
Simultaneously improving the efficiencies of organic photovoltaic devices and modules by finely manipulating the aggregation behaviors of Y-series molecules†
Yaohui
Li
abcd,
Ziyan
Jia
d,
Peihao
Huang
b,
Chuanlin
Gao
e,
Yufei
Wang
*e,
Shuangxi
Xue
a,
Shirong
Lu
 *a and
Yang
(Michael) Yang
*d
*a and
Yang
(Michael) Yang
*d
aDepartment of Material Science and Technology, Taizhou University, Taizhou 318000, P. R. China. E-mail: lushirong@cigit.ac.cn
bThin-film Solar Technology Research Center, Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, P. R. China
cUniversity of Chinese Academy of Sciences (UCAS Chongqing), Chongqing 400714, P. R. China
dState Key Laboratory of Modern Optical Instrumentation College of Optical Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: yangyang15@zju.edu.cn
eCollege of New Materials and New Energies, Shenzhen Technology University, Shenzhen 518118, P. R. China. E-mail: wangyufei@sztu.edu.cn
First published on 20th November 2024
Abstract
The introduction of an electron deficient core (e.g.: BTP, dithiophene [3.2b] pyrrolobenzothiazole) was considered to be an effective strategy for modulating the electron-vibration coupling, delocalization, and molecular stacking of high-performance Y-series non-fullerene acceptors (NFAs). However, the above means often make it difficult to achieve precise control of the various aggregation behaviors of Y-series NFAs, which is a key factor of limiting the performance improvement in the final device. In this study, we present a novel liquid additive, an electronegative alkane, which strengthens non-covalent interactions and boosts electron coupling. This promotes rapid nucleation and crystallization of the Y-series molecule, enhancing molecular stacking and aggregation. Besides, the directional induction of the BTP core in the blend active layer is well maintained, which optimizes the charge transport and reduces trap-assisted recombination of the bulk heterojunction. As a result, our strategy has substantially improved the performance of multiple Y-series NFA OPV systems, enabling thick film (≥200 nm) large-area modules (19.31 cm2) with efficiencies exceeding 14%. We believe that the broader processing window offered by the thick film is a notable advancement towards the commercialization of organic photovoltaics.
Broader contextThe development of Y-series non-fullerene acceptors (NFAs) has pushed small-area organic photovoltaics (OPVs) beyond 20% efficiency, but scaling to large areas demands precision in film uniformity and defect management to prevent performance issues like leakage currents and voltage losses. Achieving this requires controlling the active layer thickness for optimal charge transport and recombination suppression. We introduce an electronegative additive that enhances NFA aggregation and molecular packing through non-covalent interactions, maintaining these benefits in the blend layer and achieving 15.66% and 14.08% efficiencies for 120 nm and 200 nm thick films in large-area (19.3 cm2) modules, respectively, setting a new benchmark efficiency for thick-film based OPV modules. |
Introduction
Organic photovoltaics (OPVs) have attracted intensive attention for their desirable properties, such as being lightweight, flexible, semi-transparent, and suitable for large-area manufacturing.1,2 In particular, the recent advances in Y-series non-fullerene acceptors (NFAs) have driven state-of-the-art OPVs to achieve power conversion efficiencies (PCEs) nearing 20%.3,4 These NFAs, typically characterized by a planar A–DA′D–A structure, consist of a 3-dicyanomethylene-1-indanone (IC) acceptor terminal, an alkyl side chain, and a novel electron-deficient core.2,5,6 The interplay among these functional groups dictates the optoelectronic properties and photovoltaic performance.2,5Previous studies have proven that additive engineering can modify the NFA properties by inducing specific structural units and therefore enhance the device performance.7–11 Specifically, additives interact with NFA structural units to regulate molecular aggregation and stacking, thus improving electrical properties. The IC terminal unit plays an important role in promoting non-covalent intermolecular interaction and regulating the intramolecular push–pull effect.12–14 The design of additives that target the IC terminal of NFA has been extensively studied. For instance, Li et al. reported that the additive BID acts on the IC terminal unit in Y6 to induce the formation of a eutectic phase, which promotes a more ordered molecular arrangement and enhances device stability.15 Hou et al. reported that the additive DTBF can form a strong charge-quadrupole interaction with the IC terminal unit of BO4Cl, inducing a higher molecular packing density and broadening of infrared absorption.16 Similarly, Cheng et al. reported that the additive BF7 can produce an F-pi noncovalent supramolecular interaction with the Y6 terminal to optimize the size and orientation of Y6 nano-crystallites.17 Unlike the IC terminal unit, the role of the alkyl side chain is mainly focused on the stabilization of molecular conformation and the suppression of energetic disorder.5,18 At present, traditional additives such as DIO and DPE have been found to induce the alkyl side chain of NFA to form “A-to-D” type J-aggregation, creating a compact three-dimensional molecular network.19–21 These directional inductions for both the IC terminal unit and alkyl side chain of NFA can effectively enhance the device performance.
However, less explored is the impact of additive engineering on the electron-deficient core of NFAs. The typical electron-deficient core (dithienothiophen [3.2-b] pyrrolobenzothiadiazole, BTP core), can effectively reduce electron-vibration coupling for exciton dissociation, facilitating efficient charge generation with low driving forces.2,5,22,23 Designing additives targeting on the BTP core might introduce a significant impact on molecular behavior.
In this work, we introduce a novel additive 2Br (short for 1,1,2,2-tetrabromoethane) with strong electronegativity, which significantly enhances the device performance by finely manipulating the aggregation behaviors of Y-series molecules. Our findings, supported by both theoretical simulations and experimental characterizations, reveal that 2Br forms robust non-covalent interaction with the BTP core in NFA molecules. This interaction strengthens molecular packing, promotes aggregation, and boosts electronic coupling, leading to faster crystallization and improved charge mobility. Importantly, these microstructural enhancements are maintained in the blend active layer, thus leading to improved device efficiency. The integration of 2Br into Y-series NFA based OPVs has yielded remarkable results across multiple systems, with the PM6:Y6, PM6:BTP-ec9, PM6:L8BO, and PM6:D18:L8BO systems achieving efficiencies of 17.60%, 18.80%, 18.36%, and 19.51%, respectively. Extending this approach to a 200-nm-thick solar module with a 19.3 cm2 illumination area has achieved an impressive 14.08% efficiency, establishing a new benchmark for thick-film large-area modules. The thicker film offers a broader processing window for large-scale manufacturing, marking a promising advancement towards the commercialization of OPVs.
Results and discussion
Our investigation commenced with the PM6:Y6 system, a benchmark for NFA organic photovoltaics. The chemical structures of PM6, Y6 and 2Br are presented in Fig. S1 (ESI†) and Fig. 1a. The additive 2Br is a brominated alkane with a low boiling point of approximately 124 °C (Fig. S2, ESI†), it exhibits liquid properties conducive to interaction with Y6, as evidenced by its selective solubility in our experiments. It is widely recognized that the dissolution behavior of a solute can be indicative of the interactions between solvent and solute molecules.24 Herein, 2 mg PM6 and Y6 were each introduced in 0.4 mL 2Br solvent to access their chemical interactions. As shown in Fig. S3 (ESI†), 2Br selectively dissolves Y6, suggesting a strong affinity between the two. The lack of dissolution of PM6 in the 2Br solvent could be attributed to the large steric hindrance of PM6, which likely diminishes the interaction. The selective interaction is further reflected by the UV-vis absorption spectrum, as shown in Fig. S4 (ESI†). In contrast to the pristine Y6 film, the characteristic absorption peak of the 2Br-processed Y6 film shows a pronounced redshift (Δλ = 8.48 nm), indicating the occurrence of a charge transfer-mediated J-aggregation behavior among the Y6 molecules.25 | ||
| Fig. 1 (a) The chemical structure and ESP distribution of 2Br and Y6. (b) The different location of 2Br on the Y6 backbone with the respective exoergic energy in front (left) and side (right) view. Three different Y6 molecular dimer packing sketch (c) without and (d) with 2Br. | ||
Subsequently, we deeply analyzed the intermolecular interaction between Y6 and 2Br based theoretical simulation. The electrostatic potential (ESP) distribution of 2Br and Y6, computed via density functional theory (DFT),16 reveals a broad expanse of negative charge in 2Br, juxtaposed with localized positive regions in Y6, such as the BTP core unit and the Indan end-group (Fig. 1a). This complementary charge distribution predicates multiple sites of attraction.26 The optimized composite structure of the 2Br:Y6 dimer, depicted in Fig. 1b, confirms the lowest exoergic energy (−13.24 kcal mol−1) when 2Br approaches the BTP core unit, signifying the most stable configuration and a propensity for interaction at this site. To elucidate the nature of this interaction, we carried out the Fourier transform infrared (FT-IR) spectroscopy measurement of pristine Y6 and 2Br-processed Y6, as displayed in Fig. S5 (ESI†). The pristine Y6 film exhibits two stretching vibration peaks at 2920.7 and 2851.4 cm−1, respectively, attributed to the –CH2– groups within the BTP core unit.10 Upon processing with 2Br, these peaks upshift to 2926.5 and 2856.2 cm−1, respectively, confirming a significant interaction between 2Br and the BTP core unit of Y6. This strong interaction is able to enhance the non-covalent force within the core unit,11 which in turn can influence the molecular stacking and aggregation behavior.
To further gain insight into the effect of 2Br on the stacking behavior of the Y6 dimer, the optimal 3D structure of the Y6 molecular dimer was calculated by molecular dynamics simulations (Fig. 1c and d). The Y6 molecular dimer exhibits three types of J-aggregation: A-to-D type J-aggregation (S), A-to-A type J-aggregation (W1) and A-to-A′ type J-aggregation (W2).7 Among these, the A-to-D type J-aggregation Y6 (without or with 2Br) molecular dimer is the most stable with the lowest Eex (−64.16 vs. −76.90 kcal mol−1) compared with A-to-A type J-aggregation (−37.60 vs. −50.94 kcal mol−1) and A-to-A′ type J-aggregation (−36.09 vs. −46.99 kcal mol−1). The simulated intermolecular packing distances for pristine Y6 are 3.55 Å (S type), 3.35 Å (W1 type), and 3.25 Å (W2 type), which decrease to 3.06, 3.29, and 3.21 Å, respectively, for the 2Br-processed Y6. The stabilized A-to-D type J-aggregation and reduced packing distances are beneficial for forming a compact 3D molecular stacking network in Y6, which is considered to facilitate efficient charge transport pathways.27,28 In addition, the electronic coupling of the A-to-D type J-aggregation Y6 molecular dimer in its stable state was calculated to increase from 15.3 meV for pristine Y6 to 30.5 meV for 2Br-processed Y6 (Fig. S6, ESI†), indicating that the improvement of Y6 intermolecular stacking behavior and aggregation state caused by the strong interaction between 2Br and the BTP core unit of Y6 significantly enhances the ability of electron coupling.
In situ time-resolved UV-vis absorption spectroscopy measurements elucidate the phase transition dynamics of Y6 film formation. The absorption spectral characteristics of Y6 during the solvent evaporation can be categorized into three distinct phases: the liquid (wet film) phase, the drying phase, and the solid (film formation) phase.29 As shown in Fig. 3a, b and Fig. S7a (ESI†), the liquid phase duration of the Y6 film was extended from 168 to 192 ms following the incorporation of the 2Br additive. This extension can provide additional pre-aggregation time. Moreover, the transition from liquid to solid phase in the 2Br-processed Y6 film was accelerated, reducing the time to 24 ms from the original 48 ms, indicating that 2Br promotes rapid nucleation and crystallization of Y6.30 The enhanced pre-aggregation and accelerated nucleation in the 2Br-processed Y6 film are advantageous for building tighter molecular packing.
To further probe the packing behavior influenced by 2Br, grazing-incidence wide-angle X-ray scattering (GIWAXS) measurement was employed (Fig. 2e, f and Fig. S8, ESI†). The π–π stacking peak (010) located at 1.74 Å−1 in the qz direction and the lamellar stacking peak (100) located at 0.28 Å−1 in the qr direction for 2Br-processed Y6 are more pronounced than those of pristine Y6, suggesting that 2Br induces a more orderly molecular stacking. The π–π stacking distance in the out-of-plane direction was reduced from 3.81 Å in the pristine Y6 to 3.61 Å in the 2Br-processed Y6. Concurrently, the crystal coherence length (CCL) increased from 17.13 Å in pristine Y6 to 20.19 Å in 2Br-processed Y6. These findings indicate that the molecular packing of Y6 treated with 2Br is more compact, and the resulting crystallinity is enhanced. The surface roughness of 2Br-processed Y6, measured by atomic force microscopy (AFM), is slightly higher at 0.98 nm compared to the pristine Y6 at 0.96 nm. This improved molecular packing and crystallinity are expected to ensure better electronic coupling, thereby facilitating efficient charge transport in the 2Br processed Y6.31 The argument is supported by the electron mobility measurements conducted using the space charge limit current (SCLC) method. As shown in Fig. S9 (ESI†), the electron mobility of 2Br-processed Y6 is 10.43 × 10−4 cm2 V−1 s−1, which is an order of magnitude higher than that of the pristine Y6 (3.70 × 10−4 cm2 V−1 s−1).
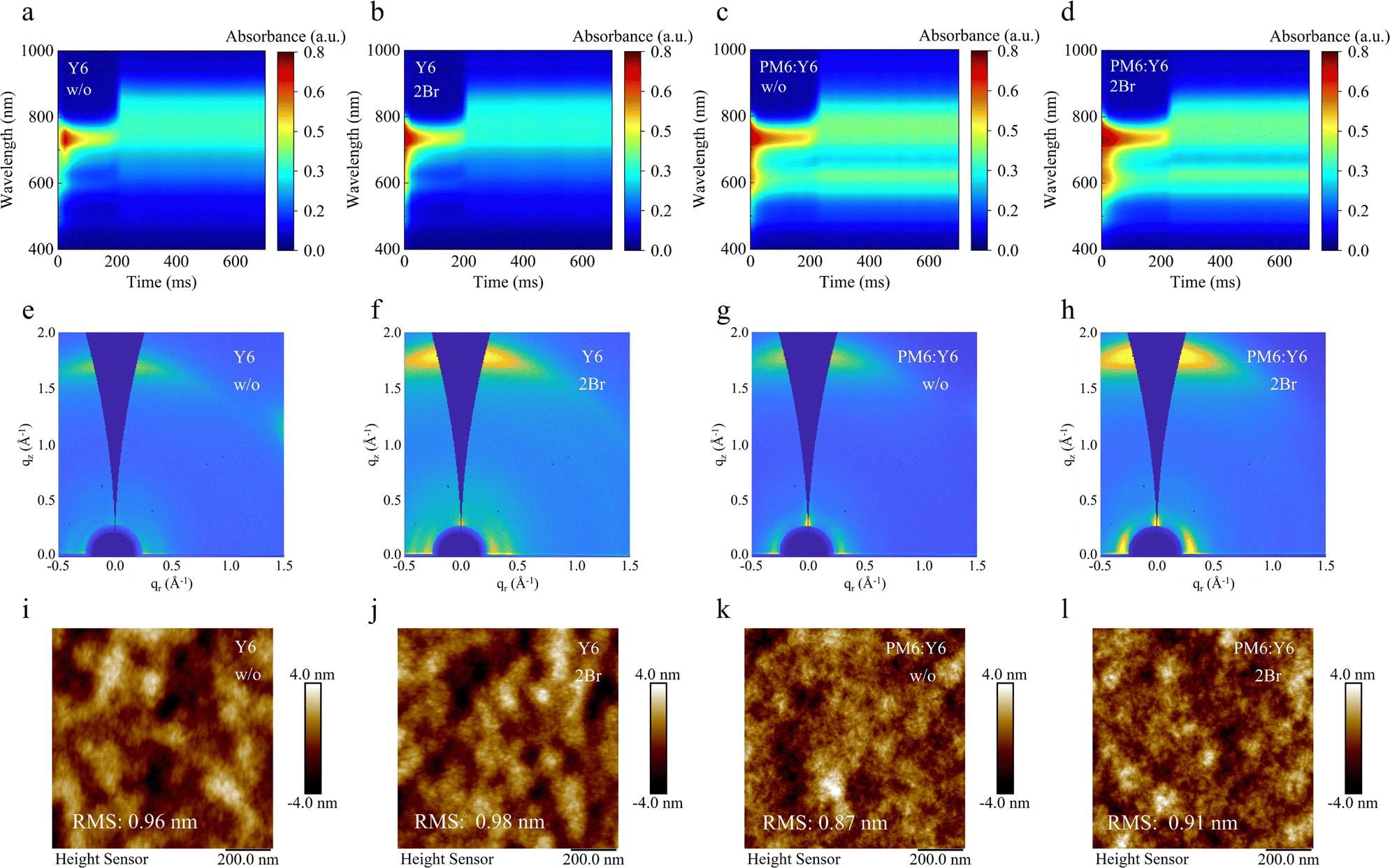 | ||
| Fig. 2 In situ UV-vis absorption spectra of (a) pristine Y6, (b) 2Br-processed Y6, (c) pristine PM6:Y6 and (d) 2Br-processed PM6:Y6. GIWAXS patterns of (e) pristine Y6, (f) 2Br-processed Y6, (g) pristine PM6:Y6 and (h) 2Br-processed PM6:Y6. AFM height image of (i) pristine Y6, (j) 2Br-processed Y6, (k) pristine PM6:Y6 and (l) 2Br-processed PM6:Y6. | ||
Notably, in situ time-resolved UV-vis absorption spectroscopy, GIWAXS and AFM analyses of 2Br-processed PM6:Y6 blended films (Fig. 2c, d, g, h, k and l) show similar trends to those of the 2Br-processed Y6 film. The 2Br-processed PM6:Y6 film exhibits a rapid phase transition, orderly and compact molecular packing, and increased surface roughness. These features indicate that the interaction between 2Br and the BTP core unit of Y6 is effectively retained within the blend. This interaction is crucial as it suggests that the microstructure improvements, driven by the strong affinity between 2Br and the BTP core unit of Y6, are likely to contribute positively to the photovoltaic performance.
Next, the application potential of the novel additive was evaluated by using a conventional device configuration of ITO/PEDOT:PSS/active layer/PNDIT-F3N/Ag (Fig. 3a). The optimal preparation conditions of 2Br additive are shown in Fig. S10 (ESI†) and Table S3 (ESI†), and the current density–voltage (J–V) curves and corresponding photovoltaic parameters are shown in Fig. 3b and Table 1, respectively. The pristine device provides a mediocre PCE of 16.03%, with an open-circuit voltage (Voc) of 0.851 V, a short-circuit current density (Jsc) of 25.84 mA cm−2 and a fill factor (FF) of 72.87%. The 2Br-processed device provides a maximum PCE of 17.60% with a Voc of 0.849, a Jsc of 26.64 mA cm−2 and a FF of 77.84%. In addition, the photovoltaic parameters of 2Br-processed devices are also better than those of traditional additives CN and DIO (ESI,† Table S4). Apparently, the improvements in Jsc and FF are the primary contributors to boost the efficiency. The increase of Jsc is confirmed by external quantum efficiency (EQE), as shown in Fig. 3c. The 2Br-processed device exhibits a higher EQE response in the wavelength range from 450 to 950 nm, causing a higher integrated current density of 25.78 mA cm−2 compared to the pristine device (24.78 mA cm−2). The integrated results are in agreement with Jsc extracted from the J–V curves, and the measurement error is about 5%. Furthermore, the exciton dissociation (Pdiss) and charge collection (Pcoll) efficiency of 2Br-processed devices were studied by plotting the correlation between photocurrent density (Jph) and effective voltage (Veff). As shown in Fig. 3d, the Pdiss and Pcoll of the 2Br-processed device are 97.7% and 89.6%, both higher than that of the pristine device (Pdiss of 96.4% and Pcoll of 87.9%), which facilitates Jsc's enhancement. In addition, charge transport characteristics of the active layer were further studied by the SCLC method. As shown in Fig. S11 (ESI†), the hole mobility of the 2Br-processed device is 8.44 × 10−4 cm2 V−1 s−1, similar to that of the pristine one (7.88 × 10−4 cm2 V−1 s−1). Moreover, the remarkably higher electron mobilities of 11.23 × 10−4 cm2 V−1 s−1 are found in the 2Br-processed PM6:Y6 compared with the device without additives (3.91 × 10−4 cm2 V−1 s−1), which is consistent with the results of GIWAXS and theoretical calculation. The ratio between hole mobility and electron mobility of 2Br-processed PM6:Y6 is 1.33, which is closer to 1 than that of pristine PM6:Y6 (2.02), suggesting that the hole and electron transport in the 2Br-processed PM6:Y6 are more balanced, which may be one of the important contributors to the large FF.32
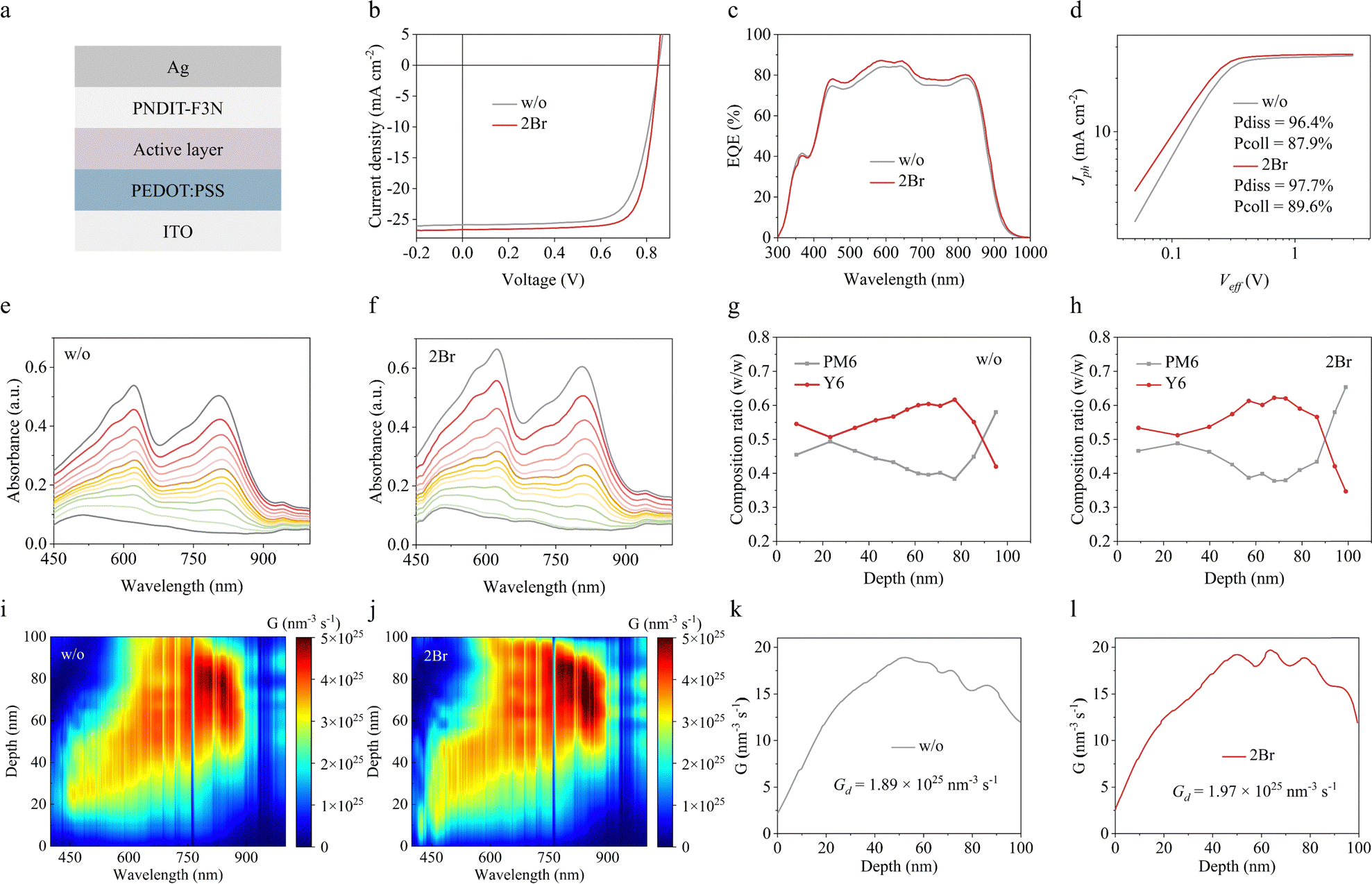 | ||
| Fig. 3 (a) Schematic illustration of the device structure. (b) J–V curves and (c) EQE spectra of the PM6:Y6-based device without and with 2Br. (d) Plot of Jphversus Veff in the PM6:Y6-based device without and with 2Br. The film-depth-dependent profiling light absorption spectra of (e) pristine and (f) 2Br-processed PM6:Y6. Composition ratio across the vertical direction of the (g) pristine and (h) 2Br-processed PM6:Y6 film. Exciton generation map across the vertical direction of the (i) pristine and (j) 2Br-processed PM6:Y6 film as a function of wavelength. Integrated generation rate in the vertical direction of the (k) pristine and (l) 2Br-processed PM6:Y6 film. | ||
| Active layer | Additive | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | J sc.int (mA cm−2) |
|---|---|---|---|---|---|---|
| PM6:Y6 | w/o | 0.851 | 25.84 | 72.87 | 16.03 (15.72) | 24.78 |
| 2Br | 0.849 | 26.64 | 77.84 | 17.60 (17.41) | 25.78 | |
| PM6:BTP-eC9 | w/o | 0.849 | 27.84 | 74.69 | 17.66 (17.39) | 26.72 |
| 2Br | 0.847 | 28.28 | 78.48 | 18.80 (18.65) | 27.19 | |
| PM6:L8BO | w/o | 0.889 | 25.73 | 75.22 | 17.22 (16.91) | 24.87 |
| 2Br | 0.889 | 25.93 | 79.67 | 18.36 (18.13) | 25.20 | |
| PM6:D18:L8BO | w/o | 0.904 | 26.62 | 75.86 | 18.25 (17.93) | 25.22 |
| 2Br | 0.903 | 26.93 | 80.24 | 19.51 (19.33) | 25.60 |
Subsequently, the vertical distribution of the active layer was investigated by measuring film-depth-dependent light absorption spectra using top-to-bottom plasma etching. Fig. 3e and f show the sublayer absorption spectrum of the PM6:Y6 film. The absorption of the 2Br-processed PM6:Y6 film is significantly enhanced with a slight redshift, signifying improved crystallinity of PM6:Y6.10,33 By analyzing the sublayer absorption, the vertical distribution of donor and acceptor materials within the active layer was determined, as shown in Fig. 3g and h. In the 2Br-processed PM6:Y6 film, the PM6 content near the PEDOT:PSS side increased from 0.58 to 0.65, while the Y6 content in the middle region increased from 0.59 to 0.61. The optimal phase composition induced by the 2Br additive is expected to reduce charge recombination in principle.
The recombination characteristic is further verified by studying the relationship between Voc and light intensity (Voc = nKT/q![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(Plight)) under open-circuit conditions, where n is the indicator of trap-assisted recombination, K is Boltzmann's constant, T is Kelvin temperature and q is the elementary charge.24 As shown in Fig. S12 (ESI†), the n for the pristine device is 1.26, which deviates from 1, meaning that the pristine device has serious trap-assisted recombination.24 After processing with 2Br additive, the n decreases to 1.07, suggesting that the trap-assisted recombination is suppressed. The transient photovoltage (TPV) measurement provided further insights into the carrier lifetime, which is indicative of charge recombination dynamics. Fig. S13 (ESI†) shows that the carrier lifetime of the 2Br-processed device is 0.47 μs, substantially longer than the 0.24 μs observed for the pristine device. This extended lifetime indicates a reduced likelihood of charge recombination, which contributes to the enhancement of Jsc and FF.
ln(Plight)) under open-circuit conditions, where n is the indicator of trap-assisted recombination, K is Boltzmann's constant, T is Kelvin temperature and q is the elementary charge.24 As shown in Fig. S12 (ESI†), the n for the pristine device is 1.26, which deviates from 1, meaning that the pristine device has serious trap-assisted recombination.24 After processing with 2Br additive, the n decreases to 1.07, suggesting that the trap-assisted recombination is suppressed. The transient photovoltage (TPV) measurement provided further insights into the carrier lifetime, which is indicative of charge recombination dynamics. Fig. S13 (ESI†) shows that the carrier lifetime of the 2Br-processed device is 0.47 μs, substantially longer than the 0.24 μs observed for the pristine device. This extended lifetime indicates a reduced likelihood of charge recombination, which contributes to the enhancement of Jsc and FF.
In addition, the exciton generation rate (G) along the vertical direction of the active layer is further extracted via the transfer matrix method.33 During the depth range of 50–80 nm (Fig. 3i and j), the G of the 2Br-processed PM6:Y6 active layer is significantly higher than that of the pristine PM6Y6. The maximum G values for the 2Br-processed and pristine PM6:Y6 were 1.97 × 1025 and 1.89 × 1025 nm−3 s−1, respectively. The higher exciton generation rate helps produce photocurrent, which is consistent with the result of the J–V curve.
Besides, the universal applicability of 2Br was validated in the two state-of-the-art Y series NFAs, named BTP-eC9 and L8BO, both of which contain a BTP core, as illustrated in Fig. 4a. Fig. 4b shows the summarized parameters of these two NFA systems. For the PM6:BTP-eC9 and PM6:L8BO solar cells, the PCE of the 2Br-processed device reaches 18.80% and 18.36%, respectively. While for the PM6:D18:L8BO solar cell, the PCE reaches 19.51% with a FF of 80.24% (Fig. S14, ESI† and Table 1). These outcomes validate the effectiveness of the strategy leveraging the interaction between the 2Br and BTP core of Y series NFAs. In addition, the strategy was extended to a large area module. In the production of the large-area module, the uniformity of the active layer thickness is crucial, as significant deviations can rapidly degrade device performance. The development of thickness-insensitive OPV has thus become a prominent area of research in recent years.34–36 A thickness exceeding 200 nm offers greater tolerance for large-area modules compared to the conventional 80–120 nm range. In this case, for the PM6:D18:L8BO system, the thickness of 200 nm can still maintain 18.05% efficiency with a small area of 0.0585 cm2 (Fig. 4c and Table S7, ESI†). When scaled up to a large area module of 19.3 cm2 (Fig. 4e), the efficiency of the 120 nm thick sample reaches 15.66%, accompanied by a Voc of 6.305 V, an Isc of 64.05 and a FF of 73.44% (Fig. 4f). When the thickness increases to 200 nm, the module efficiency still remains 14.08% (Fig. 4g), which is the highest value for a thick-film large-area module (summarized in Fig. 4h and Table S8, ESI†). This underscores the substantial potential of the strategy for fabricating efficient thick-film OPV modules.
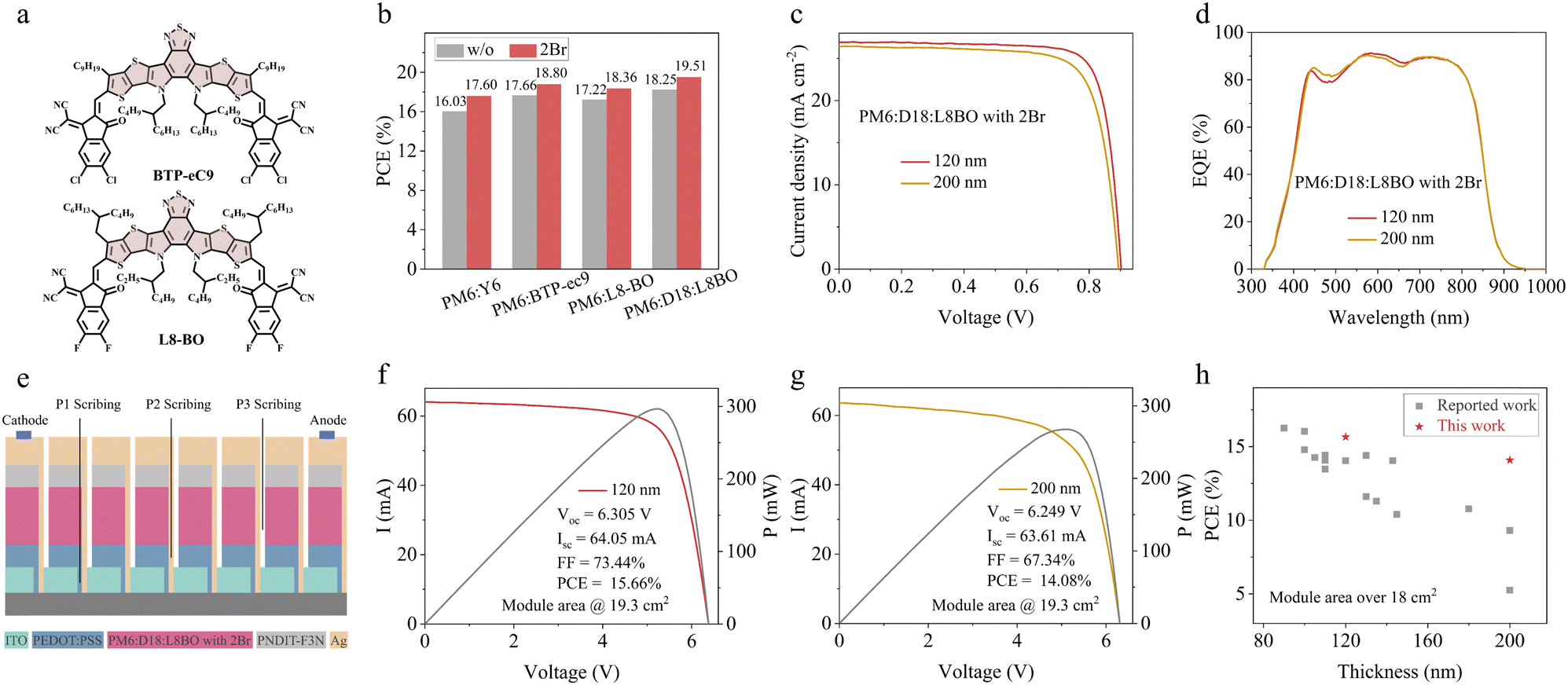 | ||
| Fig. 4 (a) The chemical structures of BTP-ec9 and L8BO. (b) PCE of OPVs with different Y series NFAs. (c) J–V and (d) EQE curves with thickness variation of the PM6:D18:L8BO system containing 2Br. (e) Schematic illustration of the OPV module. I–V and P–V curves of the PM6:D18:L8BO module with 120 nm (f) and 200 nm (g) thickness. (h) The development of PCEs with different thicknesses of the active layer for a large area module area over 18 cm2. | ||
Conclusions
In conclusion, we have introduced a novel liquid additive, 2Br, which effectively modulates the J-aggregation behavior of NFAs by strengthening the non-covalent interactions with the BTP core. The optimized stacking behavior facilitated by the 2Br additive accelerates the nucleation and crystallization of Y6, nearly doubling the electron mobility of Y-series NFAs. When integrated into the PM6:D18:L8BO blend active layer system, the 2Br additive enables a device efficiency of 19.51% on a small-area (0.0585 cm2) device and an impressive 15.66% on a large-area (19.3 cm2) module. Most notably, even when the active layer thickness is increased to 200 nm, the efficiencies for the small and large area devices remain high at 18.05% and 14.08%, respectively. The latter figure is particularly remarkable as it represents the highest efficiency reported for a thick-film large-area module. The ability to maintain high efficiency with a thicker film offers greater tolerance for the fine processing of the active layer in large-area modules, which is a notable advancement towards the practical commercialization of OPVs.Author contributions
Y. Li conceived the idea and designed the research; Y. Li and Z. Jia fabricated the small-area devices and large-area modules; P. Huang performed the theoretical simulation; C. Gao was responsible for the UV-vis absorption spectra measurement and the film-depth-dependent profiling light absorption spectra measurement with the help of Y. Wang. The manuscript was mainly written by Y. Li, Y. Wang and Y. Yang; all authors commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Data are available from the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Y. Wang thanks the National Natural Science Foundation of China (12404480) for financial support. S. Lu thanks the Natural Science Foundation of Zhejiang Province of China (LY24E030008) for financial support. Y. Yang thanks the National Natural Science Foundation of China (T2325020, 52273307) for financial support. The authors thank Hangzhou Microquanta Semiconductor Co. Ltd for the support in module fabrication.References
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef CAS PubMed
.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, 2400342 CrossRef CAS
- Y. Sun, L. Wang, C. Guo, J. Xiao, C. Liu, C. Chen, W. Xia, Z. Gan, J. Cheng, J. Zhou, Z. Chen, J. Zhou, D. Liu, T. Wang and W. Li, J. Am. Chem. Soc., 2024, 146, 12011–12019 CrossRef CAS PubMed
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS
- D. Li, N. Deng, Y. Fu, C. Guo, B. Zhou, L. Wang, J. Zhou, D. Liu, W. Li, K. Wang, Y. Sun and T. Wang, Adv. Mater., 2023, 35, 2208211 CrossRef CAS
- X. Zhang, J. Cai, C. Guo, D. Li, B. Du, Y. Zhuang, S. Cheng, L. Wang, D. Liu and T. Wang, Small, 2021, 17, 2102558 CrossRef CAS
- H. Zhang, G. Ran, X. Cui, Y. Liu, Z. Yin, D. Li, X. Ma, W. Liu, H. Lu, R. Liu, L. Cai, W. Zhang, S. Guo, H. Li, J. Yu, Y. Lin, Y. Liu, G. Lu, Z. Ma, P. Cheng and Z. Bo, Adv. Energy Mater., 2023, 13, 2302063 CrossRef CAS
- X. Song, K. Zhang, R. Guo, K. Sun, Z. Zhou, S. Huang, L. Huber, M. Reus, J. Zhou, M. Schwartzkopf, S. V. Roth, W. Liu, Y. Liu, W. Zhu and P. Müller-Buschbaum, Adv. Mater., 2022, 34, 2200907 CrossRef CAS
- J. Wang, Y. Wang, P. Bi, Z. Chen, J. Qiao, J. Li, W. Wang, Z. Zheng, S. Zhang, X. Hao and J. Hou, Adv. Mater., 2023, 35, 2301583 CrossRef CAS
- J. Jin, Q. Wang, K. Ma, W. Shen, L. A. Belfiore, X. Bao and J. Tang, Adv. Funct. Mater., 2023, 33, 2213324 CrossRef CAS
- G. Li, L.-W. Feng, S. Mukherjee, L. O. Jones, R. M. Jacobberger, W. Huang, R. M. Young, R. M. Pankow, W. Zhu, N. Lu, K. L. Kohlstedt, V. K. Sangwan, M. R. Wasielewski, M. C. Hersam, G. C. Schatz, D. M. DeLongchamp, A. Facchetti and T. J. Marks, Energy Environ. Sci., 2022, 15, 645–659 RSC
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef
- J. Fu, H. Chen, P. Huang, Q. Yu, H. Tang, S. Chen, S. Jung, K. Sun, C. Yang, S. Lu, Z. Kan, Z. Xiao and G. Li, Nano Energy, 2021, 84, 105862 CrossRef CAS
- R. Yu, H. Yao, Y. Xu, J. Li, L. Hong, T. Zhang, Y. Cui, Z. Peng, M. Gao, L. Ye, Z. Tan and J. Hou, Adv. Funct. Mater., 2021, 31, 2010535 CrossRef CAS
- K. Hung, Y. Lin, Y. Xue, H. Yang, Y. Lai, J. Chang, C. Su, A. Su, C. Hsu, U. Jeng and Y. Cheng, Adv. Energy Mater., 2022, 12, 2103702 CrossRef CAS
- Y. Jiang, Y. Li, F. Liu, W. Wang, W. Su, W. Liu, S. Liu, W. Zhang, J. Hou, S. Xu, Y. Yi and X. Zhu, Nat. Commun., 2023, 14, 5079 CrossRef CAS PubMed
- X. Zhang, H. Wang, D. Li, M. Chen, Y. Mao, B. Du, Y. Zhuang, W. Tan, W. Huang, Y. Zhao, D. Liu and T. Wang, Macromolecules, 2020, 53, 3747–3755 CrossRef CAS
- Z. Zheng, E. He, J. Wang, Z. Qin, T. Niu, F. Guo, S. Gao, Z. Ma, L. Zhao, X. Lu, Q. Xue, Y. Cao, G. T. Mola and Y. Zhang, J. Mater. Chem. A, 2021, 9, 26105–26112 RSC
- Y. Fu, L. Wang, C. Guo, D. Li, J. Cai, B. Zhou, C. Chen, C. Liu, D. Liu, W. Li and T. Wang, ACS Mater. Lett., 2022, 4, 2009–2018 CrossRef CAS
- Y. Guo, G. Han and Y. Yi, Angew. Chem., Int. Ed., 2022, 61, e202205975 CrossRef CAS
- Q. He, P. Ufimkin, F. Aniés, X. Hu, P. Kafourou, M. Rimmele, C. L. Rapley and B. Ding, SusMat, 2022, 2, 591–606 CrossRef CAS
- L. Guo, Q. Li, J. Ren, Y. Xu, J. Zhang, K. Zhang, Y. Cai, S. Liu and F. Huang, Energy Environ. Sci., 2022, 15, 5137–5148 RSC
- Y. Wang, Z. Liang, X. Liang, X. Wen, Z. Cai, Z. Shao, J. Zhang, Y. Ran, L. Yan, G. Lu, F. Huang and L. Hou, Adv. Energy Mater., 2023, 13, 2300524 CrossRef CAS
- L. Ma, H. Yao, J. Wang, Y. Xu, M. Gao, Y. Zu, Y. Cui, S. Zhang, L. Ye and J. Hou, Angew. Chem., Int. Ed., 2021, 60, 15988–15994 CrossRef CAS PubMed
- H. Lai and F. He, Adv. Energy Mater., 2020, 10, 2002678 CrossRef CAS
- G. Zhang, X.-K. Chen, J. Xiao, P. C. Y. Chow, M. Ren, G. Kupgan, X. Jiao, C. C. S. Chan, X. Du, R. Xia, Z. Chen, J. Yuan, Y. Zhang, S. Zhang, Y. Liu, Y. Zou, H. Yan, K. S. Wong, V. Coropceanu, N. Li, C. J. Brabec, J.-L. Bredas, H.-L. Yip and Y. Cao, Nat. Commun., 2020, 11, 3943 CrossRef CAS
- H. Lu, W. Liu, G. Ran, Z. Liang, H. Li, N. Wei, H. Wu, Z. Ma, Y. Liu, W. Zhang, X. Xu and Z. Bo, Angew. Chem., Int. Ed., 2023, 62, e202314420 CrossRef CAS PubMed
- X. Duan, Y. Yang, J. Yu, C. Liu, X. Li, M. H. Jee, J. Gao, L. Chen, Z. Tang, H. Y. Woo, G. Lu and Y. Sun, Adv. Mater., 2024, 36, 2308750 CrossRef CAS PubMed
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed
- J. Song, L. Zhu, C. Li, J. Xu, H. Wu, X. Zhang, Y. Zhang, Z. Tang, F. Liu and Y. Sun, Matter, 2021, 4, 2542–2552 CrossRef CAS
- C. Zhao, J. Yi, L. Wang, G. Lu, H. Huang, H. K. Kim, H. Yu, C. Xie, P. You, G. Lu, M. Qiu, H. Yan, S. Li and G. Zhang, Nano Energy, 2022, 104, 107872 CrossRef CAS
- Y. Wei, Y. Cai, X. Gu, G. Yao, Z. Fu, Y. Zhu, J. Yang, J. Dai, J. Zhang, X. Zhang, X. Hao, G. Lu, Z. Tang, Q. Peng, C. Zhang and H. Huang, Adv. Mater., 2024, 36, 2304225 CrossRef CAS
- H. Zhang, Y. Liu, G. Ran, H. Li, W. Zhang, P. Cheng and Z. Bo, Adv. Mater., 2024, 36, 2400521 CrossRef CAS
- Z. Fu, J. Qiao, F. Cui, W. Zhang, L. Wang, P. Lu, H. Yin, X. Du, W. Qin and X. Hao, Adv. Mater., 2024, 36, 2313532 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04378b |
| This journal is © The Royal Society of Chemistry 2025 |