
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heavier alkaline earth and heterobimetallic s-block “ate” complexes of a di(amido)siloxane ligand: solid-state structure and dynamic solution-phase behaviour†
Matthew D.
Haynes‡
 a,
Andrea
O'Reilly‡
b,
Alice J. M.
Poole‡
a,
Aisling F.
Roper
a,
Stefan
Thum
c,
Louis J.
Morris
a,
Martyn P.
Coles
*b,
J. Robin
Fulton
*b,
Sjoerd
Harder
c,
Zoë R.
Turner
*a and
Dermot
O'Hare
*a
a,
Andrea
O'Reilly‡
b,
Alice J. M.
Poole‡
a,
Aisling F.
Roper
a,
Stefan
Thum
c,
Louis J.
Morris
a,
Martyn P.
Coles
*b,
J. Robin
Fulton
*b,
Sjoerd
Harder
c,
Zoë R.
Turner
*a and
Dermot
O'Hare
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: zoe.turner@earth.ox.ac.uk; dermot.ohare@chem.ox.ac.uk
bSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington 6012, New Zealand. E-mail: martyn.coles@vuw.ac.nz; j.robin.fulton@vuw.ac.nz
cUniversität Erlangen-Nürnberg, Egerlandstrasse 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
First published on 12th February 2025
Abstract
The diverse solid-state structures and solution-phase dynamics of both neutral and heterometallic s-block “ate” complexes of the heavier alkaline earth metals (Ae; Ca–Ba) supported by a chelating and flexible di(amido)siloxane ligand ([NON-DippL]2− = [O(SiMe2NDipp)2]2−) are described, enabling comparison with those of closely related di(amido) ligands based on either flexible aliphatic or rigid xanthene-based backbones. Three dimeric alkaline earth complexes [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)) which feature a κ3-N,O,N′-κ1-N′-tridentate coordination mode were prepared from protonolysis reactions between NON-DippLH2 with 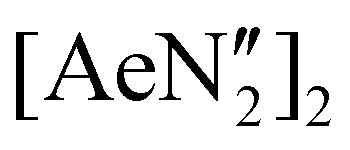 (Ae = Ca, Sr and Ba); N′′ = [N(SiMe3)2]−. In tetrahydrofuran, these complexes were readily converted into the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)). Heterometallic Ae/K amide “ate” complexes were afforded through two routes: reaction of previously reported [(NON-DippL)Mg]2 (1) with two equivalents of KN′′ at elevated temperatures resulted in [(NNO-DippL)Mg(μ-N′′)K]n (8; NNO-DippL = [OSiMe2NDippSiMe2NDipp]2−), whereas the equimolar reaction of NON-DippLH2 with
(Ae = Ca, Sr and Ba); N′′ = [N(SiMe3)2]−. In tetrahydrofuran, these complexes were readily converted into the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)). Heterometallic Ae/K amide “ate” complexes were afforded through two routes: reaction of previously reported [(NON-DippL)Mg]2 (1) with two equivalents of KN′′ at elevated temperatures resulted in [(NNO-DippL)Mg(μ-N′′)K]n (8; NNO-DippL = [OSiMe2NDippSiMe2NDipp]2−), whereas the equimolar reaction of NON-DippLH2 with 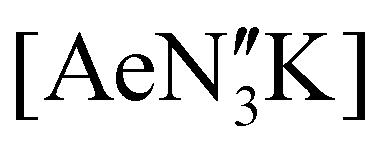 led to [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)). Complexes 8–11 exist as one-dimensional coordination polymers propagated by K+–aryl π-facial interactions in the solid-state. The mixed amide/siloxide “NNO” ligand in 8 results from a 1,3-silyl retro-Brook rearrangement of the original di(amido)siloxane ligand, while the larger Ae2+ congeners readily accommodate the coordination of KN′′ with the di(amido)siloxane ligand retaining a κ3-N,O,N′-tridentate motif in 9–11. Finally, the solution-phase behaviour of 8–11 in both toluene and thf were investigated indicating the reversible dissociation of KN′′ from 9–11 and the thermodynamic parameters of this process were elucidated.
led to [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)). Complexes 8–11 exist as one-dimensional coordination polymers propagated by K+–aryl π-facial interactions in the solid-state. The mixed amide/siloxide “NNO” ligand in 8 results from a 1,3-silyl retro-Brook rearrangement of the original di(amido)siloxane ligand, while the larger Ae2+ congeners readily accommodate the coordination of KN′′ with the di(amido)siloxane ligand retaining a κ3-N,O,N′-tridentate motif in 9–11. Finally, the solution-phase behaviour of 8–11 in both toluene and thf were investigated indicating the reversible dissociation of KN′′ from 9–11 and the thermodynamic parameters of this process were elucidated.
Introduction
Central to the development of molecular heavier alkaline earth (Ae) chemistry during the past two-decades has been the utilisation of bulky monoanionic chelating N-donor ligands, such as the ubiquitous β-diketiminate, which suppress aggregation and Schlenk-type ligand redistribution.1–3 Such ligands possess a well-defined pocket that enables delivery of a nucleophilic monodentate ligand, X (X = labile monoanionic ligand e.g. hydride, amide, alkoxide, halide etc.), towards substrates,4–8 and have also made highly reducing Ae(I) reagents (as well as low-valent “synthons”) acessible.9–11 Lacking a labile Aeδ+–Xδ− bond, neutral Ae(II) coordination complexes supported by chelating dianionic ligands, [LAe] (L = dianionic ancillary ligand, Ae = Be, Mg, Ca, Sr and Ba) have received comparatively less attention. However, alkaline earth complexes supported by various dianionic ligands, including reduced α-diimines,12–17 bora-amidinates,18 bis(phenoxyimines)19 and bis(phosphinochalcogenamides),20,21 have been shown to display reactivity resulting from the non-innocence of their ancillary ligands. Meanwhile, alkali metal magnesiates M[MgX3] (M = alkali metal) and heavier analogues based on monodentate ligands demonstrate increased reactivity as metalating reagents, bases, and catalysts relative to homometallic analogues.22–27 By analogy to the successful use of bulky, chelating monoanionic ligands in homometallic Ae chemistry, the use of chelating dianionic ligands may facilitate the wider elaboration of kinetically stabilised heterometallic s-block “ate” complexes of the type M[LAeX] featuring the heavier alkaline earth elements.28In recent years, numerous reports have exploited [LAe] as precursors for the synthesis of more reactive heterometallic species. In 2021, Hill and co-workers reported the reduction of a magnesium complex supported by a chelating ethylene-bridged bis(silylamido) ligand to form the heterometallic low oxidation state complex [Na{(CH2SiMe2NDipp)2Mg}]2 (I, Chart 1; Dipp = 2,6-diisopropylphenyl).29 The reactivity of I has since been shown to diverge from that of Mg(I) dimers supported by monoanionic ligands.30–32 Conversely, attempts to synthesise an analogous heterometallic Be(I) complex instead resulted in the C–H activation of benzene to form the polymeric alkali metal phenylberyllates [(CH2SiMe2NDipp)2Be(μ-Ph)M]n (M = Li and Na, II, Chart 1).33 In 2022, Hicks and co-workers reported the attempted reduction of magnesium and calcium complexes supported by a rigid NON-xanthene ligand ([xanth-tBuNONDipp]2− = 4,5-bis(2,6-diisopropylphenylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene), which resulted in the isolation of heterobimetallic Ae/K hydrides following C–H activation of the solvent by transient Ae(I) species (III, Chart 1).34,35 Jones and co-workers subsequently reported the isolation of a Mg/K dimer with a Mg(I)–Mg(I) bond as well as reduced Mg/K and Ca/K dinitrogen complexes (IV, Chart 1) supported by even bulkier NON-xanthene ligands.36–38
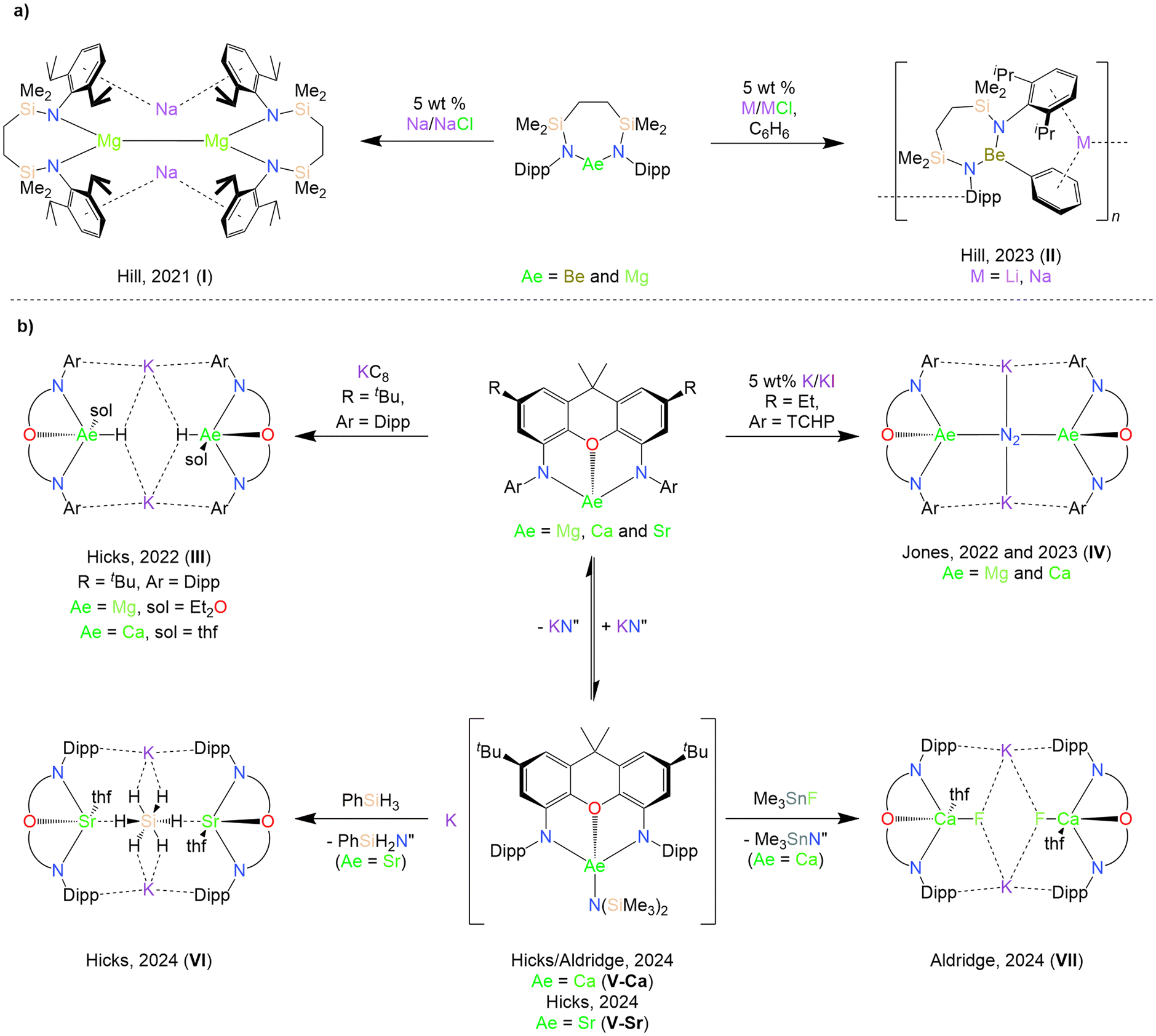 | ||
| Chart 1 Selected examples of previously reported alkaline earth complexes supported by (a) ethylene-bridged bis(silylamido) and (b) NON-xanthene ligands. | ||
More recent reports have deliberately targeted heterobimetallic heteroleptic “ate” complexes. Hicks and co-workers described attempts to rationally synthesise a heterobimetallic Sr/K hydride analogous to IIIvia the silane metathesis of a heteroleptic Sr/K amide “ate” complex (V-Sr, Chart 1b), which instead resulted in the isolation of hypervalent hydridosilicate species (VI, Chart 1b).39 Aldridge and co-workers have demonstrated the ability of the [xanth-tBuNONDipp]2− ligand to support the first reported example of a molecular calcium fluoride with a terminal Ca–F bond (VII, Chart 1b), which, unlike dimeric homometallic Ae μ-fluoride complexes, acts as a source of nucleophilic fluoride.40 The synthesis of VII employed a heterobimetallic Ca/K amide “ate” complex (also reported independently by Hicks and co-workers, V-Ca, Chart 1b), from which potassium hexamethyldisilazide, KN′′ (N′′ = [N(SiMe3)2]−) was found to reversibly dissociate in solution. Additionally, the Jones group have also described a family of both 1,3-di(amido)propane alkaline earth complexes consisting of both a thf-free magnesium dimer ([(TCHPNCN)Mg]2; [TCHPNCN] = [CH2{CH2N(TCHP)}2]2–, TCHP = 2,4,6-tricyclohexylphenyl) and a series of solvated monomeric species ([(TCHPNCN)Ae(sol)n]; sol = Et2O, Ae = Be, n = 1; sol = thf, Ae = Mg, n = 2; Ae = Ca, n = 3; Ae = Sr, n = 4),41 as well as a family of para-terphenyl bis(anilide) alkaline earth complexes.42 While both ligand systems enabled the synthesis of homometallic alkaline earth complexes, these proved unsuitable as precursors to reduced heterometallic species.
We have recently reported the synthesis of a dimeric, donor-free magnesium complex supported by a flexible di(amido)siloxane ligand, [(NON-DippL)Mg]2 ([NON-DippL]2− = [O(SiMe2NDipp)2]2−, 1).43 The reduction of 1 in diethylether resulted in the formation of low oxidation state Mg(I) centres coordinated by a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [NON-DippL]2− and [NNO-DippL]2− ligands, with the formation of the latter resulting from a previously reported 1,3-silyl retro-Brook rearrangement ([NNO-DippL]2− = [OSiMe2NDippSiMe2NDipp]2−).44,45 Recrystallisation of this mixture from tetrahydrofuran (thf) afforded [{K(thf)2}2(NNO-DippL)Mg–Mg(NNO-DippL)], verifying the formation of a Mg(I)–Mg(I) bond. These results prompted us to further investigate the development of heavier alkaline earth complexes (Ae = Ca, Sr and Ba) supported by the by the [NON-DippL]2− ligand, with a view to accessing additional reactive heterometallic species.
:1 mixture of [NON-DippL]2− and [NNO-DippL]2− ligands, with the formation of the latter resulting from a previously reported 1,3-silyl retro-Brook rearrangement ([NNO-DippL]2− = [OSiMe2NDippSiMe2NDipp]2−).44,45 Recrystallisation of this mixture from tetrahydrofuran (thf) afforded [{K(thf)2}2(NNO-DippL)Mg–Mg(NNO-DippL)], verifying the formation of a Mg(I)–Mg(I) bond. These results prompted us to further investigate the development of heavier alkaline earth complexes (Ae = Ca, Sr and Ba) supported by the by the [NON-DippL]2− ligand, with a view to accessing additional reactive heterometallic species.
Results and discussion
Donor-free dimers
We initially targeted heavier (Ae = Ca, Sr and Ba) analogues of 1 in order to assess their suitability as precursors for the synthesis of reduced heterometallic species. Gratifyingly, the 2:1 reactions of NON-DippLH2 with  (Ae = Ca, Sr and Ba) in benzene resulted in the crystallisation of the colourless dimeric complexes [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)) directly from the reaction mixture, enabling their isolation in moderate yield (66% (2), 77% (3) and 48% (4)) and characterisation through single crystal X-ray diffraction studies (SC-XRD; Scheme 1a). Complexes 2–4 each crystallise from benzene in the C2/c space group as largely isostructural N-bridged dimers (Fig. 1; Table 1).
(Ae = Ca, Sr and Ba) in benzene resulted in the crystallisation of the colourless dimeric complexes [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)) directly from the reaction mixture, enabling their isolation in moderate yield (66% (2), 77% (3) and 48% (4)) and characterisation through single crystal X-ray diffraction studies (SC-XRD; Scheme 1a). Complexes 2–4 each crystallise from benzene in the C2/c space group as largely isostructural N-bridged dimers (Fig. 1; Table 1).
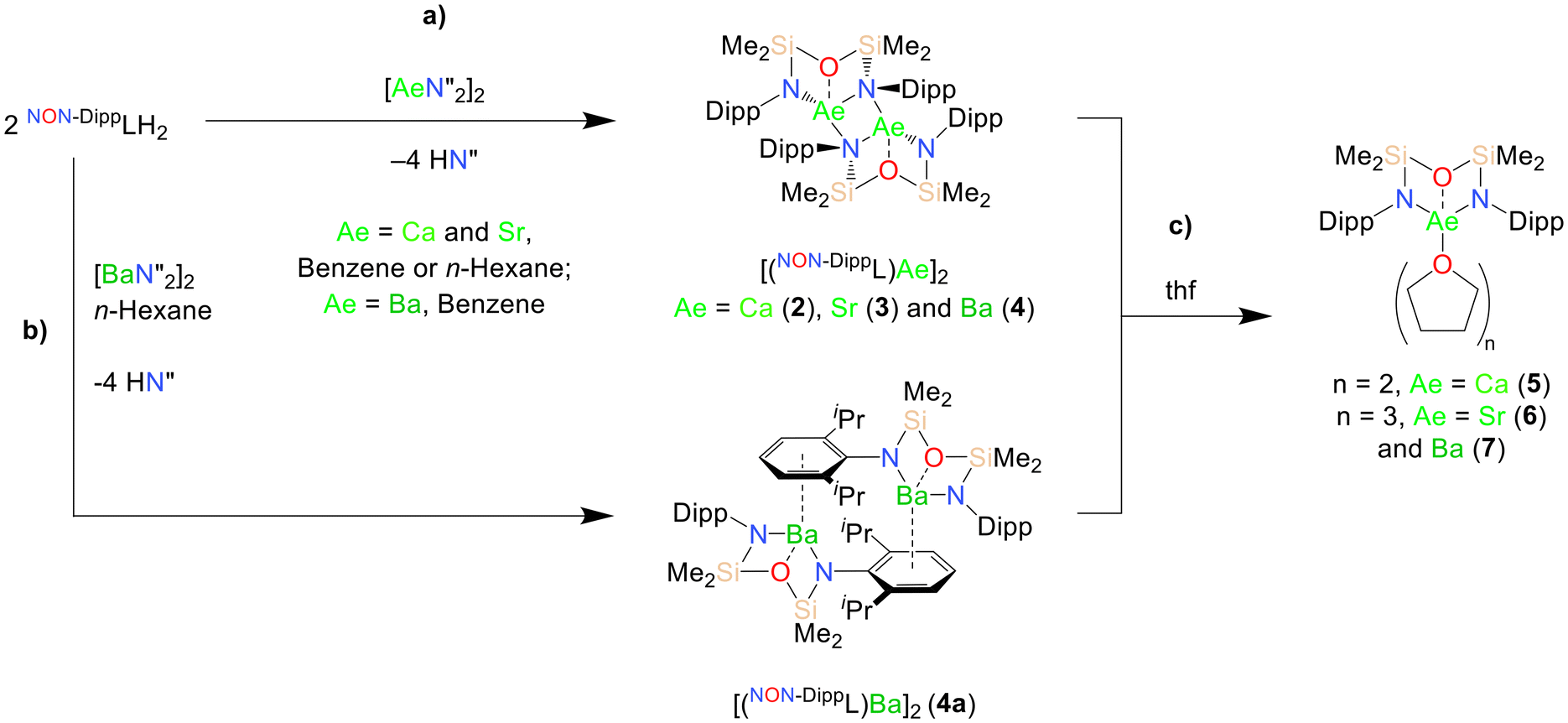 | ||
| Scheme 1 (a) Synthesis of the nitrogen-bridged dimers [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)), (b) synthesis of the aryl-bridged dimer [(NON-DippL)Ba]2 (4a) and (c) synthesis of the monomeric thf adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)). | ||
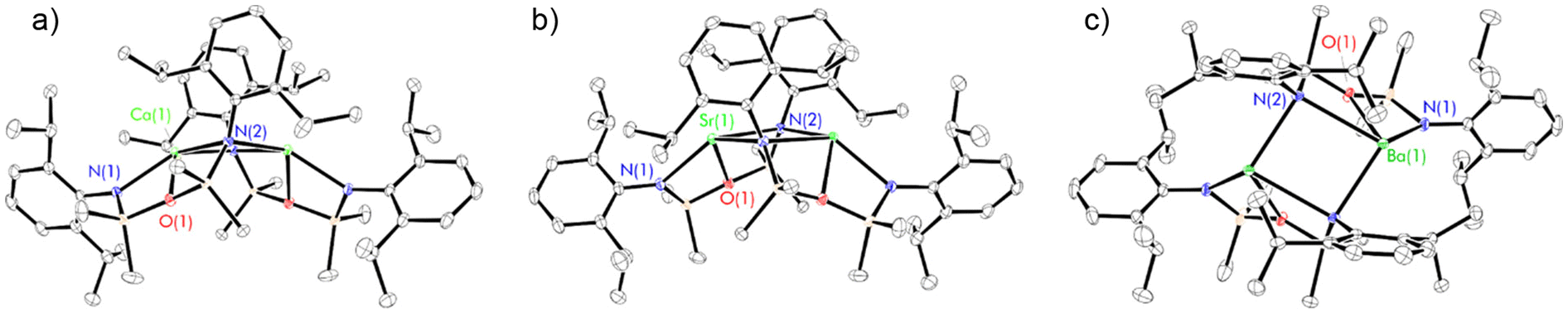 | ||
| Fig. 1 Thermal displacement ellipsoid drawings (30% probability) of (a) [(NON-DippL)Ca]2 (2), (b) [(NON-DippL)Sr]2 (3), (c) [(NON-DippL)Ba]2 (4). The full dimeric structures of 2–4 are generated by a (−X + 1, Y, −Z + 1/2) symmetry operation in each case. All hydrogen atoms have been omitted for clarity. | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Ae(1)–N(1) | 1.987(1) | 2.340(1) | 2.473(1) | 2.582(1) |
| Ae(1)–N(2) | 3.551(1) | 2.686(1) | 2.798(1) | 2.970(1) |
| Ae(1)–N(2)* | 1.999(1) | 2.3787(9) | 2.541(1) | 2.714(1) |
| Ae(1)–O(1) | 2.066(1) | 2.3494(8) | 2.5086(9) | 2.672(1) |
| Ae(1)–Ae(1)* | 4.1130(9) | 3.7151(8) | 3.9504(8) | 4.1561(8) |
| N(1)–Ae(1)–N(2) | 125.62(4) | 126.01(3) | 119.22(3) | 112.61(4) |
In each case, the asymmetric unit consists of a monomeric [(NON-DippL)Ae] fragment in which the [NON-DippL]2− ligand is coordinated to Ae2+ in a κ3-N,O,N′-tridentate manner. This contrasts with the structure of the magnesium congener 1, in which each ligand chelates the smaller Mg2+ through a bidentate N,O-bonding mode (and bridges between Mg2+ centres in a κ2-N,O-κ1-N′ manner in the full structure, Fig. S59†).43 This shows that the larger ionic radii of Ca2+, Sr2+ and Ba2+ (1.00 Å, 1.18 Å and 1.36 Å respectively)46 facilitate the coordination of an additional bulky amide donor. The resulting six-membered AeN2Si2O metallacycle consists of two four-membered AeNSiO rings; these are folded with respect to one another about the Ae–O axis, resulting in an “open-book” conformation. The full pentacyclic dimeric structures each consist of two [(NON-DippL)Ae] units bridged by a central four-membered Ae2N2 ring, with two μ2-N donors bridging between two four-coordinate Ae2+ centres. The coordination geometry at Ae2+ is constrained by the chelating ligand, and is best described as a distorted AeN3 trigonal pyramid in which the additional siloxane donor subtends the N(1)–Ae(1)–N(2) bite angle. Additional coordinative saturation is provided by both non-covalent anagostic Ae–HiPr interactions (Ca–HiPr = 2.3988(4)–2.7382(3) Å, Sr–HiPr = 2.4834(4)–2.8890(3) Å, Ba–HiPr = 2.6428(4)–3.1030(3) Å) and, in the case of 3 and 4, respective η1- and η2-π-facial Ae–aryl interactions ((Sr(1)–C(17)* = 3.029(1) Å, Ba(1)–centroidC(17),C(18)* = 3.077(1) Å); * refers to symmetry related atoms). The stabilisation of low-coordinate complexes of the heavier Ae2+ cations by anagostic and π-facial interactions has been well documented in the literature.47,48
This dimerisation mode has not been previously reported for any complexes of the [NON-DippL]2− ligand. However, related chromium and cobalt dimers bearing a less bulky N-mesityl substituted di(amido)siloxane ligand are known.44,49 Whilst lacking an additional O-donor, Jones’ magnesium 1,3-di(amido)propane complex VIII displays a similar μ-N bridged dimeric structure.41 There are also a number of related examples of N-bridged dimers with M2N2 rings supported by di(amido)siloxane ligands with less bulky N-aryl or N-alkyl substituents in which the two AeN2Si2O rings are anti with respect to one another.49–52 Nitrogen-bridged dimeric motifs are also present in various previously reported alkaline earth complexes.42,53–55
The coordination of the silylether donor to Ae2+ in 2–4 is consistent with the structure of 1, and can be rationalised by considering both the oxophilicity of the heavier Ae2+ congeners and their preference for high coordination numbers.47,48,56 Indeed, the only previously reported examples of [NON-DippL]2− complexes that feature metal–oxygen interactions feature oxophilic Zr(IV),57 U(IV)58 or Y(III)59 centres. However, silyl ether–metal interactions are generally weak,60 with the unsupported coordination of bis(trimethylsilyl)ether (which generally acts as a non-coordinating solvent) to a highly Lewis acidic cationic Mg-centred cation only recently reported.47 As such their presence in 2–4 is attributed to an entropic advantage provided by their intramolecular nature (i.e. the chelate effect). While these interactions result in relatively short Ae–O distances (2.3494(8) Å (2), 2.5086(9) Å (3) and 2.672(1) Å (4)), their weak nature is supported by a large Si–O–Si angle in each case (151.14(5)° (2), 151.32(6)° (3) and 150.32(7)° (4)).
Complexes 2–4 were targeted with a view to assessing their suitability as precursors for the synthesis of novel low-valent alkaline earth complexes. However, 2–4 each co-crystallise with a single molecule of benzene, which although non-bonding could not be removed in vacuo.61 Previous literature reports have demonstrated that the high reactivity of low-valent alkaline earth complexes can result in the reduction of benzene to form “low-valent synthons” containing the [C6H6]2− dianion, precluding the isolation of true low-valent species.62–64 Consequently, the isolation of benzene-free samples of 2–4 was targeted. The layering of a n-hexane solution of NON-DippLH2 (2 eq.) over a n-hexane solution of  (Ae = Ca, Sr and Ba) again resulted in the growth of colourless crystals of [(NON-DippL)Ae]2 (Ae = Ca (2) Sr (3) and Ba (4a), Scheme 1a and 1b). SC-XRD analysis confirmed that the calcium and strontium congeners 2 and 3 crystallise from n-hexane with benzene-free structures that are isostructural to those obtained from benzene. However, while the barium congener 4a also crystallises from n-hexane as a dimer in the C2/c space group, in this case its structure exhibits dimerisation via two Ba2+–aryl η5-π-facial interactions (Fig. 2).
(Ae = Ca, Sr and Ba) again resulted in the growth of colourless crystals of [(NON-DippL)Ae]2 (Ae = Ca (2) Sr (3) and Ba (4a), Scheme 1a and 1b). SC-XRD analysis confirmed that the calcium and strontium congeners 2 and 3 crystallise from n-hexane with benzene-free structures that are isostructural to those obtained from benzene. However, while the barium congener 4a also crystallises from n-hexane as a dimer in the C2/c space group, in this case its structure exhibits dimerisation via two Ba2+–aryl η5-π-facial interactions (Fig. 2).
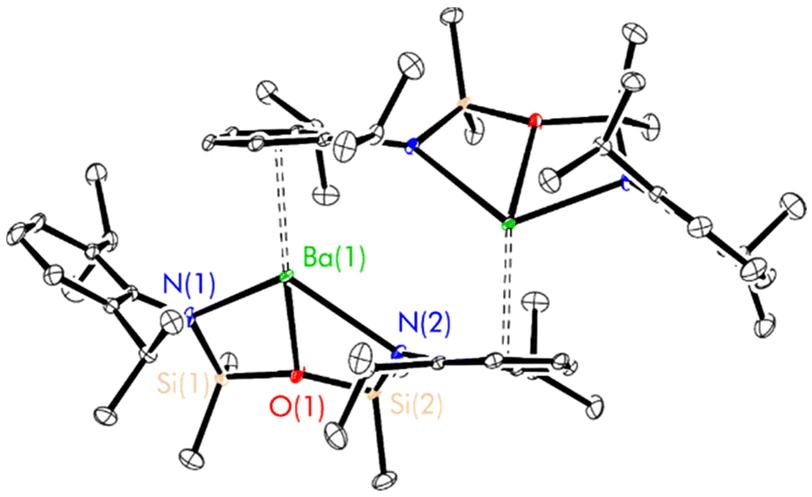 | ||
| Fig. 2 Thermal displacement ellipsoid drawing (30% probability) of the aryl-bridged complex [(NON-DippL)Ba]2 (4a) generated by a (−X + 1, Y, −Z + 3/2) symmetry operation. All hydrogen atoms have been omitted for clarity. Selected metrical parameters (* refers to symmetry related atoms): Ba(1)–N(1) = 2.569(2) Å, Ba(1)–N(2) = 2.799(1) Å, Ba(1)–O(1) = 2.651(1) Å, Ba(1)–N(2)* = 3.702(2) Å, Ba(1)–centroidC(17)–C(22)* = 2.862(1) Å, Ba(1)–Ba(1)* = 4.7644(6) Å; N(1)–Ba(1)–N(2) = 114.57(5)°, N(1)–Ba(1)–centroidC(17)–C(22)* = 112.49(4)°, N(2)–Ba(1)–centroidC(17)–C(22)* = 128.01(3)°. | ||
The Ae2N2 ring present in 2–4 is absent in 4a, resulting in marked increases in both the Ba(1)–N(2)* (2.714(1) Å (4) vs. 3.702(2) Å (4a)) and Ba–Ba* (4.1561(8) Å (4) vs. 4.7644(6) Å (4a)) distances, as well as a shortening of the Ba(1)–centroidC(17)–C(22)* distance (3.920(1) Å (4) vs. 2.862(1) Å (4a)) and planarisation of the otherwise tetrahedral nitrogen donor (Σ{∠N(2)} = 653.3(2)° (4) vs. 358.8(2)° (4a)). Besides the change in the hybridisation state of N(2) (vide supra), the [(NON-DippL)Ba] unit of 4a shows little variation from that of 4, with the κ3-N,O,N′-tridentate coordination mode of [NON-DippL]2− to Ba2+ retained. The structure of 4a is highly reminiscent of aryl-bridged Pb(II) and Sn(II) dimers supported by a related di(amido)siloxane ligand with less bulky 2,6-dimethylphenyl substituents that have been previously reported by Fulton and co-workers.51 That Ba2+ interacts with an aryl ring instead of an additional “hard” amide donor in this case reflects its greater size, electropositivity and, consequently, preference for “soft” multihaptic π-facial interactions that provide greater coordinative saturation.65 Notably, the structure of 4a does not contain any co-crystallised n-hexane (unlike crystals of 2 and 3 grown from this solvent). Furthermore, the presence of a η2-π-facial interaction in the structure of 4 suggests that there is little difference in energy with the “slipped” η5-structure of 4a. Together, these results suggest that the variation in dimerisation mode between 4 and 4a is likely induced by changes in crystal packing forces.
Monomeric thf adducts
The poor solubility of complexes 2–4 in non-donor solvents precluded a direct assessment of their solution-phase structures by NMR spectroscopy. However, 2–4 each dissolve readily in tetrahydrofuran, enabling both the structural and spectroscopic characterisation of the resulting adducts. 1H NMR spectra recorded in thf-d8 were consistent with the instantaneous conversion of the dimeric complexes 2–4 into monomeric complexes of the form [(NON-DippL)Ae(thf)n] (Ae = Ca, Sr and Ba, Scheme 1c). These each produce spectra consisting of five sharp signals attributed to the two aromatic, two isopropyl and single silylmethyl environments expected from a C2v-symmetric monomeric adduct. Increasing Ae2+ size results only in minor variations in the chemical shifts of these signals. Notably, the corresponding 29Si NMR spectra each consist of a single singlet, which shifts to lower frequency with increasing Ae2+ atomic number (δCa = −19.77 ppm > δSr = −23.74 ppm > δBa = −26.22 ppm). This trend reflects the enhanced accumulation of negative charge on the [NON-DippL]2− ligand that results from its coordination to the more electropositive heavier congeners.Structural verification was provided by SC-XRD analysis of the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Sr = 6 (6) and Ba (7)), colourless crystals of which were isolated upon the slow evaporation of concentrated thf solutions of 2–4 (Fig. 3; Table 2).
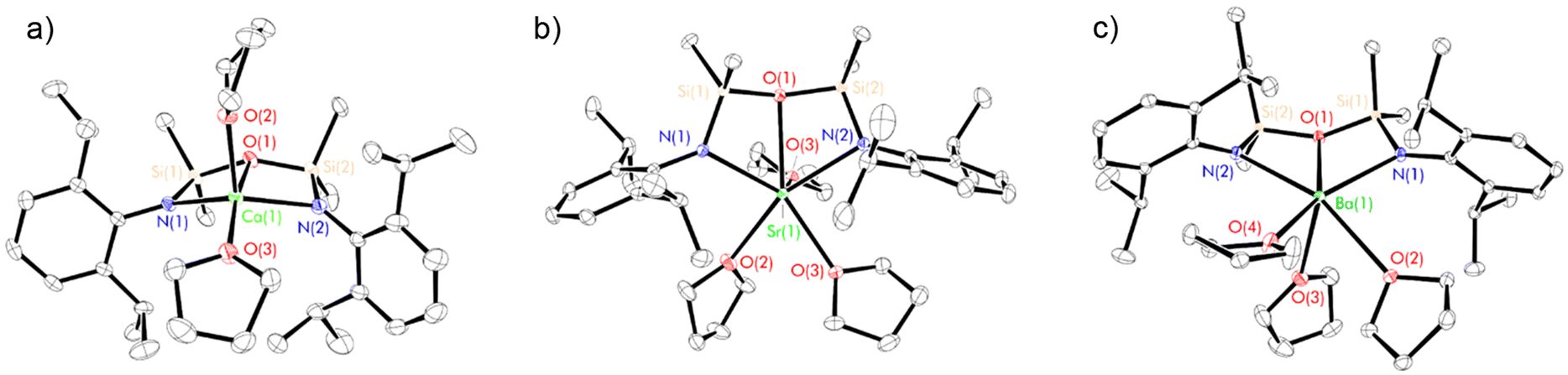 | ||
| Fig. 3 Thermal displacement ellipsoid drawings (30% probability) of (a) [(NON-DippL)Ca(thf)2] (5), (b) [(NON-DippL)Sr(thf)3] (6) and (c) [(NON-DippL)Ba(thf)3] (7). All hydrogen atoms have been omitted for clarity. | ||
| [(NON-DippL)Mg(thf)2] | 5 | 6 | 7 | |
|---|---|---|---|---|
| Ae(1)–N(1) | 2.040(2) | 2.313(1) | 2.564(2) | 2.619(2) |
| Ae(1)–N(2) | 2.041(2) | 2.285(1) | 2.507(3) | 2.676(3) |
| Ae(1)–O(1) | 2.308(2) | 2.490(1) | 2.627(2) | 2.889(3) |
| Ae(1)–O(2) | 2.065(2) | 2.337(1) | 2.557(4) | 2.838(3) |
| Ae(1)–O(3) | 2.047(2) | 2.323(1) | 2.510(3) | 2.689(2) |
| Ae(1)–O(4) | — | — | 2.591(2) | 2.772(3) |
| N(1)–Ae(1)–N(2) | 129.1(1) | 119.59(5) | 114.48(8) | 109.33(8) |
| Si(1)–O(1)–Si(2) | 143.6(1) | 147.42(8) | 148.2(2) | 151.0(2) |
In each case the κ3-N,O,N′-tridentate coordination mode of [NON-DippL]2− to Ae2+ is been retained. The structure of 5 (Fig. 3b) is very similar to that of the previously reported magnesium congener [(NON-DippL)Mg(thf)2].66 In both cases, two thf molecules are coordinated to a five-coordinate Ae2+ cation with a distorted trigonal–bipyramidal coordination geometry (τ5 = 0.70, Mg; 0.82, Ca).67 The axial positions are occupied by one thf molecule and the siloxane tether, while the equatorial plane comprises the second thf molecule and the two amide donors of a [NON-DippL]2− ligand. Contrastingly, the structures of 6 and 7 (Fig. 3b and c) each exhibit an additional coordinated thf molecule, resulting in six-coordinate Sr2+ and Ba2+ centres with pentagonal pyramidal coordination geometries. The apical position is occupied by a thf molecule in both cases, with the equatorial plane occupied by two thf molecules and the tridentate [NON-DippL]2− ligand. NMR spectra of isolated samples of 5–7 recorded in thf-d8 were fully consistent with those of 2–4, confirming the rapid full conversion of the donor-free dimers into solvated monomers upon their dissolution in thf.
Notably, 5 and 7 adopt very similar structures to the monomeric calcium and barium carbene complexes [(C{PPh2NDipp}2)Ae(thf)n] (Ae = Ca, n = 2; Ae = Ba, n = 3) reported previously by Harder and co-workers, which feature a dianionic bis(iminophosphoranomethane) ligand that is isoelectronic with the [NON-DippL]2− ligand employed in this study.68,69 While the weak nature of the Ae–O interactions in 5–7 is confirmed by large Si–O–Si angles (147.32(8)° (5), 148.2(2)° (6) and 151.0(2)° (7)),60 the respective Ca–O (2.490(1) Å) and Ba–O (2.889(3) Å) distances in 5 and 7 are shorter than the corresponding Ae–Ccarbene distances in the related monomeric carbene complexes (2.548(2) Å, Ca and 2.918(7) Å, Ba) despite the much stronger donor strength of the latter. The very similar steric profiles of the two ligands result in the coordination of the same number of thf molecules in each case (two for Ca, three for Ba).
Dimeric magnesium thf adducts
For comparison, the solution-phase behaviour of the magnesium congener 1 in thf was also investigated. The 1H NMR spectrum of 1 recorded in thf-d8 at 25 °C initially consists of broad resonances that are consistent with the formation of dimeric species with fluxional solution-phase structures (vide infra). Conversely, a subsequent spectrum recorded after heating the solution at 80 °C for 16 hours consisted of sharp signals consistent the exclusive formation of the C2v-symmetric monomeric complex [(NON-DippL)Mg(thf)2], for which very similar NMR spectra have been recorded in C6D6.66 The dynamic solution-phase behaviour of 1 was confirmed by the crystallographic characterisation of the two dimeric thf adducts [{(NON-DippL)Mg}2(thf)] (1·thf) and [(NON-DippL)Mg(thf)]2 (1·thf2), which respectively crystallised upon the slow evaporation of thf/n-hexane and thf solutions of 1 (Scheme 2 and Fig. 4).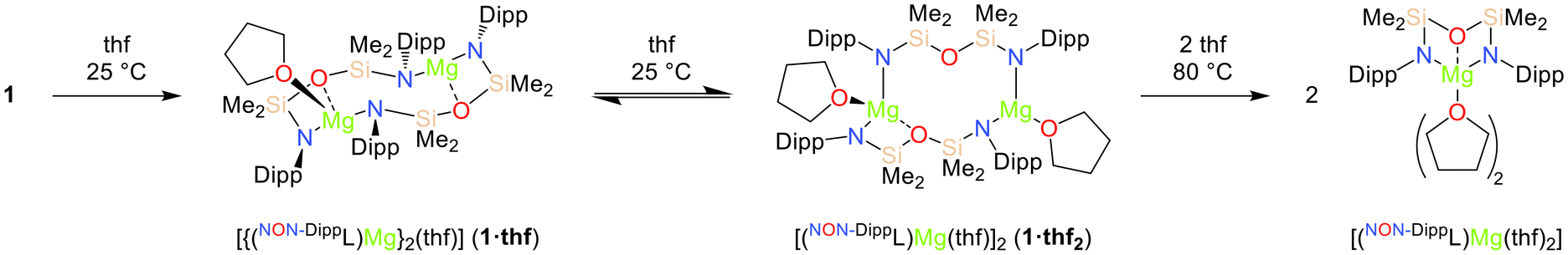 | ||
| Scheme 2 Conversion of [(NON-DippL)Mg]2 (1) into the thf adducts [{(NON-DippL)Mg}2(thf)] (1·thf), [(NON-DippL)Mg(thf)]2 (1·thf2) and [(NON-DippL)Mg(thf)2]. | ||
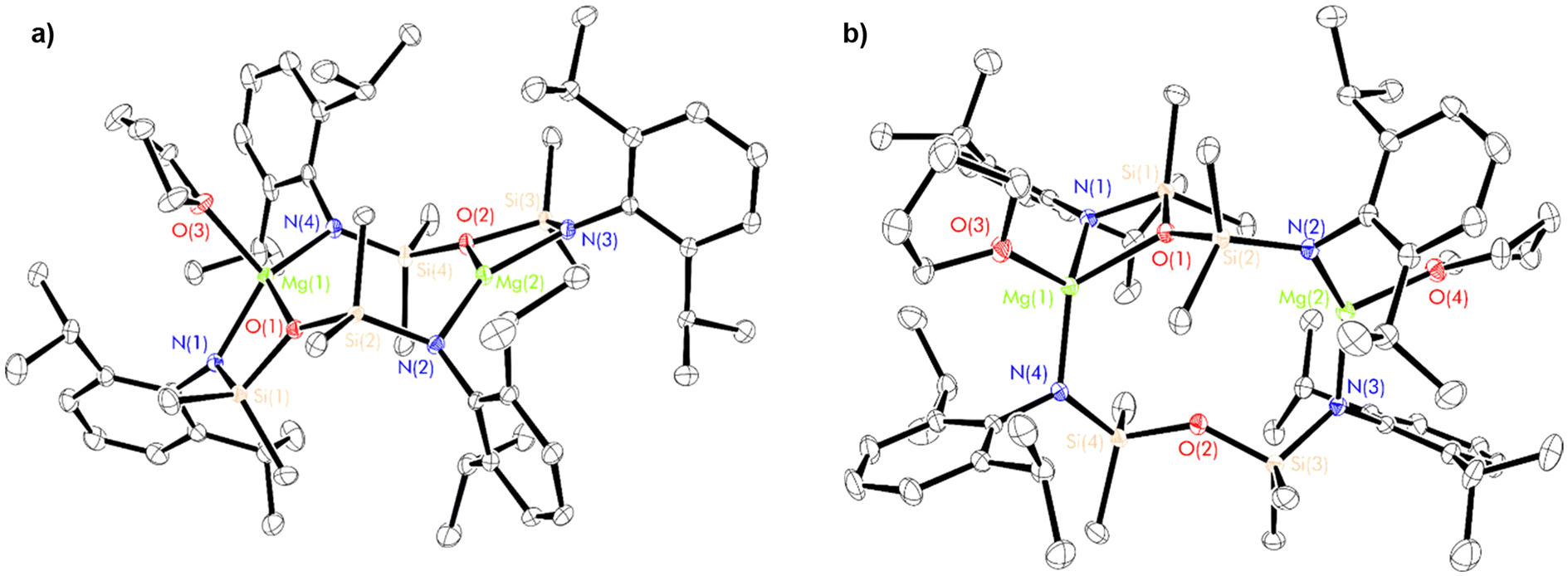 | ||
| Fig. 4 Thermal displacement ellipsoid drawing (30% probability) of (a) [{(NON-DippL)Mg}2(thf)] (1·thf) and (b) [(NON-DippL)Mg(thf)]2 (1·thf2). All hydrogen atoms have been omitted for clarity. Selected metrical parameters: Mg(1)–N(1) = 2.035(2) Å (1·thf) and 2.0626(9) Å (1·thf2), Mg(1)–N(4) = 2.019(2) Å (1·thf) and 2.999(1) Å (1·thf2), Mg(1)–O(1) = 2.128(2) Å (1·thf) and 2.133(1) Å (1·thf2), Mg(1)–O(3) Å = 2.051(2) Å (1·thf) and 2.046(1) Å (1·thf2), Mg(2)–N(3) Å = 1.950(2) Å (1·thf) and 1.992(1) Å (1·thf2), Mg(2)–N(2) = 1.969(2) Å (1·thf) and 2.003(1) Å (1·thf2), Mg(2)–O(2) = 2.040(2) Å (1·thf) and 3.549(1) Å (1·thf2), Mg(2)–O(4) = 2.018(1) Å (1·thf2), Mg(1)–Mg(2) = 4.6000(9) Å (1·thf) and 5.2386(7) Å (1·thf). | ||
The structure of 1·thf (Fig. 4a and Fig. S60†) is similar to that of 1, with the κ2-N,O-κ1-N′ coordination mode of the two [NON-DippL]2− ligands retained. The coordination sphere of the tetrahedral Mg(1) centre (Σ{∠Mg(1)} = 649.68(18)°, τ4 = 0.7270) is augmented by the coordination of a thf moiety (Mg(1)–O(3) = 2.051(2) Å), whilst Mg(2) retains a trigonal planar geometry (Σ{∠Mg(2)} = 356.88(13)°). The increased steric congestion that results from the coordination of thf to Mg(1) has resulted in the widening of the central eight-membered Mg2N2Si2O2 metallacycle. This is reflected by an increase of 0.487(1) Å in the Mg–Mg distance in 1·thf relative to 1 (Mg(1)–Mg(2) = 4.1130(9) (1) vs. 4.6000(9) Å (1·thf)). The Mg2N2Si2O2 metallacycle in 1·thf has a “chair-like” conformation with the two four-membered MgNSiO rings anti with respect to one another, in contrast with the “boat-like” structure of 1. The Mg–N and Mg–O bonds are shorter for Mg(2) than for Mg(1), reflecting the increased steric congestion that results from the coordination of thf to the latter.
Conversely, the structure of 1·thf2 (Fig. 4b and Fig. S60†) varies significantly relative to those of 1 and 1·thf. One molecule of thf is coordinated to each Mg2+ centre (Mg(1)–O(3) = 2.046(1) Å, Mg(2)–O(4) = 2.018(1) Å). While Mg(1) adopts a tetrahedral coordination geometry (Σ{∠Mg(1)} = 650.70(10)°, τ4 = 0.7670), coordination of thf to Mg(2) results in displacement of the silylether group (Mg(2)–O(2) = 3.549(1) Å ≫ Σrcov(Mg,O) = 2.02 Å71), resulting in the retention of a trigonal planar coordination geometry (Σ{∠Mg(2)} = 357.26(8)°). Consequently, 1·thf2 consists of a central 10-membered Mg2N3O2Si3 metallacycle. One [NON-DippL]2− ligand retains the κ2-N,O-κ1-N′ coordination mode observed exclusively in 1 and 1·thf, while the other instead bridges between the two Mg centres in an alternative κ1-N-κ1-N′ manner. This rearrangement likely results from the increased steric congestion that occurs upon coordination of a second equivalent of thf to 1·thf, which is relieved upon the formation of 1·thf2via ring expansion. This is reflected by a Mg(1)–Mg(2) distance in 1·thf2 (5.2386(7) Å) that is 0.639(1) Å longer than that of 1·thf. Hill and co-workers reported a similar, 14-membered metalacyclic magnesium dimer [({CH2SiMe2NDipp}2)2Mg2(thf)2] in which two [(CH2SiMe2NDipp)2]2− ligands bridge between two three-coordinate Mg2+ centres.30 Similar macrocyclic motifs have also been reported for transition metal complexes (Fe, Cr) bearing di(amido)siloxane ligands.52,72
Notably, no intermediate dimeric thf adducts analogous to 1·thf and 1·thf2 crystallised from thf solutions of 2–4. This is likely due to the similarities between the [(NON-DippL)Ae] units of the dimeric species 2–4 and their monomeric analogues 5–7, which both feature the coordination of the [NON-DippL]2− ligand in a κ3-N,O,N′-tridentate manner. Consequently, little structural rearrangement is required upon the coordination of thf, which readily outcompetes the Ae–N and π-facial interactions that facilitate the dimerisation of 2–4/4a when the strong donor solvent is absent. Conversely, the conversion of 1 into a monomeric adduct requires a more significant structural rearrangement due to the alternative κ2-N,O-κ1-N′ coordination mode of the ancillary ligand in the former. In addition, the smaller ionic radius of Mg2+ results in significantly reduced kinetic lability relative to the heavier Ae2+ congeners.3 As a result, a higher temperature of 80 °C is required to disrupt the dimeric structure of 1, which instead forms the stable dimeric intermediates 1·thf and 1·thf2 at 25 °C.
Heterometallic s-block “ate” complexes
We turned our attention to the synthesis of heterobimetallic s-block “ate” complexes supported by the [NON-DippL]2− ligand. Based on the dimeric structures of 1–4, we reasoned that the coordination of an additional monodentate [X]− ligand to Ae2+ within an anionic [(NON-DippL)Ae(X)]− unit could be accommodated, with charge balance provided by an alkali metal cation.Mg/K “ate” complex
The solubility of the magnesium compound 1 in toluene at 60 °C enabled its reaction with two equivalents of KN′′. This resulted in the formation of the potassium magnesiate complex [(NNO-DippL)Mg(μ-N′′)K]n (8), which crystallised directly from the reaction mixture upon cooling (Scheme 3a; Fig. 5).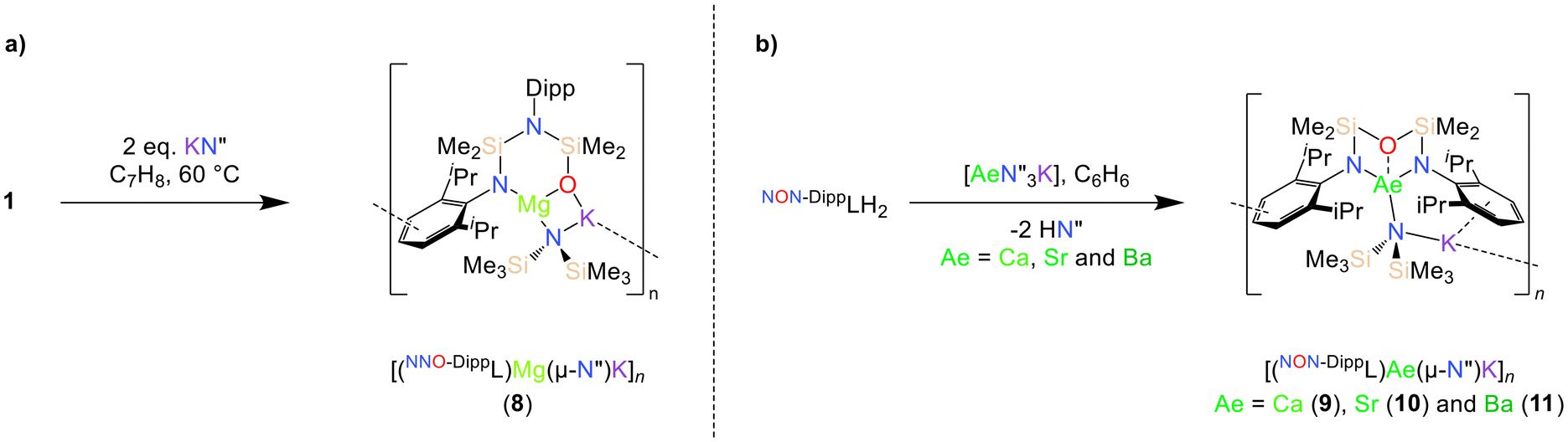 | ||
| Scheme 3 Synthesis of (a) [(NNO-DippL)Mg(μ-N′′)K]n (8) and (b) [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)). | ||
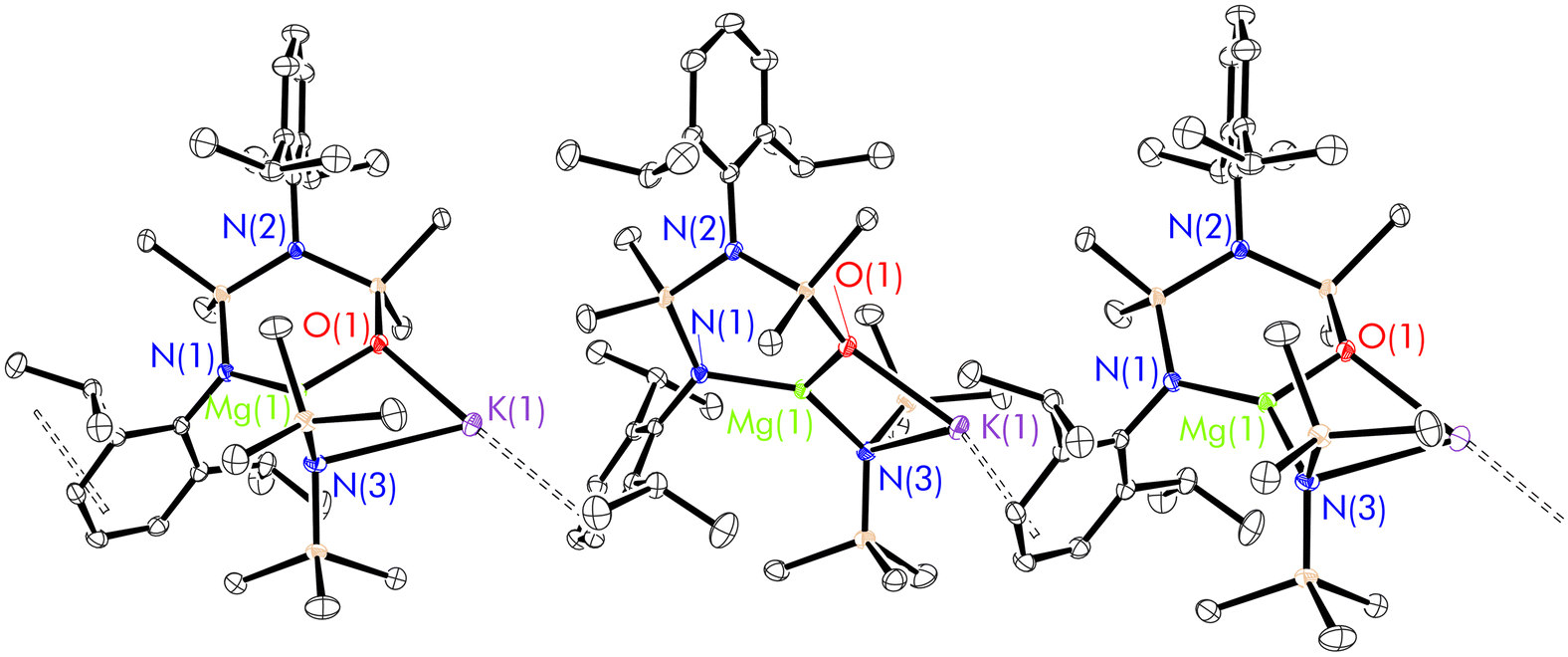 | ||
| Fig. 5 Thermal displacement ellipsoid drawing (30% probability) of the extended structure of [(NNO-DippL)Mg(μ-N′′)K]n (8). All hydrogen atoms have been omitted for clarity. Selected metrical parameters (* refers to symmetry related atoms): Mg(1)–N(1) = 2.002(1) Å, Mg(1)–O(1) = 1.899(1) Å, Mg(1)–N(3) = 2.021(1) Å, K(1)–N(3) = 3.205(1) Å, K(1)–O(1) = 2.588(1) Å, K(1)–centroidC(1)–C(6) = 3.033(1) Å, Mg(1)–K(1) = 3.4417(5) Å; N(1)–Mg(1)–O(1) = 107.64(5)°, N(1)–Mg(1)–N(3) = 138.09(6)°, O(1)–Mg(1) N(3) = 112.38(5), N(3)–K(1)–O(1) = 67.47(4)°, N(3)–K(1)–centroidC(1)–C(6) = 144.76(3)°, O(1)–K(1)–centroidC(1)–C(6) = 144.76(3)°, Mg(1)–O(1)–K(1) = 78.81(4)°, Mg(1)–N(3)–K(1) = 99.01(5)°. | ||
The crystal structure of 8 has an asymmetric unit consisting of the [NNO-DippL]2− ligand coordinated to Mg2+ in a κ2-N,O-bidentate manner, with both the siloxide donor and the [μ-N′′]− ligand bridging between Mg2+ and K+ to form a four membered metallacycle (Fig. 5). The extended structure consists of a one-dimensional coordination polymer that is propagated by η3-π-facial K+–aryl interactions between the potassium cation of one monomeric [(NNO-DippL)Mg(μ-N′′)K] unit and the aryl ring of the coordinated NDipp group of the next. The formation of this rearranged “NNO” form of the ancillary ligand is consistent with the same outcome we reported upon the reduction of 1 with KC8,43 and is attributed to a 1,3-silyl retro-Brook rearrangement that has also been reported in other cases.44,45 This rearrangement is likely favoured on both steric and electronic grounds, as the formation of the less sterically demanding siloxide donor both reduces congestion at the three coordinate Mg2+ centre and results in strong Mg–O and K–O bonds. Furthermore, the rearrangement is likely facilitated by both the temperature of 60 °C (which is required to dissolve 1 in toluene) and the coordination of an alkali metal cation. The influence of both factors was previously confirmed by a computational study of the formation of a related “NNO” aluminium complex reported by Coles and co-workers, which highlighted the ability of K+ to facilitate the dissociation of an amide donor from Al3+ and thus enable its subsequent attack on the siloxane backbone.451H and 13C{1H} NMR spectra of 8 (recorded in toluene-d8 at 80 °C due to the poor hydrocarbon solubility of 8 at ambient temperature) each contain two sets of Dipp signals and two SiMe2 signals, confirming the rearrangement of the ancillary ligand to the NNO-form. Notably, the absence of signals resulting from free KN′′ demonstrates the persistence of the Mg–N′′ bond (and therefore the [(NNO-DippL)Mg(μ-N′′)K] unit as a whole) in solution, in contrast with the dissociation of KN′′ from heavier alkaline earth “ate” complexes (vide infra) in toluene.
Heavier Ae/K “ate” complexes
The poor hydrocarbon solubility of 2–4 necessitated an alternative synthesis of the heavier “ate” congeners. The direct reaction NON-DippLH2 with the heterobimetallic precursors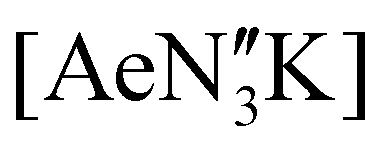 (Ae = Ca, Sr and Ba) was therefore targeted. To facilitate this protocol, the novel barium precursor
(Ae = Ca, Sr and Ba) was therefore targeted. To facilitate this protocol, the novel barium precursor 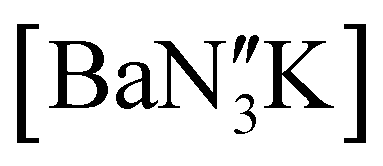 was first isolated in 60% yield via the 2:1 reaction of KN′′ with
was first isolated in 60% yield via the 2:1 reaction of KN′′ with 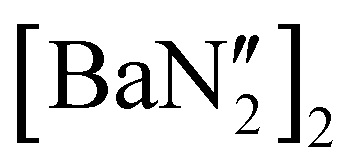 in toluene (see ESI†); the calcium and strontium congeners have been previously reported.22 Combining equimolar quantities of NON-DippLH2 with
in toluene (see ESI†); the calcium and strontium congeners have been previously reported.22 Combining equimolar quantities of NON-DippLH2 with 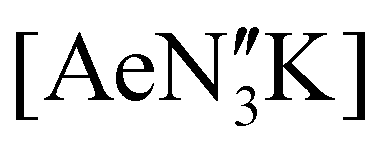 in arene solvents resulted in the crystallisation of the one-dimensional coordination polymers [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11), Scheme 3b) directly from the reaction mixture, thus enabling their isolation in moderate yield (48% (9), 55% (10) and 55% (11)) and characterisation by SC-XRD (Fig. 6 and Table 3).
in arene solvents resulted in the crystallisation of the one-dimensional coordination polymers [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11), Scheme 3b) directly from the reaction mixture, thus enabling their isolation in moderate yield (48% (9), 55% (10) and 55% (11)) and characterisation by SC-XRD (Fig. 6 and Table 3).
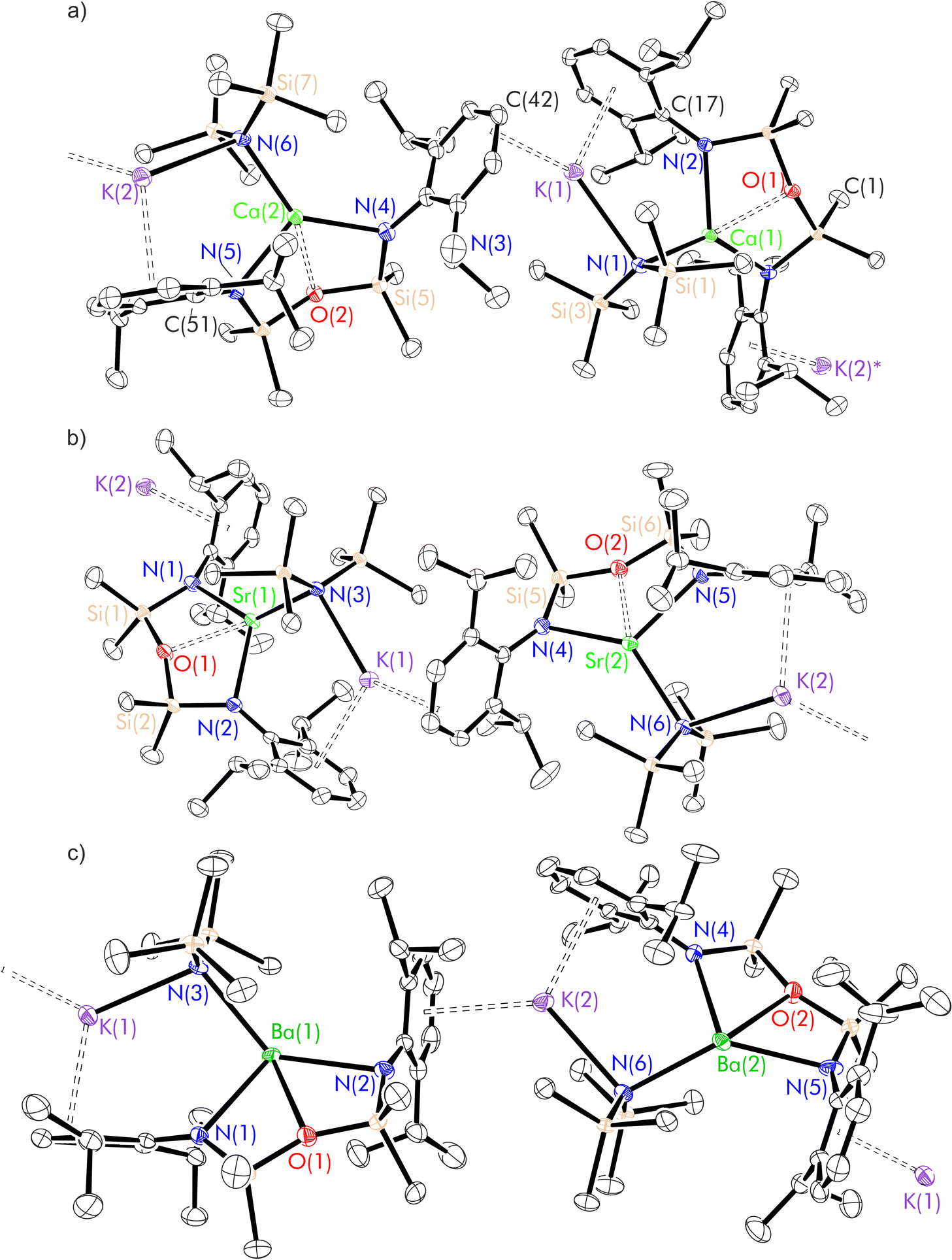 | ||
| Fig. 6 Thermal displacement ellipsoid drawing (30% probability) of the extended structures of (a) [(NON-DippL)Ca(μ-N′′)K]n (9), (b) [(NON-DippL)Sr(μ-N′′)K]n (10) and (c) [(NON-DippL)Ba(μ-N′′)K]n (11). All hydrogen atoms have been omitted for clarity. | ||
| 9 | 10 | 11 | |
|---|---|---|---|
| Ae(1)–N(1) | 2.342(2) | 2.524(3) | 2.698(8) |
| Ae(1)–N(2) | 2.365(2) | 2.533(3) | 2.741(8) |
| Ae(1)–N(3) | 2.370(2) | 2.525(3) | 2.707(7) |
| Ae(1)–O(1) | 2.4434(15) | 2.539(2) | 2.673(5) |
| K(1)–N(3) | 2.924(2) | 2.892(3) | 2.876(7) |
| K(1)–centroidC(17)–C(22) | 2.951(1) | 2.982(2) | 2.972(5) |
| K(1)–centroidC(40)–C(43) | 2.967(2) | 2.955(2) | 3.036(7) |
| Ae(1)–K(1) | 4.1338(8) | 4.2341(9) | 4.444(2) |
| N(1)–Ae(1)–N(2) | 121.23(6) | 119.77(10) | 114.2(2) |
Crystal structures of 9–11
The X-ray crystal structures of 9–11 (Fig. 6 and Table 3) are largely isostructural, and consist of [(NON-DippL)Ae(μ-N′′)K] units linked by intermolecular η3-π-facial interactions between the potassium cation of one monomeric unit and a Dipp group of the next, resulting in the formation of a one-dimensional coordination polymer. Notably, the larger ionic radii of the heavier Ae2+ congeners (relative to Mg2+) facilitate the coordination of three bulky amide donors, with the NON-form of the di(amido)siloxane ligand retained in 9–11 (in contrast to the NNO-form in the Mg/K “ate” complex 8). The asymmetric unit consists of either two (Ae = Ca and Sr) or four (Ae = Ba) crystallographically distinct [(NON-DippL)Ae(μ-N′′)K] units that exhibit similar metrical parameters. Therefore, representative metrical data from a single [(NON-DippL)Ae(μ-N′′)K] molecule are discussed in each case.The [(NON-DippL)Ae] units of 9–11 largely resemble those of 2–7, with [NON-DippL]2− coordinated to Ae2+ in a κ3-N,O,N′-tridentate manner, forming an AeN2Si2O metallacycle with an “open-book” conformation. In all cases, the Ae2+ centre has a four-coordinate distorted “see-saw” geometry (τ4 = 0.54 (9), 0.60 (10) and 0.64 (11)).70 The tetrahedral [μ-N′′]− amido donor bridges between Ae2+ and K+, and the latter is also stabilised through the formation of intramonomeric η6- and intermonomeric η3-π-facial interactions with Dipp aryl rings, resulting in a pseudo trigonal planar coordination geometry at K+ (when the relevant centroids are treated as monodentate ligands). Further coordinative saturation of K+ is provided by non-covalent anagostic interactions with the methyl protons of the [μ-N′′]− ligand. Notably, the Ae–N′′ bond lengthens with increasing Ae2+ atomic number in a manner that is consistent with the greater ionic radii of the heavier congeners (Ae(1)–N(3) = 2.3702(18) Å (9), 2.525(3) Å (10) and 2.707(7) Å (11)). Conversely, the K–N′′ bond shortens with increasing Ae2+ size (K(1)–N(3) = 2.924(2) Å (9), 2.892(3) Å (10) and 2.876(7) Å (11)). While the distances for 10 and 11 are the same within error, they are both significantly shorter than the corresponding distance in 9, suggesting that the K–N′′ bond is strengthened as the Ae–N′′ bond weakens. This supposition is consistent with the reversible dissociation of KN′′ observed in solution (vide infra).
Solution-phase behaviour of 9–11 in arenes
While 9–11 are poorly soluble in arenes at room temperature, they can each be dissolved in toluene at 70 °C. The polymeric solid-state structures of 9–11 are proposed to be disrupted at higher temperatures, resulting in the formation of more soluble monomeric [(NON-DippL)Ae(μ-N′′)K(tol)n] (Ae = Ca (9tol), Sr (10tol) and Ba (11tol)) molecules in which the K+ cation is assumed to be stabilised by π-facial interactions with the arene solvent (Scheme 4).34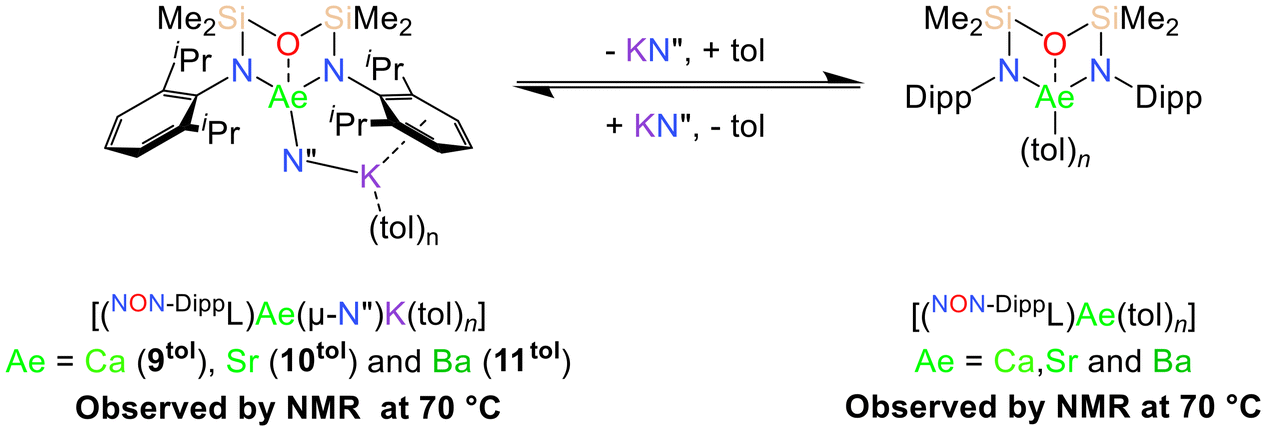 | ||
| Scheme 4 Solution-phase equilibrium between the proposed monomeric toluene adducts [(NON-DippL)Ae(μ-N′′)K(tol)n] and [(NON-DippL)Ae(tol)n] (Ae = Ca, Sr and Ba) that results from the reversible dissociation of KN′′ following the dissolution of 9–11 in toluene at 70 °C. | ||
The 1H NMR spectra of 9–11 under these conditions display the five signals expected for the [NON-DippL]2− ligand coordinated to Ae2+ in a C2v-symmetric manner, as well as an additional singlet corresponding to the [μ-N′′]− ligand. The C2v symmetry of these signals contrasts with the asymmetry of the solid-state structures of 9–11, suggesting that [(NON-DippL)Ae(μ-N′′)K(tol)n] have fluxional solution-phase structures that result from the rapid exchange of K+ between the two Dipp π-systems. While the signals assigned as [(NON-DippL)Ae(μ-N′′)K(tol)n] represent the major species in each case, NMR spectra of analytically pure crystalline samples of 9–11 reproducibly contained additional signals corresponding to a second, minor species in solution. A diagnostic singlet at δ = 0.06 ppm confirms the presence of free KN′′ in each case,73 whilst the additional signals are assigned to monomeric [(NON-DippL)Ae(tol)n] (Ae = Ca, Sr and Ba) complexes formed upon the solution-phase dissociation of KN′′ from the “ate” complex. Despite the partial dissociation of 9tol–11tol at 70 °C, they were each found to crystallise as 9–11 exclusively upon cooling to room temperature. No evidence was found for the crystallisation of the dimeric homometallic species 2–4 upon cooling, demonstrating that the proposed monomeric [(NON-DippL)Ae(tol)n] complexes formed in solution are metastable in solution at elevated temperature, and preferentially associate with KN′′ to form 9–11 upon cooling rather than dimerising to form 2–4.
Solution-phase behaviour of 8–11 in thf-d8
The “ate” complexes 8–11 are highly soluble in thf, enabling further investigation of their solution-phase behaviour by NMR spectroscopy in thf-d8 across a wide temperature range (−80 °C to 67 °C). The dissolution of the NNO-form Mg/K complex 8 in thf-d8 results in complex and temperature-dependant 1H NMR spectra, indicative of a mixture of species which could not be conclusively assigned (Scheme S1†). Conversely, 1H NMR spectra of the heavier NON-form Ae/K complexes 9–11 are simpler, and consist of signals assigned to the six proton environments that are expected for the [NON-DippL]2− and [N′′]− ligands coordinated within the C2v-symmetric and assumedly monomeric species [(NON-DippL)Ae(μ-N′′)K(thf)n] (Ae = Ca (9thf), Sr (10thf) and Ba (11thf)). In addition to the signals attributed to 9thf–11thf, these spectra also contain resonances corresponding to the monometallic thf adducts 5–7 (verified by comparison with the corresponding NMR spectra of 5–7) and free KN′′.74 This indicates the presence of solution-phase equilibria between the heterobimetallic “ate” complexes 9thf–11thf and their monometallic analogues 5–7 due to the reversible dissociation of KN′′ (Scheme 5), consistent with the same process observed in toluene-d8 (vide supra). The “ate” complex is the major species at 25 °C when Ae = Ca and Sr (9thf:5 = 2.6:1, 10thf:6 = 2.2:1); the corresponding resonances can be clearly discriminated in the 1H NMR spectra, indicating slow exchange of the respective species on the NMR timescale. However, the room temperature 1H NMR spectrum of 6/10thf contains signals that are somewhat broadened, whilst further broadening and coalescence was observed at higher temperatures (though full coalescence was not observed below 67 °C, the boiling point of thf). Furthermore, the 1H NMR spectrum of 7/11thf shows significant signal broadening and coalescence at 25 °C, consistent with higher kinetic lability of Ba2+ and rapid exchange on the NMR spectroscopic timescale. Higher temperature 1H NMR spectra of 7/11thf exhibit full coalescence and signal sharpening as 7 and 11thf enter a fast exchange regime, whilst full decoalescence is observed below 283 K (Fig. S43†). This metal-dependant behaviour qualitatively demonstrates that the rate of exchange between 5–7 and 9thf–11thf increases as expected with greater Ae2+ size and lability.
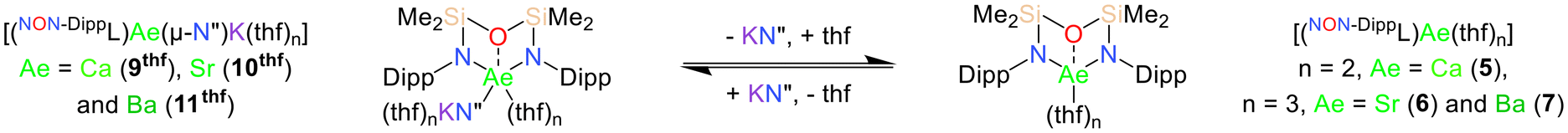 | ||
| Scheme 5 Solution phase equilibrium between [(NON-DippL)Ae(μ-N′′)K(thf)n] and [(NON-DippL)Ae(thf)n] (Ae = Ca (9thf/5), Sr (10thf/6) and Ba (11thf/7)) resulting from the reversible dissociation of KN′′. | ||
Thermodynamic analysis
The relative proportions of 5–7 and 9thf–11thf vary with temperature (Fig. 7a), with higher temperatures increasingly favouring the dissociation of KN′′ in all cases. Kd values could be calculated at 10 °C intervals using the relative integral values of 5–7, 9thf–11thf and KN′′ (normalised vs. the residual solvent signal) at each temperature, thus enabling a Van't Hoff analysis (Fig. 7b) and the calculation of ΔH, ΔS and ΔG298 K values for the forward reaction (i.e. KN′′ dissociation) in each case (Table 4). Both ΔH (ΔH = 31.58 kJ mol−1 (9thf), 12.72 kJ mol−1 (10thf) and 9.69 kJ mol−1 (11thf)) and ΔS (ΔS = 77.74 J K−1 mol−1 (9thf), 19.20 J K−1 mol−1 (10thf) and 12.60 J K−1 mol−1 (11thf)) are positive in all cases. These positive ΔH and ΔS values respectively reflect the energy required to break the ionic Ae–N′′ bond and the additional conformational degrees of freedom gained by the KN′′ moiety upon its dissociation. Notably, both values decrease in magnitude as group 2 is descended. This can be rationalised by considering the weaker Ae–N′′ bonds formed by the less charge dense heavier congeners, which therefore require less energy to break (resulting in a less positive ΔH value) and constrain the coordinated KN′′ group to a reduced degree within the “ate” complex (resulting in a less positive ΔS upon KN′′ dissociation). In all cases, ΔG298 K is also positive (ΔG298 K = 8.4 kJ mol−1 (9thf), 7.0 kJ mol−1 (10thf) and 5.9 kJ mol−1 (11thf)), consistent with the observed preference for the associated “ate” complex at 25 °C. However, ΔG298 K also becomes less positive with increasing Ae2+ size, with the extent of dissociation at 25 °C therefore increasing in the same manner. These positive ΔG298 K values contrast with a recent report from Aldridge and co-workers, which described a similar equilibrium resulting from the reversible dissociation of KN′′ from V-Ca in benzene.40 In this case, the dissociation of KN′′ was favoured at 25 °C (ΔG298 K = −2.6 kJ mol−1).40 This disparity is consistent with the greater flexibility of the di(amido)siloxane ligand, which readily facilitates the conformational adjustments required to accommodate the KN′′ moiety within a stable heterometallic framework. However, we note that direct comparisons with the thermodynamic parameters calculated in this report must be made cautiously due to the variation in solvent between the two systems (thf for 9thf–11thf, benzene for V-Ca). Together, these quantitative data provide detailed insight into the variation in the solution-state behaviour of Ae/K heterobimetallic amide “ate” complexes that results from the more labile metal–ligand bonds formed by the heavier Ae2+ congeners. We anticipate that these results will inform the elucidation of structure–activity relationships in catalysis mediated by 9–11 and heterometallic “ate” complexes more generally.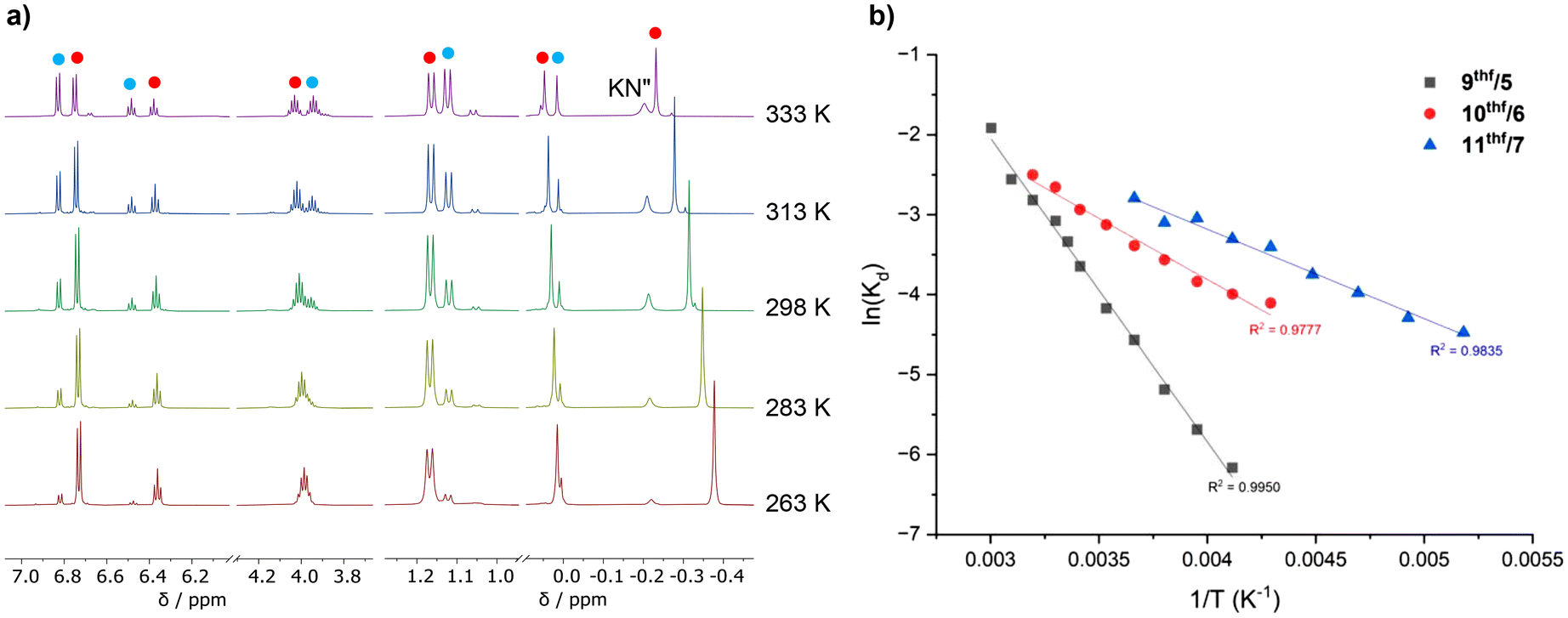 | ||
| Fig. 7 (a) Variable temperature 1H NMR spectra (499.99 MHz, thf-d8) showing the temperature-dependant mixture of 9thf (red dots), 5 (blue dots) and KN′′ formed upon the dissolution of 9 in thf-d8, and (b) Van‘t Hoff plots for the reversible dissociation of KN′′ from 9thf–11thf. Note: the relative intensities of the signals on the RHS of the spectra in (a) have been decreased relative to those on the LHS for clarity. | ||
| Equillibrium | Ae | ΔH/kJ mol−1 | ΔS/J K−1 mol−1 | ΔG298 K/kJ mol−1 |
|---|---|---|---|---|
| 9thf/5 | Ca | 31.58 | 77.74 | 8.4 |
| 10thf/6 | Sr | 12.72 | 19.20 | 7.0 |
| 11thf/7 | Ba | 9.69 | 12.60 | 5.9 |
Conclusions
The three dimeric alkaline earth complexes [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)) were successfully isolated. X-ray crystallographic analysis revealed that a κ3-N,O,N′-κ1-N′-tridentate coordination mode of the di(amido)siloxane ligand is accommodated by the large Ae2+ cations in each case, in contrast with the previously reported κ2-N,O-κ1-N′ coordination mode observed in the more sterically congested magnesium congener [(NON-DippL)Mg]2 (1) that we have reported previously. While complexes 2–4 each crystallise from benzene as N-bridged dimers, the barium congener 4a was found to exhibit an alternative aryl-bridged motif when crystallised from n-hexane.Whilst 2–4 are insoluble in non-donor solvents, they are readily converted into the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)) in thf. Conversely, the coordination of thf to the magnesium congener 1 at 25 °C resulted in the formation of the dimeric thf adducts [{(NON-DippL)Mg}2(thf)] (1·thf) and [(NON-DippL)Mg(thf)]2 (1·thf2). The requirement for heating at 80 °C to full convert 1 into the previously reported monomeric adduct [(NON-DippL)Mg(thf)2] contrasted with the rapid conversion of 2–4 into 5–7 at 25 °C, an outcome that was rationalised based on both the reduced kinetic lability of Mg2+ relative to the heavier congeners and the κ2-N,O-κ1-N′ coordination mode of the di(amido)siloxane ligand in 1.
The heterometallic Ae/K amide “ate” complexes [(NNO-DippL)Mg(μ-N′′)K]n (8) and [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)) were isolated, and exist as one-dimensional coordination polymers propagated by K+–aryl π-facial interactions in the solid-state. The mixed amide/siloxide “NNO” ligand in 8 results from a 1,3-silyl retro-Brook rearrangement of the original di(amido)siloxane ligand, and the observed preference for the “NNO” form can be rationalised on steric (less bulky siloxide donor) and electronic (strong Mg–O bond) grounds. In contrast, the larger Ae2+ congeners readily accommodate the coordination of KN′′ with the di(amido)siloxane ligand retaining a κ3-N,O,N′-tridentate motif in 9–11.
The solution-phase structures of 8–11 in both toluene-d8 (at 70 °C) and thf-d8 were subsequently probed using NMR spectroscopy. While the associated structure of 8 is stable in toluene, the reversible dissociation of KN′′ from 9–11 was found to occur in solution, resulting in the presumed formation of the metastable monomeric arene adducts [(NON-DippL)Ae(tol)n] (Ae = Ca, Sr and Ba). However, subsequent cooling resulted in the exclusive crystallisation of 9–11, demonstrating that the association of [(NON-DippL)Ae(tol)n] with KN′′ outcompetes any dimerisation of the former to form 2–4. While the behaviour of 8 in thf is complex, the dissolution of 9–11 in this donor solvent results well-defined equilibria between the “ate” complexes [(NON-DippL)Ae(μ-N′′)K(thf)n] (Ae = Ca (9thf), Sr (10thf) and Ba (11thf)) and the monometallic adducts 5–7 through reversible dissociation of KN′′, the thermodynamic parameters of which were elucidated via Van ‘t Hoff analyses of variable temperature NMR spectra. Positive ΔH and ΔS values reflect both the energy required to break the ionic Ae–N′′ bond present in the “ate” complex and the conformational degrees of freedom gained upon the dissociation of KN′′. Consequently, both ΔH and ΔS were found to decrease in magnitude with increasing Ae2+ size due to the resultant weakening of the Ae–N′′ bond. However, in all cases positive ΔG298 K > 0, with the aggregated “ate” complex therefore the major species at 25 °C.
In summary, these results provide detailed insight into the divergent solid-state structures and solution-phase dynamics of neutral Ae- and heterometallic s-block “ate” complexes supported by a chelating di(amido)siloxane ligand, enabling comparison with those of closely related di(amido) ligands based on either flexible aliphatic or rigid xanthene-based backbones. The importance of ligand design when attempting the challenging reduction of the heavier alkaline-earths cannot be underestimated.75–77 Our ongoing studies target the synthesis of reduced heterometallic complexes containing the heavier alkaline earths supported by the [NON-DippL]2− ligand (cf.III and IV). We also anticipate that the heteroleptic “ate” complexes described herein will serve as useful reagents for the further diversification of main group reactivity and catalysis mediated by heterometallic s-block complexes. We continue to explore these possibilities in our laboratories.
Author contributions
Matthew D. Haynes: Conceptualisation, investigation, formal analysis, methodology, validation, visualisation, data curation, writing – original draft, writing – review and editing. Andrea O'Reilly: Investigation, formal analysis, methodology, validation, visualisation, writing – review and editing. Alice J. M. Poole: Investigation, formal analysis, methodology. Aisling F. Roper: Formal analysis. Stefan Thum: Resources, formal analysis. Louis J. Morris: Conceptualisation, formal analysis, methodology, supervision, writing – review and editing. Martyn P. Coles: Funding acquisition, resources, conceptualisation, supervision, writing – review and editing, project administration, validation, visualisation. J. Robin Fulton: Funding acquisition, resources, conceptualisation, supervision, writing – review and editing, project administration. Sjoerd Harder: Funding acquisition, resources, conceptualisation, supervision, writing – review and editing, validation. Zoë R. Turner: Supervision, writing – review and editing, data curation, visualisation, validation. Dermot O'Hare: Funding acquisition, resources, supervision, writing – review and editing.Data availability
The data supporting this article is included as part of the ESI: General experimental details, synthetic details and characterising data for all new complexes, representative NMR spectra and crystallographic data.Conflicts of interest
The are no conflicts to declare.Acknowledgements
AO'R, MPC and JRF acknowledge funding from the Royal Society Te Apārangi (MFP-VUW2020) and from the MacDiarmid Institute for Advanced Materials and Nanotechnology. ZRT, LJM and DOH thank SCG Chemicals plc. (Thailand). MDH and AFR acknowledge funding from the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM), EP/S023828/1. SH thanks the Deutsche Forschungsgemeinschaft for support (HA 3218/11-1). The authors thank Chemical Crystallography (Oxford) for use of the diffractometers, and helpful discussions; the Rigaku XtaLAB Synergy DW diffractometer was funded by EPSRC Strategic Equipment Grant (EP/V028995/1). This research utilised a Bruker NEO 600 NMR spectrometer funded by the John Fell Oxford University Press Research Fund and an EPSRC Strategic Equipment Grant (EP/T019190/1).References
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- Y. Sarazin and J. F. Carpentier, Chem. Rec., 2016, 16, 2482–2505 CrossRef CAS PubMed.
- S. Harder and J. Langer, Nat. Rev. Chem., 2023, 7, 843–853 CrossRef PubMed.
- J. Spielmann and S. Harder, Chem. – Eur. J., 2007, 13, 8928–8938 CrossRef CAS PubMed.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–720 CrossRef PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- I. L. Fedushkin, V. A. Chudakova, A. A. Skatova, N. M. Khvoinova, Y. A. Kurskii, T. A. Glukhova, G. K. Fukin, S. Dechert, M. Hummert and H. Schumann, ZAAC, 2004, 630, 501–507 CrossRef CAS.
- I. L. Fedushkin, A. G. Morozov, V. A. Chudakova, G. K. Fukin and V. K. Cherkasov, Eur. J. Inorg. Chem., 2009, 33, 4995–5003 CrossRef.
- J. Gao, Y. Liu, Y. Zhao, X. J. Yang and Y. Sui, Organometallics, 2011, 30, 6071–6077 CrossRef CAS.
- B. Gao, D. Zhao, X. Li, Y. Cui, R. Duan and X. Pang, RSC Adv., 2014, 5, 440–447 RSC.
- M. Ma, H. Wang, J. Wang, L. Shen, Y. Zhao, W. H. Xu, B. Wu and X. J. Yang, Dalton Trans., 2019, 48, 2295–2299 RSC.
- T. K. Panda, H. Kaneko, O. Michel, K. Pal, H. Tsurugi, K. W. Törnroos, R. Anwander and K. Mashima, Organometallics, 2012, 31, 3178–3184 CrossRef CAS.
- B. Freitag, C. A. Fischer, J. Penafiel, G. Ballmann, H. Elsen, C. Färber, D. F. Piesik and S. Harder, Dalton Trans., 2017, 46, 11192–11200 RSC.
- R. L. Jones, Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2024, 43, 414–426 CrossRef CAS.
- R. K. Kottalanka, H. Adimulam, J. Bhattacharjee, H. Vignesh Babu and T. K. Panda, Dalton Trans., 2014, 43, 8757–8766 RSC.
- J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chem. – Eur. J., 2017, 23, 9319–9331 CrossRef CAS PubMed.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 CrossRef CAS.
- L. Davin, A. Hernán-Gómez, C. McLaughlin, A. R. Kennedy, R. McLellan and E. Hevia, Dalton Trans., 2019, 48, 8122–8130 RSC.
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS PubMed.
- A. J. Martínez-Martínez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O'Hara, Chem. Sci., 2013, 5, 771–781 RSC.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- A. Hernán-Gómez, T. D. Bradley, A. R. Kennedy, Z. Livingstone, S. D. Robertson and E. Hevia, Chem. Commun., 2013, 49, 8659–8661 RSC.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed.
- H.-Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. Mcmullin, E. Richards, M. S. Hill and C. L. M. C. Mullin, Angew. Chem., Int. Ed., 2022, 62, e202213670 CrossRef PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and B. L. Morrison, Chem. Commun., 2023, 59, 3846–3849 RSC.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Organometallics, 2024, 43, 879–888 CrossRef CAS PubMed.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Chem. Commun., 2023, 59, 1453–1456 RSC.
- J. S. McMullen, A. J. Edwards and J. Hicks, Dalton Trans., 2021, 50, 8685–8689 RSC.
- J. S. McMullen, R. Huo, P. Vasko, A. J. Edwards and J. Hicks, Angew. Chem., Int. Ed., 2022, 62, e2022152 Search PubMed.
- R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, 62, e202308347 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2023, 60, 1016–1019 RSC.
- R. Huo, A. J. Armstrong, G. R. Nelmes, D. J. Lawes, A. J. Edwards, C. L. McMullin and J. Hicks, Chem. – Eur. J., 2024, 30, e202400662 CrossRef CAS PubMed.
- J. J. C. Struijs, M. A. Ellwanger, A. E. Crumpton, V. Gouverneur and S. Aldridge, Nat. Chem., 2024, 16, 1473–1480 CrossRef CAS PubMed.
- C. Jones, D. T. Nguyen and C. Helling, Chem. – Asian J., 2024, 19, e202400498 CrossRef PubMed.
- C. Jones, D. T. Nguyen, C. Helling and M. J. Evans, Inorg. Chem., 2024, 63, 5718–5726 CrossRef PubMed.
- A. O'Reilly, M. D. Haynes, Z. R. Turner, C. L. McMullin, S. Harder, D. O'Hare, J. R. Fulton and M. P. Coles, Chem. Commun., 2024, 60, 7204–7207 RSC.
- F. Haftbaradaran, G. Mund, R. J. Batchelor, J. F. Britten and D. B. Leznoff, Dalton Trans., 2005, 2343–2345 RSC.
- A. O'Reilly, M. G. Gardiner, C. L. McMullin, J. R. Fulton and M. P. Coles, Chem. Commun., 2024, 60, 881–884 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC.
- Y. Sarazin and J. F. Carpentier, CHAPTER 4: Secondary (Agostic Si-H/Electrostatic C-F) interactions in alkaline earth-based catalysts, Royal Society of Chemistry, 2019 Search PubMed.
- D. B. Leznoff, G. Mund, K. C. Jantunen, P. H. Bhatia, A. J. Gabert and R. J. Batchelor, J. Nucl. Sci. Technol., 2002, 39, 406–409 CrossRef.
- A. J. Elias, H. W. Roesky, W. T. Robinson and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1993, 495–500 RSC.
- R. J. Schwamm, C. A. von Randow, A. Mouchfiq, M. J. Evans, M. P. Coles and J. R. Fulton, Eur. J. Inorg. Chem., 2021, 2021, 3466–3473 CrossRef CAS.
- A. K. Das, Z. Moatazedi, G. Mund, A. J. Bennet, R. J. Batchelor and D. B. Leznoff, Inorg. Chem., 2007, 46, 366–368 CrossRef CAS PubMed.
- P. Stegner, C. Färber, J. Oetzel, U. Siemeling, M. Wiesinger, J. Langer, S. Pan, N. Holzmann, G. Frenking, U. Albold, B. Sarkar and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 14615–14620 CrossRef CAS PubMed.
- M. J. C. Dawkins, A. N. Simonov and C. Jones, Dalton Trans., 2020, 49, 6627–6634 RSC.
- L. Lameyer, O. A. Salah, S. Deuerlein, T. Stey and D. Stalke, ZAAC, 2004, 630, 1801–1806 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- C. Wei, Y. Gao, F. Zu and C. Cui, Appl. Organomet. Chem., 2023, 37, e7048 CrossRef CAS.
- C. E. Hayes, R. H. Platel, L. L. Schafer and D. B. Leznoff, Organometallics, 2012, 31, 6732–6740 CrossRef CAS.
- M. D. Anker, M. J. Evans, S. A. Cameron and G. Laufersky, Polyhedron, 2024, 247, 116741 CrossRef CAS.
- F. Dankert and C. von Hänisch, Eur. J. Inorg. Chem., 2021, 2021, 2907–2927 CrossRef CAS.
- Prolonged drying of 2–4 in vacuo (10−2 mbar) at 25 °C did not remove co-crystallised benzene. Heating samples of 2–4 at higher temperatures in vacuo resulted in decomposition to the proligand.
- J. Mai, B. Rösch, J. Langer, S. Grams, M. Morasch, S. Harder, J. Mai, B. Rösch, J. Langer, S. Grams, M. Morasch and S. Harder, Eur. J. Inorg. Chem., 2023, 26, e202300421 CrossRef CAS.
- J. Mai, M. Morasch, D. Jędrzkiewicz, J. Langer, B. Rösch and S. Harder, Angew. Chem., Int. Ed., 2022, 62, e202212463 CrossRef PubMed.
- M. J. Evans, J. Mullins, R. Mondal and C. Jones, Chem. – Eur. J., 2024, 30, e202401005 CrossRef CAS PubMed.
- P. M. Chapple and Y. Sarazin, Eur. J. Inorg. Chem., 2020, 2020, 3321–3346 CrossRef CAS.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Orzechowski and S. Harder, Organometallics, 2007, 26, 2144–2148 CrossRef CAS.
- L. Orzechowski and S. Harder, Organometallics, 2007, 26, 5501–5506 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- G. Mund, A. J. Gabert, R. J. Batchelor, J. F. Britten and D. B. Leznoff, Chem. Commun., 2002, 24, 2990–2991 RSC.
- We note that KN′′ generally has exists as a dimer when dissolved in toluene. However, for clarity we refer to this species simply as KN′′ herein, as its nuclearity cannot be conclusively determined in this system.
- KN′′ is assumed to exist as a solvated monomeric species in thf.
- S. Harder and B. Rösch, Chem. Commun., 2021, 57, 9354–9365 RSC.
- L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef CAS.
- J. T. Boronski, Dalton Trans., 2023, 53, 33–39 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2411240–2411254. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00044k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |