
Metastable interphase induced pre-strain compensation enables efficient and stable perovskite solar cells†
Hongyu
Xu
 ab,
Yun
Xiao
*c,
Karim A.
Elmestekawy
c,
Pietro
Caprioglio
c,
Qiuyang
Li
a,
Qixuan
Zhong
a,
Yongqiang
Ji
a,
Tianyu
Huang
a,
Haoming
Yan
a,
Yingguo
Yang
d,
Laura M.
Herz
c,
Qihuang
Gong
aef,
Henry J.
Snaith
*c,
Rui
Zhu
*abef and
Lichen
Zhao
*ab
ab,
Yun
Xiao
*c,
Karim A.
Elmestekawy
c,
Pietro
Caprioglio
c,
Qiuyang
Li
a,
Qixuan
Zhong
a,
Yongqiang
Ji
a,
Tianyu
Huang
a,
Haoming
Yan
a,
Yingguo
Yang
d,
Laura M.
Herz
c,
Qihuang
Gong
aef,
Henry J.
Snaith
*c,
Rui
Zhu
*abef and
Lichen
Zhao
*ab
aState Key Laboratory for Artificial Microstructure and Mesoscopic Physics, School of Physics, Frontiers Science Center for Nano-optoelectronics & Collaborative Innovation Center of Quantum Matter, Peking University, Beijing 100871, China. E-mail: iamzhurui@pku.edu.cn; lczhao@pku.edu.cn
bSouthwest United Graduate School, Kunming 650092, China
cClarendon Laboratory, Department of Physics, University of Oxford, Oxford OX1 3PU, UK. E-mail: yun.xiao@physics.ox.ac.uk; henry.snaith@physics.ox.ac.uk
dSchool of Microelectronics, Fudan University, Shanghai 200433, China
ePeking University Yangtze Delta Institute of Optoelectronics, Nantong 226010, China
fCollaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan 030006, China
First published on 8th November 2024
Abstract
High-efficiency metal halide perovskite solar cells (PSCs) include rigid substrates with low thermal-expansion coefficients (TECs), resulting in significant TEC mismatch with the perovskites with high TECs at the buried interface. This mismatch leads to thermally induced residual tensile strain in perovskite films after annealing during film fabrication, which facilitates ion migration and defect formation, thereby compromising the performance and stability of PSCs. In this study, we present a pre-strain compensation strategy by introducing an in situ generated metastable Pb(CH3NH2)2Cl2 (PMC) phase at the buried substrate/perovskite interface, which will transform into PbCl2 upon annealing of formamidinium lead iodide (FAPbI3)-based perovskite films. This phase transformation provides a source of compressive stress for the perovskite films to counteract the adverse residual tensile strain during cooling from annealing. This strategy is demonstrated to be able to effectively reduce the defect formation and non-radiative recombination rates in the perovskite films, while enhancing the charge-carrier mobility, lowering the exciton binding energy, and weakening the electron–phonon coupling interactions. As a result, the corresponding modified n–i–p PSCs achieve a champion efficiency of 25.83% (certified at 25.36%) and exhibit improved stability.
Broader contextPhotovoltaic power generation plays a crucial role in global energy transition. Perovskite solar cells (PSCs) have garnered significant attention in the field of photovoltaics these days, and their certified power conversion efficiencies (PCEs) have surpassed 26%, demonstrating great potential for commercialization. However, high-efficiency PSCs normally utilize rigid substrates with low thermal expansion coefficients (TECs), resulting in a serious TEC mismatch between the substrate and perovskite with a high TEC. This mismatch leads to the thermally induced residual tensile strain in perovskite films after annealing during film formation, which has severe negative impacts on the performance of PSCs. In this work, we propose a pre-strain compensation strategy by introducing an in situ generated Pb(CH3NH2)2Cl2 interphase at the buried interface. During the annealing of perovskite films, the larger-volume interphase transforms into the small-volume PbCl2, providing a pre-compressive stress source to counteract the residual tensile strain during the cooling process. This strategy can effectively reduce the defect formation and non-radiative recombination rates in the perovskite films, while enhancing the carrier mobility, lowering the exciton binding energy, and weakening the electron–phonon coupling interactions. Consequently, we achieved the corresponding PSCs with a champion PCE of 25.83% (certified at 25.36%) and remarkably improved stability. |
Introduction
In recent years, the power conversion efficiencies (PCEs) of metal halide perovskite solar cells (PSCs) have rapidly increased to surpassing 26%, approaching the values of single-crystal silicon solar cells.1–3 In addition to improving the efficiency, enhancing the stability of PSCs is of another primary importance towards the commercialization of PSCs.4–6 Among the factors that affect the device stability, the residual tensile strain in the perovskite films is a worth noting concern, as it has been demonstrated to weaken the chemical bonds, lower the activation energy of ion migration, and facilitate the formation of cations and iodine vacancies.7–10 The resultant defects, acting as recombination centers for charge carriers, can thus reduce the charge transport efficiency and device PCEs, and even severely affect the long-term stability and durability of PSCs.The mismatch in thermal expansion between the substrate and the perovskite during the annealing step required for the formation of perovskite films is one of the main origins of the residual tensile strain in perovskite films.11,12 Specifically, the rigid substrates of PSCs consist of composite materials including glass, transparent conductive oxide (TCO), and/or metal oxide charge transport layers. The thermal expansion coefficients (TECs, α) of these materials are within a range (0.45 × 10−5 to 1 × 10−5 K−1),7,8,13,14 which are considerably lower than that of metal halide perovskite materials (3 × 10−5 to 8.4 × 10−5 K−1).14–18 This results in a serious mismatch in the thermal expansion behavior between the rigid substrates and the soft perovskites at the buried interface. Formamidinium lead iodide (FAPbI3) perovskite with a suitable theoretical bandgap width (∼1.50 eV) is one of the most promising absorber materials for high-efficiency single-junction PSCs.19–21 However, the fabrication of high-quality FAPbI3 perovskite films normally has to involve relatively high-temperature annealing at around 150 °C.22–24 The process of cooling to room temperature after annealing can give rise to the residual tensile strain in perovskite films due to the large TEC difference between the soft perovskite lattice and the rigid substrate.
To regulate the thermally induced residual tensile strain in the perovskite films, the researchers proposed strategies such as introducing an external compressive strain from the post-deposited hole-transport layer for rigid PSCs,8 or pre-applying a compressive strain on the flexible substrate for flexible PSCs.25 While the strategy regarding the pre-strain at the buried interface of perovskite films has rarely been concerned, where the pre-strain refers to the intentionally introduced strain into a material before the material is further processed or formally used. It serves as an effective method for adjusting the material performance and finds wide application in the field of classical materials science and engineering. The introduction of pre-strain into the processing of perovskite films for PSCs is desired to compensate for the thermally induced tensile strain, which thereby can alter the mechanical, electrical, or optical properties of perovskite films.26–28
Here, we report a pre-strain compensation strategy, where a compressive strain is introduced in advance to the buried interface of the perovskite film during high-temperature annealing to counteract the residual tensile strain that occurs when cooling to room temperature. We constructed a metastable Pb(CH3NH2)2Cl2 (PMC) intermediate phase at the buried interface of the perovskite film. The volume change of the metastable PMC phase during high-temperature annealing provides a source of compressive stress that counteracts the thermally induced residual tensile strain for the perovskite film. Benefitting from this strategy, we achieved nearly strain-free perovskite films. The release of residual tensile strain is demonstrated to suppress the formation of defects, reduce the non-radiative recombination rate, and enhance the generation and transport of charge carriers. Consequently, the highest PCE of the modified n–i–p PSCs based on the nearly strain-free perovskite films increases significantly from 23.92% to 25.83% (with a third-party certified value of 25.36%). Additionally, the modified PSC devices retain 90% of their initial performances after 1000 hours of maximum power point (MPP) tracking.
Results and discussion
We utilized glass/fluorine-doped tin oxide (FTO)/SnO2 as the substrate to deposit FAPbI3 perovskite films, which are commonly used for fabricating high-efficiency n–i–p PSCs. To facilitate the formation of α-phase FAPbI3, methylammounium chloride (CH3NH3Cl, MACl) is typically added into the perovskite precursor solution,29–31 which gradually volatilizes during sequential high-temperature annealing at ∼150 °C. For the perovskite films obtained by using the pre-strain compensation strategy, lead chloride (PbCl2) is first introduced at the substrate/perovskite interface as an interlayer (labeled as “modified”). Due to the high electronegativity of Cl−, PbCl2 is resistant to decomposition under illumination and is difficult to cause photo-degradation of the perovskite, making it a suitable modification material for the light-receiving buried interface.32–35 We first investigated the crystallization changes of the perovskite during annealing using in situ grazing-incidence wide-angle X-ray scattering (GIWAXS) (Fig. 1a–d). The measurements are conducted at a grazing incidence angle of 1.0° to ensure that the bottom of the perovskite films can be always detected during the annealing process. In the early stages of annealing, the modified perovskite film exhibits new diffraction signals at 4.62, 6.50 and 8.08 nm−1 (Fig. 1c and d and Fig. S1, ESI†), which can be attributed to the crystallization of the PMC phase (Fig. S2, ESI†).36 These diffraction signals of the PMC phase gradually disappear after 2 minutes of annealing, with the subsequent appearance of PbCl2 diffraction signals (Fig. 1d), indicating the transformation of the PMC phase to the PbCl2 phase upon annealing. Additionally, the α-phase FAPbI3 appears at the very initial annealing stage for the modified film, while the δ-phase perovskite dominates in the control sample during the first ∼2 minutes and then the α phase appears. This might be due to that the PMC phase can act as a template, lowering the formation energy of the α-phase FAPbI3 and inducing some degree of orientation in perovskite crystallization (Fig. S3, ESI†). We also characterized the GIWAXS at different grazing incidence angles on the perovskite films (Fig. S4, ESI†), and the results indicate that the resultant PbCl2 is mainly distributed at the buried interface of the perovskite films.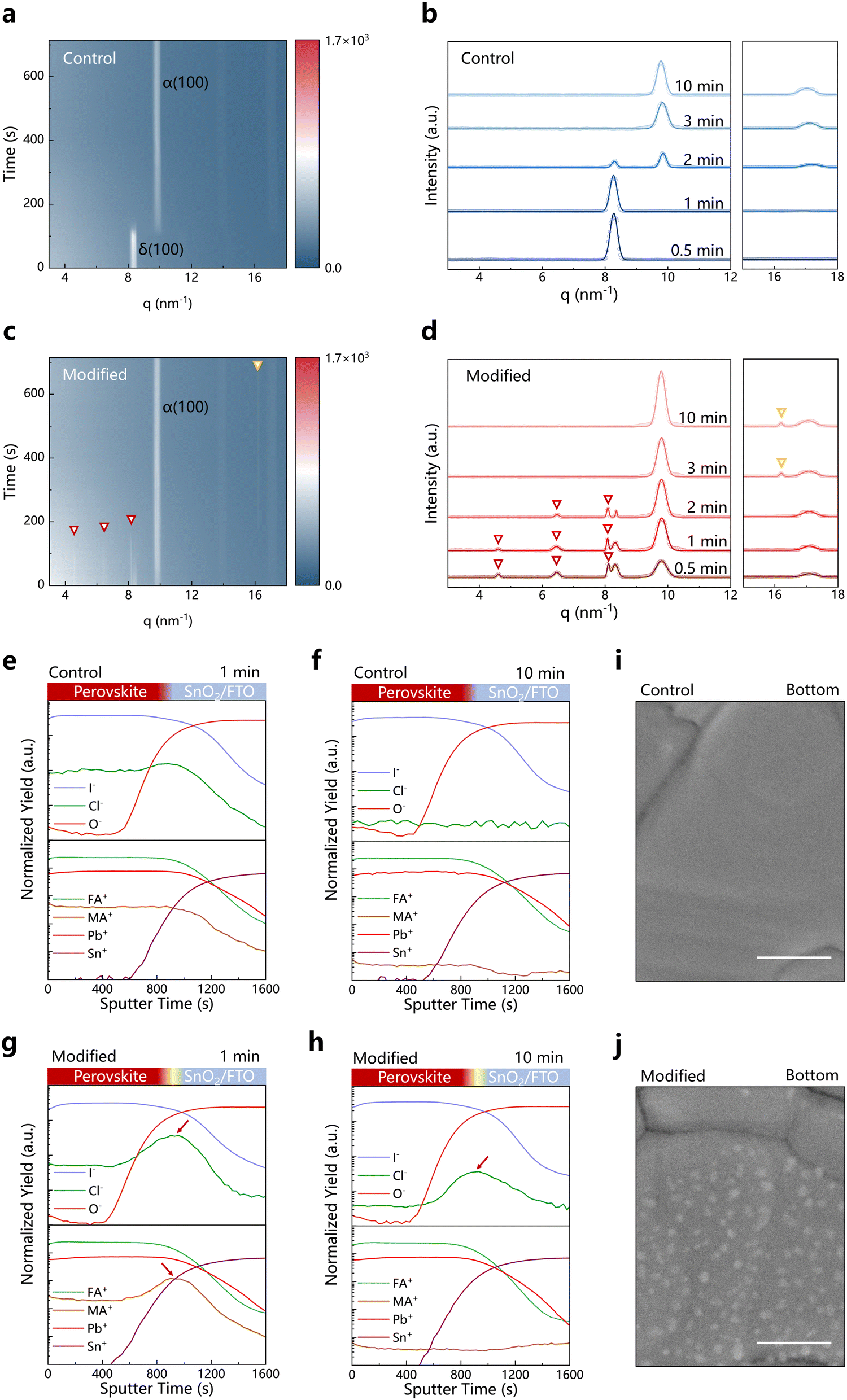 | ||
Fig. 1 The variations induced by the metastable PMC interphase. In situ GIWAXS plots of the integrated scattered intensity (color scale) versus q and time of the films annealed at 150 °C along with the time (frames): control (a) and modified (c). In situ integral GIWAXS profiles from 2D scattering images of the films at different annealing times: control (b) and modified (d). The red triangles represent the PMC interphase, and the orange triangles represent the PbCl2 phase. Time-of-flight secondary-ion mass spectrometry (ToF-SIMS) depth profiles of control (e and f) and modified (g and h) films after annealing for 1 minute (e and g) and 10 minutes (f and h), respectively. Red arrows indicate the PMC phase-enriched regions. (i) and (j) Scanning electron microscopy (SEM) images from the bottom of the perovskite films: control (i) and modified (j). Scale bars, 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm. nm. | ||
To further investigate other changes in the films during annealing induced by our strategy, we characterized the films using time-of-flight secondary ion mass spectrometry (ToF-SIMS) at different annealing durations. When the control film is annealed for just 1 minute, we observe that MA+ and Cl− ions are uniformly distributed throughout the film in the vertical direction (Fig. 1e), indicating that MA+ and Cl− remain in the film and are evenly distributed at different depths when the annealing time is short. When the control film is annealed for 10 minutes, the signals of MA+ and Cl− ions are significantly weakened to a low level, suggesting that most MA+ and Cl− are volatilized in the form of MACl (Fig. 1f). While for the modified film, the residual MA+ and Cl− are distributed throughout the film with accumulation at the buried interface after annealing for just 1 minute (Fig. 1g). After 10 minutes of annealing, the amount of MA+ ion weakens to an extremely low level (Fig. 1h), and the reduced but still existed accumulation of residual Cl− ions at the buried interface could be attributed to PbCl2 at the buried interface. Combined with the GIWAXS results, it is indicated that the metastable PMC phase formed by the interaction of PbCl2 and MACl at the buried interface hinders the early rapid volatilization of MACl. With prolonged annealing, the metastable PMC phase transforms into PbCl2. To characterize the form of PbCl2 at the buried interface, we exposed the buried interface of the perovskite film by a non-destructive peeling method (Fig. S5, ESI†). Scanning electron microscopy (SEM) results show that small PbCl2 grains with a diameter of ∼40 nm (Fig. 1i and j and Fig. S6, ESI†) are uniformly distributed at the perovskite grain bottoms in the modified polycrystalline perovskite film (Fig. S7a and b, ESI†). The surface morphologies of the control and modified perovskite films are similar (Fig. S7c and d, ESI†). The corresponding X-ray diffraction (XRD) detects a faint diffraction signal indexed to the PbCl2 (120) plane in the modified perovskite film (Fig. S8, ESI†), consistent with the GIWAXS results. In addition, for the control sample, the surface morphology of the substrate (FTO/SnO2) shows no obvious difference before and after peeling off the perovskite film (Fig. S9a and b, ESI†). However, for the modified sample, after peeling off the perovskite film, the density of small PbCl2 grains on the substrate is significantly reduced (Fig. S9c and d, ESI†), indicating that most PbCl2 grains are adhered to the buried interface of the delaminated modified perovskite film (Fig. 1j and Fig. S7b, ESI†), suggesting the tight contact between PbCl2 grains with the perovskite grain bottom.
The interfacial strain in PSCs especially the residual tensile strain in the perovskite films has a significant impact on the performance and stability of PSC devices. To investigate the stress experienced by the films, we divided the 2D GIWAXS data into different χ angle regions and integrated these sector regions (Fig. S10, ESI†) to obtain the variation of q values with different χ angles. Perovskite films are considered to be quasi-isotropic, and by maintaining a constant grazing incidence angle while varying the χ angle to obtain the strain (εχ) changes, we can obtain the residual stress in the perovskite film (Note S1, ESI†). Here, we used the grazing incidence angle of 1.0° to detect the diffraction signals at the buried interface of the perovskite films and analyzed the strain under three conditions: the control films at room temperature after cooling from the 10-minute annealing at 150 °C (type I), the modified films at 150 °C after 3-minute annealing at 150 °C (type II), and the modified films at room temperature after cooling from 10-minute annealing at 150 °C (type III). When the χ angle increases from 10° to 85°, the (100) diffraction peak of the type-I films shifts towards lower q values (Fig. 2a), indicating an increase in the interplanar spacing (d-spacing) from in-plane to out-of-plane directions, clearly demonstrating that the bottom of the control perovskite films is under the tensile strain. The residual strain is related to the lattice distortion, which can affect the carrier dynamics in the perovskite films. For type-II films, the (100) diffraction peak shifts towards higher q values while increasing the χ angle (Fig. 2b), indicating that the bottom of the modified films experiences compressive stress when annealing for a short duration such as 3 minutes. Such additional compressive strain may be caused by the transformation from the metastable PMC phase to the PbCl2 phase. The positions of the diffraction peaks of type-III films remain nearly constant at different χ angles (Fig. 2c), indicating that the bottom of the modified films after annealing for an appropriate duration of 10 minutes is almost strain-free. By fitting the variation between εχ and sin2χ (Fig. 2d–f), we obtained the residual stress (σ) at the bottom of the films under the three conditions: 26.77, −28.69, and −1.73 MPa, respectively, where the positive values indicate the tensile stress and the negative values indicate the compressive stress.
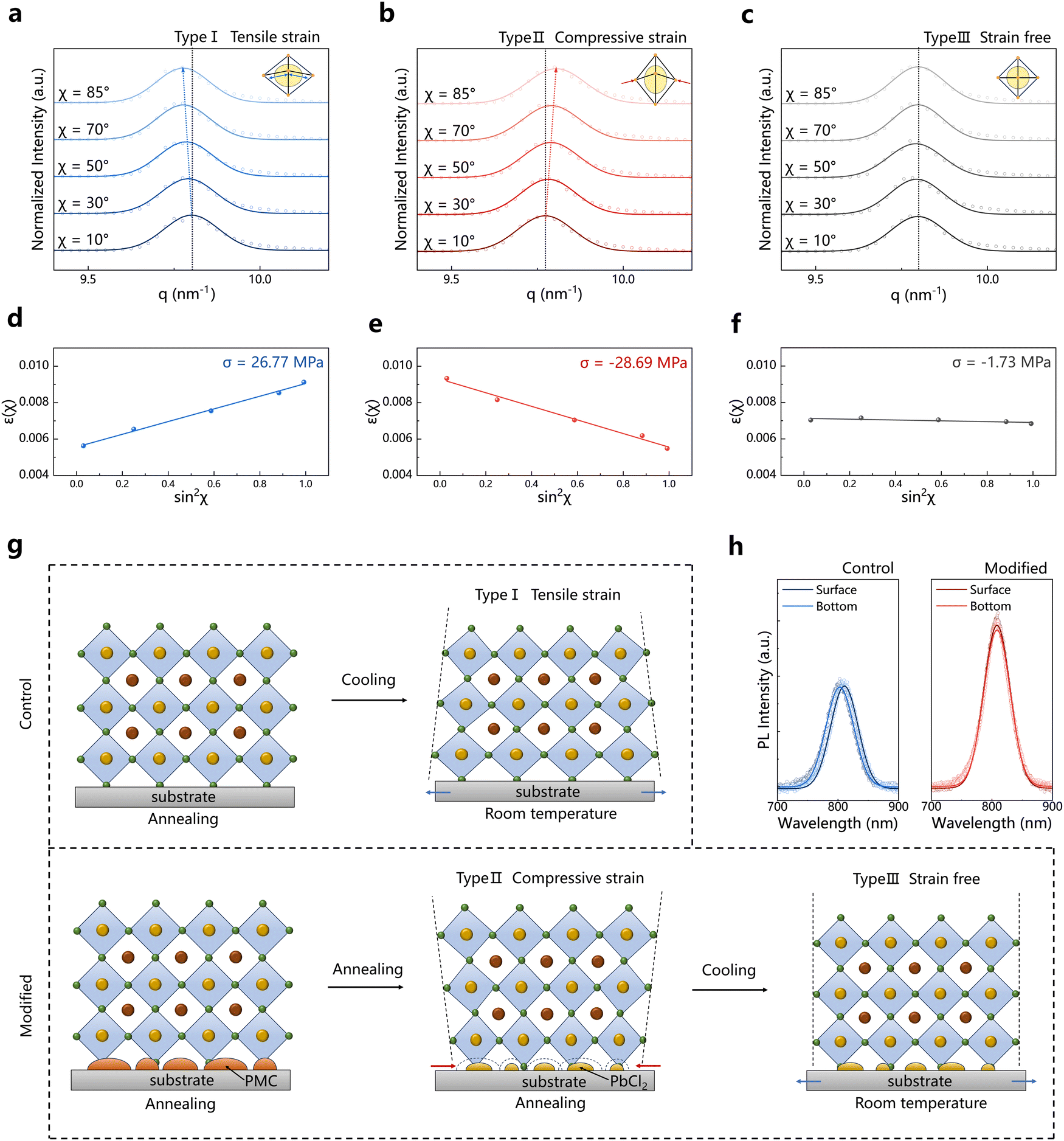 | ||
| Fig. 2 Release of the tensile strain in the perovskite films: the mechanism of the pre-strain compensation strategy. (a)–(c) The GIWAXS integral profiles of α-phase FAPbI3 (100) plane at different χ angles for type I (a), type II (b) and type III (c). (d)–(f) The variation in strain (εχ) for the films of different types with different χ angles: type I (d), type II (e) and type III (f). (g) The schematic diagram illustrating the mechanism of our pre-strain compensation strategy. (h) Steady-state photoluminescence (PL) spectra of the control and modified perovskite films from the surface and bottom side, respectively. | ||
Additionally, we performed grazing-incidence X-ray diffraction (GIXRD) at different depths of the perovskite films (Fig. S11, ESI†). By fitting the linear relationship between 2θ and sin2φ (Note S1, ESI†), the calculated stress is consistent with the GIWAXS results. The stress at depths of 0.1°, 0.5°, and 1.0° in the control perovskite film are 3.45, 10.38, and 27.3 MPa, respectively. This indicates that the residual tensile strain at the bottom of the control perovskite film is significantly larger than that at the surface, that is, the tensile strain in the in-plane direction gradually decreases away from the substrate side of the perovskite. The residual tensile strain facilitates ion migration and defect formation which are typically considered to occur primarily at the grain boundaries and interfaces of the perovskite films.37 Larger residual tensile strain at the buried interface implies that the defects are more likely to form in this region. In contrast, the modified films exhibit almost strain-free or slight compressive strain from the top surface to the buried interface, which could thereby enhance the stability of PSCs according to previous studies.38
Therefore, we proposed the mechanism of the pre-strain compensation strategy as illustrated in Fig. 2g. When the perovskite film with a relatively high TEC is deposited on the substrate with a low TEC (Fig. S12, ESI†), the contact between the two layers restricts the contraction of the perovskite film during cooling from high-temperature annealing to room temperature, thereby introducing the residual tensile strain at the buried interface of the perovskite film (upper panel in Fig. 2g). When introducing an in situ generated metastable PMC phase at the substrate/perovskite interface, the PMC phase loses MA and transforms into the PbCl2 phase upon continuous annealing (Fig. S13, ESI†). This transformation causes the unit cell volume to shrink from 2351.9 Å3 to 937.1 Å3 (Table S1, ESI†) with a reduction of approximately 60%. The shrinkage in the volume of the metastable PMC phase provides a source of compressive stress at the bottom of the perovskite film, thus pre-introducing the additional compressive strain in the in-plane direction of the perovskite film. In the subsequent cooling process after high-temperature annealing, such additional compressive strain counteracts the tensile strain induced by the substrate, resulting in a slightly compressive-strain or even strain-free perovskite film (down panel in Fig. 2g).
Furthermore, we conducted steady-state photoluminescence (PL) spectroscopy on the perovskite films (Fig. 2h). Due to the limitation of excitation depth, PL is typically used to detect the surface information of the perovskite films. We performed PL on both the surface and bottom of the perovskite films. The PL peak excited from the surface of the control film is located at 809 nm, while the PL peak excited from the bottom shifts to 801 nm. This indicates that the bottom of the control perovskite film could be under larger stress, causing more serious lattice distortion, and thereby resulting in a blue shift of the PL peak position.39 In contrast, the PL peaks excited from both the surface and the bottom of the modified perovskite films show negligible shifts, indicating homogeneity in the lattice structure across the modified film, which aligns with the previous GIXRD results.
To investigate in depth the impact of the residual tensile strain release enabled by our pre-strain compensation strategy on the perovskite films, we studied the carrier dynamics in the perovskite films. First, we conducted time-resolved photoluminescence (TRPL) spectroscopy on the perovskite films (Fig. 3a and b). We excited the perovskite films from the bottom side to clearly study the impact of the residual tensile strain release on the carrier dynamics. Since most of the photogenerated carriers are initially generated closer to the surface of the light-receiving side, and due to the higher trap-state density near the surface compared to the bulk, the PL intensity rapidly decreases early on. Subsequently, the carriers diffuse further away from the surface, spreading along the film thickness into the bulk and laterally away from the excitation point. The results show that the PL lifetime of the strain-free films is longer than that of the films with residual tensile strain under different excitation fluences. We then fitted the low excitation fluence transients with a stretched exponential function to obtain the monomolecular non-radiative recombination rate k140–42 (Note S2, ESI†). The results show that the k1 values for the control perovskite film with the residual tensile strain and the strain-free modified perovskite film are 2.30 × 106 and 0.73 × 106 s−1, respectively (Table S2, ESI†). The k1 value decreases by approximately 68%, indicating that the release of residual strain effectively suppresses the non-radiative recombination at the buried interface of the perovskite film, implying a reduction in the defects at the buried interface. This can be translated into higher photovoltage and fill factors of PSCs.
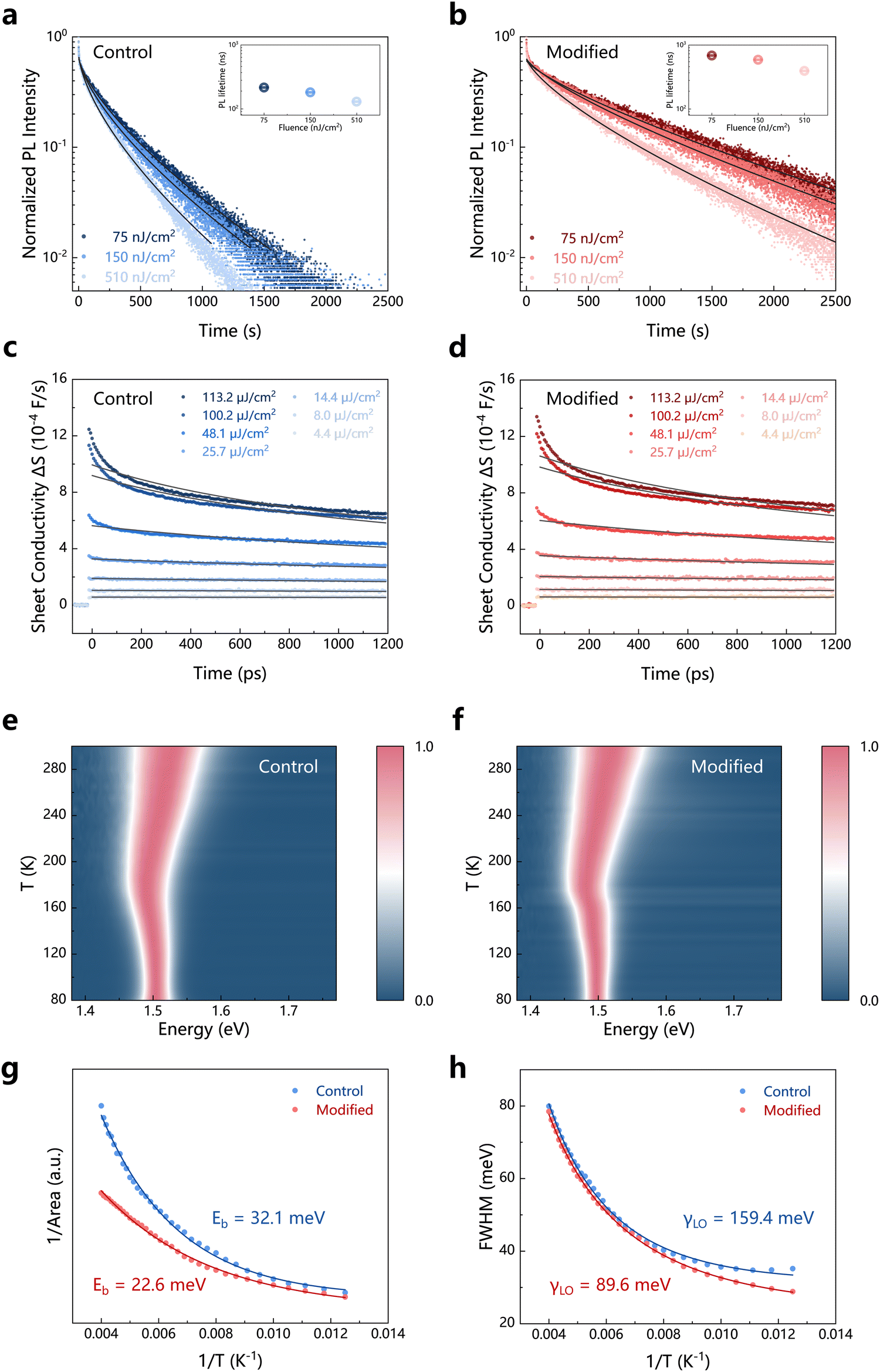 | ||
| Fig. 3 Effects of the tensile strain release by the pre-strain compensation strategy on the carrier dynamics in the perovskite films. (a) and (b) Time-resolved photoluminescence (TRPL) spectra of the control (a) and modified (b) perovskite films with different incident laser fluences. The insets show the corresponding PL lifetimes. (c) and (d) Optical-pump terahertz-probe (OPTP) spectra of the control (c) and modified (d) perovskite films. (e) and (f) Temperature-dependent PL spectra of the control (e) and modified (f) perovskite films. (g) Temperature-dependent peak area of PL spectra and the fitting curves. (h) Temperature-dependent full width at half maximum (FWHM) of PL spectra and the fitting curves. | ||
We also conducted optical-pump terahertz probe (OPTP) spectroscopy on the perovskite films (Fig. 3c and d). OPTP can be used to study the bimolecular radiative recombination and charge carrier mobility of the strain-free and tensile-strain perovskite films.42–44 By fitting the transient photoconductivity decay curves (Note S3, ESI†), we found that the mobility of the strain-free modified perovskite film is higher than that of the control perovskite film with the residual tensile strain (Table S2, ESI†). The corresponding diffusion lengths are 12.2 μm and 6.8 μm, respectively, indicating that the strain release is conducive to enhancing the carrier mobility and diffusion.
We then performed temperature-dependent PL spectroscopy on the perovskite films (Fig. 3e and f). By fitting the relationship between the PL peak area and the temperature (Fig. 3g), we obtained the exciton binding energies (Eb) of the control perovskite films with the residual tensile strain and the strain-free modified perovskite films,45,46 which are 32.1 meV and 22.6 meV, respectively (Note S4, ESI†). The UV-vis absorption spectra do not show obvious exciton peaks (Fig. S14, ESI†), which is consistent with previous research results.47,48 For the control device, stronger exciton interactions imply a larger exciton binding energy, making it more difficult for the excitons to dissociate into free electrons and holes. Excitons may remain in the material for a longer period.48 During this time, excitons are more likely to interact with defects, impurities, or other recombination centers in the material, leading to increased non-radiative recombination. This is particularly true when excitons are localized in regions such as grain boundaries or the perovskite layer/carrier transport layer interfaces, where more defects are typically present.49 In contrast, for the modified device, weaker exciton interactions can reduce this recombination, especially at the interface between the perovskite layer and the carrier transport layer, ensuring that photogenerated electron–hole pairs are promptly extracted after separation. Therefore, by weakening exciton interactions, unfavorable recombination can be effectively reduced, leading to improved device performance. Additionally, we noted that the electron–phonon coupling mechanism in metal halide perovskites is primarily attributed to the deformation potential scattering, which is the Fröhlich interaction between the electrons and the longitudinal optical (LO) mode phonons.50 This interaction arises from the Coulomb interaction between the electrons and the macroscopic electric field caused by the out-of-phase displacement of atoms with opposite charges in the LO mode phonons. This can correspond to the lattice distortion or deformation due to the strain in the crystal, and the resulting deformation potential or piezoelectric-induced electric field affects the electron energy.51 By fitting the relationship between the full width at half maximum (FWHM) of the PL spectra and the temperature (Note S5, ESI†), we obtained the electron–phonon coupling coefficients (γLO) of the perovskite films (Table S3, ESI†). The γLO of the modified perovskite film (89.6 meV) is significantly lower than that of the control perovskite film (159.4 meV) (Fig. 3h). This indicates that the interaction between carriers and LO mode phonons is weaker in the strain-free modified perovskite film, meaning that less energy is dissipated to the lattice during carrier transport, and thereby the carrier lifetime and diffusion length are prolonged. This suggests a higher photovoltaic performance of PSCs.
To validate our speculation on the impact of the tensile strain release by our strategy on PSCs, we first conducted space-charge-limited current (SCLC) measurements (Fig. S15, ESI†). The strain change occurs in the region of the film near the buried electron transport layer. Thus, the changes in the electron behavior induced by strain may have a more pronounced effect on the photovoltaic performance. Therefore, we fabricated electron-only devices for SCLC testing. The results show that the modified devices have a lower trap-filling limit voltage (VTFL), indicating that the tensile strain release suppresses the formation of defects at the buried interface and perovskite films, which is consistent with the PL results. It is beneficial for achieving higher photovoltaic performance. Finally, we fabricated the corresponding PSCs with the device structure shown in the cross-sectional SEM image (Fig. 4a). The results show that our pre-strain compensation strategy for the tensile strain release can effectively improve the photovoltaic performance of PSCs (Fig. S16, ESI†). The champion PCE of the PSC devices significantly increases from 23.92% (control) to 25.83% (modified) (Fig. 4b and Table S4, ESI†), maintaining the stable power output of 25.62% (Fig. S17, ESI†). The modified device exhibits a higher open-circuit voltage (VOC), short-circuit current density (JSC), and fill factor (FF) and suppressed hysteresis effect, which may be attributed to the suppression of ion migration by the tensile strain release. From the light intensity-dependent PL quantum yield (PLQY) results, an increase of approximately 32 mV in quasi-Fermi level splitting (QFLS) is calculated (Fig. S18, ESI†), which is consistent with the J–V results of PSCs. Moreover, to ensure the accuracy of JSC, a shading mask is used to fix the illumination area during the test, and the external quantum efficiency (EQE) also confirms this (Fig. S19, ESI†). We further sent the unencapsulated PSCs to a third-party organization and obtained a certified PCE of 25.36% with negligible hysteresis (Fig. S20, ESI†). We also fabricated larger-area PSCs of 1.05 cm2 and achieved a champion PCE of 24.22% (Fig. 4c and Table S5, ESI†).
 | ||
| Fig. 4 Tensile strain release by the pre-strain compensation strategy improves the photovoltaic performance of PSCs. (a) The cross-sectional SEM image of a PSC device. Scale bars, 1 μm. (b) The current density–voltage (J–V) curves of the control and modified PSCs under different scanning directions. (c) The J–V curves of the large-area (1.05 cm2) control and modified PSCs. (d) Stability variations after storage of unencapsulated PSCs under ∼15% RH in ambient air. (e) Stability variations of unencapsulated PSCs upon heating at 65 °C under a nitrogen atmosphere. (f) The MPP tracking of unencapsulated PSCs under a white light LED solar simulator under a nitrogen atmosphere at ∼40 °C. (g) Thermal cycling stability of unencapsulated PSCs under a nitrogen atmosphere. The rapid thermal cycle is executed by placing the PSCs on a semiconductor chilling plate of −20 °C, followed by transferring them to a hot plate of 60 °C. Each stage lasted for an extra 2 minutes to achieve thermal equilibrium. | ||
Releasing the residual tensile strain can alleviate the lattice distortion, suppress the weakening of chemical bonds, and increase the formation energy of the vacancy defects, implying that the stability of perovskite films can be improved. We thus conducted XRD tests on the perovskite films before and after being heated and aged at 65 °C for 30 hours (Fig. S21, ESI†). For the control sample, the XRD diffraction signal of α-phase perovskite becomes significantly weaker, while the PbI2 signal is enhanced, and an additional diffraction peak indexed to δ-phase perovskite appears after aging. However, the XRD diffraction signal of the modified film shows almost no change after heating for 30 hours, which indicates that the modified film has indeed better stability than the control one. We then monitored the storage stability of unencapsulated PSCs in ambient air with a relative humidity (RH) of about 15% (Fig. 4d and Table S6, ESI†). Compared to the control devices, the devices based on the strain-free perovskite films exhibit improved stability, maintaining 91% of the initial PCE after 2000 hours of storage, whereas the PCE of the control devices has dropped to 65%. We also verified the thermal stability of the PSCs (Fig. 4e and Table S7, ESI†), the modified devices show better thermal stability than the control devices because strain-free or even slightly compressive-strain perovskite films are more difficult to decompose than the perovskite films with the residual tensile strain. To evaluate the long-term operational stability, we tracked the power output at the MPP of the unencapsulated devices under one sun illumination in a nitrogen atmosphere. The results in Fig. 4f indicate that the stability of the modified device is significantly improved, maintaining 90% of the initial PCE after 1000 hours of continuous operation. Additionally, we subjected the devices to rapid thermal cycling aging between −20 °C and 60 °C (Fig. 4g and Table S8, ESI†). After 100 thermal cycles, the performance of the control device severely decreases to 56% of the initial PCE, whereas the modified device still retains 83% of the initial PCE.
Conclusion
The mismatch in TECs between the perovskite and the substrate is a significant source of the detrimental thermally induced residual tensile strain in perovskite films. Such residual tensile strain leads to decreased mobility and diffusion of charge carriers in the perovskite films, facilitates defect formation and accelerates non-radiative recombination. These factors negatively impact the performance and stability of PSCs. Our pre-strain compensation strategy has successfully released the residual tensile strain in the perovskite films. We also revealed the transformation of the larger-volume metastable PMC phase to the small-volume PbCl2 phase upon annealing and proposed the mechanism of the pre-strain compensation process. We further studied the positive effects of the tensile strain release by our strategy on the charge carrier dynamics of the perovskite films. Ultimately, the FAPbI3-based PSCs with released residual tensile strain have achieved a champion PCE of 25.83% (certified as 25.36%) and demonstrated excellent stability, retaining 90% of their initial performance after continuous operation for 1000 hours under one-sun illumination. This work provides a promising strategy for releasing the residual strain in the perovskite films and enhancing the performance and stability of perovskite optoelectronic devices.Author contributions
Y. X., H. J. S., R. Z., and L. Z. directed and supervised the project. H. X. fabricated, optimized and characterized the solar cells under the guidance of L. Z., H. X., L. Z. and Q. L. contributed to the certification of solar cells. H. X. and Y. X. designed all the experiments for characterization. K. A. E. and L. M. H. conducted the TRPL and the OPTP and analyzed the results. H. X., L. Z. and Y. Y. contributed to the GIWAXS measurement. H. X. and Y. X. analyzed and discussed GIWAXS and ToF-SIMS data. Q. Z. contributed to the temperature-dependent PL measurements. H. X. and L. Z. discussed the temperature-dependent PL data. H. Y. and H. X. conducted the SEM measurements. P. C. and Y. J. contributed to the PLQY measurements. P. C. and Y. X. analyzed and discussed the PLQY data. H. X., H. Y. and T. H. contributed to the stability tests on the devices. H. X. wrote the first draft of the paper. L. Z., Y. X., H. J. S. and R. Z. revised the paper. All authors analyzed their data and reviewed and commented on the paper.Data availability
All data are available in the main text or the ESI† and are also available from the corresponding authors on reasonable request.Conflicts of interest
Henry J. Snaith is the founder and Chief Scientific Officer of Oxford Photovoltaics, a company commercializing perovskite photovoltaics.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2021YFB3800100 and 2021YFB3800101), the National Natural Science Foundation of China (52325310, 52203208 and 52303335), the Beijing Natural Science Foundation (JQ21005), the Open Research Fund of Songshan Lake Materials Laboratory (2022SLABFK07), the Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370013), the Beijing Nova Program (contract no. 20220484148 and 20230484480), and the R&D Fruit Fund (20210001). This work was also funded by the Engineering and Physical Sciences Research Council (EPSRC), UK (EP/X038777/1). We would like to thank Tingwei He from Hebei University for GIWAXS measurements. We also thank beamlines BL17B1 at the SSRF for providing the beamtime and the User Experiment Assist System of the SSRF for their help.References
- J. Zhou, L. Tan, Y. Liu, H. Li, X. Liu, M. Li, S. Wang, Y. Zhang, C. Jiang, R. Hua, W. Tress, S. Meloni and C. Yi, Joule, 2024, 8, 1691–1706 CrossRef CAS.
- H. Chen, C. Liu, J. Xu, A. Maxwell, W. Zhou, Y. Yang, Q. Zhou, A. S. R. Bati, H. Wan, Z. Wang, L. Zeng, J. Wang, P. Serles, Y. Liu, S. Teale, Y. Liu, M. I. Saidaminov, M. Li, N. Rolston, S. Hoogland, T. Filleter, M. G. Kanatzidis, B. Chen, Z. Ning and E. H. Sargent, Science, 2024, 384, 189–193 CrossRef CAS PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef PubMed.
- N. Li, X. Niu, Q. Chen and H. Zhou, Chem. Soc. Rev., 2020, 49, 8235–8286 RSC.
- Q. Jiang, R. Tirawat, R. A. Kerner, E. A. Gaulding, Y. Xian, X. Wang, J. M. Newkirk, Y. Yan, J. J. Berry and K. Zhu, Nature, 2023, 623, 313–318 CrossRef CAS PubMed.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- D.-J. Xue, Y. Hou, S.-C. Liu, M. Wei, B. Chen, Z. Huang, Z. Li, B. Sun, A. H. Proppe, Y. Dong, M. I. Saidaminov, S. O. Kelley, J.-S. Hu and E. H. Sargent, Nat. Commun., 2020, 11, 1514 CrossRef CAS PubMed.
- X. Zheng, C. Wu, S. K. Jha, Z. Li, K. Zhu and S. Priya, ACS Energy Lett., 2016, 1, 1014–1020 CrossRef CAS.
- B. Murali, E. Yengel, W. Peng, Z. Chen, M. S. Alias, E. Alarousu, B. S. Ooi, V. Burlakov, A. Goriely, M. Eddaoudi, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 137–143 CrossRef CAS.
- H. Wang, C. Zhu, L. Liu, S. Ma, P. Liu, J. Wu, C. Shi, Q. Du, Y. Hao, S. Xiang, H. Chen, P. Chen, Y. Bai, H. Zhou, Y. Li and Q. Chen, Adv. Mater., 2019, 31, 1904408 CrossRef CAS.
- Q. Xiong, X. Huang, C. Wang, Q. Zhou, Y. Gang, T. Li, C. Hu, N. Zhang, X. Wang, J. Wu, Z. Su, X. Gao, X. Li, N. Zheng and P. Gao, Joule, 2024, 8, 817–834 CrossRef CAS.
- P. S. Peercy and B. Morosin, Phys. Rev. B: Solid State, 1973, 7, 2779–2786 CrossRef CAS.
- C. Ge, M. Hu, P. Wu, Q. Tan, Z. Chen, Y. Wang, J. Shi and J. Feng, J. Phys. Chem. C, 2018, 122, 15973–15978 CrossRef CAS.
- M. Keshavarz, M. Ottesen, S. Wiedmann, M. Wharmby, R. Küchler, H. Yuan, E. Debroye, J. A. Steele, J. Martens, N. E. Hussey, M. Bremholm, M. B. J. Roeffaers and J. Hofkens, Adv. Mater., 2019, 31, 1900521 CrossRef.
- K. Liu, B. Chen, Z. J. Yu, Y. Wu, Z. Huang, X. Jia, C. Li, D. Spronk, Z. Wang, Z. Wang, S. Qu, Z. C. Holman and J. Huang, J. Mater. Chem. A, 2022, 10, 1343–1349 RSC.
- T. J. Jacobsson, L. J. Schwan, M. Ottosson, A. Hagfeldt and T. Edvinsson, Inorg. Chem., 2015, 54, 10678–10685 CrossRef CAS PubMed.
- T. Burwig, W. Fränzel and P. Pistor, J. Phys. Chem. Lett., 2018, 9, 4808–4813 CrossRef CAS PubMed.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. Seok, Science, 2019, 366, 749–753 CrossRef CAS.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359–1364 CrossRef CAS PubMed.
- Y. Zong, N. Wang, L. Zhang, M. Ju, X. C. Zeng, X. W. Sun, Y. Zhou and N. P. Padture, Angew. Chem., Int. Ed., 2017, 56, 12658–12662 CrossRef CAS.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS.
- L. Zhao, Q. Li, C.-H. Hou, S. Li, X. Yang, J. Wu, S. Zhang, Q. Hu, Y. Wang, Y. Zhang, Y. Jiang, S. Jia, J.-J. Shyue, T. P. Russell, Q. Gong, X. Hu and R. Zhu, J. Am. Chem. Soc., 2022, 144, 1700–1708 CrossRef CAS.
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H.-B. Kim, S.-I. Mo, Y.-K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Grätzel and D. S. Kim, Science, 2022, 375, 302–306 CrossRef CAS.
- S. He, S. Li, A. Zhang, G. Xie, X. Wang, J. Fang, Y. Qi and L. Qiu, Mater. Adv., 2022, 3, 6316–6323 RSC.
- X. Zhu, X. Yang, W. Huang, W. Qiu, X. Wang, F. Guo, L. Hu and M. Gong, J. Mater. Sci. Technol., 2023, 145, 249–259 CrossRef CAS.
- H. W. Lee, T. M. Park, H.-J. Kim and J. Han, J. Mater. Sci. Technol., 2025, 206, 62–73 CrossRef.
- Y. Cui, H. Bian, Y. Li, Y. Zhao, K. Aoyagi and A. Chiba, J. Alloys Compd., 2020, 816, 152496 CrossRef CAS.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- L. Chen, M. Hu, S. Lee, J. Kim, Z.-Y. Zhao, S.-P. Han, M. S. Lah and S. I. Seok, J. Am. Chem. Soc., 2023, 145, 27900–27910 CrossRef CAS.
- F. Ye, J. Ma, C. Chen, H. Wang, Y. Xu, S. Zhang, T. Wang, C. Tao and G. Fang, Adv. Mater., 2021, 33, 2007126 CrossRef CAS PubMed.
- F. K. Aldibaja, L. Badia, E. Mas-Marzá, R. S. Sánchez, E. M. Barea and I. Mora-Sero, J. Mater. Chem. A, 2015, 3, 9194–9200 RSC.
- Y. Wang, X. Mei, J. Qiu, Q. Zhou, D. Jia, M. Yu, J. Liu and X. Zhang, J. Phys. Chem. Lett., 2021, 12, 11330–11338 CrossRef CAS PubMed.
- T. Hou, M. Zhang, X. Sun, Y. Wang, K. Chen, Z. Fu, M. He, X. Liu, Z. Liu, Y. Huang, M. A. Green and X. Hao, Adv. Energy Mater., 2024, 14, 2400932 CrossRef CAS.
- X. Qi, G. Liu, D. Wang, N. Zhu, Y. Zhang, Z. Zhang, C. Wu, X. Li, W. Luo, Y. Li, H. Hu, Z. Chen, L. Xiao, S. Wang and B. Qu, Org. Electron., 2019, 74, 52–58 CrossRef CAS.
- M. Krummer, M. Daub and H. Hillebrecht, Z. Anorg. Allg. Chem., 2022, 648, e202100176 CrossRef CAS.
- J. T.-W. Wang, Z. Wang, S. Pathak, W. Zhang, D. W. deQuilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. M. Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 RSC.
- Z. Zheng, F. Li, J. Gong, Y. Ma, J. Gu, X. Liu, S. Chen and M. Liu, Adv. Mater., 2022, 34, 2109879 CrossRef CAS.
- Y. Chen, Y. Lei, Y. Li, Y. Yu, J. Cai, M.-H. Chiu, R. Rao, Y. Gu, C. Wang, W. Choi, H. Hu, C. Wang, Y. Li, J. Song, J. Zhang, B. Qi, M. Lin, Z. Zhang, A. E. Islam, B. Maruyama, S. Dayeh, L.-J. Li, K. Yang, Y.-H. Lo and S. Xu, Nature, 2020, 577, 209–215 CrossRef CAS.
- D. C. Johnston, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184430 CrossRef.
- T. Kirchartz, J. A. Márquez, M. Stolterfoht and T. Unold, Adv. Energy Mater., 2020, 10, 1904134 CrossRef CAS.
- R. D. J. Oliver, P. Caprioglio, F. Peña-Camargo, L. R. V. Buizza, F. Zu, A. J. Ramadan, S. G. Motti, S. Mahesh, M. M. McCarthy, J. H. Warby, Y.-H. Lin, N. Koch, S. Albrecht, L. M. Herz, M. B. Johnston, D. Neher, M. Stolterfoht and H. J. Snaith, Energy Environ. Sci., 2022, 15, 714–726 RSC.
- K. Xiao, R. Lin, Q. Han, Y. Hou, Z. Qin, H. T. Nguyen, J. Wen, M. Wei, V. Yeddu, M. I. Saidaminov, Y. Gao, X. Luo, Y. Wang, H. Gao, C. Zhang, J. Xu, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2020, 5, 870–880 CrossRef CAS.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu, M. Xiao, A. Li, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- X. Yang, Y. Fu, R. Su, Y. Zheng, Y. Zhang, W. Yang, M. Yu, P. Chen, Y. Wang, J. Wu, D. Luo, Y. Tu, L. Zhao, Q. Gong and R. Zhu, Adv. Mater., 2020, 32, 2002585 CrossRef CAS PubMed.
- W. Li, X. Feng, K. Guo, W. Pan, M. Li, L. Liu, J. Song, Y. He and H. Wei, Adv. Mater., 2023, 35, 2211808 CrossRef CAS PubMed.
- K. A. Elmestekawy, B. M. Gallant, A. D. Wright, P. Holzhey, N. K. Noel, M. B. Johnston, H. J. Snaith and L. M. Herz, ACS Energy Lett., 2023, 8, 2543–2551 CrossRef CAS PubMed.
- V. D’Innocenzo, G. Grancini, M. J. P. Alcocer, A. R. S. Kandada, S. D. Stranks, M. M. Lee, G. Lanzani, H. J. Snaith and A. Petrozza, Nat. Commun., 2014, 5, 3586 CrossRef.
- Z. Ni, C. Bao, Y. Liu, Q. Jiang, W.-Q. Wu, S. Chen, X. Dai, B. Chen, B. Hartweg, Z. Yu, Z. Holman and J. Huang, Science, 2020, 367, 1352–1358 CrossRef CAS PubMed.
- R. Su, Z. Xu, J. Wu, D. Luo, Q. Hu, W. Yang, X. Yang, R. Zhang, H. Yu, T. P. Russell, Q. Gong, W. Zhang and R. Zhu, Nat. Commun., 2021, 12, 2479 CrossRef CAS PubMed.
- A. D. Wright, C. Verdi, R. L. Milot, G. E. Eperon, M. A. Pérez-Osorio, H. J. Snaith, F. Giustino, M. B. Johnston and L. M. Herz, Nat. Commun., 2016, 7, 11755 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03801k |
| This journal is © The Royal Society of Chemistry 2025 |