
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactive main group metal complexes of the neutral NNNN macrocycle, Me4TACD
Priyabrata
Ghana
 a,
Louis J.
Morris
b and
Jun
Okuda
*c
a,
Louis J.
Morris
b and
Jun
Okuda
*c
aDepartment of Chemistry, Indian Institute of Technology, Gandhinagar, Gujarat-382355, India
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52056 Aachen, Germany. E-mail: jun.okuda@ac.rwth-aachen.de
First published on 29th May 2025
Abstract
Currently, there is considerable interest in introducing molecularly defined main group metal compounds as precursors and model complexes of homogeneous catalysts for various bond cleavage and forming transformations. With a focus on the NNNN macrocyclic ligand Me4TACD (N,N′,N′′,N′′′-tetramethyl-1,4,7,10-tetraazacyclododecane), this review summarizes the versatility of the ligand Me4TACD for the stabilization of reactive main group s- and p-block (group 1, 2, 12–14) metals. Metal hydrides, hydrocarbyls and silyls are often monomeric and catalyze alkene hydrofunctionalisations. In contrast to the rich coordination chemistry of d- and f-block transition metals using a plethora of ligands, main group metals still leave room for new structures and reactivities, aligning with the current efforts to develop a systematic understanding in s- and p-block metal–ligand combinations.
1. Introduction
Commonly, reactive main group metal fragments are stabilized by anionic ligands of the general type [LlXx] (L = two-electron ligand, l = 0–4; X = one-electron ligand, x = 1–4), which exhibit sterically bulky substituents.1–10 N-Heterocyclic carbene derivatives are often employed in this regard as neutral L-type ligands.11–15 Neutral multidentate N- and O-donor ligands of the general type Ln can provide access to soluble and reactive molecular s-block complexes.16–20 Macrocyclic N-donor ligands have seen less widespread use in the main group chemistry.The macrocyclic tetraamine ligand, Me4TACD (N,N′,N′′,N′′′-tetramethyl-1,4,7,10-tetraazacyclododecane, also called 12-TMC or Me4cyclen) has been used in the coordination chemistry of 3d metals as a redox innocent supporting ligand. It was first developed in 1982 to study the effect of N-methylation of cyclic polyamines on the coordination number of late transition metals such as Ni and Cu and extensively to study reactive (di)oxygen species during O2 activation at Cr, Mn, Fe, Co, Ni, and Cu centers.21,22 This review summarizes the use of Me4TACD as a versatile supporting ligand for the study of main group metal centers featuring reactive ligands such as hydride and organyls, mostly as cations. Such species are often of low-nuclearity, often monomeric and allow to study the inherent property of main group metal–ligand interaction. Since some group 1 and 2 metals are abundant, inexpensive and non-toxic, reactions and catalysis based on molecularly defined complexes could eventually substitute some reactivity patterns so far dominated by transition metals.
2. Synthesis and properties of Me4TACD
The commonly used method for preparing the Me4TACD ligand consists of three steps. First, the Richman–Atkins cyclization assembles the macrocycle by reacting N,N′,N′′-tris(p-tolysulfonyl)diethylenetriamine-N,N′′-disodium salt (A) with tosylbis[2-(tosyloxy)ethyl]amine (B) (Scheme 1).23,24 This cyclization can also be done using diethylamine B with mesyloxy or halides (Cl–I) as the leaving group.24 The highest yield of ∼80% was obtained when the tosyloxy (OTs) group was used as a leaving group.24 This cyclization works best when carried out in N,N-dimethylformamide. The second step is the detosylation of tetra(tosyl)cyclen using concentrated sulphuric acid. Finally, the N-methylation (Eschweiler–Clarke reaction) of the parent macrocycle H4TACD employing a mixture of formic acid and formaldehyde provides Me4TACD in 45–55% yield.21,25 The main drawback of this route is its low atom economy, as it requires both tosylation and detosylation steps. Additionally, the cyclization step requires a large quantity of dry DMF. A more efficient alternative produces the parent cyclen in two steps with an overall yield up to 57% (route 2).26 The first step of this route involves the S-alkylation of dithiooxamide using excess bromoethane and subsequent reaction of the resulting bis-thioimido ester salt with triethylenetetraamine to afford the tricyclic bis-amidine (Scheme 1). The reduction of the bis-amidine with DIBALH in refluxing toluene, followed by treatment with NaF in water, resulted in the parent cyclen. Finally, N-methylation of cyclen using the Eschweiler–Clarke reaction provides Me4TACD.21,25,26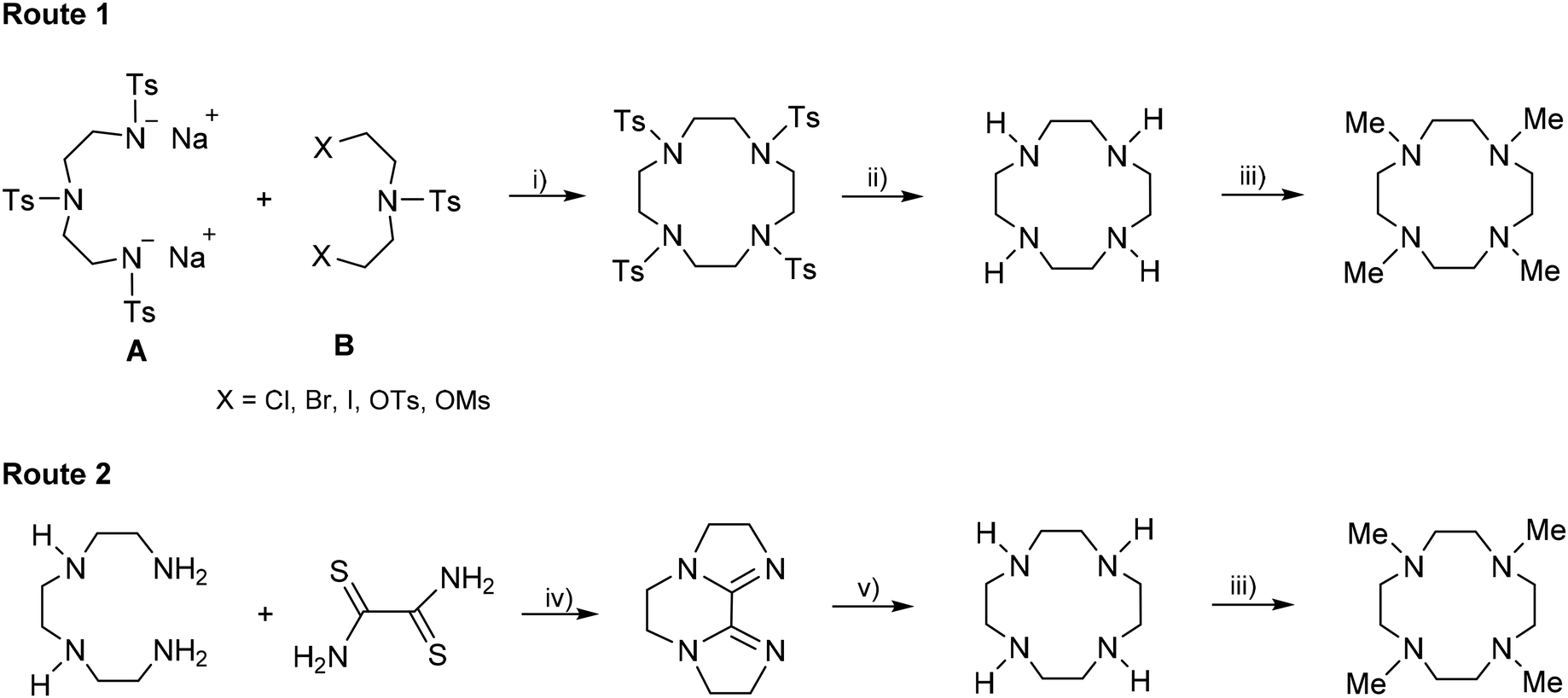 | ||
| Scheme 1 Synthetic methods of the ligand Me4TACD. (i) DMF, 100 °C. (ii) H2SO4 (conc.), NaOH. (iii) HCO2H/CH2O, 100 °C, NaOH. (iv) DIBAL-H, refluxing toluene. (v) NaF, H2O. | ||
As a ligand, Me4TACD generally binds to a metal fragment MLX in a κ4-fashion and adopts a C4-symmetric, boat-like conformation (Fig. 1a). The four nitrogen atoms of the ligand form a square planar core with the coordinated metal residing above the plane and all four NMe groups pointing towards the metal. In the solid state, the four CH2CH2 groups adopt a staggered conformation to avoid steric congestion, giving rise to two enantiomers (δδδδ, λλλλ) for an achiral metal center. The CH2 protons of the ligand are magnetically inequivalent, leading to AA′XX′-type signal sets for CH2CH2 groups in the 1H NMR spectrum. Depending on the size of the coordinated metal ion, lability of N–M bonds, and remaining coordination sphere, the AA′XX′-spin system can appear resolved, unresolved AB spin system, or collapsed broad singlet on the NMR timescale. AB-multiplets indicate rapid ring flipping between the two enantiomers but persistent macrocycle coordination with two disparate faces, whilst complete collapse can indicate fluxional ligand coordination. In two examples shown below,27,28 the Me4TACD ligand has been observed to adopt a folded conformation where one of the four methyl groups is orientated away from the metal centre (Fig. 1b). This highly strained conformation appears to relieve steric congestion for small metal cations with two strongly-bound ancillary X-type ligands (Al–H, Mg–O); weakly bound X-type ligands (e.g. Zn–I) are readily displaced by the chelating macrocycle to give auto-ionised products of the type [(κ4-Me4TACD)MX]+X−. The folded conformation in solution is diagnosed with 1H NMR spectroscopy by the presence of three methyl environments in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 integral ratio and 8-sets of magnetically inequivalent CH2 protons in the 1H NMR spectrum.28
:2:1 integral ratio and 8-sets of magnetically inequivalent CH2 protons in the 1H NMR spectrum.28
 | ||
| Fig. 1 Coordination behaviour of Me4TACD ligand: (a) κ4NNNN-bonding with boat-like conformation; (b) κ3NNN-bonding with folded conformation. | ||
3. Group 1 metals
Alkali metal compounds typically exist as saline or aggregated species, but neutral polydentate ligands help to stabilize well-defined low-nuclearity molecular species. Polyamines offer advantages over crown-ethers due to stronger N → M bonds and being less prone to nucleophilic attacks. Indeed, large aza-crowns have been used to stabilise highly reducing sodide compounds.29 A renewed interest in organoalkali compounds as useful reagents in synthetic organic chemistry17,20,30 has highlighted the impact of different chelating donor ligands on reactivity and chemoselectivity.18,19,31 While the use of Me4TACD remains limited to the examples described herein, the steric and coordinative demand and strong chelate effect (especially for lithium and sodium) make it a promising candidate for the future development of organoalkali metal chemistry.Trends in alkali metal coordination chemistry of Me4TACD complexes depend on the ionic radius and strength of M–N bonds. While lithium forms strong, monomeric complexes with 12-membered aza-crown with appreciable covalency in Li–N bonds, the heavier alkali metals exhibit more labile bonding, leading to dimeric or polymeric structures. Me4TACD has been employed to provide well-defined molecular hydridotriphenylborates, which were employed as catalysts in the hydroboration of unsaturated organic substrates. Low-nuclearity triphenyl- and trihydridosilanide complexes have also been structurally characterised.
3.1. Charge-separated lithium and sodium complexes
The first lithium complex of Me4TACD ligand, [(Me4TACD)6Li][CH(C6H5)(S-C6H5)] (1), which was obtained upon treatment of [6Li]-α-(phenylthio)benzyllithium with one equiv. of Me4TACD in THF/THF-d8, was prepared to study its structure in solution by NMR spectroscopy.32 Based on 6Li-HOESY, 1H, and 13C NMR spectroscopy, the study revealed an equilibrium between contact ion pairs and solvent-separated ion pairs at ambient temperature and with an increased proportion of solvent-separated ion pairs at lower temperatures (Scheme 2). | ||
| Scheme 2 Equilibrium between the contact ion pair and the solvent-separated ion pair of 1 in THF. | ||
Me4TACD ligated lithium and sodium complexes [(Me4TACD)M(L)][BAr4] (M = Li (2), Na (3); L = H2O, THF; Ar = C6H3-3,5-(CF3)2) were prepared from their respective borate salts [Li(H2O)4][BAr4] and [Na(THF)2][BAr4] (Scheme 3).33,34 Attempts to isolate the heavier homologues resulted in [(Me4TACD)H][BAr4], likely due to hydrolysis from traces of water present in the solvent. Unlike the sandwich structure of [12]-crown-4 complexes of group 1 metals, these complexes exhibit half-sandwich structures with five-coordinate alkali metal cations of square pyramidal geometry (Scheme 3). The Me4TACD ligand adopts a distorted boat-like conformation, positioning all four nitrogen atoms in a square planar arrangement. The metal cations are located below this N4 plane, with all NMe groups oriented toward the metal centre. In solution, the Me4TACD ligand binds the alkali metals more strongly than the corresponding crown ethers, as evidenced by a significant downfield shift of the 23Na NMR signal (δ(23Na) +12.7 ppm (3) vs. ∼0 ppm for [Na([12]-crown-4)2]+).33 In fact, the coordination of alkali metals to the aza-macrocycle is not just an electrostatic interaction but also involves a significant donation of electron density from the nitrogen's 2p-nonbonding orbitals to the alkali metal center.34
 | ||
| Scheme 3 Synthesis of the Li and Na borate complexes of Me4TACD; molecular structure of the cationic part of 3. | ||
3.2. Alkali metal hydridotriphenylborates
Me4TACD-supported alkali metal hydridotriphenylborates [(Me4TACD)M][HBPh3] (M = Li (4), Na (5), K (6)) were synthesized via two pathways.35 One approach involves a two-step, one-pot reaction where Me4TACD reacts with tetramethyldisilazides [M{N(SiHMe2)2}] (M = Li, Na, K) in THF, followed by BPh3 to obtain compounds 4–6 after elimination of (Me2HSiN–SiMe2)2 (Scheme 4). The other method involves mixing Me4TACD with the alkali metal hydridotriphenylborates [M(HBPh3)] in THF (Scheme 4, left). The structural analysis of 4–6 revealed distinct bonding geometries: while the lithium complex 4 forms a separated ion pair with a THF molecule at the lithium, the sodium (5) and the potassium (6) homologues exist as contact ion pairs due to the non-covalent M+⋯Cπ (M = Na, K) interactions along with a 3-centered-2-electron M⋯H–B bonding interaction (Scheme 4). The five-coordinate lithium cation in compound 4 adopts a distorted square-pyramidal geometry. For compounds 5 and 6, the sodium and the potassium cations are formally eight and nine coordinated and exist in a distorted square anti-prismatic and mono-capped square anti-prismatic geometry, respectively. Compounds 4–6 serve as chemoselective catalysts for carbonyls and CO2 hydroboration, with the lithium complex exhibiting the highest activity.35 While the hydridoborate provides the hydride in catalysis, the alkali metals and supporting ligand play an important role in activating the substrate by its Lewis-acidic properties. Lower denticity acyclic polyamines N,N,N′,N′-tetramethylethylenediamine (TMEDA) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) provide higher activity than Me4TACD derivatives due to easier access to the metal centre. The tetradentate, yet hemilabile ligand N,N,N′,N′,N′′,N′′-hexamethyltriethylenetetraamine (Me6TREN) provided further improved activity.36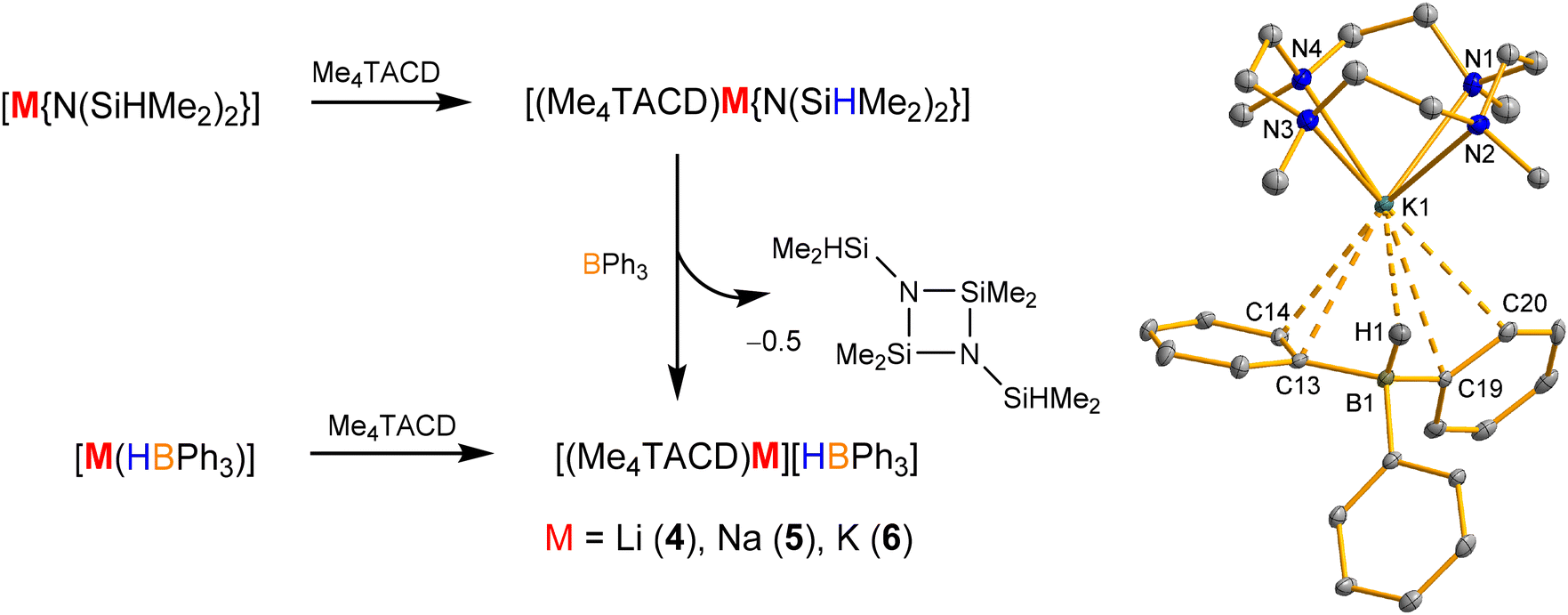 | ||
| Scheme 4 Synthesis of Me4TACD ligated alkali metal hydridotriphenylborates and the molecular structure of 6. | ||
3.3. Alkali metal silanides
Given the current interest in hydrogen storage materials, the alkali metal silanides [MSiH3]∞ (M = Li–Cs) have been widely studied recently.37–40 Normally, alkali metal silanides are thermodynamically unstable and exist as polymeric clusters. Employing the Me4TACD ligand, a series of molecular alkali metal silanides [(Me4TACD)M(SiH3)]n (M = Li (12), Na (13), K (14) and Rb (15); n = 1–2) were isolated, and exist as monomer or dimer in the solid-state.41 Compounds 12–15 were prepared from triphenylsilanides [(Me4TACD)M(SiPh3)]n (M = Li (7), Na (8), K (9a), Rb (10); n = 1 (Li–K), 2 (Rb)) and H2 or PhSiH3 (Scheme 5). Hydrogenolysis or hydrosilylation is chemoselective for the Si–C bonds to eliminate benzene or diphenylsilane, rather than heterolyzing the M–Si bond to provide the corresponding alkali metal hydride and hydrosilane. While the hydrogenolysis of 7–10 with H2 takes several days to complete, the reaction with PhSiH3 finishes within 5 min and proceeds with redistribution of the organosilane to give Ph2SiH2 and SiH4. The triphenylsilanides 7–10, along with the caesium analogue [(Me4TACD)Cs(SiPh3)]∞ (11), were synthesized from the reaction of Me4TACD, Ph3SiSiMe3, and LiCH2SiMe3 or MOtBu (M = Na–Cs). Alternatively, compounds 7 and 9–10 were prepared through the ligand exchange reaction from isolated THF adducts [M(SiPh3)(thf)n] with Me4TACD. Lithium (7) and sodium (8) triphenylsilanides exist as monomers with a direct M–Si σ-bond. The potassium complex was crystallised as a monomeric THF-adduct, [(Me4TACD)K(SiPh3)(thf)] (9b), which contains a K–Si σ-bond, but rapidly loses THF under vacuum to provide 9a, where the silanide is alternatively bound to potassium via an η6–π-facial interaction.41,42 The stability of the triphenylsilanides decreases down the group (Li: t1/2 = 14 d; Cs: t1/2 ≈ 12 h). In contrast, the trihydridosilanide 12–15 shows a reverse trend; the Na, K, and Rb homologues are stable for weeks both in solution and solid state, but the Li homologue decomposes in two days. While the light alkali metal analogues 7–9 exist as a monomer in the solid state, the heavier analogues show a more extended coordination sphere (10: dimer with bridging [SiPh3]−; 11: one-dimensional chain-like structure through Cs–CPh interactions). Similarly, trihydridosilanide 13 exists in monomeric form with square pyramidal coordination geometry around the sodium atom. The potassium (14) and rubidium (15) homologues form dimers in the solid state, with SiH3 anions bridging the two [(Me4TACD)M] fragments.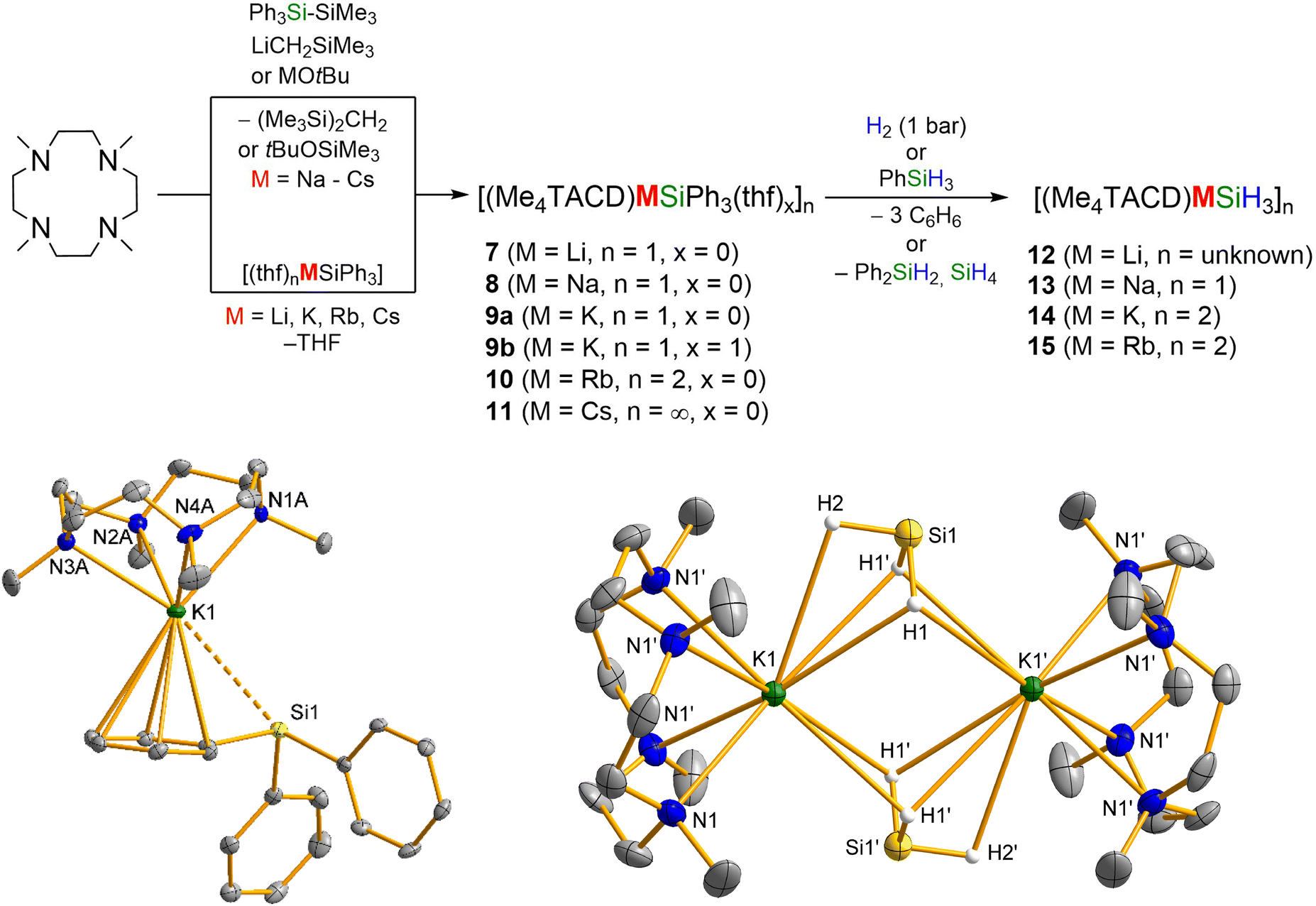 | ||
| Scheme 5 Synthesis of Me4TACD stabilized alkali metal silanides (top) and molecular structure of potassium silanide complexes 9a and 14. | ||
The complete series of alkali metal triphenylsilanide derivatives enabled comparative NMR spectroscopic analysis of the binding of the Me4TACD ligand to metal cations with increasing ionic radius. The lithium (7) and sodium (8) complexes show two multiplets for the methylene protons in their 1H NMR spectrum (THF-d8, 25 °C), consistent with time-averaged C4v-symmetry and persistent ligand coordination. In contrast, compounds 9–11 show a broad signal for the methylene environment, which is also notably sharper for Cs (11) than for K (10), indicating increasingly faster ligand dynamics for the larger metal cations. The similarity of the methylene chemical shift (δ 2.4 ppm) to that of the free ligand suggests partial ligand dissociation.
3.4. Molecular alkali metal organoperoxides
Alkali metal organoperoxides [MOOR] (M = Li, Na, K; R = hydrocarbyl), which form as unstable intermediates in the oxidation of organometallics by O2, exist in oligomeric forms. The Me4TACD ligand is suitable for stabilizing such highly reactive peroxide intermediates. The addition of Me4TACD to an n-pentane solution of LiNiPr2 and ROOH (R = tBu, CMe2Ph) at ambient temperature and subsequent cooling gave the molecular organoperoxides [(Me4TACD)Li(OOR)(ROOH)] [R = tBu (16), CMe2Ph (17)] (Fig. 2, left).43 The same reactions without the Me4TACD ligand gave the dodecameric clusters [LiOOR]12. When Me4TACD was treated with [MN(SiMe3)2] (M = Na, K) and tBuOOH in a 1:1:5 ratio in n-pentane, corresponding molecular organoperoxides [(Me4TACD)M(OOtBu)(tBuOOH)3] (M = Na (18), K (19); Fig. 2, right) were obtained in good yields. In all the syntheses, an excess of organoperoxides was necessary to maintain the homogeneity of the reaction mixtures. Single-crystal X-ray diffraction studies confirmed the mononuclear nature of compounds 17, 18, and 19, with lithium in a hexa-coordinate environment, while sodium and potassium adopt an eight-coordinate, distorted square-antiprismatic geometry. The O–H⋯O hydrogen bonding between the metal-coordinated organoperoxides and neutral peroxides plays an important role in the stability of the complexes.
 | ||
| Fig. 2 Light alkali metal organoperoxides stabilized by Me4TACD. | ||
4. Group 2 metals
The Me4TACD ligand was utilized to stabilize molecular hydrides of group 2 metals, suppressing aggregation into saline MH2. Hydride complexes were synthesised via hydrogenolysis or hydrosilanolysis of organo- and silanido-alkaline earth precursors. Structural trends reflect decreasing electronegativity and increasing ionic radius and polarizability, with increasing atomic number. While discrete dimeric (di)cations [(Me4TACD)M2Hn](4−n)+ (n = 2, 3) were structurally characterised for M = Mg, Ca, Ba, only a trimeric [(Me4TACD)3Sr3H4(thf)]2+ cluster was isolated for the Sr2+ ion. [(Me4TACD)Mg2H2]2+ exhibits hydridic reactivity towards Lewis-acidic and polar unsaturated small molecules. The larger calcium congener can access a coordinatively unsaturated state; combined with highly nucleophilic hydride ligands, this enables H/D exchange under D2 and catalytic hydrogenation/hydrosilylation of unactivated n-alkenes. The extreme nucleophilicity of Sr–H bonds led to the isolation of a rare hexahydridosilicate complex. Calcium and strontium hydrides are highly labile and undergo dynamic hydride-exchange equilibria. Neutral and cationic allyl, benzyl, and silyl derivatives of calcium, strontium, and barium have also been described as molecular Me4TACD complexes. The Me4TACD ligand has also used to stabilize related dinuclear polyhydride complexes of lanthanides, including yttrium, ytterbium and lutetium, highlighting its broad application in molecular hydride chemistry.44–464.1. Magnesium
 | ||
| Scheme 6 Me4TACD-supported magnesium hydrides 20 and 21 and the molecular structure of the dicationic part of 21. Ar = C6H3-3,5-Me2. | ||
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, affording the monomeric magnesium complexes [(Me4TACD)MgX][B(C6H3-3,5-Me2)4] (X = Cl (22), CCPh (23)) (Scheme 7).48 Lewis acids, such as BH3(thf), HBpin and DIBAL(H) gave the hydride-bridged mononuclear adducts [(Me4TACD)Mg(μ-H)3BH][B(C6H3-3,5-Me2)4] (24), [(Me4TACD)Mg(μ-H)BHpin][B(C6H3-3,5-Me2)4] (25) and [(Me4TACD)Mg(μ-H)2AliBu2][B(C6H3-3,5-Me2)4] (26), respectively (Scheme 7).47 While insertion of CO2 into the Mg–H bond gave the dimeric formate complex [(Me4TACD)2Mg2(μ-O2CH)2][B(C6H3-3,5-Me2)4]2 (27), the reactions with PhN
CH, affording the monomeric magnesium complexes [(Me4TACD)MgX][B(C6H3-3,5-Me2)4] (X = Cl (22), CCPh (23)) (Scheme 7).48 Lewis acids, such as BH3(thf), HBpin and DIBAL(H) gave the hydride-bridged mononuclear adducts [(Me4TACD)Mg(μ-H)3BH][B(C6H3-3,5-Me2)4] (24), [(Me4TACD)Mg(μ-H)BHpin][B(C6H3-3,5-Me2)4] (25) and [(Me4TACD)Mg(μ-H)2AliBu2][B(C6H3-3,5-Me2)4] (26), respectively (Scheme 7).47 While insertion of CO2 into the Mg–H bond gave the dimeric formate complex [(Me4TACD)2Mg2(μ-O2CH)2][B(C6H3-3,5-Me2)4]2 (27), the reactions with PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO and (Dipp)NCN(Dipp) (Dipp = 2,6-iPr2-C6H3) afforded the monomeric compounds [(Me4TACD)Mg(OCHNPh)][B(C6H3-3,5-Me2)4] (28) and [(Me4TACD)Mg{(DippN)2-CH}2][B(C6H3-3,5-Me2)4] (29), respectively (Scheme 7).48 Similarly, benzaldehyde and benzophenone both insert into the Mg–H bond to afford the monomeric alkoxides [(Me4TACD)Mg(OCH(R)Ph)][B(C6H3-3,5-Me2)4] (R = Me (30); Ph (31)).47 Magnesium hydride 21 also reduces pyridine to give the 1,2-dihydridopyridyl (DHP) complex [(Me4TACD)Mg(1,2-DHP)][B(C6H3-3,5-Me2)4] (32a; Fig. 3), which isomerises to the 1,4-dihyridopyridyl isomer [(Me4TACD)Mg(1,4-DHP)][B(C6H3-3,5-Me2)4] (32b) in the presence of catalytic amount of [Mg(thf)6][B(C6H3-3,5-Me2)4]2.47
CO and (Dipp)NCN(Dipp) (Dipp = 2,6-iPr2-C6H3) afforded the monomeric compounds [(Me4TACD)Mg(OCHNPh)][B(C6H3-3,5-Me2)4] (28) and [(Me4TACD)Mg{(DippN)2-CH}2][B(C6H3-3,5-Me2)4] (29), respectively (Scheme 7).48 Similarly, benzaldehyde and benzophenone both insert into the Mg–H bond to afford the monomeric alkoxides [(Me4TACD)Mg(OCH(R)Ph)][B(C6H3-3,5-Me2)4] (R = Me (30); Ph (31)).47 Magnesium hydride 21 also reduces pyridine to give the 1,2-dihydridopyridyl (DHP) complex [(Me4TACD)Mg(1,2-DHP)][B(C6H3-3,5-Me2)4] (32a; Fig. 3), which isomerises to the 1,4-dihyridopyridyl isomer [(Me4TACD)Mg(1,4-DHP)][B(C6H3-3,5-Me2)4] (32b) in the presence of catalytic amount of [Mg(thf)6][B(C6H3-3,5-Me2)4]2.47
 | ||
| Scheme 7 Reactivity of magnesium hydride 21; [Mg] = [(Me4TCD)Mg]2+; Ar = C6H3-3,5-Me2. | ||
 | ||
| Fig. 3 Molecular structure of the cationic part of 1,2-dihydropyridyl complex 32a.47 | ||
Dihydridopyridyl complexes 32a and 32b undergo slow exchange with pyridine-d5 at 70 °C to give fully deuterated species 32a-d6 and 32b-d6via partially deuterated species 32a-d5 and 32b-d5. Due to the reversibility of the 1,2-insertion, gradual generation of the deuteride 21-d accounts for the fully deuterated species. Compounds 32a and 32b catalysed the hydroboration of pyridine using pinacolborane, providing a mixture of regioisomers.47 Compound 21 reacts with diphenyl disulfide to give the thiophenolate complex [(Me4TACD)Mg(SPh)][B(C6H3-3,5-Me2)4] (33) after H2 elimination.48
The dimeric formate complex 27 crystallized as a co-crystalline mixture of two conformers. The minor conformer displayed a rare folded conformation of the Me4TACD ligands (Fig. 1b), with one of the NMe groups pointing away from the metal.48 However, according to NMR spectroscopy, the ligand in solution adopts its usual C4-symmetric, boat-like conformation.
4.2. Calcium
 | ||
| Scheme 8 Synthesis of Me4TACD ligated calcium benzyl and silanide complexes 34–38. | ||
Dibenzyl 34 reacts with the weak Brønsted acid [NEt3H][BAr4] (Ar = C6H4-4-tBu, C6H3-3,5-Me2, C6H4-4-nBu) to yield the cationic benzyl complex [(Me4TACD)Ca(CH2Ph)(thf)][BAr4] (36a, Ar = C6H4-4-tBu; 36b, Ar = C6H3-3,5-Me2; 36c, Ar = C6H4-4-nBu) (Scheme 8). Loss of THF provides access to the η6-benzyl complex [(Me4TACD)Ca(CH2Ph)][B(C6H3-3,5-Me2)4] (36d).42 The related red ytterbium(II) analogues [(Me4TACD)Yb(CH2Ph)2] (34-Yb) and [(Me4TACD)Yb(CH2Ph)][B(C6H3-3,5-Me2)4] (36d-Yb) have also been synthesised and crystallographically characterised; similar to calcium, both benzyl moieties are η1-bonded for 34-Yb, but purple 36d-Yb crystallises as an η6-benzyl complex without coordinated THF.44 Addition of two equiv. of [NEt3H][BAr4] to 34 provided the dicationic bis(borate) salt [(Me4TACD)Ca(thf)2][BAr4] (37a, Ar = C6H4-4-tBu;4937b, Ar = C6H3-3,5-Me2).52 In THF-solution, 36b was found to exist in equilibrium with the zwitterionic compound [(Me4TACD)Ca{CH2(C6H3-3-BAr3-5-Me)}] (38, Ar = C6H3-3,5-Me2) via deprotonation of one of the meta-methyl groups of the borate anion and elimination of toluene.49 Notably, in contrast to 34, the putative neutral dibenzyl complex of the pentadentate aza-macrocycle Me5PACP (Me5PACP = N,N′,N′′,N′′′,N′′′′-pentamethyl-1,4,7,10,13-pentaazacyclopentadecane) is unstable to ligand decomposition.53
Cationic calcium derivatives can be accessed by protonolysis of neutral bis(organo)calcium or bis(silanido)calcium precursors using cationic conjugate acid of Me4TACD (Scheme 9).42,53–55 Reaction of [(Me4TACD)H][B(C6H3-3,5-Me2)4] with a THP slurry of THF-free dibenzylcalcium [Ca(CH2Ph)2], provided direct access to the η6-benzyl complex 36d.42 Similarly, bis(allyl)calcium reacts with [(Me4TACD)H][B(C6H3-3,5-Me2)4] in THF to yield the cationic allyl complex [(Me4TACD)Ca(η3-C3H5)(thf)][B(C6H3-3,5-Me2)4] (39) under elimination of propene.5439 adopts a distorted pseudo-trigonal prismatic geometry with the allyl ligand coordinating in an η3-manner. Protonolysis of bis(triphenylsilanido)calcium with [(Me4TACD)H][B(C6H3-3,5-Me2)4] yielded a cationic silanide complex, 40, characterised by NMR spectroscopy.42
 | ||
| Scheme 9 Direct access to cationic calcium η6-benzyl (36d), η3-allyl (39), and triphenylsilanide (40) complexes by protonolysis using [(Me4TACD)H][BAr4]. The cationic part of the crystal structure of compound 39. | ||
 | ||
| Scheme 10 Hydrogenolysis of compound 35 and molecular structure of dicalcium trihydride silanide salt 41a. | ||
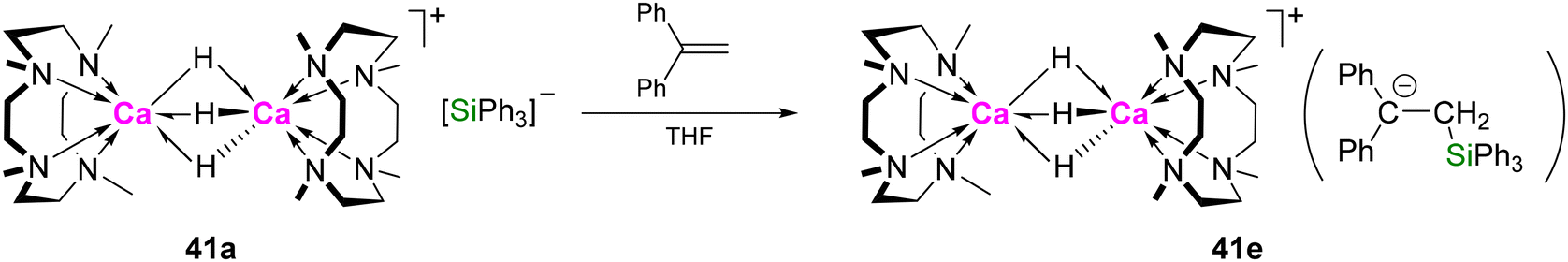
Hydrogenolysis of 34 provided an insoluble precipitate, likely CaH2, along with free Me4TACD and decomposition products. However, using triphenylsilane as a hydride source produced a dimeric dicalcium dihydride cation, which crystallised as its triphenylsilylbenzyl salt [(Me4TACD)2Ca2(μ-H)2][PhCHSiPh3]2 (42a) (Scheme 11).49 The crystal structure of 42a consists of a dicationic Ci-symmetric dimer with two six-coordinate calcium centres bridged by two μ-hydride ligands. 42a decomposes in THF-d8 (t1/2 = 6 h) via ligand degradation with formation of PhCH2SiPh3 amongst other species. Due to the reactive silylbenzyl anion, 42a activates H2 in an FLP-like manner to yield the dimeric trihydride cation [(Me4TACD)2Ca2(μ-H)3][PhCHSiPh3] (41b) under elimination of PhCH2SiPh3. 41b can also be formed directly by hydrogenolysis of 34 in the presence of PhCH2SiPh3.
 | ||
| Scheme 11 Synthesis of dicalcium di- and trihydrides 42a and 41bvia hydrogenolysis and silanolysis of benzyl precursors in THF. Molecular structure of compound 42a with selected hydrogen atoms shown. | ||
Molecular calcium di- and trihydride dimers can be accessed as relatively robust tetraarylborate salts via hydrogenolysis of 36 or a 1:1 mixture of 36 and 34 (Scheme 12), respectively providing [(Me4TACD)Ca2(μ-H)2][BAr4]2 (42b, Ar = C6H4-4-tBu; 42c, Ar = C6H3-3,5-Me2) or [(Me4TACD)Ca2(μ-H)3][BAr4] (41c, Ar = C6H4-4-tBu; 41d, Ar = C6H3-3,5-Me2).4941c can be converted to 42cvia protonolysis by addition of [NEt3H][B(C6H4-4-tBu)4], or through hydride-redistribution by combining with calcium bis(borate) 37a. Hydrogenolysis of 3642,53 or silanolysis of either 3642,53 or 3954 with RSiH3 (R = n-octyl or Ph), followed by crystallisation from THF/n-pentane provides reliable access to the dimeric dihydride dication as a THF-solvate [(Me4TACD)2Ca2(μ-H)2(thf)][BAr4]2 (42d, Ar = C6H3-3,5-Me2;5442e, Ar = C6H3-4-nBu).53
 | ||
| Scheme 12 Synthesis of dimeric calcium hydrides 41c,d, and 42b–e in THF. | ||
The crystal structures of 42d (Fig. 4),54 and 42e53 reveal a dimeric dication with six- and seven-coordinate calcium centres bridged by two μ-hydrides. Compared to 42a, the seven-coordinate calcium centre exhibits longer Ca–H distances, leading to an elongated Ca–Ca separation (3.6306(11) Å vs. 3.4650(10) Å). The red Yb(II) congener [(Me4TACD)2Yb2(μ-H)2(thf)][B(C6H3-3,5-Me2)4]2 is isostructural to 42d, and was prepared similarly by hydrogenolysis of the corresponding cationic Yb(II) benzyl complex.44
 | ||
| Fig. 4 Molecular structure of the dicationic part of compound 42d. | ||
In THF-d8 solution, the anions have minimal effect on the hydride resonances of trihydride complexes 41a–d, with silanide, silylbenzyl, and tetraarylborate salts all displaying a singlet at δ 4.71–4.73 ppm in their 1H NMR spectra.49,50 However, the dihydride silylbenzyl salt 42a exhibits a significantly downfield-shifted hydride resonance (δ 4.70 ppm)49 compared to borate salts 42b–e (δ 4.49–4.54 ppm).42,49,53,54 This indicates that whilst compounds 41a–d and 42b–e exist as charge-separated species in THF, a significant anion–cation interaction may exist for 42a. THF coordination to the dicationic core in 42 is highly labile, as 42c and 42d exhibit identical NMR spectra with time-averaged Ci-symmetry, and THF resonates at the same shift as free solvent. Coordination of THF to the unsolvated dimer [(Me4TACD)2Ca2H2]2+ is mildly exothermic (ca. 33 kJ mol−1),54 consistent with Lewis acidity of the relatively large and coordinatively unsaturated metal cation.
Molecular calcium hydrides commonly adopt a dimeric dihydride structure when supported by bulky or highly coordinating ligands to suppress further aggregation. The dimeric trihydride motif [(L)2M2H3]+, however, is unique in alkaline-earth chemistry to cationic complexes supported by neutral macrocycles, observed only in compounds 41a–d and the related strontium hydride [(Me5PACP)2Sr2(μ-H)3][B(C6H3-3,5-Me2)4].52 The ability to accept a third hydride reflects the electrophilic and coordinatively unsaturated nature of the metal cations in the dimeric dihydride. The combination of nucleophilic hydride and electrophilic metal centre is crucial to the observed reactivity of such complexes towards kinetically inert substrates such as ethylene and carbon monoxide (vida infra), and in the catalytic hydrogenation and hydrosilylation of olefins. The kinetic stabilisation of low-nuclearity calcium hydrides is dependent on ligand bulk and coordination strength. Employing the smaller Me3TACN (N,N′,N′′-trimethyl-1,4,7-triazacyclononane) ligand results in the isolation of tetranuclear [(Me3TACN)4Ca4(μ-H)6][B(C6H3-3,5-Me2)4]2.55 Discrete calcium hydride clusters can be isolated via silanolysis of amide precursors.56,57 Cluster nuclearity can be controlled by the size of supporting amine and/or amide ligands, with smaller clusters exhibiting higher activity as hydrogenation catalysts.58,59
Dihydrogen and hydrosilanes. The silanide salt of dicalcium trihydride 41a exchanges with D2 forming 41a-d3 and HD via mixed isotopomers (Scheme 13). This was proposed to involve the silyl anion, as carrying out the reaction with D2 in the presence of HSiPh3 also resulted in H/D exchange to give DSiPh3. Dimer-dissociation is implied by the rapid equilibration of 41a and 41a-d3 to a mixture of isotopomers.5042b also exchanges with D2; formation of HD was observed by NMR spectroscopy, and complete deuteration occurred after 8 h (1 bar pressure), via intermediate formation of the monodeutero isotopomer (Scheme 13). Exposing 42b to an equimolar mixture of D2 and H2 or HD resulted in rapid equilibration to a mixture of isotopomers after 5 min with respective formation of HD or H2 and D2.49
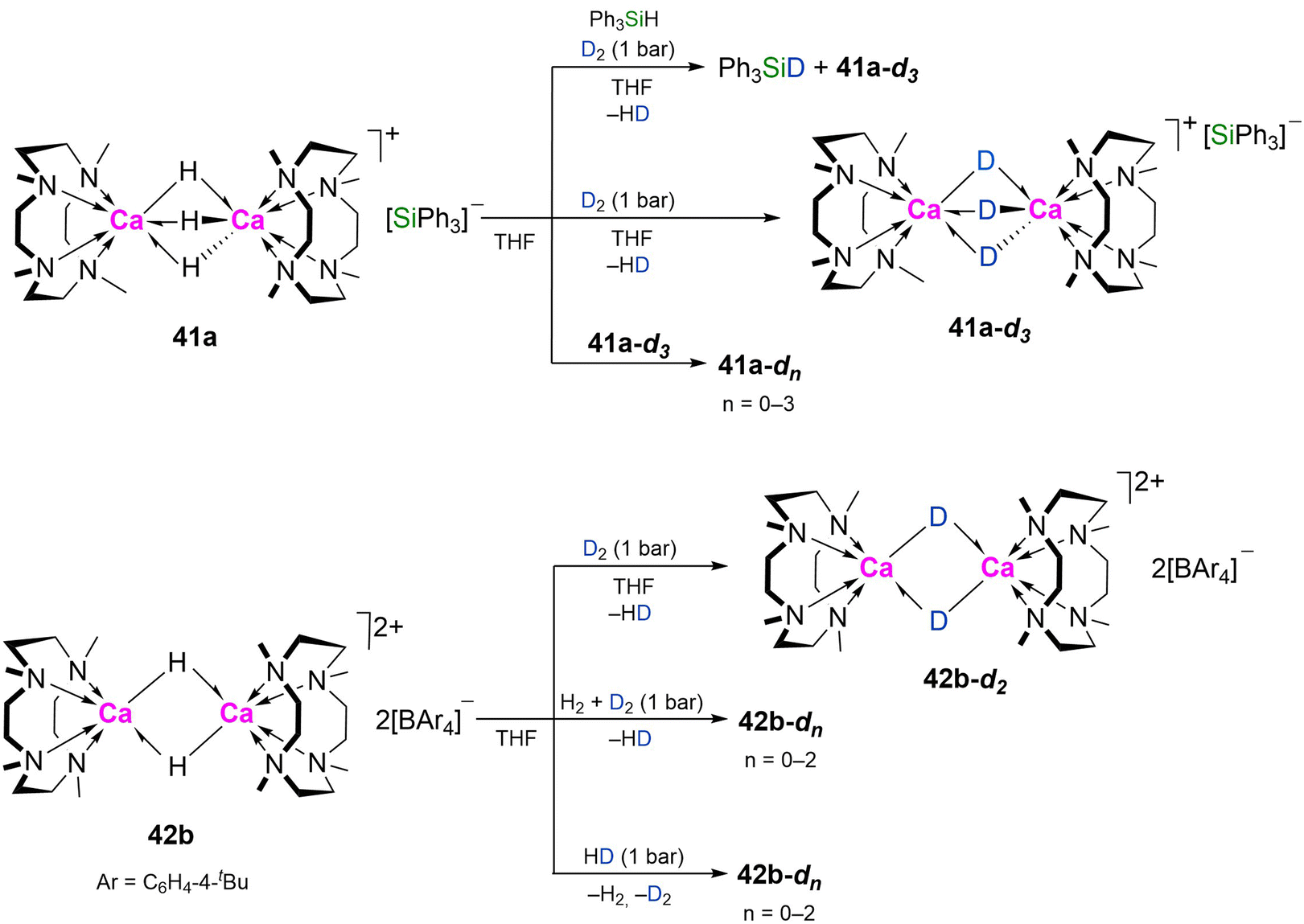 | ||
| Scheme 13 Isotopic exchange of 41a and 42b. | ||
42d catalyses organosilane redistribution, converting PhSiH3 into Ph2SiH2 and SiH4 (Scheme 14). Exposing a solution of 42d and RSiH3 (R = n-octyl, Ph) to D2 produces HD, 42d-dn, and an isotopomeric mixture of hydrosilanes.54 Broad SiH and CaH resonances in the 1H NMR spectrum suggest mutual exchange according to EXSY NMR experiments, while NOESY NMR reveals NOE correlations between the silicon hydrides and NCH3 resonances. The reaction likely involves hypervalent silicates formed via nucleophilic hydride attack on silane. Similar propositions have been made for [(BDIdipp)Ca(μ-H)]2 (BDIdipp = HC{C(CH3)N(C6H3-2,6-iPr2)}2) synthesis using PhSiH3 as a hydride source.3 Hypervalent silicates have been experimentally and computationally implicated as intermediates in alkaline-earth catalysed Si–E dehydrocoupling (E = N, O) reactions, in preference to concerted σ-bond metathesis.60–62 Further, crystallographically characterised calcium and strontium hydridosilicate complexes derived from the reaction of organo- and amido-alkaline earth complexes with hydrosilanes have also been isolated (vide infra).63,64
 | ||
| Scheme 14 Calcium-mediated redistribution and deuteration of organosilanes. | ||
Insertion of unsaturated C–X multiple bonds. The nucleophilic hydrides of 42d readily insert CO2, forming the dimeric formate complex [(Me4TACD)2Ca2(μ-OCHO)2(thf)2][B(C6H3-3,5-Me2)4]2 (43, Scheme 15),65 and react with CO to yield the cis-ethenediolate complex [(Me4TACD)2Ca2(OC(H)C(H)O)2][B(C6H3-3,5-Me2)4]2 (44, Scheme 15). Monomerization of 42d was calculated to be only slightly endothermic; DFT calculations suggest that CO insertion occurs at the monomeric hydride [(Me4TACD)Ca(H)(thf)x]+, providing a formyl intermediate that subsequently dimerizes into 44 (Scheme 15).65 The relative ease by which 42d can monomerize in THF solution carries important mechanistic implications in hydrofunctionalisation catalysis. Dimeric β-diketiminato magnesium and calcium hydrides [(BDIdipp)M(μ-H)(thf)n]2 (M = Mg, n = 0; M = Ca, n = 1) also react with CO to form cis-enediolates.66–68 In these cases, however, the dimer is proposed to remain intact throughout the reaction, with a calculated mechanism involving initial two-fold hydride insertion to give an oxomethylene intermediate, followed by insertion of a second CO molecule and subsequent 1,2-hydride shift.
 | ||
| Scheme 15 Reactivity of compound 42d towards small molecules, and molecular structure of cis-enediolate 44. Ar = C6H3-3,5-Me2. | ||
Ethylene readily inserts into the Ca–H bonds of 42d, forming the unstable ethyl-calcium complex [(Me4TACD)CaEt(thf)x]+ (45), observed via1H NMR spectroscopy. This species decomposes rapidly (t1/2 = 10 min) into an intractable mixture.54 Similar to [(BDIdipp)Ca(μ-H)]2 and [(BDIdipep)Sr(μ-H)]2 (BDIdipep = HC{C(CH3)N(C6H3-2,6-(C(H)Et2)2)}2),3,6945 also reacts further with ethylene to generate labile calcium n-alkyl species. The nuclearity of 45 remains unclear; given the low energy barrier to hydride monomerization, which is proposed to occur during hydrogenation and hydrosilylation catalysis,49,53,65,70 and the isolation of a related mononuclear seven-coordinate calcium ethyl cation [(Me5PACP)CaEt(thf)][B(C6H3-3,5-Me2)4],53 it is plausible that Me4TACD-supported calcium n-alkyl complexes also exist as monomers in THF solution.
Nucleophilic and Brønsted-basic reactivity. The nucleophilic hydride of 42d reacts with alkynyl silanes Me3SiC
CSiMe3 and HMe2SiCCSiMe3via nucleophilic substitution, forming dimeric alkynyl calcium complex [(Me4TACD)2Ca2(μ-CCSiMe3)2][B(C6H3-3,5-Me2)4]2 (46a) after elimination of Me3SiH or Me2SiH2, respectively (Scheme 16).65 Similarly, reaction with N(SiMe2H)3, MeSiH(OMe)2, or O(SiMe2H)2 resulted in nucleophilic substitution to form [(Me4TACD)Ca(N(SiHMe2)2)][B(C6H3-3,5-Me2)4] (47), [(Me4TACD)2Ca2(μ-OMe)2][B(C6H3-3,5-Me2)4]2 (48), or [(Me4TACD)Ca(OSiMe2H)][B(C6H3-3,5-Me2)4] (49) with elimination of Me2SiH2 or MeSiH2OMe.54,65 Whereas 48 crystallises as a dimer with bridging methoxide ligands, 47 is monomeric with an anagostic interaction between calcium and one of the Si–H bonds. Similarly, reactions with Me3SiI and Me3SiN3 provide monomeric iodide, and dimeric azide complexes, [(Me4TACD)CaI(thf)2][B(C6H3-3,5-Me2)4] (50) and [(Me4TACD)2Ca2(μ-N3)2][B(C6H3-3,5-Me2)4]2 (51) after eliminating Me3SiH.65 Hydridic reactivity of 42d is also observed in the reaction with BH3·THF leading to tetrahydridoborate complex [(Me4TACD)Ca(BH4)(thf)2][B(C6H3-3,5-Me2)4] (52).65 The strong nucleophilicity of 42d enables nucleophilic aromatic substitution with fluorobenzene, cleaving the Csp2–F bond to form the dimeric fluoride complex [(Me4TACD)2Ca2(μ-F)2(thf)][B(C6H3-3,5-Me2)4]2 (53) with benzene elimination.65 Structurally, 53 resembles the parent hydride, with two fluoride ligands bridging six- and seven-coordinate calcium centres. As a strong Brønsted base, 42d deprotonates RCCH (R = SiMe3, C3H5) to yield dimeric acetylides, 46a and [(Me4TACD)2Ca2(μ-CCC3H5)2][B(C6H3-3,5-Me2)4]2 (46b), and reacts with trans,trans-1,4-diphenylbutadiene to provide the dinuclear butadienyl complex [(Me4TACD)2Ca2(μ2–η4-1,4-Ph2C4H2)][B(C6H3-3,5-Me2)4]2 (54) (Fig. 5). Anisole and 1,3-dimethoxybenzene are deprotonated, forming aryl-calcium complexes [(Me4TACD)Ca(κ2-O,C-C6H4-6-OMe)(thf)][B(C6H3-3,5-Me2)4] (55a) and [(Me4TACD)Ca(κ2-O,C-C6H4-2,6-(OMe)2)(thf)][B(C6H3-3,5-Me2)4] (55b).42 Compound 55b undergoes σ-bond metathesis with nOctSiH3 to regenerate 42d under elimination of nOct(C6H3-2,6-(OMe)2)SiH2.
 | ||
| Scheme 16 Reactivity of calcium hydride 42d. [Ca] = [(Me4TACD)Ca]2+; Ar = C6H3-3,5-Me2. | ||
 | ||
| Fig. 5 Molecular structure of the dicationic part of the butadienyl calcium complex 54.65 | ||
 | ||
| Scheme 17 Silanide-centred reactivity of 41a towards 1,1′-diphenylethene. | ||
The calcium complex 41e catalyses the hydrogenation of 1,1′-diphenylethylene at 60 °C within 24 h under 1 bar H2, likely via Ca–H insertion and subsequent σ-bond metathesis (Scheme 18a). The borate-salt 41c catalysed the same reaction in 12 h at 25 °C, whilst the dicationic dihydride 42b is more active, achieving 98% conversion in 6 h under ambient conditions.4942b also catalyses the hydrogenation of styrene (10 h, 25 °C), 1,2-diphenylethylene (24 h, 60 °C), triphenyl- (16 h, 60 °C) and (trimethyl)vinylsilane (36 h, 60 °C). Catalytic activity was not restricted to activated alkenes; 1-hexene, 1-octene, 3-vinylcyclohexene, 1,5-hexadiene, and 1,9-decadiene were also hydrogenated (Scheme 18b). Dihydride 42b displayed higher activity than trihydride 41c. No hydrogenation of internal double bonds (3-vinylcyclohexene, cyclohexene) was observed. For 1-hexene, 1-octene, 1,5-hexadiene, and 1,9-decadiene, 5% 2-alkene was observed in the product mixture; for 1,5-hexadiene, 4% of the cyclisation product methylcyclopentane was also formed.
 | ||
| Scheme 18 Hydrogenation of (a) activated, (b) unactivated alkenes catalysed by calcium hydrides 41c,e, and 42b. | ||
The activity of compounds 41c,e, and especially 42b in alkene hydrogenation compares well with other alkaline-earth catalysts. Bulky cyclopentadienyl-derivatives [(η5-C5R5)2M2(μ-H)2(L)] (M = Ca, Sr, Ba; R = C6H3-3,5-iPr; L = THF or DABCO) also catalyse alkene hydrogenation, including unactivated n-alkenes at 30 °C under 6 bar H2, with activity increasing with metal size.71 By comparison, a magnesium PNP pincer complex [{C5H3N(C(H)PtBu2)(CH2PtBu2)}2Mg2Et2(μ-1,4-dioxane)] was reported to catalyse the hydrogenation of alkenes including unactivated 1-dodecene, 1-octene, 3-(ortho-methoxy)phenyl-prop-1-ene, 4-phenyl-but-1-ene, and 3-(trimethylsilyl)-prop-1-ene, but requires harsh conditions (120 °C, 5 bar).72 A related calcium-based system was limited to activated substrates like 1,1′-diphenylethene and styrene.73 The β-diketiminato derivative [(BDIdipp)Ca(μ-H)(thf)]2 catalyses the hydrogenation of 1,1′-DPE and styrene derivatives at 60 °C under 20 bar H2 pressure.74 In contrast, the unsolvated analogue [(BDIdipp)Ca(μ-H)]2 catalyses the hydrogenation of unactivated n-alkenes at ambient conditions,75 although catalysis was limited to room temperature with slow conversion (21 d), due to competitive nucleophilic alkylation of benzene at elevated temperature.3,75 The Me4TACD-based systems are thermally robust, permitting mildly elevated temperatures and appreciable rates. High activity compared to [(BDIdipp)Ca(μ-H)(thf)]274 was attributed to a high degree of Lewis acidity and facile monomerization due to cationic charge. Notably, heavier alkaline-earth amides [Ae(NRR′)2]n (R = SiMe3, SiiPr3; R′ = SiMe3, SiiPr3, C6H3-2,6-iPr2) act as pre-catalysts for the efficient hydrogenation and transfer hydrogenation of challenging unactivated n-alkenes, as well as internal secondary alkenes and aromatic rings under relatively mild conditions (up to 120 °C, 1–6 bar H2).58,59,76,77 Here, multinuclear (amido)alkaline-earth hydride clusters are proposed as active species, with bulkier amides leading to lower nuclearity and higher activity.
Hydride 42d also catalysed alkene hydrosilylation.54 Ethylene was hydrosilylated by various aromatic and aliphatic hydrosilanes at 70 °C in under 60 min (Scheme 19a). Primary and secondary hydrosilanes yielded di- and monoethylated silanes. Longer-chain aliphatic n-alkenes were hydrosilylated more slowly (24 h, 70 °C, conversion 70–96%) with anti-Markovnikov regioselectivity (Scheme 19b). Markovnikov products were observed for aryl-substituted olefins (Scheme 19c), as the result of a π-interaction of the phenyl group with the Lewis-acidic Ca centre. A mixture of Markovnikov and anti-Markovnikov products was obtained for the hydrosilylation of triphenyl(vinyl)silane with n-octylsilane. Internal double bonds were not hydrosilylated. Arylsilanes undergo scrambling reactions promoted by the electrophilic calcium hydride (Scheme 14), providing SiH4 and Ph2SiH2 in the case of phenylsilane, and alkoxy and siloxy calcium derivatives (e.g., 48, 49, Scheme 16)54,65 from alkoxy- and siloxy-substituted hydrosilanes, thus making aliphatic hydrosilanes preferable. Attempted hydrosilylation of terminal alkyne HCCSiMe3 with nOctSiH3 using 5 mol% 42d instead provided dehydrocoupling products (Me3SiCC)(nOct)SiH2 and (Me3SiCC)2(nOct)SiH in a 2:1 ratio (Scheme 19e).42
 | ||
| Scheme 19 Hydrosilylation of olefins catalysed by 42b: (a) hydrosilylation of ethylene; (b) anti-Markovnikov selective hydrosilylation of aliphatic 1-alkenes; (c) Markovnikov selective hydrosilylation of styrene derivatives; (d) hydrosilylation of triphenyl(vinyl)silane with mixed selectivity; (e) dehydrocoupling of trimethylsilylacetylene and n-octyl silane. | ||
Monomerization of [(Me4TACD)2Ca2(μ-H)2(thf)x]2+ to give a reactive terminal hydride species [(Me4TACD)Ca(H)(thf)y]+ was suggested to precede alkene insertion and catalytic turnover (Scheme 20). While monomeric hydride or alkyl-derivatives were not isolated for Me4TACD, a mononuclear terminal ethyl complex [(Me5PACP)CaEt(thf)][B(C6H3-3,5-Me2)4], was crystallised for the larger 15-membered macrocycle, Me5PACP.53 Further, kinetic studies on the hydrosilylation of 1-octene by n-octylsilane showed a 1/2-order dependence on dimeric hydride pre-catalyst for both 42d and [(Me5PACP)2Ca2(μ-H)2][B(C6H4-4-nBu)4]2, implying a monomeric active species in both cases.53 Despite increased steric and coordinative demand, the Me5PACP derivative was more active than 42d, likely due to easier access to mononuclear species for the larger ligand. Catalysis is first-order in 1-octene and pseudo-zeroth order in n-octylsilane, suggesting rate-limiting alkene insertion and rapid σ-bond metathesis of the n-alkyl intermediate with hydrosilane.
 | ||
| Scheme 20 Contrast in proposed mechanisms for olefin hydrogenation and hydrosilylation mediated by Me4TACD/Me5PACP-calcium and BDIdipp-calcium/magnesium catalysts. | ||
When compared with the similarly dimeric pre-catalyst [(BDIdipp)Ca(μ-H)]2 (Scheme 20), dimeric alkyl-hydride species [(BDIdipp)2Ca2(μ-H)(μ-R)] (R = n-alkyl) were observed by in situ NMR spectroscopy as a resting-state in the catalytic run, indicating a persistent dimeric active species, slow insertion of alkene into the second μ-H, and rate-limiting σ-bond metathesis with H2.75 Dimeric dialkyl insertion products [(BDIdipp)Ca(μ-R)]2 could be isolated in the absence of H2.3,75,78 Conversely, the hydride resonance in the 1H NMR spectrum of 42b-mediated n-alkene hydrogenation remained unchanged in catalysis, and attempts to isolate insertion products of 1-hexene or 3-vinylcyclohexane failed, suggesting reversible and rate-limiting alkene insertion/β-hydride elimination and rapid σ-bond metathesis with H2.49,53
The hydrosilylation of unactivated n-alkenes using phenylsilane has also been reported for the dimeric magnesium pre-catalyst [(BDIdipp)Mg(μ-H)]2.79 Near-quantitative conversion was achieved in over 4 d at 60 °C, with bulkier or aryl alkenes like styrene showing reduced activity. Similar to the calcium analogue, n-alkene insertion was calculated to occur at the intact dimeric μ-hydride complex rather than through mononuclear species, although ready dissociation of the resultant mixed hydride-alkyl species [(BDIdipp)2Mg2(μ-H)(μ-R)] implicates mononuclear on-cycle intermediates during catalysis (Scheme 20). The faster reaction rates observed for 42b compared to [(BDIdipp)Mg(μ-H)]2 are likely due to calcium's lower electronegativity, stronger polarization, and lower steric hindrance at the metal centre, facilitating more efficient alkene activation and hydrosilylation. The cationic charge of the active species may also play a role in promoting the coordination and polarisation of alkene and silane during catalysis.
The mechanism of the hydrogenation of 1-alkenes mediated by [(Me4TACD)2Ca2(μ-H)2]2+ as pre-catalyst was studied computationally.70 A mechanism involving an intact μ-H bridged dimer was computed to be more energetically favourable for H2 isotope exchange than with a mononuclear terminal hydride. Anti-Markovnikov addition of aliphatic 1-alkenes was competitive for either hydride-bridged dimer or mononuclear terminal hydride [(Me4TACD)Ca(H)(thf)]+, whilst Markovnikov addition of styrene was observed for the mononuclear hydride, influenced by Ca+–Ph cation–π interactions, which directed regioselectivity.
In summary, it appears that the ability for chelating polydentate aza-macrocycles to infer relatively high stability towards highly Lewis acidic mononuclear calcium cations may play an important role in the comparatively high activity of calcium hydride complexes 42b,d as pre-catalysts for hydrogenation and hydrosilylation of unactivated alkenes.
4.3. Strontium
 | ||
| Scheme 21 Synthesis of dibenzyl strontium complexes 56a,b, cationic benzyl complexes 57a–c, and strontium bis(borate) salt 58. Molecular structure of the cationic part of 57c, with benzylic hydrogen atoms shown. | ||
Whilst hydrogenolysis or silanolysis of 36 provides access to dimeric calcium dihydride, hydrogenolysis of 57a in THF leads to a complex mixture.80 A single species repeatedly crystallised from these solutions, and was structurally characterised as the trinuclear cluster dication, [(Me4TACD)3Sr3(μ-H)4(thf)][B(C6H3-3,5-Me2)4]2 (59), which can be rationally synthesised by hydrogenating a 2:1 mixture of 57a and 56b (Scheme 22).
 | ||
| Scheme 22 Synthesis of trinuclear strontium hydride 59, isotopic exchange with D2, and proposed formation of dimeric [L2Sr2H2]2+ species in solution. | ||
The trinuclear dication of 59 (Fig. 6) consists of three seven-coordinate strontium centres and can be described as an adduct of [(Me4TACD)SrH2(thf)], and [(Me4TACD)2Sr2(μ-H)2]2+ units, or of two [(Me4TACD)SrH2] units and a [(Me4TACD)Sr(thf)]2+ dication. Sr1 bridges Sr2 and Sr3 via a hydride ligand and coordinates to a THF ligand, while Sr2 and Sr3 share two bridging hydrides. The 1H NMR spectrum reveals two distinct {(Me4TACD)Sr} environments, with a single hydride resonance at δ 5.98 ppm, indicating rapid hydride exchange between the distinct strontium centres even at −60 °C. Complex 59 rapidly exchanges with D2, providing the fully deuterated isotopologue after 30 min at room temperature (Scheme 22). Combining 59 and 58 resulted in an additional hydride resonance at δ 5.92 ppm in the 1H NMR spectrum, which was tentatively assigned to the elusive [(Me4TACD)2Sr2(μ-H)2(thf)x]2+, although attempts to isolate it remained unsuccessful. Me4TACD lacks sufficient steric and coordinative demand to prevent redistribution. A stable [Sr2H2]2+ dication, [(Me5PACP)2Sr2(μ-H)2][B(C6H3-3,5-Me2)4]2, was isolated using the larger 15-membered macrocycle Me5PACP.52
 | ||
| Fig. 6 Molecular structure of the cationic part of trinuclear strontium hydride 59.80 | ||
Hydrogenation of an equimolar THF solution of 57a and 36b provided the heterobimetallic hydride complex [(Me4TACD)2CaSr(μ-H)2(thf)][B(C6H3-3,5-Me2)4]2 (60; Scheme 23), which is isostructural to 42d.54 The crystal structure of 60 revealed a Ca(μ-H)2Sr core, where the calcium is six-coordinate and the strontium is seven-coordinate with an additional THF ligand. The 1H NMR analysis of 60 in THF-d8 showed a major hydride resonance at δ 5.10 ppm, intermediate between homometallic Sr hydride 59 (δ 5.98 ppm) and Ca hydrides 41 (δ 4.72 ppm) and 42 (δ 4.45 ppm).80 Notably, resonances corresponding to calcium hydride dimers 41 and 42, and several broad resonances between δ 5.98 and 5.92 ppm for strontium hydrides, were also observed in the spectrum. 1H–1H EXSY experiments confirmed rapid exchange between these species, suggesting facile dissociation and recombination of 60 in solution. Thermodynamic studies indicated that Ca–H bonds are more favourable than Sr–H bonds, as excess [(L)Ca]2+ shifted the equilibrium toward calcium hydrides. Mixing calcium bis-borate 37b with calcium–strontium hydride 60 resulted in the selective formation of calcium hydride dimer 41 and strontium bis-borate 58. Conversely, combining 58 and 60 resulted in the persistence of 60 as the major species, with minor quantities of 41 and 42 and traces of postulated [(Me4TACD)2Sr2(μ-H)2(thf)x][B(C6H3-3,5-Me2)4]2. Complexes 59 and 60 catalyse n-alkene hydrogenation,80 but quantitative activity comparison with homometallic calcium hydride dimers 41 and 42 is not appropriate due to the aforementioned hydride lability and poorly-defined solution-state speciation.
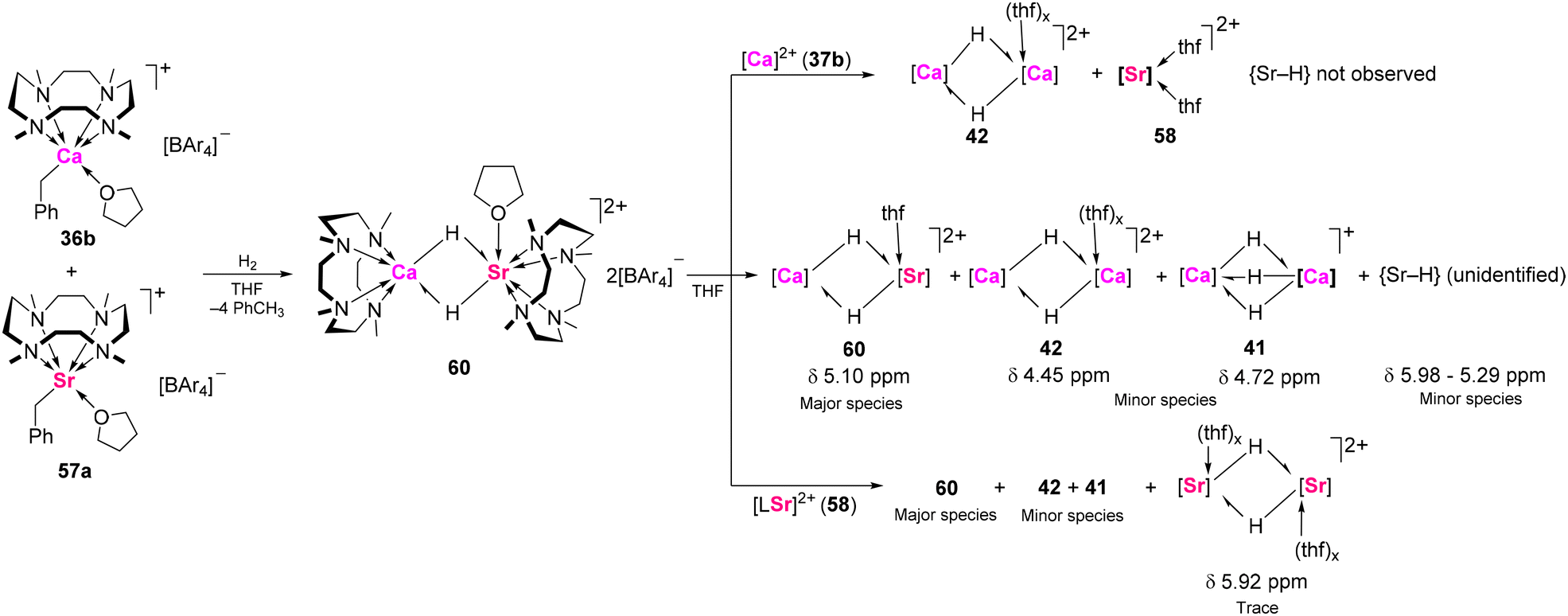 | ||
| Scheme 23 Synthesis of Sr/Ca hydride 60 and hydride exchange reactions with 37b and 58 in solution. 1H NMR chemical shifts are shown for diagnostic hydride resonances. Ar = C6H3-3,5-Me2. | ||
:1 mixture of 57a and 57b with excess phenylsilane at room temperature produced the trinuclear hydride-silanide cluster, [(Me4TACD)3Sr3(μ-H)3(μ3-SiH3)2][B(C6H3-3,5-Me2)4] (63). The observed evolution of H2 in this reaction also suggests the dehydrogenation of a hypervalent silicate species.
 | ||
| Scheme 24 Synthesis of strontium hydridosilicate complexes 61a,b and 64, and silanide complexes 62a,b and 63. | ||
Similarly, the synthesis and isolation of a carbozolido barium silanide complex via metathesis of a hexamethyldisilazide precursor with PhSiH3 has been reported; the {SiH3}− moiety was found to act either as nucleophilic silanide, or as a hydride surrogate.83 Heavy alkaline-earth elements facilitate hydrido(aryl)silanes redistribution, but using nOctSiH3 as hydride source suppresses such redistribution reactions, yielding the trinuclear hydride-hydridosilicate cluster, [(Me4TACD)3Sr3(μ-H)3(μ3-SiH5(nOct))][B(C6H3-3,5-Me2)4] (64) with the n-alkyl group intact. Compound 64 was rationally synthesised in 65% yield by reacting a 2:1 mixture of 56b and 57a with a ten-fold excess of nOctSiH3 (Scheme 24). These complexes likely result from two-fold nucleophilic addition of highly reactive [(Me4TACD)SrH]+ units to either SiH4 (derived from [(Me4TACD)SrH]+ mediated organosilane scrambling), or nOctSiH3. This interpretation corroborates with the broadened and exchanging hydride resonances observed for calcium hydride 42d and PhSiH3 mixture, where Ph2SiH2 and SiH4 are also observed, implicating hydridosilicate species.54
Similar hydridosilicate complexes were not isolated for the analogous reaction of either 36, 39,42,54 or [(Me5PACP)Sr(CH2Ph)(thf)][BAr4] (Ar = C6H3-3,5-Me2 or C6H4-4-nBu)52 with RSiH3, suggesting that the kinetic stability of 61 depends on the high nucleophilic character of the Sr–H bonds, and the moderate size of the Me4TACD ligand relative to the ionic radius of Sr2+. Previously, a dinuclear ruthenium hexahydridosilicate complex [{(PhB(CH2PPh2)3)Ru}2{μ–η4,η4-SiH6}] was reported.84 In this case, the {SiH6} unit binds to the ruthenium centres via covalent 3-centre-2-electron bonding interactions. By contrast, the interaction between Sr and {SiH6} in 61 is predominantly electrostatic, as indicated by NMR and vibrational spectroscopy, as well as DFT calculations. Recently, a comparable calcium–potassium hexahydridosilicate complex [(NON)2Ca2K2(μ4–κ3:κ3:κ2:κ2-SiH6)(thf)2] (NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethyl-xanthene) was reported, and similarly reacts as a masked metal hydride under SiH4 elimination.64 Unlike 61, [(NON)2Ca2(μ4–κ3:κ3:κ2:κ2-SiH6)(thf)2] is stable in benzene at room temperature, probably due to a more robust binding cavity offered by the tetranuclear Ca2K2 assembly compared to the labile dinuclear structure of 61. A strontium–potassium pentahydrido(aryl)silicate complex [(NON)2Sr2K2(μ4–κ3:κ3:κ2:κ2-PhSiH5)(thf)2] reminiscent of 64 was also reported.64
Single crystal X-ray diffraction revealed the cationic part of 61 to formally contain an [SiH6]2− dianion sandwiched in a μ–κ3:κ3-fashion between two [(Me4TACD)Sr(thf)2]2+ dications (Fig. 7). The dinuclear core exhibits flexibility around the Sr–Sr axis. In the solid-state, 61a contains a non-centrosymmetric dication with a gauche arrangement of the neutral ligands on each Sr, whilst 61b is crystallographically centrosymmetric, with an anti-periplanar arrangement.
 | ||
| Fig. 7 The molecular structure of dication [61b]2+, with only silicon-bound hydrogen atoms.54 | ||
Compound 61 was characterised by NMR spectroscopy at low temperatures due to its instability in THF above 0 °C. In the 1H NMR spectrum, the hydrides appeared as a singlet at δ 5.39 ppm, sharpening when cooled to −40 °C. Direct 29Si NMR measurement of 61 was unsuccessful, but at −40 °C, a signal was observed by 29Si–1H HSQC and HMBC experiments at δSi −172.6 ppm, appearing as a doublet with a coupling constant J(29Si–1H) = 118 Hz. These spectroscopic features indicate largely ionic Sr–H bonding interactions and thus a relatively unperturbed [SiH6]2− dianion compared to [{(PhB(CH2PPh2)3)Ru}2{μ–η4,η4-SiH6}], whose more covalent Ru–H bonding results in smaller 29Si–1H coupling constant, upfield shifted 29Si resonance in the NMR spectrum, and longer Si–H distances.84 Similarly, [(NON)2Ca2(μ4–κ3:κ3:κ2:κ2-SiH6)(thf)2] exhibited hydride resonances at δ 5.38 ppm, and a 29Si signal at δ −261.2 ppm.64 A Si–H stretching absorption was observed in the ATR-IR spectrum of 61a at ν = 1717 cm−1, similar to previously reported values for K2SiH6 and [{(PhB(CH2PPh2)3)Ru}2{μ–η4,η4-SiH6}]. In contrast, the ATR-FTIR spectrum of [(NON)2Ca2(μ4–κ3:κ3:κ2:κ2-SiH6)(thf)2] showed three bending modes at 1053, 1013, and 976 cm−1, and three stretching modes at 1620, 1558, and 1503 cm−1.64
The crystal structure of 64 features a triangular Sr3 core with each metal bridged by a μ-H ligand, while an octahedral [nOctSiH5] unit bridges all three metals via four equatorial hydrides and one terminal hydride (Fig. 8). 1H NMR spectroscopy shows rapid hydride exchange at room temperature, giving a broad resonance at δ 5.66 ppm, which splits into a complex pattern of resonances at −80 °C. At −80 °C, a resonance at δSi 112.4 ppm was observed in a 29Si–1H HSQC experiment. The crystal structure of 63 contains a similar μ-H bridged triangular Sr3 core to that in 64, with the trigonal bipyramidal cluster capped by two μ3-SiH3 ligands.
 | ||
| Fig. 8 Cationic part of the molecular structure of compound 64 with hydrogen atoms omitted except for hydride ligands (left); the {Sr3H3(RSiH5)} core of 64 without ligands and n-octyl chain (right).54 | ||
Compound 61 acts as a masked equivalent of the elusive dimeric strontium hydride [(Me4TACD)2Sr2(μ-H)2(thf)x]2+; reacting with Brønsted acid [NEt3H][B(C6H3-3,5-Me2)4], CO2, or oxidant 1,3,5,7-cyclooctatetraene (COT), to eliminate SiH4 and yield strontium hydride-based products (Scheme 25). Specifically, [NEt3H][B(C6H3-3,5-Me2)4] yielded the bis(borate) salt 58, CO2 provided the dimeric formate [(Me4TACD)2Sr2(μ-OCHO)(thf)2][B(C6H3-3,5-Me2)4]2 (65), and COT was reduced under loss of H2 to form the dinuclear inverse sandwich compound [(Me4TACD)2Sr2(μ–η8:η8-COT)][B(C6H3-3,5-Me2)4]2 (66).63 Based on literature precedent for the formation of [(BDIdipp)2Ca2COT],8566 is most likely formed through an even-electron insertion–deprotonation sequence, rather than via two-fold single electron reduction of the conjugated tetraene. It is noteworthy that calcium hydride complex 42d is unreactive towards COT; formation of [(Me4TACD)2Ca2(μ–η8:η8-COT)][B(C6H3-3,5-Me2)4]2 (67) was instead achieved by in situ reduction with caesium metal and salt metathesis of iodide complex 50.42
 | ||
| Scheme 25 Reactivity of hexahydridosilicate complex 61a to give bis(borate) 58, formate 65, and cyclooctadienyl complex 66. For comparison, calcium hydride 42d is unreactive towards 1,3,5,7-cyclooctadiene; calcium cyclooctadienyl complex 67 is accessed from iodide 50via caesium-mediated reduction and salt-metathesis. Dicationic part of the crystal structure of complex 66. Ar = C6H6-3,5-Me2. | ||
4.4. Barium
The molecular coordination chemistry of barium is far less studied than that of lighter group 2 elements, especially for organo- and hydridobarium complexes. The large size, extremely low electronegativity, and dominance of undirectional ionic bonding make isolating well-defined, low-nuclearity, and heteroleptic complexes a significant challenge.86Neutral and cationic Me4TACD barium benzyl complexes were prepared using a method similar to that of lighter homologues. Adding Me4TACD to a THF-suspension of [Ba(CH2Ph)2] yields a yellow solution from which the neutral dibenzyl complex [(Me4TACD)Ba(CH2Ph)2] (68) crystallised (Scheme 26).87 Comparing its crystal structure to related calcium (34) and strontium (56a) complexes highlights differences in coordination. Whereas 34 contains a six-coordinate calcium centre and two η1-benzyl ligands, the larger Sr2+ cation accommodates an additional THF molecule, with both benzyl ligands remaining η1-bound. Whilst the still-larger Ba2+ is expected to adopt a yet-higher coordination number via ligation of additional solvent molecules, 68 is THF-free, and the coordination sphere is satisfied by slipped η2 and η3-bound benzyl ligands. This is consistent with the high lability of Ba-THF bonds and π-acidity of the softer metal cation. Protonolysis of 68 with [NEt3H][B(C6H3-3,5-Me2)4] provided the cationic benzyl complex [(Me4TACD)Ba(CH2Ph)(thf)x][B(C6H3-3,5-Me2)4] (69), which was not crystallographically characterised, but shows downfield shifted CH2 resonance in the 1H NMR, suggesting increased benzyl hapticity.
 | ||
| Scheme 26 Synthesis of barium benzyl complexes 68 and 69 and their respective hydrogenolysis to give BaH2 and 70. Ar = C6H6-3,5-Me2. | ||
Hydrogenolysis of 68 resulted in the release of free Me4TACD, toluene, and the precipitation of [BaH2]n (Scheme 26). The in situ1H NMR spectrum displayed a broad resonance at δ 9.4 ppm, attributed to soluble oligomeric barium hydride clusters. Hydrogenolysis of 69 resulted in the formation of the THF insoluble dimeric dihydride cation [(Me4TACD)2Ba2(μ-H)2(thf)4][B(C6H3-3,5-Me2)4]2 (70), which crystallised directly from the reaction mixture (Scheme 26). Complex 70 represents a rare crystallographically characterised molecular barium hydride. Heptanuclear and tetradecanuclear clusters [Ba7H7(N(SiMe3)2)7]·2C6H6 and [Ba14H12{N(SiMe3)2}12{(Me3Si)(Me2SiCH2)N}4] were also structurally characterised.88 The only other examples of dinuclear species are [(TpAd,iPr)2Ba2(μ-H)2] (TpAd,iPr = hydrotris(3-adamantyl-5-isopropyl-pyrazolyl)borate)89 and [(η5-C5R5)2Ba2(μ-H)2(DABCO)] (R = C6H3-3,5-iPr),71 which are supported by extremely bulky 5-electron donor L2X-type ligands. Although insolubility precluded characterisation of 70 by NMR spectroscopy, the crystal structure reveals the dicationic part to consist of two eight-coordinate barium centres each bound to the κ4-Me4TACD, two μ-hydrides, and two THF molecules (Fig. 9). The higher coordination number compared to that of the lighter metals in 60 and 42 reflects the large size of the Ba2+ cation and its tendency to adopt higher coordination geometries.
 | ||
| Fig. 9 Dicationic part of the crystal structure of barium hydride 70 with hydrogen atoms omitted except for barium-bound hydrides.87 | ||
The isolation of the barium hydride 70 is notable given the extreme lability and inaccessibility of the putative strontium congener [(Me4TACD)2Sr2(μ-H)2(thf)x]2+. Its isolation, likely aided by its insolubility, prevents ligand redistribution in solution. Given the success in employing the large 15-membered NNNNN macrocycle Me5PACP to strontium,52,53 and the often-higher activity of barium catalysts compared to the lighter group 2 elements,58,71,76,77,86 soluble complexes with [Ba2H2]2+ moiety supported by large aza-macrocycles may be of interest. Indeed, preliminary results suggest that a soluble molecular barium hydride can be accessed by hydrogenolysis of a benzyl barium cation supported by a very large 18-membered NNNNNN macrocycle, [(Me6HACO)Ba(CH2Ph)][B(C6H3-3,5-Me2)4] (Me6HACO = N,N′,N′′,N′′′,N′′′′,N′′′′′-hexamethyl-1,4,7,10,13,16-hexaazacyclooctadecane), although isolation and characterisation of the proposed hydride product remains unrealised.90
5. Group 12 metals
Although part of the d-block, zinc is considered a main-group element due to the stable closed-shell 3d10 electronic configuration. While zinc(II) shares some chemical similarities with magnesium(II), its higher electronegativity and increased covalent contributions lead to notable differences. Whilst no Me4TACD complexes of highly toxic cadmium and mercury are known to date, the solution-state binding of Me4TACD to Zn2+ and Cd2+ as aqueous nitrate salts has been studied alongside other aza-macrocyclic ligands.91,92 The steric and coordinative demand of Me4TACD on the relatively small Zn2+ cation (five-coordinate effective ionic radius 0.68 Å)93 is such that the invariably five-coordinate metal centre is coordinatively over-saturated. The fifth coordination site is occupied by a Lewis-basic L-type ligand in a dicationic complex or by an X-type ligand (X = halide, hydride) in a monocationic complex. [(Me4TACD)ZnX][HBPh3] salts are active catalysts in the hydroboration and hydrosilylation of polar organic substrates, and the [(Me4TACD)ZnH]+ cation displays hydride-centred nucleophilicity towards electrophilic CO2. The resolute coordinative saturation, however, restricts more complex metal-centred reactivity compared to heavier alkaline-earth derivatives or related [LnZnX]+ cations supported by di- and tridentate ligands. Me4TACD has also recently been employed as a ligand for dizinc(I) complexes.5.1. Zinc(II)
All crystallographically characterised Me4TACD zinc complexes reported to date adopt a distorted square pyramidal geometry within a five-coordinate (di)cation structure. Complexation of zinc dihalides with Me4TACD in THF leads to auto-ionisation, forming charge-separated salts [(Me4TACD)ZnX]X (71a, X = Cl; 71b, X = Br; 71c, X = I), in high yields (Scheme 27).94 Anion exchange with Na[BPh4] or K[HBPh3] provided access to cationic halide complexes [(Me4TACD)ZnX][HBPh3] (72a, X = Cl; 72b, X = Br; 72c, X = I)94 and [(Me4TACD)ZnCl][BPh4] (73)95 in high yields (Scheme 27). A two-fold halide abstraction was carried out by reacting Me4TACD with ZnI2 and two equiv. Ag[BF4] or Ag[PF6] in one pot, yielding dicationic acetonitrile complexes [(Me4TACD)Zn(NCCH3)][A]2 (74a, [A] = BF4; 74b, [A] = PF6; Scheme 27).96 Compounds 73 and 74 were employed as diamagnetic diluents in magnetic relaxation studies of isostructural Cu(II) congeners.95–97 Hydridotriphenylborate complexes 72a–c react with CO2via insertion into the H–B bond to provide formatotriphenylborate salts [(Me4TACD)ZnX][HCO2BPh3] (75a, X = Cl; 75b, X = Br; 75c, X = I; Scheme 27).94 | ||
| Scheme 27 Complexation of zinc dihalides by Me4TACD, anion-exchange derivatives, and reactivity of hydridoborate derivatives towards CO2. Cationic part of the crystal structure of compound 73 (H-atoms omitted).94–96 | ||
Cationic amido zinc complex [(Me4TACD)Zn(N(SiMe2H)2)][HBPh3] (72d) was prepared by BPh3 mediated hydride abstraction of [Zn(N(SiMe2H)2)2]2 in the presence of Me4TACD, with elimination of the cyclic silazane [(Me2HSiN)2(SiMe2)2] (Scheme 28).94 Like its halide analogues, 72d reacts with CO2 through rapid insertion into the B–H bond, forming formatoborate salt [(Me4TACD)Zn(N(SiMe2H)2)][HCO2BPh3] (75d).
 | ||
| Scheme 28 Synthesis of amidozinc hydridoborate complex, and its reactivity towards CO2.94 | ||
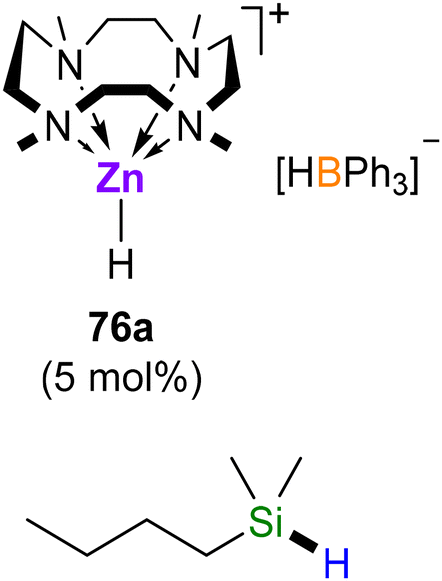
Zinc dihydride [ZnH2]n does not react with Me4TACD alone but reacts with Lewis acids in the presence of neutral N-donors to provide molecular complexes. Reaction of [ZnH2]n with Me4TACD in the presence of BPh3 yields a five-coordinate zinc hydride cation [(Me4TACD)ZnH][HBPh3] (76a; Scheme 29).98 A similar, four-coordinate charge-separated species [(PMDTA)ZnH][HBPh3] was obtained using tridentate PMDTA, but bidentate N-donors provide four-coordinate dihydrides [(L)Zn(H)(μ2-H)BPh3], with a μ2-hydride bridging the zinc and boron centres.98 The cationic zinc hydride cation was also obtained as tetraarylborate salts, [(Me4TACD)ZnH][BAr4] (Ar = C6H3-3,5-Me2 (76b), C6H3-3,5-(CF3)2 (76c)) by combining zinc dihydride with Brønsted acidic [(Me4TACD)H][BAr4] in THF.99
 | ||
| Scheme 29 Reaction of [ZnH2]n with Me4TACD in the presence of Lewis acids BPh3 or CO2, reaction of [ZnH2]n with [(Me4TACD)H]+, and onward reactivity of hydridozinc cations toward CO2 and BH3·thf.98,99 Crystal structure of compound 77a (H-atoms omitted except for H1 and H14).98 | ||
CO2 rapidly inserts into both B–H and Zn–H bonds of 76a to provide charge-separated zinc formate-formatoborate complex [(Me4TACD)Zn(κO-OCHO)][(OCHO)BPh3] (77a),98 or into the Zn–H bond of 76b to form formatozinc cation [(Me4TACD)Zn(κO-OCHO)][BAr4] (77b; Scheme 29).99 The terminal formate ligand in 77a and 77b binds zinc in a κO manner. The direct reaction of [ZnH2]n with CO2 in the presence of Me4TACD yielded the charge-separated diformate [(Me4TACD)Zn(κO-OCHO)][OCHO] (77c), unlike acyclic bi- and tri-dentate polyamines, which form monomeric diformate complexes [(Ln)Zn(OCHO)2] (Ln = TMEDA, TEEDA, TMPDA, PMDTA).98
Compound 76b also reacts with BH3·thf to form the mononuclear tetrahydridoborate [(Me4TACD)Zn(μ-H)2BH2][BAr4] (78, Ar = C6H3-3,5-Me2).99 The η2 binding mode of the tetrahydridoborate moiety in 78 contrasts to the η3-coordinated tetrahydridoborate in the congeneric magnesium complex 24.47
| Precatalyst (loading), reducing agent | Substrate | Products |
|---|---|---|

|

|

|
| CO2 |

|
|

|

|
|

|

|
|

|
|
|
|
CO2 |

|
The tetraarylborate derivative 76b is inactive in the hydrosilylation of CO2. Crystals of the zinc formate-hydridoborate salt [(Me4TACD)Zn(κO-OCHO)][HBPh3] (77d, Fig. 10) were obtained from a concentrated reaction solution of the hydrosilylation of CO2 with n-butyldimethylsilane and catalyst 77a, which may imply that the zinc formate cation is itself incapable of turnover.99 Notably, zinc hydride cations [(Ln)ZnH][BAr4] supported by acyclic ligands (Ln = TMEDA, TEEDA, PMDTA, Me6TREN; Ar = C6H3-3,5-(CF3)2) are far more active in the catalytic hydrosilylation and hydroboration of CO2.101,102 Thus, combined hydricity and Lewis acidity is essential for active catalysis; the steric and coordinative demand of the strongly chelating Me4TACD macrocycle quenches Lewis acidity and coordinative availability of the zinc centre and precludes zinc-mediated catalysis.
 | ||
| Fig. 10 Zinc formate hydridotriphenylborate 77d, isolated from the catalytic reaction mixture of CO2 hydrosilylation by nBuMe2SiH catalysed by 76a. | ||
5.2 Zinc(I)
The unsymmetrical dizinc(I) cation [Cp*ZnZn(Me4TACD)][BAr4] (79; Ar = C6H3-3,5-Me2), synthesised from [Cp*2Zn2] and [(Me4TACD)H][BAr4] under elimination of Cp*H (Scheme 30),103 is unreactive towards further equivalents of [(Me4TACD)H][BAr4]. The dizinc(I) dication [(Me4TACD)2Zn2]2+ was instead accessed by reacting 79 with HBpin, providing [(Me4TACD)2Zn2][BAr4] (80) in 30% yield (Scheme 30).103 The Zn–Zn bond in 79 is somewhat longer (2.3510(3) Å) compared to that in [Zn2Cp*2] (2.302(1) Å), whilst 80 shows an elongated Zn–Zn bond of 2.4860(6) Å, significantly longer than those in analogous [Zn2L6]2+ dications (2.36–2.41 Å; L = THF, DMAP).104,105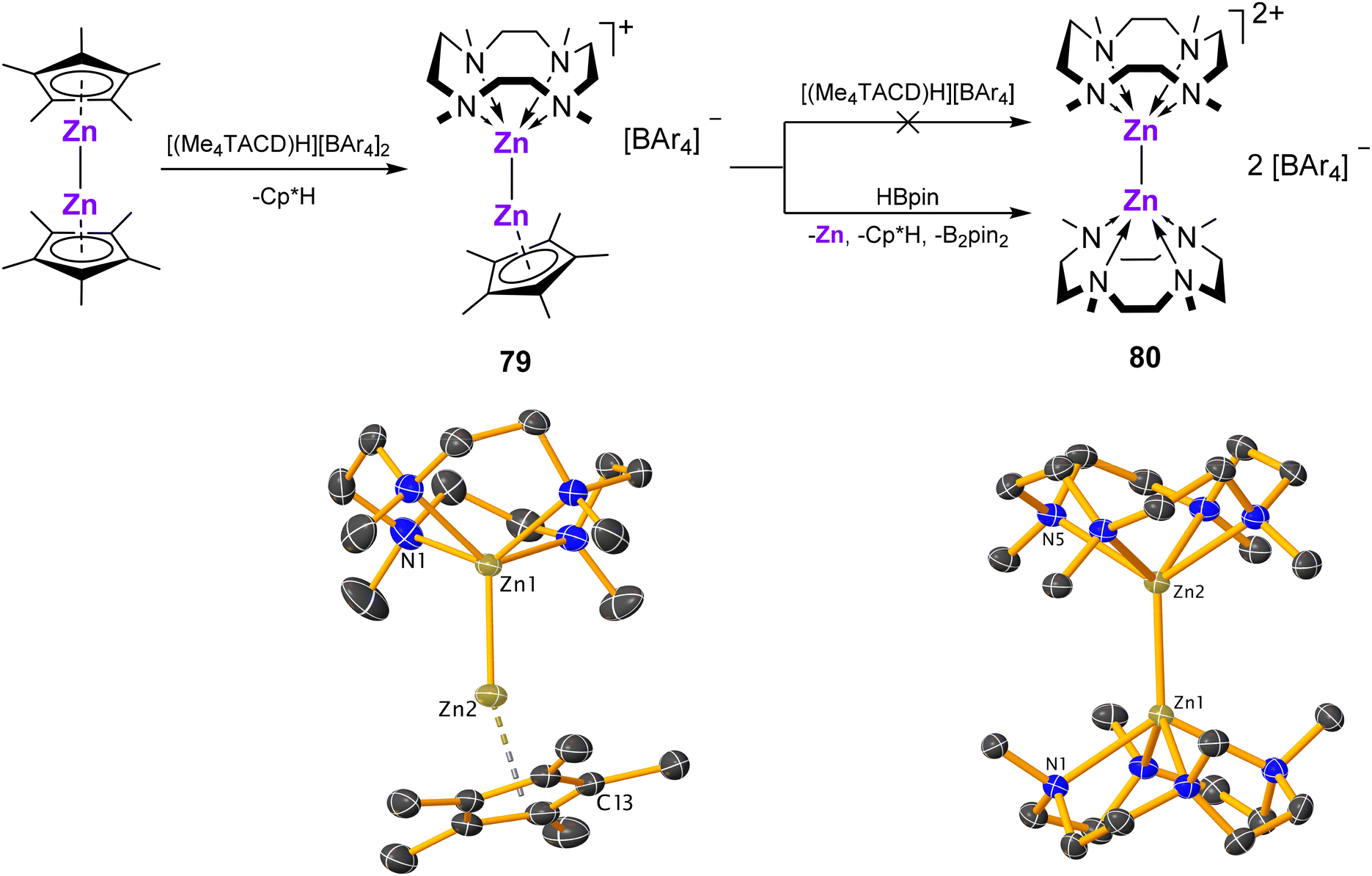 | ||
| Scheme 30 Synthesis of dizinc(I) complexes 79 and 80 and the (di)cationic part of their crystal structures (H-atoms omitted).103 Ar = C6H3-3,5-Me2. | ||
Compound 79 mediates the heterolysis of activated C–H bonds in NCCH3 and PhCCH in the presence of catalytic Me4TACD (10 mol%) as a Brønsted base, providing organozinc(II) complexes [(Me4TACD)ZnR][BAr4] (81a, b; R = H2CCN, CCPh; Scheme 31).103
 | ||
| Scheme 31 Reaction of dizinc(I) compound 79 with acetonitrile and with phenylacetylene. Ar = C6H3-3,5-Me2. | ||
6. Group 13 metals
Protonolysis of organo- and hydrido-triel precursors with tetra-aryl borate salts of [(Me4TACD)H]+ yielded (di)cationic hydride complexes [(Me4TACD)AlH2]+ and [(Me4TACD)MH]2+ (M = Al, Ga), and univalent cations [(Me4TACD)M]+ (M = Ga, In, Tl). The small ionic radius of Al3+ and Ga3+ resulted in an unusual, folded ligand conformation for [(Me4TACD)AlH2]+ and Brønsted acidic reactivity of the Ga–H bond for coordinatively saturated [(Me4TACD)GaH]2+. The acid–base chemistry of the Ga(I)–(III) couple was studied.6.1. Aluminium
The dihydridoaluminium cation [(Me4TACD)AlH2][BAr4] (82a, Ar = C6H3-3,5-Me2; 82b, Ar = C6H3-3,5-(CF3)2) was synthesised in near quantitative yields by reacting Et3N·AlH3 with [(Me4TACD)H][BAr4] (Scheme 32).28 The macrocyclic ligand in 82 adopts a rare syn–syn–syn–anti configuration in both solid and solution states, with one methyl group pointing towards the distal face of the complex, away from the metal centre. | ||
| Scheme 32 Synthesis of aluminium hydrides 82a,b. | ||
The crystal structure of 82b reveals a slightly distorted mer-octahedral geometry, with the two hydride ligands cis to one another (Fig. 11). Due to macrocyclic strain, the Al1–N1 distance (2.466(3) Å) is significantly longer than the other Al–N distances (2.091(3), 2.096(3), 2.145(3) Å), with NBO analysis indicating a low Wiberg Bond Index (WBI) for this Al–N interaction compared to the others. This folded conformation was previously observed only as a minor crystalline component in the solid-state structure of 27, which exists in an exclusively all-syn conformation in solution.27 Complex 82 represents the steric limit of the 12-membered macrocycle in chelating a small ion (effective ionic radius for six-coordinate aluminium, 0.535 Å)93 whilst accommodating two hydride ligands. The related 14-membered macrocycle Me4cyclam reacts with Me3N·AlH3 by auto-ionization to form [(Me4cyclam)AlH2][AlH4], which adopts an octahedral trans-dihydride motif with the larger macrocycle encircling the equatorial plane.106 The folded ligand conformation of 82 is retained in solution up to 60 °C, as evidenced by 1H NMR spectroscopy showing a complex array of multiplets corresponding to four pairs of magnetically inequivalent methylene protons, and three methyl environments in a 1:2:1 integral ratio.
 | ||
| Fig. 11 Crystal structure (H-atoms omitted except H1 and H2) of the cationic part of the of 82b in oblique (left) and side (right) views.28 | ||
Complex 82 reacts with the weak Brønsted acid [Et3NH][BAr4], yielding the dicationic hydride complexes [(Me4TACD)AlH][BAr4]2 (83a, Ar = C6H3-3,5-Me2; 83b, Ar = C6H3-3,5-(CF3)2; Scheme 33).281H NMR spectra confirmed that upon removal of one hydride, the macrocycle reverts to a time-averaged C4-symmetric boat-like conformation. Depending on the counter-ion, the methylene environments in THF-d8 appear either as a broad, unresolved signal (Ar = C6H3-3,5-Me2), or a well-defined AA′XX′ multiplet (Ar = C6H3-3,5-(CF3)2), suggesting non-negligible ion pairing for the non-fluorinated anion. Compound 83 was also obtained via protonolysis of tetrameric aluminium(I) reagent [Cp*Al]4 with [(Me4TACD)H][BAr4] at 70 °C (Scheme 33).28 Due to the formation of a bis-borate salt, eight equivalents of conjugate acid were required to achieve quantitative conversion to an equimolar mixture of Me4TACD, Cp*H, and 83. Four equiv. of conjugate acid resulted in the incomplete conversion of the aluminium(I) reagent. The reaction of [(Me4TACD)H][BAr4] with [Cp*Al] leads to protonation of the Cp* ligand, forming Cp*H, and formal two-electron oxidation of Al(I) to Al(III) hydride dication. The reaction likely proceeds via protonation of transient [(Me4TACD)Al]+. Isolable mononuclear aluminium(I) cations remain elusive, but the related tetranuclear monovalent cluster cation [(κ3-Me3TACN)Al{κ3-(AlCp*)3}][Al(ORF)4] (ORF = OC(CF3)3), has been reported.107
 | ||
| Scheme 33 Synthesis of aluminium hydride dication 83a,b from protonolysis of 82a,b, or protonolysis-protonation of [Cp*Al]4. | ||
6.2. Gallium
Unlike Et3N·AlH3, protonolysis of Me3N·GaH3 with equimolar [(Me4TACD)H][B(C6H3-3,5-(CF3)2)4] yielded gallium(I) cation [(Me4TACD)Ga][B(C6H3-3,5-(CF3)2)4] (84a, Scheme 34),28 which likely forms through spontaneous dehydrogenation of a short-lived gallium(III) dihydride cation [(Me4TACD)GaH2]+. The spontaneous dehydrogenation of transient base-free [GaH2]+ has also been described.108 Consistent with higher stability of the gallium(I) cation compared to its lighter congener, [(Me4TACD)Ga][B(C6H3-3,5-Me2)4] (84b) was synthesised via protonolysis of [Cp*Ga] with [(Me4TACD)H][B(C6H3-3,5-Me2)4] (Scheme 34).28 Subsequent protonation of 84b with [Et3NH][B(C6H3-3,5-Me2)4] provided the dicationic hydride [(Me4TACD)GaH][B(C6H3-3,5-Me2)4]2 (85) after release of triethylamine (Scheme 34).28 This observation is consistent with the protonation of proposed [(Me4TACD)Al]+ in the synthesis of 83 from [Cp*Al]4. Similar protonation with oxidative addition of H–P bond to [Ga]+ was reported in the reaction of [(PhF)nGa][Al(ORF)4] with [HPPh3][Al(ORF)4] in the presence of triphenylphosphine.109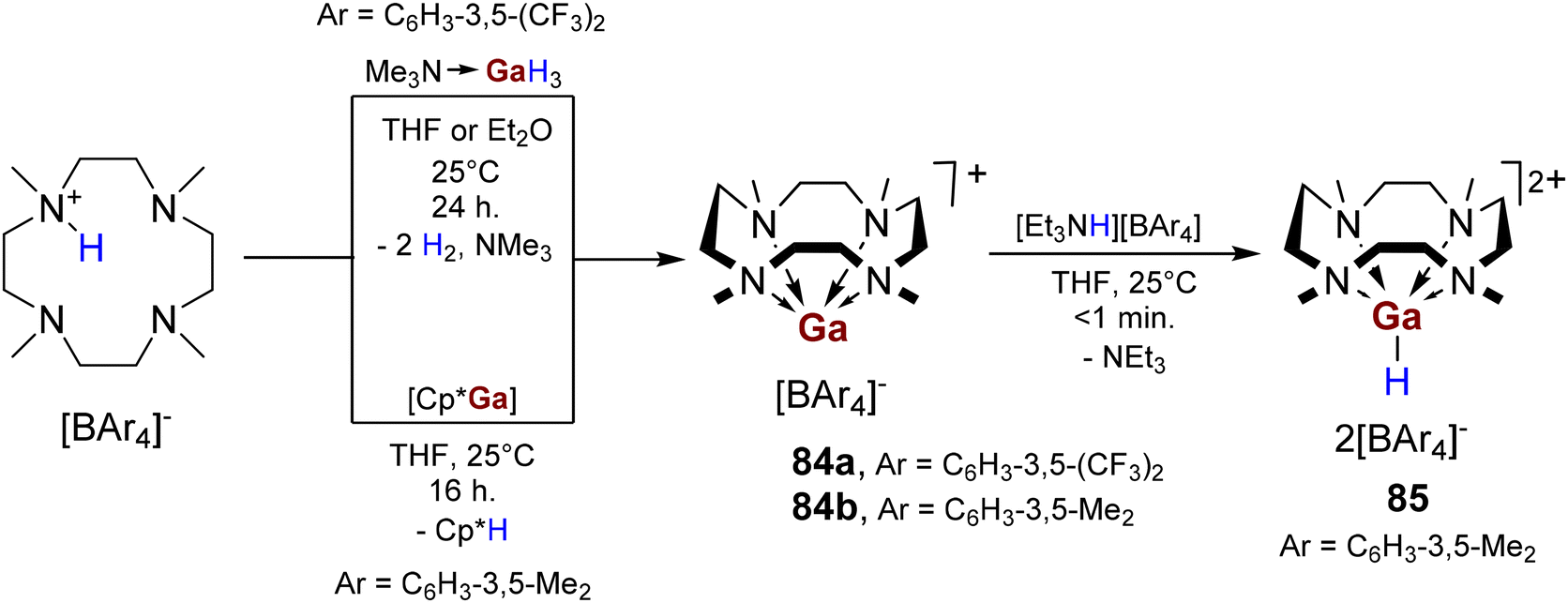 | ||
| Scheme 34 Synthesis of gallium(I) complexes 84a,b by protonolysis/dehydrogenation of Me3N·GaH3 or protonolysis of [Cp*Ga]; synthesis of gallium(III) hydride dication 85. | ||
The gallium(I) cation 84 forms a charge-separated ion-pair in the solid-state, with a four-coordinate gallium cation and a pseudo C4-symmetric macrocyclic ligand (Fig. 12a).28 The Ga–N distances are notably shorter than the Ga–O distances in the related [(12-crown-4)Ga][A] (A = [GaCl4]−, [B(C6F5)4]−)110 and the metal centre projected less from the basal plane of the four donor atoms. 84 is formally isoelectronic to the germanium dication of [(Me4TACD)Ge]X2 (90a, X = CF3SO3−; 90b, X = GeCl3−) (vide infra), which displays significantly shorter M–N bonds due to the higher nuclear charge of the tetrel dication.111 The 1H NMR spectrum confirms stable macrocycle coordination, displaying a resolved methylene AA′XX′ spin system at room temperature. The 71Ga NMR spectrum (122 MHz, 298 K) of 84 contains a broad signal at δ −173 ppm (84b, acetonitrile-d3) or −188 ppm (84a, THF-d8), significantly downfield-shifted compared to Ga[Al(ORF)4] coordinated by arene or fluoroarene solvents (δ −756 ppm, C6H5F; −520 ppm, C7H8)112 or ethers (δ −448 ppm, Ga[Al(ORF)4] in THF;112 −471 ppm, [(12-crown-4)Ga][GaCl4] in toluene).110 Unlike 84, the 1H NMR spectrum of [(Me3TACN)Ga][Al(ORF)4]107 shows a single broad singlet for the methylene protons and no detectable 71Ga resonance, suggesting ligand lability. In the crystal structure of 85 (Fig. 12d), the metal centre adopts a square pyramidal geometry (τ5 = 0.03) with a terminal hydride in the apical position. Reflecting the higher formal oxidation state of +3, the Ga–N bonds are contracted by 0.33 Å, and the metal is pulled much closer to the basal N4-plane compared to [(Me4TACD)Ga]+.
 | ||
| Fig. 12 Crystal structures (H-atoms omitted except for the gallium-bound hydride) of the (di)cationic parts from (a) 84b; (d) 85; (b) calculated HOMO, and (c) LUMO of [(Me4TACD)Ga]+; (e) calculated HOMO, and (f) LUMO of [(Me4TACD)GaH]2+.28 | ||
The macrocycle size and the nature of donor atoms significantly impact the frontier molecular orbitals of [(Ln)Ga]+. NBO analysis of 84 revealed a HOMO comprising of an out-of-phase combination of a metal-localized lone pair and Ga–N σ-bonds, while the LUMO is an empty 4p orbital (Fig. 12b and c).28 [(12-crown-4)Ga]+ displayed a similar directional lone pair (HOMO) and p-like LUMO, but the weaker donor properties of the crown-ether resulted in a lower energy HOMO compared to 84.110 This suggests reduced reactivity, though no experimental studies have confirmed this effect. The 18-crown-6 complex cation [(18-crown-6)Ga]+ displays a non-directional lone-pair that is only slightly influenced by weakly bound axial solvent molecules, owing to near-coplanarity of the larger macrocycle and metal centre.113–115 On account of a stabilised ns-orbital, “naked” Ga+ and In+ cations, ligated by weakly-bound solvent molecules and supported by weakly-coordinating anions react as oxidants and soft Lewis acids.116–118 Coordination of σ-donor ligands raises the energy of the HOMO, leads to a directional lone-pair, and promotes reductive chemistry but often leads to disproportionation into zero- and trivalent products.109,110,119–123 The Me4TACD ligand leads to activation of the 4s orbital, leading to Brønsted basic reactivity towards [Et3NH]+ whilst imparting remarkable kinetic stability to the monovalent cation; 84b is stable up to at least 60 °C in THF. No oxidation event could be determined for 84b by cyclic voltammetry up to +1.1 V (vs. Fc/Fc+ in acetonitrile), but an irreversible reduction at −2.5 V was tentatively ascribed to the GaI/Ga0 couple.124 [Ga(o-C6H4F2)n][Al(ORF)4] gives a scan-rate dependent, partially reversible event at about +3.0 V (GaI/Ga0vs. Fc/Fc+ in o-C6H4F2).116
NBO analysis of 85 showed that its HOMO (Fig. 12e) is primarily composed of Ga–N bonding orbitals, while the LUMO is dominated by the Ga–H σ* antibonding orbital (Fig. 12f).28 Accordingly, 85 is readily deprotonated at room temperature by IMe4 (1,3,4,5-tetramethyl-imidazol-ylidene) to return to 84b and eliminate the imidazolium salt [IMe4H][B(3,5-Me2-C6H3)4] (Table 2). Treating 85 with equimolar DBU (1,8-diazabicyclo[5.4.0]undec-7-ene, pKa(CH3CN) = 24.3) resulted in an equilibrium mixture of conjugate acid–base pairs, which allows estimation of the Brønsted acidity of the gallium hydride as pKa(CH3CN) = 24.5 using NMR spectroscopy. Brønsted acidity is appreciable for heavier tetravalent group 14 hydrides,4,125,126 but the reactivity of group 13 hydrides is normally characterised by strong Brønsted basicity and nucleophilicity. Brønsted acidity is more reminiscent of late-transition metal hydrides.
28
|
|
||
|---|---|---|
| Base | K eq | Solvent |
| NEt3 | ≫1 (quantitative protonation) | THF |

|
0.77 | Acetonitrile |

|
0 (quantitative deprotonation) | THF |

Unlike isoelectronic [(Me4TACD)ZnH]+ (76a,b), which rapidly inserts CO2 to give [(Me4TACD)Zn(OCHO)]+ (77a,b), 85 is inert towards CO2 under ambient conditions. However, 84b reacts with CO2 (1 bar) in THF or acetonitrile solution to provide the cationic carbonate complex [(Me4TACD)Ga(κ2-O2CO)][B(C6H3-3,5-Me2)4] (86) with CO extrusion (Scheme 35). This reaction likely proceeds via a putative oxido gallium cation [(Me4TACD)GaO]+ through CO extrusion from a transient CO2 complex [(Me4TACD)Ga(CO2)]+. Although [(Me4TACD)GaO]+ remained elusive, oxidation of 84b with N2O in the presence of BPh3 produces [(Me4TACD)GaO·BPh3][B(C6H3-3,5-Me2)] (87), which itself reacts with CO2 to form a gallium carbonate. 84b acts as a pre-catalyst for the CO2 hydroboration to form pinBOC(H)O using HBpin. Although the mechanistic details remain obscure, the formation of a Me4TACD-containing intermediate with a 1H NMR typical for gallium(III) (downfield shifted AA′XX′ multiplet) and CO during catalyst activation suggests initial oxidation of 84b by CO2.124
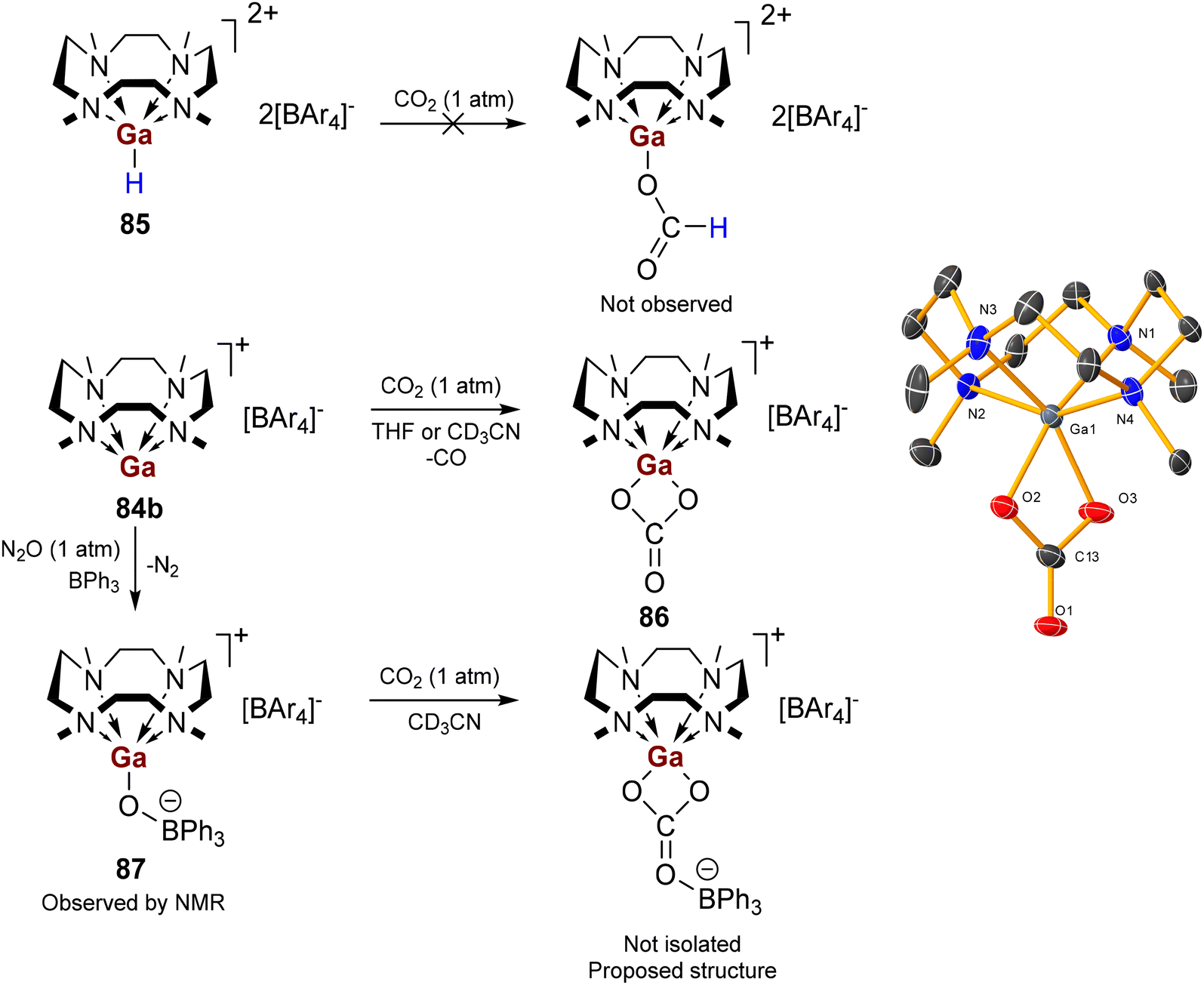 | ||
| Scheme 35 Reactivity of 84b and 85 towards CO2 and N2O (Ar = C6H3-3,5-Me2). Cationic part of the crystal structure of 86 (H-atoms omitted).124 | ||
6.3. Indium and thallium
Similar to the synthesis of 84b, the heavier monovalent cations [(Me4TACD)M][B(C6H3-3,5-Me2)4] (8 M = In; 89, M = Tl) were prepared by protonolysis of [Cp*M]n (M = In, n = 6; M = Tl, n = ∞) with [(Me4TACD)H][B(C6H3-3,5-Me2)4] (Scheme 36). | ||
| Scheme 36 Synthesis of compounds 88 and 89. | ||
The compounds 84, 88, and 89 are structurally similar, with metal centre moving further from the basal N4-plane with increasing atomic number (1.3007(15) Å, 84; 1.501(2)/1.521(2) Å, 88; 1.616(5) Å, 89), with a larger increase in difference between Ga and In (ca. 0.2 Å) than between In and Tl (0.1 Å) due to the lanthanide contraction. The crystal structure of 88 is shown in Fig. 13a. Calculated Wiberg Bond Indices (WBIs) for the M–N bonds decreased down group 13, reflecting increased size- and hard–soft mismatch between ligand and metal. Alternative polydentate ligands with softer sulphur, phosphorus, and arsenic donors have been explored for the heavier group 14 elements,127 and may be well-suited to heavy univalent group 13 cations. Indeed, acyclic monodentate and chelating phosphines have already been employed for univalent gallium and indium cations.128–132
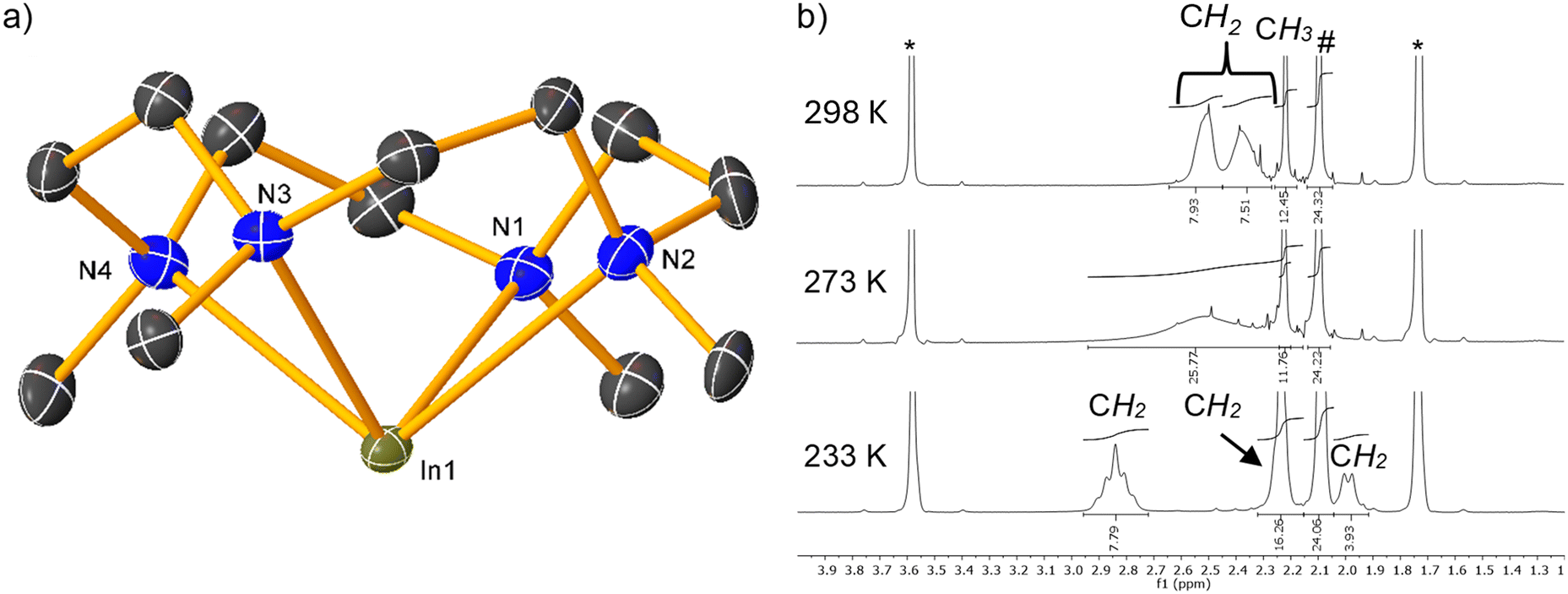 | ||
| Fig. 13 (a) The crystal structure of the cationic part of 88 (30% level, H-atoms excluded); (b) stacked 1H NMR spectra (400 MHz, THF-d8 (*)) of 88 at different temperatures (# borate anion).28 | ||
The 1H NMR spectra of 88 and 89 resemble that of 84b but lack a resolved methylene spin system. Similar signal broadening was observed for increasingly heavy alkali metal silanides, but this was attributed to the lability of the macrocycle.41 Ligand resonances in the 1H NMR spectrum of [(Me3TACN)Tl][Al(ORF)4] shift on addition of excess Me3TACN,107 indicating labile coordination to soft Tl(I) cation. However, for 88 and 89, persistent coordination is observed at elevated temperatures as their methylene resonances split into two unresolved multiplets. At low temperatures, the time-averaged ring-twisting motion is “frozen-out”, as indicated by 1H and 13C NMR spectroscopy showing four inequivalent C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2 (ABMX spin system) and two inequivalent
2 (ABMX spin system) and two inequivalent ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2 resonances indicative of a static C4-symmetric structure (Fig. 13b). The methyl resonances in the 1H and 13C{1H} NMR spectra of 89 appeared as doublets at room temperature, due to temperature-dependent J-coupling to the 205Tl nucleus (S = 1/2), although the corresponding 205Tl NMR spectrum was not recorded.
H2 resonances indicative of a static C4-symmetric structure (Fig. 13b). The methyl resonances in the 1H and 13C{1H} NMR spectra of 89 appeared as doublets at room temperature, due to temperature-dependent J-coupling to the 205Tl nucleus (S = 1/2), although the corresponding 205Tl NMR spectrum was not recorded.
Neither 88 nor 89 react with [Et3NH][B(C6H3-3,5-Me2)4], suggesting that the putative conjugate acids [(Me4TACD)MH]2+ have a pKa(CH3CN) ≫ 18.83 and that the univalent cations are less basic than NEt3. The lone pairs (HOMO) of 88 and 89 were calculated to reside −3.1 kcal mol−1 and −23.1 kcal mol−1 lower in energy than that of 84, suggesting [(Me4TACD)InH]2+ to be a synthetically viable target.28
7. Group 14 metals
Me4TACD supported germanium dications [Ge(Me4TACD)]X2 (90a, X = CF3SO3−; 90b, X = GeCl3−) were isolated as colourless solids by reacting GeCl2(dioxane) with Me4TACD in 1:1 and 1:3 ratios, respectively, in the presence of Me3SiO3SCF3 for the former compound (Fig. 14).111 Both compounds exist as charge-separated ion pairs in the solid state, with no significant cation–anion interactions. Whilst computational studies show that the HOMO of cryptand encapsulated Ge2+ dication [(222-crypt)Ge][O3SCF3]2 and related [12]-crown-4 sandwich dication [([12]-crown-4)2Ge][A]2 ([A] = GeCl3, O3SCF3) is an essentially non-directional 4s orbital,133–135 the stronger coordinating ability of the aza-macrocycle and ‘half-sandwich’ structure of 90a and 90b likely induces some directionality to the 4s lone pair. This effect has been computationally demonstrated for thia-macrocycle complexes of Ge(II),136 as well as formally isoelectronic 84 and [(12-crown-4)Ga]+.28,110 Similar to 90, the 14-membered macrocycle Me4cyclam provides a charge-separated Ge2+ complex [(Me4cyclam)Ge][GeCl3]2,134 whilst 12-membered Me3TACN permits closer interaction with triflate and chloride anions, though bromide remains fully charge separated.111,134
 | ||
| Fig. 14 Me4TACD supported germanium and lead cations 90a,b, and 91 and the molecular structure of the cationic part of 90a.111 | ||
The aqueous coordination chemistry of Pb(NO3)2 has been studied for several aza-macrocyclic ligands, including Me4TACD, although the resulting lead complex 91 was not structurally characterised.91,92
8. Conclusions
The macrocycle Me4TACD is a versatile neutral ligand that has been employed to stabilise low-nuclearity molecular complexes of various main group metals. Compared to crown ethers, it offers greater robustness against degradation by nucleophilically induced ring-opening and stronger coordination due to the superior σ-donating properties of its amine functionality. Its inherent flexibility allows it to accommodate main group metal centres with remarkably varying ionic radii and reactive bonds such as terminal hydride.138Me4TACD efficiently encapsulates small metal cations like Li+, Mg2+, Zn2+, Al3+, and Ga3+, leading to coordinative saturation and limiting reactivity. For example, complexes such as [(Me4TACD)ZnH]+ (ref. 98) or [(Me4TACD)2Mg2(μ-H)2]2+ (ref. 47) react only with highly activated polar electrophiles, but not with apolar substrates such as H2 or alkenes. On the other hand, blocking access to the metal can induce unusual chemical reactivity, such as Brønsted acidity of the gallium(III) hydride dication [(Me4TACD)GaH]2+.28
Predominantly electrostatic interactions with the larger and less-well encapsulated group 1 cations K–Cs lead to labile coordination of the macrocycle, as indicated by NMR spectra of the triphenylsilanide series [(Me4TACD)MSiPh3] (M = Li–Cs).41 Heavier monovalent group 13 cations Ga–Tl are more strongly coordinated, such that the macrocycle engenders a degree of basic and reducing reactivity to the otherwise low-lying 4s lone-pair of Ga+.28 The tuning of frontier molecular orbital energies is important in developing transition-metal-like reactivity in low-valent p-block chemistry.
Me4TACD effectively stabilizes cationic calcium hydride complexes,137 where it strikes a balance between suppressing aggregation and ligand redistribution (Schlenk equilibria), and allowing facile access of substrates to the coordinatively unsaturated metal centre. Dimeric calcium hydrides salts [(Me4TACD)2Ca2(μ-H)2(thf)x]2+ and [(Me4TACD)2Ca2(μ-H)3]+ serve as active catalysts for alkene hydrogenation and hydrosilylation. However, its ability to stabilise analogous complexes of strontium instead leads to trinuclear hydride [(Me4TACD)3Sr3(μ-H)4(thf)]2+ (ref. 80) and di- or trinuclear hydridosilicate species. Dimeric strontium complexes are better stabilised by the larger 15-membered NNNNN macrocycle Me5PACP. Whilst a dimeric barium dihydride dication [(Me4TACD)2Ba2(μ-H)2(thf)4]2+ has been crystallographically characterised, its solution-state chemistry remains unexplored.87
Although the elaboration of reaction chemistry and catalysis for very small or very large main group metals may be limited by respective coordinative saturation and lability, as a supporting ligand for reactive cations, Me4TACD has the potential to offer significant opportunities in the future development of s-block mediated catalysis and low-valent p-block chemistry alongside its more widely established cousins such as Me3TACN, crown-ethers and acyclic polyamines. We hope this review stimulates future research activity in this regard.
Author contributions
P. G., L. M. and J. O. conceptualised and wrote the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. O. acknowledges the Deutsche Forschungsgemeinschaft for financial support and all the former coworkers for their hard work. Professor Shigehiro Yamaguchi, Nagoya University, kindly provided facilities at the Research Center for Material Science to complete this manuscript.References
- C. M. Cui, H. W. Roesky, H. G. Schmidt, M. Noltemeyer, H. J. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- P. M. Chapple, S. Kahlal, J. Cartron, T. Roisnel, V. Dorcet, M. Cordier, J.-Y. Saillard, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2020, 59, 9120–9126 CrossRef CAS PubMed.
- B. Rosch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, 62, e202308347 Search PubMed.
- F. Dankert, J. Messelberger, U. Authesserre, A. Swain, D. Scheschkewitz, B. Morgenstern and D. Munz, J. Am. Chem. Soc., 2024, 146, 29630–29636 CrossRef CAS PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 Search PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 Search PubMed.
- Y. Wang and G. H. Robinson, J. Am. Chem. Soc., 2023, 145, 5592–5612 CrossRef CAS PubMed.
- J. L. Dye, Philos. Trans. R. Soc., A, 2015, 373, 20140174 CrossRef PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- N. Davison, P. G. Waddell and E. Lu, J. Am. Chem. Soc., 2023, 145, 17007–17012 CrossRef CAS PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2024, 63, e202313556 CrossRef CAS PubMed.
- J. Coates, D. Hadi and S. Lincoln, Aust. J. Chem., 1982, 35, 903–909 CrossRef CAS.
- J. Cho, R. Sarangi and W. Nam, Acc. Chem. Res., 2012, 45, 1321–1330 CrossRef CAS PubMed.
- J. E. Richman and T. J. Atkins, J. Am. Chem. Soc., 1974, 96, 2268–2270 CrossRef CAS.
- T. J. Atkins, J. E. Richman and W. F. Oettle, Org. Synth., 1978, 58, 86 CrossRef CAS.
- J. F. Desreux, E. Merciny and M. F. Loncin, Inorg. Chem., 1981, 20, 987–991 CrossRef CAS.
- G. R. Weisman and D. P. Reed, J. Org. Chem., 1996, 61, 5186–5187 Search PubMed.
- L. E. Lemmerz, D. Mukherjee, T. P. Spaniol, A. Wong, G. Menard, L. Maron and J. Okuda, Chem. Commun., 2019, 55, 3199–3202 Search PubMed.
- L. J. Morris, P. Ghana, T. Rajeshkumar, A. Carpentier, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2022, 61, e202114629 CrossRef CAS PubMed.
- R. Riedel, A. G. Seel, D. Malko, D. P. Miller, B. T. Sperling, H. Choi, T. F. Headen, E. Zurek, A. Porch, A. Kucernak, N. C. Pyper, P. P. Edwards and A. G. M. Barrett, J. Am. Chem. Soc., 2021, 143, 3934–3943 CrossRef CAS PubMed.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202308766 CrossRef PubMed.
- S. Schade and G. Boche, J. Organomet. Chem., 1998, 550, 359–379 CrossRef CAS.
- J. Dyke, W. Levason, M. E. Light, D. Pugh, G. Reid, H. Bhakhoa, P. Ramasami and L. Rhyman, Dalton Trans., 2015, 44, 13853–13866 RSC.
- H. Bhakhoa, L. Rhyman, E. P. Lee, D. K. W. Mok, P. Ramasami and J. M. Dyke, Dalton Trans., 2017, 46, 15301–15310 Search PubMed.
- H. Osseili, D. Mukherjee, T. P. Spaniol and J. Okuda, Chem. – Eur. J., 2017, 23, 14292–14298 CrossRef CAS PubMed.
- D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790–10793 CrossRef CAS PubMed.
- R. Janot, W. S. Tang, D. Clémençon and J. N. Chotard, J. Mater. Chem., 2016, 4, 19045–19052 RSC.
- W. S. Tang, J.-N. Chotard, P. Raybaud and R. Janot, J. Phys. Chem. C, 2014, 118, 3409–3419 CrossRef CAS.
- W. S. Tang, J.-N. Chotard, P. Raybaud and R. Janot, Phys. Chem. Chem. Phys., 2012, 14, 13319–13324 RSC.
- J.-N. Chotard, W. S. Tang, P. Raybaud and R. Janot, Chem. – Eur. J., 2011, 17, 12302–12309 CrossRef CAS PubMed.
- D. Schuhknecht, V. Leich, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Chem. – Eur. J., 2020, 26, 2821–2825 CrossRef CAS PubMed.
- D. Schuhknecht, Doctoral Thesis, RWTH Aachen University, 2020.
- H. Osseili, K.-N. Truong, T. P. Spaniol, D. Mukherjee, U. Englert and J. Okuda, Chem. – Eur. J., 2017, 23, 17213–17216 CrossRef CAS PubMed.
- D. Schuhknecht, K.-N. Truong, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2018, 54, 11280–11283 RSC.
- S. Arndt, M. U. Kramer, W. Fegler, Y. Nakajima, I. Del Rosal, R. Poteau, T. P. Spaniol, L. Maron and J. Okuda, Organometallics, 2015, 34, 3739–3747 CrossRef CAS.
- W. Fegler, A. Venugopal, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2013, 52, 7976–7980 CrossRef CAS PubMed.
- L. E. Lemmerz, D. Mukherjee, T. P. Spaniol, A. Wong, G. Ménard, L. Maron and J. Okuda, Chem. Commun., 2019, 55, 3199–3202 RSC.
- L. E. Lemmerz, A. Wong, G. Ménard, T. P. Spaniol and J. Okuda, Polyhedron, 2020, 178, 114331 CrossRef CAS.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 4794–4797 CrossRef CAS PubMed.
- S. Harder, S. Müller and E. Hübner, Organometallics, 2004, 23, 178–183 CrossRef CAS.
- T. Höllerhage, T. P. Spaniol, A. Carpentier, L. Maron and J. Okuda, Inorg. Chem., 2022, 61, 3309–3316 CrossRef PubMed.
- T. Höllerhage, D. Schuhknecht, A. Mistry, T. P. Spaniol, Y. Yang, L. Maron and J. Okuda, Chem. – Eur. J., 2021, 27, 3002–3007 CrossRef PubMed.
- D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 310–314 CrossRef CAS PubMed.
- D. Schuhknecht, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Chem. Commun., 2019, 55, 14837–14839 RSC.
- J. Martin, J. Eyselein, J. Langer, H. Elsen and S. Harder, Chem. Commun., 2020, 56, 9178–9181 RSC.
- B. Maitland, M. Wiesinger, J. Langer, G. Ballmann, J. Pahl, H. Elsen, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 11880–11884 CrossRef CAS PubMed.
- J. Martin, C. Knüpfer, J. Eyselein, C. Färber, S. Grams, J. Langer, K. Thum, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 9102–9112 CrossRef CAS PubMed.
- H. Bauer, M. Alonso, C. Fischer, B. Rösch, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 15177–15182 CrossRef CAS PubMed.
- E. Le Coz, Z. Zhang, T. Roisnel, L. Cavallo, L. Falivene, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2020, 26, 3535–3544 CrossRef CAS PubMed.
- C. Bellini, V. Dorcet, J.-F. Carpentier, S. Tobisch and Y. Sarazin, Chem. – Eur. J., 2016, 22, 4564–4583 CrossRef CAS PubMed.
- C. Bellini, J.-F. Carpentier, S. Tobisch and Y. Sarazin, Angew. Chem., Int. Ed., 2015, 54, 7679–7683 CrossRef CAS PubMed.
- T. Höllerhage, P. Ghana, T. P. Spaniol, A. Carpentier, L. Maron, U. Englert and J. Okuda, Angew. Chem., Int. Ed., 2022, 61, e202115379 CrossRef PubMed.
- R. Huo, A. J. Armstrong, G. R. Nelmes, D. J. Lawes, A. J. Edwards, C. L. McMullin and J. Hicks, Chem. – Eur. J., 2024, 30, e202400662 CrossRef CAS PubMed.
- D. Schuhknecht, T. P. Spaniol, Y. Yang, L. Maron and J. Okuda, Inorg. Chem., 2020, 59, 9406–9415 CrossRef CAS PubMed.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- Z.-W. Qu, H. Zhu and S. Grimme, Chem. – Eur. J., 2023, 29, e202202602 CrossRef CAS PubMed.
- X. Shi, G. Qin, Y. Wang, L. Zhao, Z. Liu and J. Cheng, Angew. Chem., Int. Ed., 2019, 58, 4356–4360 CrossRef CAS PubMed.
- Y. Liang, U. K. Das, J. Luo, Y. Diskin-Posner, L. Avram and D. Milstein, J. Am. Chem. Soc., 2022, 144, 19115–19126 CrossRef CAS PubMed.
- Y. Liang, I. Efremenko, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2024, 63, e202401702 CrossRef CAS PubMed.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed.
- A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- H. Bauer, K. Thum, M. Alonso, C. Fischer and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 4248–4253 CrossRef CAS PubMed.
- H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef CAS.
- A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- T. Höllerhage, A. Carpentier, T. P. Spaniol, L. Maron, U. Englert and J. Okuda, Chem. Commun., 2021, 57, 6316–6319 RSC.
- C. N. de Bruin-Dickason, T. Sutcliffe, C. Alvarez Lamsfus, G. B. Deacon, L. Maron and C. Jones, Chem. Commun., 2018, 54, 786–789 RSC.
- D. Mukherjee, T. Höllerhage, V. Leich, T. P. Spaniol, U. Englert, L. Maron and J. Okuda, J. Am. Chem. Soc., 2018, 140, 3403–3411 CrossRef CAS PubMed.
- X. Sun and A. Hinz, Inorg. Chem., 2023, 62, 10249–10255 CrossRef CAS PubMed.
- M. C. Lipke and T. D. Tilley, Angew. Chem., Int. Ed., 2012, 51, 11115–11121 CrossRef CAS PubMed.
- M. S. Hill, M. F. Mahon, A. S. S. Wilson, C. Dinoi, L. Maron and E. Richards, Chem. Commun., 2019, 55, 5732–5735 RSC.
- P. M. Chapple and Y. Sarazin, Eur. J. Inorg. Chem., 2020, 3321–3346 CrossRef CAS.
- T. Höllerhage, T. P. Spaniol, U. Englert and J. Okuda, Z. Anorg. Allg. Chem., 2023, 649, e202200315 CrossRef.
- M. Wiesinger, B. Maitland, C. Färber, G. Ballmann, C. Fischer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 16654–16659 CrossRef CAS PubMed.
- X. Shi, C. Hou, C. Zhou, Y. Song and J. Cheng, Angew. Chem., Int. Ed., 2017, 56, 16650–16653 CrossRef CAS PubMed.
- T. Höllerhage, T. P. Spaniol, U. Englert and J. Okuda, Inorg. Chim. Acta, 2022, 543, 121198 CrossRef.
- R. D. Hancock, P. W. Wade, M. P. Ngwenya, A. S. De Sousa and K. V. Damu, Inorg. Chem., 1990, 29, 1968–1974 CrossRef CAS.
- M. T. S. Amorim, S. Chaves, R. Delgado and J. J. R. F. da Silva, J. Chem. Soc., Dalton Trans., 1991, 3065–3072 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- D. Mukherjee, A. K. Wiegand, T. P. Spaniol and J. Okuda, Dalton Trans., 2017, 46, 6183–6186 RSC.
- H. H. Cui, W. Lv, W. Tong, X. T. Chen and Z. L. Xue, Eur. J. Inorg. Chem., 2019, 4653–4659 CrossRef CAS.
- H. H. Cui, J. Wang, X. T. Chen and Z. L. Xue, Chem. Commun., 2017, 53, 9304–9307 RSC.
- M. X. Xu, Z. Liu, B. W. Dong, H. H. Cui, Y. X. Wang, J. Su, Z. X. Wang, Y. Song, X. T. Chen, S. D. Jiang and S. Gao, Inorg. Chem., 2019, 58, 2330–2335 Search PubMed.
- F. Ritter, L. J. Morris, K. N. McCabe, T. P. Spaniol, L. Maron and J. Okuda, Inorg. Chem., 2021, 60, 15583–15592 Search PubMed.
- F. Ritter, Doctoral Thesis, RWTH Aachen University, 2021.
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC.
- F. Ritter, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 23335–23342 Search PubMed.
- R. Chambenahalli, R. M. Bhargav, K. N. McCabe, A. P. Andrews, F. Ritter, J. Okuda, L. Maron and A. Venugopal, Chem. – Eur. J., 2021, 27, 7391–7401 CrossRef CAS PubMed.
- P. Mahawar, T. Rajeshkumar, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2024, 60, 11359–11362 RSC.
- S. Schulz, D. Schuchmann, I. Krossing, D. Himmel, D. Bläser and R. Boese, Angew. Chem., Int. Ed., 2009, 48, 5748–5751 CrossRef CAS PubMed.
- H. Banh, C. Gemel, R. W. Seidel and R. A. Fischer, Chem. Commun., 2015, 51, 2170–2172 RSC.
- J. L. Atwood, K. D. Robinson, C. Jones and C. L. Raston, J. Chem. Soc., Chem. Commun., 1991, 1697–1699 RSC.
- P. Dabringhaus and I. Krossing, Chem. Sci., 2022, 13, 12078–12086 RSC.
- R. J. Wehmschulte, R. Peverati and D. R. Powell, Inorg. Chem., 2019, 58, 12441–12445 CrossRef CAS PubMed.
- M. Schorpp, R. Tamim and I. Krossing, Dalton Trans., 2021, 50, 15103–15110 RSC.
- J. L. Bourque, R. A. Nanni, M. C. Biesinger and K. M. Baines, Inorg. Chem., 2021, 60, 14713–14720 CrossRef CAS PubMed.
- M. Everett, A. Jolleys, W. Levason, M. E. Light, D. Pugh and G. Reid, Dalton Trans., 2015, 44, 20898–20905 RSC.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228–3231 Search PubMed.
- J. T. Boronski, M. P. Stevens, B. van Ijzendoorn, A. C. Whitwood and J. M. Slattery, Angew. Chem., Int. Ed., 2021, 60, 1567–1572 CrossRef CAS PubMed.
- C. G. Andrews and C. L. B. Macdonald, Angew. Chem., Int. Ed., 2005, 44, 7453–7456 CrossRef CAS PubMed.
- A. Higelin, C. Haber, S. Meier and I. Krossing, Dalton Trans., 2012, 41, 12011–12015 RSC.
- A. Barthelemy, K. Glootz, H. Scherer, A. Hanske and I. Krossing, Chem. Sci., 2022, 13, 439–453 RSC.
- Z. L. Li, G. Thiery, M. R. Lichtenthaler, R. Guillot, I. Krossing, V. Gandon and C. Bour, Adv. Synth. Catal., 2018, 360, 544–549 CrossRef CAS.
- Z. L. Li, S. W. Yang, G. Thiery, V. Gandon and C. Bour, J. Org. Chem., 2020, 85, 12947–12959 Search PubMed.
- P. Dabringhaus, A. Barthelemy and I. Krossing, Z. Anorg. Allg. Chem., 2021, 647, 1660–1673 CrossRef CAS.
- K. Glootz, D. Kratzert, D. Himmel, A. Kastro, Z. Yassine, T. Findeisen and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 14203–14206 CrossRef CAS PubMed.
- M. R. Lichtenthaler, F. Stahl, D. Kratzert, L. Heidinger, E. Schleicher, J. Hamann, D. Himmel, S. Weber and I. Krossing, Nat. Commun., 2015, 6, 8288 CrossRef CAS PubMed.
- C. Dohmeier, D. Loos and H. Schnockel, Angew. Chem., Int. Ed. Engl., 1996, 35, 129–149 CrossRef CAS.
- J. C. Beamish, A. Boardman and I. J. Worrall, Polyhedron, 1991, 10, 95–99 Search PubMed.
- L. J. Morris, P. Mahawar and J. Okuda, J. Org. Chem., 2022, 80, 5090–5096 Search PubMed.
- M. Auer, F. Diab, K. Eichele, H. Schubert and L. Wesemann, Dalton Trans., 2022, 51, 5950–5961 RSC.
- J. J. Maudrich, F. Diab, S. Weiss, M. Widemann, T. Dema, H. Schubert, K. M. Krebs, K. Eichele and L. Wesemann, Inorg. Chem., 2019, 58, 15758–15768 CrossRef CAS PubMed.
- K. R. Cairns, R. P. King, R. D. Bannister, W. Levason and G. Reid, Dalton Trans., 2023, 52, 2293–2308 RSC.
- A. Higelin, U. Sachs, S. Keller and I. Krossing, Chem. – Eur. J., 2012, 18, 10029–10034 Search PubMed.
- A. Barthélemy, H. Scherer, H. Weller and I. Krossing, Chem. Commun., 2023, 59, 1353–1356 RSC.
- A. Barthélemy, H. Scherer, M. Daub, A. Bugnet and I. Krossing, Angew. Chem., Int. Ed., 2023, 62, e202311648 Search PubMed.
- A. Barthélemy, H. Scherer and I. Krossing, Chem. – Eur. J., 2022, 28, e202201369 CrossRef PubMed.
- P. Dabringhaus, H. Scherer and I. Krossing, Nat. Synth., 2024, 3, 732–743 Search PubMed.
- P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360–1363 CrossRef CAS PubMed.
- F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5152–5154 CrossRef CAS PubMed.
- P. A. Rupar, R. Bandyopadhyay, B. F. T. Cooper, M. R. Stinchcombe, P. J. Ragogna, C. L. B. Macdonald and K. M. Baines, Angew. Chem., Int. Ed., 2009, 48, 5155–5158 CrossRef CAS PubMed.
- R. P. King, J. M. Herniman, W. Levason and G. Reid, Inorg. Chem., 2023, 62, 853–862 Search PubMed.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS PubMed.
- M. M. D. Roy, A. A. Omana, A. A. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |