
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic evidence of the interaction of titanium(IV) coordination complexes with a phosphate head group in phospholipids†
Matthieu
Scarpi-Luttenauer
a,
Zahia
Boubegtiten-Fezoua
 *a,
Petra
Hellwig
*a,
Alain
Chaumont
a,
Bruno
Vincent
b,
Laurent
Barloy
*a and
Pierre
Mobian
*a
*a,
Petra
Hellwig
*a,
Alain
Chaumont
a,
Bruno
Vincent
b,
Laurent
Barloy
*a and
Pierre
Mobian
*a
aUniversité de Strasbourg, CNRS, UMR 7140, F-67000 Strasbourg, France. E-mail: mobian@unistra.fr
bUniversité de Strasbourg, CNRS, UAR 2042, F-67000 Strasbourg, France
First published on 30th December 2024
Abstract
Biological membranes are potentially involved in the transport of metal ions, such as Ti(IV), and, sometimes, their associated ligands. Understanding the interactions of Ti(IV) ions and complexes with biological membranes provides a basis for elucidating the action mechanism of titanium anticancer drugs. Herein, we investigated the interactions of two neutral titanium(IV) complexes, viz. [H2Ti(Cat)3] (1) and [H2Ti(Napht)3] (2), incorporated into DOPC multi-bilayers using ATR-FTIR spectroscopy. Infrared results showed that complexes 1 and 2, when interacting with DOPC multi-bilayers, highly affected the hydration of the lipid phosphate group and its mobility, revealing that the phosphate group is the main group involved in the interactions of complexes 1 and 2 with DOPC phospholipids. NMR studies involving complex 1 and DOPC dissolved in deuterated DMSO solution were performed, and interactions between the Ti complex and DOPC phosphate group could be evidenced. DFT calculations of model complexes were in good agreement with experimental data, and the stability of three model complexes was estimated. On the basis of the obtained data, it can be suggested that the oxygen atom(s) of the phosphato group of the DOPC ligand acted as donor atoms for Ti.
Introduction
Titanium is usually characterized by its low toxicity towards living matter, making it a valid target for applications in bioinorganic chemistry, especially in the domain of new anticancer therapies.1–7 These novel treatments aim to diminish the side effects of the already marketed metal-based chemotherapies. About 40 years ago, interest in titanocene dichloride and budotitane in this field was demonstrated.8,9 Although these compounds have not yet succeeded in entering the antitumor market owing to their lack of stability in the human body, numerous analogues have been synthesized since then. For compounds based on Ti(IV), the issue of their uptake into the intracellular medium and thus their transport across cell membranes has been raised.10–13 It was considered that the metal is transported when associated with proteins adapted for the uptake of FeIII-like transferrin or serum albumin.14–20 The interaction of metal complexes with the biological membrane presents considerable challenges and provides a basis for elucidating the mechanism of penetration of titanium anticancer drugs into cells.Cell membranes perform a variety of functions in cell biology and typically consist of a lipid bilayer with a zwitterionic phosphocholine (PC) lipid as the major component. In previous studies on the interaction of lipids with titanium, TiO2 solid surfaces were used.21–24 A stable lipid bilayer supported on titanium oxide represents an attractive platform for several applications, including biosensor development and device fabrication. An interesting example of the fusion and adsorption of the inverse phosphocholine liposome DOCP on the TiO2 surface was demonstrated,24 where phosphate groups directly bound to the TiO2 surface, forming strong chemical interactions. In addition, DNA adsorption on TiO2 nanoparticles was described to occur mainly via the phosphate backbone.25
To the best of our knowledge, very few studies on the interaction of titanium(IV) complexes with lipid membranes have been reported thus far. One specific investigation highlighted the ability of a Ti(IV)–metallocene species to enhance the permeability of a liposomal membrane. However, this study did not specify the mode of binding between the organometallic Ti(IV)-species compound and phospholipids.26 Additionally, it should be noted that several studies devoted to the description of the interaction of Ti(IV) complexes with phosphoesters on nucleotides have been reported.27,28 Besides, we have recently been interested in the involvement of Ti complexes in biology; in this regard, we recently synthesized monomeric Ti(IV) compounds based on a TiO2N4 core containing a luminescent diimine ligand for biological applications.29 We address the issue of the interaction of Ti complexes with a lipid model membrane made of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) multi-bilayers using polarized attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy. To determine which molecular group of the lipid represents the driving force for the interaction with the Ti(IV) compounds, further investigations using solution NMR spectroscopy, mass spectrometry and DFT calculations are implemented.
Results and discussion
Two titanium(IV) complexes were used, namely the catecholato and naphtalenediolato Ti complexes [H2Ti(Cat)3] (complex 1)30 and [H2Ti(Napht)3] (complex 2),31 which were synthesized by the reaction of Ti(OiPr)4 with catechol (H2Cat) and 2,3-dihydroxynaphthalene (H2Napht), respectively (Chart 1). These two compounds are selected for the present investigation because catecholato-based ligands are known to form Ti(IV) species, displaying good stability in aqueous media.32–34 Additionally, catechol (CatH2) associated with Ti(IV) is labile enough to be displaced from the coordination sphere of the metal ion,35 permitting the reaction of the Ti(IV) ion with coordinating biological molecules. Therefore, complexes 1 and 2 are relevant candidates for mimicking the general behaviour of Ti(IV) complexes in biological matter.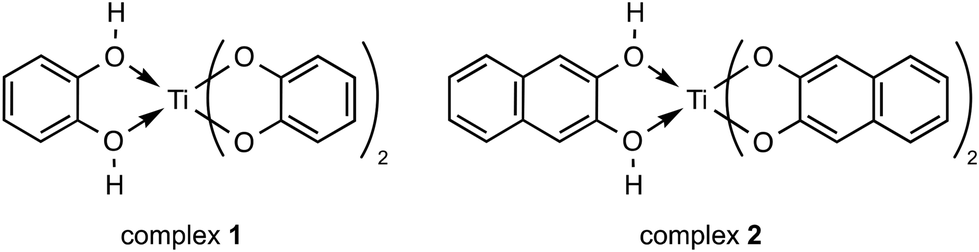 | ||
| Chart 1 Structure of complex 1 and 2. | ||
Polarized ATR-FTIR study of titanium(IV) complexes in interaction with DOPC multi-bilayers
ATR-FTIR spectra were recorded for the samples of complexes 1 and 2, both as dried films from a solution in methanol. The data obtained for the dry films are shown in the ESI (see Fig. S1a and b†). Two strong bands at 1251 and 1477 cm−1 were observed in the IR spectrum of complex 1, which can be tentatively assigned to the C–O stretching vibration (1251) of the coordinated catecholate and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the catecholate ring (1477) (see Fig. S1a†). For complex 2, bands at 1255 and 1458 cm−1 were detected and could be tentatively attributed to the νC–O and νCC modes, respectively (see Fig. S1b†). Similar bands for the νC–O and νCC modes have been previously reported36,37 for analogous titanium(IV) and copper(II) complexes containing a catecholato ligand.
C stretching vibration of the catecholate ring (1477) (see Fig. S1a†). For complex 2, bands at 1255 and 1458 cm−1 were detected and could be tentatively attributed to the νC–O and νCC modes, respectively (see Fig. S1b†). Similar bands for the νC–O and νCC modes have been previously reported36,37 for analogous titanium(IV) and copper(II) complexes containing a catecholato ligand.
We further investigated the interactions of complexes 1 and 2 with a model phospholipid membrane. We used a model membrane system made of single zwitterionic lipids, DOPC, as it constitutes the main lipid in mammal membranes.38 We performed polarized ATR-FTIR experiments to analyse the structure of the Ti complexes in DOPC multi-bilayers and the changes induced by the complexes upon interaction with the lipids. The critical complex/lipid molecular ratio allowing the visualization of changes in the lipid head groups and acyl chain vibrations upon interaction with Ti complexes was found to be around 1/20. Initially, we recorded the polarized ATR spectra of DOPC multi-bilayers hydrated with H2O (Fig. S2†). In the spectral range between 1850 and 950 cm−1 (Fig. S2a†), four main bands were observed at 1728, 1465, 1226 and 1086 cm−1 and assigned to the ν(CO), δ(CH2) and νas,s(PO2−) vibrational modes of the lipid, respectively.39 Within the 3000–2800 cm−1 spectral region (Fig. S2b†), two bands of interest originating from the vibrations of the lipid hydrocarbon chains that form the hydrophobic core of the lipid multi-bilayers were found. These bands near 2923 and 2854 cm−1 were assigned to the methylene antisymmetric stretching (νas(CH2)) and methylene symmetric stretching (νs(CH2)) modes of the lipid, respectively. The frequencies of these bands are known to be sensitive to changes in the conformation of the acyl chains, in chain mobility and packing.40 The observed values of both νas,s(CH2) bands of DOPC multi-bilayers are characteristic of a conformational disorder of hydrocarbon chains, indicating that these multi-bilayers were in a phase of fluid lipids at room temperature (22 °C). The DOPC multi-bilayers comprise unsaturated dioleyl chains (C18:1) with a negative phase transition (−15 °C to −20 °C). The lipid bilayers are oriented at the surface of the ATR crystal by shearing (see Materials and methods), and the dichroic ratio (RATR) of the νs(CH2) band reflects the acyl chain orientation. The RATR value of the νs(CH2) band (1.47 on average) of the DOPC multi-bilayers (Fig. S2b†) indicates the presence of an oriented system.34 Upon the interaction of Ti(IV) complexes with the bilayers at a 1/20 complex/lipid ratio (Fig. S3b and S4b†), there is a downshift of the νs(CH2) by 2 cm−1 in the case of complex 1 (Fig. S3b†), indicating an increase in the acyl chain order. This is confirmed by the evolution of the dichroic ratio (RATR) of the νs(CH2) band of the lipid chains, which is measured on the polarized ATR spectra (Fig. S3b and S4b†).41,42 Indeed, because the transition moment for the νs(CH2) mode lies perpendicular to the chain axis, the introduction of order or disorder alters the direction of the main chain with respect to the normal of the ATR crystal.42 The observed RATR of the νs(CH2) band decreases in the complex 1/DOPC system (1.16 in average) (Fig. S3b†) compared to the lipid alone (1.47 in average) (Fig. S2b†), while the RATR of the 2854 cm−1 feature does not change considerably between the lipid alone and complex 2 in DOPC (Fig. S4b†) (from RATR = 1.47 to RATR = 1.33). This corresponds to a reorientation of the lipid chains from approximately 45° compared to the normal to the interface (the average C–C–C angle of the lipid carbon chains) for the lipid alone to 38° and 42° in the presence of complexes 1 and 2, respectively.42
The ester carbonyl stretching (ν(CO)) band is one of the most intense signals from the lipid polar head group. Its frequency is sensitive to hydrogen bonding and thus provides information about the state of hydration of the lipid.43,44 A small increase in the frequency of the ν(CO) band from 1728 cm−1 in hydrated DOPC multi-bilayers (Fig. S2a†) to 1730 cm−1 in the complex 1/DOPC mixture (Fig. S3a†) spectra was obtained. This change indicates a small decrease in the hydration of the interfacial carbonyl groups of the lipid molecules; these frequencies were assigned previously to hydrogen-bound carbonyl groups.43,44 For the νs(CH2) band, the dichroic ratio of ν(CO) ester band of the lipids decreases from 1.70 for the DOPC alone to around 1.27 and 1.13 in the presence of complexes 1 and 2, respectively (see Fig. S2a, S3a and S4a†). This indicates that the orientation of the CO ester carbonyl head groups was perturbed by the Ti complexes. These slight changes in the hydration and orientation of the lipid carbonyl groups may be explained by their close proximity to the Ti(IV) complexes.
Finally, we report the ATR-FTIR spectra in the range 1300–950 cm−1 where symmetric and antisymmetric PO2− stretching vibrations originating from the phosphate head group of the lipid are typically observed (Fig. 1). The νas(PO2−) and νs(PO2−) vibrations of the hydrated DOPC multi-bilayers yield bands at 1226 and 1086 cm−1, respectively (Fig. 1, black curve).45 Interaction of Ti complexes with the lipid bilayers induces broadening and splitting of the νas(PO2−) band into two bands located at 1226, 1246 and 1226, 1258 cm−1 in complex 1/DOPC and complex 2/DOPC mixtures, respectively (Fig. 1, blue and red curves). Because the νas(PO2−) vibration is sensitive to hydrogen bond formation,46,47 its frequency indicates some degree of hydration of the phosphate group in the lipid layers. In mixtures of DOPC and Ti complexes, there are two DOPC species: one with a hydrated phosphate group (band at 1226 cm−1) and one with a dehydrated phosphate group (band at 1246 cm−1 in the presence of complex 1 and at 1258 cm−1 in the presence of complex 2). These new band features at 1246 cm−1 and 1258 cm−1 in the presence of complexes 1 and 2, respectively, may be associated with the formation of a DOPC–Ti complex involving the phosphate head group of DOPC. In addition to its interaction with the phosphate group, Ti(IV) causes the dehydration of this portion of the lipid head group. In agreement with this hypothesis, similar shifts towards higher wavenumbers for the νas(PO2−) band feature were previously observed for phosphatidylcholine45,48 and phosphatidylserine.49–51 Casal et al.51 established a correlation between the state of hydration of the phosphate group and its mobility in the presence of Ca2+ by infrared and 31P NMR. Furthermore, the marked changes in the contours and the position of the νas(PO2−) absorption band may indicate conformational changes that alter the orientation of the phosphate head group of the lipid in the mixed Ti complex/DOPC bilayers. The dichroic ratio of the νas(PO2−) band at 1226 cm−1 of the lipid changes slightly between DOPC and Ti complexes/DOPC bilayers (from RATR = 1.4 to RATR = 1.17 (or 1.28)) (see Fig. S2a, S3a and S4a†). This small decrease in RATR of νas(PO2−) band in the mixed Ti complexes/DOPC bilayers reveals that the average orientation of the phosphate group is slightly altered, which probably results from the favoured access of Ti(IV) to the phosphate group in the phosphatidylcholine head group of DOPC. In addition to the phosphate stretching bands, the spectra of the Ti complex/DOPC mixtures contained bands at 1174 cm−1 for complex 1 and 1172 cm−1 for complex 2 owing to the antisymmetric CO–O–C stretching vibration. Bands at 1066 and 971 cm−1 due to the symmetric CO–O–C stretching and antisymmetric (CH3)3N+ stretching vibrations were also observed for both mixtures (Fig. 1, blue and red curves). The shifts of these vibrational modes appeared small for both mixture samples compared with those of DOPC alone (Fig. 1, black curve).
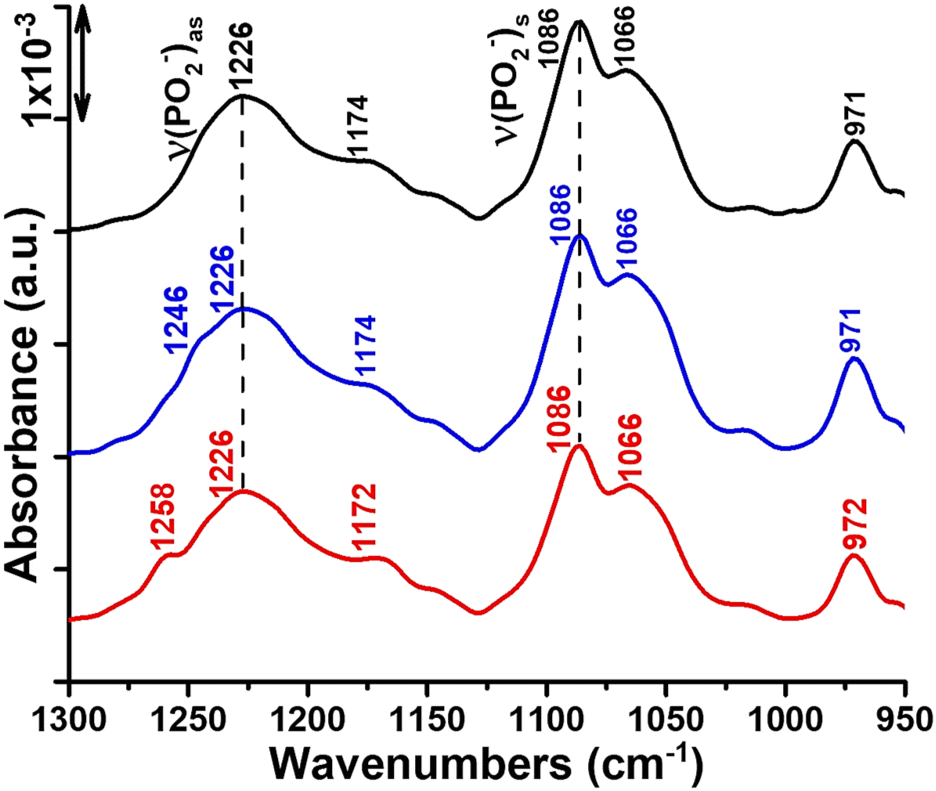 | ||
| Fig. 1 ATR-FTIR spectra highlighting the interaction of titanium complexes 1 and 2 with DOPC multi-bilayers at room temperature. Black curve: spectrum of DOPC multi-bilayers hydrated with H2O. Blue and red curves: spectra of complexes 1 and 2, respectively, inserted into DOPC multi-bilayers at a complex/lipid ratio of 1/20 and hydrated by H2O. | ||
To highlight the small but clear changes in the lipid head group vibrations induced by the interaction of Ti complexes with the DOPC bilayers, the difference spectra were generated by subtracting the DOPC spectrum from the Ti complexes/DOPC spectra (Fig. 2a and b, black curves). For the subtraction, the spectra are normalized with respect to the lipid band at 971 cm−1 to compensate for variations in the lipid concentration in the samples. Indeed, very small changes occur in the contour, intensity and position of the ν((CH3)3N+)as an absorption band when we directly compare the DOPC and Ti complexes/DOPC sample spectra (Fig. 1).
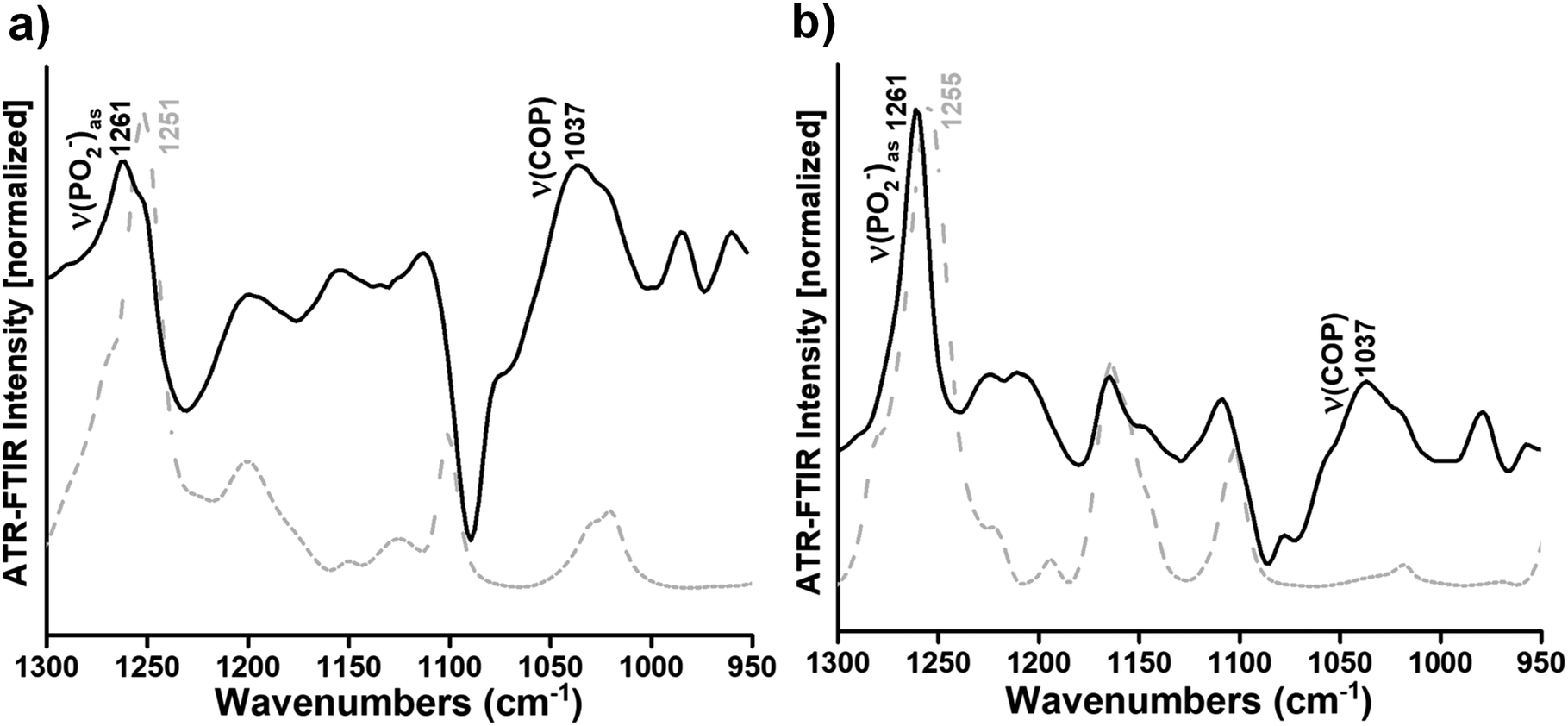 | ||
| Fig. 2 Gray dashed curves: ATR-FTIR spectra of complexes 1 and 2 as dry films from solutions in methanol (a and b, respectively). Black curves: difference ATR-FTIR spectra calculated by subtracting the spectrum of DOPC multi-bilayers from the spectra of complex 1/DOPC and complex 2/DOPC multi-bilayers (a and b). Both spectra in a and b were normalized to the same intensity for better visualization of the characteristic contributions of complexes 1 and 2 in the respective difference spectra. | ||
In the difference spectra, only the vibrational modes of the lipid that changed due to the interaction are observed. The band observed at 1261 cm−1 revealed the perturbation of the PO2− groups of DOPC due to the interaction with complex 1 or complex 2 (Fig. 2a and b, black curves). However, the absorption band arising from the C–O stretching mode in complexes 1 and 2 spectra (i.e. the bands at 1251 and 1255 cm−1, respectively) (Fig. 2a and b, gray dashed curves) seemed to interfere with the phosphate absorption band of the lipid (i.e. the νas(PO2−) band at 1261 cm−1) (Fig. 2a and b, black curves). Overall, it seems that the complexes mainly affect the phosphate group of the phospholipid upon interaction. Moreover, the appearance of a large band at around 1037 cm−1 in the presence of complex 1 or complex 2 attributed to the ν(COP) mode (Fig. 2a and b, black curves) also indicates a modification of the orientation of the C–O–P ester group after interaction between titanium(IV) complexes and the phosphate group of the lipid. However, the difference spectra in the spectral range of the CO ester carbonyl stretching were calculated between the Ti complexes/DOPC spectra and the DOPC spectrum, as described above. Three bands centred around 1737, 1733 and 1730 cm−1 in the presence of complex 1 and three bands at 1739, 1730, and 1726 cm−1 in the presence of complex 2 were shown (see Fig. S5a and b†). The bands at 1737 and 1739 cm−1 were attributed to free hydrogen carbonyl groups,44 while the bands at 1730 and 1726 cm−1 (see Fig. S5a†) as well as 1733 and 1730 cm−1 (see Fig. S5b†) were assigned to hydrogen-bound carbonyl groups in Ti complex/DOPC mixtures.44 The appearance of these different subpopulations of carbonyl groups was previously observed52 and suggested a different structural arrangement and environment of the interfacial carbonyl groups of the lipid. Hence, Ti complexes affect the interfacial carbonyl groups, which are probably located close to the lipid phosphate group that interacts with the metal complexes.
Study of the interaction of a Ti(IV) complex with DOPC by solution NMR spectroscopy and mass spectrometry
We monitored the reaction of complex 1 with racemic DOPC in solution at room temperature by 1H and 31P{1H} NMR. As previous studies performed with related Ti(IV) complexes required a prior dilution in DMSO before using these complexes in biological media to assess their cytotoxicity,29 DMSO was also employed in the present investigation to dissolve the Ti complex.First, we assigned the 1H resonances of pure DOPC in DMSO-d6 following the atom-numbering scheme represented in Chart 2 (vide infra experimental data in Materials and methods). The 1H NMR spectrum of a solution of complex 1 in DMSO-d6 exhibited two aromatic signals at 6.45 and 6.13 ppm (Fig. S6†).
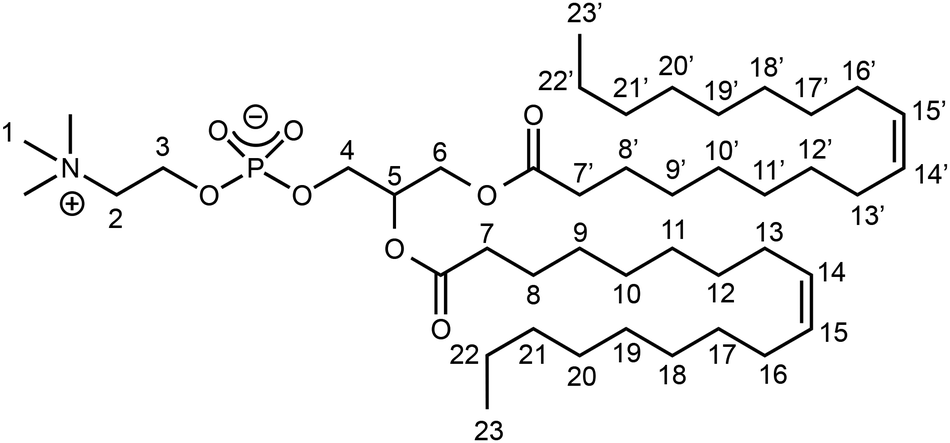 | ||
| Chart 2 Atom-numbering scheme for racemic DOPC. | ||
Then, in a preliminary experiment, we allowed DOPC to react with a roughly stoichiometric amount of complex 1 in DMSO-d6 at RT, and we recorded the NMR spectra of the resulting solution A. The 31P{1H} NMR spectrum of solution A (Fig. S8b†) shows a new singlet at −7.32 ppm, in addition to the phosphate signal of DOPC (δ = −1.07 ppm, see Fig. S8c†). We observed on the 1H NMR spectrum of A (Fig. S9†) the signals of complex 1 and free DOPC together with those of a novel species (compound 3); however, the conversion of DOPC into compound 3 was rather low (ca. 35% estimated from the deconvolution of the NMe3+ signals, see Fig. S10 and S11†).
At the next stage, we sought to increase the conversion of free DOPC into novel compound 3; for that purpose, a large excess of complex 1 (ca. 8 equiv.) was allowed to react with DOPC in DMSO-d6 (final solution B). Under these conditions, the species identified in solution B by 1H NMR were as follows: complex 1, present in large excess; compound 3, for which the NMR signals could be fully identified and their integrations determined (Fig. S12 and S13†); and a small amount of unreacted DOPC. From the deconvolution of the NMe3+ signals, the conversion of DOPC into complex 3 was estimated to be ca. 95% (Fig. S10 and S14†). In agreement with this statement, the 31P NMR singlet at −7.32 ppm was very intense, while the free DOPC signal was much weaker (Fig. S8a†).
From the 1H NMR signals of compound 3 in the aliphatic and olefinic regions (0–5.5 ppm), we noted that the structure of the DOPC was preserved. The chemical shifts of the protons belonging to the hydrocarbon chains were roughly the same for DOPC and 3. However, we observed that the chain protons closest to the ester functions (and therefore to the coordination sphere) were slightly shielded in 3, in comparison with DOPC (H7 in α position and H8 in β position: Δδ = −0.02 ppm). The resonances of the choline protons H2 and H3 were shifted to a higher frequency by ca. 0.1–0.3 ppm, as well as the glycerol protons H4 (Δδ = +0.21 ppm). Moreover, the resonances of the glycerol H6 protons shifted to a lower frequency by ca. 0.1 ppm. The trimethylammonium resonance was almost unaffected (Δδ = +0.01 ppm).
In the aromatic region of the spectrum of compound 3, 2 catecholato broad signals at 6.37 and 6.05 ppm, each integrating for 4 protons, were observed (Fig. S12 and S13†); this established that two catecholato ligands coordinated only the metal center. These signals were slightly shielded with regard to those of complex 1.
All these NMR data suggest that compound 3 is a [Ti(Cat)2]–DOPC adduct. In analogy with the literature concerning the interactions of phospholipids with titanium,22,24 and considering the coordinatively unsaturated sites on a [Ti(Cat)2] fragment, we can reasonably hypothesize that compound 3 is a coordination complex of titanium, where DOPC acts as a ligand of Ti through its phosphato donor group. Notably, several titanium(IV) complexes bearing phosphato ligands have been reported in the literature, but to the best of our knowledge, they are only related to multinuclear (mostly binuclear) complexes with bridging phosphates.53,54
To check the nuclearity of complex 3, a 1H DOSY NMR analysis of solution B was carried out. A partial view of the DOSY 2D map depicted in Fig. 3 focuses on the lowest measured values of the diffusion coefficient, i.e. the species in solution that have the highest hydrodynamic radii values. The aromatic signals at 6.45 and at 6.13 ppm of complex 1, which was present in excess in solution B, corresponded to a diffusion coefficient D = 2.5 × 10−10 ± 10% m2 s−1 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −9.60; see Fig. 3b). This value was in perfect agreement with a control DOSY analysis of the sole complex 1 observed after dissolution of in DMSO-d6. With regard to complex 3, characteristic DOSY spots could be observed with D ≈ 1.7 × 10−10 ± 10% m2 s−1 (logD ≈ −9.77). They corresponded to the catecholato signals at 6.37 and 6.05 ppm, and to some of the most intense 1D 1H NMR resonances of the DOPC part: viz. the hydrocarbon chain protons (H7–23 and H7′–23′, i.e. olefinic at 5.32 ppm, aliphatic in the range 2.30–0.80 ppm), and the trimethylammonium protons at 3.13 ppm. A control DOSY analysis of DOPC was also recorded in DMSO-d6, which led to the value D = 1.7 × 10−10 ± 10% m2 s−1 (Fig. S15†). Therefore, no clear distinction could be made between the DOSY data of DOPC and complex 3. Overall, this DOSY study shows that (1) one DOPC and two catecholato ligands are incorporated within the same species (i.e., Ti complex 3), and their respective signals correspond to the same diffusion coefficient; (2) the size of the complex in the solution is similar to the size of the DOPC, and we can deduce that complex 3 is monomeric.
D = −9.60; see Fig. 3b). This value was in perfect agreement with a control DOSY analysis of the sole complex 1 observed after dissolution of in DMSO-d6. With regard to complex 3, characteristic DOSY spots could be observed with D ≈ 1.7 × 10−10 ± 10% m2 s−1 (logD ≈ −9.77). They corresponded to the catecholato signals at 6.37 and 6.05 ppm, and to some of the most intense 1D 1H NMR resonances of the DOPC part: viz. the hydrocarbon chain protons (H7–23 and H7′–23′, i.e. olefinic at 5.32 ppm, aliphatic in the range 2.30–0.80 ppm), and the trimethylammonium protons at 3.13 ppm. A control DOSY analysis of DOPC was also recorded in DMSO-d6, which led to the value D = 1.7 × 10−10 ± 10% m2 s−1 (Fig. S15†). Therefore, no clear distinction could be made between the DOSY data of DOPC and complex 3. Overall, this DOSY study shows that (1) one DOPC and two catecholato ligands are incorporated within the same species (i.e., Ti complex 3), and their respective signals correspond to the same diffusion coefficient; (2) the size of the complex in the solution is similar to the size of the DOPC, and we can deduce that complex 3 is monomeric.
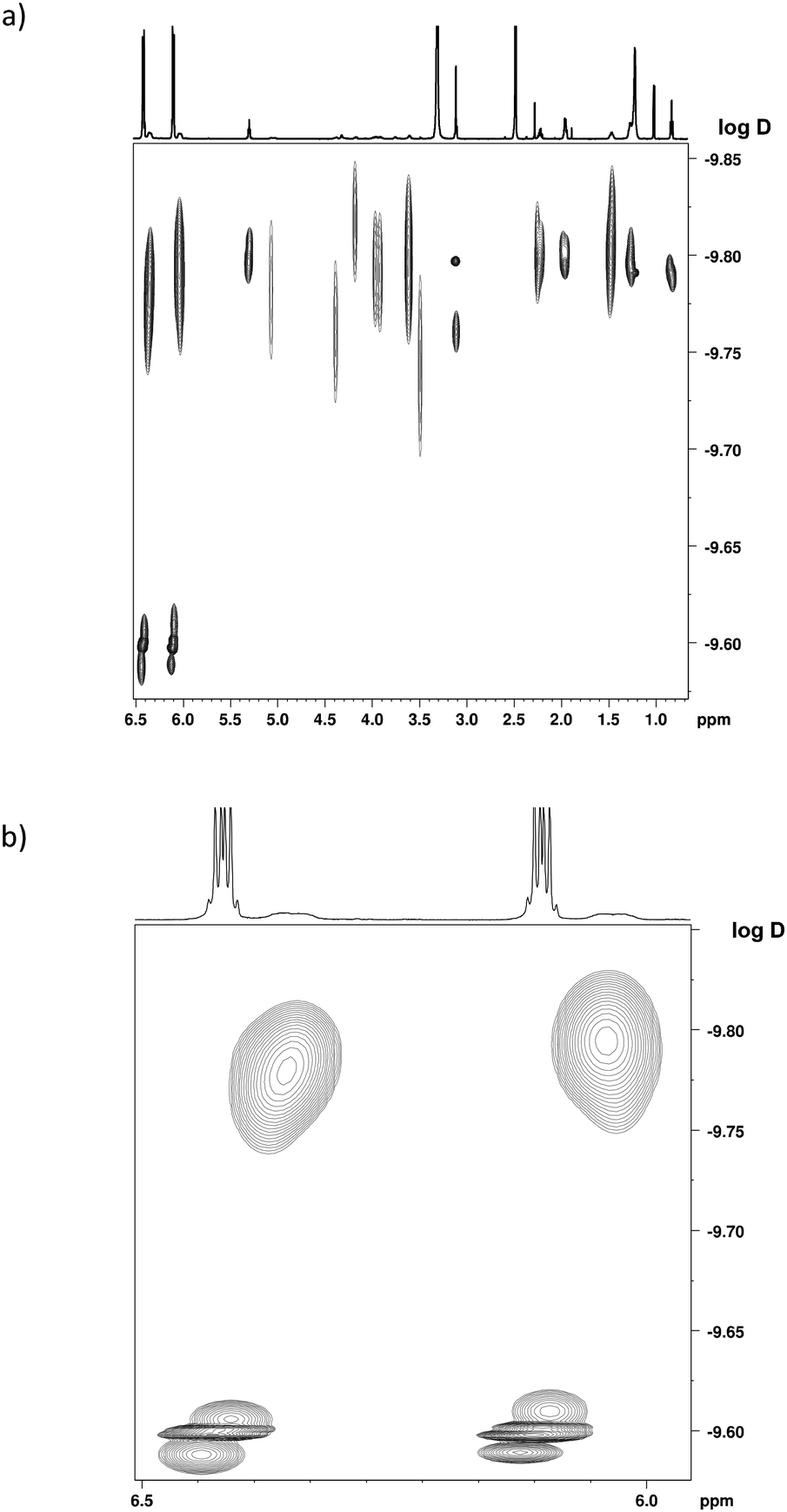 | ||
| Fig. 3 1H NMR DOSY spectrum of solution B, exhibiting the signals of complexes 1 and 3 in DMSO-d6 (600 MHz). (a) Section from 0.7 to 6.5 ppm. (b) Zoom on the catecholato signals. | ||
A 1H–1H NMR ROESY analysis of solution B revealed inter-ligand proton–proton spatial proximity within complex 3 (Fig. 4). Specifically, NOE cross-peaks were observed, as shown in Fig. 4a and b, between the catecholato H24 proton and some of the DOPC protons (H1–6), which is consistent with the representation of complex 3 proposed in Fig. 4c.
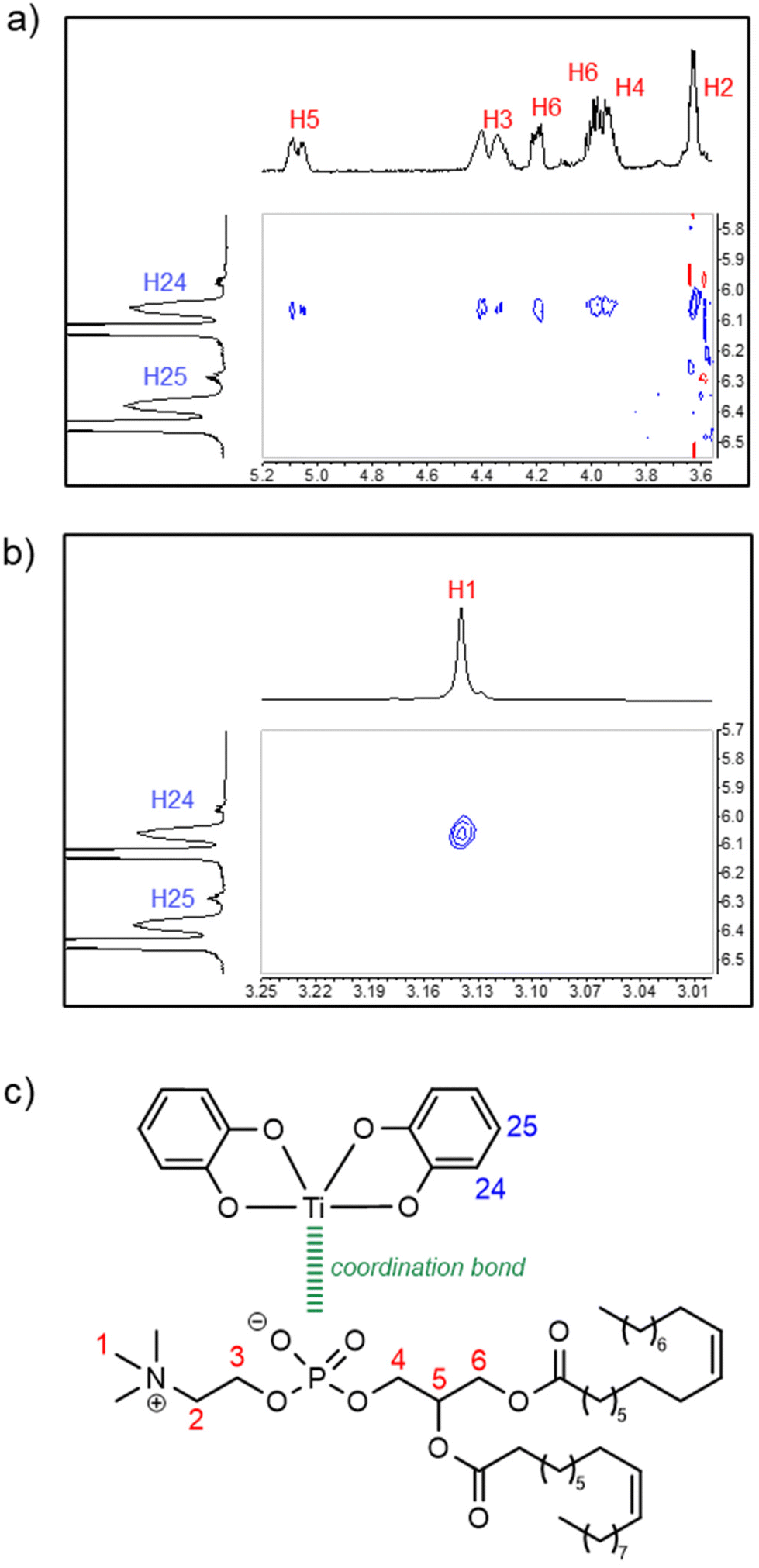 | ||
| Fig. 4 (a and b) Sections of the 1H–1H NMR ROESY spectrum of solution B in DMSO-d6 (500 MHz), exhibiting signals of complex 3. (c) The proposed structural formula for complex 3. The cross-peaks reveal a close spatial contact between one of the catecholate protons (represented in blue) and the DOPC protons (represented in red). | ||
If we consider that Ti is an asymmetric center with Δ or Λ configuration, the fact that only two catechol 1H NMR signals are observed on the spectrum of 3 (vide supra) indicates that both stereoisomers interconvert rapidly. This statement is supported by the literature related to tris-catecholato-55 or bis-catecholato-coordinated56 Ti complexes. Besides, we noticed that a few 1H and 13C NMR signals of complex 3 were split: for example, in the 1H NMR spectrum, the H6 doublet of doublets (Fig. 5a), and in the 13C NMR spectrum, the C6 singlet and C4 doublet (Fig. 5b). This can be interpreted as the result of diastereomers of 3 arising from two other stereocenters, which are the carbon atom C5 and the phosphorus atom of DOPC in all likelihood.57 This implies that the phosphato group coordinates the metal with only one of its free oxygen atoms (Fig. 5c), and it excludes a κ2O,O′-coordination where P is not an asymmetric center.
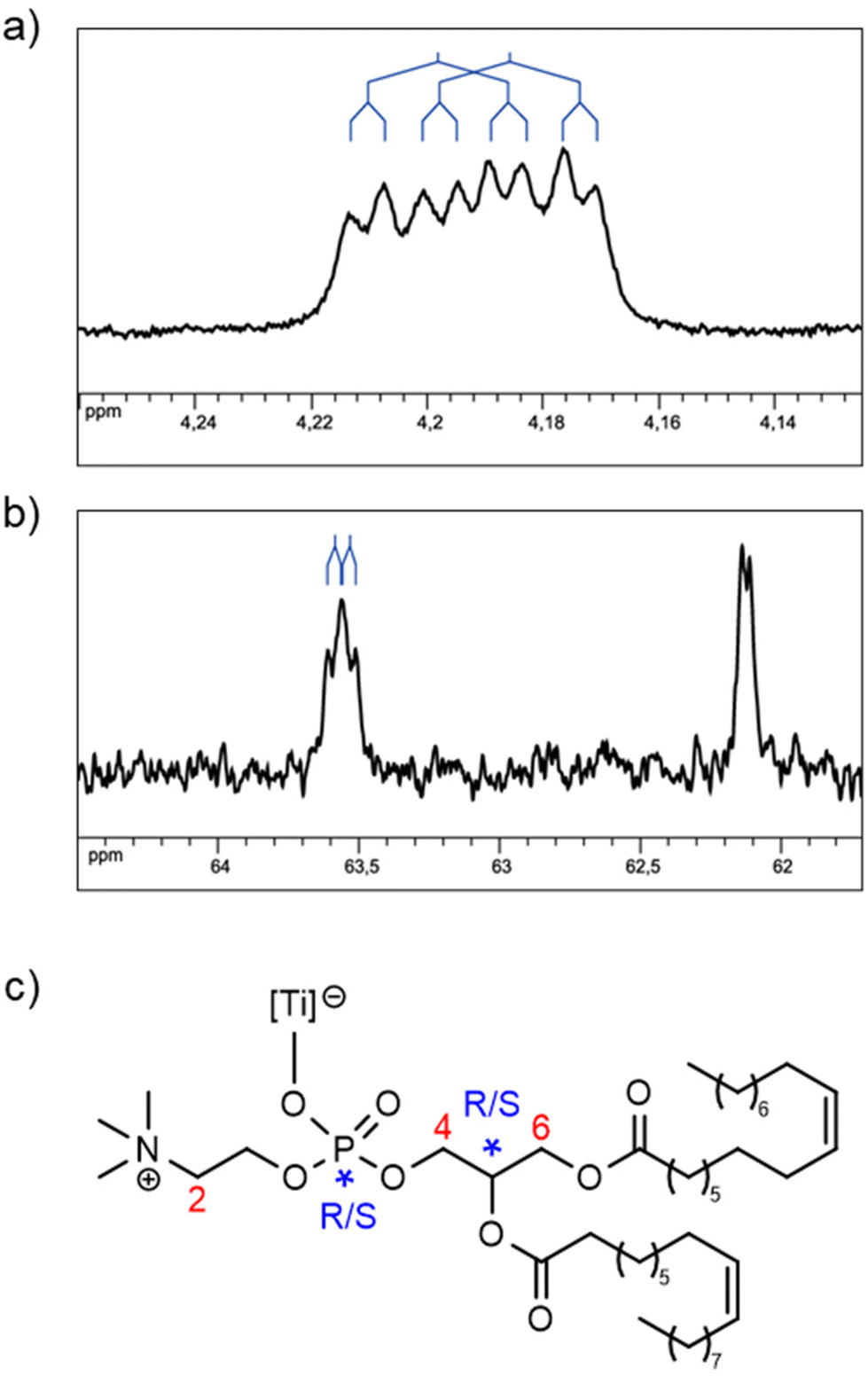 | ||
| Fig. 5 (a and b) Sections of 1D NMR spectra of solution B in DMSO-d6, exhibiting a splitting of some of the signals of complex 3. (a) 1H NMR (500 MHz): one of the H6 signals. (b) 13C{1H} NMR (125 MHz): C4 and C6 signals (decreasing chemical shift). (c) The proposed partial formula for complex 3 with stereogenic centers and partial numeration of the protons and carbons. | ||
To confirm these findings with negative-mode electrospray ionization mass spectrometry (ESI−-MS), we mixed complex 1 with DOPC (ca. 1 equiv.) in methanol instead of DMSO.58 The result was rather disappointing, as long as [Ti(Cat)2(OMe)]− was the overwhelmingly major detected species at m/z = 295.01. However, a signal was detected at m/z = 1080.60, corresponding to [Ti(Cat)2(DOPC)(OMe)]− with the proper pattern (although with a very low intensity: 0.4%; Fig. S16†). This may support our conclusions drawn from the NMR data, i.e. the presence of catecholato and DOPC ligands in the coordination sphere of titanium. However, we are aware that one has to remain cautious with such data obtained from different solvents: it has been reported that the outcome of the reaction of titanium(IV) complexes with phosphates is solvent dependent.54
Computational investigations
Density functional theory (DFT) calculations have been implemented to obtain a more accurate idea of the nature of complex 3. 1,2-Diacetyl-sn-glycero-3-phosphocholine (DAcPC)59 was used as a model for DOPC to reduce computational costs by avoiding the sampling of numerous conformers due to the long alkyl chains present in DOPC. The optimized geometry of the pentacoordinated complex [Ti(Cat)2(κ1-DAcPC)] 4 and the hexacoordinated complexes trans-[Ti(Cat)2(κ1-DAcPC)(DMSO)] 5 and cis-[Ti(Cat)2(κ1-DAcPC)(DMSO)] 6 (Chart 3) were obtained via the GAUSSIAN 09 (Rev. D01)60 software using the wB97XD61 functional and a def2-TZVP basis set.62 Note that for each complex, different starting conformations have been considered, sometimes leading to different minimum structures. Herein, we discuss only the optimized structure of each complex with the lowest energy. The XYZ structure files of the optimized complexes are given in the ESI.†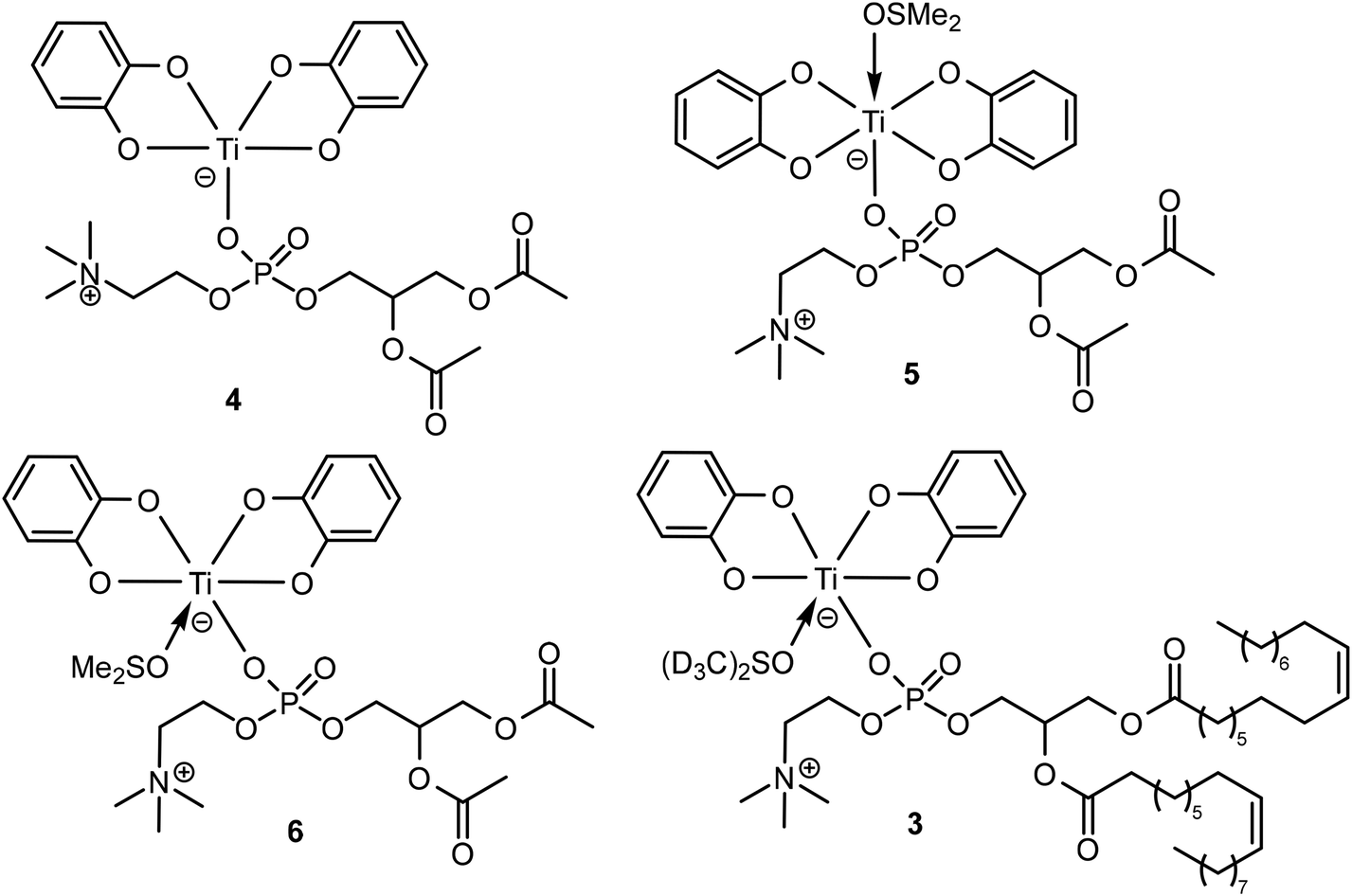 | ||
| Chart 3 Schematic representation of the proposed structural formula of complex 4–6 investigated by DFT, and of complex 3. | ||
The computed energies of the three complexes are 5E(4) = −3082.59634069 Hartree, E(5) = −3635.85448309 Hartree and E(6) = −3635.87640519 Hartree. Thus, a comparison between the isomeric DMSO-coordinated complexes 5 and 6 reveals an energy difference of 57.5 kJ mol−1, 6 being by far the most stable isomer. We now consider the insertion of the DMSO ligand into the coordination sphere of titanium, starting from pentacoordinated 4 (eqn (1)).
| complex 4 + DMSO → complex 6. | (1) |
An energy (ΔE) of −115.3 kJ mol−1 is determined for this reaction (with E(DMSO) = −553.236144767 Hartree), which is in favor of complex 6.
Therefore, these DFT data allow us to propose a formula for complex 3 (Chart 3), viz. cis-[Ti(Cat)2(κ1-DOPC)(DMSO-d6)], being analogous to 6. In support of this statement, we also observe that some of the interligand NOE cross-peaks of complex 3 represented in Fig. 4 are consistent with the short H⋯H distances calculated for complex 6 (Fig. S19b†).
Considering this formulation for complex 3, we estimated the binding Ti–DOPC constant from the NMR analysis of solution B (vide supra) to be 7.0 ± 5% (see details in the ESI†).
Conclusion
This study aims to determine the nature of the interactions of a phospholipid with a discrete titanium(IV) complex, first by the FTIR analysis of Ti complexes in DOPC multi-bilayers, and second by the NMR analysis of solutions in DMSO-d6. The latter results were supported by DFT calculations. For the first time, we demonstrated that the interaction of both Ti(IV) complexes probably occurred with the phosphate head group of the DOPC model membrane using ATR-FTIR spectroscopy. We observed significant changes in the phosphate vibrational modes of the phospholipid, which could mainly indicate the formation of DOPC–Ti complexes. We therefore hypothesize that this result can be correlated with the interaction mechanism proposed for the adsorption of zwitterionic liposomes with TiO2 nanoparticles.24 Moreover, small modifications in the vibrational signals of CH2 and CO functional groups of DOPC were observed in the Ti complex/DOPC mixture spectra, resulting from the effect of the interaction between the phospholipid and the Ti(IV) complexes.
Subsequently, NMR analyses performed on DMSO-d6 solutions resulting from mixing 1 with DOPC revealed the presence of mononuclear Ti complex 3, comprising two catecholato ligands and one DOPC ligand. Further, DFT calculations using DAcPC as a model of DOPC allowed us to propose the formula cis-[Ti(Cat)2(κ1-DOPC)(DMSO-d6)] for complex 3.
It is interesting to note that although the experimental conditions for both spectroscopic analyses were radically different (medium and initial Ti/DOPC ratio), they led to convergent conclusions. Overall, this study demonstrates that titanium complexes can interact with phospholipids and, hence, must perturb the integrity of the cellular membrane. Thus, this feature may explain the anticancer properties of certain types of Ti(IV) coordination compounds and permit the elucidation of their modes of action.
Materials and methods
Chemicals
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) powder with a purity of ∼99% was purchased from Avanti Polar Lipids Inc. A 1 mM solution of DOPC was prepared by dissolving the powder in chloroform of HPLC grade. Ultrapure water (Milli-Q, Millipore) was used for all the experiments. CDCl3 and DMSO-d6 were obtained from Eurisotop. For titanium complexes, complex 1 was synthesized following a reported procedure;30 and complex 231 was obtained following the same procedure by replacing catechol with 2,3-dihydroxynaphthalene (see ESI†).
ATR-FTIR spectroscopy of multi-bilayers
Stock solutions of DOPC in chloroform were mixed with titanium complexes 1 or 2 (10 mM in methanol), followed by vortexing for 10 min. Solvents were removed by argon evaporation and kept overnight in a lyophilizer. The gel was obtained by rehydration of the pure lipid or complex 1 (or 2)/lipid mixtures with H2O at the molecular complex to lipid ratio, Ri = 1/20. Hydration ratios were higher than 70%. Multi-bilayers were obtained by shearing a pure DOPC or mixed complex 1 (or 2)/lipid gels at the diamond ATR crystal surface (Harrick Crystal, Bruker, Germany) with a Teflon stick.63The ATR-FTIR spectra of multi-bilayers39,40,64,65 were recorded using a Vertex 70 FTIR spectrometer (Bruker, Germany) equipped with an MCT (mercury cadmium telluride) detector and an ATR (attenuated total reflection) Harrick crystal Diamond Prism. The spectrometer was purged with dry air to avoid contributions from humidity in the spectra. Because ATR spectroscopy is sensitive to the orientation of the structures,64,65 spectra were recorded with parallel (p) and perpendicular (s) polarization of the incident light with respect to the ATR plate. The orientational information is then contained in the dichroic ratio RATR = Ap/As, where Ai represents the absorbance of the considered band for the p or s polarization of the incident light. The ATR-FTIR spectra were recorded in the spectral range from 4000 to 800 cm−1 with a scan velocity of 40 kHz. Generally, three spectra with a resolution of 4 cm−1 (256 co-added scans) were averaged for each sample of multi-bilayers.
The ATR-FTIR spectra of complex 1 and complex 2 samples were performed as dry films from solutions in methanol. 2 μl of each sample (10 mM) was deposited to obtain a thin film on the diamond crystal. All experiments were carried out at 22 °C.
The ATR-FTIR spectra were recorded with Opus software from Bruker (Opus. 6.0) and analysed with the Origin program (OriginLab. 8.5).
NMR spectroscopy and mass spectrometry
Bruker Avance III HD-500 and Avance III-600 spectrometers were used for the solution NMR analyses performed at 25 °C. The 1H NMR spectra were recorded at 500.13 or 600.13 MHz and referenced to SiMe4. The 31P{1H} NMR spectra (broadband decoupled) were recorded at 125.51 or 202.50 MHz and referenced to 85% aqueous H3PO4. The 13C{1H} NMR spectra (broadband decoupled) were recorded at 125.77 MHz and referenced to SiMe4. The chemical shifts are reported in ppm. Multiplicity: s = singlet, d = doublet, m = multiplet. The 13C{1H} signals are singlets unless otherwise stated. The NMR assignments were supported by 2D NMR analyses; for the atom numbering scheme, see Chart 2 and Fig. 4c. Self-diffusion measurements were performed at 600.13 MHz with a 5 mm PABBI 1H–19F/X z-gradient probe, providing 300 G cm−1 (30 G cm−1 A−1), and applying a PFGSTE pulse sequence using bipolar gradients. For each experiment, a total relaxation time of 5 s between each scan was obtained, and 40 gradient strength increments from 5 to 95% were acquired. Diffusion time and gradient length were set up at 30 ms, and between 800 and 1100 μs, respectively, so that less than 10% of the original signal remains at the end. The DOSY spectra were generated with Bruker's Dynamics Center software via multi exponential fitting of the data.Negative-mode electrospray ionization mass spectra (ESI−-MS) were performed using a MicroTOF or a MicroTOF II focus spectrometer (Bruker) equipped with an electrospray source.
:1.
1H NMR (DMSO-d6, 500 MHz): δ 6.40–6.34 (m, 4H, H25), 6.08–6.02 (m, 4H, H24), 5.35–5.28 (m, 4H, H14 + H14′ + H15 + H15′), 5.12–5.02 (m, 1H, H5), 4.45–4.27 (m, 2H, H3), 4.20 and 4.19 (2 dd, 1H, 2JHH = 12.2 Hz, 3JHH = 3.0 Hz, H6), 4.03–3.88 (m, 3H, H4 + H6), 3.65–3.59 (m, 2H, H2), 3.13 (s, 9H, H1), 2.28–2.20 (m, 4H, H7 + H7′), 2.00–1.95 (m, 8H, H13 + H13′ + H16 + H16′), 1.53–1.44 (m, 4H, H8 + H8′), 1.33–1.19 (m, 40H), 0.85 (m, 6H, H23 + H23′). 31P{1H} NMR (DMSO-d6, 202 MHz): δ −7.32 (s). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 172.50, 172.29, 172.26, 159.37, 159.32, 129.67, 129.66, 129.58, 117.95, 110.27, 110.21, 70.04 (d, 3JPC = 7 Hz), 69.99 (d, 3JPC = 7 Hz), 65.22 (d, 3JPC = 7 Hz), 63.59 (d, 2JPC = 6 Hz), 63.53 (d, 2JPC = 6 Hz), 62.13, 62.11, 59.17 (d, 2JPC = 5 Hz), 59.13 (d, 2JPC = 5 Hz), 53.10, 33.55, 33.39, 31.32, 29.17, 29.15, 29.13, 28.87, 28.74, 28.69, 28.65, 28.63, 28.58, 28.55, 28.47, 28.44, 26.65, 26.63, 26.60, 24.48, 24.42, 13.98.
Abbreviations
| H2Cat | Catechol |
| H2Napht | 2,3-Dihydroxynaphthalene |
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| ATR-FTIR | Attenuated total reflection-Fourier transform infrared |
| R ATR | Dichroic ratio |
| NMR | Nuclear magnetic resonance |
| DOSY | Diffusion-ordered NMR spectroscopy |
| ESI-MS | Electrospray ionization mass spectrometry |
| DFT | Density functional theory |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We warmly thank Prof. M. Henry for fruitful discussions, and J. Xhema for technical assistance. Financial support of the University of Strasbourg and of the CNRS is gratefully acknowledged. This work was supported by the Interdisciplinary Thematic Institute SysChem via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d'Avenir to PH. PH acknowledges the Institut Universitaire de France (IUF). This is an open access article under the terms of the Creative Commons Attribution Non-Commercial NoDerivs License, which permits use and distribution in any medium, provided the original work is properly cited, the use is non-commercial and no modifications or adaptations are made.References
- M. Tacke, L. T. Allen, L. Cuffe, W. M. Gallagher, Y. Lou, O. Mendoza, H. Müller-Bunz, F. J. K. Rehmann and N. J. Sweeney, J. Organomet. Chem., 2004, 689, 2242–2249 CrossRef.
- N. J. Sweeney, O. Mendoza, H. Müller-Bunz, C. Pampillón, F. J. K. Rehmann, K. Strohfeldt and M. Tacke, J. Organomet. Chem., 2005, 690, 4537–4544 CrossRef.
- M. Shavit, D. Peri, C. M. Manna, J. S. Alexander and E. Y. Tshuva, J. Am. Chem. Soc., 2007, 129, 12098–12099 CrossRef.
- T. A. Immel, U. Groth and T. Huhn, Chem. – Eur. J., 2010, 16, 2775–2789 CrossRef PubMed.
- A. Kumar Singh, A. Kumar, H. Singh, P. Sonawane, P. Pathak, M. Grishina, J. Pal Yadav, A. Verma and P. Kumar, Chem. Biodiversity, 2023, 20, e202300061 CrossRef PubMed.
- T. Zhao, P. Wang, X. Zhang, N. Liu, W. Zhao, Y. Zhang, P. Yuan, S. Li, M. Yang, Z. Yang and T. Huhn, Curr. Top. Med. Chem., 2023, 23, 1835–1849 CrossRef.
- S. Parveen, F. Arjmand and S. Tabassum, Eur. J. Med. Chem., 2019, 175, 269–286 CrossRef.
- H. Köpf and P. Köpf-Maier, Angew. Chem., Int. Ed. Engl., 1979, 18, 477–478 CrossRef PubMed.
- B. K. Keppler, C. Friesen, H. G. Moritz, H. Vongerichten and E. Vogel, in Bioinorganic Chemistry. Structure and Bonding, Springer, Berlin, Heidelberg, 1991, 78, 97–127 Search PubMed.
- C. W. Schwietert and J. P. McCue, Coord. Chem. Rev., 1999, 184, 67–89 CrossRef.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 32, 3474–3482 RSC.
- K. M. Buettner and A. M. Valentine, Chem. Rev., 2012, 112, 1863–1881 CrossRef PubMed.
- S. A. Loza-Rosas, M. Saxena, Y. Delgado, K. Gaur, M. Pandrala and A. D. Tinoco, Metallomics, 2017, 9, 346–356 CrossRef PubMed.
- E. Y. Tshuva and J. A. Ashenhurst, Eur. J. Inorg. Chem., 2009, 15, 2203–2218 CrossRef.
- A. D. Tinoco, E. V. Eames, C. D. Incarvito and A. M. Valentine, Inorg. Chem., 2008, 47, 8380–8390 CrossRef.
- T. B. Parks, Y. M. Cruz and A. D. Tinoco, Inorg. Chem., 2014, 53, 1743–1749 CrossRef PubMed.
- A. Tzubery, N. Melamed-Book and E. Y. Tshuva, Dalton Trans., 2018, 47, 3669–3673 RSC.
- M. Guo, H. Sun, H. J. McArdle, L. Gambling and P. J. Sadler, Biochemistry, 2000, 39, 10023–10033 CrossRef.
- M. Cini, T. D. Bradshaw and S. Woodward, Chem. Soc. Rev., 2017, 46, 1040–1051 RSC.
- M. Saxena, S. A. Loza-Rosas, K. Gaur, S. Sharma, S. C. Pérez Otero and A. D. Tinoco, Coord. Chem. Rev., 2018, 363, 109–125 CrossRef PubMed.
- N. J. Cho and C. W. Frank, Langmuir, 2010, 26, 15706–15710 CrossRef.
- F. Wang and J. Liu, Small, 2014, 10, 3927–3931 CrossRef PubMed.
- N. J. Cho, J. A. Jackman, M. Liu and C. W. Frank, Langmuir, 2011, 27, 3739–3748 CrossRef PubMed.
- F. Wang and J. Liu, J. Am. Chem. Soc., 2015, 137, 11736–11742 CrossRef PubMed.
- X. Zhang, F. Wang, B. Liu, E. Y. Kelly, M. R. Servos and J. Liu, Langmuir, 2014, 30, 839–845 CrossRef.
- P. Ghosh, A. T. Kotchevar, D. D. DuMez, S. Ghosh, J. Peiterson and F. M. Uckun, Inorg. Chem., 1999, 38, 3730–3737 CrossRef.
- M. Guo, Z. Guo and P. J. Sadler, J. Biol. Inorg. Chem., 2001, 6, 698–707 CrossRef PubMed.
- R. P. Eberle, Y. Hari and S. Schürch, J. Am. Soc. Mass Spectrom., 2017, 28, 1901–1909 CrossRef.
- G. Khalil, C. Orvain, L. Fang, L. Barloy, A. Chaumont, C. Gaiddon, M. Henry, N. Kyritsakas and P. Mobian, Dalton Trans., 2016, 45, 19072–19085 RSC.
- Y. Chi, P.-F. Hsu, J.-W. Lan, C.-L. Chen, S.-M. Peng and G.-H. Lee, J. Phys. Chem. Solids, 2001, 62, 1871–1879 CrossRef.
- A. Sugawara, Jpn. Pat, 2006335835A, 2006 Search PubMed.
- G. Dehaen, S. V. Eliseeva, K. Kimpe, S. Laurent, L. Vander Elst, R. N. Muller, W. Dehaen, K. Binnemans and T. N. Parac-Vogt, Chem. – Eur. J., 2012, 18, 293–302 CrossRef.
- S. L. Hancock, R. Gati, M. F. Mahon, E. Y. Tshuva and M. D. Jones, Dalton Trans., 2014, 43, 1380–1385 RSC.
- I. Rodríguez, L. Fernández-Vega, A. N. Maser-Figueroa, B. Sang, P. González-Pagán and A. D. Tinoco, Antibiotics, 2022, 11, 158–175 CrossRef.
- N. Hewage, C. Mastriano, C. Brückner and M. Zeller, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2022, 78, 385–391 CrossRef.
- B. A. Borgias, S. R. Cooper, Y. B. Koh and K. N. Raymond, Inorg. Chem., 1984, 23, 1009–1016 CrossRef.
- T. Plenge, R. Dillinger, L. Santagostini, L. Casella and F. Tuczek, Z. Anorg. Allg. Chem., 2003, 629, 2258–2265 CrossRef.
- M.-C. Giocondi, D. Yamamoto, E. Lesniewska, P.-E. Milhiet, T. Ando and C. Le Grimellec, Biochim. Biophys. Acta, 2010, 1798, 703–718 CrossRef.
- R. N. A. H. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, 2013, 1828, 2347–2358 CrossRef.
- Z. Fezoua-Boubegtiten, B. Hastoy, P. Scotti, A. Milochau, K. Bathany, B. Desbat, S. Castano, R. Oda and J. Lang, Biochim. Biophys. Acta, 2019, 1861, 670–676 CrossRef PubMed.
- A. Agopian and S. Castano, Biochim. Biophys. Acta, 2014, 1838, 117–126 CrossRef PubMed.
- S. Castano and B. Desbat, Biochim. Biophys. Acta, 2005, 1715, 81–95 CrossRef.
- K. Leberle, I. Kempf and G. Zundel, Biophys. J., 1989, 55, 637–648 CrossRef PubMed.
- A. Blume, W. Hubner and G. Messner, Biochemistry, 1988, 27, 8239–8249 CrossRef.
- J. L. R. Arrondo, F. M. Goñi and J. M. Macarulla, Biochim. Biophys. Acta, 1984, 794, 165–168 CrossRef.
- K. Cieślik-Boczula, B. Czarnik-Matusewicz, M. Perevozkina and M. Rospenk, RSC Adv., 2015, 5, 40455–40464 RSC.
- M. Hereć, A. Islamov, A. Kuklin, M. Gagoś and W. Gruszecki, Chem. Phys. Lipids, 2007, 147, 78–86 CrossRef.
- A. Mellier and A. Diaf, Chem. Phys. Lipids, 1988, 46, 51–56 CrossRef.
- R. A. Dluhy, D. G. Cameron, H. H. Mantsch and R. Mendelsohn, Biochemistry, 1983, 22, 6318–6325 CrossRef.
- H. L. Casal, H. H. Mantsch and H. Hauser, Biochim. Biophys. Acta, 1989, 982, 228–236 CrossRef.
- H. L. Casal, H. H. Mantsch, F. Paltauf and H. Hauser, Biochim. Biophys. Acta, 1987, 919, 275–286 CrossRef PubMed.
- H. H. Matsch, D. G. Cameron, P. A. Tremblay and M. Kates, Biochim. Biophys. Acta, 1982, 689, 63–72 CrossRef PubMed.
- R. Murugavel, A. Choudhury, M. G. Walawalkar, R. Pothiraja and C. N. R. Rao, Chem. Rev., 2008, 108, 3549–3655 CrossRef PubMed.
- G. A. Bhat, S. Verma, A. Rajendran and R. Murugavel, Inorg. Chem., 2018, 57, 7644–7654 CrossRef PubMed.
- See for instance: A. V. Davis, T. K. Firman, B. P. Hay and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 9484–9496 CrossRef PubMed.
- See for instance: S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058–10059 CrossRef PubMed.
- We thank one of the referees for this valuable interpretation.
- DMSO was not an appropriate solvent for MS analyses.
- R. Subramaniam, R. Jagadeesan, I. Mathew, Y. Cen and S. Balaz, Org. Process Res. Dev., 2017, 21, 177–181 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- W. Hubner and H. H. Mantsch, Biophys. J., 1991, 59, 1261–1272 CrossRef CAS.
- E. Goormaghtigh, V. Cabiaux and J.-M. Ruysschaert, Eur. J. Biochem., 1990, 193, 409–420 CrossRef CAS PubMed.
- E. Goormaghtigh, V. Raussens and J.-M. Ruysschaert, Biochim. Biophys. Acta, 1999, 1422, 105–185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ATR-FTIR, NMR and mass spectra, experimental procedures, geometry of complexes obtained by DFT calculations. See DOI: https://doi.org/10.1039/d4dt02966f |
| This journal is © The Royal Society of Chemistry 2025 |