
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergetic effects of cation and anion of Mg(NO3)2 as electrolyte additives in stabilizing Li metal anode†
Hyerim
Kim‡
a,
Jimin
Park‡
a,
Hyokyeong
Kang
a,
Sunggun
Lim
b,
Shivam
Kansara
 a,
Zhaowei
Sun
*c,
Shizhao
Xiong
*c,
Yun-Chae
Jung
d,
Chihyun
Hwang
d,
Marco
Agostini
e and
Jang-Yeon
Hwang
*ab
a,
Zhaowei
Sun
*c,
Shizhao
Xiong
*c,
Yun-Chae
Jung
d,
Chihyun
Hwang
d,
Marco
Agostini
e and
Jang-Yeon
Hwang
*ab
aDepartment of Energy Engineering, Hanyang University, Seoul 04763, Republic of Korea. E-mail: jangyeonhw@hanyang.ac.kr
bDepartment of Battery Engineering, Hanyang University, Seoul 04763, Republic of Korea
cFaculty of Material Science and Engineering, Kunming University of Science and Technology, Kunming, 650093, China. E-mail: 20230231@kust.edu.cn; shizhao.xiong@kust.edu.cn
dBatteries Research Center, Korea Electronics Technology Institute, Seongnam, Gyeonggi 13509, Republic of Korea
eDepartment of Chemistry and Drug Technologies, Sapienza University of Rome, P.le Aldo Moro 5, Rome 00185, Italy
First published on 2nd April 2025
Abstract
The fabrication of a uniform solid electrolyte interface (SEI) with high robustness and ionic conductivity has long been necessary for Li metal anodes with high-performance; however, realizing such a design remains challenging. In this work, we applied magnesium nitrate (Mg(NO3)2) as an additive and γ-butyrolactone (GBL) as a solubility mediator to optimize an ester-based electrolyte for Li metal anodes. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) and X-ray photoelectron spectroscopy (XPS) results revealed that the Mg(NO3)2 in the 0.1 Mg(NO3)2-GBL electrolyte spontaneously reacted with the Li metal anodes to form a continuous Li–Mg alloy with Li3N phases dominating the SEI on the anode. The formed SEI induced a homogenous Li plating/stripping behavior on the anode surface, which improved the charge/discharge performance of the anode and inhibited Li dendritic growth. Furthermore, owing to the high ionic conductivity and large young's modulus of the inorganic phases in the optimized SEI, the Li metal anode maintained a stable and flat surface morphology even after cycling for 3000 h without any dendrite-induced issues, thus guaranteeing a long lifespan for the corresponding battery system. In addition, the symmetrical cell using only 10 μL of 0.1 Mg(NO3)2-GBL electrolyte maintained constant voltage hysteresis in the range of 12–18 mV over 620 h before the voltage swelled, suggesting such a superior electrolyte under a restricted condition.
Broader contextWith a high specific capacity of 3860 mA h g−1 and a low potential of −3.04 V (vs. SHE), the lithium metal anode (LMA) has long been regarded as the best candidate for high energy density in future batteries. However, the use of lithium metal batteries (LMBs) remains challenging because of severe and uncontrollable issues including capacity fading and safety concern. In general, these issues are related to the inhomogeneous interfacial reaction behavior of Li+ on the LMA causing the growth of dendrites. To overcome these issues, the most effective strategy is to modify the composition of commercial carbonate electrolytes, thereby regulating the interfacial reaction processes of LMAs. In this work, we applied magnesium nitrate (Mg(NO3)2) as an additive and γ-butyrolactone (GBL) as a solubility mediator for stabilizing LMA in carbonate electrolyte. The Mg2+ and NO3− ions were not only able to adjust the solvation structure of Li+, but could also introduce favorable inorganic SEI components such as Li3N and Li–Mg alloy on the LMA. In systems with such an electrolyte, the LMA can maintain a densely packed surface morphology and show stable cycling in LMBs. This study may inspire future electrolyte designs and promote LMA application in commercial batteries. |
Introduction
With the rapid development of society, industries for energy-consuming items like portable devices and electric vehicles are growing quickly, which drives an urgent need for high-performance electric energy storage batteries.1,2 However, the commonly applied commercial Li ion battery is approaching its theoretical upper performance limits and cannot meet market demand.3 The necessity of a breakthrough in improving electrochemical performance has motivated many attempts to fabricate new battery systems.4Among the currently proposed alternatives, Li metal batteries have attracted widespread industrial attention, mainly because Li metal anodes have demonstrated high gravimetric capacity (3860 mA h g−1) and low redox potential (−3.04 V vs. standard hydrogen electrode), showing great promise for high-energy batteries.5,6 However, the transfer of laboratory Li metal batteries to industrial applications is very challenging owing to the inherent issues of Li metal anodes, including rapid capacity loss and Li dendrite-induced problems.7 These two major issues mainly arise from uneven Li plating/stripping behavior on the anode surface. Because Li metal anodes store and release energy by direct Li plating/stripping on the anode surface,8 any unevenness leads to the local overgrowth of Li and a strong deformation force, thus damaging the passivating solid electrolyte interface (SEI) and inducing uncontrollable dendrite growth and side-reactions.9 In addition, active Li metal can react spontaneously with non-aqueous carbonate electrolytes forming an inhomogeneous and loose SEI dominated by harmful Li2CO3 and organic Li salts with poor strength and low Li ion conductivity,10,11 which in turn worsens the structural integrity of the SEI and interfacial reaction uniformity of the anode.12–14
To overcome these issues related to uneven plating/stripping,15 the interfacial reaction behavior must be optimized and the structural stability of the SEI must be improved.16 Researchers have proposed many approaches,9,17 which can be divided into the four categories of applying three-dimensional hosts,18 using solid-state-electrolytes,19 designing creditable artificial surface layers,20 and modifying the electrolyte components.21 Among these strategies, modifying the electrolyte components seems to be the most effective solution, inspiring extensive research on different points. For example, many previous studies have concentrated on optimizing the Li ion flux along the anode surface,22 aiming to realize a relatively homogenous Li ion distribution on the anode during cycling. Some researchers promoted moderate spontaneous reactions between the Li metal anode and sacrificial additives, which can introduce beneficial inorganic phases (Li halides, Li alloys, etc.) into the SEI structure and reduce the content of harmful organic Li salts,23 thus improving the ionic conductivity and structural stability of the SEI. Other works have emphasized points such as improving the solvated structure of Li ions,24 adjusting the local charge field in the Helmholtz double layer,25 and so on. All these strategies have effectively improved the SEI structure and properties, harmonized the charge field, and eliminating Li dendrite formation, thus providing inspiration for future development of Li metal batteries.
Despite these remarkable achievements, current electrolytes still face three obstacles (Fig. 1a). First, owing to the high reducibility of Li metal, spontaneous reactions between Li and the modified carbonate electrolyte are inevitable and induce the over-production of unfavorable organic Li salts in the SEI.26 Second, even the best SEI produced by the reaction between Li and additives cannot withstand high-rate charging/discharging conditions.27 Finally, realizing a homogeneous SEI is difficult owing to the uncontrollable synchronous reactions between Li and complicated electrolyte components.28 Therefore, the development of electrolytes that can form inorganic-phase rich, uniform, and durable SEI on Li metal anodes is necessary for future electrolyte development.29
 | ||
| Fig. 1 Schematic illustrations of the different SEI formed in baseline or 0.1 Mg(NO3)2-GBL electrolytes. (a) The inhomogeneous SEI with defects and abundant organic Li salts produced by spontaneous reactions between Li and the baseline electrolyte, which results in uneven deposition of Li that cannot block the penetration of dendrites. (b) Mg(NO3)2 additive contributes to the formation of an SEI containing plentiful favorable inorganic phases, which helps maintain relatively homogeneous interfacial reactions on the anode surface and suppresses dendritic growth. | ||
In our previous study,30 we discovered that a small amount of Magnesium nitrate (Mg(NO3)2) additive added to a carbonate electrolyte could spontaneously react with the Li metal anode to introduce beneficial inorganic Li–Mg alloy31 and Li3N32 phases (Table S1, ESI†) to the SEI, thereby improving the general performance of the anode. However, owing to the poor solubility of Mg(NO3)2 due to Gutmann donor number (DN) of NO3− (ref. 33–35) in carbonate, the modified electrolyte was supersaturated, which weakened the stability of the electrolyte and battery. In addition, the supersaturated electrolyte was inhomogeneous and formed an uneven SEI on the anode, thus reducing the advantages of the Mg(NO3)2 additive. To fully exploit the merits of the Mg(NO3)2 additive and produce a stable and high-performance electrolyte, we herein demonstrate a Mg(NO3)2 dissolved in carbonate electrolyte, in which Mg(NO3)2 functions as a sacrificial agent and γ-butyrolactone (GBL) serves as a solubility mediator, named the 0.1 Mg(NO3)2-GBL electrolyte. GBL solvents can help improve the solubility of NO3− in ester electrolyte due to the higher DN value of 18 kcal mol−1 (ref. 36) and high polarity37 compared with FEC35 and EMC38 solvents (Fig. S1, ESI†). Owing to the assistance of GBL, the 0.1 mol L−1 (M) Mg(NO3)2 additive was dissolved completely, forming a stable and uniform electrolyte. Therefore, the Mg2+ and NO3− could be reduced homogeneously on the surface of the Li metal anode, forming an SEI containing plentiful Li–Mg alloy, Li3N, and other beneficial inorganic phases (Fig. 1b). Furthermore, the strong interaction between Li+ and NO3− and coordination between Mg2+ and solvent molecules could decrease the number of solvent molecules surrounding the Li+,39 leading to facile Li+ desolvation during plating, which not only improved the interfacial reaction kinetics, but also added the favorable LiF phase40 to the SEI. Because of the high robustness and Li+ conductivity of the inorganic phases, the structural stability and mass transfer of the SEI were significantly improved. The combined merits of the 0.1 Mg(NO3)2-GBL electrolyte greatly improved the performance of the Li metal anode and the asymmetric cell based on the 0.1 Mg(NO3)2-GBL electrolyte showed a promising rate performance and cyclic stability and withstood a high current density of 12 mA cm−2 at a capacity loading of 4 mA h cm−2 for as long as 3000 h with no sign of dendrite-induced short circuiting. Furthermore, when paired with a sulfurized polyacrylonitrile (SPAN) cathode, the full battery using 0.1 Mg(NO3)2-GBL electrolyte exhibited a high coulombic efficiency of 99.98%, along with a low capacity decay rate of 0.82% after 150 cycles, demonstrating the practical value of the Mg(NO3)2-modified carbonate electrolyte with GBL.
Results and discussion
To investigate the advantages of the 0.1 Mg(NO3)2-GBL electrolyte in this study, a baseline electrolyte was prepared by dissolving 1 M lithium hexafluorophosphate (LiPF6) and 0.05 M lithium difluoro(oxalato)borate (LiDFOB) in a solution of ethyl methyl carbonate (EMC) and fluoroethylene carbonate (FEC) at a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The baseline electrolyte was clear and uniform, indicating a homogeneous solution. However, after adding 0.1 M Mg(NO3)2 to the baseline electrolyte, the liquid immediately became opaque, owing to the poor solubility of Mg(NO3)2 in carbonate. This non-uniform suspension proved to be ineffective for forming a homogeneous and intact SEI on the Li metal anode.41 However, after adding only a small amount of GBL to the Mg(NO3)2-containing baseline electrolyte, the solution soon became transparent, which can be attributed to the solubility-mediating function of GBL that effectively improves the solubility of Mg(NO3)2 in carbonate (Fig. 2a). Therefore, the uniformity and stability of the 0.1 Mg(NO3)2-GBL electrolyte were improved. The solubility-mediating function of GBL was also demonstrated by Raman spectra, as the curve of Mg(NO3)2 in GBL mixture depicted a more intense NO3− peak at 1040 cm−1 than that of the GBL solvent (Fig. S2, ESI†), suggesting that GBL facilitated the dissociation of Mg(NO3)2.
:1. The baseline electrolyte was clear and uniform, indicating a homogeneous solution. However, after adding 0.1 M Mg(NO3)2 to the baseline electrolyte, the liquid immediately became opaque, owing to the poor solubility of Mg(NO3)2 in carbonate. This non-uniform suspension proved to be ineffective for forming a homogeneous and intact SEI on the Li metal anode.41 However, after adding only a small amount of GBL to the Mg(NO3)2-containing baseline electrolyte, the solution soon became transparent, which can be attributed to the solubility-mediating function of GBL that effectively improves the solubility of Mg(NO3)2 in carbonate (Fig. 2a). Therefore, the uniformity and stability of the 0.1 Mg(NO3)2-GBL electrolyte were improved. The solubility-mediating function of GBL was also demonstrated by Raman spectra, as the curve of Mg(NO3)2 in GBL mixture depicted a more intense NO3− peak at 1040 cm−1 than that of the GBL solvent (Fig. S2, ESI†), suggesting that GBL facilitated the dissociation of Mg(NO3)2.
 | ||
| Fig. 2 Analysis of electrolytes. (a) Optical images of electrolytes: baseline, Mg(NO3)2 only (without GBL solvent), and 0.1 Mg(NO3)2-GBL. NMR spectra of (b) 7Li and (c) 13C corresponding to FEC and EMC solvation changes, respectively. (d) Schematic illustration of the diverse Li+ solvation structures of corresponding to the baseline and 0.1 Mg(NO3)2-GBL electrolytes. Raman spectra of (e) FEC and PF6− in the range of 710–760 cm−1, and (f) EMC in the range of 910–960 cm−1. | ||
To investigate the effect of Mg(NO3)2 on the Li+ solvation structure, 7Li and 13C of nuclear magnetic resonance (NMR) spectra were obtained to analyze the baseline and 0.1 Mg(NO3)2-GBL electrolytes (Fig. 2b, c and Fig. S3, ESI†). As shown in Fig. 2b, the 7Li curve of the 0.1 Mg(NO3)2-GBL electrolyte has a weaker and broader peak at −0.86 ppm compared to the 7Li curve of the baseline electrolyte (−0.92 ppm), suggesting a denser extranuclear electron cloud and a decreased number of solvent molecules surrounding the Li ion.42 Similarly, the 13C curve of EMC and FEC (Fig. 2c) in the 0.1 Mg(NO3)2-GBL electrolyte also depicted characteristic peaks shifting upfield compared to those of the baseline electrolyte, indicating a lower-density extranuclear electron cloud on the carbon atoms.43 These phenomena were due to the Mg(NO3)2 additive, which changed the electron configurations of the solvent components and thus that of the Li+ solvation structure. Owing to the higher positive-charge density on the valence layer, Mg2+ exhibited stronger adsorption to the solvent molecules than Li+; therefore, the FEC and EMC components in the electrolyte were more likely to form a solvation shell surrounding Mg2+, thus decreasing the number of solvent molecules around Li+. In addition, NO3− also had a strong interaction with Li+,42 thus contributing to the downfield effect of 7Li and weakening the dipolar interaction between EMC/FEC and the cations.44,45 Furthermore, as demonstrated by density functional theory (DFT) calculation results, the interaction between Li+ and NO3− (−1.083 eV per atom) was much stronger than that between Mg2+ and NO3− (−0.636 eV per atom, Table S2, ESI†); therefore, NO3− could replace the solvent molecules surrounding the Li+ and contribute to changing the solvation structure of Li+, while pushing PF6− anions away from Li+ as well.
Simplified models based on the Raman results are displayed in Fig. 2d to provide a clearer description of the diversity of the different electrolytes. The effectiveness of the Mg(NO3)2 additive on the carbonate solvent molecules was further verified by the Raman shift (Fig. 2e, f and Fig. S4, ESI†). The Raman spectra of the 0.1 Mg(NO3)2-GBL electrolyte showed that the percentages of solvated FEC (51%) and EMC (72%) increased with the addition of Mg(NO3)2, compared to those in the baseline electrolyte (36.4% FEC and 70% EMC), suggesting a strong coordination effect of Mg2+ on the solvated molecules and Li+ with NO3−. According to previous studies, the weak solvation structure of Li+ helps promote its desolvation on the anode surface, thus enhancing the interfacial reaction kinetics and improving the reaction uniformity on the anode. The DFT calculation results also showed that the interaction between Li+ and NO3− (−1.083 eV per atom) is stronger than that between Li+ and PF6− (−0.982 eV per atom, Table S2, ESI†), indicating that PF6− anions would be pushed away from Li+ by NO3− in the 0.1 Mg(NO3)2-GBL electrolyte. Therefore, the PF6− anions may be more electrochemically active and more likely to react with Li metal to form beneficial LiF phases in the SEI.
To recognize the function of the different electrolyte components in the formation of the SEI, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels were calculated using DFT. The results (Fig. 3a) showed that LiPF6, LiDFOB, and Mg(NO3)2 possessed relative lower LUMO energy levels of −1.797, −1.545, and −1.511 eV, respectively, compared with GBL, FEC, and EMC. The results suggest that the salt in the 0.1 Mg(NO3)2-GBL electrolyte reacts preferentially on the anode surface, whereas the reduction of organic components on the anode surface is suppressed; accordingly, the formation of favorable inorganic phases is promoted and that of harmful organic phases is restricted. From this perspective, the SEI formed in the 0.1 Mg(NO3)2-GBL electrolyte contained more inorganic phases than the SEI in the baseline electrolyte. The preferential reduction of Mg(NO3)2 on the Li metal anode was demonstrated using linear sweep voltammetry (LSV). The test was performed based on asymmetric cells with Li foil as the working electrode and Cu foil as the reference/counter electrode, using either the baseline electrolyte or the 0.1 Mg(NO3)2-GBL electrolyte. The LSV test was performed at a scan rate of 0.5 mV s−1 within the voltage range of 0.1–3.5 V. As shown in Fig. 3b, the curve of the cell using the 0.1 Mg(NO3)2-GBL electrolyte shows an obvious reduction peak at 1.67 V, which corresponds to the reduction of NO3− anions on the Li metal anode. In contrast, the curve of the cell using the baseline electrolyte shows only weak peaks corresponding to the reduction of the solvent species (GBL, FEC, and EMC) at relatively low potentials of 0.74–1.46 V. The LSV results proved the preferential reduction behavior of Mg(NO3)2 additives on the Li metal anode, which promoted the production of inorganic phases such as Li3N, Li2O, and the Li–Mg alloys on the anode surface, thus forming an inorganic phase-rich layer and optimizing the structure and performance of the SEI. The reaction behavior of the Li–Mg alloys was confirmed by X-ray diffraction (XRD) analysis. As shown in Fig. S4,† the XRD pattern of the Li metal anode after reaction with the 0.1 Mg(NO3)2-GBL electrolyte included a unique characteristic peak at 2θ = 65°, which was attributed to the Li3Mg7 alloy phase. Different Li plating behaviors in the different electrolytes were observed using scanning electron microscopy (SEM). After Li deposition onto Li metal anode at a capacity loading of 4 mA h cm−2 and current density of 12 mA cm−2, the Li metal anode in baseline electrolytes exhibited an uneven surface consisting of twisted Li dendrites (Fig. 3c), suggesting poor homogeneity of Li, guided by the spontaneous SEI formation in the carbonate electrolyte. In contrast, the Li metal anode in the 0.1 Mg(NO3)2-GBL electrolyte showed a relatively uniform morphology with no visible dendritic growth or mossy structures resulting from uniform Li+ flux distribution of the SEI consisting of Li–Mg alloy, Li3N (Fig. 3d). A similar phenomenon is observed in the cross-sectional SEM images (Fig. S6, ESI†), after plating at a capacity loading of 4 mA h cm−2 and current density of 12 mA cm−2. With the baseline electrolyte, the anode had a loose structure with a thick Li deposition layer (78 μm), while that in the 0.1 Mg(NO3)2-GBL electrolyte displayed a dense Li deposition layer (23 μm), further supporting the ability of the 0.1 Mg(NO3)2-GBL electrolyte to regulate the Li plating/stripping process on the Li metal anode.
 | ||
| Fig. 3 Calculations and SEM observations. (a) The HOMO and LUMO energy levels of the salts and solvents. (b) LSV curves of Li/Cu asymmetric cells with baseline and 0.1 Mg(NO3)2-GBL electrolytes. Top-view SEM images of (c) baseline and (d) 0.1 Mg(NO3)2-GBL electrolytes after deposition at a capacity loading of 4 mA h cm−2 and current density of 12 mA cm−2. | ||
To obtain a deeper understanding of the different SEI formed in the electrolytes with and without additives, time-of-flight secondary ion mass spectrometry (TOF-SIMS) was used to analyze the different SEI films. The detected Li−, Mg−, C2HO−, and F− were sourced from the anode samples after the reaction in the baseline and 0.1 Mg(NO3)2-GBL electrolytes (Fig. 4a). According to the three-dimensional profiles, the SEI induced by 0.1 Mg(NO3)2-GBL exhibited a homogeneous Mg− signal, suggesting that the formed Li–Mg alloy was dispersed uniformly in the SEI, which would benefit both the mechanical strength and Li+ transference of the SEI. Moreover, the F− profile indicates that the SEI induced by 0.1 Mg(NO3)2-GBL contained more of the favorable LiF phase, attributed to the presence of NO3−, which contributed to the separation of Li+ and PF6− and thus promoted the reduction of PF6− on the Li metal anode. As discussed previously, the LiF phase can effectively enhance the structural integrity and ionic conductivity of the SEI, leading to optimized interfacial reactions. Moreover, according to the C2HO− profile, the SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte had a reduced content of harmful organic phases. In contrast, the SEI formed in the baseline electrolyte yielded a non-uniform F− signal, suggesting heterogeneous LiF dispersion in the SEI, which may arise from the inhomogeneous reaction between PF6− and the Li metal anode in the baseline electrolyte (Fig. 4a and Fig. S7, ESI†). Furthermore, as demonstrated by the C2HO− profile, the SEI induced by the baseline electrolyte contained more harmful organic compounds, which may weaken its structural stability and integrity. The high content of organic compounds may result from further reactions between the Li dendrites and solvent molecules. In summary, the SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte is dominated by inorganic phases, which are beneficial for improving the general properties of the SEI and realizing a uniform Li plating/stripping processes, whereas the SEI induced by the baseline electrolyte contains many organic compounds that reduce the performance and stability of the SEI, thus inducing uneven Li plating/stripping behavior and uncontrollable dendritic growth. This result is consistent with the deposition observations shown in Fig. 3c, d and Fig. S6 (ESI†).
 | ||
| Fig. 4 Characterization of SEI formed in different electrolytes. (a) 3D rendering of TOF-SIMS of SEI layers with ion fragments of Li−, Mg−, C2HO−, and F− using the baseline and 0.1 Mg(NO3)2-GBL electrolytes. XPS spectra of SEI layers on Li metal anode using electrolytes, showing the (b and e) O 1s, (c and f) N 1s, and (d and g) F 1s energy levels. (b–d: baseline and e–g: 0.1 Mg(NO3)2-GBL electrolytes). | ||
In addition to the TOF-SIMS observations, X-ray photoelectron spectroscopy (XPS) was performed to obtain a comprehensive understanding of the composition and structure of the SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte. According to the O 1s spectra, the SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte exhibited a weaker signal from Li2CO3 (532.6 eV) and organic Li salts (533.1 eV, Fig. 4b) than that induced by the baseline electrolyte (Fig. 4e), suggesting that the 0.1 Mg(NO3)2-GBL electrolyte is beneficial for reducing harmful organic Li salts and Li2CO3 phases in the SEI. However, according to the N 1s and F 1s spectra, the SEI formed in the 0.1 Mg(NO3)2-GBL electrolyte contained more Li3N (397.5 eV) and LiF (685.4 eV) in SEI layer than that formed in the baseline electrolyte (Fig. 4c, d, f and g). As previously reported, the Li3N and LiF are prominent which can improve the structural stability and ion transference of the SEI. Therefore, the XPS results suggest that the SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte possessed improved properties than the baseline in all respects and was more capable of maintaining uniform Li plating/stripping behavior and suppressing Li dendrites. These results are consistent with the TOF-SIMS results and the performances of cells with the different electrolytes. In other words, the TOF-SIMS and XPS results again proved that the 0.1 Mg(NO3)2-GBL electrolyte was much more effective to inhibition of Li dendritic growth than the baseline electrolyte in forming a robust SEI and optimizing the properties of the Li metal anode.
To determine the advantages of the 0.1 Mg(NO3)2-GBL electrolyte, average coulombic efficiency (CE) tests were conducted using a two-electrode Li/Cu asymmetric cell. For comparison, the 0.1 Mg(NO3)2-GBL and baseline electrolytes were separately used in different cells. According to the CE results (Fig. 5a), the Li/Cu asymmetric cell with the 0.1 Mg(NO3)2-GBL electrolyte exhibited a relatively high CE of 96.26%, which was obviously superior to that of the Li/Cu asymmetric cell with the baseline electrolyte (95.57%). The different CE values indicate better reversibility of the Li plating in the 0.1 Mg(NO3)2-GBL electrolyte, which can be attributed to the optimized interfacial reactions and the modified SEI induced by the 0.1 Mg(NO3)2-GBL electrolyte, which restricted the capacity loss resulting from side reactions and Li dendritic growth. In addition, the cell with the 0.1 Mg(NO3)2-GBL electrolyte showed a much smaller inner resistance than the cell with the baseline electrolyte, which can be mainly attributed to the uniform and the higher ionic conductivity of the SEI induced by the optimized electrolyte. To better describe the different performances between the 0.1 Mg(NO3)2-GBL and baseline electrolytes, only 10 μL of the electrolyte was added to each cell. The cell with the baseline electrolyte demonstrated fluctuating voltage profiles after only 142 h of cycling, whereas the cell using the 0.1 Mg(NO3)2-GBL electrolyte maintained constant voltage hysteresis within the range of 12–18 mV over 620 h (Fig. 5c), which was also shown by the inner resistance test results (Fig. 5b and Fig. S8, ESI†). Furthermore, we prepared a special electrolyte with only GBL added to the baseline electrolyte (Fig. S9, ESI†). The Li/Li symmetric cell using this electrolyte performed no better than the baseline electrolyte, confirming that the improvement in performance was due to the addition of Mg(NO3)2. The improvement in cyclic stability was due to the 0.1 Mg(NO3)2-GBL electrolyte, which guided the homogeneous Li plating/stripping process and preserved the stable inner structure of the cell. To verify the proper concentration of the Mg(NO3)2 additive, we prepared two extra electrolyte samples with 0.2 and 0.3 M Mg(NO3)2 to examine the cyclic properties of cells using these electrolytes with different amounts of Mg(NO3)2 (Fig. 5c). Surprisingly, the symmetric cells failed more quickly with increasing amounts of Mg(NO3)2. Based on our previous work, we concluded that this phenomenon was caused by the exacerbation of side reaction between the Li metal anode and the enhanced amount of Mg(NO3)2 additive, as well as the thickened SEI layer on the anode, which led to poor interfacial reaction kinetics and extra consumption of the Mg(NO3)2-rich electrolyte. Ionic conductivities with different electrolytes have been further investigated to recognize the optimized concentration of Mg(NO3)2 additives (Fig. S10, ESI†). Although high concentration of nitrate additive is requested in the electrolyte to form inorganic and ionic conductive SEI phase on anode surface, increasing concentration of salt in electrolyte causes increase of viscosity and decrease of ionic conductivity, further resulting in declined cycle performance especially under extreme working conditions.41,46 While the baseline and 0.1 Mg(NO3)2-GBL show similar ionic conductivities, adding more Mg(NO3)2 exhibited decrease of ionic conductivities implying poor cyclability at high current density. Based on the physical property and cyclic test results, we suggest that 0.1 M Mg(NO3)2 is the most appropriate formula for the Mg(NO3)2-GBL electrolyte. The cyclic properties and dendritic growth risks of the different electrolytes were further demonstrated using fast charge-performance tests. The symmetric cells were fabricated using Li metal as electrodes and 50 μL of the baseline or 0.1 Mg(NO3)2-GBL electrolyte, respectively. As shown by the voltage profiles in Fig. 5d and Fig. S11(ESI†), the cell with the 0.1 Mg(NO3)2-GBL electrolyte exhibited a relatively stable voltage profile with steady voltage hysteresis, whereas the cell with the baseline electrolyte showed an increased polarization, indicating failure of the inner structure of the Li metal anode and the corresponding cell.
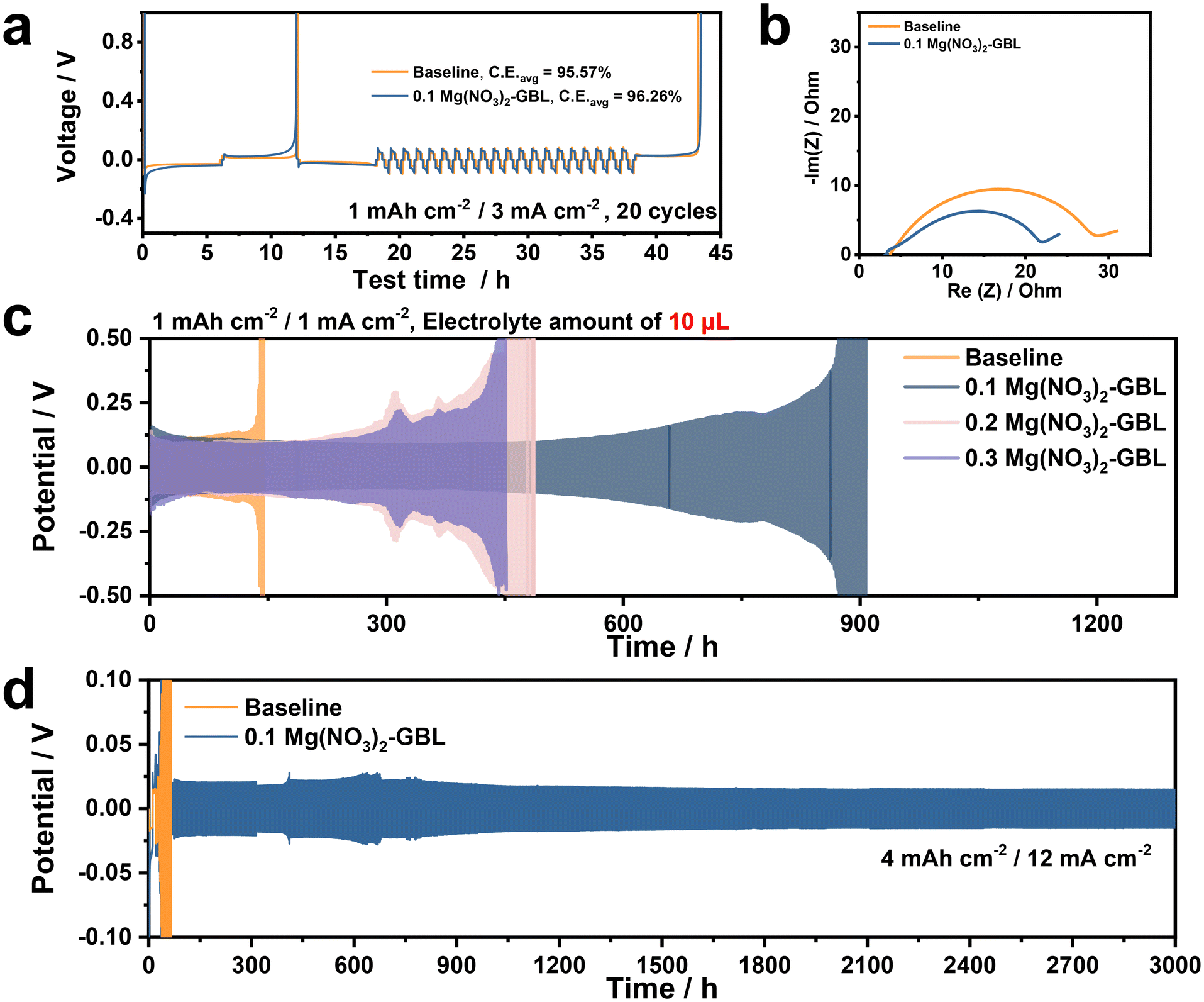 | ||
| Fig. 5 Cyclic tests of cells with different electrolytes. (a) Average coulombic efficiency of Li metal anodes in Li/Cu asymmetric cells using baseline and 0.1 Mg(NO3)2-GBL electrolytes. (b) AC impedance spectra in Li/Li symmetric cells after 20 cycles at a capacity loading of 1 mA h cm−2 and current density of 3 mA cm−2 using the baseline and 0.1 Mg(NO3)2-GBL electrolytes. Galvanostatic test for Li/Li symmetric cells (c) with a small volume (10 μL) of the baseline and modified electrolytes according to the content of Mg(NO3)2 additive (0.1, 0.2 and 0.3 M Mg(NO3)2) at a capacity loading of 1 mA h cm−2 and current density of 1 mA cm−2 (0.2 and 0.3 M of Mg(NO3)2 additives are denoted 0.2 Mg(NO3)2-GBL and 0.3 Mg(NO3)2-GBL, respectively) and (d) 50 μL of electrolytes at a capacity loading of 4 mA h cm−2 and high current density of 12 mA cm−2 corresponding to 3 C. | ||
The practical value of the 0.1 Mg(NO3)2-GBL electrolyte was further determined through full-battery tests. The full battery used an Li metal anode and SPAN cathode at a capacity loading of 4 mA h cm−2. The cells with the 0.1 Mg(NO3)2-GBL electrolyte were repeatedly charged/discharged at 1 C in a voltage window of 1.0–3.0 V. The Li/SPAN full cell with the 0.1 Mg(NO3)2-GBL electrolyte exhibited a low decay rate of 0.82% and a high CE value of 99.98% after 150 cycles (Fig. 6a and b), demonstrating the advantage of the 0.1 Mg(NO3)2-GBL electrolyte and the related relatively homogeneous SEI. This result shows the potential of the 0.1 Mg(NO3)2-GBL electrolyte as a design material for future high-energy Li metal batteries.
 | ||
| Fig. 6 Full battery tests using 0.1 Mg(NO3)2-GBL electrolyte. (a) Voltage hysteresis of Li/SPAN batteries with 0.1 Mg(NO3)2-GBL electrolyte in the voltage range of 1.0–3.0 V. Sulfur loading in the SPAN cathode is 4 mA h cm−2. (b) Cyclic stability of full cell for cycling at 1 C. | ||
Conclusions
In this study, we adopted a simple but effective strategy to optimize an ester-based electrolyte using Mg(NO3)2 as an additive and GBL as a solubility mediator. The high-concentration Mg(NO3)2 additive reacts preferentially with Li metal anode to fabricate an inorganic-phase-dominated, relatively uniform SEI on the anode surface containing large amounts of highly Li ion-conductive and robust Li3N and Li–Mg alloy. Owing to the relatively uniform structure and ion conductivity of the as-formed SEI, homogeneous interfacial reactions occur on the anode, leading to stable Li plating/stripping behavior and charging/discharging properties of the Li metal anodes. In addition, due to the high robustness of the inorganic phases in the SEI, the Li metal anode showed its densely packed and intact surface morphology after electrodeposition and maintained stable cycling even over 3000 h of cycling at a capacity loading of 4 mA h cm−2 and high current density of 12 mA cm−2 in an unfavorable ester-based electrolyte, showing no harmful dendritic growth or mossy-like structures. Our discoveries in this work will provide inspiration for future ester-based electrolyte designs and help realize practical lithium metal battery systems.Author contributions
H. Kim: writing – original draft, visualization, formal analysis, conceptualization. J. Park: writing – original draft, visualization, formal analysis, conceptualization. H. Kang: formal analysis. S. Kansara: formal analysis. S. Lim: data curation. Z. Sun: writing – review & editing and visualization. S. Xiong: writing – review & editing, supervision and visualization. Y.-C. Jung: formal analysis. C. Hwang: data curation. M. Agostini: data curation. J.-Y. Hwang: supervision, conceptualization, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was also supported by Korea Institute for Advancement of Technology (KIAT) grant funded by the Korea Government (MOTIE) (grant no. RS-2024-00417730, HRD Program for Industrial Innovation) and the National Supercomputing Center with supercomputing resources including technical support (KSC-2024-CRE-0109). We also appreciate financial support from the National Natural Science Foundation of China (22409077 and 22479067).References
- M. Mishra and M. Kumar, Phys. Scr., 2024, 99, 125929 CrossRef CAS.
- Y. Yang, Z. Wu, J. Yao, T. Guo, F. Yang, Z. Zhang, J. Ren, L. Jiang and B. Li, Energy Rev., 2024, 3, 100068 CrossRef.
- H. Zhao, X. Bo, H. Xu, L. Wang, W. Daoud and X. He, Energy Storage Mater., 2024, 72, 103696 CrossRef.
- S. Yan, H. Liu, X. Chen, Y. Lu, Q. Cao and K. Liu, Sci. China: Chem., 2024, 67, 4116–4124 CrossRef CAS.
- D.-H. Liu, Z. Bai, M. Li, A. Yu, D. Luo, W. Liu, L. Yang, J. Lu, K. Amine and Z. Chen, Chem. Soc. Rev., 2020, 49, 5407 RSC.
- Y. Chen, Z. Ma, Y. Wang, P. Kumar, F. Zhao, T. Cai, Z. Cao, L. Cavallo, H. Cheng, Q. Li and J. Ming, Energy Environ. Sci., 2024, 17, 5613–5626 RSC.
- D. Wang, J. Qiu, N. Inui, R. Hagiwara, J. Hwang and K. Matsumoto, ACS Energy Lett., 2023, 8, 5248–5252 CrossRef CAS.
- J. H. Ji Hyun and S.-H. Yu, Adv. Energy Mater., 2021, 11, 2003004 CrossRef.
- X. Duan, L. Wang, G. Li, X. Liu, M. Wan, J. Du, R. Zhan, W. Wang, Y. Li, S. Tu, Y. Shen, Z. W. Seh, L. Wang and Y. Sun, Adv. Funct. Mater., 2023, 33, 2210669 CrossRef CAS.
- A. Begoña, M. C. Maria, G. Elena, I. Ruiz de Larramendi, A. Villaverde, J. Rikarte and L. Fallarino, Adv. Energy Mater., 2023, 13, 2203744 CrossRef.
- J.-L. Li, Y.-N. Wang, S.-Y. Sun, Z. Zheng, Y. Gao, P. Shi, Y.-J. Zhao, X. Li, Q. Li, X.-Q. Zhang and J.-Q. Huang, Adv. Energy Mater., 2024, 15, 2403845 CrossRef.
- Y. Liao, H. Zhang, Y. Peng, Y. Hu, J. Liang, Z. Gong, Y. Wei and Y. Yang, Adv. Energy Mater., 2024, 14, 2304295 CrossRef CAS.
- S.-Y. Sun, X.-Q. Zhang, Y.-N. Wang, J.-L. Li, Z. Zheng and J.-Q. Huang, Mater. Today, 2024, 77, 39–65 CrossRef CAS.
- Y.-Y. Wang, Y.-N. Wang, N. Yao, S.-Y. Sun, X.-Q. Ding, C.-X. Bi, Q.-K. Zhang, Z. Zheng, C.-B. Jin, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, J. Energy Chem., 2024, 95, 644–650 CrossRef CAS.
- H. Yuan, X. Ding, T. Liu, J. Nai, Y. Wang, Y. Liu, C. Liu and X. Tao, Mater. Today, 2022, 53, 173–196 CrossRef CAS.
- J. Tan, L. Ma, Z. Li, Y. Wang, M. Ye and J. Shen, Mater. Today, 2023, 69, 287–332 CrossRef CAS.
- G. Lu, J. Nai, D. Luan, X. Tao and X. W. D. Lou, Sci. Adv., 2023, 9, eadf1550 CrossRef CAS PubMed.
- Y. Cheng, J. Chen, Y. Chen, X. Ke, J. Li, Y. Yang and Z. Shi, Energy Storage Mater., 2021, 38, 276–298 CrossRef.
- Y. Zhong, X. Yang, R. Guo, L. Zhai, X. Wang, F. Wu, C. Wu and Y. Bai, Electrochem. Energy Rev., 2024, 7, 30 CrossRef CAS.
- H. Zhou, S. Yu, H. Liu and P. Liu, J. Power Sources, 2020, 450, 227632 CrossRef CAS.
- S. Li, Z. Luo, L. Li, J. Hu, G. Zou, H. Hou and X. Ji, Energy Storage Mater., 2020, 32, 306–319 CrossRef.
- J. Huang, J. Liu, J. He, M. Wu, S. Qi, H. Wang, F. Li and J. Ma, Angew. Chem., 2021, 133, 20885–20890 CrossRef.
- G. Wang, Q.-K. Zhang, X.-Q. Zhang, J. Lu, C. Pei, D. Min, J.-Q. Huang and H. S. Park, Adv. Energy Mater., 2025, 15, 2304557 CrossRef CAS.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- W. Zhang, Y. Lu, L. Wan, P. Zhou, Y. Xia, S. Yan, X. Chen, H. Zhou, H. Dong and K. Liu, Nat. Commun., 2022, 13, 2029 CrossRef CAS PubMed.
- Q. Li, J. Zhang, Y. Zeng, Z. Tang, D. Sun, Z. Peng, Y. Tang and H. Wang, Chem. Commun., 2022, 58, 2597–2611 RSC.
- Q. Wang, T. Lu, Y. Xiao, J. Wu, L. Guan, L. Hou, H. Du, H. Wei, X. Liu, C. Yang, Y. Wei, H. Zhou and Y. Yu, Electrochem. Energy Rev., 2023, 6, 22 CrossRef CAS.
- O. B. Chae and B. L. Lucht, Adv. Energy Mater., 2023, 13, 2203791 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS.
- S. H. Lee, J.-Y. Hwang, J. Ming, Z. Cao, H. A. Nguyen, H.-G. Jung, J. Kim and Y.-K. Sun, Adv. Energy Mater., 2020, 10, 2000567 CrossRef CAS.
- B. Guo, P. Guo, G. Zhao, S. Liu, J. Shi, M. Huang, Z. Shi, H. Wang and Z. Yan, Sustainable Energy Fuels, 2022, 6, 4137–4145 RSC.
- M. S. Kim, Z. Zhang, J. Wang, S. T. Oyakhire, S. C. Kim, Z. Yu, Y. Chen, D. T. Boyle, Y. Ye, Z. Huang, W. Zhang, R. Xu, P. Sayavong, S. F. Bent, J. Qin, Z. Bao and Y. Cui, ACS Nano, 2023, 17, 3168–3180 CrossRef CAS PubMed.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- Y. Jie, X. Liu, Z. Lei, S. Wang, Y. Chen, F. Huang, R. Cao, G. Zhang and S. Jiao, Angew. Chem., Int. Ed., 2020, 59, 3505–3510 CrossRef CAS PubMed.
- C. Zhao, Y. Lu, K. Yan, Y. Guan, S. Jiang, J. Wang, S. Guo, M. Cao, N. Li, Y. Su, F. Wu and L. Chen, Adv. Energy Mater., 2024, 14, 2304532 CrossRef CAS.
- R. A. Miranda-Quintana and J. Smiatek, J. Mol. Liq., 2021, 322, 114506 CrossRef CAS.
- C.-C. Su, M. He, R. Amine, T. Rojas, L. Cheng, A. T. Ngo and K. Amine, Energy Environ. Sci., 2019, 12, 1249–1254 RSC.
- L. Zhang, J. Li, Y. Huang, D. Zhu and H. Wang, Langmuir, 2019, 35, 3972–3979 CrossRef CAS PubMed.
- G. Huang, Y. Liao, X. Zhao, X. Jin, Z. Zhu, M. Guan and Y. Li, Adv. Funct. Mater., 2023, 33, 2211364 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- Y. Liang, W. Wu, D. Li, H. Wu, C. Gao, Z. Chen, L. Ci and J. Zhang, Adv. Energy Mater., 2022, 12, 2202493 CrossRef CAS.
- W. Wahyudi, V. Ladelta, L. Tsetseris, M. M. Alsabban, X. Guo, E. Yengel, H. Faber, B. Adilbekova, A. Seitkhan, A.-H. Emwas, M. N. Hedhili, L.-J. Li, V. Tung, N. Hadjichristidis, T. D. Anthopoulos and J. Ming, Adv. Funct. Mater., 2021, 31, 2101593 CrossRef CAS.
- S. Liu, J. Xia, W. Zhang, H. Wan, J. Zhang, J. Xu, J. Rao, T. Deng, S. Hou, B. Nan and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210522 CrossRef CAS PubMed.
- Q. Li, G. Liu, P. Kumar, F. Zhao, Y. Wang, T. Cai, Y. Chen, H. Xie, W. Wahyudi, Z. Ma and J. Ming, Adv. Funct. Mater., 2025, 35, 2416714 CrossRef CAS.
- Q. Li, G. Liu, Y. Chen, J. Wang, P. Kumar, H. Xie, W. Wahyudi, H. Yu, Z. Wang, Z. Ma and J. Ming, Adv. Funct. Mater., 2025, 35, 2420327 CrossRef.
- C. Zhao, Y. Lu, K. Yan, Y. Guan, S. Jiang, J. Wang, S. Guo, M. Cao, N. Li, Y. Su, F. Wu and L. Chen, Adv. Energy Mater., 2024, 14, 2304532 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00025d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |