
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interpreting solvent effects on complex dihydrogenphosphate binding using amidobenzimidazole functionalised emissive ruthenium diimine complexes†
Andrew J.
Stocker
 ,
Chloe L.
Howells
and
Nicholas C.
Fletcher
*
,
Chloe L.
Howells
and
Nicholas C.
Fletcher
*
Department of Chemistry. Lancaster University, Bailrigg, Lancaster LA1 4YB, UK. E-mail: n.fletcher@lancaster.ac.uk
First published on 16th May 2025
Abstract
The photophysical behaviour of two amidobenzimidazole complexes, [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 (where bbiab = 4,4′-bis(N-1H-benzimidazol-2-yl-carboxamidyl)-2,2′-bipyridine) in a variety of solvents suggest that the 3MLCT state is sensitive to the introduction of small aliquots of solvent (dilution) resulting in distinct incremental changes to the absorption spectra, and quenching in emission. This solvent dependency is more pronounced in polar media and is proposed to be a consequence of “dimerization” of these complexes, which is supported by 1H-DOSY-NMR spectroscopy and gas phase DFT studies on the free ligands. The pH behaviour of these complexes indicates that protonation of the imidazole results in changing the emissive state from the bpy and phen ligands to bbiab. Significant quenching in both complexes is seen on the introduction of acetate, which 1H-NMR spectroscopy suggests is association with the bbiab imidazole groups. On the introduction of HSO4− and H2PO4− salts, the photophysical response suggests several distinct interactions are present as anion concentration increases, with both the amide and imidazole groups able to act as hydrogen-bond donors and acceptors. These effects are solvent dependent implying a complex set of equilibria, ranging from protonation of the imidazole in non-protic solvents, dissociation of the complex dimer, a simple one-to-one species, and a combined bis(dihydrogenphosphate)-complex anion, with speciation reliant on complex charge, associated ligand hydrophobicity, and solvent.
Introduction
The recognition and speciation of biologically relevant phosphate-based anionic species, such as orthophosphate, pyrophosphate and phosphonucleotides, remain critical areas of medicinal, industrial and environmental significance. Phosphate anions are implicated in enzymatic dysregulation,1 cancers,2 and several other medical conditions including arthritis.3 And it plays a ubiquitous role in industrial and agricultural wastewater pollution.4 Phosphate anions are characterized by a high hydration enthalpy (ΔGhyd of H2PO4− approx. −465 kJ mol−1),5 a tetrahedral geometry and a variable protonation state that is dependent on pH. At pH 7, phosphate exists in an equilibrium between the mono- and dihydrogen phosphate species, each with distinct charge states. At higher concentrations, phosphates can form oligomeric species, aided by the formation of anti-electrostatic hydrogen bonds, further complicating speciation.6 The high hydration enthalpy also presents a significant challenge in sensing applications, where the solvent participation can determine the host–guest equilibria, which results in a lower enthalpic gain than predicted in solvent-free environments, such as DFT calculations.7 In biological media, extensive hydrogen bonding occurs between the solvent, the host and guest molecules, leading to competition between the solvent and the guest for the binding cavity, assuming that other non-covalent interactions are involved.8 This competition can extend to host–host dimers, oligomers or self-assembling macromolecules. Consequently, binding interactions in polar aqueous media may differ considerably from those in non-competitive or aprotic solvents.9To study binding modes and establish stability constants, non-competitive, or weakly competitive solvents such as acetonitrile (ACN) or dichloromethane (DCM) have been routinely used. But these conditions do not reflect a complex environmental or biological system, and tends towards an artificial binding strength, with the solvents being incapable of participating in non-covalent interactions. As a more biologically relevant alternative, solvent mixtures like aquated DMSO are increasingly being employed, offering closer chemical similarities to water, while maintaining good solvation.10 Yet even this remains an imperfect solution. Additionally, solvatochromism or solvent-induced fluorescence quenching is frequently observed in solvents such as water or methanol which can complicate the results of a photophysical study.11 The choice of solvent then becomes crucial when studying emissive complexes, and when trying to interpret anion binding modes, particularly in a biological context.12
Amide-based anion receptors have been at the fore in anion recognition. Pascal prepared the first in 1986,13 followed closely by Beer14 and Reinhoudt15 in 1993, Raposo in 1995,16 Beer again17 and then Anslyn in 199718 who facilitated the partial encapsulation of tetrahedral anionic species.19 These studies show that amide groups exhibit a strong tendency to bind anions20 including tetrahedral oxoanions via strong directional hydrogen bonds. And the partially positive charge of the amide NH group may enhance anion association through electrostatic interactions.21 Introducing an amide group to a ligand system is synthetically straightforward, increases conformational flexibility and rotational freedom, allowing for optimal hydrogen bond alignments that promote stronger anion binding.22 This also facilitates better encapsulation of the tetrahedral geometry, potentially tuning the sensors selectivity towards phosphate over competing anions, such as acetate, nitrate and sulfate.19,22,23 More recent work has explored the use of amide functionalities for the detection of acetate, carboxylates, and halides,20 yet there has been little focus on its application for the speciation of, and between, phosphates in an aqueous biological environment.12b,24
Recently we reported a series of photo-emissive complexes containing 4,4′-bis(benzimidazol-2-yl)-2,2′-bipyridine (bbib),25 based on the premise that incorporation of both a hydrogen donor, and an acceptor group may permit speciation of protonated oxo-anions, such as phosphate in a pH range of 4 to 9. The use of imidazole functions for anion recognition is well documented,26 but has given limited selectivity. Our initial ideas proposed using a rigid “cleft”, which proved to interact with acetate, in a typical one-to-one fashion, and more effectively with dihydrogenphosphate, with a high degree of selectivity over a range of other common anions in 10% aquated DMSO. The interaction with dihydrogenphosphate, is however complicated with several different binding modes being identified. Subtle changes in the protic environment can disturb the equilibria, and potentially the dominant species, which appear to include a dimeric two complex to one guest, the anticipated one-to-one species, the “anti-electrostatic” one-to-two form, and a surprising two-to-two dimer. Here we explore these ideas in more depth, by the inclusion of amidic linkages to the benzimidazole complexes, in species reminiscent of the early work by Beer,27 illustrating the solvent impact, the water solubility and the selectivity for phosphate salts over other commonly encountered environmental and biologically relevant anions.
Results and discussion
Synthesis
A series of emissive ruthenium complexes were prepared using standard literature procedures. 4,4′-Dicarboxylic acid-2,2′-bipyridine was converted to the diacyl chloride, followed by condensation with 2-aminobenzimidazole. Precipitation in basic aqueous solution resulted in 4,4′-bis(N-1H-benzimidazol-2-yl-carboxamidyl)-2,2′-bipyridine (bbiab) as a pale brown amorphous solid in 52% yield (Scheme 1). The product was characterized by 1H-NMR spectroscopy (Fig. S1†) and mass spectrometry. The 1H-NMR signal attributed to the H3 on the pyridine ring is in a significantly downfield position at 9.17 ppm presumably arising from the adjacent amido functionality forming a weak hydrogen bond, and the proximity to the imidazole unit. The product was very insoluble in most common solvents, including DMSO preventing analysis by 13C-NMR spectroscopy. | ||
| Scheme 1 The synthesis of complexes used in this study, conditions (a) (i) SOCl2, (ii) 2-amino-benzimidazole, NEt3, toluene, (b) (i) [Ru(bpy)Cl2] or [Ru(phen)Cl2], CF3SO3H, in ethylene glycol, (ii) KPF6 (aq). | ||
The ligand bbiab was coordinated to ruthenium(II) by heating it with either [Ru(bpy)2Cl2] or [Ru(phen)2Cl2] in ethylene glycol with a small quantity of triflic acid to improve ligand solubility. The products [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 were realised as dark red solids following precipitation from aqueous solution as hexafluorophosphate salts and subsequently purified using size exclusion chromatography (Sephadex® LH20) in 14 and 23% yield, respectively. The two products were characterized by 1H-NMR spectroscopy (Fig. S2 and S3†) and fully assigned via COSY and NOESY techniques. Consistent with the bbiab ligand, the signal arising from the H3 proton adjacent to the imidazole function is observed to be significantly downfield at 9.3 ppm, and like the NH signal (typically at 12 to 14 ppm) was very susceptible to the presence of base.28 There is a notable broadening of the aromatic signals related to the benzimidazole function. The identity was further confirmed by mass spectroscopy, with the two products identified from corresponding divalent cation less both associated anions. Despite repeated attempts, crystals suitable for X-ray crystallographic analysis were elusive, potentially due to irregular solvation and unusual packing configurations which appear to be extremely solvent dependant as discussed subsequently.
Complex photophysical characterisation
The absorbance spectra of [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 (Fig. 1, Table 1) consist of a series of well-defined bands in the range 200–520 nm with considerable similarity to the previously reported analogous bbib complexes.25 As anticipated [Ru(bpy)2(bbiab)](PF6)2 has a strong absorption band at 288 nm consistent with ancillary bpy π–π* transition present in [Ru(bpy)3]2+. But in addition, there is a significant shoulder at 305 nm assigned to bbiab π–π*, and a broad band at 361 nm which appear to be benzimidazole transitions. The metal-to-ligand-charge-transfer (MLCT) transitions (410 to 520 nm) present as a very broad band and are attributed to a complex manifold of energy transitions including metal 4d to both πbpy* (around 450 nm), and πbbiab* (490 nm). These assignments were confirmed by DFT optimization of the complex showing that the LUMO is located on the bpy-amide moiety, 0.172 eV below LUMO+1 on the ancillary bpy ligands (Fig. S4†). It is noteworthy that the HOMO to HOMO−6 are localised in the π orbitals on the benzimidazole function giving a strong indication of the electron rich nature of this group. Similarly, [Ru(phen)2(bbiab)](PF6)2 also exhibits the anticipated phen π–π* transition at 263 nm, two resolved absorptions at absorptions at 292 and 306 nm and a broad band at 369 nm assumed to be related to bbiab π–π* transitions, in addition to the MLCT from 410 to 520 nm. And similarly, DFT optimization again highlights that the LUMO is located on the bpy-amide moiety, 0.126 eV below the phen centred LUMO+1 (Fig. S5†). Both [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 proved to be remarkably emissive, having higher quantum yields than [Ru(bpy)3](PF6)2. | ||
| Fig. 1 Photophysical characterization of [Ru(bpy)2(bbiab)](PF6)2 (blue) [Ru(phen)2(bbiab)](PF6)2 (orange) compared to [Ru(bpy)3](PF6)2 (dashed) (a) UV-Vis spectra under an ambient atmosphere in acetonitrile at 298 K and a concentration of 1 × 10−5 M; (b) the emission normalized with an absorption of 0.1 at excited 450 nm. | ||
| Absorptiona | Emissiona | |||
|---|---|---|---|---|
| λ max | (ε × 105) | λ max |
Φ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
|
| Complex | ±2 nm | dm−3 mol−1 cm−1 | ±2 nm | |
| a Recorded under an ambient atmosphere in acetonitrile at 298 K, excited at 450 nm. b Emission quantum yields (Φem) were calculated relative to emission quantum yield calculated relative to [Ru(bpy)3](PF6)2 (Φem = 0.040) in acetonitrile.29 | ||||
| [Ru(bpy)2(bbiab)](PF6)2 | 230 | (439) | 639 | 0.058 |
| 254 | (254) | |||
| 288 | (582) | |||
| 305 | (362) | |||
| 361 | (253) | |||
| 469 | (179) | |||
| [Ru(phen)2(bbiab)](PF6)2 | 225 | (790) | 634 | 0.074 |
| 263 | (728) | |||
| 292 | (297) | |||
| 309 | (299) | |||
| 369 | (258) | |||
| 441 | (184) | |||
| 469 | (202) | |||
Complex pH behaviour
To consider the availability of the benzimidazole group to act as both hydrogen donor and acceptor, the photophysical behaviour of both [Ru(bpy)2(bbiab)]2+ and [Ru(phen)2(bbiab)]2+, was considered over a pH range from 2 to 12 using an aqueous Britton–Robinson buffer and adjusted by addition of an aqueous NaOH solution (Fig. S6–S9†). The UV-Vis absorbance spectra for both complexes demonstrate a small variation in the pH over a broad range from pH 7 to 12, and above this there is a drop in absorption in the region of 320 to 380 nm, confirming the assignment that this region is associated with benzimidazole groups, and presumably relates to the sequential deprotonation of the two imidazole groups. Subtle changes are also observed in the MLCT region, with the peak at 450 nm becoming more pronounced. The ground state DFT studies suggests this can be related to increasing the energy level of the πbpy-bbiab* orbital to LUMO+2, lying higher than the ancillary πbpy* (Fig. S10†). Upon protonation, as the pH changes from 7 to 2.5, there are more significant changes in these two identified regions, most noticeably below a pH of 4. In the 480 to 520 nm region there is a redshift in the MLCT metal 4d to πbpy-bbiab* absorbance. Again, DFT studies suggests that this is related to a drop in the πbpy-bbiab* LUMO, which upon protonation lies 1.58 eV below the ancillary πbpy* orbital (Fig. S11†).With [Ru(bpy)2(bbiab)]2+ there is very little variation in both the quantum yield, and the emissive wavelength, under basic conditions, suggesting that the complex is not readily deprotonated, but the primary emission in basic media (the 3MLCT) is located on the ancillary πbpy*. Upon protonation though, there is a significant red shift, alongside a quenching in the emission, which suggests that the long-lived emissive state switches to a πbpy-bbiab* orbital. These effects are more pronounced with [Ru(phen)2(bbiab)]2+, where in addition to the notable effects seen on protonation, in basic media, there is a significant quenching observed, which cannot readily be explained, but may be related to the dimerization ideas discussed subsequently.
The precise determination of the pKA and pKB was not easily undertaken given that there are evidently the sequential removal and addition of protons to the two imidazole units over a wide pH range. But it is noteworthy that the points of inflection that relate to the protonation are estimated to be in very different places when determined through the ground state absorption (∼pH 2.5), and the excited state emission (∼pH 4.0). This provides further evidence that the imidazole function is involved in the MLCT process. However, the control of the pH requires the presence of a Britton–Robinson buffer, which contains a cocktail of anions including borate, acetate and phosphate. So it is possible that the observed changes, whilst related to the evident changes in pH, are not independent of the presence of these anions as discussed subsequently.
Solvent dependent behaviour
To ascertain the dilution effect in the subsequent anions studies, the addition of small aliquots of solvent to samples of both [Ru(bpy)2(bbiab)]2+ and [Ru(phen)2(bbiab)]2+ resulted in a marked perturbation to both absorption and emission spectra (Fig. 2). This is not witnessed when performed with the previously reported [Ru(phen)2(bbib)](PF6)2 (Fig. S12a†) under identical conditions. A decrease in absorption intensity from 290–410 nm (bpy) and 260–405 nm (phen) was observed, with a significantly larger decrease at 310 nm and 363 nm corresponding to bbiab π–π* and bpy/phen π–π* transitions respectively. A small increase in the 410–540 nm (bpy) and 405–540 nm (phen) MLCT d–π* transition can also be seen, which are more pronounced with the phen co-ligands. This was also evident with a remarkable quenching in emission (48% for [Ru(bpy)2(bbiab)]2+ and 59% for [Ru(phen)2(bbiab)]2+). This decrease is far too large to be ascribed purely to dilution and it does not follow a Beer–Lambert relationship. It was also noted that the absorbance changes in the UV-Vis regions are primarily associated with the bbiab functionality, suggesting specific chemical interactions are taking place whereby the dilution is changing the local arrangement around this extended functionality. The same experiment was repeated with a vacuum dried sample of [Ru(phen)2(bbiab)](PF6)2 dissolved in, and subsequently diluted in, SPS dried acetonitrile. This resulted in only a 15% loss in emission intensity (Fig. S12c†) suggesting that the presence of stoichiometric amounts of water in the HPLC grade solvent is probably responsible for the observed quenching. | ||
| Fig. 2 The UV-Vis absorption spectra (a) and emission spectra (b) excited at 450 nm, at 298 K of [Ru(phen)2(bbiab)](PF6)2 (4.61 μM), showing the dilution by addition of solvent (up to 500 μL) by 10 μL increments to solutions under an ambient atmosphere in acetonitrile. | ||
To understand the impact of the solvent, a series of ‘blank’ UV-Vis and emission dilution titrations were subsequently performed with both [Ru(phen)2(bbiab)](PF6)2 and [Ru(bpy)2(bbiab)](PF6)2 in DMSO, 10% aquated DMSO (Fig. 3), acetone, methanol and dichloromethane (Fig. S13†). As with HPLC grade acetonitrile, a considerable quenching in emission was observed with DMSO (33% and 56% respectively), and even with 10% aquated DMSO (34% and 31% respectively, Fig. 3). DMSO has a similar polarity to acetonitrile, so they may support the formation of hydrogen bonds with the amido-imidazole groups, allowing for an enhanced ability to stabilize ionic species in solution. An increase in water concentration introduces competitive hydrogen bonds, which may disrupt the assumed structure of the complexes. If water content is important, then it would be reasonable to observe a smaller overall emissive decrease in aquated DMSO compared to DMSO and acetonitrile. No perturbation of UV-Vis spectra, and very little peak movement was observed in the emission titrations with [Ru(phen)2(bbiab)](PF6)2 in acetone and methanol. And an increase in emission and for all absorption bands is observed in dichloromethane, suggesting that the photophysical response is not only related to the addition of water to the system, but the solvent itself.
 | ||
| Fig. 3 The emission spectra excited at 450 nm, at 298 K under an ambient atmosphere of [Ru(phen)2(bbiab)](PF6)2 (4.61 μM), showing the dilution by addition of (a) HPLC grade DMSO and (b) 10% aquated DMSO (up to 500 μL) by 10 μL increments to solutions in (a) DMSO and (b) 10% aquated DMSO. | ||
The photophysical character of [Ru(phen)2(bbiab)](PF6)2 in each of the solvents was also informative (Table 2). Taking the relative emission under an ambient atmosphere using HPLC grade acetonitrile of [Ru(bpy)3](PF6)2 to be the standard (with a quantum yield 0.040),29 the observed quantum yield in dichloromethane, acetone and HPLC grade acetonitrile suggest they are comparable, being 228%, 221% and 185% more emissive than the standard. However, in solvents with a protic environment, as seen with methanol and 10% aquated DMSO, the relative emission decreases significantly, suggesting it could adopt a different structure in the solution, although this may be caused by quenching enabled by specific solvent–relaxation interactions, as well as the water content in the solvent (particularly with DMSO). However, the bimodal and switchable behaviour does indicate that solution speciation is non-trivial.
| Absorptiona | Emissiona | |||
|---|---|---|---|---|
| λ max | (ε × 105) | λ max |
Φ
emb |
|
| Solvent | ±2 nm | dm−3 mol−1 cm−1 | ±2 nm | (% of standard) |
| a Recorded under ambient atmosphere in acetonitrile at 298 K, excited at 450 nm. b Emission quantum yield in acetonitrile (Φem) were calculated relative to the emission quantum yield calculated relative to [Ru(bpy)3](PF6)2 (Φem = 0.040) under ambient atmosphere in acetonitrile.29 | ||||
| Acetonitrile | 263 | (728) | 633 | 0.074, 185% |
| 292 | (297) | |||
| 309 | (299) | |||
| 369 | (258) | |||
| 441 | (184) | |||
| 469 | (202) | |||
| DMSO | 266 | (512) | 648 | 0.040, 99% |
| 305 | (284) | |||
| 316 | (314) | |||
| 380 | (277) | |||
| 441 | (179) | |||
| 475 | (200) | |||
| 10% H2O DMSO | 266 | (664) | 650 | 0.022, 56% |
| 296 | (312) | |||
| 302 | (312) | |||
| 314 | (320) | |||
| 378 | (268) | |||
| 447 | (192) | |||
| 473 | (206) | |||
| Acetone | 373 | (209) | 632 | 0.088, 221% |
| 441 | (145) | |||
| 470 | (158) | |||
| Methanol | 264 | (831) | 637 | 0.053, 133% |
| 312 | (257) | |||
| 376 | (179) | |||
| 442 | (134) | |||
| 470 | (140) | |||
| 264 | (887) | |||
| Dichloromethane | 290 | (388) | 616 | 0.091, 228% |
| 312 | (301) | |||
| 351 | (228) | |||
| 377 | (260) | |||
| 441 | (203) | |||
| 463 | (239) | |||
The initial premise to account for the resulting behaviour observed on the introduction of small quantities of solvent, is the extent of association of the monomeric dicationic form. Aggregated species, potentially dimers, would be less exposed to solvent and oxygen, so would presumably have higher quantum yields. Given that the amido-imidazole ligand is involved in this association, potentially held together around a small halide salt (as discussed later) or solvent through appropriately orientated hydrogen bonds on the imidazole group, and that this function also hosts the excited state SOMO, a change in the “protic nature” of the local environment would result in considerable variation in the absorption spectra. In acetone and dichloromethane, it is assumed that the aggregation is maintained on dilution. Whilst in methanol, as a protic solvent, the competitive hydroxyl groups ensure complete dissociation. However, in acetonitrile and in DMSO, the addition of a small amount of water inherent in the solvent is sufficient that a stoichiometric addition leads to partial dissociation, and potentially a change in protonation, as suggested by the systematic changes to the absorbance spectra.
DOSY-NMR examination
To investigate whether aggregation, potentially dimerization, is a realistic possibility, 1H DOSY-NMR spectroscopy was applied to both ruthenium complexes [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 (1 mM in D6-DMSO), showing a dynamic molecular mass of 1715 and 1816 (±15%) respectively (calculated via SEGWE,30Table 3) which closely resembles that anticipated for the dimeric species {[M]}24+ (1776 and 1872 amu) in solution, with the assumption that these complexes exist as solvated ions, and that they are not necessarily associated to the PF6− counterions. Whilst less accurate, 1H DOSY-NMR spectra for [Ru(phen)2(bbiab)](PF6)2 in D6-acetone, shows a comparable value with the monomer molecular weight lying outside the 95% confidence range suggesting the presence of a “dimer”. In D4-methanol, a considerably lower mass of 1224 (±15%) suggest it is a solvated monomeric unit.| Diffusion constant | DOSY determined weight | Estimated hydrodynamic radius | |
|---|---|---|---|
| Complex | ×10−10 m2 s−1 | g mol−1 ± 15% | ×10−12 m |
| Diffusion constants obtained from DOSY-NMR of a variety of metal complexes, taken in D6-DMSO, 298 K. Diffusion constants calculated from dynamics centre and consequent estimates for the values of hydrodynamic radius and molecular weight acquired using SEGWE.30 | |||
| [Ru(bpy)2(bbiab)](PF6)2D6-DMSO | 1.25 | 1715 | 1027 |
| [Ru(phen)2(bbiab)](PF6)2 D6-DMSO | 1.22 | 1816 | 1047 |
| [Ru(phen)2(bbiab)](PF6)2D6-Acetone | 8.20 | 1699 | 1024 |
| [Ru(phen)2(bbiab)](PF6)2 D4-Methanol | 4.82 | 1224 | 918 |
To see whether anion association impacts the perceived molecular weight, DOSY spectra were also recorded with [Ru(phen)2(bbiab)](PF6)2 in D6-DMSO, in the presence of 0, 0.5, 1, 2, and 5 equivalents of TBA H2PO4. Addition of H2PO4− anions however, up to two equivalents, did not appear to affect the diffusion constant suggesting that the possible “capture” of the anion did not result in the formation of “dimers”. At the addition of five equivalents, precipitation occurred, and a red solid assumed to be [Ru(phen)2(bbiab)](H2PO4)2, is formed.
Optical anion binding studies in acetonitrile
The exploration of the anion binding behaviour of these potential hosts is complicated by the observed solvent induced quenching in both acetonitrile and DMSO. To ensure consistency, rather than using dry solvent, that could then absorb moisture during the titration, a standard fresh bottle of HPLC grade acetonitrile was selected to make up stock solutions of a series of anion solutions, as their TBA salts and both [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2, and a subsequent “subtraction” applied to the UV-Vis absorption and emission data to account for the quenching observed on dilution titration. Consequently, the processed data (Fig. 4, Fig. S14–S16†) suggests that both ruthenium complexes are largely invariant to the increasing presence of Cl−, Br−, NO3− and ClO4− with a small, but insignificant emissive increase upon addition of Cl−, and a slight decrease with Br−, NO3− and ClO4−. There is, however, significant quenching in the emission on the introduction of a stoichiometric quantity of AcO−, and a commensurate blueshift and flattening of the absorption bands in the region 300–390 nm and a small blueshift and large increase in absorption band in region 390–540 nm corresponding to the MLCT. This behaviour is consistent with the pH titration data suggesting a deprotonation event is taking place, rather than a simple binding event. | ||
| Fig. 4 The ‘blank’ adjusted change in emissive behaviour in acetonitrile of (a) [Ru(bpy)2(bbiab)](PF6)2 and (b) [Ru(phen)2(bbiab)](PF6)2 on the addition of TBA chloride (green), TBA bromide (maroon), TBA nitrate (blue), TBA acetate (red), TBA hydrogensulfate (yellow), TBA dihydrogenphosphate (orange) and TBA perchlorate (purple). | ||
The UV-Vis spectra on the addition of both TBA HSO4− and H2PO4− are remarkably similar, showing a linear decrease in absorption with increasing anion concentration in the region 260–500 nm, encompassing all bbiab/imidazole ligand and MLCT attributed bands. This effect is slightly less pronounced with H2PO4−. Further, precipitation was noted to occur with HSO4− once five equivalents had been added. In the emission spectra, the addition of both HSO4− and H2PO4− show unusual binding isotherms, suggestive of a multi-step processes. Adjusting for the changes in emission arising from the introduction of the solvent alone, on the addition of HSO4− to both complexes, an increase in emission is observed with 1.0 equivalent, with an associated blueshift of approximately 10 nm. Following this, there is a gradual decrease in emission and a redshift of up to 16 nm, being more pronounced with [Ru(phen)2(bbiab)](PF6)2. The emission spectra are similarly complex with the addition of H2PO4−, showing a similar, but less pronounced pattern.
The similarity of the observed interactions between both complexes and TBA HSO4− and H2PO4− suggest a similar, yet complex binding relationship of these anions with the hosts. This is complicated by the fact that introducing the damp solvent to the sample is resulting in potentially a loss of the assumed complex–complex dimeric structure. So, the enhanced emission, particularly in the case of HSO4−, appears to suggest that the anion is “preventing” the water induced disaggregation, i.e. has the potential to act as “glue”, inhibiting dissociation, in what would appear to be a one-to-one (or even a two-to-two) stoichiometry. For H2PO4−, there is potentially a further reaction towards a one-to-two stoichiometry which then results in a competitive dissociation and hence drop in emission.
Attempts were made to determine appropriate stability constants using BindFit31 using both the using UV-Vis absorption and emission date in acetonitrile, exploring a two-to-one, a one-to-one and a one-to-two host to guest binding models. Unfortunately though, in each case, the competitive binding of water, in effect negates the validity of the determined values.
UV-Vis and emission anion studies in DMSO
UV-Vis and emission spectra of both [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 in DMSO, and in a 10% water/DMSO mixture were also invariant to TBA Cl−, Br−, NO3− and ClO4− compared to that of the effects of the ‘blank’ titration (Fig. S17–S24†). Slight overall changes to emission can be explained by a relatively weak association of anions. Unlike the behaviour in acetonitrile, the addition of HSO4− to the complexes in DMSO does not induce a complex binding isotherm, rather a simple weak one-to-one binding stoichiometric effect is seen, with only a small increase in the emission evident over and above that observed from the quenching assumed to arise from the addition of the solvent. A similar result is seen again in a 10% water/DMSO mixture with an initial drop in emission with up to one equivalent of HSO4−, then the emission increases once more, suggesting further complexity is present as concentration increases.In DMSO, addition of both AcO− and H2PO4− caused precipitation with three to four equivalents of the anion, with a significant quenching of emission. This does not occur in the presence of 10% water in the DMSO, where addition of AcO− results in the expected quenching in emission, alongside subtle changes in the absorption spectra consistent with those observed in acetonitrile. Addition of H2PO4− to both complexes results in a quenching of emission in what appears to be a less complex binding mode than seen in acetonitrile, and a similar change in the absorption as to that seen with the addition of AcO−. These results do tend towards a highly complex set of binding modes between the anions and the potential hosts, where the presence of “protic” media, in particular stoichiometric quantities of water can vary the binding behaviour with the anions, the complex itself, and the localised environment. This establishes the need to consider more than just the interactions between a complex and anions when determining binding modes and association constants. Attempts were made to determine the binding constants, but the system proved to be far too complex to provide reliable and reproducible values.
Effects of solvent on phosphate binding
A series of TBA H2PO4− titrations was performed in acetone, methanol and dichloromethane to further investigate the effect of solvent on the association with [Ru(phen)2(bbiab)](PF6)2. As noted above, H2PO4− association to both reported complexes is highly sensitive to the solvent environment (Fig. S25†). Methanol, being protic, readily forms hydrogen bonds which can potentially influence the molecular interactions (implied from the DOSY-NMR data). [Ru(phen)2(bbiab)](PF6)2 likely presents as a monomer in methanol, suggesting that it disrupts an “aggregation” of the cations significantly. On the addition of H2PO4− anions, there is no significant change in either the UV-Vis absorption or emission spectra, suggesting that there is no significant interaction with the anion.However, in acetone there are large perturbations noted in the absorption spectra in both the regions associated with the benzimidazole, and the MLCT transitions, and a significant drop in emission, with a redshift with 1 equivalent of H2PO4− representative of initial dissolution of the dimer and association to a two-to-two or a one-to-one complex. This is followed by almost complete emissive quenching, and significant perturbations and “flattening” of the absorption spectra consistent with protonation (see earlier). A similar result is observed for the same interaction in DCM (Fig. 5), although this time with a small initial blueshift in the emission with one equivalent of the anion, and a much larger initial quenching. On further addition, there is a further decrease in emission up to two equivalents, and then a slight increase in emission. This suggests that there is a stepwise set of binding equilibria, enabled by the absence of competitive hydrogen-bond formation with the solvent, in particular water. The similarity in the appearance of the UV-Vis absorbance spectra of [Ru(phen)2(bbiab)](PF6)2 in both acetone and DCM to the pH titrations reported above, particularly in the acidic region, suggest that H2PO4− transfers a proton to the bbiab function, unlike AcO− where deprotonation of an amide can be observed in non-protic media.
 | ||
| Fig. 5 The UV-Vis absorption spectra (a) and emission spectra (b) excited at 450 nm, at 298 K of [Ru(phen)2(bbiab)](PF6)2 (4.61 μM), showing the addition of TBA dihydrogenphosphate (up to 10 equiv.) in dichloromethane. | ||
1H NMR studies
To explore the binding behaviour of both [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2, 1H-NMR titration studies were conducted. Initial attempts to undertake these studies in DMSO alone, at an appropriate concentration for reasonable NMR studies resulted in an almost stoichiometric precipitation, but more success was had in a 10% D2O mixture in D6-DMSO mixture, although the imidazole and amide NH peaks in the region 11–15 ppm are absent. Each complex (at 1 mM) was screened by addition of the TBA salts of Cl−, Br−, NO3−, AcO−, HSO4−, H2PO4− and ClO4− (Fig. S26–S39†), showing no significant interaction with Cl−, Br−, NO3− and ClO4−. Interestingly, no significant perturbations were observed with HSO4− despite showing interactions in the UV-Vis and emission spectra. This may be explained that at higher concentrations, HSO4− can form dimers via AEHB interactions,32 and that these species are less responsive to the ruthenium complexes, although the bbiab-H3 signals (9.17 and 9.19 ppm for [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 respectively) were noted to sharpen significantly.Upon the addition with AcO− (Fig. 6a) to both complexes, the bbiab-H3 signals resolve significantly, and both move down-field by 0.085 ppm with two equivalents. All of the benzimidazole aromatic peaks shift up-field, with HAr4/5 having a larger shift than the bbiab-H3 signal. Stability constants were determined using a full spectrum analysis with BindFit,31 which modelled well to a one-to-one host to guest scenario with a binding coefficient for both complexes of pKstab = 3.29 ± 3.0%.
 | ||
| Fig. 6 1H-NMR titration of [Ru(phen)2(bbiab)](PF6)2 (1 mM) with up to 10 equiv. of (a) TBA acetate and (b) TBA dihydrogenphosphate in 10% D2O in DMSO-D6. | ||
Similarly, the introduction of H2PO4− to both complexes (Fig. 6b) has a significant effect on the bbiab-H3 proton, with a downfield shift of 0.37 and 0.38 ppm with two equivalents. But contrary to observation with AcO−, the imidazole bbiab-ArH3/6 protons at 7.49 and 7.48 ppm show a small down-field perturbation, while the bbiab-ArH4/5 protons at 7.26 and 7.25 ppm show a small up-field shift suggesting a “deeper” interaction with H2PO4− into the bbiab cavity, and potentially a different binding mode to that observed with AcO−, with a one-to-one host to guest binding coefficient of 3.56 and 3.50 ± 1.9% for the two complexes respectively. However, given the complexity of the equilibria, this could be more representative of a two-to-two binding mode.25
Discussion of binding modes and molecular modelling
Surprisingly, both the complexes [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2 do not have an observed interaction with many of the anions studied. That does not suggest that they have no interaction, just that they do not induce a discernible perturbation in the applied spectroscopic technique. To explore this further, optimised DFT studies (B3LYP/cc-pVDZ using Gaussian 09, Revision E01)33 with the bbiab ligand alone were undertaken assuming a gas phase configuration. This resulted in effectively a planar structure for the ligand (Scheme 2), with very little energy variation between the various considered configurations (Fig. S40†). It was observed that the space between the two “arms” of the ligand, where the amidic protons are turned inward, offer a smaller binding cavity than with the previously reported bbib ligand.25 And the imidazole NH group, assumed to be responsible for the “anion binding” forms hydrogen bonds to the amidic carbonyl group. The DFT studies indicate that with anions in the small bbiab cavity, that the cleft is ideally suited to bind halides (Fig. S41†), which is optimal for bromide. The binding of halides does not appear to affect the photophysical, or even the NMR behaviour despite an optimal fit. The modelling studies did reveal that a second bbiab ligand could readily coordinate around the halide centre, suggesting that a two-to-one binding motif is possible. This “dimeric” system is likely to be more emissive than the one-to-one unit, given the exclusion of solvent and oxygen from the low lying LUMO (Fig. S3 and 4†). | ||
| Scheme 2 Overview of the potential binding modes to the generic bbiab ligand system with dihydrogenphosphate, and associated DFT optimized structures.33,34 | ||
When the modelled halide is replaced by water in the bbiab cleft, the DFT calculations optimise again to form a stable planar orientation as seen with halides, reinforced by additional hydrogen bonds. And like the behaviour with halides, a strong dimeric structure can readily nucleate around the water molecule. This could account for the observed quenching in the presence of small amounts of water extracted from the solvent and would result in disruption of the emissive “dimeric” form. Placing a larger oxo-anions in the system (Fig. S42†) results in a degree of non-planarity, with nitrate and acetate significantly distorting the structure, the latter placing the methyl group pointing out of the plane of the ligand. With these two anions, there is not an easy way to visualise the binding of two anions to the ligand, without forcing the ligand to fold outwards which when bound to the metal centre with large co-ligands offers a significant steric challenge explaining the observed one-to-one selectivity for acetate (Scheme 2).
The experimental behaviour with HSO4− differs considerably to that with AcO−. The DFT studies (Fig. S43†) indicate that in the absence of solvent, that the most optimal structures, have the proton transfer from the hydrogen sulfate to the imidazole reinforced by the resulting external NH–O amide interaction. However, the sulfate unit itself is again too large to sit within the cavity, and forces a degree distortion, although this is optimised by inverting one of the amidic groups, so that the CO group points in towards the cleft, giving a remarkably stable configuration. It is likely that this orientation gives rise to the good one-to-one binding of hydrogensulfate in acetonitrile, breaking apart the assumed dimeric unit. However, in a protic media, this configuration will be compromised by competitive hydrogen bonding, both between the anion, and the host, and the surrounding solvent. Further DFT studies also indicated that hydrogen sulfate cannot readily act as a “nucleating” unit to coordinate two hosts around the one anion given its preference to sit outside of the plane of the cleft in either orientation.
DFT modelling studies between the ligand with H2PO4− added further complexity (Scheme 3) to the perceived binding mode. As with HSO4−, the anion bound in the “cleft”, but in preference, there was a clear indication of proton transfer to the two imidazoles, as suggested by the UV-Vis absorption studies (complex protonation), forming a one-to-one complex (Fig. S44†). But unlike the hydrogen sulfate situation, where the inverted amide gave an additional degree of stability, this was not the case with H2PO4−. It is unlikely that there is sufficient space to form a good two-to-one complex. However, it was noted that a stable configuration was observed by binding two dihydrogen phosphates to the host, as previously seen with more open systems,35 adopting anti-electrostatic hydrogen bonds,32 and this may indicate, that as with the previously reported system with the smaller bbib ligand,25 that two complexes can “dimerise”.
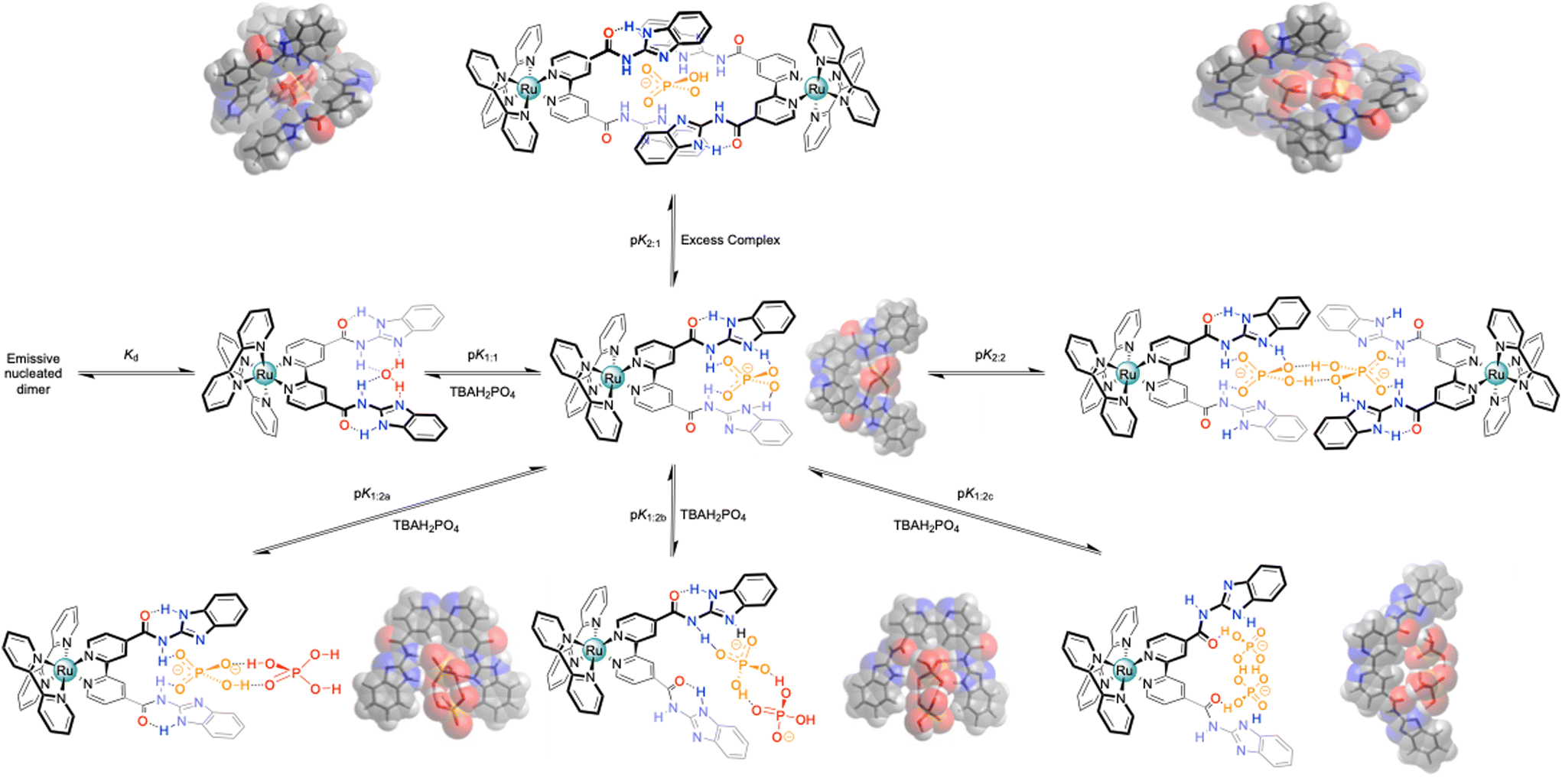 | ||
| Scheme 3 Overview of the potential binding modes to the generic bbiab ligand system with dihydrogenphosphate, and associated DFT optimized structures.33,34 | ||
Conclusions
The bbiab ligand system, whilst effectively insoluble in most media, will coordinate to form emissive ruthenium(II) trischelate diimine complexes. However, the resulting species show very subtle and unanticipated solvent effects where the introduction of small amounts of water into a hydrophobic environment cause considerable quenching in emission. This suggests that these species could potentially exist normally as dimeric units, nucleated around either a halide, or even water itself in dry non-protic environments. This is supported by both DOSY and DFT studies. But on the introduction of competing species these separate, most significantly with acetate and dihydrogenphosphate, but even small amounts of water will result in this under the right conditions and concentrations. In the presence of an aquated or protic solution however, there is a degree of selectivity for the anions as exemplified by 1H NMR titration studies.In our preceding studies we had already become aware of the complexity, and potential opportunities that the introduction of a hydrogen donor/and acceptor can bring for the selectivity of dihydrogen phosphate through the inclusion of an imidazole function.25 Naively we had assumed that by the inclusion of additional amidic functions we could encourage selectivity towards a straightforward one-to-one, or even a one-to-two host guest interaction, following on from the data previously reported by Beer et al.14,17,19,27,36 However, as evidenced here, this simple idea has introduced an additional layer of complexity to the system, where the extent to which intermolecular hydrogen bonding occurs, speciates between a number of different configurations, where subtle changes in the protic nature of the solvent can have a significant effect on this intricate dance, selecting different combinations of partners as conditions change, far exceeding the simple direct coupling of a host with its guest. Thus, the direct determination of equilibria constants is far from simple and can only be considered in the most extreme “forced” conditions. However, there does appear to be a dominant preference for binding to dihydrogen phosphate with the systems presented here, and interestingly, not by the abstraction of an acidic proton from the host in organic media (as typified in the interactions with acetate), but via the transfer of a proton from the protonated salt to the host in combination with providing hydrogen donor groups. With this understanding, we will continue to explore the selective binding of biological phosphate with these and related systems.
Experimental
Physical measurements
NMR spectra were recorded using Bruker Avance III 400 spectrometer at 298 K and referenced against solvent. Absorbance spectra were recorded using a 1 cm path length quartz cuvette on an Agilent Carey 60 UV-Vis spectrometer; and fluorescence spectra obtained on a Cary Eclipse spectrometer. Quantum yields were determined by normalization against [Ru(bpy)3]2+ under an ambient atmosphere in water (Φem = 0.018) and acetonitrile (0.040).29 E.S.I. mass spectroscopy was recorded using a Shimadzu LCMS-IT-TOF mass spectrometer at 298 K. Elemental analysis were recorded using a Vario MICRO Cube CHNS analysis instrument.pH studies
The pH was adjusted systematically over a pH range of 2.5 to 12.5 by the addition of a 0.1 M aqueous NaOH solution containing 10 μM aqueous solution of either [Ru(bpy)2(bbiab)]2+ and [Ru(phen)2(bbiab)]2+ (pH 13) to a 0.1 M Britton–Robinson aqueous buffer (pH 2.5) solution containing 10 μM aqueous solution of the appropriate metal complex. The raw data was standardised for scatter by normalisation at 600 nm, and concentration standardised at the assumed isosbestic points at 479 and 420 nm ([Ru(bpy)2(bbiab)]2+) and 479 and 400 nm ([Ru(phen)2(bbiab)]2+).Anion binding studies
UV-Vis and emission titrations were conducted by preparing a 10 mL stock solution of the metal complex in the micromolar (μM) range. To this is titrated sub-stoichiometric quantities of the guest tetrabutylammonium chloride, bromide, nitrate, acetate, hydrogensulfate, dihydrogen phosphate or perchlorate salt, starting at 10 μL, or 0.2 equivalent increments, to 10 equivalents, taking a UV-Vis and emission spectra after each sequential addition. The resulting data is plotted as a binding isotherm, accounting for dilution by normalizing data, and subtracting the quenching of emission observed for each equivalent in a dilution titration from that observed in subsequent anion titrations.1H NMR titrations were conducted by preparing a 0.5 mL stock solution (1 × 10–3 M) of the metal complex. To this is titrated sub-stoichiometric quantities of the guest tetrabutylammonium chloride, bromide, nitrate, acetate, hydrogensulfate, dihydrogen phosphate or perchlorate, starting at 0.2 equivalent increments, to 10 equivalents directly into the tube and recording 1H NMR spectrum after each sequential addition.
Association constants (Ka) were calculated using BindFit31 for non-linear curves fitting, plotting changes in absorption, emission or NMR chemical shift upon the addition of tetrabutylammonium chloride, bromide, nitrate, acetate, hydrogen sulphate, dihydrogenphosphate or perchlorate salts to [Ru(bpy)2(bbiab)](PF6)2 and [Ru(phen)2(bbiab)](PF6)2. Curves were fit according to 1:1, 1:2 and 2:1 binding modes, and converted to log(Ka) values.
Materials
All reagents were purchased from either Merck or Fisher Scientific and used as supplied unless otherwise specified. Deuterated NMR solvents were acquired from Fluorochem. [Ru(bpy)2Cl2],37 [Ru(phen)2Cl2]38 and 4,4′-dicarboxylic acid-2,2′-bipyridine39 were prepared by literature procedures. Dry acetonitrile was isolated from a solvent purification system (SPS) using HPLC grade, sparged with N2 for 45 minutes prior to fitting to reservoir, and passed through two columns of activated alumina on an Innovative Technology Pure Solv SPS tower (Model No.: PS-MD-5EN, Serial No.: PS-13-247B), under inert gas pressure.4,4′-Bis(N-1H-benzimidazol-2-yl-carboxamidyl)-2,2′-bipyridine (bbiab)
4,4′-Dicarboxylic acid-2,2′-bipyridine (1.00 g, 4.10 mmol) SOCl2 (∼25 mL) were heated under reflux for 24 h under a N2 atmosphere. The remaining SOCl2 was removed by distillation and the resulting acyl chloride dried in vacuo. This was then suspended in dry toluene (∼30 mL) before 2-aminobenzimidazole (1.09 g, 8.19 mmol) and triethylamine (5 mL) were added, and the mixture refluxed under N2 for 48 h. After cooling, the solution was slowly poured into saturated aqueous NaHCO3 (∼250 mL, pH 10) and stirred for 30 minutes. The resulting brown precipitate was collected by filtration, washed with water (3 × 30 mL) and dried; yield = 1.01 g, 2.13 mmol, 52%. Elemental analysis: calculated for C26H18N8O2, 4.5 H2O, 0.45 S2Cl2, 0.28 C6H16ClN: C, 50.77; H, 4.85; N, 17.71; S, 4.41%, found C, 51.04; H, 4.53; N, 17.50; S, 4.09%. IR νmax/cm−1: 3185br, 3090br and 3060br (NH), 2965br, 2896br, 2810br, 2770br and 2690br (CH), 1626m (CO), 1564s and 1544s (CC), 1477m, 1423s and 1361s (CH), 1294m, 1270m, 1253m and 1233m (CN), 740s (CH). 1H-NMR (400 MHz, δ (ppm), DMSO-D6,): 12.70 (2H, br, imidazole bbiabNH), 9.17 (2H, s, bbiabH3), 8.91 (2H, d, J = 4.9 Hz, bbiabH6), 8.10 (2H, dd, J = 4.9, 1.3 Hz, bbiabH5), 7.47 (4H, m, bbiabHAr4,Ar7), 7.23 (4H, m, bbiabHAr5,Ar6). HRMS (ESI+): m/z [M + H]+ calculated for C26H18N8O2: 475.1625, found: 475.1612.[Ru(bpy)2(bbiab)](PF6)2
[Ru(bpy)2(Cl)2] (0.161 g, 0.33 mmol), 4,4′-bis(N-1H-benzimidazol-2-yl-carboxamidyl)-2,2′-bipyridine (bbiab) (0.153 g, 0.32 mmol) and trifluoromethane sulfonic acid (0.5 mL) in ethylene glycol (30 mL) were stirred at 140 °C under a N2 atmosphere for 4 h. After cooling, the crude product was precipitated by addition of excess saturated aqueous KPF6 and the resulting red solid isolated by filtration, washed with water (3 × 30 mL) and dried. The crude product was purified using a LH-20 Sephadex® size exclusion chromatography, eluting with 7:3 MeOH/ACN mixture, followed by a second column, eluted at 9:1 MeOH/ACN mixture and recrystallized from acetone (2 mL) and addition of aqueous KPF6 (∼0.5 g in 100 mL) as a red microcrystalline solid; yield = 0.054 g, 0.046 mmol, 14%. Elemental analysis: calculated for C46H34F12N12O2P2Ru, 4.7 (CH3)2CO, 1 KPF6: C, 44.15; H, 3.83; N, 10.28; S, 0.00%, found C, 43.79; H, 4.2; N, 10.17; S, 0.26%. IR νmax/cm−1: 3380br and 3200br (NH), 2955m, 2922m and 2853m (CH), 1720w and 1660w (CO), 1617w, 1565m and 1535m (CC), 1455m, 1420m and 1395m (CH), 1345m, 1265m, 1160m, 1095m and 1020m (CN), 815s and 765s (CH). 1H-NMR (400 MHz, δ (ppm), D6-DMSO): 12.77 (4H, br s, imidazole/amide bbiabNH), 9.16 (2H, s, bbiabH3), 8.88 (4H, dd, J = 8.3, 3.2 Hz, bpyH3,3′), 8.22 (2H, ddd, J = 8.0, 8.0, 1.4 Hz, bpyH4), 8.20 (2H, ddd, J = 8.0, 8.0, 1.4 Hz, bpyH4′), 8.10 (2H, dd, J = 5.8, 1.6 Hz, bbiabH5), 7.96 (2H, d, J = 5.8 Hz, bbiabH6), 7.83 (2H, d, J = 5.6 Hz, bpyH6), 7.76 (2H, dd, J = 5.6, 1.2 Hz, bpyH6′), 7.58 (2H, ddd, J = 7.7, 5.6, 1.2 Hz, bpyH5′), 7.54 (2H, ddd, J = 7.7, 5.8, 1.3 Hz, bpyH5), 7.50 (4H, m, bbiabHAr4,Ar7), 7.27 (4H, m, bbiabHAr5,Ar6). HRMS (ESI+): m/z [M]2+ calculated for C46H34N12O2Ru: 444.0986, found: 444.0971.
[Ru(phen)2(bbiab)](PF6)2
Prepared according to the procedure for [Ru(bpy)2(bbib)](PF6)2 using [Ru(phen)2(Cl)2] (0.162 g, 0.30 mmol) and bbiab (0.142 g, 0.30 mmol); yield = 0.084 g, 0.07 mmol, 23%. Elemental Analysis: calculated for C50H34F12N12O2P2Ru, 6H2O, 2 (CH3)2CO: C, 46.38; H, 4.03; N, 11.59; S, 0.00%, found: C, 46.05; H, 3.64; N, 11.24; S, 0.17%. IR νmax/cm−1: 3590br and 3320br (NH), 3090w, 2955w, 2920m and 2851w (CH), 1723m and 1654m (CO), 1565m (CC), 1430m and 1405m (CH), 1315m, 1260m, 1245m, 1140m, 1100m, 1090m and 1020m (CN), 810s, 765s and 715s (CH). 1H-NMR (400 MHz, δ (ppm), D6-DMSO): 12.76 (4H, s, imidazole/amide bbiabNH), 9.20 (2H, s, bbiabH3), 8.86 (2H, dd, J = 8.3, 1.2 Hz, phenH4), 8.78 (2H, dd, J = 8.3, 1.2 Hz, phenH7), 8.42 (2H, dd, J = 8.5, 1.1 Hz, phenH5), 8.41 (2H, dd, J = 8.5, 1.1 Hz, phenH6), 8.34 (2H, dd, J = 5.3, 1.2 Hz, bbiabH5), 7.99 (2H, dd, J = 5.8, 1.6 Hz, phenH9), 7.98 (2H, dd, J = 5.8, 1.6 Hz, phenH2), 7.95 (2H, dd, J = 8.3, 5.3 Hz, phenH3), 7.92 (2H, d, J = 8.3, 5.3 Hz, bbiabH6), 7.75 (2H, dd, J = 5.9, 3.2 Hz, phenH8), 7.49 (4H, m, bbiabHAr4,Ar7), 7.26 (4H, m, bbiabHAr5,Ar6). HRMS (ESI+): m/z [M]2+ calculated for C50H34N12O2Ru: 468.0987, found: 468.0977.DFT studies
All calculations were performed as optimized on the Gaussian 09 Software using B3LYP as a functional.33 For free ligand and anion guest interactions the correlation consistent basis set cc-pVTZ was used, and for the metal complex a split 6-31G*/LanL2DZ (Ru) basis set was applied. Visualisation and analysis was undertaken using Avogadro 1.2.0.34Author contributions
AJS, CLH and NCF (project lead) share equal responsibility for conceptualisation, data acquisition, curation, and formal analysis. NCF undertook the computational studies, and the manuscript has been prepared by AJS and NCF.Data availability
The data supporting this article have been included as part of the ESI.† The original spectra are available via the authors institution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. S. and C. L. H. acknowledges Ph.D. funding from Lancaster University. This research has been supported by Lancaster University.References
- A. K. Hirsch, F. R. Fischer and F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 338–352 CrossRef CAS PubMed.
- (a) R. B. Brown, P. Bigelow, J. A. Dubin and J. G. Mielke, Nutrients, 2023, 15, 3735 CrossRef CAS PubMed; (b) R. B. Brown, P. Bigelow, J. A. Dubin and E. Neiterman, J. Appl. Toxicol., 2024, 44, 17–27 Search PubMed.
- M. Doherty, C. Belcher, M. Regan, A. Jones and J. Ledingham, Ann. Rheum. Dis., 1996, 55, 432–436 CrossRef CAS PubMed.
- (a) S. S. Ali, A. Gangopadhyay, A. K. Pramanik, S. K. Samanta, U. N. Guria, S. Manna and A. K. Mahapatra, Analyst, 2018, 143, 4171–4179 RSC; (b) X. Zhu and J. Ma, TrAC, Trends Anal. Chem., 2020, 127, 115908 CrossRef CAS; (c) N. A. Esipenko, P. Koutnik, T. Minami, L. Mosca, V. M. Lynch, G. V. Zyryanov and P. Azenbacher, Jr., Chem. Sci., 2013, 4, 3617–3623 RSC; (d) C. Warwick, A. Guerreiro and A. Soares, Biosens. Bioelectron., 2013, 41, 1–11 CrossRef CAS PubMed.
- Y. Marcus, Biophys. Chem., 1994, 51, 111–127 CrossRef CAS.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- F. Biedermann and H.-J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
- S. B. Khan and S. L. Lee, Molecules, 2021, 26, 3995 CrossRef CAS PubMed.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS PubMed.
- F. Wurthner, J. Org. Chem., 2022, 87, 1602–1615 Search PubMed.
- N. C. Fletcher, M. D. Ward, S. Encinas, N. Armaroli, L. Flamigni and F. Barigelletti, Chem. Commun., 1999, 2089–2090 RSC.
- (a) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 Search PubMed; (b) S. Kubik, ChemistryOpen, 2022, 11, e202200028 CrossRef CAS; (c) S. C. Patrick, P. D. Beer and J. J. Davis, Nat. Rev. Chem., 2024, 8, 256–276 CrossRef.
- R. A. Pascal, J. Spergel and D. Van Engen, Tetrahedron Lett., 1986, 27, 4099–4102 CrossRef CAS.
- P. D. Beer, C. A. P. Dickson, N. C. Fletcher, A. J. Goulden, A. Grieve, J. Hodacova and T. Wear, J. Chem. Soc., Chem. Commun., 1993, 1240 Search PubMed.
- S. Valiyaveettil, J. F. J. Engbersen, W. Verboom and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1993, 32, 900–901 CrossRef.
- C. Raposo, N. Pérez, M. Almaraz, M. L. Mussons, M. C. Caballero and J. R. Moran, Tetrahedron Lett., 1995, 36, 3255–3258 CrossRef CAS.
- P. D. Beer, N. C. Fletcher and T. Wear, Polyhedron, 1996, 15, 1339–1347 CrossRef CAS.
- A. P. Bisson, V. M. Lynch, M. K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 2003, 36, 2340–2342 CrossRef.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 Search PubMed.
- U. Manna and G. Das, Coord. Chem. Rev., 2021, 427, 213547 CrossRef CAS.
- (a) N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed; (b) E. A. Katayev, J. L. Sessler, V. N. Khrustalev and Y. A. Ustynyuk, J. Org. Chem., 2007, 72, 7244–7252 Search PubMed.
- L. S. Yakimova, D. N. Shurpik and I. I. Stoikov, Chem. Commun., 2016, 52, 12462–12465 RSC.
- (a) D. Pelleteret, N. C. Fletcher and A. P. Doherty, Inorg. Chem., 2007, 46, 4386–4388 CrossRef CAS PubMed; (b) S. Ghosh, K. Goswami and K. Ghosh, Supramol. Chem., 2017, 29, 946–952 CrossRef CAS; (c) C. M. G. Dos Santos, T. McCabe, G. W. Watson, P. E. Kruger and T. Gunnlaugsson, J. Org. Chem., 2008, 73, 9235–9244 CrossRef CAS PubMed; (d) L.-J. Kuo, J.-H. Liao, C.-T. Chen, C.-H. Huang, C.-S. Chen and J.-M. Fang, Org. Lett., 2003, 5, 1821–1824 Search PubMed; (e) A. Jain, V. Gupta and J. Raisoni, Talanta, 2006, 69, 1007–1012 CrossRef CAS PubMed; (f) N. H. Evans, C. J. Serpell, K. E. Christensen and P. D. Beer, Eur. J. Inorg. Chem., 2012, 2012, 939–944 CrossRef CAS; (g) C. R. Rice, C. Slater, R. A. Faulkner and R. L. Allan, Angew. Chem., Int. Ed., 2018, 57, 13071–13075 CrossRef CAS; (h) J. A. Thomas, Dalton Trans., 2011, 40, 12005–12016 Search PubMed.
- (a) M. L. Korczak, K. Maslowska-Jarzyna and M. J. Chmielewski, RSC Adv., 2024, 14, 29883–29889 Search PubMed; (b) S. Dey, S. Ghosh, A. Das, R. N. Yadav, R. Chakrabarty, S. Pradhan, D. Saha, A. K. Srivastava and M. F. Hossain, J. Fluoresc., 2024, 34, 1829–1840 CrossRef CAS PubMed; (c) P. Zhao, Y. Liu, C. He and C. Duan, Inorg. Chem., 2022, 61, 3132–3140 Search PubMed.
- C. L. Howells, A. J. Stocker, J. N. Lea, N. R. Halcovitch, H. Patel and N. C. Fletcher, Chem. – Eur. J., 2024, 30, e202401385 CrossRef CAS PubMed.
- (a) Y. Cui, H. J. Mo, J. C. Chen, Y. L. Niu, Y. R. Zhong, K. C. Zheng and B. H. Ye, Inorg. Chem., 2007, 46, 6427–6436 CrossRef CAS PubMed; (b) Y. Cui, Y. L. Niu, M. L. Cao, K. Wang, H. J. Mo, Y. R. Zhong and B. H. Ye, Inorg. Chem., 2008, 47, 5616–5624 Search PubMed; (c) J. C. Freys, G. Bernardinalli and O. S. Wenger, Chem. Commun., 2008, 4267–4269 RSC; (d) H. J. Mo, Y. L. Niu, M. Zhang, Z. P. Qiao and B. H. Ye, Dalton Trans., 2011, 40, 8218–8225 Search PubMed; (e) B. Chowdhury, S. Khatua, R. Dutta, S. Chakraborty and P. Ghosh, Inorg. Chem., 2014, 53, 8061–8070 CrossRef CAS PubMed; (f) S. Das, S. Karmakar, S. Mardanya and S. Baitalik, Dalton Trans., 2014, 43, 3767–3782 RSC; (g) S. A. Rommel, D. Sorsche and S. Rau, Dalton Trans., 2016, 45, 74–77 RSC; (h) T. K. Ghosh, S. Mondal, S. Bej, M. Nandi and P. Ghosh, Dalton Trans., 2019, 48, 4538 RSC; (i) A. Paul, M. Bar, T. Ahmed and S. Baitalik, Polyhedron, 2020, 190, 114772 Search PubMed; (j) S. Naithani, T. Goswami, F. Thetiot and S. Kumar, Coord. Chem. Rev., 2023, 475, 214894 Search PubMed.
- P. D. Beer, S. W. Dent and T. J. Wear, J. Chem. Soc., Dalton Trans., 1996, 2341–2346 Search PubMed.
- E. C. Constable and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1982, 34–46 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 Search PubMed.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 3987–3994 Search PubMed.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0, 2012 Search PubMed.
- A. L. Blackburn, N. C. A. Baker and N. C. Fletcher, RSC Adv., 2014, 4, 18442–18452 RSC.
- P. D. Beer, S. W. Dent, N. C. Fletcher and T. Wear, Polyhedron, 1996, 15, 2983–2996 CrossRef CAS.
- P. A. Lay, A. M. Sargeson and H. Taube, Inorg. Synth., 1986, 24, 291–299 Search PubMed.
- N. A. Al-Rawashdeh, S. Chatterjee, J. A. Krause and W. B. Connick, Inorg. Chem., 2014, 53, 294–307 CrossRef CAS PubMed.
- J. Muldoon, A. E. Ashcroft and A. J. Wilson, Chem. – Eur. J., 2010, 16, 100–103 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The original data for all spectroscopic titrations. See DOI: https://doi.org/10.1039/d5dt00842e |
| This journal is © The Royal Society of Chemistry 2025 |