
Molecular polarity regulation of polybromide complexes for high-performance low-temperature zinc–bromine flow batteries†
Ming
Zhao
ab,
Tao
Cheng
ab,
Tianyu
Li
 ac,
Shuo
Wang
a,
Yanbin
Yin
*ac and
Xianfeng
Li
*ac
ac,
Shuo
Wang
a,
Yanbin
Yin
*ac and
Xianfeng
Li
*ac
aDivision of Energy Storage, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: yinyanbin@dicp.ac.cn; lixianfeng@dicp.ac.cn
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cKey Laboratory of Long-Duration and Large-Scale Energy Storage, Chinese Academy of Sciences, China
First published on 26th November 2024
Abstract
Frigid environments notably impair the electrochemical performance of zinc–bromine flow batteries (ZBFBs) due to polybromide solidification, restricting their widespread deployment in cold regions. Here, two independently used complexing agent cations, n-propyl-(2-hydroxyethyl)-dimethylammonium (N[1,1,3,2OH]+) and diethyl-(2-hydroxyethyl)-methylammonium (N[1,2,2,2OH]+), are proposed to enable ZBFBs to exhibit excellent performance at both low and room temperatures, through the precise regulation of the molecular polarity of polybromide complexes. Benefiting from the optimized design of the carbon number and position on the skeleton, the molecular polarity of the N[1,1,3,2OH]+- and N[1,2,2,2OH]+-polybromide complexes is appropriately reduced compared with that of choline, which is conductive to the enhancement of bromine capture capability. Interestingly, the intermolecular hydrogen bonding effect of their hydroxyl groups is not significantly enhanced, ensuring that the formed complexes maintain good fluidity at low temperatures. Thus, ZBFBs with a single complexing agent not only demonstrate an impressive average Coulombic efficiency of >95% across 1600 cycles at room temperature, but also can sustain operation with a high current density of 40 mA cm−2 for 250 cycles at −20 °C. This research significantly advances the comprehensive understanding of the mechanisms involved, thereby substantially contributing to the development of enhanced low-temperature complexing agents.
Broader contextZinc–bromine flow batteries (ZBFBs) have advanced to the demonstration phase for projects with a 100 kW h capacity, indicating promising application prospects. One critical concern is their low-temperature operation, which affects reliability, potential applications, and geographical deployment. This issue requires urgent resolution but has not been adequately addressed. The key point of entry lies in the research and development of complexing agents which are currently the most crucial factor influencing the battery system's operational characteristics. Here, we propose two types of single-component bromide complexing agents that can enable ZBFBs to perform well at both room temperature and low temperatures, thereby enhancing their overall performance. A thorough analysis of the relationship between the battery's structure and its properties establishes a robust foundation for developing superior complexing agents. Remarkably, achieving operation at −20 °C represents significant progress for any aqueous flow battery. We successfully operated a ZBFB at −20 °C using these novel single-component complexing agents, marking a substantial breakthrough. The continued advancement and success of ZBFBs will significantly propel energy storage technology development and support the large-scale, efficient use of green renewable energies. |
Introduction
In response to the target of low carbon emission, the development of stationary energy storage is of significant importance for promoting the wide deployment of renewable energies and then enhancing grid stability.1–5 Zinc–bromine flow batteries (ZBFBs), with the advantages of high energy density, low cost, and intrinsic safety, are regarded as highly promising energy storage/conversion technologies.6–8 However, the suboptimal performance of batteries at low temperatures constitutes a significant impediment to their broader utilization. This challenge imperatively calls for the formulation of efficacious strategies to enhance their performance under frozen conditions.9–13Generally, a typical ZBFB must use a bromine complexing agent (BCA) to complex with the cathode charging products to form an oil phase.14 This method immobilizes the bromine at the cathode, thereby maintaining a high Coulombic efficiency (CE) and suppressing the corrosive nature of free Br2. N-methyl-N-ethylpyrrolidinium bromide (MEP) is the most widely used BCA.15–18 However, polybromides with MEP will preferentially solidify over the bulk electrolyte at low temperatures, thereby severely hindering the mass transport and causing rapid battery failure. Therefore, the key to the low-temperature operation of ZBFBs lies in the development of advanced BCAs to maintain polybromides consistently in a liquid state. Utilizing mixed BCAs is a valid strategy, and some viable solutions have been proposed.19–21 By fully considering the interactions between components and optimizing their proportions, it can effectively regulate the low-temperature state of polybromides. However, the inherent complexity of the mixed BCA strategy may pose substantial challenges for subsequent in-depth investigations into its underlying mechanisms and complicate efforts aimed at optimizing designs for more advanced structures. Moreover, the development approach based on the diversified combination of multi-component chelating agents may hinder the structural innovation of low-temperature BCAs. Choline chloride (ChCl) proposed by our group in the mixed BCA study demonstrates exceptional ability to maintain polybromides in the liquid state even at −40 °C, showcasing significant research values and promising application prospects.21 Meanwhile, the disadvantage of the low CE of ChCl at room temperature (RT) limits its independent function in ZBFBs. Therefore, it is highly necessary to develop a single-component complexing agent with excellent comprehensive performances for scientific research and technological advancement. And establishing meaningful structure–property relationships will provide invaluable experience for the design of future high-performance low-temperature complexing agents without having to rely on trial and error.
Herein, benefiting from precise regulation of molecular polarity of polybromide complexes, n-propyl-(2-hydroxyethyl)-dimethylammonium (N[1,1,3,2OH]+) and diethyl-(2-hydroxyethyl)-methylammonium (N[1,2,2,2OH]+) are proposed as single complexing agent cations to enable effective control over the state of polybromide complexes. To thoroughly analyze the mechanism and the effect of structures on properties, a series of choline derivatives are systematically synthesized and investigated. Thanks to the optimal design of the cation structures, the molecular polarities of N[1,1,3,2OH]+- and N[1,2,2,2OH]+-polybromide complexes are appropriately reduced, while the role of hydroxyl does not show significant changes, compared with choline. These two proposed cations demonstrate both excellent bromine capture capability at RT and exceptionally fluid state at low temperatures. Consequently, N[1,2,2,2OH]+ and N[1,1,3,2OH]+ not just endow the ZBFBs with a high average CE at RT, but also enable them to exhibit a long cycle life even at −20 °C with high current density.
Results and discussion
To systematically validate the beneficial effects of N[1,1,3,2OH]+ and N[1,2,2,2OH]+ and investigate the underlying mechanisms in depth, three series of choline derivatives were designed and successfully synthesized. Series 1: elongating the carbon chain with the hydroxy group; Series 2: elongating the carbon chain on the one methyl group; and Series 3: increasing the number of ethyl groups. The structures and corresponding abbreviations of these derivatives are displayed in Fig. 1 (N[1,1,1,2OH]+ and N[1,1,2,2OH]+ maybe shown with different colours for the comparative study). C5–C8 are used to denote the derivatives containing 5, 6, 7 and 8 carbon atoms, respectively. The structure and purity of the as-prepared choline derivatives were identified by 1H nuclear magnetic resonance (NMR) spectroscopy. The 1H peak numbers, chemical shifts and splitting characteristics of these products are consistent with those reported in the literature, indicating that the designed choline derivatives are successfully synthesized.22–27 Additionally, no impurity peaks were observed in the 1H NMR spectra, suggesting high purity (Fig. S1, ESI†). The composition of polybromide complexes was identified by Raman spectroscopy (Fig. S2, ESI†). There are two peaks on the spectra, 166 and 264 cm−1, which represent the symmetrical stretching vibration of Br3− and Br5−,28,29 respectively. The results demonstrate that all polybromide anions are composed of Br3− and Br5−.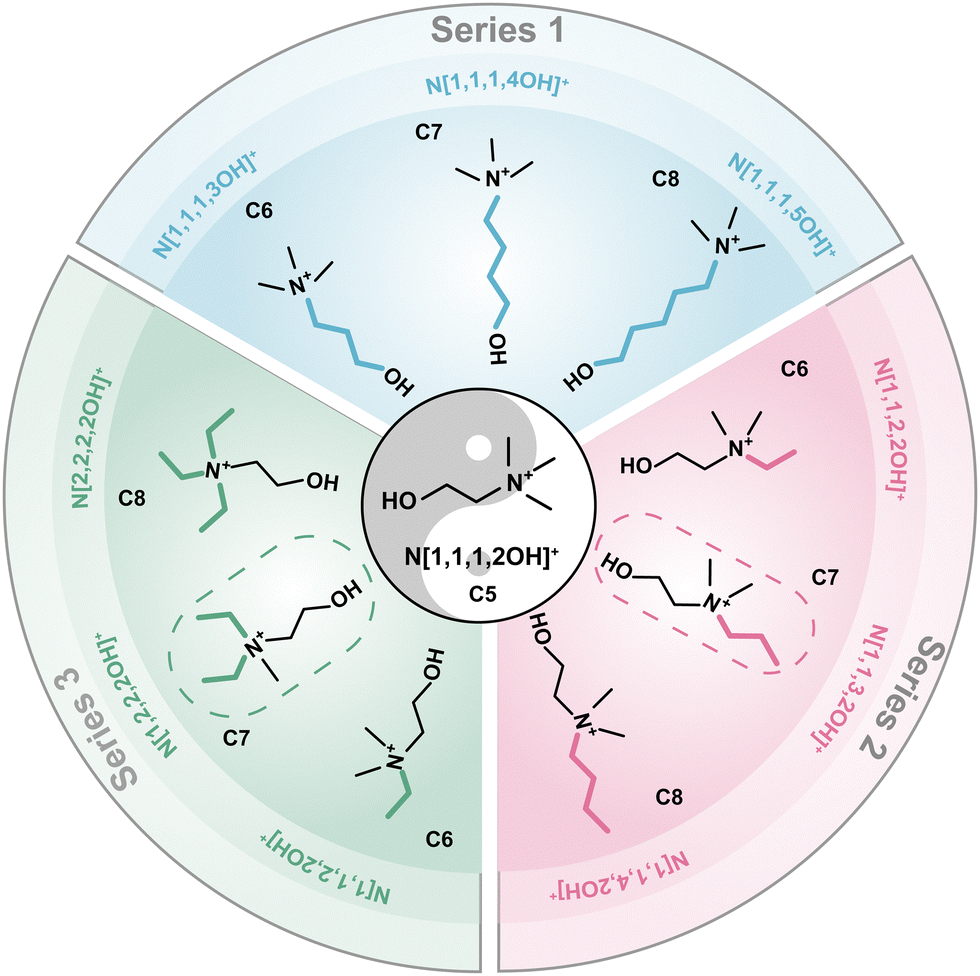 | ||
| Fig. 1 The structures and names of three series of choline derivatives. | ||
The main merit of the choline cation (N[1,1,1,2OH]+) lies in the excellent liquid retention of its polybromide complex at low temperatures. For N[1,1,3,2OH]+ and N[1,2,2,2OH]+, inheriting this necessary feature is a critical prerequisite for their feasibility. Firstly, we conducted visualization experiments to directly observe the impact of the synthesized derivatives on the state of polybromide complex phases at different temperatures. It can be found that the complexes are still solid when using N[1,1,1,4OH]+ or N[1,1,1,5OH]+ even at RT (Fig. S3, ESI†). Furthermore, the N[2,2,2,2OH]+-polybromide complex also transitioned into a solid phase as the temperature decreased to −20 °C (Fig. 2a). These results indicate that they can be eliminated as the BCA in low-temperature ZBFBs. Interestingly, the complexes of N[1,1,3,2OH]+ and N[1,2,2,2OH]+ can remain in the liquid state at −40 and −20 °C, respectively, endowing ZBFBs with excellent low-temperature performance and making them competitive candidates. Although some derivatives also exhibit similar behaviours, this prompts us to further investigate and compare their other properties. Differential scanning calorimetry (DSC) was used to further investigate the phase transformation behaviour of the polybromide complexes with the derivatives. N[1,1,1,4OH]+, N[1,2,2,2OH]+ and N[2,2,2,2OH]+-polybromides demonstrated distinct phase transition at around 34.6, −26.8 and 11.6 °C (Fig. S4, ESI†). Nevertheless, the curves of the other derivatives displayed the characteristics of glass transition, verifying that the solidification of N[1,1,1,3OH]+- and N[1,1,4,2OH]+-polybromide phases is due to the increase in viscosity as the temperature decreases. To deeply analyse the properties of the three series of the derivatives, we carried out viscosity tests on the formed polybromide complexes. The data in Fig. 2b demonstrate a pronounced trend of escalating viscosity in correlation with an increase in the number of carbon atoms in each series, suggesting that as the carbon count rises, the intermolecular interactions within these polybromide complexes become more robust.
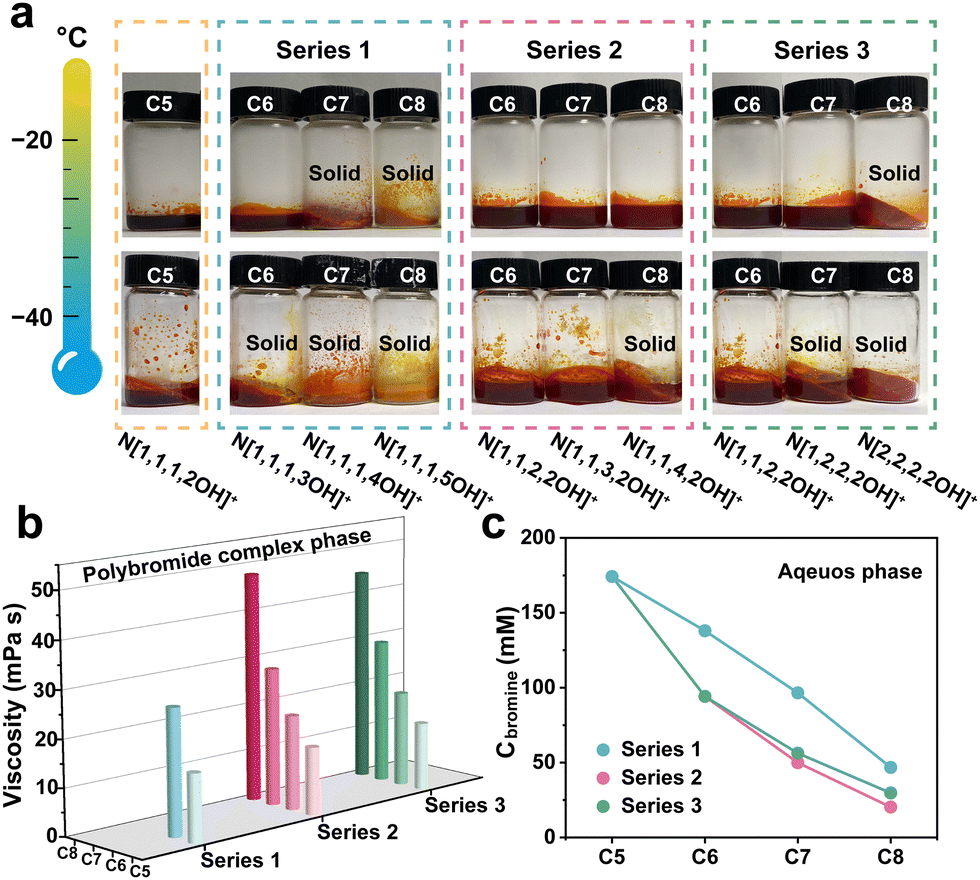 | ||
| Fig. 2 (a) The digital photographs of the polybromide complex phase with different derivatives at −20 and −40 °C. (b) Viscosities of the polybromide complexes with various derivatives (N[1,1,1,4OH]+- and N[1,1,1,5OH]+-polybromide complexes were missing because they are solid at RT). (c) The concentration of bromine in the aqueous phase in different catholytes for simulated charging. | ||
Moreover, the lower the concentration of bromine in the aqueous phase, the more favourable it should be for improving the CE of ZBFBs. In the catholyte under a simulated charging state with various derivatives (Fig. S5, ESI†), the colour of the aqueous phase becomes lighter with a higher molecular mass of the derivatives, especially Series 2 and 3, indicating that there are less dissociative active bromine substances. We further quantified the concentration of bromine dissolved in the aqueous phase by UV-Vis spectroscopy (Fig. 2c). As expected, the bromine content decreases with the increased molecular weight of the derivative. It is worth noting that Series 2 and 3 have more significant effects on reducing the amount of free bromine. Therefore, the current strategy for the structural optimization can notably improve the CE at room temperature, while maintaining low-temperature performance as much as possible. It is noteworthy that in terms of viscosity and bromine concentration, both N[1,1,3,2OH]+ (Series 2) and N[1,2,2,2OH]+ (Series 3) are at moderate levels in their respective series, which will help to illustrate the precision of the precise regulation of molecular polarity.
A thorough investigation into the relationship between the structures and properties of these derivatives is of paramount importance for enhancing our comprehension of the modulation of the interactions and developing more effective omnipotent BCAs. Electrostatic potential (ESP) of the cations and its formed polybromide molecule was calculated using Multifwn30–32 and VMD33 programs based on the results of density functional theory (DFT) calculations. The ESP range of the cations corresponding to the colour scale is from 0 to 6 eV (Fig. 3a). It is evident that, for Series 1, the electron density of the O atom on the hydroxy group intensifies with the increase of carbon as the group gradually moves away from the positive charge centre, while the other two series do not display any significant trend variations. Likewise, the results of the derivatives’ cations incorporating polybromide anions (Br3− and Br5−), respectively, demonstrate analogous trends (Fig. S6–S8, ESI†). We conjecture that these significant changes in the ESP of the polybromide complexes will indicate the variations in molecular polarity, which would impact the inter-/intra-molecular forces, thereby influencing the aggregation state of complexes and the battery performances.
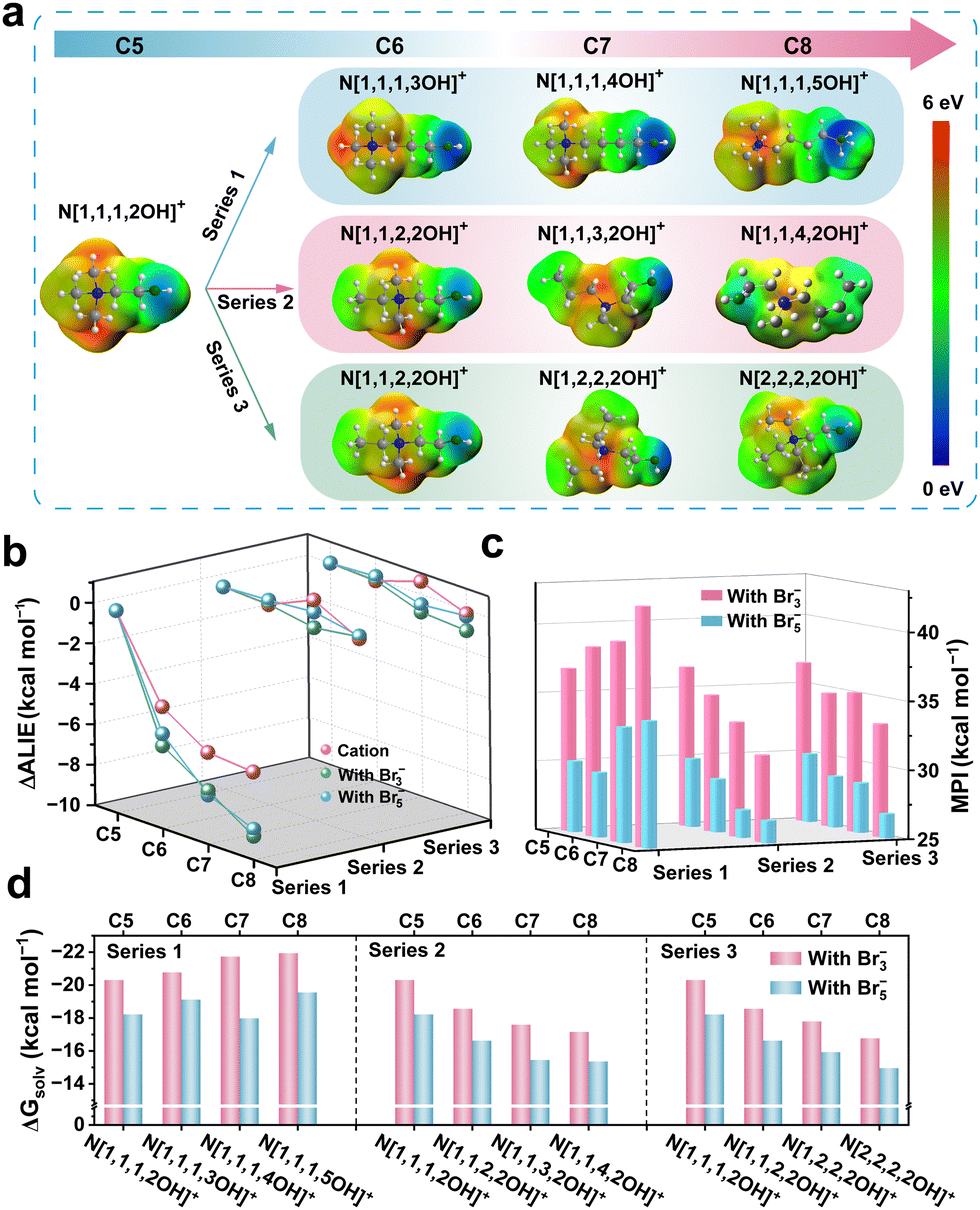 | ||
| Fig. 3 (a) The ESP mapping of different cations. (b) The ΔALIE of the derivative cations and their polybromide complexes (N[1,1,1,2OH]+ was set as the reference). (c) MPI and (d) ΔGsolv of the polybromide complexes with different derivatives. | ||
Furthermore, we employed average local ionization energy (ALIE)34,35 to conduct a quantitative assessment of the trends exhibited by the electronic activity of oxygen atoms. The lower ALIE represents the higher average energy of a local electron here, which means the stronger electron activity and weaker electron bounded. And a highly active electron possesses stronger nucleophilic properties, which tends to attract positively charged atoms. ΔALIE refers to the ALIE difference between the derivatives and N[1,1,1,2OH]+. As shown in Fig. 3b, no significant change of ΔALIE was observed in Series 2 and Series 3 with the addition of –CH2–, while Series 1 exhibits a rapid decrease in ΔALIE, whatever cations or polybromide complexes. These results align with the findings from the ESP analysis indicating that the modulation of oxygen electronegativity within cations is expected to significantly impact the properties of complexes and should thus be a focus of attention. Compared with N[1,1,1,2OH]+-polybromide, ALIEs of both N[1,1,3,2OH]+- and N[1,2,2,2OH]+-polybromides do not show significant changes.
Molecular polarity can influence the interactions between polybromide and water, especially when hydroxyl groups are present in the derivatives. To further elucidate the correlation mechanism between molecular polarity and structures, a molecular polarity index (MPI)36 was also calculated. The MPI of the polybromide complex molecule with the derivatives in Series 1 increases with the elongated carbon chain, whereas the MPI in Series 2 and Series 3 exhibits a significant negative correlation (Fig. 3c). In addition, combined analysis of Fig. 3b and c, it can be revealed that the increase in polarity of the complex molecules in Series 1 originates from the enhanced influence of the electronic activity of oxygen atoms on the hydroxyl group. Upon calculating the solvation free energy (ΔGsolv), we found that their trends are consistent with those of MPIs (Fig. 3d). The solubility of the derivatives in Series 1 will increase in polar aqueous solutions with the increasing number of –CH2–, whereas the solubility of those in Series 2 and Series 3 will decrease. Compared with the pristine structure (N[1,1,1,2OH]+), the solubility of N[1,1,3,2OH]+- and N[1,2,2,2OH]+-polybromides is moderately reduced, which may contribute to an improvement of the CE in ZBFBs. However, considering the trends of the bromine concentration mentioned above (Fig. 2c), only Series 2 and 3 are in great agreement with the theoretical calculation of ΔGsolv. In fact, the polybromide complexes are phase-separated from the electrolyte, and intermolecular interactions should be another important factor to consider. In terms of the molecular structure, the existent of –OH suggests that there may be the presence of hydrogen bonds (HBs) between polybromide complexes, which thus affect the actual dissolution of them.
For polybromide complexes, intra- and inter-molecular HB interactions should significantly influence their solubility and low-temperature phase transition behaviour. Infrared spectroscopy (IR) was adopted to further investigate the influence of various structures in the three series of derivatives on intra/inter-molecular HBs produced by the hydroxyl group. In these IR spectra, the broad peak located at 3300–3600 cm−1 can be attributed to the stretching vibration absorption peak of –OH (Fig. S9, ESI†). And the spectra could be deconvoluted into three subpeaks:37,38 ∼3300 cm−1 corresponded to the HB between the cations of the derivatives (C–C HB), attributed to the inter-molecular HB;39 ∼3500 cm−1 assigned to the –OH stretching mode along the HB from the cation to the anion (C–A HB), indicating the HB between –OH and Brn−, which belongs to the intra-molecular HB;40,41 ∼3600 cm−1 representing a quasi-free OH vibrational mode (Fig. 4a). In Series 1, the ratio of C–C HBs increased and C–A HBs decreased significantly with the elongation of the carbon chain with hydroxyl, suggesting that OH trended to interact with another OH on choline derivative cations (Fig. 4b). In contrast, in Series 2 and Series 3, only minor fluctuations can be observed. The C–A HBs still dominates, which is in favour of enhancing the binding affinity between the derivative cations and polybromide anions along with Coulomb-attraction of them (Fig. 4c and d). In addition, the C–C HB peak ratio of N[2,2,2,2OH]+ increases slightly with the declining C–A HB peak ratio, indicating that there were more C–C HBs in the N[2,2,2,2OH]+-polybromide complex which might be the reason for the increased solidification temperature (11.6 °C). Combined with the results from the visualization experiment at −40 °C in Fig. 2a, the complexes with the derivatives in Series 1 demonstrate stronger propensity for solidification, compared with Series 2 and 3. This phenomenon should be derived from the formation of a continuous network structure at low temperatures, which results from stronger intermolecular HBs. N[1,1,3,2OH]+- and N[1,2,2,2OH]+-complexes exhibit slightly weaker C–C HB interactions and stronger C–A HB interactions compared with the original structure (N[1,1,1,2OH]+), suggesting that they can effectively prevent solidification caused by C–C HBs and reduce the solubility of polybromides.
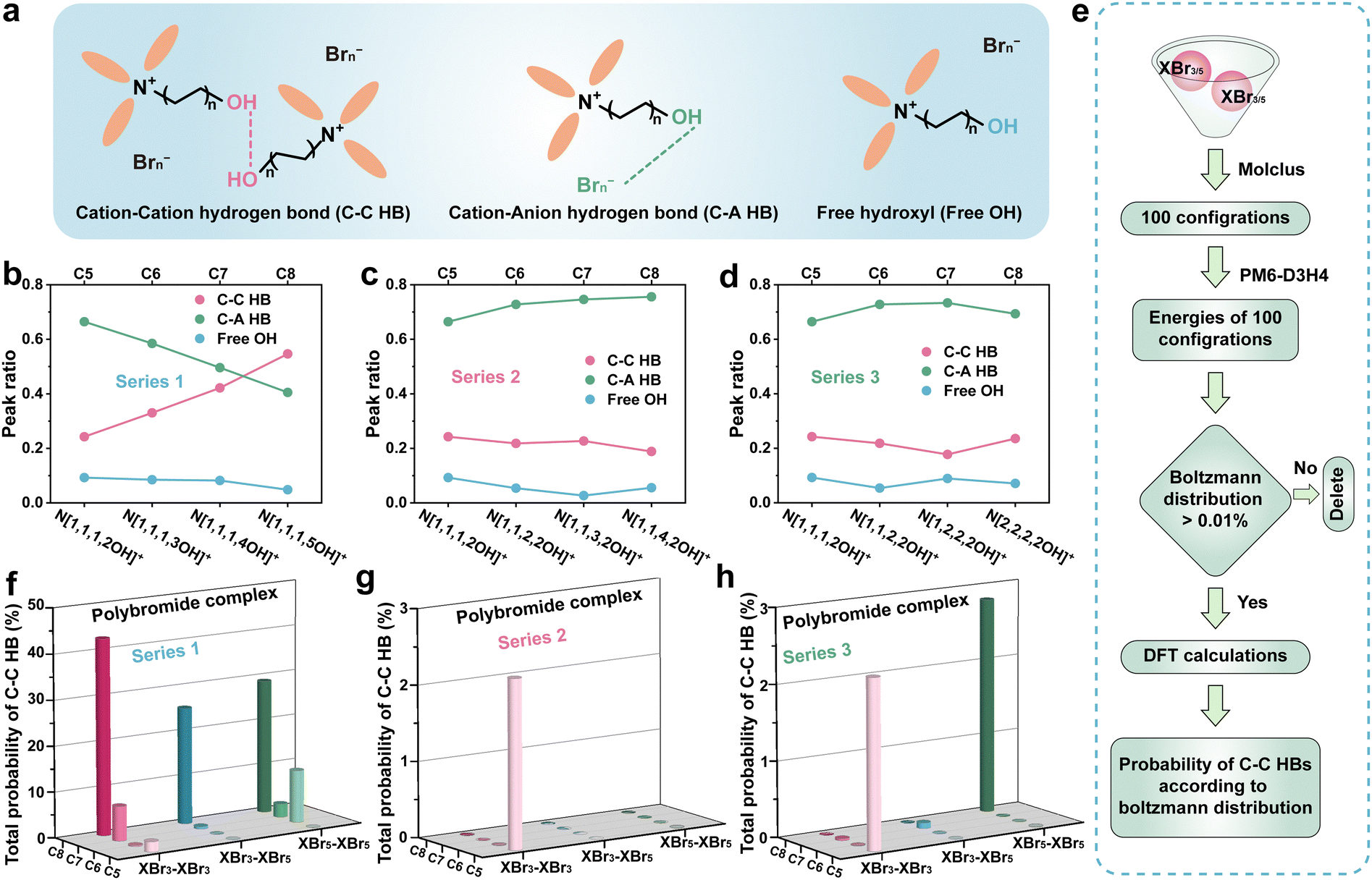 | ||
| Fig. 4 (a) The scheme of the HB types in the polybromide complex. (b)–(d) The IR peak area ratio of polybromide complexes with different series choline derivatives. (e) The illustration of the calculation process on intermolecular HBs between polybromide complexes. The total probabilities of HBs with different choline derivatives in (f) Series 1, (g) Series 2, and (h) Series 3, respectively. | ||
DFT calculations were further adopted to study the C–C HB between the polybromide complexes with different choline derivatives. As shown in Raman spectra (Fig. S2, ESI†), the polybromide complexes consisted of XBr3 and XBr5 (X represents the choline derivative cation). So, three kinds of intermolecular interactions, XBr3–XBr3, XBr3–XBr5 and XBr5–XBr5, should be taken into consideration. Firstly, as shown in Fig. 4e, 100 initial cluster configurations with two XBrn (n = 3 or 5) molecules were randomly generated using Molclus software,42 and their energies were preliminarily calculated by a semi-empirical approach at the PM6-D3H443–46 precision level. Next, the Boltzmann distributions of them were calculated at 298 K according to these energies, and the configurations with a probability of less than 0.01% were removed. The residual configurations were performed with high-precision calculations to obtain more accurate space structures and energies. Similarly, the Boltzmann distributions were calculated again for the remaining configurations. Finally, the total probability of the configuration with C–C HBs would be calculated (Tables S1–S27, ESI†). The calculation results are summarized in Fig. 4f–h, as the elongation of the hydroxyl-containing chain, the probability of the configurations with C–C HBs is gradually increased. In contrast, in Series 2 and 3, the probabilities decrease with the elongation of carbon chains (Fig. 4g and h). The results of these calculations also indicate that the intermolecular HB interactions of N[1,1,3,2OH]+- and N[1,2,2,2OH]+-complexes are weak and insignificant. These results suggest that the differences in the properties of Series 1 may stem from the enhanced C–C HB interactions induced by chain elongation, compared with Series 2 and 3.
Based on the above analysis, structural regulation of the derivative leads to corresponding property variations as shown in Fig. 5. In Series 1, the molecular polarity of the complexes exhibits an increasing trend, while Series 2 and Series 3 are decreased. Judging by the polybromide concentration in the aqueous phase, the strengthened intermolecular interactions from C–C HBs play a more critical role than the impact of increased ΔGsolv and decreased C–A HB interactions in Series 1, resulting in a declining trend in the solubility of polybromides. However, the obviously enhanced intermolecular forces from C–C HBs lead to the easy solidification of the polybromide complexes at low temperatures. In Series 2 and Series 3, the role of intermolecular C–C HBs is not manifested, ΔGsolv are reduced, and the effect of C–A HBs is slightly enhanced, thereby strengthen the bromine capture capacity. It can be speculated that the solubility of the complexes with these derivatives in Series 2 and Series 3 depends on ΔGsolv and C–A HB interactions and does not readily cause polybromide solidification at low temperatures. In the complexes with N[1,1,3,2OH]+ and N[1,2,2,2OH]+, the C–C HB interactions that lead to poor low-temperature performance do not significantly increase, the molecular polarities are appropriately reduced and the C–A interactions are enhanced, compared with the choline cation. These behaviors should be beneficial for the performance of ZBFBs at both room temperature and low temperatures.
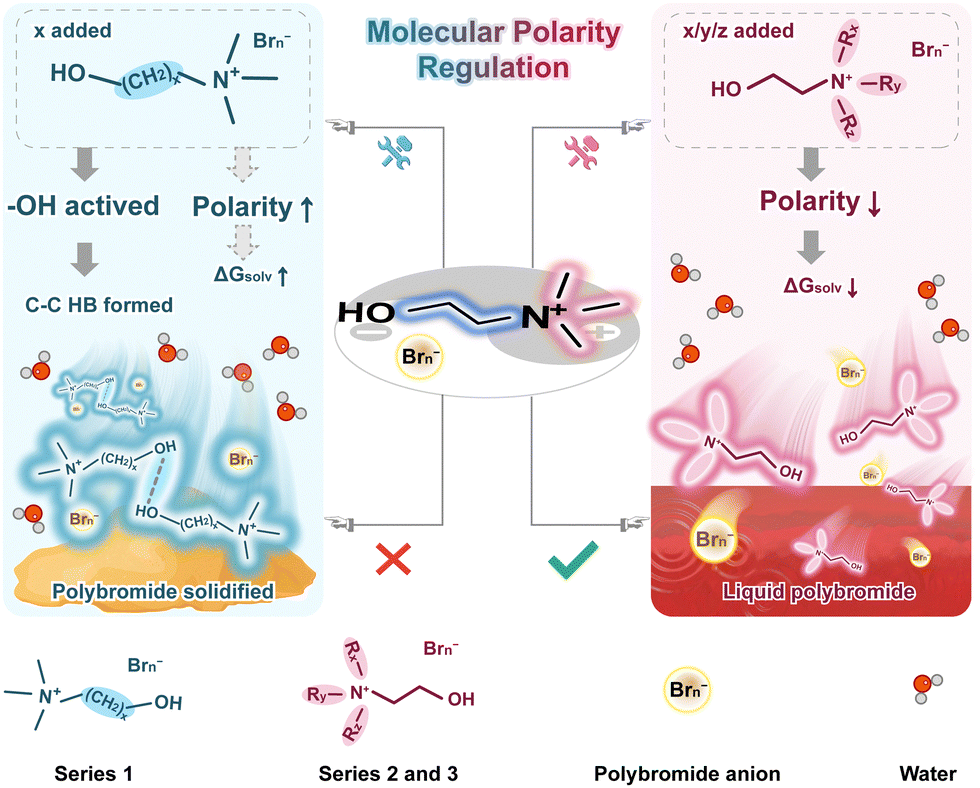 | ||
| Fig. 5 The scheme of the mechanism of molecular polarity regulation. | ||
To validate the above theoretical analysis and the actual effects of N[1,1,3,2OH]+ and N[1,2,2,2OH]+, the electrochemical performances of ZBFBs using the electrolyte with different derivatives (Table S28, ESI†) were tested at RT and −20 °C. Firstly, the average CE of 500 cycles was used as preliminary determination. As displayed in Fig. 6a and Fig. S10 and S11 (ESI†), including N[1,1,3,2OH]+ and N[1,2,2,2OH]+, the average CEs of ZBFBs using N[1,1,4,2OH]+- and N[2,2,2,2OH]+-electrolytes also exceed 95%, which are higher than that using N[1,1,1,2OH]+ (around 90%). However, by analyzing the images in Fig. 2a, we can predict that the N[2,2,2,2OH]+-electrolyte may not be suitable at −20 °C, due to the polybromide solidification. Upon testing the ZBFBs with different electrolytes at −20 °C, it was confirmed that the battery with the N[2,2,2,2OH]+-electrolyte indeed could not operate at a current density of 40 mA cm−2 (Fig. S12d, ESI†). According to the average energy efficiency (EE) of 100 cycles at −20 °C, N[1,1,3,2OH]+ (∼67%) and N[1,2,2,2OH]+ (∼68%) demonstrate strong competitiveness at low temperatures (Fig. 6b and Fig. S12a and b, ESI†). Meanwhile, the battery using the N[1,1,4,2OH]-electrolyte experiences rapid performance degradation and maintains only an EE of ∼58.0% (Fig. S12c, ESI†), owing to the high viscosity of its polybromide complex. After a comprehensive performance evaluation, the ZBFBs with N[1,1,4,2OH]+ are at a competitive disadvantage (Fig. S13, ESI†). Due to the decreased diffusion rate and the reactivity of bromine species at low temperatures, the CEs of the available ZBFBs with the choline derivates are all around 99% at −20 °C, which should not be the evaluation criterion (Fig. S14, ESI†).
 | ||
| Fig. 6 (a) The average CE for 500 cycles of ZBFBs with different choline-based complexing agents at RT. (b) The average EE for 100 cycles of ZBFBs with different choline-based complexing agents at −20 °C. (c) and (d) The long cycle life of ZBFBs with N[1,1,3,2OH]- and N[1,2,2,2OH]-electrolytes at RT, respectively. (e) and (f) The low temperature performance of ZBFBs with different choline derivative-electrolytes at −20 °C. (g) and (h) The rate performance of the ZBFBs with different current densities at −20 °C. (i) The performance comparison of zinc–bromine batteries using various BCAs at room temperature.18,21,29,47–56 (j) The performance comparison of zinc-based flow batteries at low temperatures.5,9,19–21,57,58 | ||
As expected, the ZBFBs show excellent cycling life over 1600 cycles at RT using N[1,1,3,2OH]+ (an average CE of 95.7%) and N[1,2,2,2OH]+ (an average CE of 96.8%), respectively (Fig. 6c and d). Interestingly, at −20 °C, the batteries can operate stably with an average CE over 98.0% and an EE over 60.0% for 250 cycles at a current density of 40 mA cm−2 (Fig. 6d and f). It is worth noting that the derivatives in Series 1, along with those of C6 and C8 in Series 2 and Series 3, do not demonstrate excellent performance at both room temperature and low temperatures, which reflects the crucial role and significance of precise control over molecular polarity of the polybromide complexes. In addition, the rate performance of the ZBFBs with N[1,1,3,2OH]+ and N[1,2,2,2OH]+ was further characterized at −20 °C. The CE data of the battery using N[1,1,3,2OH]+ and N[1,2,2,2OH]+ exhibit excellent cycling stability at various current densities of 40–120 mA cm−2 (Fig. 6g and h). Even at 120 mA cm−2, the batteries retain the ability to withstand charging/discharging cycles, indicating the prospect of employing N[1,1,3,2OH]+ and N[1,2,2,2OH]+ as complexing agent cations in low-temperature ZBFBs. We further conducted an extensive comparison of recent studies on zinc–bromine batteries18,21,29,47–56 and other low-temperature zinc-based flow batteries,5,57,58 aiming to provide a comprehensive assessment of the performance achieved in this work. In contrast to other zinc–bromine batteries, the ZBFBs with N[1,1,3,2OH]+ and N[1,2,2,2OH]+, respectively, exhibit superior overall performance at both room temperature and low temperatures (Fig. 6i and j). Remarkably, even when compared with low-temperature counterparts, such as low-temperature zinc-iodide flow battery5,57 and low-temperature zinc-ferricyanide flow battery,58 the proposed ZBFBs manifest marked advantages with respect to frost resistance, current density and CE (Fig. 6j). These outcomes not only demonstrate that our complexing agents are competitive, but also fully illustrate the excellent application potential of the ZBFB system.
Conclusions
In summary, based on the precise control of the molecular polarity of polybromide complexes, N[1,1,3,2OH]+ and N[1,2,2,2OH]+ were proposed and synthesized as complexing agent cations, which demonstrate both excellent bromine capture capability at RT and exceptionally fluid state at low temperatures. Confirmation was obtained that the elongation of the carbon chain without –OH in the choline cation significantly diminishes the molecular polarity of polybromide complexes. This results in a slight strengthening of the C–A HB interaction and a reduction in both the ΔGsolv and the solubility of the complexes. Interestingly, compared with extending the carbon chain with –OH, this approach can effectively prevent the adverse effects on the low-temperature fluidity of the complexes caused by the enhancement of C–C HB interactions. Adding two carbons to the carbon chain without a hydroxyl group is optimal. As a result, the targets N[1,1,3,2OH]+ and N[1,2,2,2OH]+ not only endow ZBFBs with a high average CE at room temperature but also enable them to exhibit a prolonged cycle life and excellent rate capability at −20 °C. This research provides important theoretical and practical insights for the development of high-performance, low-temperature BCAs in future ZBFBs.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the CAS Strategic Leading Science & Technology Program (A) (XDA0400201), the Energy Revolution S&T Program of Yulin Branch, the Dalian National Laboratory for Clean Energy, CAS (DNL-YLEC202201), and the Youth Innovation Promotion Association, CAS (2022184).References
- X. Jiang, L. Wu, L. Che, M. Wang and J. Peng, J. Energy Storage, 2024, 80, 110147 CrossRef.
- J. Mao, M. Jafari and A. Botterud, Energy, 2022, 238, 121668 CrossRef.
- B. Zakeri, G. C. Gissey, P. E. Dodds and D. Subkhankulova, Energy, 2021, 236, 121443 CrossRef.
- J. T. Kim, A. Rao, H.-Y. Nie, Y. Hu, W. Li, F. Zhao, S. Deng, X. Hao, J. Fu, J. Luo, H. Duan, C. Wang, C. V. Singh and X. Sun, Nat. Commun., 2023, 14, 6404 CrossRef CAS.
- L. Zhang and G. Yu, Angew. Chem., Int. Ed., 2021, 60, 15028–15035 CrossRef CAS PubMed.
- S. Biswas, A. Senju, R. Mohr, T. Hodson, N. Karthikeyan, K. W. Knehr, A. G. Hsieh, X. Yang, B. E. Koelb and D. A. Steingart, Energy Environ. Sci., 2017, 10, 114–120 RSC.
- Y. Cho, J. G. Kim, D. H. Kim and C. Pak, Chem. Eng. J., 2024, 490, 151538 CrossRef CAS.
- Z. Li and Y.-C. Lu, Adv. Mater., 2020, 32, 2002132 CrossRef CAS PubMed.
- E. Lancry, B. Z. Magnes, I. Ben-David and M. Freiberg, ECS Trans., 2013, 53, 107–115 CrossRef.
- F. Ai, Z. Wang, N.-C. Lai, Q. Zou, Z. Liang and Y.-C. Lu, Nat. Energy, 2022, 7, 417–426 CrossRef CAS.
- H. Zhang, J. Chen, Z. Li, Y. Peng, J. Xu and Y. Wang, Adv. Funct. Mater., 2023, 33, 2304433 CrossRef CAS.
- F. Yue, Z. Tie, S. Deng, S. Wang, M. Yang and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 13882–13886 CrossRef CAS PubMed.
- S. Chen, T. Wang, L. Ma, B. Zhou, J. Wu, D. Zhu, Y. Y. Li, J. Fan and C. Zhi, Chem, 2022, 9, 497–510 Search PubMed.
- D. J. Eustace, J. Electrochem. Soc., 1980, 127, 528–532 CrossRef CAS.
- Y. Yin, Z. Yuan and X. Li, Phys. Chem. Chem. Phys., 2021, 23, 26070–26084 RSC.
- M. E. Easton, A. J. Ward, B. Chan, L. Radom, A. F. Masters and T. Maschmeyer, Phys. Chem. Chem. Phys., 2016, 18, 7251–7260 RSC.
- P. M. Hoobin, K. J. Cathor and J. O. Niere, J. Appl. Electrochem., 1989, 19, 943–945 CrossRef CAS.
- L. Amit, D. Naar, R. Gloukhovski, G. J. La and M. E. Suss, ChemSusChem, 2021, 14, 1068–1073 CrossRef CAS.
- K. J. Cathro, K. Cedzynska, D. C. Constable and P. M. Hoobin, J. Power Sources, 1986, 18, 349–370 CrossRef CAS.
- K. Cedzynska, Electrochim. Acta, 1995, 40, 971–976 CrossRef CAS.
- M. Zhao, T. Cheng, T. Li, R. Bi, Y. Yin and X. Li, Small, 2024, 20, 2307627 CrossRef CAS PubMed.
- C. Roche, M. Pucheault, M. Vaultier and A. Commerçon, Tetrahedron, 2010, 66, 8325–8334 CrossRef CAS.
- K. Ukawa, E. Imamiya, H. Yamamoto, K. Mzuno, A. Tasaka, Z.-I. Terashita, T. Okutanl, H. Nomura, T. Kasukabe, M. Hozumi, I. Kudo and K. Inoue, Chem. Pharm. Bull., 1989, 37, 1249–1255 CrossRef CAS PubMed.
- C. I. Melo, A. I. Rodrigues, R. Bogel-Łukasik and E. Bogel-Łukasik, J. Phys. Chem. A, 2012, 116, 1765–1773 CrossRef CAS PubMed.
- L.-R. Nie, S. Yao, B. Dong, X.-L. Li and H. Song, J. Mol. Liq., 2017, 240, 152–161 CrossRef CAS.
- U. Tonellato, J. Chem. Soc., Perkin Trans. 2, 1977, 821–827 RSC.
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef.
- Y. Wu, P.-W. Huang, J. D. Howe, Y. Yan, J. Martinez, A. Marianchuk, Y. Zhang, H. Chen and N. Liu, Angew. Chem., Int. Ed., 2019, 58, 15228 CrossRef CAS.
- X. Li, T. Li, P. Xu, C. Xie, Y. Zhang and X. Li, Adv. Funct. Mater., 2021, 31, 2100133 CrossRef CAS.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS PubMed.
- P. Sjoberg, J. S. Murray, T. Brinck and P. Politzer, Can. J. Chem., 1990, 68, 1440–1443 CrossRef CAS.
- P. Politzer, J. S. Murray and F. A. Bulat, J. Mol. Model., 2010, 16, 1731–1742 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2021, 171, 514–523 CrossRef CAS.
- A. Knorr, K. Fumino, A.-M. Bonsa and R. Ludwig, Phys. Chem. Chem. Phys., 2015, 17, 30978–30982 RSC.
- A. E. Khudozhitkov, A. G. Stepanov, D. I. Kolokolov and R. Ludwig, J. Phys. Chem. B, 2023, 127, 9336–9345 CrossRef CAS PubMed.
- S. P. Verevkin, D. H. Zaitsau and K. V. Zherikova, J. Mol. Liq., 2023, 382, 121938 CrossRef CAS.
- Y. Chen, A. Li, X. Li, L. Tu, Y. Xie, S. Xu and Z. Li, Adv. Mater., 2023, 35, 2211917 CrossRef CAS.
- L. A. Sheakh, T. Niemann, A. Villinger, P. Stange, D. H. Zaitsau, A. Strate and R. Ludwig, Chem. Phys. Chem., 2021, 22, 1850–1856 CrossRef.
- T. Lu, Molclus program, Version 1.12, can be found under https://www.keinsci.com/research/molclus.html (accessed 2024, 3, 12).
- A. Pecina, J. Fanfrlík, M. Lepšík and J. Řezáč, Nat. Commun., 2024, 15, 1127 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- J. Řezáč and P. Hobza, Chem. Phys. Lett., 2011, 506, 286–589 CrossRef.
- J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2011, 8, 141–151 CrossRef.
- H. Deng, X. Wang, Z. Wei, W. Liao, S. Li, W. Xu and S. Shi, J. Energy Storage, 2024, 81, 110449 CrossRef.
- L. Hu, C. Dai, Y. Zhu, X. Hou, Z. Liu, X. Geng, H. Wang, J. Chen, N. Sun, Q. Rong, Y. Zhu, X. He and Y. Lin, Energy Environ. Sci., 2024, 17, 5552–5562 RSC.
- H. Park, G. Park, S. Kumar, H. Yoon, J. Baek, T. Tamulevičius, S. Tamulevičius and H.-J. Kim, J. Power Sources, 2023, 580, 233212 CrossRef CAS.
- J. S. Cha, J. Lee, N.-U. Seo, D. K. Kim, Y.-C. Kang and J. H. Yang, J. Power Sources, 2023, 553, 232243 CrossRef CAS.
- S. H. Han, S. Kim, H. Y. Lim, S. Park, K. Shin, S. Kim, H.-T. Kim, S. K. Kwak, C. Yang and N.-S. Choi, Chem. Eng. J., 2023, 464, 142624 CrossRef.
- P. Xu, T. Li, Q. Zheng, H. Zhang, Y. Yin and X. Li, J. Energy Chem., 2022, 65, 89–93 CrossRef CAS.
- C. Dai, L. Hu, X. Jin, Y. Wang, R. Wang, Y. Xiao, X. Li, X. Zhang, L. Song, Y. Han, H. Cheng, Y. Zhao, Z. Zhang, F. Liu, L. Jiang and L. Qu, Sci. Adv., 2022, 8, eabo6688 CrossRef CAS PubMed.
- Y. Lee, D. Yun, J. Park, G. Hwang, D. Chung, M. Kim and J. Jeon, J. Power Sources, 2022, 547, 232007 CrossRef CAS.
- U. Jiménez-Blasco, E. Moreno, M. Cólera, P. Díaz-Carrasco, J. C. Arrebola, A. Caballero, J. Morales and Ó. A. Vargas, Int. J. Mol. Sci., 2021, 22, 9288 CrossRef.
- Z. Pang, Y. Gong, M. Yuan and X. Li, Batteries, 2020, 6, 27 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS.
- L. Zhi, C. Liao, P. Xu, G. Li, Z. Yuan and X. Li, Angew. Chem., Int. Ed., 2024, e202412559 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04046e |
| This journal is © The Royal Society of Chemistry 2025 |