
Achieving advanced hydrogen evolution under large current density using an amorphous/crystalline core–shell electrocatalyst of a-NiCoP/Co2P†
Xiaodong
Chen
a,
Zhi
Cheng
a,
Jiao
Li
a,
Hongyu
Chen
b,
Siyuan
Liu
 a,
Shuxian
Wei
b,
Zhaojie
Wang
*a and
Xiaoqing
Lu
*a
a,
Shuxian
Wei
b,
Zhaojie
Wang
*a and
Xiaoqing
Lu
*a
aSchool of Materials Science and Engineering, China University of Petroleum, Qingdao, 266580, PR China. E-mail: wangzhaojie@upc.edu.cn; luxq@upc.edu.cn
bCollege of Science, China University of Petroleum, Qingdao, 266580, PR China
First published on 10th January 2025
Abstract
Non-precious transition metal-based electrocatalysts with high activities are promising candidates for substituting Pt- or Ru-based electrocatalysts in hydrogen evolution. In this study, we propose core–shell engineering to combine the amorphous NiCoP and crystalline Co2P (a-NiCoP/Co2P@NF), which requires an ultra-low overpotential of only 26 mV to achieve the benchmark current density of 10 mA cm−2. Furthermore, it achieves an industrial-level hydrogen evolution current density of 500 mA cm−2 with excellent stability. The superior catalytic performance and stability can be attributed to the hierarchical amorphous/crystalline interface and the electron-rich interfacial Co sites. The amorphous NiCoP shell can not only protect the internal Co2P from corrosion, but also provide a larger electrochemically active area. Together, the Co2P core provides fast electron transport and promotes H2 emission from the interfacial electron-rich Co sites. This work provides inspiration to the rational design of an advanced core–shell structure between amorphous and crystalline states.
1. Introduction
Due to the excessive exploitation and use of fossil fuels, energy and environmental issues are becoming more and more severe.1–4 Hydrogen, as a green and renewable energy carrier, has great potential in the near future.5,6 Among all the methods to produce hydrogen, water electrolysis driven by renewable energy represents the most promising technology.7,8 However, the feasibility of the hydrogen evolution reaction (HER) still highly depends on efficient and stable electrocatalysts. Although platinum (Pt) group metals have been the most active HER catalysts until now, their high cost and scarcity hinder their large-scale industrial application.9–13 Recently, numerous transition metal phosphides,14,15 carbides,16 and selenides17 with Pt-like properties have been developed. Among them, transition metal phosphides (TMPs) such as nickel phosphides, cobalt phosphides, and iron phosphides have been proposed as new HER electrocatalysts. For example, phosphorus species in cobalt phosphides can grab electrons from cobalt atoms and exhibit affinity to protons during the HER. However, these electrocatalysts with different morphologies, e.g. nanoparticles or nanoarrays, tend to collapse or aggregate at high current density and in long-term tests for the HER, resulting in poor stability and low activity irrespective of the pH value.18–23Interfacial engineering, such as coating a protective shell with rich active sites and good conductivity, is an effective measure to improve the HER performance.24,25 Amorphous electrocatalysts, particularly amorphous metal phosphide electrocatalysts, have attracted the attention of researchers with their structures of long-range disordering with short/medium-range ordering. In other words, amorphous structures are composed of disordered atomic arrangement and expose dangling bonds and high surface areas with more active sites.5,26 Moreover, initially amorphous electrocatalysts can retain their amorphous states during/after electrochemical reactions, exhibiting corrosion resistance, and they have been extensively studied. Therefore, to simultaneously realize good activity and stability of HER electrocatalysts, initially amorphous assisted crystalline hybrid materials are a significant requirement for HER technology.
Recent research studies have proposed some heterojunctions of crystalline and amorphous states, such as, Ni–P/Ni(OH)2,27 CoP/CeOx,28 and CeO2/Co4N,29 which exhibit better HER performances than crystalline/crystalline hybrids. However, there are still some problems that need to be considered: (i) it is difficult to precisely control the amorphous parts of morphology and proportion; and (ii) the synthesis processes are generally completed under harsh conditions. Finding a simple preparation method under mild reaction conditions to obtain heterostructures that have excellent performance is the current research focus.30–32 It is accepted that their catalytic properties are determined by the synergy of the electronic and geometric features that originate at the interface between the core and the shell layer.
In this study, we designed and synthesized a core–shell heterostructure of an amorphous NiCoP layer coating on Co2P nanoneedles (a-NiCoP/Co2P@NF) with outstanding HER activity and stability. In 1 M KOH, only 26 and 123 mV ultralow overpotentials are required to achieve 10 and 500 mA cm−2, respectively, which is superior to the recently reported TMP electrocatalysts. The experimental characterization and DFT calculations confirm that the crystalline/amorphous heterostructure results in interfacial electron redistribution. Electron-rich interfacial Co active sites serve to weaken hydrogen adsorption and accelerate H2 emission. Meanwhile, the corrosion resistance of amorphous and strong interfacial electronic interactions endow the electrocatalyst with excellent stability in electrochemical reactions. This work provides a versatile strategy to improve the performance of TMP electrocatalysts.
2. Experimental section
2.1. Chemicals and materials
Cobalt nitrate hexahydrate (Co(NO3)2·6H2O) was purchased from Aladdin Co., Ltd. Sodium hypophosphite (NaH2PO2) was purchased from Macklin Co., Ltd. Urea (CH4N2O), ammonium fluoride (NH4F), nickel(II) chloride hexahydrate (NiCl2·6H2O), cobalt(II) chloride hexahydrate (CoCl2·6H2O), boric acid (H3BO3), hydrochloric (HCl), acetone (C3H6O), sodium chloride (NaCl), and potassium hydroxide (KOH) were purchased from Sinopharm Chemical Reagents. The chemicals were used directly without further treatment.2.2. Synthesis of cobalt carbonate hydroxide (Co–CH@NF)
Co–CH@NF was synthesized using a hydrothermal method according to ref. 33. Firstly, a piece of nickel foam (NF, 3 × 2 cm2) was washed under ultrasonication with acetone, 1 M HCl, deionized water, and ethanol for 20 min in turn and then dried under vacuum at 60 °C for 6 h. Secondly, a mixture of Co(NO3)2·6H2O (1.164 g), urea (0.6 g), and NH4F (0.185 g) was dissolved in 30 ml of DI water before being transferred into a Teflon-lined autoclave. Then, clean NF was added to the above solution. Finally, the autoclave was sealed and maintained at 120 °C for 5 h. After cooling down to room temperature, the NF-supported product was washed several times with DI water and ethanol, and dried in a vacuum oven at 60 °C for 6 h.2.3. Synthesis of cobalt phosphide (Co2P@NF)
To obtain Co2P@NF, Co–CH@NF and 1 g of NaH2PO2 were put into the downstream and upstream quartz boats, respectively. Then, the tube furnace was annealed to 350 °C at a heating rate of 2 °C min−1 and maintained for 120 min under a N2 atmosphere.2.4. Synthesis of amorphous nickel cobalt phosphides (a-NiCoP/Co2P@NF and a-NiCoP@NF)
a-NiCoP/Co2P@NF and a-NiCoP@NF were synthesized by one-step electrodeposition, in which Co2P@NF and NF were used as the working electrodes, respectively. Ag/AgCl electrode and Pt plate were employed as the reference electrode and counter electrode, respectively. The electrolyte was prepared with 50 ml of uniform aqueous solution containing NiCl2·6H2O (2.38 g), CoCl2·6H2O (2.38 g), H3BO3 (0.46 g), NaCl (0.5844 g), and NaH2PO2 (1.32 g).34,35 Amorphous NiCoP was electrodeposited onto the substrate at a potential of −1.2 V.36 Then, the products were washed with DI water and ethanol several times to remove the residual electrolyte and dried in a vacuum oven at 60 °C for 12 h.2.5. Preparation of Pt/C@NF
To prepare the Pt/C working electrode, 5 mg of the commercial Pt/C catalyst was dispersed in 370 μl of ethanol, 600 μl of DI water and 30 μl of Nafion (5 wt%) solution under sonication for 1 h to form a homogeneous ink. It was dripped onto clean NF (1 × 1 cm) with a loading amount of ∼5 mg cm−2. Finally, the electrode was dried at 60 °C for 6 h.2.6. Characterization of materials
The morphology and microstructure of the fabricated electrodes were studied via scanning electron microscopy (SEM, HITACHI Regulus100) and transmission electron microscopy (TEM, JEM-2100 PLUS). The composition and structure of the sample were analyzed using X-ray diffraction (XRD, Regaku Miniflex 600). The chemical composition of samples was studied by EDS. More information on the crystal structure was acquired by high-resolution transmission electron microscopy (HRTEM). The oxidation state and chemical bindings were characterized via X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha).2.7. Electrochemical characterization
Electrochemical tests were carried out with a three-electrode system on a CHI 660E electrochemical workstation. The resultant NF supported electrodes were cut into 1 cm × 1 cm as working electrodes before the measurement. A graphite rod and an Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. The electrocatalytic performance of the prepared catalyst was tested in 1.0 M KOH by linear sweep voltammetry (LSV) with a scan rate of 5 mV s−1. The electrochemical double-layer capacitance (Cdl) was calculated according to the CV curves, and the electrochemical surface area (ECSA) was calculated using eqn (1).| ECSA = Cdl/Cs | (1) |
2.8. Computational method
Density functional theory (DFT) calculations were performed using the Dmol3 module in the Materials Studio program of Accelrys. The generalized gradient approximation method using the Perdew–Burke–Ernzerhof functional was applied for the exchange–correlation functional. The core was treated using the effective core potential, and the double-numerical-polarization functions basis set was employed. The correction of van der Waals interaction was included using the DFT-D method of Grimme. The structures were optimized with 1 × 10−5 Hartree for energy change, 0.02 Hartree per Å for maximum force, and 0.05 Å for maximum displacement, respectively. For all of the calculations of slab models, a 2 × 3 × 1 Monkhorst–Pack grid k-point mesh was employed in the Brillouin zone. To avoid periodic interactions, a vacuum space of 20.0 Å was used along the normal direction to the catalyst surface.3. Results and discussion
As schematically illustrated in Scheme 1, a-NiCoP/Co2P@NF was synthesized by a hydrothermal reaction followed by low-temperature phosphorization and electrodeposition. The optical images of the as-prepared samples obtained in each step are shown in the Fig. S1.† First, the Co–CH@NF precursor was deposited on the surface of clean NF through a hydrothermal method. Next, after the typical phosphorization treatment under a N2 atmosphere, Co2P@NF was obtained. The following electrodeposition process realized the in situ growth of the a-NiCoP shell on the Co2P surface (a-NiCoP/Co2P@NF). In this process, Co2+ and Ni2+ ions accepted electrons and induced co-deposition of P generated through the reduction of H2PO2−.34,37 The X-ray diffraction (XRD) was applied to track the crystal structure evolution from Co–CH@NF to a-NiCoP/Co2P@NF. XRD patterns of the NF skeleton and the Co–CH@NF precursor are shown in Fig. S2 and S3,† respectively. As illustrated in Fig. S3,† except for the diffraction peaks of the NF substrate, all other peaks correspond well with the standard PDF card of orthorhombic Co(CO3)0.5OH·0.11H2O (PDF #48-0083). After the phosphorization procedure, the diffraction peaks of Co2P@NF (Fig. S4†) can be readily indexed to hexagonal Co2P (PDF #54-0413). Notably, the XRD pattern of a-NiCoP/Co2P@NF coincides with the NF substrate and hexagonal Co2P, without any obvious crystal peaks of NiCoP (Fig. S5†). This implies possible heterogeneous interfaces consisting of amorphous NiCoP and crystalline Co2P, which can be verified from the XRD pattern of a-NiCoP (Fig. S6†). | ||
| Scheme 1 Schematic illustration of the fabrication of a-NiCoP/Co2P@NF. | ||
The morphological evolution of samples is characterized by scanning electron microscopy (SEM). Fig. S7† presents a SEM image of the NF skeleton with a smooth surface. After the hydrothermal reaction, Co–CH@NF with a nanoneedle array structure was successfully obtained (Fig. 1a), which was well-maintained after phosphorization (Fig. 1b). After the electrodeposition process, the increased average diameter suggests that the Co2P nanoneedle is wrapped by a layer of rough shell (Fig. 1c). In addition, for comparison, irregular cellular amorphous NiCoP was prepared directly on NF using a similar electrodeposition process (Fig. S8†).
 | ||
| Fig. 1 SEM images of (a) Co–CH@NF, (b) Co2P@NF, and (c) a-NiCoP/Co2P@NF. (d) Low and (e) high magnification TEM images of a-NiCoP/Co2P. (f) HRTEM image of a-NiCoP/Co2P. Inset in (f) shows the SAED pattern. (g) EDX elemental mappings images of a-NiCoP/Co2P. | ||
To further study the morphology and microstructure of a-NiCoP/Co2P@NF, transmission electron microscopy (TEM) was applied to characterize the nanoneedles. As shown in Fig. 1d and e, we can see that a thin layer with several nanometers is closely coated on the surface of cobalt phosphide nanoneedles, which further confirms the core–shell structure of a-NiCoP/Co2P@NF. The HRTEM image was used to further confirm the amorphous/crystalline heterostructure in a-NiCoP/Co2P. As shown in Fig. 1f, the interface between crystalline Co2P and amorphous NiCoP can be clearly identified. The lattice fringes with spacings of 0.29 nm and 0.21 nm can be assigned to the (110) and (111) crystal planes of hexagonal Co2P, respectively. However, no lattice fringe can be observed in the shell of a-NiCoP, which demonstrates that there is no long-range ordered structure. The inset in Fig. 1f shows the SAED pattern of the core–shell a-NiCoP/Co2P. The simultaneous existence of diffraction and dispersion rings further confirms the successful construction of crystalline and amorphous heterostructure. The energy dispersive X-ray (EDX) elemental mappings (Fig. 1g) show that the Co, Ni, P elements are uniformly dispersed in a-NiCoP/Co2P@NF and the Co element is the most densely distributed one. It can be seen from above, the amorphous shell obtained by electrodeposition combines closely with the internal Co2P core. The synergistic effect of core–shell structure is expected to improve the HER catalytic performance of the catalyst.
X-ray photoelectron spectroscopy (XPS) was used to characterize the chemical state and interfacial electron redistribution of a-NiCoP/Co2P. Fig. 2a presents XPS survey spectra of a-NiCoP/Co2P and Co2P. In detail, the high-resolution XPS spectra of Ni 2p, Co 2p, and P 2p are deconvoluted. The existence of Ni2+ species can be verified by the main peak at 855.92 eV of Ni 2p in Fig. 2b.38,39 The lower binding energy at 852.22 eV corresponds to Niδ+ species (δ is close to 0). As for Co2P, the binding energy at 781.48 eV in Co 2p spectra is attributed to Co2+, while the lower binding energy at 778.99 eV is Coδ+ species (Fig. 2c).40,41 After undergoing the electrodeposition procedure, the typical peaks of Co 2p in a-NiCoP/Co2P@NF were clearly shifted to lower binding energies, indicating that an electron-rich state of Co was attained after electron redistribution. With regard to the P 2p spectrum in a-NiCoP/Co2P (Fig. 2d), the peak at 129.43 eV can be assigned to P bonded with Ni or Co (metal phosphide). Moreover, a higher peak at 134.06 eV is recognized as the oxidized phosphate species (P–O), which are probably induced by the partial oxidation of metal phosphides in air.33,42 The P 2p binding energy of a-NiCoP/Co2P@NF shifted towards higher binding energy, confirming the electron-deficient state of P. Fig. S9 and S10† reveal that the strong interaction between a-NiCoP and Co2P leads to a significant redistribution of electrons at the amorphous/crystalline heterogeneous interface (Fig. 2e). The resulting electron-rich metal sites are regarded as potential high-quality active sites for the HER.
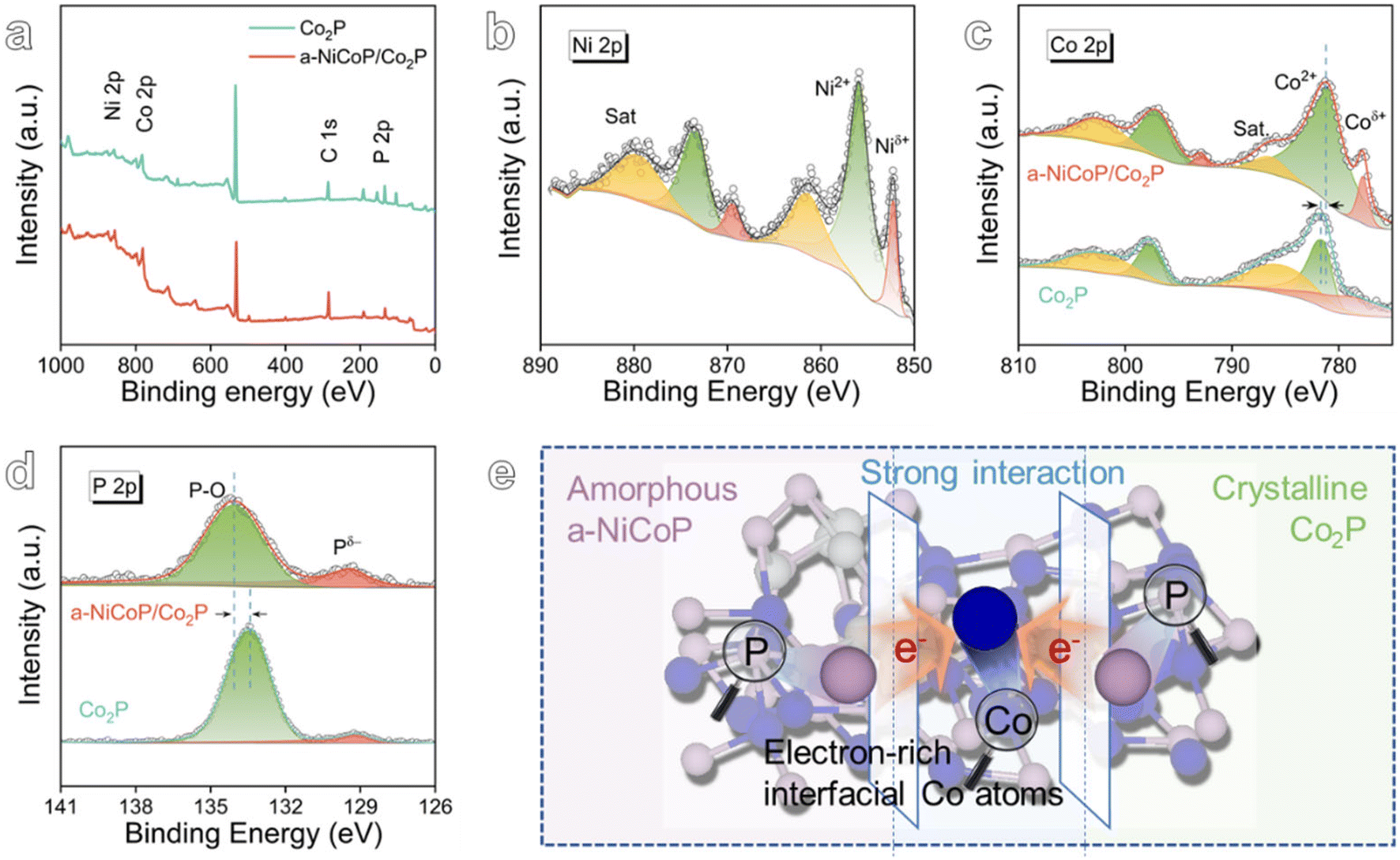 | ||
| Fig. 2 (a) XPS survey of a-NiCoP/Co2P. (b) High-resolution XPS spectra of Ni 2p. High-resolution XPS spectra of (c) Co 2p, (d) P 2p of Co2P and a-NiCoP/Co2P. (e) Schematic illustration of electronic structure modulation. | ||
The impact of tailored electron-rich metal sites on the HER performance of a-NiCoP/Co2P@NF electrocatalysts was assessed using a standard three-electrode system in 1 M KOH solution at room temperature (Fig. S11†). The Ag/AgCl reference electrode was pre-calibrated relative to the reversible hydrogen electrode (RHE) prior to electrochemical testing (Fig. S12†).43Fig. 3a presents the linear sweep voltammetry (LSV) curves of the bare NF substrate, commercial Pt/C@NF and as-prepared catalysts. All of the curves were corrected with an iR compensation of 100%. The bare NF substrate shows negligible HER catalytic activity. It is evident that the presence of electron-rich metal sites significantly enhances the HER activity compared to a-NiCoP@NF and Co2P@NF. Specifically, a-NiCoP/Co2P@NF electrocatalysts require an overpotential of only 26 mV to obtain a current density of 10 mA cm−2, respectively. In addition, the HER kinetics was assessed through the corresponding Tafel plots. As illustrated in Fig. 3b, the Tafel slope for a-NiCoP/Co2P@NF is 52 mV dec−1 at low current density, which is comparable to that of Pt/C (32 mV dec−1) and lower than the values for other as-prepared catalysts. Notably, a-NiCoP/Co2P@NF exhibits significantly accelerated HER kinetics (49 mV dec−1) at high current density, superior to that of standard Pt/C catalysts (91 mV dec−1).44 The superior performance indicators of a-NiCoP/Co2P@NF due to the interfacial electron-rich Co sites compared to other samples are presented in Fig. 3c. Furthermore, such an HER performance of a-NiCoP/Co2P@NF even exceeds those of most of the recently reported catalysts, as summarized in Fig. 3d and Table S1.†
 | ||
| Fig. 3 The electrochemical measurements of the samples in 1 M KOH. (a) Linear sweep voltammetry curves with iR-corrected. (b) Tafel slopes of as-prepared catalysts. (c) Comprehensive comparison of the overpotential at a current density of 10 mA cm−2, Tafel slopes at low and high current density. (d) Comparison of the HER performance with recent work. (e) The electrochemical double layer capacitances (Cdl) of a-NiCoP/Co2P, Co2P and a-NiCoP. (f) EIS of the as-prepared catalysts. (g) Chronopotentiometry curve of a-NiCoP/Co2P. | ||
In order to find out the reasons for the excellent HER performance of a-NiCoP/Co2P@NF, we first compared the electrochemical double layer capacitances (Cdl) of a-NiCoP/Co2P@NF, Co2P@NF and a-NiCoP @NF. We performed cyclic voltammetry experiments in the non-Faraday potential range at different scan rates and calculated the Cdl values proportional to the electrochemically active surface area (ECSA) (Fig. S13†).45 As shown in Fig. 3e, the Cdl value of a-NiCoP/Co2P@NF is 50 mF cm−2, which is about 1.3 and 1.7 times those of Co2P@NF and a-NiCoP @NF, respectively. The ECSA value of a-NiCoP/Co2P@NF (1250 cm2) is larger than those of Co2P@NF (950 cm2) and a-NiCoP@NF (725 cm2) (Table S2†), which indicates that plenty of active sites are introduced into a-NiCoP/Co2P@NF on account of the construction of a heterogeneous structure. In addition, the charge transfer behavior of the as-prepared catalysts was evaluated by electrochemical impedance spectroscopy (EIS). The charge transfer resistance was analyzed by fitting the EIS results to the equivalent circuit model shown in Fig. S14.† As illustrated in Fig. 3f, the charge transfer resistance (Rct) of a-NiCoP/Co2P@NF is 0.88 Ω, which is significantly lower than those of a-NiCoP@NF (1.53 Ω) and Co2P@NF (1.71 Ω). This indicates that a-NiCoP/Co2P@NF facilitates faster charge transfer during HER electrocatalysis. The stability of the catalyst is another fundamental criterion for evaluating the catalysts in addition to catalytic activity. As shown in Fig. 3g, the chronopotentiometry response shows that a-NiCoP/Co2P@NF can operate stably for 30 h at a current density of 10 mA cm−2 with negligible potential change, signifying the robust HER stability.
To evaluate the practicability of the a-NiCoP/Co2P@NF catalyst, the HER activity and long-term stability were studied at a high current density. As presented in Fig. 4a, the a-NiCoP/Co2P@NF catalyst exhibits the best performance with an overpotential of only 123 mV at 500 mA cm−2. The superior HER performance of the a-NiCoP/Co2P@NF catalyst at high current density exceeds those of the vast majority of recently reported Co-based catalysts (Table S1,†Fig. 4b and S15†). Notably, the excellent HER dynamics of a-NiCoP/Co2P@NF at high currents may originate from the faster H2 emission from the interfacial electron-rich Co sites (Fig. 4c). Additionally, the chronoamperometry results are presented in Fig. 4d, which indicate that the Co2P catalyst exhibits reduced stability at a high current density of 500 mA cm−2 in alkaline media, with an activity decrease of approximately 8% within 20 h. In contrast, the a-NiCoP/Co2P catalyst was able to retain its original catalytic activity for more than 50 h at the same high current density, demonstrating its robust stability for HER electrolysis. As can be seen from XRD and SEM images of the samples after the stability test in Fig. S16 and S17,† respectively, the structure and morphology of the a-NiCoP/Co2P@NF catalyst remained almost unchanged. In contrast, the performance of Co2P@NF decreased significantly after 20 h of stability testing. This confirms the excellent stability of a-NiCoP/Co2P@NF, indicating the effective protection of the internal active sites by the amorphous shell (Fig. 4e).
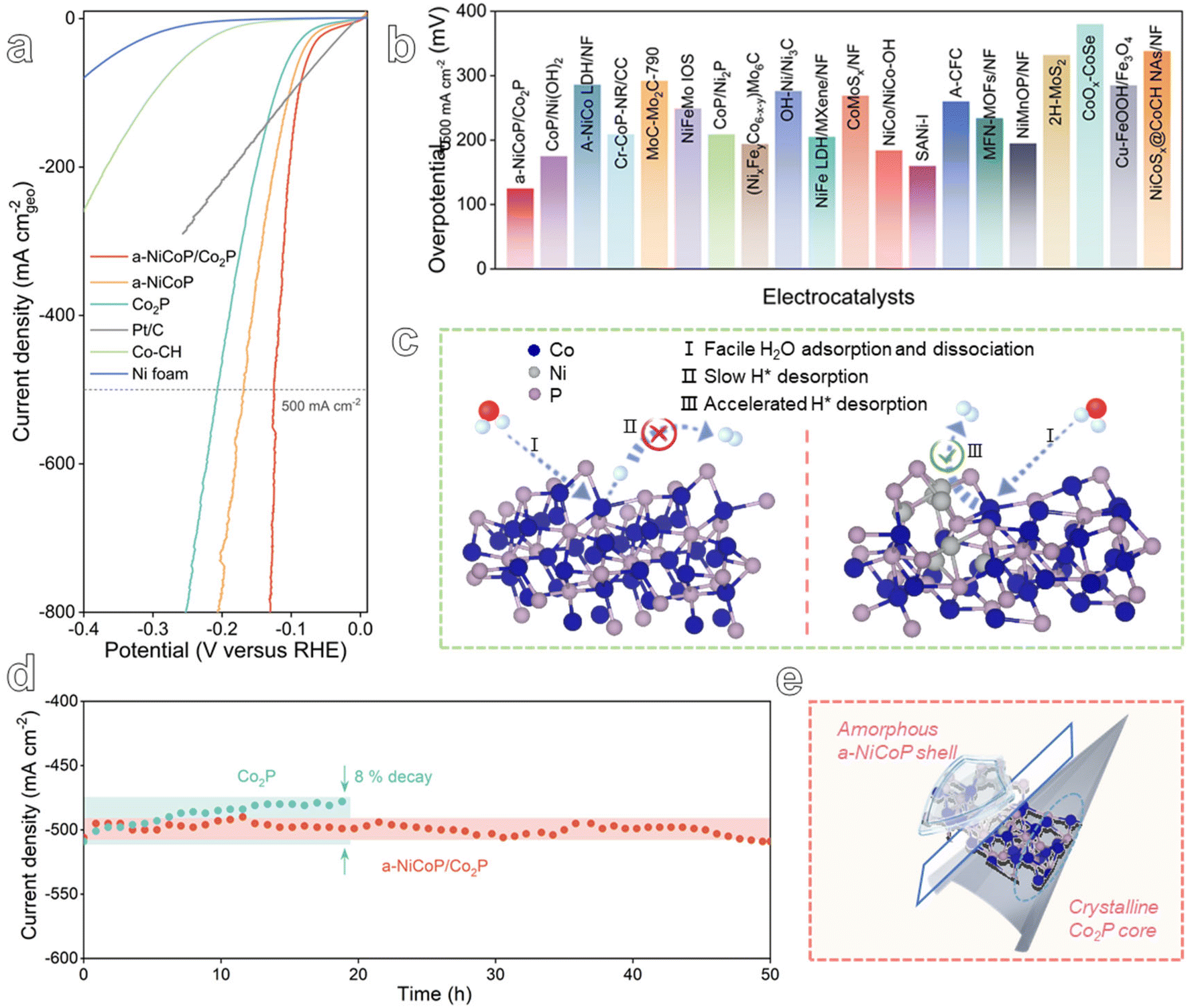 | ||
| Fig. 4 (a) LSV curves of the as-prepared catalysts at a scan rate of 5 mV s−1. (b) Comparison of the HER performance at 500 mA cm−2 with recent work. (c) Analysis of the HER mechanism for Co2P (left) and a-NiCoP/Co2P (right). (d) Chronoamperometry curves of Co2P and a-NiCoP/Co2P. (e) Schematic illustration of the excellent stability. | ||
In order to further elucidate the enhanced HER performance of a-NiCoP/Co2P@NF, we designed the theoretical models of Co2P, a-NiCoP and a-NiCoP/Co2P (Fig. 5a–c). The optimized structural models and adsorption of different reaction intermediates are shown in Fig. S18.† As shown in Fig. 5c, three adsorption sites of a-NiCoP/Co2P were selected to compare the adsorption ability for H2O and it was found that site II (interfacial Co site) exhibits the lowest adsorption energy of H2O molecules (−0.39 eV) compared with sites I (−0.10 eV) and III (Ni 0.02 eV) (Table S3†). Therefore, the interfacial Co site in a-NiCoP/Co2P can be regarded as the main electrocatalytically active site. The densities of states (DOS) of Co2P, a-NiCoP, and a-NiCoP/Co2P were calculated and compared (Fig. S19†). It can be concluded that the DOS of Co atoms makes a major contribution near the Fermi level, which may play a critical role in the catalytic activity for the HER. Free energy diagrams of the HER on Co2P, a-NiCoP, and a-NiCoP/Co2P in Fig. 4d show the reaction pathway of the HER process. It can be concluded that hydrogen desorption in the HER process is the potential determining step (PDS) for Co2P (ΔG = 0.50 eV) and a-NiCoP (ΔG = 0.34 eV). Moreover, a-NiCoP/Co2P exhibits a continuous downhill step for the HER and exhibits a superior electrocatalytic HER performance to Co2P and a-NiCoP, which is consistent with the experimental results. The construction of a heterojunction structure effectively regulated the electronic structure of active sites. Therefore, the combination of Co2P and a-NiCoP plays an important role in promoting hydrogen desorption and enhancing electrocatalytic activity. These results can further confirm the feasibility of the core–shell heterostructure. And electron-rich interfacial cobalt active sites serve to accelerate the desorption of H* intermediates.
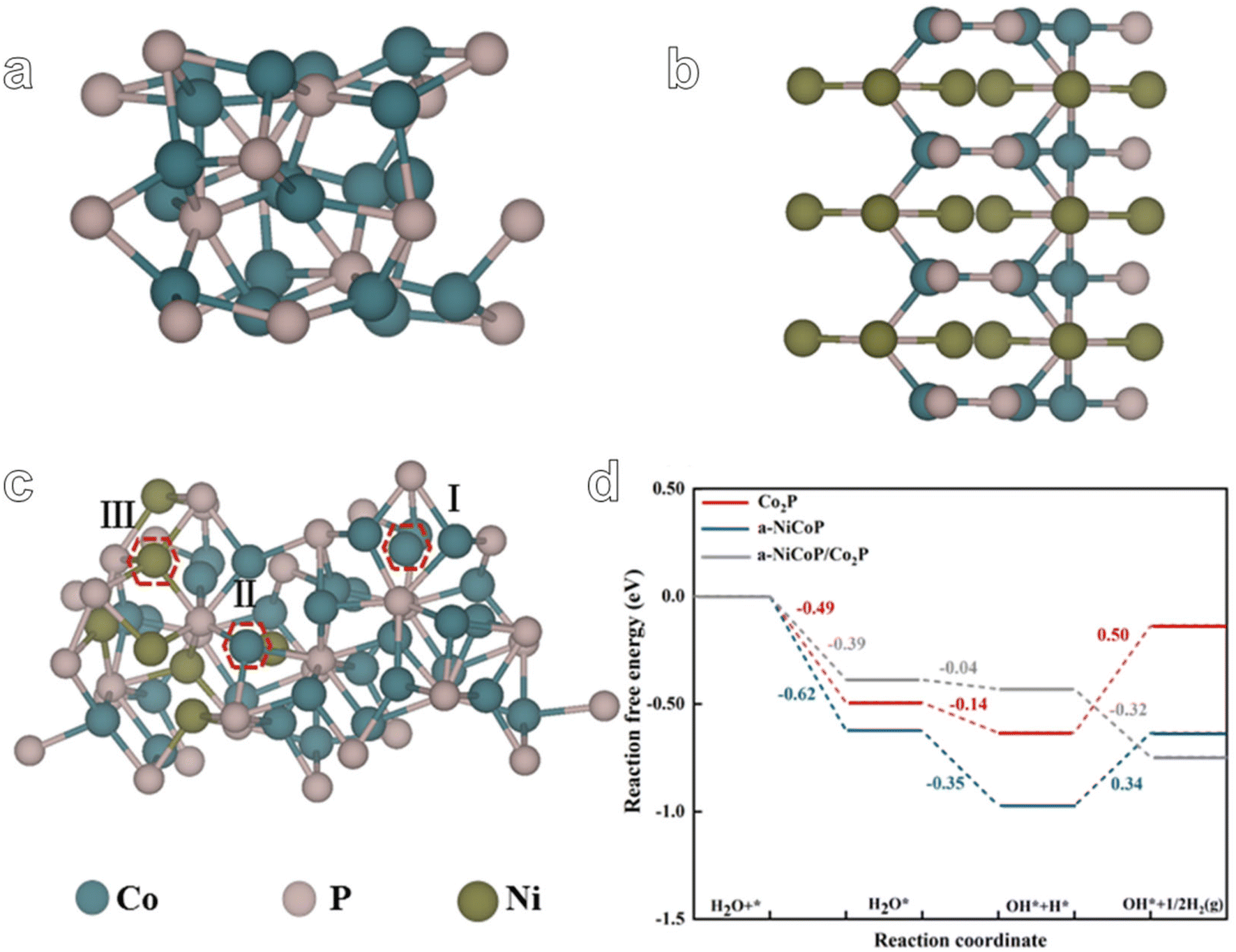 | ||
| Fig. 5 The theoretical model of (a) Co2P, (b) a-NiCoP, and (c) a-NiCoP/Co2P. (d) The reaction pathway of the HER process for Co2P, a-NiCoP and a-NiCoP/Co2P. | ||
In brief, the excellent catalytic performance of a-NiCoP/Co2P@NF in the hydrogen evolution reaction can be attributed to the unique crystalline and amorphous core–shell structure, and mainly includes the following aspects: (1) the 3D core–shell structure provides a larger specific surface area and exposes more active sites; (2) the amorphous NiCoP shell closely coated on Co2P can protect the Co2P core from KOH corrosion with its corrosion resistance properties; and (3) the strong interaction between a-NiCoP and Co2P induces interfacial electron redistribution, resulting in electron-rich Co sites that weaken hydrogen adsorption and accelerate H2 emission.
4. Conclusions
In summary, we designed and prepared a unique amorphous/crystalline core–shell structure of a-NiCoP/Co2P@NF with excellent HER activity and stability at industrial current density. It requires only 26 and 123 mV ultralow overpotential to obtain 10 and 500 mA cm−2, respectively. Experimental results and theoretical calculations reveal that the interfacial electron redistribution leads to electron-rich interfacial Co sites, which weaken the hydrogen adsorption and accelerate H2 emission. Meanwhile, the corrosion resistance of the amorphous shell and interfacial strong interaction endow the electrocatalyst with outstanding stability in electrocatalysis. This work provides a promising strategy for designing corrosion-resistant and efficient catalysts in multiphase catalysis.Author contributions
Xiaodong Chen: research design, investigation, and writing – original draft. Zhi Cheng: investigation and writing – original draft. Jiao Li: formal analysis and methodology. Hongyu Chen: methodology and investigation. Siyuan Liu: investigation and writing – review & editing. Shuxian Wei: methodology. Zhaojie Wang: research design, writing – review & editing, and funding acquisition. Xiaoqing Lu: conceptualization, supervision, writing – review & editing, and funding acquisition.Data availability
Data available on request from the authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22471289, 22478430, 22101300, and 22275210), the Shandong Natural Science Foundation (ZR2022ME105 and ZR2023ME004), the Qingdao Natural Science Foundation (23-2-1-232-zyyd-jch), and the Fundamental Research Funds for the Central Universities (24CX06042A, 24CX06070A, 22CX03010A, and 22CX01002A-1).References
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- J. K. Das, A. K. Samantara, K. A. Sree Raj, C. S. Rout and J. N. Behera, Dalton Trans., 2019, 48, 15955–15961 RSC.
- J. K. Das, A. Padhy, S. Parida, R. M. Pathi and J. N. Behera, Dalton Trans., 2022, 51, 11526–11535 RSC.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2015, 15, 197–203 CrossRef PubMed.
- J. K. Das, A. K. Samantara, S. Satyarthy, C. S. Rout and J. N. Behera, RSC Adv., 2020, 10, 4650–4656 RSC.
- S. Shen, Z. Wang, Z. Lin, K. Song, Q. Zhang, F. Meng, L. Gu and W. Zhong, Adv. Mater., 2022, 34, e2110631 CrossRef PubMed.
- X. Chen, S. Cao, X. Lin, X. Wei, Z. Wang, H. Chen, C. Hao, S. Liu, S. Wei, D. Sun and X. Lu, Nano Res., 2023, 16, 12253–12262 CrossRef CAS.
- S. Anantharaj and S. Noda, Energy Environ. Sci., 2022, 15, 1461–1478 RSC.
- A. Eftekhari, Int. J. Hydrogen Energy, 2017, 42, 11053–11077 CrossRef CAS.
- S. Lu and Z. Zhuang, Sci. China Mater., 2016, 59, 217–238 CrossRef CAS.
- K. Ojha, S. Saha, P. Dagar and A. K. Ganguli, Phys. Chem. Chem. Phys., 2018, 20, 6777–6799 RSC.
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009–2004031 CrossRef CAS.
- J. K. Das, N. Sahu, P. Mane, B. Chakraborty and J. N. Behera, Sustainable Energy Fuels, 2023, 7, 4110–4119 RSC.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, e1802880 CrossRef PubMed.
- X. Liu, Y. Li, Z. Cao, Z. Yin, T. Ma and S. Chen, J. Mater. Chem. A, 2022, 10, 1617–1641 RSC.
- S. M. El-Refaei, P. A. Russo and N. Pinna, ACS Appl. Mater. Interfaces, 2021, 13, 22077–22097 CrossRef CAS PubMed.
- S. Jeoung, B. Seo, J. M. Hwang, S. H. Joo and H. R. Moon, Mater. Chem. Front., 2017, 1, 973–978 RSC.
- S. Lai, C. Lv, S. Chen, P. Lu, X. She, L. Wan, H. Wang, J. Sun, D. Yang and X. Zhao, J. Alloys Compd., 2020, 817, 152727–152734 CrossRef CAS.
- H. Pei, L. Zhang, G. Zhi, D. Kong, Y. Wang, S. Huang, J. Zang, T. Xu, H. Wang and X. Li, Chem. Eng. J., 2022, 433, 133643–133653 CrossRef CAS.
- Y. Yan, X. R. Shi, M. Miao, T. He, Z. H. Dong, K. Zhan, J. H. Yang, B. Zhao and B. Y. Xia, Nano Res., 2018, 11, 3537–3547 CrossRef CAS.
- T. Liu, X. Ma, D. Liu, S. Hao, G. Du, Y. Ma, A. M. Asiri, X. Sun and L. Chen, ACS Catal., 2016, 7, 98–102 CrossRef.
- X. Xu, T. Wang, W. Lu, L. Dong, H. Zhang and X. Miao, ACS Sustainable Chem. Eng., 2021, 9, 4688–4701 CrossRef CAS.
- J. Wu, R. Zhao, H. Xiang, C. Yang, W. Zhong, C. Zhang, Q. Zhang, X. Li and N. Yang, Appl. Catal., B, 2021, 292, 120200–120209 CrossRef CAS.
- Y. Liu, Q. Li, R. Si, G. D. Li, W. Li, D. P. Liu, D. Wang, L. Sun, Y. Zhang and X. Zou, Adv. Mater., 2017, 29, 1606200–1606207 CrossRef PubMed.
- F. Zhao, H. Liu, H. Zhu, X. Jiang, L. Zhu, W. Li and H. Chen, J. Mater. Chem. A, 2021, 9, 10169–10179 RSC.
- X. Z. Song, W. Y. Zhu, J. C. Ni, Y. H. Zhao, T. Zhang, Z. Tan, L. Z. Liu and X. F. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 33151–33160 CrossRef CAS PubMed.
- X. Chen, Q. Li, Q. Che, Y. Chen and X. Xu, ACS Sustainable Chem. Eng., 2018, 7, 2437–2445 CrossRef.
- S. Anantharaj and S. Noda, Small, 2020, 16, e1905779 CrossRef PubMed.
- W. Xu and H. Wang, Chin. J. Catal., 2017, 38, 991–1005 CrossRef CAS.
- H.-M. Zhang, J.-J. Wang, Y. Meng and J. Sun, Int. J. Hydrogen Energy, 2022, 47, 36084–36097 CrossRef CAS.
- H. Liu, X. Ma, H. Hu, Y. Pan, W. Zhao, J. Liu, X. Zhao, J. Wang, Z. Yang, Q. Zhao, H. Ning and M. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 15528–15536 CrossRef CAS PubMed.
- K. S. Anuratha, Y.-Z. Su, M.-K. Huang, C.-K. Hsieh, Y. Xiao and J.-Y. Lin, J. Alloys Compd., 2022, 897, 163031–163039 CrossRef CAS.
- G. B. Darband, M. Aliofkhazraei, S. Hyun, A. S. Rouhaghdam and S. Shanmugam, J. Power Sources, 2019, 429, 156–167 CrossRef CAS.
- X. Li, X. Qian, Y. Xu, F. Duan, Q. Yu, J. Wang, L. Chen, Y. Dan and X. Cheng, J. Alloys Compd., 2020, 829, 154535–154543 CrossRef CAS.
- V. G. Dileepkumar, C. Pratapkumar, R. Viswanatha, B. M. Basavaraja, R. R. Maphanga, M. Chennabasappa, N. Srinivasa, S. Ashoka, Z. Chen, S. Rtimi, K. Jayaramulu, R. S. Varma, G. Szekely and M. Sridhar Santosh, Chem. Eng. J., 2021, 426, 131315–131324 CrossRef CAS.
- J. Zhang, Y. Gu, Y. Lu, C. Zhu, G. Liu, C. Wang, D. Sun, Y. Tang and H. Sun, Appl. Catal., B, 2023, 325, 122316–121324 CrossRef CAS.
- N. A. Saguì, P. Ström, T. Edvinsson and İ. Bayrak Pehlivan, ACS Catal., 2022, 12, 6506–6516 CrossRef PubMed.
- Y. Wang, B. Li, W. Xiao, X. Wang, Y. Fu, Z. Li, G. Xu, J. Lai, Z. Wu and L. Wang, Chem. Eng. J., 2023, 452, 139175–139182 CrossRef CAS.
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. D. Lou, Adv. Mater., 2019, 31, e1902339 CrossRef PubMed.
- F. S. Zhang, J. W. Wang, J. Luo, R. R. Liu, Z. M. Zhang, C. T. He and T. B. Lu, Chem. Sci., 2018, 9, 1375–1384 RSC.
- X. Chen, Z. Wang, S. Cao, X. Lin, C. Hao, X. Zhang, H. Chen, J. Wang, J. Liu, S. Liu, S. Wei, X. Lu and D. Sun, Appl. Catal., B, 2024, 357, 124295–124306 Search PubMed.
- H. Q. Fu, M. Zhou, P. F. Liu, P. Liu, H. Yin, K. Z. Sun, H. G. Yang, M. Al-Mamun, P. Hu, H. F. Wang and H. Zhao, J. Am. Chem. Soc., 2022, 144, 6028–6039 CrossRef CAS PubMed.
- D. Kong, C. Meng, Y. Wang, X. Chen, J. Zhang, L. Zhao, J. Ji, L. Zhang and Y. Zhou, Appl. Catal., B, 2024, 343, 123578–123588 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03258f |
| This journal is © The Royal Society of Chemistry 2025 |