, 47, 6675–6678 RSC
- A. Bashall, E. L. Doyle, C. Tubb, S. J. Kidd, M. McPartlin, A. D. Woods and D. S. Wright, Chem. Commun., 2001, 1, 2542–2543 RSC
- A. Nordheider, K. Hull, K. S. Athukorala Arachchige, A. M. Z. Slawin, J. D. Woollins, R. Thirumoorthi and T. Chivers, Dalton Trans., 2015, 44, 5338–5346 RSC
- Y. X. Shi, R. Z. Liang, K. A. Martin, D. G. Star, J. Diaz, X. Y. Li, R. Ganguly and F. Garcia, Chem. Commun., 2015, 51, 16468–16471 RSC
-
(a) P. Kommana and K. C. Kumara Swamy, Inorg. Chem., 2000, 39, 4384–4385 CrossRef CAS
-
(a) M. K. Pandey, S. Sheokand and M. S. Balakrishna, Dalton Trans., 2023, 52, 6420–6425 RSC
- G. S. Ananthnag, S. Kuntavalli, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2012, 51, 5919–5930 CrossRef CAS PubMed
- D. Bawari, B. Prashanth, K. Jaiswal, A. R. Choudhary and S. Singh, Eur. J. Inorg. Chem., 2017, 4123–4130 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, HRMS and X-ray diffraction data. CCDC 2403424–2403428. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03229b |
| This journal is © The Royal Society of Chemistry 2025 |