
Polyoxometalate-based complex@graphene composite electrodes for efficient nitrate reduction to ammonia†
Nan
Zhao
a,
Xinming
Wang
 *a,
Shuang
Rong
b,
Qiushuang
Jiang
a,
Haoyun
Li
a,
Haijun
Pang
*a and
Huiyuan
Ma
a
*a,
Shuang
Rong
b,
Qiushuang
Jiang
a,
Haoyun
Li
a,
Haijun
Pang
*a and
Huiyuan
Ma
a
aKey Laboratory of Green Chemical Engineering and Technology of College of Heilongjiang Province, School of Materials Science and Chemical Engineering, Harbin University of Science and Technology, Harbin 150040, P. R. China. E-mail: wangxinming20@126.com
bHeilongjiang Electric Power Research Institute, State Grid, Harbin 150030, P. R. China
First published on 21st May 2025
Abstract
To replace the energy-intensive and polluting traditional ammonia synthesis process, in this study, we designed two polyoxometalate (POM)-based nickel/cobalt metal–organic composite catalysts, namely (HNCP)2[Ni(H2O)4]2[NiMo12(HPO4)4(PO4)4O30] (Ni-P4Mo6) and (HNCP)2[Co(H2O)4]2[CoMo12(HPO4)4(PO4)4O30] (Co-P4Mo6). These catalysts utilized {P4Mo6} as the structural unit, nickel/cobalt as the metal node, and π-conjugated organic ligands as the linkers, and they were loaded onto graphene oxide (GO) to enhance the conductivity and reaction contact area. Experimental results showed that in acidic electrolytes, Ni-P4Mo6/GO achieved an ammonia yield of 2.62 mg h−1 mg−1cat. at −0.6 V (vs. RHE) with a faradaic efficiency (FE) of 83.9% at −0.5 V, outperforming Co-P4Mo6/GO (1.63 mg h−1 mg−1cat. at −0.7 V; FE 85.3% at −0.3 V). Under neutral conditions, both the catalysts exhibited significantly improved performances (Ni-P4Mo6/GO: 11.6 mg h−1 mg−1cat. yield, 88.4% FE; Co-P4Mo6/GO: 11.1 mg h−1 mg−1cat. yield, 78.5% FE), surpassing most comparable catalysts. This work provides a novel strategy for developing efficient electrocatalysts for nitrate reduction to ammonia (e-NO3RR).
1. Introduction
As an important carrier and energy storage intermediate in the carbon neutralization industry, ammonia is considered to be a future fuel substitute and plays a vital role in various industries such as leather, papermaking and agricultural fertilizer production.1–6 The production of ammonia has been mainly dependent on a macroscale synthesis technology using the Haber–Bosch technique since 1913; however, its thermodynamic constraints—requiring extreme conditions (400–500 °C, 15–25 MPa) to overcome the high activation energy barrier of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond dissociation—result in massive energy consumption.7–11 In contrast, the electrocatalytic nitrate reduction reaction (e-NO3RR) for the synthesis of ammonia uses renewable electricity to drive the eight-electron reduction process, which can operate at room temperature and atmospheric pressure, and it is more thermodynamically inclined to choose nitrate rather than inert N2 as the nitrogen source.12–16 The electrocatalytic reduction of NO3− to NH3 is an efficient process for transforming nitrate into the more valuable compound, NH3. The conversion of NO3− can not only directly mitigate the problem of NO3− water pollution but also realize the green synthesis of ammonia. As a result, this reaction is regarded as one of the most potential methods for the eco-friendly and continuous production of ammonia in the future.17–22 However, the e-NO3RR faces enormous challenges such as reduced catalytic activity, low selectivity, and low ammonia production rate.23 Therefore, developing cost-effective catalysts with high efficiency is crucial for overcoming these bottlenecks.24–27
N bond dissociation—result in massive energy consumption.7–11 In contrast, the electrocatalytic nitrate reduction reaction (e-NO3RR) for the synthesis of ammonia uses renewable electricity to drive the eight-electron reduction process, which can operate at room temperature and atmospheric pressure, and it is more thermodynamically inclined to choose nitrate rather than inert N2 as the nitrogen source.12–16 The electrocatalytic reduction of NO3− to NH3 is an efficient process for transforming nitrate into the more valuable compound, NH3. The conversion of NO3− can not only directly mitigate the problem of NO3− water pollution but also realize the green synthesis of ammonia. As a result, this reaction is regarded as one of the most potential methods for the eco-friendly and continuous production of ammonia in the future.17–22 However, the e-NO3RR faces enormous challenges such as reduced catalytic activity, low selectivity, and low ammonia production rate.23 Therefore, developing cost-effective catalysts with high efficiency is crucial for overcoming these bottlenecks.24–27
So far, various catalysts have been developed for the e-NO3RR. Among them, non-metallic catalysts have the characteristics of cost-effectiveness, strong electronic structure, adjustability and durability. However, the catalytic activity of non-metallic catalysts used in the e-NO3RR is not satisfactory.28 Noble metal catalysts are the most common high-activity electrocatalysts including ruthenium (Ru), rhodium (Rh) and platinum (Pt).29–32 Nevertheless, the high cost and scarcity of resources have limited their further application. In contrast to noble metals, transition metal catalysts, along with their various compounds, such as oxides, sulfides, carbides, phosphides, and nitrides, provide unique performance advantages.33–35 These materials have gained significant attention, especially in the field of e-NO3RR.36–38 For example, Cu/2D-CuOx, Co/TiO2 NSs and Ni(OH)2@Ni show excellent electrocatalytic performance.39–42
Polyoxometalates (POMs) are a prominent category of inorganic compounds composed of numerous metal atoms interconnected through oxygen bridges, which are usually composed of highly oxidized transition metals (mainly W, V and Mo) with d0 or d1 electronic configurations.43–47 Because of its reversible redox properties, diverse and controllable structural composition, it has great potential in catalysis, photochemistry, nanotechnology, medicine and other fields.48,49 Nevertheless, the limited conductivity, tendency to agglomerate, and poor water solubility of POMs restrict their use in electrocatalysis. The construction of POMs–based metal–organic complexes can effectively ameliorate the aforementioned problems.50 The Khalaji-Verjani team synthesized a PMo11-Ru (acac)3 electrocatalyst, showing high HER activity and stability.51 Yang et al. developed two novel polyoxometalate-based metal–organic complexes through a self-assembly approach to link polyoxometalate clusters with CuCTC, which served as purpose catalysts, enabling both the electrochemical sensing and photocatalytic conversion of Cr(VI). The rates of Cr(VI) photoreduction achieved with these complexes outperformed those of Cu3 and most other reported catalysts.52
In the family of POMs, the [P4MoV6O31]12− ({P4Mo6}) anion has a stable structure with minimal steric hindrance and a rich array of active oxygen atoms. It can have a good synergistic effect with metal active sites and organic ligands, which gives {P4Mo6} good topological scalability. Notably, the molybdenum in the structure is in pentavalent reduced state (MoV), which is an excellent electron donor. This unique characteristic can be leveraged as a potential “electron-donating unit” to pair with catalytically active species.53–55 Therefore, the integration of {P4Mo6} with composite materials could potentially establish an optimized catalytic framework, enhancing electron transport dynamics while improving both reaction efficiency and product specificity in nitrate reduction (NO3RR) for ammonia synthesis.
Based on the above analysis, we selected the {P4Mo6}-type anion as the basic structural unit, nickel (Ni) and cobalt (Co) metal ions as nodes, and 2-(4-(4-carboxyphenyl)phenyl)imidazo(4,5-f)-(1,10)-phenanthroline (HNCP) with a large π-conjugated system as the organic ligand to construct two {P4Mo6} polyoxometalate-based metal–organic complexes, (HNCP)2[Ni(H2O)]2[NiMo12(H2PO4)6(PO4)2(OH)6O24] (Ni-P4Mo6) and (HNCP)2[Co(H2O)]2[CoMo12(H2PO4)6(PO4)2(OH)6O24] (Co-P4Mo6), which was anchored onto functionalized graphene oxide (GO) substrates to obtain Ni-P4Mo6/GO and Co-P4Mo6/GO composites. The structural integration of graphene within the composite architecture significantly boosts the electrical conductivity and surface area, enhances the charge transfer kinetics for improved composite dispersion, and optimizes active site utilization to maximize the electrocatalytic performance.56–59 Ni-P4Mo6/GO and Co-P4Mo6/GO composites show superior electrocatalytic efficacy in nitrate-to-ammonia conversion and stability, reaching the apex of NH3 yield of 11.6 mg h−1 mg−1cat and 11.1 mg h−1 mg−1cat. and Faraday efficiency of 88.4% and 78.5% at −1.0 V in neutral electrolytes, respectively. Our findings propose a rational design framework for polyoxometalate-integrated catalytic systems, achieving unprecedented faradaic efficiency in sustainable ammonia synthesis via nitrate electroreduction.
2. Experimental section
2.1 Preparation of (HNCP)2[Ni(H2O)4]2[NiMo12(HPO4)4(PO4)4O30] (Ni-P4Mo6)
A mixture of [(NH4)6Mo7O24·4H2O] (0.85 g), (NiCl2·6H2O) (0.40 g), HNCP (0.05 g) and CH3CH2OH (10 mL) was stirred for 10 min, and then deionized water (40 mL) and H3PO4 were added, respectively, with continuous stirring for 60 min. The pH value was adjusted to 1.0–1.5 with 1.0 M HCl or NaOH, and then the suspension was transferred to a 20 mL Teflon-lined autoclave and kept at 180 °C for 5 days. After slow cooling to room temperature at a rate of 10 °C h−1, yellow diamond crystals were obtained (yield 65% based on Mo).2.2 Preparation of (HNCP)2[Co(H2O)4]2[CoMo12(HPO4)4(PO4)4O30] (Co-P4Mo6)
The compound Co-P4Mo6 was prepared following the same procedure as that of compound Ni-P4Mo6 except that NiCl2·6H2O (0.40 g) was replaced with CoCl2·6H2O. Following gradual cooling at a rate of 10 °C h−1 until reaching room temperature, yellow-brown rhombic crystals of Co-P4Mo6 were obtained (yield 55% based on Mo).2.3 Preparation of Ni-P4Mo6/GO
A mixture of Ni-P4Mo6 (0.50 g) and 50 mL deionized water was ultrasonicated for 3 h. Then, GO (0.25 g) and polyethyleneimine (C2H5N) (10 μL) were added and stirred for 30 min. The dispersion was washed with deionized water and centrifuged at 8000 rpm. After vacuum freeze-drying, a black fluffy powder was obtained (yield 40% based on Mo).2.4 Preparation of Co-P4Mo6/GO
Co-P4Mo6/GO was prepared following the same procedure as that of Ni-P4Mo6/GO except that Ni-P4Mo6 (0.50 g) was replaced with Co-P4Mo6 (0.50 g). After vacuum freeze-drying, a black fluffy powder was obtained (yield 40% based on Mo).2.5 Characterization
Fourier transform infrared (FTIR) spectra were recorded using a Bruker Tensor II spectrometer (Bruker, Germany) in the range of 4000–400 cm−1 by a KBr tablet method. X-ray powder diffraction (PXRD) was performed using a Rigaku D/max-TTR-III diffractometer with Cu-Kα-ray irradiation. The scanning speed was 5° s−1 and the measurement range was 5–50°. X-ray photoelectron spectroscopy (XPS) spectra were recorded at room temperature using a 12 kV and 20 mA Al Kα X-ray source by VG ESCALAB MK (VK, UK). The surface structure and morphology of the samples were characterized by transmission electron microscopy (TEM) (JEOL, JEM-2010, 200 kV) and field emission scanning electron microscopy (FE-SEM) (Hitachi, SU 8000). Energy-dispersive spectroscopy (EDS) spectra were recorded by ensemble measurements using an SEM and a TEM. Ultraviolet–visible (UV-vis) spectra were recorded using a Shimadzu UV-1601 spectrometer in the wavelength range of 550–800 nm. Electrochemical tests were performed using a three-electrode system in a CHI 760E electrochemical workstation (Shanghai Chenhua).3. Results and discussion
3.1 Preparation of composite catalyst materials
Ni-P4Mo6/GO and Co-P4Mo6/GO composite catalysts were synthesized for the e-NO3RR (Scheme 1). The composite materials were mainly synthesized in two steps. First, crystal materials Ni-P4Mo6 and Co-P4Mo6 were prepared via a one-pot hydrothermal approach, and they were loaded onto graphene oxide (GO) to obtain Ni-P4Mo6/GO and Co-P4Mo6/GO. | ||
| Scheme 1 Synthetic procedure for Ni-P4Mo6/GO and Co-P4Mo6/GO composites. | ||
3.2 Morphology and structure characterization of samples
The morphology and composition of the two prepared catalysts were analyzed by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The TEM result confirmed that the particles of the complex were distributed on the surface of the lamellar layers (Fig. 1 and Fig. S1†). The SEM images showed that the overall morphologies of Ni-P4Mo6/GO and Co-P4Mo6/GO are similar, both showing the lamellar morphology of GO (Fig. S2a and S3a†). The energy-dispersive X-ray spectroscopy (EDS) spectra of Ni-P4Mo6/GO and Co-P4Mo6/GO (Fig. 1 and Fig. S1–S3†) showed that C, P, Mo, N and Ni/Co elements were present and uniformly distributed on the surface of Ni-P4Mo6/GO or Co-P4Mo6/GO, which indicates that the crystalline materials were successfully loaded onto the surface of GO. | ||
| Fig. 1 (a–b) TEM of Ni-P4Mo6/GO. (c–h) Element mapping of images of Ni-P4Mo6/GO. | ||
The Ni-P4Mo6/GO and Co-P4Mo6/GO powders showed the same characteristic peaks in the FT-IR spectrum (Fig. 2a). The characteristic bands in the range of 594–1080 cm−1 were assigned to the v(Mo–O–Mo), v(Mo–O) and v(P–O) stretching vibrations of {P4Mo6}, respectively. The characteristic peak at 1200–1400 cm−1 was attributed to the v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibration in the ligand. The characteristic absorption peaks at 1500–1600 cm−1 and 1693 cm−1 were attributed to the v(CC) stretching vibration and v(CO) stretching vibration of GO and the ligand in the compound, respectively.
N) stretching vibration in the ligand. The characteristic absorption peaks at 1500–1600 cm−1 and 1693 cm−1 were attributed to the v(CC) stretching vibration and v(CO) stretching vibration of GO and the ligand in the compound, respectively.
 | ||
| Fig. 2 (a) XRD spectra of Ni-P4Mo6/GO and Co-P4Mo6/GO. (b) FTIR spectra of Ni-P4Mo6/GO and Co-P4Mo6/GO. | ||
Single-crystal X-ray diffraction analysis (Table S1†) showed that Ni-P4Mo6 and Co-P4Mo6 were isomorphic crystals. Ni-P4Mo6/GO and Co-P4Mo6/GO were characterized by powder X-ray diffraction (PXRD) (Fig. 2b). The experimental diffraction peak of the composites is consistent with the simulated diffraction peaks of Ni-P4Mo6 and Co-P4Mo6, respectively, and the characteristic peak at 26.18° was attributed to GO, indicating that the crystalline material was successfully compounded with GO. In addition, the elemental composition and valence state of Ni-P4Mo6/GO and Co-P4Mo6/GO composites were measured by X-ray photoelectron spectroscopy (XPS) (Fig. 3 and Fig. S4†). The XPS full spectra (Fig. 3a) confirmed the presence of Ni/Co, Mo, C, P, O, and N in Ni-P4Mo6/GO and Co-P4Mo6/GO, respectively. From the XPS fine spectrum of Ni-P4Mo6/GO (Fig. 3b), it can be seen that in the Ni 2p spectrum, the 856.5 eV and 873.5 eV peaks were attributed to Ni 2p3/2 and Ni 2p1/2, and the 862.5 eV and 880.5 eV characteristic peaks were attributed to Ni 2p3/2 and Ni 2p1/2 satellite peaks,60 indicating that the Ni element in Ni-P4Mo6/GO is Ni2+. The fine spectrum of Mo 3d in Fig. 3c shows that the characteristic peaks at 231.7 eV and 234.7 eV belong to Mo 3d5/2 and Mo 3d1/2, indicating that the Mo element in Ni-P4Mo6/GO is Mo5+.61 The characteristic peaks at 398.3 eV and 399.5 eV in the N 1s spectrum belonged to the imidazole nitrogen in the organic ligand (Fig. 3d). The C 1s spectrum showed three peaks, 284.8, 286.4, and 288.9 eV corresponding to C–C/CC, C–O, and O–CO, respectively (Fig. 3e). The characteristic peaks of P 2p at 132.8 and 134.2 eV were attributed to P 2p3/2 and P 2p5/2 (Fig. 3f).
 | ||
| Fig. 3 (a) XPS survey spectra of Ni-P4Mo6/GO and Co-P4Mo6/GO. High-resolution XPS spectra of (b) Ni 2p, (c) Mo 3d, (d) N 1s, (e) C 1s, and (f) P 2p. | ||
The nitrogen adsorption–desorption isotherms of Ni-P4Mo6/GO, Co-P4Mo6/GO and GO samples were measured (Fig. 4). The results indicate that the specific surface areas of Ni-P4Mo6/GO and Co-P4Mo6/GO are 23.3 m2 g−1 and 18.0 m2 g−1, respectively, which are significantly higher than the specific surface area of GO (1.26 m2 g−1), which proves the successful loading of the sample. Simultaneously, the higher specific surface area obtained after loading can increase the contact area between the catalyst and the electrolyte, which improves the electrocatalytic performance of the material.62
 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms and corresponding pore-size distribution curves of (a) Ni-P4Mo6/GO, (b) Co-P4Mo6/GO, and (c) GO. | ||
3.3 Electrochemical e-NO3RR measurement
The e-NO3RR study was carried out in a typical three-electrode system with an H-type electrolytic cell. Prior to all measurements, high-purity Ar was continuously purged through the cathode compartment at a flow rate of 150 mL min−1 for a minimum of 30 minutes to eliminate N2 from both the electrolyte and the reactor. The measured potentials in this study were calibrated to the RHE scale using the following equation: E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 + 0.059 × pH, and all the potentials were referenced to reversible hydrogen electrode (vs. RHE) potentials. Ammonia production was analyzed by indophenol blue spectrophotometry,63 and by-product nitrite was analyzed by naphthalene ethylenediamine hydrochloride spectrophotometry (Fig. S5–S8†).64 | ||
| Fig. 5 NH3 yield rate and FE of (a) Ni-P4Mo6/GO and (b) Co-P4Mo6/GO. | ||
 | ||
| Fig. 6 (a) LSV curves of Ni-P4Mo6/GO and Co-P4Mo6/GO. NH3 yield and FE of (b) Ni-P4Mo6/GO and (c) Co-P4Mo6/GO. | ||
The best NH3 yield rate and FE of Ni-P4Mo6/GO in electrolyte with different NO3− concentrations are shown in Fig. 7 and Table S2.† At a nitrate concentration of 10 mM, an NH3 yield of Ni-P4Mo6/GO is 1.05 mg h−1 mg−1cat. (−1.2 V vs. RHE) and the FE is only 45.2% (−0.9 V vs. RHE) (Fig. 7a). This may be attributed to the fact that when the concentration of nitrate is too low, the e-NO3RR is inhibited and the performance of the catalyst is reduced.
 | ||
| Fig. 7 (a–g) NH3 yield rate and FE of Ni-P4Mo6/GO with different NO3− concentrations. (h) FE corresponding to the optimal voltage of Ni-P4Mo6/GO at each concentration. | ||
Then, the electrocatalytic test of Ni-P4Mo6/GO was further compared in an electrolyte containing 0.1 to 0.6 M of nitrate (Fig. 7b–g). It is important to highlight that as the nitrate concentration increases, the ammonia production rate and FE of Ni-P4Mo6/GO show a trend of gradually increasing first (0.1 M to 0.5 M) and then decreasing (0.6 M). When the nitrate concentration is 0.5 M, the highest NH3 yield is 11.6 mg h−1 mg−1cat. (−1.3 V vs. RHE), and the best FE is 88.4% (−1.1 V vs. RHE), which is at a high level in the current reported electrocatalytic nitrate reduction yield (Table S3†). When the nitrate concentration is 0.6 M, ammonia production and FE all decrease (Fig. 7g), because the concentration of NO3− in the solution continues to increase, the current density increases, but the adsorption ability of Ni-P4Mo6/GO for NO3− tends to saturate. Meanwhile, the transfer of hydrogen is somewhat limited, resulting in a decrease in NH3 production and FE.66
For comparison, the e-NO3RR tests of Co-P4Mo6/GO and GO were also conducted in 0.5 M NaNO3/Na2SO4 electrolyte (Fig. 8). The results indicate that Co-P4Mo6/GO also has positive e-NO3RR performance (the NH3 yield of 11.1 mg h−1 mg−1cat. (−1.1 V vs. RHE) and FE of 78.5% (−1.0 V vs. RHE)) (Fig. 8c). The NH3 yield of 0.55 mg h−1 mg−1cat. (−1.1 V vs. RHE) and the FE of 39.2% (−0.9 V vs. RHE) are obtained for GO, which exhibits almost minor e-NO3RR activity compared with the composite (Fig. 8f).
 | ||
| Fig. 8 (a) Chronoamperometric (j–t) curves of Co-P4Mo6/GO. (b) UV-vis absorption spectra of Co-P4Mo6/GO. (c) NH3 yield rate and FE of Co-P4Mo6/GO. (d) Corresponding chronoamperometric (j–t) curves of GO. (e) UV-vis absorption spectra of GO. (f) NH3 yield rate and FE of GO. | ||
Since the e-NO3RR to NH3 is an intricate process requiring an eight-electron transfer, the by-products include NO2−, which is also studied by hydrochloric acid naphthalene ethylenediamine spectroscopy analysis (Fig. S12†). We observe that the FE values for NO2− are below 10% (Fig. 9a), with the maximum yield reaching only 0.3 mg h−1 mg−1cat., suggesting that Ni-P4Mo6/GO demonstrates strong selectivity during the nitrate-to-ammonia conversion process.
 | ||
| Fig. 9 (a) NO2− yield rate and FE of Ni-P4Mo6/GO. (b) Electrochemical impedance spectra of Ni-P4Mo6/GO, Co-P4Mo6/GO and GO. | ||
Electrochemical impedance spectroscopy (EIS) is widely acknowledged as an effective approach for investigating catalytic kinetics. The Nyquist plots67 and Bode plots of Ni-P4Mo6/GO, Co-P4Mo6/GO and GO were performed (Fig. 9b and Fig. S13†). From the Nyquist plots, the linear slope of GO itself is the smallest, indicating that the diffusion resistance of GO is large, which is not conducive to the e-NO3RR dynamic process. After the crystal material was loaded, the slope increases and the diffusion resistance decreases, which is consistent with the high e-NO3RR performance of Ni-P4Mo6/GO and Co-P4Mo6/GO. Ni-P4Mo6/GO demonstrates minimal charge transfer resistance and optimal electron transport efficiency, confirming that the Faraday process between it and the electrolyte interface was faster. The Cdl value of Ni-P4Mo6/GO is 3.7 mF cm−2, and the Cdl value of Co-P4Mo6/GO is 3.1 mF cm−2, indicating that Ni-P4Mo6/GO has better electrochemical performance (Fig. S14†).
Good stability is essential for catalysts. After 24 hours of continuous electrolysis, the XRD pattern, FTIR spectrum and XPS spectra of the Ni-P4Mo6/GO composite showed that the structure of the material did not change (Fig. 10a, b and Fig. S15†). This reveals that the material structure did not change. Under the optimal conditions for ammonia synthesis (−1.1 V vs. RHE, 0.5 M NaNO3 + 0.1 M Na2SO4 electrolyte), the current density of the catalyst remained basically unchanged during continuous electrolysis for 24 h (Fig. 10). The NH3 yield and FE of Ni-P4Mo6/GO changed in a small range after 24 cycles of electrocatalytic tests under the same conditions, which indicates that the catalyst Ni-P4Mo6/GO exhibits good stability.
 | ||
| Fig. 10 (a) Comparison of the XRD patterns of Ni-P4Mo6/GO after continuous electrolysis for 24 hours. (b) FT-IR spectra of Ni-P4Mo6/GO before and after continuous electrolysis for 24 hours. (c ) Corresponding chronoamperometric (j–t) curves for 24 h continuous electrolysis at −1.1 V vs. RHE and NH3 yield rate and FE with different cycles. | ||
A set of control experiments were conducted to verify the origin of the produced NH3: (1) Ni-P4Mo6/GO served as the working electrode in an Ar-saturated blank solution lacking NaNO3, and can hardly detect ammonia after electrolysis (Fig. 11a); (2) using bare carbon cloth as the working electrode, very little ammonia was produced after electrolysis for 1 h in a 0.1 M Na2SO4 solution with NaNO3. To verify the origin of NH3, N-isotope labeling experiments were conducted. When Na14NO3 served as the nitrogen source, the 1H NMR spectrum of the electrolyte exhibited three distinct characteristic peaks of 14NH4+ following the e-NO3RR test. When Na15NO3 was used as the nitrogen source, the 1H NMR spectrum of the electrolyte after the electrocatalytic test showed two characteristic peaks of 15NH4+ (Fig. 11b).68 The above-mentioned findings provide strong evidence that all the ammonia is derived from the conversion of NO3− on Ni-P4Mo6/GO.
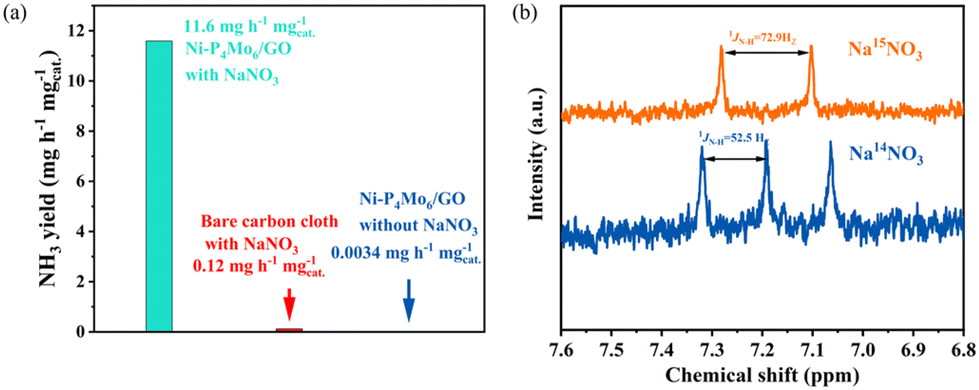 | ||
| Fig. 11 (a) NH3 yield rate of Ni-P4Mo6/GO in 0.1 M Na2SO4 with or without NaNO3, respectively, and blank carbon paper in 0.1 M Na2SO4 solution with NaNO3. (b) 1H NMR spectra of the electrolyte after 1 h of NO3RR testing at −1.1 V vs. RHE with 14NO3− and 15NO3− as the N source, respectively. | ||
4. Conclusions
In this work, two crystalline materials were synthesized using a one-pot mode and loaded with GO to obtain two composites (Ni-P4Mo6/GO and Co-P4Mo6/GO). The systematic optimization of electrolyte composition and nitrate feed concentrations was conducted to elucidate their synergistic influence on electrocatalytic activity, selectivity, and stability metrics in the nitrate reduction reaction. In the neutral electrolyte, the catalytic activity of Ni-P4Mo6/GO and Co-P4Mo6/GO was significantly improved and superior to some similar catalysts owing to the combined influence of the electron-donating capacity of the {P4Mo6} unit, good catalytic activity of Ni/Co, and the excellent charge transport ability of the graphene substrate. It is worth noting that the NH3 yield (11.6 mg h−1 mg−1cat.) and FE (88.4%) of Ni-P4Mo6/GO are higher than those of Co-P4Mo6/GO (NH3 yield of 11.1 mg h−1 mg−1cat., FE of 78.5%). This work establishes a transformative design paradigm for polyoxometalate-derived electrocatalysts, offering mechanistic insights and synthetic protocols that advance the frontier of e-NO3RR technologies.Author contributions
Nan Zhao: data curation, formal analysis, investigation methodology, software, writing – original draft. Xinming Wang: conceptualization, data curation, formal analysis, funding acquisition, methodology, resources, supervision, writing – review & editing. Shuang Rong: software, validation. Qiushuang Jiang: conceptualization, methodology. Haoyun Li: data curation, formal analysis. Haijun Pang: funding acquisition, software, validation. Huiyuan Ma: conceptualization, data curation, formal analysis, methodology, resources, supervision.Data availability
All relevant data are available within the manuscript and its additional files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171059), the ZR2022MB096 project funded by the Shandong Provincial Natural Science Foundation, and Heilongjiang Provincial Natural Science Foundation of China (LH2024B015).References
- F. Lü, S. Zhao, R. Guo, J. He, X. Peng, H. Bao, J. Fu, L. Han, G. Qi, J. Luo, X. Tang and X. Liu, Nano Energy, 2019, 61, 420–427 Search PubMed.
- Z. Mo, J. Mu and B. Liu, J. Electroanal. Chem., 2024, 969, 118533 CrossRef CAS.
- H. Wang, J. Huang, J. Cai, Y. Wei, A. Cao, B. Liu and S. Lu, Small Methods, 2023, 7, 00169 Search PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- S. Feng, W. Gao, H. Cao, J. Guo and P. Chen, Acta Chim. Sin., 2020, 78, 916 CrossRef CAS.
- Z. Ni, N. Liu, C. Zhao and L. Mi, Polyoxometalates, 2024, 3, 9140044 CrossRef.
- Z. Fang, P. Wu, Y. Qian and G. Yu, Angew. Chem., Int. Ed., 2020, 60, 4275–4281 CrossRef PubMed.
- Q. Song, M. Li, J. Li, S. Zhang, L. Yang, X. Chen, F. Meng and X. Liu, Chem. Eng. J., 2022, 430, 133152 CrossRef CAS.
- J. Zheng, Y. Lyu, M. Qiao, R. Wang, Y. Zhou, H. Li, C. Chen, Y. Li, H. Zhou, S. P. Jiang and S. Wang, Chem, 2019, 5, 617–633 CAS.
- X. Wang, M. Yang, C. J. Gómez-García, X. Cao, Z. Jin, H. Ma, H. Pang and G. Yang, ACS Sustainable Chem. Eng., 2025, 13, 1708–1718 CrossRef CAS.
- S. P. Wallbridge, K. Lawson, A. E. Catling, C. A. Kirk and S. E. Dann, Dalton Trans., 2022, 51, 18010–18023 RSC.
- G. Duan, Y. Chen, Y. Tang, K. A. M. Gasem, P. Wan, D. Ding and M. Fan, Prog. Energy Combust. Sci., 2020, 81, 100860 Search PubMed.
- H. Jiang, T. Li, Y. Gao, J. Fan, D. Gan, S. Yuan, L. Hong, Y. Feng, J. Sun, Q. Song, T. Zhang, A. R. Jalili, P. J. Cullen and R. Zhou, J. Energy Chem., 2025, 105, 630–668 CrossRef CAS.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 Search PubMed.
- A. Ma, J. Gui, Y. Huang and Y. Yu, Nano Res., 2024, 17, 7824–7829 CrossRef CAS.
- Y. Zhang, J. Xiong, B. Liu and S. Yan, Cell Rep. Phys. Sci., 2024, 5, 101994 Search PubMed.
- J.-Y. Fang, J.-L. Fan, S.-B. Liu, S.-P. Sun and Y.-Y. Lou, Materials, 2023, 16, 4000 Search PubMed.
- Q. Zhou, X. Wang, S. Rong, G. Li, Q. Jiang, H. Pang and H. Ma, Inorg. Chem., 2025, 64, 5291–5301 CrossRef CAS PubMed.
- X. Lu, J. Yu, J. Cai, Q. Zhang, S. Yang, L. Gu, G. I. N. Waterhouse, S.-Q. Zang, B. Yang and S. Lu, Cell Rep. Phys. Sci., 2022, 3, 100961 Search PubMed.
- Y. Xu, Y. Sheng, M. Wang, T. Liu, H. Yu, K. Deng, Z. Wang, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 16290–16296 RSC.
- K. Wu, C. Sun, Z. Wang, Q. Song, X. Bai, X. Yu, Q. Li, Z. Wang, H. Zhang, J. Zhang, X. Tong, Y. Liang, A. Khosla and Z. Zhao, ACS Mater. Lett., 2022, 4, 650–656 CrossRef CAS.
- J. Yan, P. Liu, J. Li, H. Huang, S. Tong and W. Song, Chem. Eng. J., 2024, 498, 155108 CrossRef CAS.
- M. Wang, T. Hu, C. Wang, F. Du, H. Yang, W. Sun, C. Guo and C. M. Li, Sci. China Mater., 2023, 66, 2750–2758 CrossRef CAS.
- D. Hao, Z.-g. Chen, M. Figiela, I. Stepniak, W. Wei and B.-J. Ni, J. Mater. Sci. Technol., 2021, 77, 163–168 CrossRef CAS.
- Y. Xu, M. Wang, K. Ren, T. Ren, M. Liu, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2021, 9, 16411–16417 Search PubMed.
- Z. A. Jonoush, A. Rezaee and A. Ghaffarinejad, J. Cleaner Prod., 2020, 242, 118569 CrossRef CAS.
- M. Teng, J. Yuan, Y. Li, C. Shi, Z. Xu, C. Ma, L. Yang, C. Zhang, J. Gao and Y. Li, J. Colloid Interface Sci., 2024, 654, 348–355 CrossRef CAS PubMed.
- E. Lacasa, P. Cañizares, J. Llanos and M. A. Rodrigo, J. Hazard. Mater., 2012, 213–214, 478–484 CrossRef CAS PubMed.
- T. Zhang, S. Liu, F. Wang, W. Liu, X. He, Q. Liu, X. Zhang and X. Liu, Microstructures, 2024, 4, 2024043 CAS.
- J. Wang, T. Feng, J. Chen, V. Ramalingam, Z. Li, D. M. Kabtamu, J.-H. He and X. Fang, Nano Energy, 2021, 86, 106088 CrossRef CAS.
- M. Jiang, J. Su, X. Song, P. Zhang, M. Zhu, L. Qin, Z. Tie, J.-L. Zuo and Z. Jin, Nano Lett., 2022, 22, 2529–2537 CrossRef CAS PubMed.
- H.-Y. Kwon, S. E. Braley, J. P. Madriaga, J. M. Smith and E. Jakubikova, Dalton Trans., 2021, 50, 12324–12331 RSC.
- S. Li, Z. Zhao, T. Ma, P. Pachfule and A. Thomas, Angew. Chem., Int. Ed., 2021, 61, e202112298 CrossRef PubMed.
- S. Li, C. Cheng, A. Sagaltchik, P. Pachfule, C. Zhao and A. Thomas, Adv. Funct. Mater., 2018, 29, 1807419 CrossRef.
- C.-Y. Sun, W. Li, K. Wang, W.-J. Zhou and H.-Q. Wang, Rare Met., 2024, 43, 1845–1866 Search PubMed.
- L. Wang, M. Li, X. Liu, C. Feng, N. Chen, X. Ma and G. Ding, J. Electrochem. Soc., 2017, 164, E326–E331 CrossRef CAS.
- D. He, Y. Li, H. Ooka, Y. K. Go, F. Jin, S. H. Kim and R. Nakamura, J. Am. Chem. Soc., 2018, 140, 2012–2015 CrossRef CAS PubMed.
- D. Zhao, J. Yao, C. Ma, A. Wang, H. Xie, J. Zhao, J. Yan, K. Zhu, Y. Zhu, D. Cao and G. Wang, J. Power Sources, 2024, 592, 233945 CrossRef CAS.
- H. Li, M. Tu, Y. Fang, T. Hao and B. Wang, ACS Appl. Nano Mater., 2023, 6, 18238–18246 CrossRef CAS.
- Y.-T. Xu, Y. Han, D. K. Sam and Y. Cao, J. Mater. Chem. A, 2022, 10, 22390–22398 Search PubMed.
- W. Zheng, L. Zhu, Z. Yan, Z. Lin, Z. Lei, Y. Zhang, H. Xu, Z. Dang, C. Wei and C. Feng, Environ. Sci. Technol., 2021, 55, 13231–13243 Search PubMed.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 018–0112 Search PubMed.
- M. Yang, X. Wang, C. J. Gómez-García, Z. Jin, J. Xin, X. Cao, H. Ma, H. Pang, L. Tan, G. Yang and Y. Kan, Adv. Funct. Mater., 2023, 33, 2214495 Search PubMed.
- M. R. Horn, A. Singh, S. Alomari, S. Goberna-Ferrón, R. Benages-Vilau, N. Chodankar, N. Motta, K. Ostrikov, J. MacLeod, P. Sonar, P. Gomez-Romero and D. Dubal, Energy Environ. Sci., 2021, 14, 1652–1700 RSC.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 Search PubMed.
- G. Yang, H. Liu, S. Zhang, Y. Yang, K. Li, H. Li, Y. Liu and H. Zang, Inorg. Chem., 2024, 63, 22955–22961 Search PubMed.
- G. Yang, J. Cao, Y. Xiao, Q. Hu, H. Liu, X. Cao, K. Li, Y. Zhang and X. Li, Inorg. Chem., 2025, 64, 1041–1047 CrossRef CAS PubMed.
- J.-X. Liu, X.-B. Zhang, Y.-L. Li, S.-L. Huang and G.-Y. Yang, Coord. Chem. Rev., 2020, 414, 213260 CrossRef CAS.
- Q. Hu, K. Li, X. Chen, Y. Liu and G. Yang, Polyoxometalates, 2024, 3, 9140048 CrossRef.
- L. Guo, L. He, Q. Zhuang, B. Li, C. Wang, Y. Lv, J. Chu and Y. F. Song, Small, 2023, 19, 07315 Search PubMed.
- M. Khalaji-Verjani and M. Masteri-Farahani, ACS Appl. Energy Mater., 2024, 7, 6612–6620 CrossRef CAS.
- L. Yang, Z. Yuan, L. He, L. Han, B. Li and Y. Xu, Inorg. Chem., 2024, 63, 12564–12571 CrossRef CAS PubMed.
- H.-L. Guo, X.-X. Xing, S.-X. Mao, T. Feng, Y.-H. Fan, Z.-J. Qin, J.-Y. Pang, Y. Bai and D.-B. Dang, Dalton Trans., 2022, 51, 18090–18098 RSC.
- J.-Q. Niu, W.-T. An, X.-J. Zhang, Y.-Y. Ma and Z.-G. Han, Chem. Eng. J., 2021, 418, 129408 CrossRef CAS.
- H.-X. Bi, M.-S. Guo, J. Du, Y.-Y. Ma and Z.-G. Han, Coord. Chem. Rev., 2024, 518, 216092 CrossRef CAS.
- J. Wang, S. Wang, Z. Zhang and C. Wang, J. Environ. Manage., 2020, 276, 111357 CrossRef CAS PubMed.
- X. Ma, M. Li, X. Liu, L. Wang, N. Chen, J. Li and C. Feng, Chem. Eng. J., 2018, 348, 171–179 CrossRef CAS.
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- N. M. Mahmoodi, M. Oveisi and E. Asadi, J. Cleaner Prod., 2019, 211, 198–212 CrossRef CAS.
- Q. Sheng, Y. Wang, P. Zhang, Y. Jiang, J. Zhang, Q. Liu, Q. Chang, Y. Yao, X. Liao, Z. Li, S. Lyu and Y. Qin, Appl. Surf. Sci., 2024, 666, 160369 CrossRef CAS.
- Y.-Q. Zhang, L.-Y. Zhou, Y.-Y. Ma, K. Dastafkan, C. Zhao, L.-Z. Wang and Z.-G. Han, Chem. Sci., 2021, 12, 1886–1890 Search PubMed.
- Y. Hou, D. Chai, B. Li, H. Pang, H. Ma, X. Wang and L. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 20845–20853 CrossRef CAS PubMed.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Sci., 2019, 6, 02109 Search PubMed.
- Q.-H. Wang, L.-J. Yu, Y. Liu, L. Lin, R.-g. Lu, J.-p. Zhu, L. He and Z.-L. Lu, Talanta, 2017, 165, 709–720 CrossRef CAS PubMed.
- L. Bai, F. Franco, J. Timoshenko, C. Rettenmaier, F. Scholten, H. S. Jeon, A. Yoon, M. Rüscher, A. Herzog, F. T. Haase, S. Kühl, S. W. Chee, A. Bergmann and R. C. Beatriz, J. Am. Chem. Soc., 2024, 146, 9665–9678 CrossRef CAS PubMed.
- Q. Hu, K. Yang, O. Peng, M. Li, L. Ma, S. Huang, Y. Du, Z.-X. Xu, Q. Wang, Z. Chen, M. Yang and K. P. Loh, J. Am. Chem. Soc., 2023, 146, 668–676 CrossRef PubMed.
- Z. Feng, G. Li, X. Wang, C. J. Gómez-García, J. Xin, H. Ma, H. Pang and K. Gao, Chem. Eng. J., 2022, 445, 136797 CrossRef CAS.
- K. Kamiya, K. Hashimoto and S. Nakanishi, ChemElectroChem, 2014, 1, 858–862 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00597c |
| This journal is © The Royal Society of Chemistry 2025 |