
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodegradation of naphthalene-derived particle oxidation products†
Félix
Sari Doré‡
 a,
Cecilie
Carstens
a,
Jens
Top
b,
Yanjun
Zhang
a,
Clément
Dubois§
a,
Sébastien
Perrier
a,
Imad
El Haddad
b,
David M.
Bell
*b and
Matthieu
Riva
*a
a,
Cecilie
Carstens
a,
Jens
Top
b,
Yanjun
Zhang
a,
Clément
Dubois§
a,
Sébastien
Perrier
a,
Imad
El Haddad
b,
David M.
Bell
*b and
Matthieu
Riva
*a
aUniversité Claude Bernard Lyon1, CNRS, IRCELYON, UMR 5256, 69100 Villeurbanne, France
bPSI Center for Energy and Environmental Sciences, Paul Scherrer Institute, Villigen, Switzerland
First published on 6th January 2025
Abstract
While photochemical aging is known to alter secondary organic aerosol (SOA) properties, this process remains poorly constrained for anthropogenic SOA. This study investigates the photodegradation of SOA produced from the hydroxyl radical-initiated oxidation of naphthalene under low- and high-NOx conditions. We used state-of-the-art mass spectrometry (MS) techniques, including extractive electrospray ionization and chemical ionization MS, for the in-depth molecular characterization of gas and particulate phases. SOA were exposed to simulated irradiation at different stages, i.e., during formation and growth. We found a rapid (i.e. >30 min) photodegradation of high-molecular-weight compounds in the particle-phase. Notably, species with 20 carbon atoms (C20) decreased by 2/3 in the low-NOx experiment which was associated with particle mass loss (∼12%). Concurrently, the formation of oligomers with shorter carbon skeletons in the particle-phase was identified along with the release of volatile products such as formic acid and formaldehyde in the gas-phase. These reactions are linked to photolabile functional groups within the naphthalene-derived SOA products, which increases their likelihood of being degraded under UV light. Overall, photodegradation caused a notable change in the molecular composition altering the physical properties (e.g., volatility) of naphthalene-derived SOA.
Environmental significanceNaphthalene is a major anthropogenic volatile organic compound (VOC) and an important contributor to secondary organic aerosol (SOA) formation in urban environments. However, the evolution of the chemical composition of naphthalene-derived SOA remains uncertain. In this study, SOA were generated from the OH-initiated oxidation of naphthalene and exposed to UV radiation to investigate the influence of photochemical aging on the chemical composition. We observed that photodegradation induced a loss of SOA mass and high-molecular-weight dimers that resulted in the production of smaller unsaturated dimers and the emission of volatile species into the atmosphere. These observations underline the importance of photochemistry on urban SOA physicochemical properties and lifetime. |
Introduction
Organic aerosols (OA) are nefarious to human health,1–3 influence the global climate by scattering and absorbing solar radiation,4,5 and modify the lifetime and albedo of clouds by acting as cloud condensation nuclei.6–10 OA are a key component of atmospheric aerosols and account for 20 to 90% of the tropospheric submicron aerosol mass.11,12 The OA sources include anthropogenic sources; i.e., traffic emissions, industrial activities, energy production,13,14 and biogenic sources such as biomass burning and emissions from vegetation.15–18From these important emission sources, the oxidation of volatile organic compounds (VOCs) leads to oxidation products of lower volatility than the precursor VOC and greatly participates in SOA formation and growth. This growth occurs either by the reaction of an oxidized gas molecule with a particle after collision (i.e., reactive uptake),19,20 by absorptive partitioning21,22 or by the condensation of low-vapor pressure compounds.12,18,23,24 Due to the importance of SOA on our environment and health, investigating the chemical processes involved in SOA growth and aging is necessary, especially given that organic species experience substantial chemical changes over their lifetime (usually a couple of days).25
SOA aging is influenced by a variety of chemical processes, such as oxidative processes, (e.g. heterogeneous oxidation from ozone (O3), N2O5, or hydroxyl radicals (OH), which can be formed in the aerosol itself),26 or non-oxidative processes, e.g., hydrolysis, aldolisation, esterification, peroxyhemiacetal/hemiacetal formation.27,28 These chemical reactions lead to different outcomes (e.g., fragmentation, functionalization, or oligomerization) in the condensed phase.12,29 Such changes not only alter the SOA chemical composition but also ultimately influence their optical and physical properties (e.g., volatility, hygroscopicity, viscosity).30–32 Functionalization and oligomerization tend to decrease the volatility of the molecules by increasing their polarity or carbon number, thus changing their lifetime in the aerosol phase. Fragmentation and carbon bond cleavage result in higher vapor pressure products, which can cause a reduction of the SOA mass if fragmentation continues unabated.31,33 Photochemical processes have also been shown to greatly impact the SOA chemical composition during aging,34–38 and are hypothesized to be a key driver of SOA physicochemical properties. More specifically, previous work showed that photochemical aging of biogenic-derived SOA alters their morphology from well-mixed fresh to phase-separated aerosols.39 This process may be caused by an increase in oligomer content increasing particle viscosity. In addition to impacting SOA composition and morphology, photodegradation is a major SOA removal mechanism that needs to be considered.30,40–44 This mass loss is associated with the presence of photolabile functional groups that are susceptible to be photodegraded and form volatile species that can outgas from the condensed phase. Among them, organic peroxides and carbonyls represent an important fraction of the SOA mass45–50 and have been identified as photochemically active species.40,42–44 Krapf et al.40 suggested that carbonyls determine the photolability of α-pinene SOA rather than peroxides while Pospisilova et al.30 showed that moderately oxygenated molecules are the most photolabile species. These uncertainties underline the need to perform additional investigations on the SOA evolution under various environmental conditions.
In urban atmospheres, polycyclic aromatic hydrocarbons (PAHs) represent an important group of anthropogenic precursors capable of forming SOA.51–54 Composed of fused aromatic rings, PAHs can be rapidly oxidized by the different atmospheric oxidants, predominantly OH radicals.55–59 PAH-derived SOA are also known to constitute a considerable health hazard because of their deleterious effect on human health (carcinogenic, embryotoxic, oxidative potential).2–4,60–64 Studies have shown that PAH-derived SOA are sensitive to photochemistry.65 As a result, evaluating the impact of photodegradation on PAH-derived SOA is important to understand the evolution of aerosol particles notably in urban environments.
In this study, we investigated the formation and evolution of secondary organic aerosol (SOA) produced from the oxidation of naphthalene, one of the most abundant gaseous PAH in the atmosphere,63,66 by OH radicals under various UV and environmental conditions. Experiments were conducted in a Teflon atmospheric simulation chamber. Gas and particle-phase chemical characterization was achieved using state-of-the-art online mass spectrometry techniques.
Experimental section
Experiments were conducted at the Paul Scherrer Institute in Switzerland in a 9 m3 atmospheric chamber made of Teflon® fluorocarbon film described by Platt et al.67 They were performed in batch mode (i.e., static), with no dilution once the particle formation started, except during the first part of the low-NOx experiment, in which an airflow was unintentionally kept open for 2.5 hours before being closed. The container housing the chamber was temperature-regulated and maintained at a temperature of 24 °C (±2 °C). The chamber was flushed overnight at a flow rate of 35 standard liter per minute (SLPM) with dry purified air obtained from an AADCO (AADCO 250 series, AADCO Instruments, Inc., USA) system. At the start of the experiments, humid air was produced by bubbling dry clean air through ultrapure water. Several instruments were used to monitor the experimental conditions. O3 was obtained by passing pure air over a UV light source inside a stainless-steel cylinder and was measured by a UV photometric O3 analyzer (model 49C, Thermo). A dew point hygrometer (SC-05, Rotronic) was used to monitor the temperature and relative humidity (RH). NOx concentrations were not available, but significant differences in NOx levels were expected depending on the OH production process. OH radicals were produced within the chamber by either the ozonolysis of tetramethylethylene (TME) (IUPAC name: 2,3-dimethyl-2-butene), a reaction yielding OH, HO2 radicals and acetone with low NOx levels,68 or by the photolysis of nitrous acid (HONO), resulting in OH radicals and nitric oxide (NO).69 In the dark experiment, NOx concentrations would remain below limit of detection since there was no NOx injections into the chamber. Historically, generating OH radicals with HONO results in NOx concentrations below 2 ppb without the injection of either NO or NO2.69,70 Photodegradation was initiated using a bank of black light (set of 40 Cleo Performance solarium lamps from Philips with a power of 90–100 W each, emission spectrum available in Fig. S1†). Another study using this chamber estimated the UV-A lights JNO2 to be 3 × 10−3 s−1.30 OH radical concentrations were estimated by monitoring the decay of butanol-d9 (1 μL–30 ppb) as previously described by Barmet et al.71 Naphthalene was injected into the chamber by passing an airflow through the headspace of solid naphthalene and was monitored with a total hydrocarbon counter (THC monitor APHA-370, Horiba).The impact of aging and photochemistry-driven aging on naphthalene-derived SOA was investigated by performing two types of experiments: (i) particles grown in the dark before being exposed to the lights, (ii) particles grown with the lights on and aged in the dark. SOA formation in the dark was performed by mixing naphthalene with OH radicals generated from TME ozonolysis, resulting in oxidation under low-NOx levels. Therefore, this experiment is referred to as the low-NOx experiment. This experiment was conducted at high relative humidity (RH = 71%) with a concentration of 1.25 ppm and 106 ppb of O3 and naphthalene, respectively. 10 μL of TME (227 ppb) was injected twice through a septum into a heated line which was flushed with dry air to introduce the VOC into the chamber. The first injection was diluted by the airflow that remained open. After closing the airflow, the second injection was performed to form more SOA. During the high-NOx experiment, OH radicals were formed from the photolysis of HONO. This experiment was conducted at high relative humidity (RH ∼ 82%) in the presence of 131 ppb of naphthalene with a background level of 0.09–0.1 ppm of O3. HONO was obtained by passing an airflow through a reactor where both sulfuric acid (H2SO4) and sodium nitrite (NaNO2) were added dropwise.72
The chemical composition of gaseous species was retrieved with an atmospheric pressure chemical ionization (CI) inlet coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific, USA), hereafter referred to as CI-Orbitrap, and a Vocus proton transfer reaction time-of-flight mass spectrometer (Vocus 2R PTR-TOF, Tofwerk), later referred to as Vocus. An in-depth description and characterization of the inlet was performed by Riva et al.,73 so the CI inlet was only described succinctly here. A coaxial sample flow of 10 SLPM was supplied to the instrument and was kept in the center of the drift tube by adding a 20–40 SLPM sheath flow controlled by a mass flow controller (MFC) to minimize wall losses. By flushing 2 standard cubic centimeters per minute (sccm) of dry pure air over a 1% liquid ammonia solution, ammonia vapors (NH3) were generated and mixed with the sheath flow. NH3 vapors passed through a soft X-ray photoionizer (Hamamatsu, L9491) to generate ammonium ions (NH4+). NH4+ was chosen as the reagent ion due to its low selectivity (i.e., suitable for a large variety of oxidation products74).
The Vocus reagent ion source consisted of a hollow cathode producing a plasma between two conical surfaces.75 Like the CI-Orbitrap, the Vocus was operated in ammonia mode. The generated NH4+ ions entered the focusing ion-molecule reactor (FIMR), which replaced the drift tube of traditional PTR-TOF.75–79 NH4+ ions and the analytes were mixed within the FIMR where adduct ions were formed. Using both instruments allowed us to study compounds with an extended volatility range74,80 and to be more robust toward possible RH variations. Indeed, the NH4+-CI-Orbitrap has exhibited a RH dependence, which was not the case for the Vocus-PTR.74,75
Particles in the experiment were monitored using a scanning mobility particle sizer (SMPS, TSI Incorporated, USA) over a 15–638 nm diameter range. Particles were chemically characterized using an extractive electrospray ionization inlet coupled to an atmospheric pressure interface time-of-flight mass spectrometer (APi-TOF, Tofwerk AG, Thun, Switzerland). This instrument was previously described by Lopez-Hilfiker and referred to as EESI-TOF from henceforth.81 The aerosol flow (0.8–1.0 SLPM) before entering the EESI-TOF went through a multi-channel carbon denuder (Ionicon) to remove >99.9% of gas-phase compounds.81,82 Periodical blanks (∼every 5–15 min) were performed by passing the aerosols through a HEPA filter to obtain a background-corrected signal. The EESI electrospray solution consisted of ultrapure water doped with 100 ppm of sodium iodide (NaI), pushed through an untreated fused silica capillary (360 μm outer diameter, 75 μm internal diameter, BGB Analytik AG, Boeckten, Switzerland) at 100–200 mbar. The fluid flow was controlled by a high-precision pressure regulator (MFCS-EZ 1000 mbar, Fluigent, Inc., Lowell, MA, USA) coupled to the electrospray liquid bottle. 2700–2900 V was applied to ensure spray stability and particle extraction.81,83 The instrument was operated in positive mode, resulting in the formation of [analyte + Na]+ adduct ions. The EESI-TOF and Vocus signal were normalized using the method detailed in the ESI.†
Results and discussion
Particle formation and growth
Fig. 1 shows the SOA formation and the evolution of the mass concentration and chamber conditions throughout the experiments, with particle densities assumed to be 1.5 g cm−3 (Kautzman et al. 2010).84 Both the total SOA mass concentration (orange circles) and the mass loadings for individual particle sizes (color scale) are indicated. An overview of the experimental conditions was compiled in Table S1.† While SOA can form from naphthalene ozonolysis,85 particles were not observed before the production of OH radicals in both experiments. Presumably, this was due to the weak naphthalene reactivity with ozone. The first injection of TME resulted in particle formation up to ∼102 μg m−3. Due to the unintentional dilution flow in the chamber, OH levels were not estimated and SOA mass rapidly decreased. Therefore, a second injection of 10 μL of TME was performed after ∼2.75 hours (t = −1 hour in Fig. 1A) to increase particle mass, with RH = 33% and 0.6 ppm of O3 present in the chamber. This second injection resulted in an OH radical concentration of 2.7 × 107 molecules per cm3, which was significantly higher than tropospheric OH levels (1.1 × 106 molecules per cm3).86 This high OH level rapidly increased particle mass concentration to 122 μg m−3. While the first injection raised the number of particles from 646 particles per cm3 (i.e., background level) to 1.2 × 106 particles per cm3, the 2nd injection only showed an increase from 5.4 × 104 to 2 × 105 particles per cm3. This difference indicates that existing particles acted as a condensation sink for gaseous oxygenated species produced from naphthalene oxidation, and inhibited part of the new particle formation. The fast SOA formation from the 1st injection was followed by a significant mass concentration decrease, from ∼102 to 42 μg m−3 (59% decrease). | ||
| Fig. 1 Evolution of SOA mass concentration measured by the SMPS and chamber conditions for (A) low-NOx and (B) high-NOx experiment. The mass loadings of the various sizes of particles are indicated by the color scale. t = 0 hours marked the start of the UV lights, and dotted white bars indicate the period when UV lights were turned on. The chamber conditions monitored include the total SOA mass (wall-loss corrected, orange circles), RH (blue), ozone (green), and non-methane hydrocarbon (NMHC, yellow). NMHC was used to monitor the naphthalene concentrations as well as the TME injected. The measurement unit was ppmC, meaning that the real concentrations of naphthalene and TME were respectively 10 times and 6 times lower than what the NMHC suggests. | ||
The 2nd injection raised the mean mass geometric diameter to ∼140 nm, indicating that gaseous species condensed onto preexisting particles instead of participating in new particle formation and allowed to produce sufficient SOA mass to study aging processes. The mean diameter stayed within 140–155 nm for the rest of the experiment. Lights were turned on ∼50 min after the second injection for 65 min to study the impact of the photodegradation processes on dark-grown SOA. When lights were turned on, a net decrease of both the mean mass geometric diameter (from 150 to 140 nm) and particle mass concentration (12% loss after wall-loss corrections, details about the correction in Pospisilova et al.)87 was observed. This is indicative that photodegradation led to particle mass reduction due to a release of volatile compounds, as observed in previous studies.30,41–44
During the high-NOx experiment, UV lights were turned on for 1.5 hours. OH radicals concentration reached 4.1 × 108 molecules per cm3 and up to 1.8 × 105 particles per cm3 were formed. SOA formation peaked at 254 μg m−3, with a geometric mean diameter of 160 nm. The mass loss associated with photodegradation cannot be observed in this case, as it was overshadowed by the SOA formation. Over the next 5 hours, particles continued to grow due to either the slow condensation of organic vapors or coagulation, and the geometric mean diameter increased by ∼20% up to 190 nm.
Gas- and particle-phase species identification
Naphthalene oxidation from OH radicals occurred mostly (∼95%) from the OH addition pathway,88 with the remaining fraction of the reaction taking place through the hydrogen abstraction pathway. The resulting RO2 radicals were C10H7Oy, C10H9Oy, and C10H11Oy, and the main dimers formed from the bimolecular reaction of these RO2 radicals were C20H16Oy, C20H18Oy and C20H20Oy.88–90 The suggested reaction pathways were given in Fig. S2,† and the resulting closed-shell oxidation products observed were compiled in Fig. S3 and Table S2.† The contribution of the oxidation pathways to SOA formation was estimated by comparing the ratios of C10H6 oxidation products (formed through the H abstraction pathway) to C10H10 compounds (formed through the OH addition pathway). At the peak of SOA formation of the low-NOx experiment, particle-phase ratio amounted to 0.07 while gas-phase
ratio amounted to 0.07 while gas-phase  ratio was 0.25, showing that C10H10 species were more abundant (relatively to C10H6) in the particle-phase than in the gas-phase. Similar observations from the beginning of SOA formation in the high-NOx experiment were noted, with particle-phase and gas-phase
ratio was 0.25, showing that C10H10 species were more abundant (relatively to C10H6) in the particle-phase than in the gas-phase. Similar observations from the beginning of SOA formation in the high-NOx experiment were noted, with particle-phase and gas-phase  amounting to 0.11 and 0.84, respectively. This suggests that species produced from the OH addition pathway partitioned more efficiently towards particle-phase, although it is important to stress that these observations were qualitative.
amounting to 0.11 and 0.84, respectively. This suggests that species produced from the OH addition pathway partitioned more efficiently towards particle-phase, although it is important to stress that these observations were qualitative.
In the low-NOx experiment, the SOA chemical composition remained similar across the two injections (Fig. S3-A†). Moreover, most of the gaseous and particulate compounds detected were consistent with what was observed in previous studies on naphthalene oxidation.52,65,84,91 Table S2† depicts both N-containing and non-N-containing species identified in previous studies. However, to ease the comparison between the different experiments, this section focuses on the non-N-containing compounds. Important non-N-containing oxidation products were identified such as phthaldialdehyde (C8H6O2), 1,4-naphthoquinone (C10H6O2), and 2-formyl-cinnamaldehyde (C10H8O2). Generally, condensed-phase non-N-containing compounds were highly oxidized (i.e., O ≥ 4 for monomers and ≥6 for dimers), with no visible difference between the low- and high-NOx conditions. As shown in Fig. S3-A and Table S2,† some of the most abundant particle-phase compounds identified in the low-NOx experiment included C8H6O4 (m/z = 166.026, number 7 in Fig. S3 and Table S2†), C10H8O3 (m/z = 176.047, number 10), C10H8O4 (m/z = 192.042, number 13), C10H10O3 (m/z = 178.062, number 11), C10H10O4 (m/z = 194.057, number 14), and C10H10O5 (m/z = 210.052, number 17). The three former compounds (C8H6O4, C10H8O3, and C10H8O4) were ring-opening products. Contrariwise, C10H10O3 was a ring-retaining product and C10H10O4 and C10H10O5 were also most likely ring-retaining compounds. High-NOx particulate oxidation products (Fig. S3-C and Table S2†) exhibited a lesser fraction of non-N-containing ring-retaining species, suggested by the larger concentration of C10H8O4 compared to C10H10O4. The same trend was observed for the gas-phase oxidation products, where C10H10O2 (m/z = 160.052, number 6) dominated in the low-NOx environment (Fig. S3-B and Table S2†) while C10H8O2 (m/z = 162.068, number 5) dominated in the high-NOx environment (Fig. S3-D and Table S2†). These results agree with previous studies,52,84 which noted a higher relative intensity in gas-phase ring-retaining products for low-NOx conditions compared to high-NOx conditions.
EESI signal variations
Fig. S4-A† compares the variation of the wall-loss corrected SOA mass recorded by the SMPS with the evolution of the normalized total signal recorded by the EESI for C8–10HxOy and C18–20HxOy species during the low-NOx experiment. The light period caused a 12% loss of the particle mass loadings, while the EESI normalized signal originally did not show a considerable drop. After that, the particle mass remained constant, while the EESI signal continued to decay quickly. A possible explanation can be the dependency of the EESI sensitivity as a function of the particle size. Indeed, as shown by Lee et al.92 the EESI signal depends not only on the mass loading but also on the geometric mean aerosol diameter. This is caused by smaller particles diffusing and coalescing more efficiently with the electrospray droplets, therefore resulting in greater extraction efficiency. However, this effect is mainly important for particles with a diameter lower than 100 nm. In our experiment, naphthalene-derived SOA exhibited a mean diameter of ∼150 nm (Fig. 1A). The variation in the mean diameter during the low-NOx irradiation period was roughly 3%, i.e., 145 nm. This variation was negligible and should not have influenced the extraction efficiency. Furthermore, Pospisilova et al.30 observed a similar trend, i.e., a larger decrease in the EESI signal compared to the SOA mass loading. In their experiment, the mean diameter of the SOA did not vary significantly under irradiation, which was consistent with our observations. Hence, the EESI signal decrease was most likely not caused by the EESI sensitivity dependence on the particles' diameter. Instead, Pospisilova et al.30 proposed that the EESI-TOF was less sensitive to the composition of the aged α-pinene SOA, which were composed of a higher fraction of highly oxygenated compounds after irradiation. Lopez-Hilfiker et al.,81 who characterized the EESI-TOF, showed that the instrument was the most sensitive for species with a relatively low oxygen number (O = 3–4 for carboxylic acids, 5–6 for saccharides and 4–6 for α-pinene SOA), while more oxygenated species were detected less efficiently. Bell et al.93 also found that the EESI-TOF had a positive bias towards the less oxygenated monomers, and a negative bias towards highly oxygenated monomers and dimers. A possible explanation is that more oxygenated species are less volatile, which makes them less likely to evaporate and be measured by the instrument. On the other hand, less oxygenated species could more readily evaporate from the EESI droplets. Therefore, the formation of more oxygenated species at the expense of lower oxygen-containing monomers could explain the loss of EESI-TOF sensitivity. In contrast to the photodegradation period, the decay rates of the mass and EESI signal during the dark aging period were consistent, both decreasing by ∼10% by the end of the low-NOx experiment and by ∼18% by the end of the high-NOx experiment (Fig. S4†). Dark aging did not change the chemistry of the SOA enough to cause a change in the EESI sensitivity, underlying the importance of photochemical aging.Photolysis rates of bulk SOA
During the low-NOx experiment, photolysis rates of the total wall loss-corrected particle mass and the total EESI signal were estimated to be 3.5 × 10−3 s−1 and 2.7 × 10−3 s−1, respectively. These photolysis rates were obtained using the method presented by Pospisilova et al.30 In that previous study, photolysis rates of 6.5–7.6 × 10−4 s−1 for the particle mass and 4.1–5.6 × 10−4 s−1 for the EESI total signal were estimated. The differences in photolysis rates are likely caused by the fact that α-pinene-derived SOA are more photorecalcitrant than naphthalene-derived SOA. This is further supported by Romonosky et al.94 who showed that naphthalene-derived SOA were more likely to be lost through photodegradation than α-pinene-derived SOA. Additionally, naphthalene-derived SOA in our low-NOx experiment were formed under higher RH than Pospisilova et al.30 Hence, naphthalene-derived SOA photolytic losses were most likely enhanced by the higher RH, as shown by Wong et al.44 Indeed, photodissociation products in less viscous SOA (high RH) could more efficiently diffuse out of the particle, while recombination could have more time to happen in more viscous particles (low RH).Global impact of photodegradation on SOA chemical composition
The time evolution of SOA species is depicted in Fig. 2A and B while the evolution of gaseous species is presented in Fig. 2C and D (t = 0 hours indicates when UV lights were turned on). The time scale during the low-NOx experiment showed species formed in the dark (Fig. 2A and C starting at t = −0.7 hours) before they were exposed to UV lights (t = 0 hours). During the high-NOx experiment, the time scale of the experiment started after irradiation began (Fig. 2B and D starting at t = 0.4 hours), as species could not be detected by the EESI-TOF before that point. EESI-TOF figures (Fig. 2A and B) show the evolution of the relative intensity of the species, with all compounds combined amounting to 1. Gas-phase figures (Fig. 2C and D) show the evolution of the signals of compounds measured by the CI-Orbitrap and the Vocus. The compounds were grouped as C8–9HxOy, C10HxOy, C18–19HxOy, and C20HxOy, the first two groups being monomers and the last two groups being dimers. These compounds are expected to contain functionalities such as a peroxide group or an ester group as the linking functionality between the two monomeric units.18,95 Dimeric compounds such as ester accretion products can also be produced from particulate-phase reactions.95,96 Unfortunately, the instruments used in this study do not provide information about the chemical structures. | ||
| Fig. 2 Time evolution of the relative intensity of species (the sum of all compounds amounting to 1) and wall-corrected organic mass measured by the EESI-TOF and SMPS, respectively, for (A) the low-NOx experiment (t = −0.7 hours to 2.6 hours) and (B) the high-NOx experiment (t = 0.4 hours to 6.6 hours). The evolution of the signal for the gaseous species, measured by the CI (full lines) and the Vocus (dotted lines), was shown in (C) and (D) for the low-NOx and high-NOx experiments, respectively. The irradiation period started at t = 0 hours and is delimited by the yellow lines, ending just over t = 1 hour for (A) and (C), and ending at t ∼1.5 hours for (C) and (D). | ||
Fig. 2A shows that photodegradation-induced SOA mass loss correlated with a change in particle composition during the low-NOx experiment. This change was seen for dimeric species, which evolved differently under irradiation depending on their carbon numbers. The main dimers produced from gas-phase chemistry, i.e., C20HxOy species, experienced a net loss during photodegradation. On the other hand, the fraction of dimeric compounds with lower carbon numbers, i.e., C18–19HxOy species, increased when the lights were on. The difference in dimer composition (C20 → C18,19) suggest that dimer photodegradation altered specifically C20HxOy species. Similar results were observed in Fig. 2B for the high-NOx experiment. It should be noted that Lee et al.91 showed a decrease in the double bond equivalent (DBE) which they associated with the fragmentation of larger naphthalene-derived SOA compounds.
Regarding the monomers during the low-NOx experiment, the main species (C10HxOy) initially increased when the lights were turned on and then slowly decreased under irradiation, while the C8–9HxOy molecules steadily increased with irradiation. It is worth mentioning that the fraction of monomers and dimers remained at similar levels (55% and 45% respectively) before and after the lights were on. This indicates that accretion reactions were not the main reaction pathway. The gaseous monomers and dimers species also increased, as seen in Fig. 2C. Comparing the evolution of gaseous C20 dimers was not possible since the instruments were not able to detect C20HxOy. A possible reason is the prompt formation of new particles which reached high mass loadings and provided an additional condensation sink that could have favored the partitioning of species with low volatility, such as C20HxOy oxidation products. However, the evolution of the gaseous monomers did not show significant differences between C8–9HxOy and C10HxOy where the gas-phase species were delayed for those formed in the particulate phase.
Finally, dimeric species also behaved differently than monomers in the dark. In the dark, both experiments showed that the fraction of particulate C10HxOy and C8–9HxOy started decreasing once irradiation stopped, while the fraction of dimers increased. This process was probably not linked to partitioning, as the gas-phase monomers remained stable or decreased in the dark. Species detected by the Vocus seemed to decrease faster than those measured by the CI-Orbitrap, most likely due to the instrument's selectivity toward different species.74,80
Photodegradation of CxHy compounds
To elucidate which compounds were optically active or formed from the photodegradation, oxidation products were sorted out according to their carbon and hydrogen numbers as well as their evolution. Before photodegradation in the low-NOx experiment, C20H16–22Oy had high relative signals (see Fig. 3 or Table S3† for numeric values). As shown in Fig. S2,† such compounds can be expected from bimolecular RO2 reactions and can rapidly condense and partition to the particle-phase due to their low vapor pressure. However, these species could also have been formed from particle-phase reactions. The current data does not allow to confirm the dominance of one pathway to another. Nevertheless, these species increased in the dark before being depleted by the photodegradation process which led to the production of species with a different number of carbon, hydrogen, and oxygen atoms.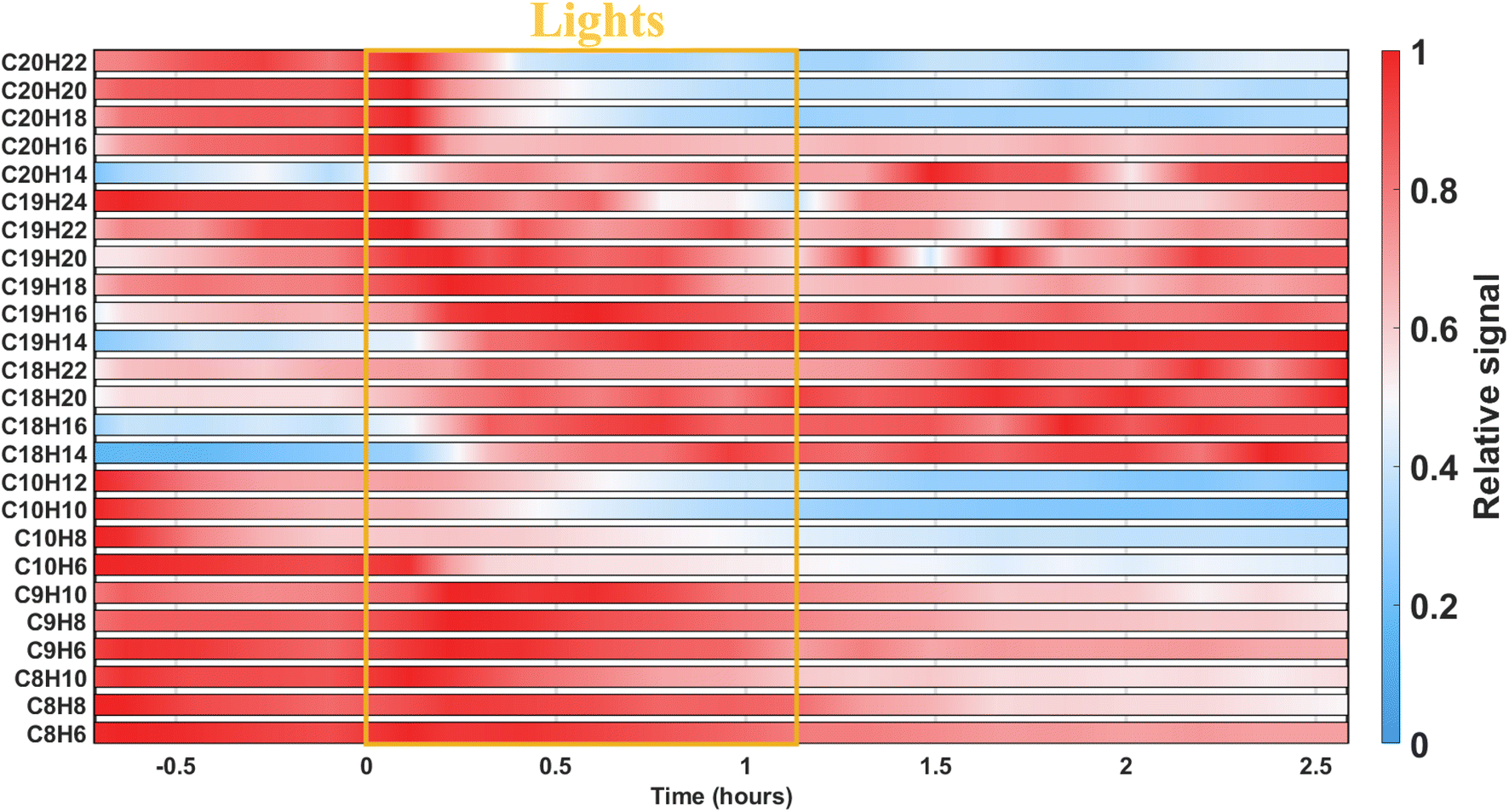 | ||
| Fig. 3 Evolution of the relative signal of particle-phase species during the low-NOx experiment, with compounds grouped by carbon and hydrogen numbers. Red indicates that the species are close to their maximum relative intensity, while blue signals a low relative intensity. UV lights are indicated by the yellow box. | ||
During the low-NOx experiment, photodegradation was not uniform across all dimeric compounds and appeared to be dependent on the hydrogen number. The most saturated C20 species (i.e., C20H18–22Oy), which were expected from radical reactions as described before, experienced a greater loss under irradiation than the compounds having larger DBE (e.g., C20H14,16Oy). Similarly, the increase of relative signal for C18–19 species was more important for the most unsaturated species (e.g., C18–19H14–16Oy) than for the ones with a higher hydrogen-content. This indicated that the unsaturated species were mainly produced when the UV lights were on. It is worth noting that they were moderately formed from gas-phase chemistry and then remained constant in the condensed phase when the UV lights were off.
This difference in reactivity could also be explained by the nature of the functional groups present in the molecule. It is known that carbonyl and peroxy moieties are photolabile and likely to be depleted under irradiation.97 It has also been shown that photodegradation results in an increase of species carrying a carboxylic acid group.30,65,97 Replacing an organic peroxy functional group with a carboxylic acid would be associated with the loss of at least 2 hydrogen atoms, which aligns with the increase of unsaturated dimers. Additionally, dimers with a high H number probably did not bear functional groups associated with a reduced H content, such as carbonyls and carboxylic acids. Instead, they were more likely to bear organic peroxide moieties or hydroxyl groups. Finally, similar observations can be made for monomeric species such as C10HxOy, decreasing under the UV lights due to photodegradation, with the most saturated species (i.e., C10H10–12Oy) decreasing more than the ones with lower H content (C10H6–8Oy). C8HxOy behaved similarly, although the variations were not as important as for C10HxOy. Interestingly, these changes appeared to be exclusive to the particulate phase, as all gas-phase monomers increased homogeneously under irradiation as depicted in Fig. S6.† Similar results were found in the high-NOx experiment, as shown in Fig. S7,† for both non-N-containing and N-containing dimers. This suggests that autoxidation reactions resulted in oxidation products bearing photolabile functional groups (such as carbonyls)40,42–44 under both low- and high-NOx levels.
Evolution of individual species under irradiation
After having determined how the families of species evolved under UV lights, variations of the individual particulate monomers and dimers during the low-NOx experiment were further explored as shown in Fig. 4A and B respectively. This was done using stacked general Kendrick analysis (GKA) plots. GKA plots are related to traditional Kendrick mass defect analysis but use a tunable parameter to contract or expand the mass scale and a base unit that determines the horizontal alignment of the species. In this study, the base unit chosen was oxygen, so species were plotted horizontally according to their oxygen content. To generate the stacked figures, GKA values were calculated for different experimental times using the method presented by Alton et al.,98 and were then stacked on top of each other to represent the time evolution of the relative intensity of the compounds. Many low-oxygenated species (O = 2–4), both for C8–9HxOy and C10HxOy, decreased under irradiation. This suggests that photolabile functional groups were already present even for low-oxygenated species. Therefore, the OH-initiated oxidation of naphthalene must have resulted in at least one photolabile moiety early on. This was supported by previous studies showing monomers with a low oxygen content bearing carbonyl functional groups.84,85,99 However, it cannot be ruled out that the loss of C8–10HxO2–4 could also be due to repartitioning from the particle-phase to the gas-phase, as proposed by Pospisilova et al.30 As a result, the less oxygenated particulate monomers were then possibly depleted both by photodegradation and/or evaporation. More oxidized species should also have been sensitive to photodegradation but seemed to increase (e.g., C8H8O3–5, C9H6O4–5, C9H8O4–6, and C9H10O5–7) or remain stable during irradiation, as also observed in previous studies.30,97 The lack of loss could be explained by the presence of photorecalcitrant functional groups such as carboxylic acids or through the production of oxidants in the particle-phase during irradiation producing highly oxygenated monomers.26 Another source of those species would be gas-phase compounds condensing after being formed under UV lights (Fig. S8-B†). The decay in the dark of the compounds within one family seemed to also depend slightly on the O content of the species, as the most oxidized particulate compounds decayed more slowly (e.g., C9H10O7 compared to C9H10O2). Higher oxygenated compounds have lower volatility, meaning that even with photodegradation they could continue to have sufficiently low volatility to remain in the particle-phase.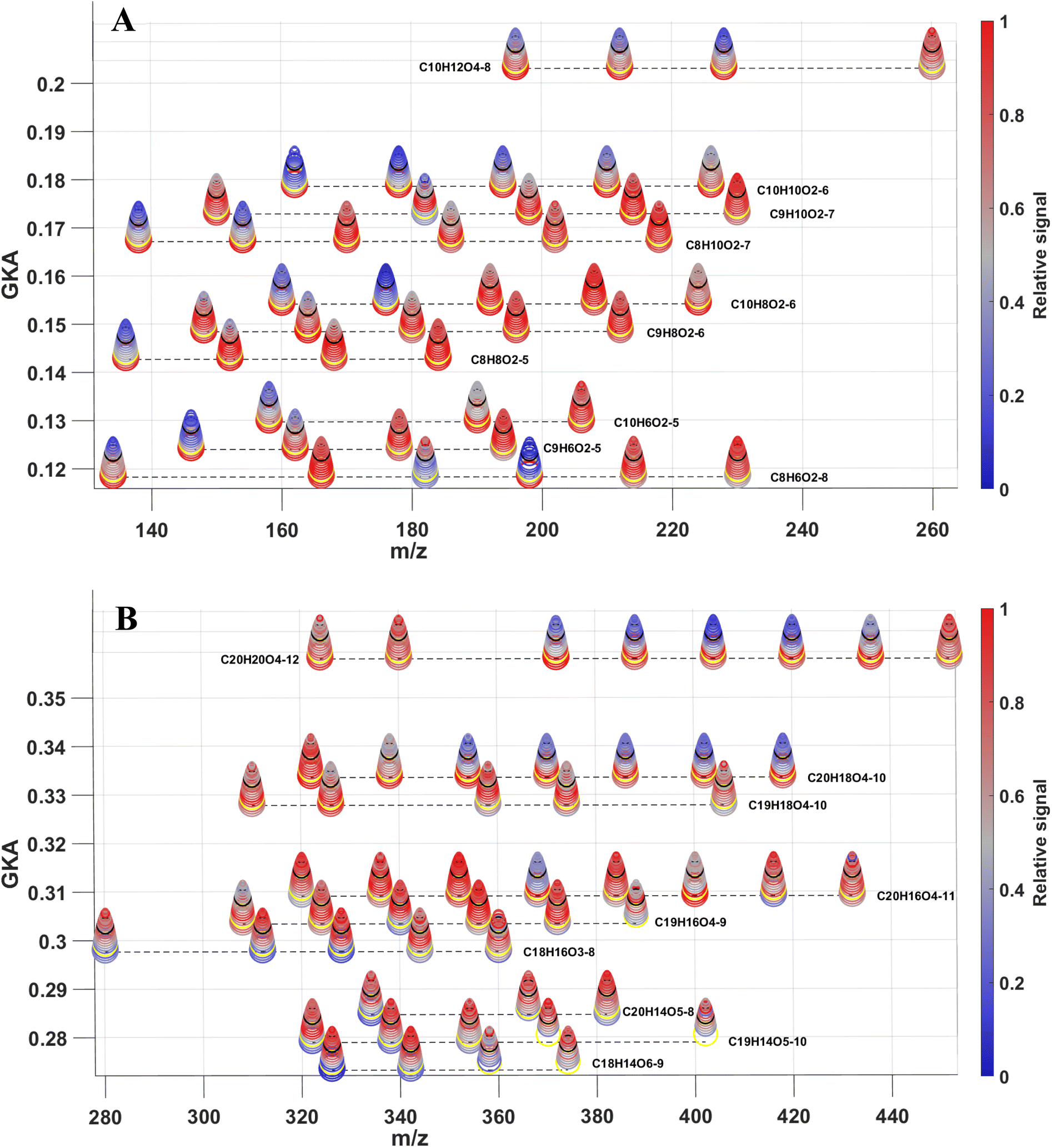 | ||
| Fig. 4 Stacked generalized Kendrick analysis plots for (A) monomers and (B) dimers during the low-NOx experiment. The multiple rings show the elapsed time from the start of the experiment (largest and lowest ring) to the end of the experiment (smallest and highest ring). The relative intensity of each compound is indicated by the color (blue = low intensity, red = high intensity, grey = medium intensity). The yellow and black rings indicate the beginning and the end of the irradiation period respectively. | ||
The evolution of the dimers is presented in Fig. 4B. Similarly to the monomers, the photodegradation efficiency of the C20H16–20Oy compounds was dependent on the oxygen number. However, whereas many low-oxygenated monomers were lost upon photodegradation, this was not the case for the dimers, the low-oxidized dimers seeming less inclined to be photodegraded. Contrariwise to the monomers, dimers with a low oxygen content (O = 3–5) were less likely to evaporate from the particle phase. Additionally, C20HxOy species with a low number of oxygen atoms were less photodegraded. A possible explanation is that less oxygenated C20HxOy species possessed fewer functional groups, which decreased the chance of carrying a photolabile functional group. Furthermore, low-oxygenated dimers could also have been formed from the photodegradation of the more oxygenated dimers. Almost all the C18–19H14–18Oy showed an increase in production during irradiation. The clearest increase concerned C18–19H14–16O≤7 species, i.e., unsaturated low-oxidized dimers. This was consistent with the results of the C20H16–20Oy, whose photodegradation would have resulted in C18–19HxOy. Many of the C18–19H14–16O>7 compounds were not present at first, but were formed during irradiation. Overall, this showed that the photodegradation of the particles was not only dependent on the hydrogen and carbon numbers but also on the oxygen number of a molecule. The global O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio slightly increased after the photodegradation (from 0.408 to 0.417), which was similar to the observations in Bateman et al.,97 who showed an increased O:C ratio after the irradiation of D-limonene-derived SOA. Results from high- and low-NOx experiments were consistent as presented in Fig. S10.†
:C ratio slightly increased after the photodegradation (from 0.408 to 0.417), which was similar to the observations in Bateman et al.,97 who showed an increased O:C ratio after the irradiation of D-limonene-derived SOA. Results from high- and low-NOx experiments were consistent as presented in Fig. S10.†
Emission of volatile species from particle- to gas-phase following photodegradation
The photodegradation of C20H16–20Oy to C18–19H14–16Oy would most likely result in the emission of formic acid (HC(O)OH), formaldehyde (HCHO), acetic acid (CH3C(O)OH), or acetaldehyde (CH3CHO) to the gas-phase, as proposed in Fig. 5A. The emission of these species is a distinctive sign of photodegradation that has been shown in multiple SOA photodegradation studies.35–37,44,97 We investigated the production of these species using the Vocus and found that formic acid, acetic acid, and acetaldehyde increased during photodegradation (Fig. 5B and C). This confirms the photodegradation pathway and has important atmospheric implications. Indeed, these species are also highly important in the atmosphere due to their abundance, the acids' ability to contribute to the free atmospheric acidity, and the aldehydes' role as transition species leading to the formation of HO2 or CO in the atmosphere.100,101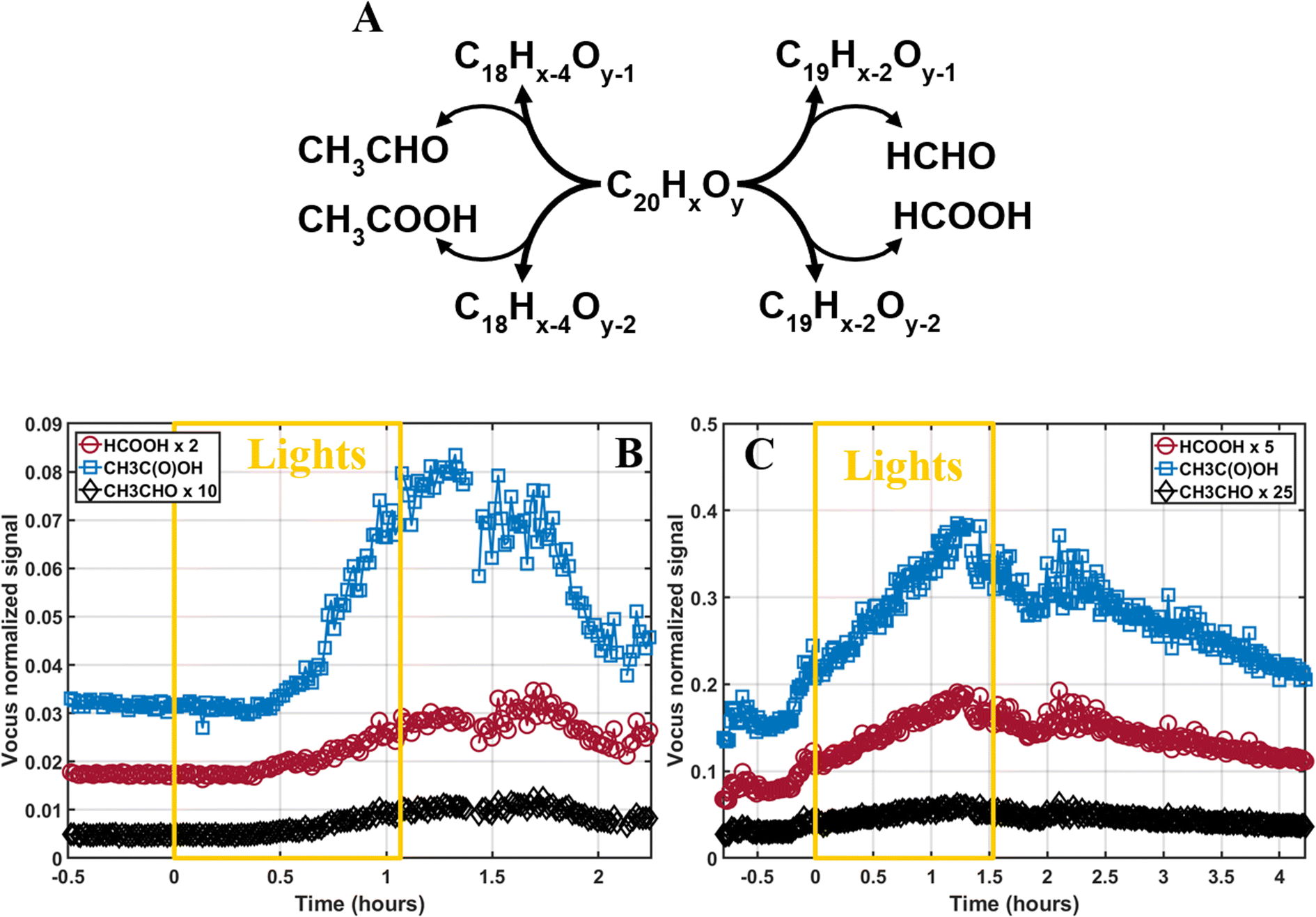 | ||
| Fig. 5 (A) Photodegradation mechanism of C20HxOy dimers. The time evolution of formic acid (HC(O)OH), acetic acid (CH3C(O)OH), and acetaldehyde (CH3CHO) are shown during (B) low- and (C) high-NOx experiments. The intensity of the HC(O)OH and CH3CHO signals was adjusted for easier visualization. t = 0 hours indicate the start of lights, and the yellow box indicates the irradiation period. | ||
To estimate the concentrations of acetic acid, formic acid, and acetaldehyde emitted, previous experiments from the same campaign using known amounts of monoterpene injected as well as the Vocus sensitivity towards these monoterpenes were used. Furthermore, we estimated the sensitivity of the Vocus towards the photodegradation volatile species by comparing the differences in instrumental sensitivity between monoterpene and organic acids/carbonyls from another study.102 The resulting concentrations are shown in Table 1 and reached in the low-NOx experiment up to 3 ppb, 0.5 ppb, and 0.07 ppb of acetic acid, formic acid, and acetaldehyde, respectively. During the high-NOx experiment, emissions reached up to 10 ppb, 1 ppb, and 0.08 ppb for acetic acid, formic acid, and acetaldehyde, respectively. The amounts of volatile products emitted were consistent with the loss of SOA mass in the low-NOx experiment, but it should be noted that these results remain qualitative.
| ppb | μg m−3 | SOA mass loss | % Of SOA loss from the volatile species | |
|---|---|---|---|---|
| Low-NOx | Acetic acid: 3 | Acetic acid: 7.5 | 14 μg m−3 | Acetic acid: 54% |
| Formic acid: 0.5 | Formic acid: 0.93 | Formic acid: 7% | ||
| Acetaldehyde: 0.07 | Acetaldehyde: 0.12 | Acetaldehyde: 0.9% | ||
| High-NOx | Acetic acid: 10 | Acetic acid: 25 | ✗ | ✗ |
| Formic acid: 1 | Formic acid: 2.3 | |||
| Acetaldehyde: 0.08 | Acetaldehyde: 0.15 |
Evolution of the volatility pre-to post-photodegradation
To characterize the change in the particles' physical properties that is expected from the changed chemical composition, the effective saturation mass concentration (C*) of each particulate oxidation product has been calculated. The estimation was made using the equation from Li et al.103 and followed their structural and formulaic estimations. Particle-phase oxidation products were categorized as intermediate volatile organic compounds (IVOCs), semi-volatile organic compounds (SVOCs), low volatile organic compounds (LVOCs), extremely low volatile organic compounds (ELVOCs), and ultra-low volatile organic compounds (ULVOCs) according to their estimated C*. Fig. 6 shows how the fraction of these species evolved during the low-NOx experiment pre- (Fig. 6A) to post-photodegradation (Fig. 6B). The fraction of the species during the high-NOx experiment at the beginning of the photodegradation and after photodegradation ended are displayed in Fig. 6C and D, respectively. Both experiments showed that species with low volatilities (i.e. LVOCs, ELVOCs and ULVOCs) slightly increased in both experiments after the particles were photodegraded. Additionally, the mean C* was calculated and showed that irradiated particles were less volatile after photodegradation (from 0.74 to 0.67 μg m−3 for low-NOx and from 1.53 to 1.04 μg m−3 for high-NOx). Low-NOx particles globally showed a lower C* than high-NOx particles, but low-NOxC* also did not decrease as much as high-NOxC* after irradiation. This was consistent with the larger decrease of IVOC post-photodegradation for the high-NOx experiment. Although C* variations were not very significant, they still agreed with the results from Pospisilova et al.30 who showed that α-pinene SOA presented lower volatility after its exposure to UV lights by performing isothermal evaporation experiments. However, our results may have been influenced by changes in the parametrizations used to calculate C*. Indeed, these parametrizations may have differed from pre-photodegradation to post-photodegradation as the functional groups in the particle-phase compounds could have changed. The fraction of non-N-containing and N-containing species in the high-NOx experiment remained unaffected by photodegradation. It went from a 0.76/0.24 ratio for non-N-containing and N-containing species respectively at the beginning of photodegradation (t = 0.4 hours) to a 0.75/0.25 ratio after photodegradation (t = 1.7 hours). | ||
| Fig. 6 (A) Volatility distribution of the particulate oxidation products for the low-NOx experiment before photodegradation (t = −0.1 h) and (B) after photodegradation (t = 1.13 h). (C) Volatility distribution for the high-NOx experiment at the beginning of photodegradation (t = 0.37 h) and (D) after photodegradation (t = 1.73 h). Colors indicate the fraction of ultra-low volatilite (ULVOCs), extremely low volatilite (ELVOCs), low volatilite (LVOCs), semi-volatile (SVOCs) and intermediate volatilite (IVOCs) organic compounds respectively. | ||
Conclusion
This study investigated the composition of naphthalene SOA and the influence of photodegradation as a marker for anthropogenic VOC/PAH aerosol photodegradation due to its abundance and role in urban environments. Our results showed that the changes observed in particles aging under irradiation were significantly greater than the variations caused by other chemical processes. These other processes were controlled by letting particles age under a similar timescale in the dark. Prominent degradation (2/3 loss) of the C20 dimers during photodegradation was revealed through an in-depth molecular characterization of naphthalene-derived SOA. The degradation of the dimers yielded particulate-phase fragmentation products with C18–19 while releasing C1–2 gaseous compounds. Photodegradation was found to be dependent on the chemical composition and functional groups with hydroperoxide and carbonyl species being the most photolabile groups. SOA physical properties were also affected by photodegradation, as particles showed a decrease in volatility. Further work coupling the EESI-TOF to a thermal denuder as done by Bell et al.93 would provide more accurate information about the particles changing volatilities.The photochemical process was accompanied by a loss of particle mass under irradiation (−12%) which corresponded to the release of volatile products. Among them, three products playing a major role in global atmospheric chemistry were identified: formic acid, acetic acid, and acetaldehyde. These results highlight the need to consider photochemical activity as one of the main drivers of SOA aging as naphthalene-derived SOA and other anthropogenic-sourced aerosols are an important factor affecting health, visibility, and the environment. Predicting their impact on any of these subjects requires information about their chemical composition and physical properties, both of which are modified due to UV irradiation, showcasing the complexity of considering SOA photodegradation in broader atmospheric issues.
Data availability
The data that support the findings of this study are available on request from the corresponding authors, D. B. at: E-mail: david.bell@psi.ch; and/or M. R. at: matthieu.riva@ircelyon.univ-lyon1.fr.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC-StG MAARvEL; no. 852161), the European Union’s Horizon 2020 research and innovation program through the ATMO-ACCESS Integrating Activity under grant agreement no. 101008004, as well the Swiss National Foundation (SNF – grant no. 200021 213071) for their financial support. PSI’s atmospheric simulation chamber is a facility of ACTRIS ERIC and receives funding from the Swiss State Secretariat for Education, Research and Innovation (SERI). We thank as well the Orbitool team for developing the tools to analyze Orbitrap mass spectra data.References
- R. Burnett, H. Chen, M. Szyszkowicz, N. Fann, B. Hubbell, C. A. Pope, J. S. Apte, M. Brauer, A. Cohen, S. Weichenthal, J. Coggins, Q. Di, B. Brunekreef, J. Frostad, S. S. Lim, H. Kan, K. D. Walker, G. D. Thurston, R. B. Hayes, C. C. Lim, M. C. Turner, M. Jerrett, D. Krewski, S. M. Gapstur, W. R. Diver, B. Ostro, D. Goldberg, D. L. Crouse, R. V. Martin, P. Peters, L. Pinault, M. Tjepkema, A. Van Donkelaar, P. J. Villeneuve, A. B. Miller, P. Yin, M. Zhou, L. Wang, N. A. H. Janssen, M. Marra, R. W. Atkinson, H. Tsang, T. Quoc Thach, J. B. Cannon, R. T. Allen, J. E. Hart, F. Laden, G. Cesaroni, F. Forastiere, G. Weinmayr, A. Jaensch, G. Nagel, H. Concin and J. V. Spadaro, Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9592–9597 CrossRef CAS.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Pöschl, S. Takahama, A. Takami, H. Tong, B. Weber, A. Yoshino and K. Sato, Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- T. Déméautis, M. Delles, S. Tomaz, G. Monneret, O. Glehen, G. Devouassoux, C. George and A. Bentaher, Pathogenic Mechanisms of Secondary Organic Aerosols, Chem. Res. Toxicol., 2022, 35(7), 1146–1161 Search PubMed.
- U. Pöschl, Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- J. Haywood and O. Boucher, Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review, Rev. Geophys., 2000, 38, 513–543 CrossRef CAS.
- Clouds in the Perturbed Climate System: Their Relationship to Energy Balance, Atmospheric Dynamics, and Precipitation, ed. J. Heintzenberg and R. J. Charlson, MIT Press, Cambridge, Mass, 2009 Search PubMed.
- U. Lohmann and J. Feichter, Global indirect aerosol effects: a review, Atmos. Chem. Phys., 2005, 5(3), 715–737 CrossRef CAS.
- S. Twomey, The nuclei of natural cloud formation part II: The supersaturation in natural clouds and the variation of cloud droplet concentration, Geofis. Pura Appl., 1959, 43, 243–249 CrossRef.
- B. A. Albrecht, Aerosols, Cloud Microphysics, and Fractional Cloudiness, Science, 1989, 245, 1227–1230 CrossRef CAS PubMed.
- R. Pincus and M. B. Baker, Effect of precipitation on the albedo susceptibility of clouds in the marine boundary layer, Nature, 1994, 372, 250–252 CrossRef CAS.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. V. Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5(4), 1053–1123 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS.
- J. G. Watson, J. C. Chow and E. M. Fujita, Review of volatile organic compound source apportionment by chemical mass balance, Atmos. Environ., 2001, 35, 1567–1584 CrossRef CAS.
- A. Kansal, Sources and reactivity of NMHCs and VOCs in the atmosphere: A review, J. Hazard. Mater., 2009, 166, 17–26 CrossRef CAS PubMed.
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. Mckay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, A global model of natural volatile organic compound emissions, J. Geophys. Res.:Atmos., 1995, 100, 8873–8892 CrossRef CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J.-F. Müller, U. Kuhn, P. Stefani and W. Knorr, Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 2014, 14, 9317–9341 CrossRef.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9(14), 5155–5236 CrossRef CAS.
- Y.-H. Lin, Z. Zhang, K. S. Docherty, H. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton, T. E. Kleindienst, A. Gold and J. D. Surratt, Isoprene Epoxydiols as Precursors to Secondary Organic Aerosol Formation: Acid-Catalyzed Reactive Uptake Studies with Authentic Compounds, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS.
- P. Mettke, M. Brüggemann, A. Mutzel, R. Gräfe and H. Herrmann, Secondary Organic Aerosol (SOA) through Uptake of Isoprene Hydroxy Hydroperoxides (ISOPOOH) and its Oxidation Products, ACS Earth Space Chem., 2023, 7, 1025–1037 CrossRef CAS.
- I. Riipinen, J. R. Pierce, T. Yli-Juuti, T. Nieminen, S. Häkkinen, M. Ehn, H. Junninen, K. Lehtipalo, T. Petäjä, J. Slowik, R. Chang, N. C. Shantz, J. Abbatt, W. R. Leaitch, V.-M. Kerminen, D. R. Worsnop, S. N. Pandis, N. M. Donahue and M. Kulmala, Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 2011, 11, 3865–3878 CrossRef CAS.
- X. Zhang, S. N. Pandis and J. H. Seinfeld, Diffusion-Limited Versus Quasi-Equilibrium Aerosol Growth, Aerosol Sci. Technol., 2012, 46, 874–885 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- L. Gerritz, J. Wei, T. Fang, C. Wong, A. L. Klodt, S. A. Nizkorodov and M. Shiraiwa, Reactive Oxygen Species Formation and Peroxide and Carbonyl Decomposition in Aqueous Photolysis of Secondary Organic Aerosols, Environ. Sci. Technol., 2024, 58, 4716–4726 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- A. Zare, K. M. Fahey, G. Sarwar, R. C. Cohen and H. O. T. Pye, Vapor-Pressure Pathways Initiate but Hydrolysis Products Dominate the Aerosol Estimated from Organic Nitrates, ACS Earth Space Chem., 2019, 3, 1426–1437 CrossRef CAS.
- H. J. Chacon-Madrid and N. M. Donahue, Fragmentation vs. functionalization: chemical aging and organic aerosol formation, Atmos. Chem. Phys., 2011, 11, 10553–10563 CrossRef CAS.
- V. Pospisilova, D. M. Bell, H. Lamkaddam, A. Bertrand, L. Wang, D. Bhattu, X. Zhou, J. Dommen, A. S. H. Prevot, U. Baltensperger, I. El Haddad and J. G. Slowik, Photodegradation of α-Pinene Secondary Organic Aerosol Dominated by Moderately Oxidized Molecules, Environ. Sci. Technol., 2021, 55, 6936–6943 CrossRef CAS PubMed.
- T. Tritscher, J. Dommen, P. F. DeCarlo, M. Gysel, P. B. Barmet, A. P. Praplan, E. Weingartner, A. S. H. Prévôt, I. Riipinen, N. M. Donahue and U. Baltensperger, Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber, Atmos. Chem. Phys., 2011, 11, 11477–11496 CrossRef CAS.
- T. Feng, Y. Wang, W. Hu, M. Zhu, W. Song, W. Chen, Y. Sang, Z. Fang, W. Deng, H. Fang, X. Yu, C. Wu, B. Yuan, S. Huang, M. Shao, X. Huang, L. He, Y. R. Lee, L. G. Huey, F. Canonaco, A. S. H. Prevot and X. Wang, Impact of aging on the sources, volatility, and viscosity of organic aerosols in Chinese outflows, Atmos. Chem. Phys., 2023, 23, 611–636 CrossRef CAS.
- Y. Zhang, M. Cheng, J. Gao and J. Li, Review of the influencing factors of secondary organic aerosol formation and aging mechanism based on photochemical smog chamber simulation methods, J. Environ. Sci., 2023, 123, 545–559 CrossRef CAS.
- M. L. Walser, J. Park, A. L. Gomez, A. R. Russell and S. A. Nizkorodov, Photochemical Aging of Secondary Organic Aerosol Particles Generated from the Oxidation of d-Limonene, J. Phys. Chem. A, 2007, 111, 1907–1913 CrossRef CAS PubMed.
- S. A. Mang, D. K. Henricksen, A. P. Bateman, M. P. S. Andersen, D. R. Blake and S. A. Nizkorodov, Contribution of Carbonyl Photochemistry to Aging of Atmospheric Secondary Organic Aerosol, J. Phys. Chem. A, 2008, 112, 8337–8344 CrossRef CAS PubMed.
- X. Pan and J. S. Underwood, Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry, Atmos. Chem. Phys., 2009, 9(12), 3851–3865 CrossRef CAS.
- K. T. Malecha and S. A. Nizkorodov, Photodegradation of Secondary Organic Aerosol Particles as a Source of Small, Oxygenated Volatile Organic Compounds, Environ. Sci. Technol., 2016, 50, 9990–9997 Search PubMed.
- C. Wu, D. M. Bell, E. L. Graham, S. Haslett, I. Riipinen, U. Baltensperger, A. Bertrand, S. Giannoukos, J. Schoonbaert, I. El Haddad, A. S. H. Prevot, W. Huang and C. Mohr, Photolytically induced changes in composition and volatility of biogenic secondary organic aerosol from nitrate radical oxidation during night-to-day transition, Atmos. Chem. Phys., 2021, 21, 14907–14925 CrossRef CAS.
- J. E. Shilling, M. A. Zawadowicz, J. Liu, R. A. Zaveri and A. Zelenyuk, Photochemical Aging Alters Secondary Organic Aerosol Partitioning Behavior, ACS Earth Space Chem., 2019, 3, 2704–2716 Search PubMed.
- M. Krapf, I. El Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. H. Prévôt, U. Baltensperger and J. Dommen, Labile Peroxides in Secondary Organic Aerosol, Chem, 2016, 1, 603–616 Search PubMed.
- M. A. Zawadowicz, B. H. Lee, M. Shrivastava, A. Zelenyuk, R. A. Zaveri, C. Flynn, J. A. Thornton and J. E. Shilling, Photolysis Controls Atmospheric Budgets of Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 2020, 54, 3861–3870 Search PubMed.
- S. A. Epstein, S. L. Blair and S. A. Nizkorodov, Direct Photolysis of α-Pinene Ozonolysis Secondary Organic Aerosol: Effect on Particle Mass and Peroxide Content, Environ. Sci. Technol., 2014, 48, 11251–11258 CrossRef CAS PubMed.
- K. M. Henry and N. M. Donahue, Photochemical Aging of α-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 2012, 116, 5932–5940 CrossRef CAS PubMed.
- J. P. S. Wong, S. Zhou and J. P. D. Abbatt, Changes in Secondary Organic Aerosol Composition and Mass due to Photolysis: Relative Humidity Dependence, J. Phys. Chem. A, 2015, 119, 4309–4316 Search PubMed.
- D. E. Romonosky, A. Laskin, J. Laskin and S. A. Nizkorodov, High-Resolution Mass Spectrometry and Molecular Characterization of Aqueous Photochemistry Products of Common Types of Secondary Organic Aerosols, J. Phys. Chem. A, 2015, 119, 2594–2606 CrossRef CAS PubMed.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O3, Environ. Sci. Technol., 2005, 39, 4049–4059 Search PubMed.
- J. D. Surratt, S. M. Murphy, J. H. Kroll, N. L. Ng, L. Hildebrandt, A. Sorooshian, R. Szmigielski, R. Vermeylen, W. Maenhaut, M. Claeys, R. C. Flagan and J. H. Seinfeld, Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 2006, 110, 9665–9690 CrossRef CAS PubMed.
- T. B. Nguyen, A. P. Bateman, D. L. Bones, S. A. Nizkorodov, J. Laskin and A. Laskin, High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 2010, 44, 1032–1042 Search PubMed.
- P. Mertes, L. Pfaffenberger, J. Dommen, M. Kalberer and U. Baltensperger, Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP), Atmos. Meas. Tech., 2012, 5, 2339–2348 Search PubMed.
- S. Aimanant and P. J. Ziemann, Development of Spectrophotometric Methods for the Analysis of Functional Groups in Oxidized Organic Aerosol, Aerosol Sci. Technol., 2013, 47, 581–591 Search PubMed.
- K. M. Shakya and R. J. Griffin, Secondary Organic Aerosol from Photooxidation of Polycyclic Aromatic Hydrocarbons, Environ. Sci. Technol., 2010, 44, 8134–8139 Search PubMed.
- A. W. H. Chan, K. E. Kautzman, P. S. Chhabra, J. D. Surratt, M. N. Chan, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 2009, 9(9), 3049–3060 CrossRef CAS.
- H. O. T. Pye and G. A. Pouliot, Modeling the Role of Alkanes, Polycyclic Aromatic Hydrocarbons, and Their Oligomers in Secondary Organic Aerosol Formation, Environ. Sci. Technol., 2012, 46, 6041–6047 Search PubMed.
- D. S. Tkacik, A. A. Presto, N. M. Donahue and A. L. Robinson, Secondary Organic Aerosol Formation from Intermediate-Volatility Organic Compounds: Cyclic, Linear, and Branched Alkanes, Environ. Sci. Technol., 2012, 46, 8773–8781 CrossRef CAS.
- M. Tsapakis and E. G. Stephanou, Diurnal Cycle of PAHs, Nitro-PAHs, and oxy-PAHs in a High Oxidation Capacity Marine Background Atmosphere, Environ. Sci. Technol., 2007, 41, 8011–8017 Search PubMed.
- L. Wang and L. Wang, Atmospheric oxidation mechanism of acenaphthene initiated by OH radicals, Atmos. Environ., 2020, 243, 117870 Search PubMed.
- M. Riva, R. M. Healy, P.-M. Flaud, E. Perraudin, J. C. Wenger and E. Villenave, Gas- and particle-phase products from the photooxidation of acenaphthene and acenaphthylene by OH radicals, Atmos. Environ., 2017, 151, 34–44 Search PubMed.
- Y. Bedjanian, M. L. Nguyen and G. Le Bras, Kinetics of the reactions of soot surface-bound polycyclic aromatic hydrocarbons with the OH radicals, Atmos. Environ., 2010, 44, 1754–1760 CrossRef CAS.
- R. Atkinson and J. Arey, Atmospheric chemistry of gas-phase polycyclic aromatic hydrocarbons: formation of atmospheric mutagens, Environ. Health Perspect., 1994, 102, 117–126 CAS.
- K. Ravindra, R. Sokhi and R. Vangrieken, Atmospheric polycyclic aromatic hydrocarbons: Source attribution, emission factors and regulation, Atmos. Environ., 2008, 42, 2895–2921 Search PubMed.
- Y. Yang, P. Guo, Q. Zhang, D. Li, L. Zhao and D. Mu, Seasonal variation, sources and gas/particle partitioning of polycyclic aromatic hydrocarbons in Guangzhou, China, Sci. Total Environ., 2010, 408, 2492–2500 CrossRef CAS.
- K.-H. Kim, S. A. Jahan, E. Kabir and R. J. C. Brown, A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects, Environ. Int., 2013, 60, 71–80 CrossRef CAS PubMed.
- N.-D. Dat and M. B. Chang, Review on characteristics of PAHs in atmosphere, anthropogenic sources and control technologies, Sci. Total Environ., 2017, 609, 682–693 CrossRef CAS PubMed.
- A. Nel, Air Pollution–Related Illness Effect of Particles, Science, 2005, 308, 804–805 CrossRef CAS PubMed.
- M. Riva, E. S. Robinson, E. Perraudin, N. M. Donahue and E. Villenave, Photochemical Aging of Secondary Organic Aerosols Generated from the Photooxidation of Polycyclic Aromatic Hydrocarbons in the Gas-Phase, Environ. Sci. Technol., 2015, 49, 5407–5416 CrossRef CAS.
- J. Arey, R. Atkinson, B. Zielinska and P. A. McElroy, Diurnal concentrations of volatile polycyclic aromatic hydrocarbons and nitroarenes during a photochemical air pollution episode in Glendora, California, Environ. Sci. Technol., 1989, 23, 321–327 CrossRef CAS.
- S. M. Platt, I. El Haddad, A. A. Zardini, M. Clairotte, C. Astorga, R. Wolf, J. G. Slowik, B. Temime-Roussel, N. Marchand, I. Ježek, L. Drinovec, G. Močnik, O. Möhler, R. Richter, P. Barmet, F. Bianchi, U. Baltensperger and A. S. H. Prévôt, Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 2013, 13, 9141–9158 CrossRef.
- A. T. Lambe, J. Zhang, A. M. Sage and N. M. Donahue, Controlled OH Radical Production via Ozone-Alkene Reactions for Use in Aerosol Aging Studies, Environ. Sci. Technol., 2007, 41, 2357–2363 CrossRef CAS PubMed.
- V. Kumar, J. G. Slowik, U. Baltensperger, A. S. H. Prevot and D. M. Bell, Time-Resolved Molecular Characterization of Secondary Organic Aerosol Formed from OH and NO3 Radical Initiated Oxidation of a Mixture of Aromatic Precursors, Environ. Sci. Technol., 2023, 57, 11572–11582 CrossRef CAS PubMed.
- L. Stirnweis, C. Marcolli, J. Dommen, P. Barmet, C. Frege, S. M. Platt, E. A. Bruns, M. Krapf, J. G. Slowik, R. Wolf, A. S. H. Prévôt, U. Baltensperger and I. El-Haddad, Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from a-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 2017, 17, 5035–5061 CrossRef CAS.
- P. Barmet, J. Dommen, P. F. DeCarlo, T. Tritscher, A. P. Praplan, S. M. Platt, A. S. H. Prévôt, N. M. Donahue and U. Baltensperger, OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 2012, 5, 647–656 CrossRef CAS.
- M. Taira and Y. Kanda, Continuous generation system for low-concentration gaseous nitrous acid, Anal. Chem., 1990, 62, 630–633 CrossRef CAS.
- M. Riva, M. Ehn, D. Li, S. Tomaz, F. Bourgain, S. Perrier and C. George, CI-Orbitrap: An Analytical Instrument To Study Atmospheric Reactive Organic Species, Anal. Chem., 2019, 91, 9419–9423 CrossRef CAS PubMed.
- D. Li, D. Wang, L. Caudillo, W. Scholz, M. Wang, S. Tomaz, G. Marie, M. Surdu, E. Eccli, X. Gong, L. Gonzalez-Carracedo, M. Granzin, J. Pfeifer, B. Rörup, B. Schulze, P. Rantala, S. Perrier, A. Hansel, J. Curtius, J. Kirkby, N. M. Donahue, C. George, I. El-Haddad and M. Riva, Ammonium CI-Orbitrap: a tool for characterizing the reactivity of oxygenated organic molecules, Atmos. Meas. Tech., 2024, 17, 5413–5428 CrossRef CAS.
- J. Krechmer, F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, D. Worsnop, K. Fuhrer, M. Gonin and J. De Gouw, Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 2018, 90, 12011–12018 CrossRef CAS PubMed.
- A. Hansel, A. Jordan, R. Holzinger, P. Prazeller, W. Vogel and W. Lindinger, Proton transfer reaction mass spectrometry: on-line trace gas analysis at the ppb level, Int. J. Mass Spectrom. Ion Processes, 1995, 149–150, 609–619 CrossRef.
- R. S. Blake, P. S. Monks and A. M. Ellis, Proton-Transfer Reaction Mass Spectrometry, Chem. Rev., 2009, 109, 861–896 CrossRef CAS PubMed.
- M. Graus, M. Müller and A. Hansel, High resolution PTR-TOF: Quantification and formula confirmation of VOC in real time, J. Am. Soc. Mass Spectrom., 2010, 21, 1037–1044 Search PubMed.
- A. Jordan, S. Haidacher, G. Hanel, E. Hartungen, L. Märk, H. Seehauser, R. Schottkowsky, P. Sulzer and T. D. Märk, A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 2009, 286, 122–128 CrossRef CAS.
- M. Riva, P. Rantala, J. E. Krechmer, O. Peräkylä, Y. Zhang, L. Heikkinen, O. Garmash, C. Yan, M. Kulmala, D. Worsnop and M. Ehn, Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 2019, 12, 2403–2421 CrossRef CAS.
- F. D. Lopez-Hilfiker, V. Pospisilova, W. Huang, M. Kalberer, C. Mohr, G. Stefenelli, J. A. Thornton, U. Baltensperger, A. S. H. Prevot and J. G. Slowik, An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 2019, 12, 4867–4886 CrossRef CAS.
- P. Eichler, M. Müller, B. D'Anna and A. Wisthaler, A novel inlet system for online chemical analysis of semi-volatile submicron particulate matter, Atmos. Meas. Tech., 2015, 8, 1353–1360 CrossRef CAS.
- M. Surdu, V. Pospisilova, M. Xiao, M. Wang, B. Mentler, M. Simon, D. Stolzenburg, C. R. Hoyle, D. M. Bell, C. P. Lee, H. Lamkaddam, F. Lopez-Hilfiker, L. R. Ahonen, A. Amorim, A. Baccarini, D. Chen, L. Dada, J. Duplissy, H. Finkenzeller, X.-C. He, V. Hofbauer, C. Kim, A. Kürten, A. Kvashnin, K. Lehtipalo, V. Makhmutov, U. Molteni, W. Nie, A. Onnela, T. Petäjä, L. L. J. Quéléver, C. Tauber, A. Tomé, R. Wagner, C. Yan, A. S. H. Prevot, J. Dommen, N. M. Donahue, A. Hansel, J. Curtius, P. M. Winkler, M. Kulmala, R. Volkamer, R. C. Flagan, J. Kirkby, D. R. Worsnop, J. G. Slowik, D. S. Wang, U. Baltensperger and I. E. Haddad, Molecular characterization of ultrafine particles using extractive electrospray time-of-flight mass spectrometry, Environ. Sci.: Atmos., 2021, 1, 434–448 CAS.
- K. E. Kautzman, J. D. Surratt, M. N. Chan, A. W. H. Chan, S. P. Hersey, P. S. Chhabra, N. F. Dalleska, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Chemical Composition of Gas- and Aerosol-Phase Products from the Photooxidation of Naphthalene, J. Phys. Chem. A, 2010, 114, 913–934 Search PubMed.
- J. Liu, S. Zhu, T. Guo, B. Jia, L. Xu, J. Chen and P. Cheng, Smog chamber study of secondary organic aerosol formation from gas- and particle-phase naphthalene ozonolysis, Atmos. Environ., 2023, 294, 119490 Search PubMed.
- M. Li, E. Karu, C. Brenninkmeijer, H. Fischer, J. Lelieveld and J. Williams, Tropospheric OH and stratospheric OH and Cl concentrations determined from CH4, CH3Cl, and SF6 measurements, npj Clim. Atmos. Sci., 2018, 1, 29 Search PubMed.
- V. Pospisilova, F. D. Lopez-Hilfiker, D. M. Bell, I. El Haddad, C. Mohr, W. Huang, L. Heikkinen, M. Xiao, J. Dommen, A. S. H. Prevot, U. Baltensperger and J. G. Slowik, On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 2020, 6, eaax8922 Search PubMed.
- R. Atkinson and J. Arey, Mechanisms of the gas-phase reactions of aromatic hydrocarbons and PAHs with OH and NO3 radicals, Polycyclic Aromat. Compd., 2007, 27, 15–40 Search PubMed.
- A. Shiroudi and M. S. Deleuze, Theoretical Study of the Oxidation Mechanisms of Naphthalene Initiated by Hydroxyl Radicals: The H Abstraction Pathway, J. Phys. Chem. A, 2014, 118, 3625–3636 CrossRef CAS PubMed.
- A. Shiroudi, M. S. Deleuze and S. Canneaux, Theoretical Study of the Oxidation Mechanisms of Naphthalene Initiated by Hydroxyl Radicals: The OH-Addition Pathway, J. Phys. Chem. A, 2014, 118, 4593–4610 CrossRef CAS.
- H. J. (Julie) Lee, P. K. Aiona, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 2014, 48, 10217–10226 Search PubMed.
- C. P. Lee, M. Surdu, D. M. Bell, H. Lamkaddam, M. Wang, F. Ataei, V. Hofbauer, B. Lopez, N. M. Donahue, J. Dommen, A. S. H. Prevot, J. G. Slowik, D. Wang, U. Baltensperger and I. El Haddad, Effects of aerosol size and coating thickness on the molecular detection using extractive electrospray ionization, Atmos. Meas. Tech., 2021, 14, 5913–5923 CrossRef CAS.
- D. M. Bell, J. Zhang, J. Top, S. Bogler, M. Surdu, J. G. Slowik, A. S. H. Prevot and I. El Haddad, Sensitivity Constraints of Extractive Electrospray for a Model System and Secondary Organic Aerosol, Anal. Chem., 2023, 95, 13788–13795 Search PubMed.
- D. E. Romonosky, N. N. Ali, M. N. Saiduddin, M. Wu, H. J. (Julie) Lee, P. K. Aiona and S. A. Nizkorodov, Effective absorption cross sections and photolysis rates of anthropogenic and biogenic secondary organic aerosols, Atmos. Environ., 2016, 130, 172–179 CrossRef CAS.
- O. Peräkylä, T. Berndt, L. Franzon, G. Hasan, M. Meder, R. R. Valiev, C. D. Daub, J. G. Varelas, F. M. Geiger, R. J. Thomson, M. Rissanen, T. Kurtén and M. Ehn, Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 2023, 145, 7780–7790 CrossRef PubMed.
- C. M. Kenseth, N. J. Hafeman, S. P. Rezgui, J. Chen, Y. Huang, N. F. Dalleska, H. G. Kjaergaard, B. M. Stoltz, J. H. Seinfeld and P. O. Wennberg, Particle-phase accretion forms dimer esters in pinene secondary organic aerosol, Science, 2023, 382, 787–792 Search PubMed.
- A. P. Bateman, S. A. Nizkorodov, J. Laskin and A. Laskin, Photolytic processing of secondary organic aerosols dissolved in cloud droplets, Phys. Chem. Chem. Phys., 2011, 13, 12199 Search PubMed.
- M. W. Alton, H. J. Stark, M. R. Canagaratna and E. C. Browne, Generalized Kendrick analysis for improved visualization of atmospheric mass spectral data, Atmos. Meas. Tech., 2023, 16, 3273–3282 CrossRef CAS.
- V. Lannuque and K. Sartelet, Development of a detailed gaseous oxidation scheme of naphthalene for secondary organic aerosol (SOA) formation and speciation, Atmos. Chem. Phys., 2024, 24, 8589–8606 CrossRef CAS.
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Atmospheric formic and acetic acids: An overview, Rev. Geophys., 1999, 37, 227–248 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley, 2016 Search PubMed.
- L. Xu, M. M. Coggon, C. E. Stockwell, J. B. Gilman, M. A. Robinson, M. Breitenlechner, A. Lamplugh, J. D. Crounse, P. O. Wennberg, J. A. Neuman, G. A. Novak, P. R. Veres, S. S. Brown and C. Warneke, Chemical ionization mass spectrometry utilizing ammonium ions (NH4+ CIMS) for measurements of organic compounds in the atmosphere, Atmos. Meas. Tech., 2022, 15, 7353–7373 Search PubMed.
- Y. Li, U. Pöschl and M. Shiraiwa, Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 2016, 16, 3327–3344 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00125g |
| ‡ Present address: Department of Chemistry and Molecular Biology, Atmospheric Science, University of Gothenburg, SE-41390, Gothenburg, Sweden. |
| § Present address: Department of Chemistry, Aarhus University, DK-8000, Aarhus C, Denmark. |
| This journal is © The Royal Society of Chemistry 2025 |