
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethane pyrolysis for hydrogen production: navigating the path to a net zero future
Alireza Lotfollahzade
Moghaddam
 a,
Sohrab
Hejazi
a,
Moslem
Fattahi
a,
Md Golam
Kibria
*b,
Murray J.
Thomson
*c,
Rashed
AlEisa
d and
M. A.
Khan
*ad
a,
Sohrab
Hejazi
a,
Moslem
Fattahi
a,
Md Golam
Kibria
*b,
Murray J.
Thomson
*c,
Rashed
AlEisa
d and
M. A.
Khan
*ad
aDepartment of Chemical and Materials Engineering, University of Alberta, Donadeo Innovation Centre for Engineering (ICE), 116 Street, Edmonton (T6G 1H9), Canada. E-mail: mohd3@ualberta.ca
bDepartment of Chemical and Petroleum Engineering, University of Calgary, 2500 University Drive, NW, Calgary, Alberta T2N 1N4, Canada. E-mail: md.kibria@ucalgary.ca
cDepartment of Mechanical and Industrial Engineering, University of Toronto, Ontario, Canada. E-mail: murray.thomson@utoronto.ca
dSustainable Fuels and Chemicals Team, Saudi Aramco Research and Development Center, Dhahran, 31311, Saudi Arabia
First published on 14th February 2025
Abstract
The global push to keep global warming to less than 1.5 °C, will require us to quickly adopt zero-emission energy carriers. Hydrogen, a versatile energy vector, is pivotal in this transition, especially for sectors that are challenging to electrify. Methane pyrolysis is emerging as a promising route for producing hydrogen with minimal greenhouse gas emissions. In this review, we provide a comprehensive overview of methane pyrolysis, and explore its potential to contribute to a net-zero future. Current hydrogen production methods, including steam methane reforming and water electrolysis, are also discussed in terms of efficiency, emissions, and costs for comparison with methane pyrolysis. The review then delves into the various technologies under development for methane pyrolysis, categorizing them into catalytic and non-catalytic routes. Key aspects such as reactor design, catalyst performance, and economic viability are critically examined. We also analyze the importance of the carbon co-product produced in the process, and its market potential. Finally, by evaluating industrial activities around methane pyrolysis, this paper underscores its role in the global energy transition, emphasizing the requirements to overcome current challenges and achieve large-scale deployment.
Broader contextMethane pyrolysis offers a promising pathway to produce hydrogen, a clean energy carrier, without direct carbon dioxide emissions, aligning with global efforts to achieve net-zero greenhouse gas emissions. This method generates “turquoise” hydrogen while producing solid carbon as a by-product. Thus, advancing this technology could significantly reduce the environmental footprint of hydrogen production, currently dominated by carbon-intensive processes, and promote exploration into the efficient utilization and storage of solid carbon by-products. Moreover, the development of this technology will stimulate interdisciplinary research among academia, industry, and government to scale up and commercialize this promising technology, accelerating the global shift towards sustainable energy solutions. In this article, we present a comprehensive review of all aspects of methane pyrolysis, including reaction mechanisms, catalyst & technology development, reactor designs, carbon products, process economics, greenhouse gas emissions, and scale up activities. Finally, we outline future research directions, policy needs, and investment opportunities that could support the wider adoption of this technology. |
1. Introduction
To limit the increase in global warming to less than 1.5 °C, many nations have pledged to achieve net-zero greenhouse gas (GHG) emissions by 2050.1 The extraction, refinement, distribution, and combustion of fossil fuels account for over 70% of global GHG emissions,2 a major effort is required to transition to lower-carbon alternatives such as electricity, hydrogen, and biofuels.In the transition to net-zero emissions, the electrification of end-use energy demand presents a notable advantage owing to its higher system efficiencies and the pre-existence of much of the value chain infrastructure (e.g., electrical grid) and conversion technologies (e.g., heaters, heat pumps, motors, electric cars). Nonetheless, certain sectors pose challenges in electrification, particularly those designated as hard-to-abate, such as heavy industry (e.g., iron/steel, chemicals), space heating in cold climates, and heavy-duty transport. In such instances, hydrogen emerges as the preferred zero-emission fuel.3
At present, globally we generate more than 75 million tonnes of pure hydrogen per year and an additional 45 million tonnes per year as a gaseous mixture,4,5 utilized as an industrial feedstock for bitumen upgrading, oil refining, ammonia production, and the synthesis of various chemicals. The predominant method for producing this hydrogen involves natural gas reforming, a process that releases carbon dioxide (CO2) into the atmosphere as a greenhouse gas (GHG) byproduct. This resulting hydrogen, often termed ‘gray’ hydrogen, is linked with emissions averaging between 9 to 10 kilograms of CO2 per kilogram of hydrogen (kgCO2 kgH2−1), alongside an additional 1.5 to 2 kilograms of CO2 equivalent per kilogram of hydrogen (kgCO2eq. per kgH2) attributed to the retrieval and upgrading of the natural gas.6
Transitioning to a net-zero energy system where hydrogen is used as a clean fuel (as depicted in Fig. 1) will require the production of hydrogen with minimal or no GHG emissions and the creation of new value chains that make hydrogen available at a reasonable cost at widely distributed locations. One strategy involves the centralized production of low-GHG hydrogen via the production of ‘blue’ hydrogen, where the by-product carbon dioxide from natural gas reforming is captured and sequestered underground. Jurisdictions which possess extensive fossil fuel resources and porous rock formations suitable for permanent CO2 storage can make ‘blue’ hydrogen. With low cost natural gas (<4 $ per GJ), centralized production using steam methane reforming (SMR) can lead to low hydrogen production costs (≤2 $ per kgH2).7 However, processing and delivery of low-density hydrogen is complex and costly, in particular when the demand is low, adding ∼3–5 $ per kgH2 to production costs.7 The hydrogen transportation costs can be reduced through large-scale pipeline operations. Nonetheless, it will take significant capital investment (billions of $) and many years to build long-distance transmission pipelines for delivering pure hydrogen at scale and at a competitive cost. Moreover, there are numerous remote locations that are far away from centralized hydrogen production sites where the demand is insufficient to justify the construction of a pipeline.
 | ||
| Fig. 1 Potential role of hydrogen in a net-zero future: the figure shows the required hydrogen value chains to support traditional demand as chemical feedstock and emerging fuel demand for heavy-duty vehicles, heat & power, heavy industry, and export. | ||
Alternatively, one could design fuel hydrogen value chains around distributed production. Low-carbon ‘green’ hydrogen can be produced in a distributed manner from water electrolysis powered by low-carbon electricity. The production costs of green hydrogen from water electrolysis are influenced by various technical and economic factors, including the capital cost (CAPEX) of the electrolyzer, its conversion efficiency (kW h kgH2−1), electricity costs, and annual operating hours. There are numerous studies published in the literature, that demonstrate the challenge for economically viable green hydrogen production is its need for near-continuous access (ideally 6000+ h per year) to low-cost (<30 $ per MW h), low-carbon electricity.7 A key consideration is not just the expense and carbon intensity of clean electricity, but also its availability in substantial amounts as producing 1 kg of H2 requires approximately 42–55 kW h of electrical energy.8
In the past decade, distributed hydrogen production via methane pyrolysis has come into the limelight, as an attractive technology that could have far-reaching implications.9 Numerous companies around the world are pursuing the development of this technology. This process decomposes methane into its elemental components, hydrogen, and solid carbon (CH4 → C + 2H2). The carbon is not combusted within this process, so neither a CO2 separation step nor its subsequent storage is needed, and the hydrogen produced is often referred to as ‘turquoise’ hydrogen. The solid carbon (e.g. carbon black) can be used to produce rubber tires or paints, used as a soil amendment, or permanently stored (e.g., landfill) in a location where it will not be oxidized to CO2. There is also interest in using carbon black to make carbon fiber, graphite, graphene, or a form of carbon that will store electricity.
Methane pyrolysis technologies to produce ‘turquoise’ hydrogen could be deployed anywhere with access to a natural gas supply, making the requirement for CCS capabilities unnecessary. This technology is attractive for all regions that have natural gas pipelines, making it possible to produce hydrogen where and when it is needed, therefore avoiding the high costs associated with delivery and storage of hydrogen. This could rapidly expand the availability of fuel hydrogen for transportation, building, or heat and power markets by utilizing the existing natural gas infrastructure. However, this technology possesses a low technology readiness level (TRL) since it is mainly in the R&D phase. Current research is spread across understanding reaction mechanisms, optimizing heat source, reactor design, and catalyst performance.
In this paper we provide a comprehensive review on methane pyrolysis, exploring its potential to contribute to a net-zero future. A significant focus is placed on methane pyrolysis as a complementary technology bridging steam methane reforming (SMR) and water electrolysis (Section 2). By systematically comparing process design, efficiencies, CO2 emissions, and cost of hydrogen production from these methods, this review presents a nuanced perspective on how methane pyrolysis can fulfill hydrogen demand in an economically viable and sustainable manner until electrolysis becomes more accessible.
Thereafter, an in-depth assessment of various pyrolysis methods (Section 3), including solid catalysts, molten metals, molten salts and alloys, plasma, thermal, and microwave-assisted processes. The different technologies are classified into catalytic and non-catalytic routes and discussed in detail. Catalytic pyrolysis is discussed with a focus on the selection of catalysts, their lifetime, and the technical challenges such as deactivation due to coking. Non-catalytic pyrolysis is explored in terms of its temperature requirements and its potential to produce a wider range of by-products.
In Section 4, the production of different forms of carbon co-products, and their market opportunities in advancing a circular economy are discussed. In Section 5, a thorough analysis of the technoeconomic aspects of methane pyrolysis across various scales is presented, identifying key barriers to commercial deployment. In Sections 6 and 7, we review current state of commercialization, highlighting advancement from pilot projects to industrial applications, and government policies, investments, and support mechanisms for hydrogen production and utilization in North America, which could potentially be applicable to the deployment of methane pyrolysis technologies. Finally, in the conclusion we map out future research directions, policy needs, and investment opportunities.
To the best of our knowledge, this is the first review to comprehensively address the diverse approaches to methane pyrolysis, advancing the understanding of pyrolysis technologies while identifying critical gaps in policy, commercialization, and investment strategies to enable its deployment.
2. Current hydrogen production and storage
2.1. Natural gas reforming
The primary method for hydrogen production worldwide has been natural gas reforming, constituting approximately 75% of the annual global hydrogen production of 70 million tonnes.10 This process consumes around 205 billion cubic meters of natural gas, equivalent to 6% of global natural gas usage, and results in the release of over 700 million tonnes of CO2 annually.11 Key players in the merchant hydrogen production sector include companies such as Air Liquide, Linde/Praxair, and Air Products.When natural gas reforming is coupled to carbon capture and utilization/storage (CCUS), the product is called ‘blue’ hydrogen. The two major technologies for blue hydrogen production are steam methane reforming (SMR) with CCS and auto-thermal reforming (ATR) with CCS. Table 1 presents a summary of the findings from a literature review of operating and planned hydrogen production facilities with CCUS.12 Currently, only a few hydrogen production facilities are equipped with CO2 capture (mostly with less than 90% capture rate) but several new projects are in development with much higher capture rates (>90%).
| Condition | Plant/project name | Location | Hydrogen production (kN m3 h−1) | Technology | CO2 capture rate (%) |
|---|---|---|---|---|---|
| Operating | Air Products Port Arthur | USA | 220 | SMR | 60 |
| Air Liquide Port Jerome | France | 50 | SMR | 60 | |
| Shell Quest | Canada | 210 | SMR | 50 | |
| Announced/under development | H-Vision | Netherlands | 700 | ATR | 88 |
| HyNet | United Kingdom | 100 | ATR | 97.2 | |
| H21 | United Kingdom | 3200 from 9 units | ATR | 94.2 | |
| Acorn | Scotland | 53 | ATR | 98.7 | |
| H2 Teesside | United Kingdom | 275 | TBD | 98 | |
| Air Products Alberta | Canada | >695 | ATR | 95 | |
| Air Products Louisiana | USA | >837 | ATR/partial oxidation | 95 | |
Herein, we provide an overview of the SMR integrated with CCS process. For more in-depth information, readers are directed to the National Energy Technology Laboratory (NETL) report.12 A SMR plant is typically designed to be supplied by pipelined natural gas at a pressure of 31 bar and a temperature of 15 °C (Fig. 2). The natural gas is treated in a sulfur polishing unit to remove the mercaptan additive using a zinc oxide sulfur guard bed. A pre-reformer is utilized to reform the C2+ hydrocarbons, reducing carbon deposition on the downstream catalyst, enhancing carbon capture ability, and improving product recovery.
 | ||
| Fig. 2 Block flow diagram of a Steam Methane Reforming Plant with CO2 capture. Source: adapted from reference (DOE Public Access).12 | ||
The partially reformed gas and steam from the pre-reformer undergo further reforming in the primary reformer using a nickel-based catalyst at high temperatures (700–900 °C) and pressures 2–3 MPa. The reforming reaction is strongly endothermic, with energy supplied by firing the reformer on the outside of the catalyst tubes with recycled syngas from the downstream hydrogen purification process plus supplemental natural gas. The SMR unit is also equipped with an integrated heat recovery unit, enabling the convective heat recovery from the reformer flue gas. This is used to supply heat to various sections of the plant, improving plant efficiency and minimizing the requirement to burn natural gas. Lastly, a water gas shift reactor (WGSR) is used to react the synthesis gas (CO and H2) to form CO2 and hydrogen-rich syngas, to maximize hydrogen yield and CO2 separation. The overall following reaction is as follows:
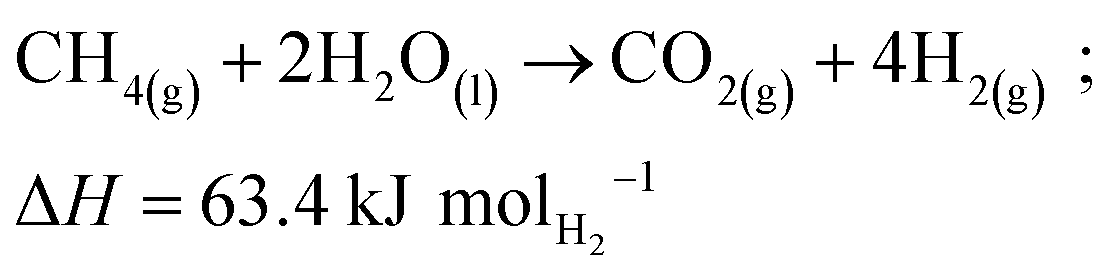 | (R1) |
In SMR + CCS plants, methyl diethanolamine (MDEA) is typically used for CO2 capture. CO2 is extracted from the WGSR syngas using circulating lean amine, with rich amine regenerated in a stripper column via a steam boiler, adding a significant steam load compared to SMR-only plants. Unlike a gray hydrogen production plant, the SMR + CCS plant uses all the steam generated within the plant due to additional demands of the acid gas removal (AGR) units. If the CO2 were only captured from the shifted syngas stream, the overall CO2 recovery would be about 62%. To boost the overall carbon recovery over 90%, a second CO2 removal process based upon the Cansolv system is utilized in the reformer heater stack to remove CO2 resulting from fuel gas combustion. The Cansolv solvent is optimized for low CO2 content environments, enhancing recovery. Further details on the Cansolv system can be found in the NETL report.12 The majority of SMR plants produce between 100 and 200 million standard cubic feet hydrogen per day (MMSCFD), equivalent to between ∼236 and 472 tH2 per day.12
2.2. Water electrolysis
Another way to produce hydrogen is via water electrolysis. Water electrolysis is an electrochemical process that splits water into hydrogen and oxygen, requiring 9 kg of water to make 1 kg of hydrogen. The overall following reaction is as follows: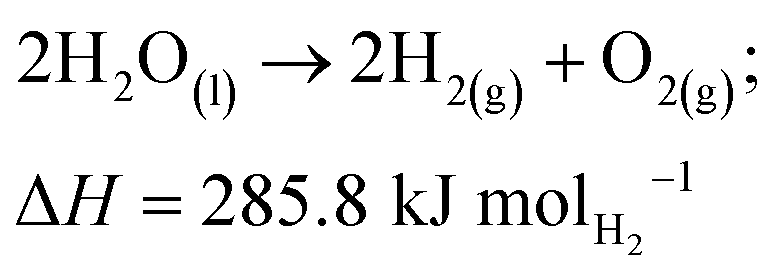 | (R2) |
Less than 0.1% of global hydrogen production comes from water electrolysis today,11 and the hydrogen produced is mostly used in markets where high-purity hydrogen is required (for example, electronics and polysilicon). With the reduction in costs for low-GHG renewable electricity from solar PV and wind, interest in electrolytic hydrogen is growing and there have been several demonstration projects in recent years.
Presently, the two commercial electrolyzer technologies are alkaline electrolysis and proton exchange membrane (PEM) systems. Alkaline electrolysis, established since the 1920s, has been widely used for hydrogen production in industries like fertilizers and chlorine.11 While electrolyzers up to 165 MW were built in the 20th century, most were decommissioned during the 1970s when steam methane reforming (SMR) became dominant. Alkaline electrolyzers are distinguished by their lower capital costs compared to PEM systems, primarily due to the absence of precious metal catalysts.13,14
While alkaline electrolysis systems exhibit high overall efficiencies (approximately 55–70% based on lower heating value, LHV), their low current density (<0.45 A cm−2) and operating pressures (<30 bar) have a negative impact on system size and hydrogen production costs.15 Additionally, their limited dynamic operation range (25–100% of nominal load) can reduce efficiency and gas purity during frequent start-ups or with variable power input.16
PEM water electrolysis originated in the early 1950s with pioneering work by Grubb and further development led by General Electric Co. in the 1960s.11 PEM systems use pure water as the electrolyte, eliminating the need to recover and recycle corrosive potassium hydroxide. They are favored for their compact design, high efficiency (52–69% LHV) at high current density (1–2 A cm−2), rapid response, dynamic operation (0–160% of nominal load), low operating temperatures (20–80 °C), and ability to produce ultrapure hydrogen at high pressures of 30–80 bar.14,16,17 Recent cost reductions in PEM stacks position it as the leading technology for sustainable hydrogen production by 2030.11,14
A large-scale PEM electrolysis plant comprises electrolyzer stacks and mechanical and electrical balance of plant (BoP) components, depicted in Fig. 3. The mechanical BoP includes auxiliary components such as water purification systems, deionizers, pumps, heat exchangers, and the temperature swing adsorption (TSA) system. One critical requirement of PEM water electrolysis is the necessity for high purity water, with ASTM Type II deionized (DI) water (resistivity >1 MΩ cm) as a minimum standard, and ASTM Type I DI water (>10 MΩ cm) preferred.18 Such high-purity water can be produced through using technologies like reverse osmosis (RO), multi-stage flash distillation (MSF), electrodialysis (ED), or multiple-effect distillation (MED), often supplemented with ion exchange or electrodeionization (EDI). Knockout pots or liquid–vapor separators ensure hydrogen and oxygen purity above 99.9%.19 The electrical BoP encompasses the AC to DC rectifier, control systems, sensors, circuit breakers, and other components.
 | ||
| Fig. 3 Block flow diagram of PEM water electrolysis plant. Source: adapted from reference (DOE Public Access).20 | ||
Currently, large-scale water electrolysis plants are either operational or in the process of being commissioned, with capacities reaching up to 20 MW. Notably, several multi-megawatt PEM electrolyzer plants are planned under European initiatives such as Haeolus (Hydrogenics; 2.5 MW), H2Future (6 MW), and REFHYNE (ITM; 10 MW initially, scaling up to 100 MW in phase 2). Additionally, as of January 2021, a 20 MW PEM electrolyzer (manufactured by Hydrogenics) has been commissioned in Canada.21 In the period spanning from 2020 to 2025, numerous planned projects are awaiting final financing decisions, with capacities ranging from 50 MW to over 250 MW.
2.3. Current hydrogen production costs
There have been various studies published on the technoeconomic analysis (TEA) of gray or blue hydrogen production.22,23 The most comprehensive process description and TEA using SMR or SMR + CCS was presented in the NETL report mentioned earlier.12 However, the costs presented in this report were for a fixed large size plant producing 434 tH2 per day.Various studies have offered scaling factors for the Steam Methane Reforming (SMR) process to estimate scaling costs. Yang and Ogden utilized a scaling factor (f; eqn (1)) of 0.70 concerning the “capital cost of SMR plants”, which was dependent on hydrogen production capacity.24 Jiang applied scaling factors of 0.67 for the pre-reformer and main reformer units, and 0.55 for the PSA unit.25 Meanwhile, Hamelinck employed scaling factors of 0.60 for reformer units, 0.85 for water gas shift unit, and 0.70 for the PSA unit in their analyses.26
 | (1) |
The most comprehensive approach was introduced by Elnigoumi et al.27 where the capital cost estimate of SMR plants was based on information from previously constructed plants which are then adjusted using appropriate cost indices to account for inflation. Using the methodology presented by Elnigoumi et al.,27 the plant costs in 2020 US$ for SMR and SMR + CCS plants as function of production capacity (kgH2 per day) is shown in Fig. 4(a). The economies of scale using this approach (0.73–0.74) is consistent with other literature sources as well. In addition to the capital costs, the annual operating costs (OPEX) for a SMR or SMR + CCS plant consists of the raw materials costs (chemicals, catalysts, PSA adsorbent), feedstock costs (natural gas), utility costs (cooling water, electricity), and annual operation and maintenance costs (labor, maintenance, insurance, taxes).
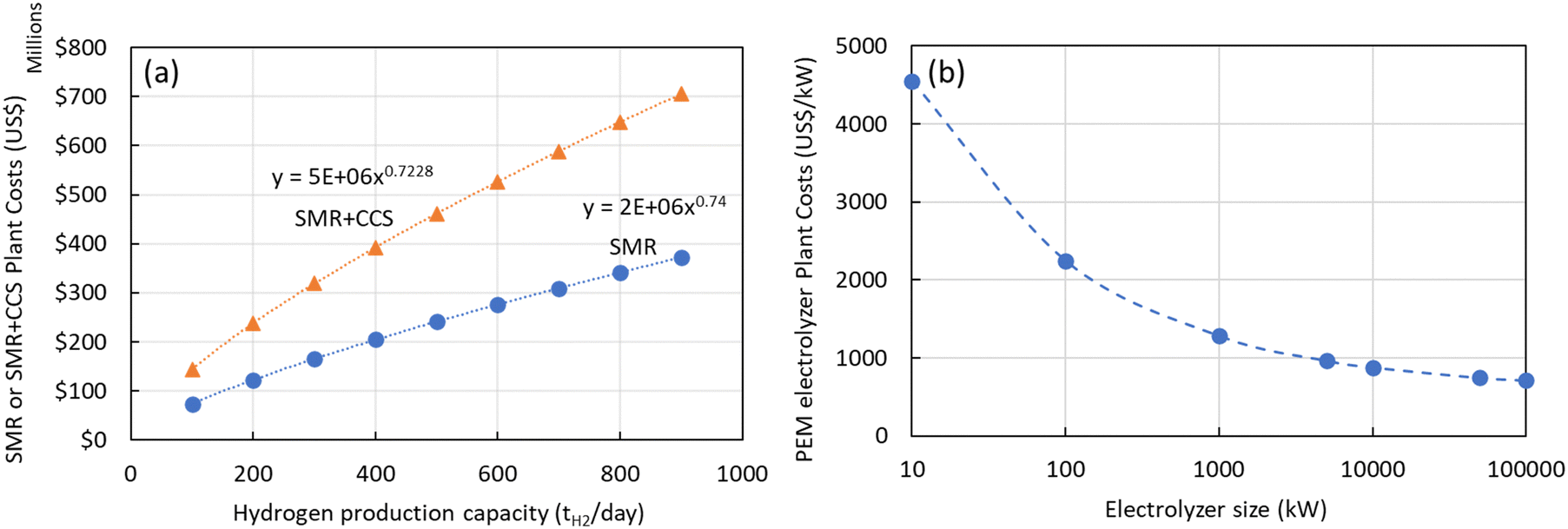 | ||
| Fig. 4 (a) Plant costs (2020 US$) for SMR or SMR + CCS plants versus plant size (kgH2 per day) production (b) plant costs (2020 US$) for PEM electrolyzers versus electrolyzer size (kW). | ||
Similarly, there have been various TEA studies on the cost of green hydrogen production via PEM water electrolysis. The hydrogen production costs of PEM water electrolysis are determined by various technical and economic factors, including the capital cost (CAPEX) of the electrolyzer, its conversion efficiency (kW h kgH2−1), electricity costs, and annual operating hours.28 The most comprehensive capital costs (CAPEX) model that includes the impact of electrolyzer size and technology was published by Reksten et al.29 These costs are based on (i) detailed bottom-up cost estimates or (ii) based on quotes/inquiries from electrolyzer manufacturers. The resulting plant cost (in $ per kW) for PEM electrolyzers is shown in Fig. 4(b) and was based on the following co-relation (eqn (2)):
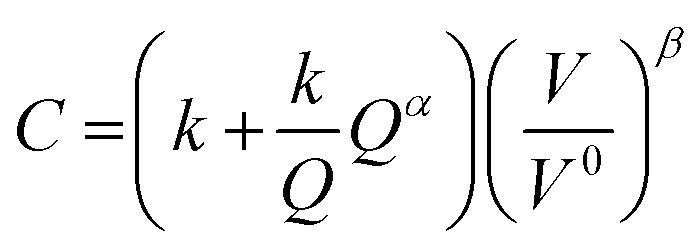 | (2) |
Using the technoeconomic parameters summarized in Table 2, the levelized cost of hydrogen (LCOH) for both blue and green hydrogen was calculated. Fig. 5(a) shows the levelized cost of blue hydrogen production from a SMR + CCS plant as a function of plant size (100–800 tH2 per day) and natural gas price (2–9 $ per GJ). These costs exclude the cost of CO2 transport and sequestration. The cost of CO2 storage in a deep saline formation will vary depending on the geological formation. The CO2 storage costs were estimated using the NETL CO2 storage cost model ranges between 11–26 C$ per tCO2.30 Depending on natural gas price and plant size, the cost of blue hydrogen production from a SMR + CCS plant will vary between 1.2–3 $ per kgH2. The key driver to the cost of blue hydrogen production is the price of natural gas due to the high consumption at ∼0.196 GJNG kgH2−1. The natural gas feedstock contributes between 40–60% of total costs but given the high thermal efficiency (∼69%) and mature technology, the cost of blue hydrogen with SMR + CCS is <3 $ per kgH2, even with a high natural gas price of >8 $ per GJ.
| Techno-economic parameter | SMR + CCS | PEM electrolysis |
|---|---|---|
| CO2 capture rate (%) | 96 | — |
| Capacity factor (%) | 90 | 11.4–91.3 |
| Electricity price ($ per kW h) | 0.07 | 0.02–0.09 |
| Natural gas price ($ per GJ) | 2–9 | — |
| Discount rate (%) | 10 | 10 |
| Plant lifetime (years) | 20 | 20 |
| Taxes (%) | 35 | 35 |
| Contingency (%) | 15 | 20 |
| Working capital (%) | 15 | 15 |
 | ||
| Fig. 5 (a) Levelized cost of blue hydrogen ($ per kgH2) produced via SMR + CCS as a function of natural gas price ($ per GJ) and plant size (tH2 per day). (b) Levelized cost of green hydrogen ($ per kgH2) produced via PEM electrolysis as a function of electricity price ($ per MW h) and annual operating hours (hours per year). | ||
On the other hand, the cost of green hydrogen is significantly influenced by the price of low-carbon electricity and annual operating hours which are dictated by the capacity factor of the electricity source. Fig. 5(b) shows the levelized cost of green hydrogen from the PEM electrolysis plant as a function of annual operating hours (1000–8000 hours per year) and electricity price (20–80 $ per MW h). Currently, at any reasonable electricity price (∼40–70 $ per MW h) and annual operating hours (4000–6000 h per year), green hydrogen will be expensive at 5–7 $ per kgH2. Today, the production of low-cost green hydrogen (<3 $ per kgH2), will need extremely cheap electricity (<30 $ per MW h) with high-capacity factors (>80%).
Future cost projections will be influenced by technology development. Experts predict that R&D investments aimed at developing low-cost electrodes and membranes, system level optimization, and economies of scale in the manufacturing processes will lead to significant CAPEX reductions for PEM electrolyzers. However, given that electricity costs account for more than 70% of the cost of producing green hydrogen, and only 20–25% of the cost is due to capital costs, green hydrogen production will always need extremely cheap electricity to drive economic feasibility.
2.4. Current hydrogen storage methods
Methane pyrolysis offers a promising solution to the challenges of large-scale hydrogen storage by facilitating on-demand, on-site hydrogen production. Before evaluating the technologies for methane pyrolysis, it is important to address the challenges of hydrogen storage to appreciate the benefits of such on-site production technologies. This section provides a brief overview of the thermodynamic and material challenges for hydrogen storage. For a comprehensive assessment, the readers are encouraged to refer to previously published articles.31–35Hydrogen, the smallest and lightest molecule, has a very low gaseous density (0.082 kg m−3 at 1 atm, 0 °C), requiring advanced storage technologies to achieve energy densities comparable to fossil fuels.36 For example, 1 liter of hydrogen at 0 °C and 1 atm contains only 0.01 MJ of energy, compared to 33.3 MJ for 1 liter of gasoline at 15 °C and 1 atm. Storage technologies depicted in Fig. 6 can be broadly categorized into two main categories: (1) physical storage of pure hydrogen (liquid, compressed, and cryo-compressed hydrogen), and (2) material-assisted routes for solid-state hydrogen storage.34
 | ||
| Fig. 6 Different hydrogen storage methods. Adapted from ref. 34 with permission from the Elsevier, copyright 2022. | ||
Physical storage of hydrogen via compression or liquefaction is the currently the most mature technology but requires significant energy. Hydrogen compression stores gas in aboveground tanks or underground geological structures.37 The energy required for compression primarily depends on the molar flow rate and the compressibility factor (Z) of the gas. Hydrogen's low molar energy density (∼0.066 kW h per mole vs. ∼0.248 kW h per mole for natural gas) is a disadvantage. Along Moreover, a deviation from the ideal gas law, leads to a higher Z value for hydrogen below 600 bar compared to gases like CH4, O2, and CO2.38,39 For example, when compressed from 1 bar to 700 bar, hydrogen's density increases only 477-fold (from 0.0898 to 42.9 kg m−3), leading to higher compression energy requirements due to the direct influence of Z. The theoretical minimum energy for isothermal compression from 20 to 700 bar at 20 °C is 1.35 kW h kgH2−1.40 However, due to compressor inefficiencies, energy consumption for hydrogen compression typically ranges between 2.9 and 3.1 kW h kgH2−1.40
The costly storage vessels are another barrier for aboveground storage of compressed hydrogen. Storage vessels made from materials such as austenitic stainless steel, aluminum, and carbon fiber-reinforced polymers and classified as Type I to Type IV, and selected based on the required pressure and storage scale. The choice of material is constrained by the risk of hydrogen embrittlement, which limits the use of certain metals under high-pressure conditions.32
Underground storage of compressed hydrogen in depleted natural gas reservoirs, aquifers, and salt caverns offers a potentially lower-cost option for large-scale storage.41 This approach requires relatively lower compression pressures, typically between 100 and 200 bar. However, it is feasible only in locations with an impermeable caprock to safely contain the hydrogen. While successful hydrogen storage has been demonstrated in salt caverns, it has not yet been implemented in depleted gas fields or aquifers.42 A significant limitation of this method is the scarcity of suitable underground structures in specific regions, posing a major challenge to its broader adoption.
Liquid hydrogen supply is often preferred due to its higher density (70.8 kg m−3) compared to gaseous hydrogen (0.0898 kg m−3 at 1 bar) and compressed hydrogen (42.9 kg m−3 at 700 bars). However, liquefying hydrogen requires significantly more energy than compression, with a theoretical minimum of 3.3 kW h kgH2−1, or 3.9 kW h kgH2−1 when converting to para-hydrogen.43 Hydrogen exists as ortho and para isomers, distinguished by nuclear spin alignment: ortho-H2 has aligned spins, while para-H2 has opposite spins. At ambient conditions, hydrogen is ∼25% para and ∼75% ortho, but liquid hydrogen is >99% para.44 The conversion to para (and liquid) occurs at hydrogen's boiling point (20 K), but the process is very slow in the absence of a catalyst. Therefore, modern liquefiers use catalysts inside heat exchangers to accelerate the conversion. Today's industrial liquefiers entail high capital costs and are energy-intensive processes, typically ranging from 8 to 12 kW h kgH2−1,45 representing ∼30% of hydrogen LHV. A recent liquid hydrogen production plant serving the California market was reported to cost US$150 million for a 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kg per day capacity.38
000 kg per day capacity.38
The challenges associated with hydrogen's low volumetric density and high energy required for compression and liquefaction have encouraged the exploration of solid-state hydrogen storage using nanostructured materials and hydrides to bond with hydrogen. This approach can generally be classified into two categories: chemically and physically bound hydrogen storage.
Metal hydrides, spanning elemental hydrides (e.g.: MgH2, AlH3), borohydrides (M(BH4)n), and other complex hydrides, have been investigated for chemically bound hydrogen storage. However, challenges such as high operating temperatures, energy-intensive regeneration processes, and material costs remain significant. MgH2 is attractive due to its low cost and large theoretical hydrogen storage capacity of 7.6 wt% but requires high energy for desorption (75 kJ mol−1 H2) and operates at high temperatures of approximately 300 °C.46 AlH3 has a higher theoretical storage capacity of 10.1 wt%, but it demands very high pressures for hydrogen absorption. Hydrogen storage in M(BH4)n compounds (e.g., LiBH4) and complex metal hydrides (e.g., NaAlH4, LiBH4) is also an attractive area of research due to their very high storage capacities (15–20 wt%).
Liquid organic hydrogen carriers (LOHC) are another attractive route that absorbs and releases hydrogen via chemical reactions. Some of the most promising LOHCs identified recently include toluene/methylcyclohexane,47 naphthalene/decalin,48 and benzene/cyclohexane.49 These LOHCs remain in a liquid state at standard temperature and pressure, offering a high volumetric energy density, which simplifies the equipment and infrastructure required for storage and transport.
Overall, high hydrogenation/dehydrogenation temperatures, slow reaction kinetics, and reversibility should be addressed in future studies on chemical hydrogen storage.50 Catalyst development is critical, as current catalysts can degrade over time or may lack sufficient activity and selectivity under milder conditions. Furthermore, the recycling of carrier molecules and addressing potential degradation products is essential for ensuring the economic and environmental sustainability of this technology.51
Finally, adsorption-based storage using Metal–Organic Frameworks (MOFs) or carbon-based materials, relies on weaker van der Waals forces to bind hydrogen molecules.52–54 These materials feature substantial surface area and rapid kinetics but the requirement of cryogenic conditions to maximize hydrogen adsorption hinders widespread adoption. In summary, hydrogen storage technologies, each with unique safety risks, economic considerations, and varying levels of maturity, face notable challenges. Therefore, the development of on-demand, on-site hydrogen production technologies could bypass the challenges associated with large-scale hydrogen storage and transport, playing a pivotal role in the advancement of a future hydrogen economy.
3. Methane pyrolysis
Methane pyrolysis is the thermal decomposition of methane into hydrogen and solid carbon (R3).55 According to standard reaction enthalpies, 37.5 kJ of energy is required to produce one mole of hydrogen via methane pyrolysis as shown in Fig. 7.55,56 This is considerably lower than SMR (R1) or water electrolysis (R2) which require 63.4 kJ and 285.8 kJ per mole of hydrogen, respectively.57 Steinberg calculated the overall thermal efficiency of SMR, SMR + CCS, and methane pyrolysis at 75%, 60%, and 58%.58 This demonstrates methane pyrolysis as comparable to SMR coupled with CCS, albeit with significantly lower emissions.58| CH4 → C(s) + 2H2; ΔH = 37.5 kJ mol−1 H2 | (R3) |
 | ||
| Fig. 7 Enthalpy diagrams of (A) SMR (from liquid water), (B) water electrolysis, and (C) methane pyrolysis for hydrogen production. Reproduced from ref. 56 with permission from the American Chemical Society (CC-BY 4.0), copyright 2021. | ||
Methane pyrolysis is an endothermic reaction and requires high reaction temperatures of >1000 °C in the absence of catalysts. This is necessary due to the stable non-polar tetrahedral molecular geometry of CH4 and high C–H bond energy ΔHC–H = 439.3 kJ mol−1. The equilibrium conversion of the pyrolysis process is influenced by temperature and pressure as shown in Fig. 8.59 Based on Le Chatelier's principle, an increase in temperature leads to an increase in equilibrium conversion. Similarly, since there are three moles of product (2H2 and 1C) formed for every mole of reactant (CH4), enhancement in pressure will lead to a decrease in conversion.
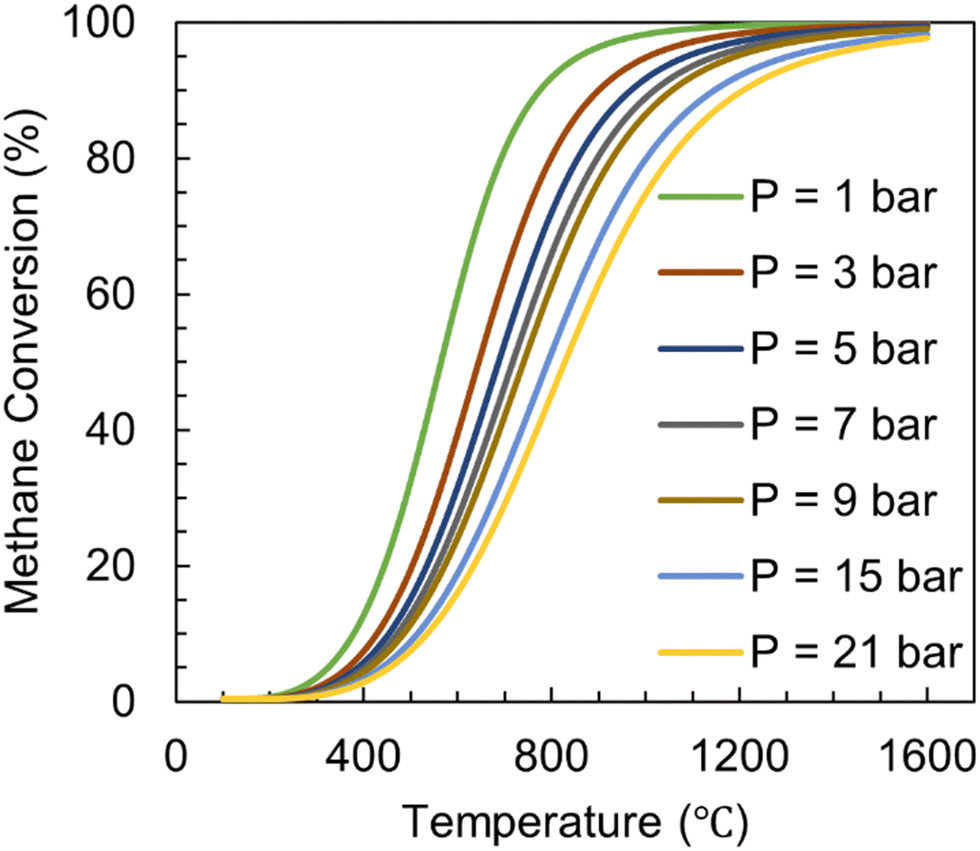 | ||
| Fig. 8 Equilibrium conversion of methane pyrolysis calculated versus temperature at different pressures for pure CH4 as feed. | ||
To lower the reaction temperature and improve the hydrogen yield, different solid metals (Ni, Fe, Co, etc.) and C-based catalysts have been developed. These solid catalysts have been effective in reducing temperatures compared to non-catalytic pyrolysis, but suffer from deactivation and risk carbon byproduct contamination. Recently, the use of liquid catalysts and heat transfer media such as molten metals (Pb, Sn, Bi), molten metal alloys (Ni–Bi, Cu–Bi), and molten salts (KBr, NaBr, NaCl, NaF, MnCl2, KCl, FeCl2) has attracted attention.
The catalytic and non-catalytic pyrolysis processes vary based on the energy source, heat transfer medium, and reactor design such as fluidized-bed reactors, packed-bed reactors, tubular reactors, microwave reactors, plasma reactors, molten metal/salt reactors, and others as shown in Fig. 9. These technologies vary in energy efficiency, hydrogen yield, operating temperature, carbon byproduct quality, and hydrogen production costs. The following sections provide a comprehensive overview of these different pyrolysis technologies.
 | ||
| Fig. 9 Different routes for hydrogen production through methane pyrolysis. First column: categorization of methane pyrolysis routes. Second column: subcategories within each type. Third column: catalysts under investigation. Fourth column: types of reactors utilized. | ||
3.1. Catalytic methane pyrolysis
Several reaction mechanisms have been postulated to explain the catalytic pyrolysis of methane. A majority of studies have proposed a molecular adsorption mechanism (Fig. 10(a)),60,61 and a few others discuss a dissociative adsorption model (Fig. 10(b)).62,63 | ||
| Fig. 10 (a) Molecular adsorption mechanism. (b) Dissociative adsorption mechanism for catalytic methane pyrolysis. The red arrows indicate the rate-determining steps. Adapted from ref. 56 with permission from the American Chemical Society (CC-BY 4.0), copyright 2021. | ||
In the molecular adsorption mechanism, methane is first adsorbed on the catalyst surface and then dissociated following a series of stepwise surface dehydrogenation reactions.61 In most studies, the rate-limiting step (RLS) is often considered to be the abstraction of the first hydrogen atom from molecularly adsorbed methane to form an adsorbed methyl group.64 However, a few studies also suggest the RLS be either methane adsorption over the surface, or the removal of the second hydrogen from the adsorbed methyl fragment.65
In the dissociative adsorption mechanism, methane dissociates upon adsorption on the catalytic active sites generating chemisorbed CH3 and H fragments.66 Subsequent surface dissociation reactions mirror those in the molecular adsorption mechanism. As with molecular adsorption, different steps are considered as RLS. Most studies confirm that methane dissociation into CH3 and H controls the overall mechanism.67,68
Zhang and Smith reported that molecularly adsorbed CH4 over Group VIII metal catalysts would have higher activation energy (CH4S + S → CH3S + HS, where S represents an active site) versus gas phase (CH4 + 2S → CH3S + HS).69 Saraswat et al. analyzed both pathways, considering different steps as RLS,70 and concluded that molecular adsorption with the first step as RLS is suitable mechanism for Ni–Cu–Zn/Al2O3 catalyst. Both pathways primarily yield hydrogen catalytically, whereas the non-catalytic route can produce a variety of compounds such as C2H6, C2H4, C2H2, and C6H6.71–73 This is because catalysts play dual roles by reducing activation energy for faster reactions and enabling selectivity. On the other hand, non-catalytic methane pyrolysis can follow different reaction pathways based on operating conditions, discussed in Section 3.2.
In recent years, various solid and liquid catalysts have been studied for methane pyrolysis. Solid catalysts, categorized as metal-based and carbon-based, are covered in Sections 3.1.1 and 3.1.2. Liquid metals and molten salts, serving as both heat transfer media and catalysts, are discussed in Section 3.3.
| Catalyst | Flow (mL min−1) | Temperature (°C) | CH4 molar ratio in feed (%) | Conversion (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|
| 7% Ni/TiO2 | 60 | 600 | 100 | 60–50 | 13 | 74 |
| 7% Ni/Al2O3 | 60 | 600 | 100 | 50–30 | 2 | 74 |
| 7%Ni/CaCO3 | 60 | 600 | 100 | 50–20 | 2 | 74 |
| 20% Ni/TiO2 | 60 | 600 | 100 | 62–74 | 15 | 74 |
| 7% Ni/(TiO2 + Al2O3) | 60 | 600 | 100 | 68–60 | 15 | 74 |
| Ni/TiO2 | 7.5 | 600 | 100 | 40–38 | 8 | 76 |
| Co–Cu (75/25) | 25 | 700 | 25 | 30 | 2 | 75 |
| Ni–Cu–Co (70/5/25) | 25 | 750 | 25 | 60 | 3 | 75 |
| Ni–Cu–Co (50/25/25) | 25 | 750 | 25 | 80 | 5 | 75 |
| Ni–Cu–Co (50/25/25) | 25 | 800 | 25 | 84 | Fast deactivation | 75 |
| Ni–Al (75/25) | 70 | 700 | 28.6 | 83 | Fast deactivation | 77 |
| Ni–Fe–Al (25/50/25) | 70 | 700 | 28.6 | 58 | 1 | 77 |
| Ni–Fe–Al (25/50/25) | 70 | 700 | 28.6 | 70 (40) | 2 (210) | 77 |
| Fe/Al2O3 | 70 | 700 | 30 | 58–25 | 1 | 78 |
| Fe/SiO2 | 70 | 700 | 30 | 50–15 | 1 | 78 |
| Fe/H-ZSM-5 | 70 | 700 | 30 | 36–14 | 1 | 78 |
| Ni–Cu–SiO2 | 35 | 750 | 100 | 41 | Fast deactivation | 79 |
| Co/SiO2 | 250 (5000) | 800 | 100 | 33–25 | 5 | 80 |
| Ni/SiO2 | 250 (5000) | 800 | 100 | 59–23 | 5 | 80 |
| Fe/SiO2 | 250 (5000) | 800 | 100 | 43–27 | 5 | 80 |
| Ni/CeO2 | 150 (4500) | 700 | 100 | 45–30 | 6 | 81 |
| Ni/ZrO2 | 150 (4500) | 700 | 100 | 44–32 | 6 | 81 |
| Ni/La2O3 | 150 (4500) | 700 | 100 | 41–33 | 6 | 81 |
| Ni/La2O3 | 15 | 700 | 100 | 80–60 | 4 | 82 |
| Fe/CeO2 | 150 (4500) | 800 (700) | 100 | 47 (39)–49 (35) | 6 | 83 |
| Fe/La2O3 | 150 (4500) | 800 (700) | 100 | 50 (41)–33 (23) | 6 | 83 |
| Ni–Co/SBA-15 | 150 (4500) | 700 | 100 | 39–22 | 5 | 84 |
| Ni–Fe/SBA-15 | 150 (4500) | 700 | 100 | 34–20 | 5 | 84 |
| Fe–Co/SBA-15 | 150 (4500) | 700 | 100 | 33–28 | 5 | 84 |
The catalytic activity reported varies by metal, with Ni > Co > Fe. Ni demonstrates the highest initial activity (CH4 conversion and H2 yield) but suffers from fast deactivation due to carbon coking and poisoning. Kang and Lee reported a high conversion of ∼ 90% and 80% at 850 °C and 750 °C, respectively, using Ni catalysts, nearing equilibrium conversion,85 But the conversion dropped to below 60% in less than 4 hours. Salipira et al. reported 60% conversion at 600 °C and showed that the activity is directly proportional to Ni concentration.74 Deactivation accelerated with temperature increase, starting within 3 hours at 650 °C and 1 hour at 700 °C, due to faster carbon formation than diffusion in Ni particles, which blocks active sites and halts methane conversion.86
To address catalyst deactivation, other catalysts have been investigated. Co, while the second most active for methane decomposition, is costly and toxic, making it less suitable.87 Fe catalysts, being cheaper and non-toxic, offer a viable alternative. Pudukody et al. explored Ni, Co, and Fe catalysts on various supports such as CeO2, ZrO2, La2O3, and SBA-15 for methane pyrolysis.81,83,84 The Fe/CeO2 sample demonstrated the best stability and performance, achieving 35% conversion (52% H2 yield) after 6 hours.83 Fe's carbon diffusion rate, three orders of magnitude higher than Ni's, accounts for its greater stability.88
Combining Ni and Fe aims to leverage Ni's activity and Fe's stability.89 Tezel et al. demonstrated that 4.46%Ni–1.1%Fe/Al2O3 catalysts had lower activity (57% conversion) than 5.87%Ni/Al2O3 catalysts (66% conversion) but improved stability.90 Wang et al. studied CH4 pyrolysis at 600 °C using Ni3−xFexAl catalysts.91 Catalyst lifetime increased with Fe content up to x = 0.6 (Ni2.4Fe0.6Al, 7 h) before declining at higher Fe content. Fe's higher diffusion coefficient (100 times Ni) reduces carbon accumulation, leading to higher catalytic life and carbon yield.
Ni has also been combined with other metals to improve catalytic activity.92 Li et al. found that adding 10 wt% Cu to Ni–SiO2 increased conversion from 12% to 22%, while raising Cu to 20 wt% reduced initial conversion (22% to 17%) but improved stability (200 to 350 minutes).79 Shen and Lua reported that Ni–Cu on carbon nanotubes (78% Ni, 22% Cu) achieved 55% CH4 conversion at 700 °C with over 20 hours of stability, though at a lower feed flow rate (7.5 mL min−1).93 Cu has also been used with mixed metal Ni–Fe catalysts offering several benefits such as higher reducibility, improved dispersion, and hindering carbon encapsulation.94,95
For Fe-based catalysts, reducibility is a significant factor whereby metallic iron (Fe0) is required for methane decomposition.96 Al is reported to be the most effective promotor for Fe compared to Co, Mg, Ce, Ti, and Ca.96 An Fe–Al2O3 sample (65 wt% Fe) prepared by co-precipitation reached >70% conversion at 750 °C, where Al2O3 played a key role in enhancing Fe crystallization, and increasing Fe0 surface exposure for graphitic carbon to deposit. The synthesis method also has a big impact on the catalytic performance with co-precipitation preferred to impregnation to ensure better dispersion.97 Ibrahim et al. demonstrated this for Fe–Al2O3 catalysts prepared via co-precipitation, achieving a maximum hydrogen yield of 77.2%, stable over 4 hours.98
The catalyst support also plays a key role, influencing metal particle dispersion, reducibility and mechanical stability.99 Weak metal–support interactions cause leaching, while strong interactions can reduce dispersion. Wang et al. investigated the impacts of different supports such as Al2O3, SiO2, and H-ZSM-5 on Fe catalysts.78 They found that Fe/Al2O3 provided superior performance (58% conversion) than the two others (50% and 36% conversion for ZSM-5 and SiO2, respectively), attributed to better Fe dispersion and reducibility with Al2O3. Salipira et al. observed that TiO2 and Al2O3–TiO2 supports provided the highest hydrogen production rate and stability for Ni catalysts.74 Takenaka tested supports like SiO2, TiO2, MgO, ZrO2, and Al2O3 for Ni catalysts, finding Ni/TiO2 had the highest initial methane conversion at 500 °C, while Ni/SiO2 performed better over time.100 The K-edge XANES spectra showed Ni predominantly as metal in Ni/TiO2 and Ni/SiO2, whereas it appeared as NiO in other supports. Awadallah et al. demonstrated via temperature programmed reduction (TPR) studies that strong metal–support interaction in Ni/MgO required high reduction temperatures (912 °C), forming MgxNi(1−x)O solid solutions that reduced catalytic activity.101
In summary, advances have been made in developing solid metal catalysts for methane pyrolysis, but carbon deposition remains a major challenge, causing catalyst deactivation and reactor clogging. Deactivation occurs when the metal catalysts are covered by carbon or due to loss of the metal particles which get detached from the support.102 These two mechanisms will be discussed in more detail in Section 4. Meanwhile, significant research has focused on carbon-based catalysts as a cheaper and more stable alternative, discussed in the next section.
| Catalyst | VHSVa (mL h−1 gcat−1) | Temperature (°C) | CH4 molar ratio in feed (%) | Conversion (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|
| a VHSV = volumetric hourly space velocity. | ||||||
| Biochar | 6000 | 900 | 90 | 51–20 | 6 | 106 |
| Biochar | 6000 | 700 | 90 | 25–0 | 0.5 | 106 |
| Activated char | 6000 | 900 | 90 | 58–21 | 6 | 106 |
| Activated char | 6000 | 700 | 90 | 30–0 | 0.5 | 106 |
| Carbon black | 15000 |
1120 | — | 80 | 2.5 | 107 |
| Carbon black | 15000 |
970 | — | 62 | 2.5 | 107 |
| Carbon black | 15000 |
900 | 62.5 | 12 | 2 | 108 |
| Carbon black | 13000 |
900 | — | 52–15 | 10 | 109 |
| Activated carbon | 13000 |
900 | — | 67–15 | 2 | 109 |
| Carbon nanofiber | 13000 |
900 | — | 15 | 10 | 109 |
| Mesoporous carbon | 13000 |
900 | — | 45–15 | 3 | 109 |
| Trimodal carbon | 24000 |
900 | 25 | 52–15 | 10 | 110 |
| Ni/CNT | 26000 |
575 | 100 | 35–0 | 20 | 93 |
| 78%Ni-22%Cu/CNT | 26000 |
700 | 100 | 55–47 | 20 | 93 |
| 58%Ni-42%Cu/CNT | 26000 |
700 | 100 | 53–40 | 20 | 93 |
| 74%Ni–26%Cu/CNT | ∼35000 |
700 | — | 80 | 1 | 111 |
| 78%Ni–22%Cu/CNT | 9000 | 700 | 100 | 50 | 20 | 112 |
| Ni/CNT | 9000 | 600 | 100 | 55 | Fast | 112 |
Among carbon-based catalysts, amorphous carbons such as activated carbons and carbon blacks have demonstrated promising activity.113–115 The reported activation energy of activated carbon and carbon black is 160–201 kJ mol−1 and 205–236 kJ mol−1, respectively.116,117 Activated carbons show high initial activity (conversion rates) but are less stable than carbon black. Nishii et al. demonstrated a high initial conversion of 67% using activated carbon at 900 °C, but it dropped to 15% in less than 2 hours.109 In contrast, carbon black had a lower initial conversion rate of 52% but was more stable at over 30% for more than 7 hours.
Several factors impact the stability and activity of carbon-based catalysts for methane pyrolysis. Surface area is critical for initial activity, as highlighted by Muradov et al., who demonstrated a direct correlation between surface area and initial conversion, with activated carbon (up to 2208 m2 g−1) achieving the highest initial activity.109 However, beyond this initial phase, porosity and particle size come into play and can affect methane conversion. Carbon deposition during decomposition can block surface pores, causing rapid deactivation, as seen with activated carbon. In contrast, carbon black, with more accessible active sites, maintains longer activity. In another research, mesoporous carbon structures were found to be more active than microporous carbon due to their facilitation of mass transfer barriers.118
The crystal structure and lattice defects of the carbon catalysts also significantly influences their catalytic activity.119,120 Raman spectra peaks at 1580 cm−1 (G band) and 1350 cm−1 (D band) correspond to graphitic lattice vibrations and symmetry-reducing lattice defects, respectively.121,122 The intensity ratio (ID/IG) measures the degree of graphitization, and literature suggests that higher ID/IG values (indicating greater lattice defects) correlate with higher activity, as observed in the comparison between carbon black and mesoporous carbon.123 Both carbon black and activated carbon, classified as amorphous carbons, feature abundant high-energy sites (defects, vacancies, edges) and large surface areas, resulting in high catalytic activity for methane pyrolysis.124
It is important to note that the carbon formed during the reaction is also reported to exhibit catalytic activity, but its effectiveness depends on the surface area and crystal structure. Nishii et al. found stable ∼17% conversion across catalysts like activated carbon, carbon black, mesoporous carbon, and carbon nanofibers, driven by the activity of the deposited carbon.109 However, this was significantly lower than the initial conversion of the catalysts which was in the range of 45 to 70%. This was primarily due to the lower surface area of the produced carbon at 10 m2 g−1versus the carbon catalysts which had specific surface areas of >1500 m2 g−1.109 Furthermore, the carbon formed at temperatures of 800–1470 °C, characterized by graphitic and nanotube structures (as will be discussed in Section 4), shows reduced methane decomposition efficiency.125
To summarize, various catalysts such as Ni, Fe, Co, and C have been used for methane pyrolysis. Fig. 11 demonstrates that the reported operating temperatures which is in order of Ni < Co < Fe < carbon-based. It must be noted that conversion efficiency for C-based catalysts is typically lower than other systems as summarized in Table 4. The required reaction temperature with catalytic systems (600–1000 °C) is significantly lower than non-catalytic methane pyrolysis. However, the main drawback of current catalysts (metal or carbon) is the requirement of a regeneration step which is discussed next.
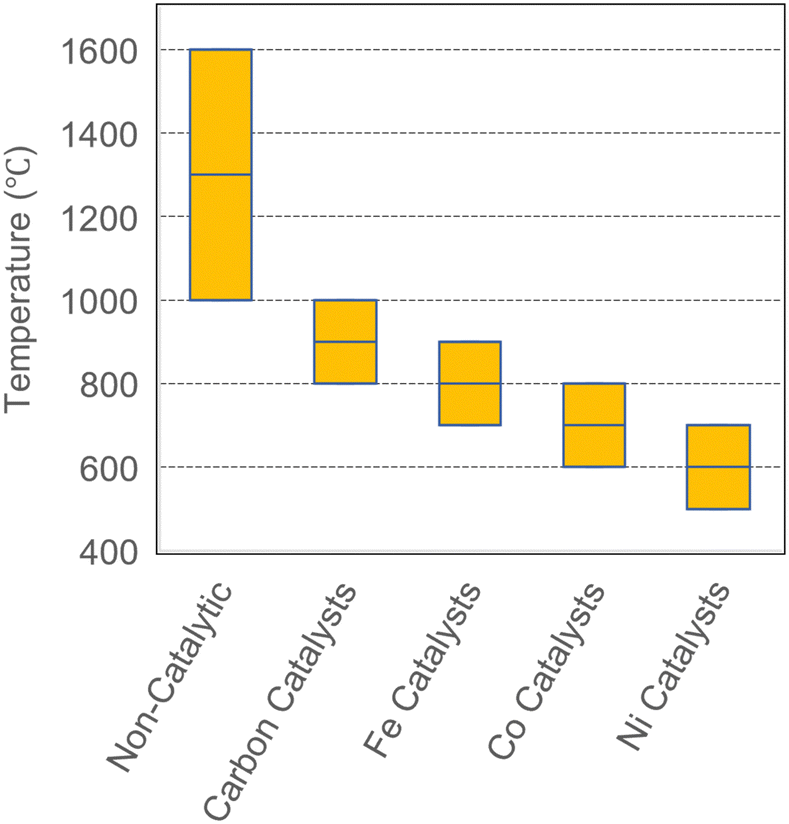
Villacampa et al. studied Ni/Al2O3 catalysts regenerated with O2 at 600 °C and found that sintering of Ni particles reduced hydrogen production in subsequent cycles (1.5 vs. 0.5 mmol H2 gcat−1 min−1 after 20 minutes for fresh and regenerated catalysts, respectively).128 Additionally, the carbon produced over catalysts dropped from >0.3 gC gcat−1 to 0.1 gC gcat−1 after the first regeneration cycle, corresponding to more than 66% reduction in activity.
Takenaka et al. utilized CO2 gasification to regenerate Ni catalysts on three different supports: TiO2, SiO2, and Al2O3.127 In general, regeneration led to particle agglomeration for all three catalysts (for Ni/SiO2:40–100 nm → 200 nm) which inversely affected activity. However, for the TiO2 and Al2O3 support, the first few regeneration cycles led to an improvement in catalytic activity, declining only after additional cycles. This improvement was attributed to the small initial Ni particle size on these supports, where early regeneration cycles (e.g., 3 cycles for Ni/TiO2) allowed Ni particles to reach an optimum size.
Ammendola utilized all three methods of regeneration for Cu/Al2O3 catalysts (Fig. 12) to compare them in a similar process.129 The regeneration was performed at 800 °C under air flow for combustion, under 5.5% CO2, and 3% steam in the air for CO2 and steam gasification, respectively. While the combustion in the air was completed in 15 minutes, the gasification with CO2 and water required at least 80 and 70 minutes to remove 45% of the deposited carbon. As a result of carbon removal, the catalyst regenerated by combustion in air restored its complete activity after two cycles. However, two other options were not as effective in regenerating the catalyst.
 | ||
| Fig. 12 The activity of Cu/Al2O3 in producing H2 in fresh and after regenerated by (A) air, (B) CO2, and (C) steam. Adapted from ref. 129 with permission from Taylor & Francis, copyright 2008. | ||
Similar to metal catalysts, carbon-based catalysts can undergo gasification and combustion with CO2, steam, and O2 to be regenerated.130,131 However, in this scenario, regeneration involves not only removing deposited carbon but also losing part of the initial carbon catalyst.132 This loss is more significant with O2 combustion, as the initial carbon is more active than the deposited carbon.133 Consequently, combustion is not considered a suitable option for regenerating carbon-based catalysts and gasification is preferred.
Pinilla investigated the regeneration of spent carbon catalysts through CO2 gasification at temperatures of 700–950 °C and analyzed the specific surface area, amount of oxygenated groups created, and weight loss of the catalyst.132 It was observed that the specific surface area and the oxygenated groups increased as the regeneration conditions became more severe. More importantly, it was observed that multiple deactivation-regeneration cycles led to a decrease in weight, surface area, and oxygenated groups. This was attributed to the removal of carbon from the initial catalyst, which is less resistant to gasification, rather than the graphite-like carbon deposited during methane pyrolysis at 850–1000 °C, known for its gasification resistance.
Investigations into catalyst reactivation via gasification with H2O showed promising results.134 Regenerating the catalyst through steam treatment at 950 °C for 0.5 hours restored 92% of the initial surface area.116 Although the first cycle demonstrated superior activity restoration compared to CO2 gasification, further studies are needed to evaluate the effects of both gasification methods (CO2 and H2O) on the same deactivated sample. It's crucial to acknowledge that while regenerating of catalysts through combustion or gasification may result in stable systems, it also leads to the release of COx, which contradicts the aim of zero-emission hydrogen production.
Fixed-bed reactors, commonly studied on a laboratory scale, were the focus of early methane pyrolysis research. Kim et al. used a quartz-tube flow reactor with activated carbon of varying particle sizes, finding methane conversions of 8–12%, higher with smaller particles due to intraparticle mass transport effects. However, all catalysts deactivated rapidly within 100–120 min due to pore blockage by deposited carbon. Similarly, Lee et al. achieved 100% methane conversion using carbon black in a fixed-bed reactor at 1020–1170 °C, though stability was not reported.107 In another study, Pinilla et al. utilized a fixed-bed reactor with Fe-based catalysts at 900 °C, resulting in a 93% hydrogen concentration in the output gas.138 The carbon was deposited as multiwall carbon nanotubes which slowly led to catalyst deactivation over 180 min. Fixed-bed reactors face drawbacks such as hot spots, thermal gradient, pressure drops, and challenges with carbon coking, limiting their practical application.139 These issues become more significant in methane pyrolysis due to the substantial solid carbon yield (75% wt. of CH4), which can lead to rapid reactor blockage. Coupled reactors could address this barrier by allowing simultaneous pyrolysis in one reactor and regeneration in another (Fig. 13(a)).
 | ||
| Fig. 13 Methane pyrolysis for a continuous process using (a) fixed-bed configuration with a parallel pyrolysis reactor, and (b) fluidized-bed reactor coupled with a regeneration reactor. Adapted from ref. 140 with permission from the Elsevier, copyright 2001. (CAT-CARB: deactivated catalysts with solid carbon, Catalyst: regenerated catalyst). | ||
On the other hand, fluidized bed reactors are known to be more effective for enhancing mixing, heat, and mass transfers.116,135,136,141–145 In these reactors, the solid catalyst particles are suspended by the upward flow of the gases or liquids. This fluidization improves mixing and contact between the catalyst and the reactants, leading to improved heat and mass transfer.
Łamacz and Łabojko studied methane decomposition over Ni/CeZrO2 in a fluidized-bed reactor under varying gas hourly space velocities (GHSV), particle sizes, and temperatures equipped with H2O regeneration.146 Over four cycles, this reaction/regeneration process demonstrated promising results. The results revealed an inverse relationship between GHSV and particle size with conversion and stability, respectively. While fluidized bed reactors are more robust to carbon blockages than packed bed reactors, catalyst particles still grow due to carbon deposition and eventually cause blockage. Lee et al.147 and Dunker et al.139 also demonstrated this issue in fluidized bed reactosr for methane pyrolysis reactions with activated carbon and carbon black catalysts, respectively. In both studies, the catalyst activity significantly dropped within 120–1000 min due to blockage from carbon produced during reaction.
To maintain a continuous process, a regeneration reactor can be integrated with the pyrolysis reactor, as illustrated in Fig. 13(b). Carbon-covered catalysts are continuously transferred to the regeneration reactor for carbon removal or combustion, and the reactivated catalyst is recycled back into the pyrolysis reactor. Alternatively, two parallel decomposition reactors can be used, allowing the redirection of the stream between them, and facilitating the regeneration. Nevertheless, as discussed, besides GHG formation, catalyst regeneration also leads to diminished activity compared to fresh catalysts, thereby reducing the efficiency of the process. This necessitates frequent catalyst replacement, increasing overall process costs.
Moving-bed reactors can also be used for either catalytic or non-catalytic methane pyrolysis. They can be designed for concurrent (same direction) or countercurrent (opposite direction) flow of feed gas and solid particles (catalysts/heat transfer media).148,149 Countercurrent designs (Fig. 14) face challenges from gas flow distortion caused by significant drag from solid flow,150 but enhance heat transfer, hence making it commonly preferred method for this process.151,152 Moving-bed reactors also provide advantages such as low-pressure drops, reduced maintenance costs, and improved gas–solid surface contact.153
 | ||
| Fig. 14 Moving-bed reactor schematic and flows.152 | ||
Carbonaceous materials like carbon black are often used as solid heat transfer materials due to their similar composition to the produced carbon during the pyrolysis process.154 The carbon stream can be directly heated by electricity, enabling the temperature of the reactor to reach >1400 °C, required for a reasonable conversion.155 BASF has been developing the moving carbon bed reactor for methane pyrolysis since 2013, as discussed further in Section 6.137,156 The moving bed reactor can also incorporate catalyst particles (for example Ni catalysts) equipped with an additional regeneration reactor.157 Further research is needed to investigate process designs for utilizing the energy from the hot gas exiting the reactor to support heat integration processes and hydrogen purification.
3.2. Non-catalytic methane pyrolysis
Non-catalytic methane pyrolysis requires temperatures exceeding 927 °C due to the strong C–H bond,158,159 and typically leads to multiple reactions based on temperature and residence time, producing H2, C2H2, C2H4, C2H6, C4H6, C6H6, tar, and polycyclic aromatic hydrocarbons (PAHs).160–163 Most studies propose that the non-catalytic reaction involves a free radical scheme with the RLS being the dissociation of methane into a methyl radical and a hydrogen atom.160 Chen et al. proposed that C–H bond cleavage generates methyl radicals, which react with intermediate hydrocarbons to release hydrogen.164 The CH3 radical plays a central role in this mechanism, dimerizing to form C2H6, the primary product of methane pyrolysis. Key intermediate species include ethane, ethylene, acetylene, and propylene. The summarized net reactions are as follows:161| 2CH4 → C2H6 + (H2) → C2H4 + (2H2) → C2H2 + (3H2) → 2C + 4H2 | (R4) |
Olsvik and Billaud provided the step reaction as illustrated in Fig. 15.
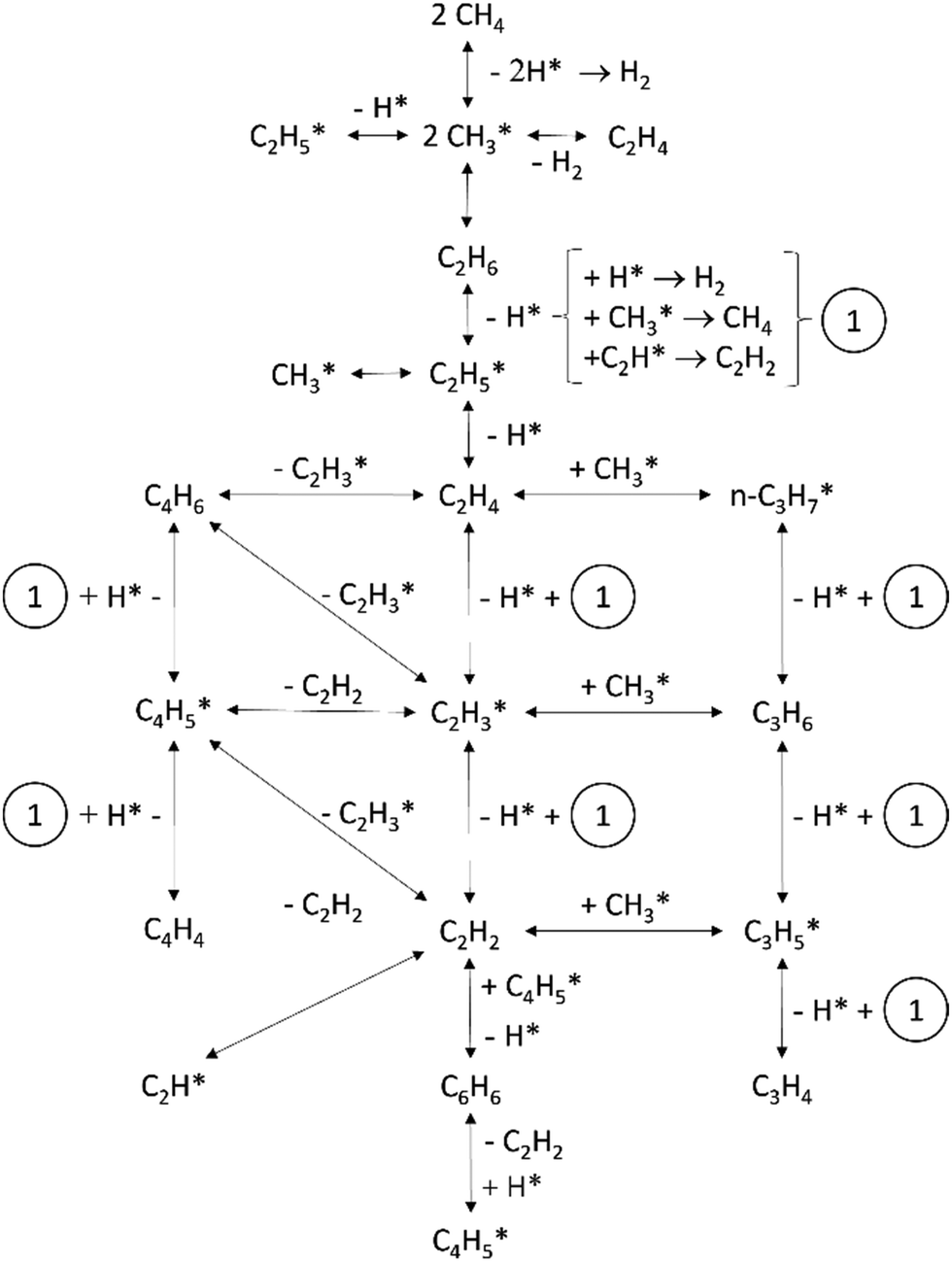 | ||
| Fig. 15 Proposed reaction series in non-catalytic methane pyrolysis. Adapted from ref. 160 with permission from Elsevier, copyright 1994. | ||
An alternate mechanism has been proposed to involve the dissociation of methane directly into methylene (CH2) radicals and H2 molecules, as suggested by Kassel.165,166 The CH2 radicals lead to methane conversion into ethane, which undergoes dehydrogenation to ethylene, then acetylene, and ultimately to hydrogen and carbon.167 The process is highly temperature-dependent, with CH4 dissociation to CH2, occurring at temperatures exceeding 1390 °C.165 In the following sections, we will summarize the two most common non-catalytic configurations i.e., microwave and plasma pyrolysis. It is important to mention that while the focus of this section is primarily on non-catalytic pyrolysis, the catalytic variations reported for these configurations are also described briefly.
For methane pyrolysis, the non-polar nature of CH4 requires a highly efficient microwave-absorbing material to utilize radiation energy for breaking C–H bonds. As illustrated in Fig. 16, the process consists of a microwave source, a packed-bed or fluidized-bed reactor, and solid materials acting as wave receptors to achieve high temperatures and facilitate methane decomposition. A magnetron is used to generate microwave radiation when a large potential difference is applied to the anode and cathode.
 | ||
| Fig. 16 Microwave driven methane decomposition, Reproduced from ref. 173 with permission from Elsevier, copyright 2023. | ||
Various catalytic and non-catalytic materials such as Ni, Pd, Cu, CNT's, and carbon nanofibers have been used to absorb and transfer the microwave radiation energy (Table 5).173,174 The selection of materials depends on their microwave absorption efficiency and structural stability under radiation.175 Metals due to their free electrons can be heated by induction heating from the microwave's magnetic field and have been demonstrated for production of value-added carbon products. For instance, Galanov et al. investigated catalytic microwave methane pyrolysis with metal catalysts such as Ni, Fe, Mo, TiNi, and AlNi,176 finding that Mo and Ni catalysts exhibited highest methane conversion (77.6 and 79.5%), and led to the formation of single-walled and multi-walled carbon nanotubes. Similarly, Fidalgo et al. demonstrated that metal catalysts such as Fe and Ni on carbon support can act as nucleation sites leading to the growth of carbon nanofilaments during the microwave-assisted methane pyrolysis reaction,177 a phenomenon not observed with conventional heating in an electric furnace.
| Solid material | Flow mL min−1 (GHSV 1/h) | Temperature (°C) | Pressure (bar) | Power | H2 selectivity | CH4 conversion | Ref. |
|---|---|---|---|---|---|---|---|
| 10Ni–1Pd (wt%) supported on CNT | 30 (30% CH4, 70% N2) | 550 | 1.01 | 5.85–6.65 GHz | — | 68 | 174 and 178 |
| 10Ni–1Cu/CNT | 30 (30% CH4, 70% N2) | 550 | — | 5.85–6.65 GHz | — | 37 | 174 and 178 |
| Granular | — | 1216 | — | 600 W, 2.45 GHz | 98% | 90 | 173 |
| Ni | 94% CH4, 6% C2 | — | — | 2.45 GHz | 84.9% | 79.8% | 176 |
In addition to metals, carbon-based materials such as activated carbon, graphite, and CNTs possess free electrons and are highly conductive which can be used as carrier energy to receive microwave energy, heat up, and transfer the heat to CH4 molecules.179 Jiang et al. explored a microwave-assisted process with CNT-supported Ni–Pd and Ni–Cu catalysts.174 Compared to conventional heating, these catalysts heated more efficiently in microwave pyrolysis, reducing the apparent activation energy from 45.5 kJ mol−1 (conventional convective heating) to 24.8 kJ mol−1. This efficiency is attributed to the unique electrical properties of CNTs, derived from their sp2 carbon atom network.
Carbon-based materials are often used both as microwave receptors and as catalysts. Fidalgo et al. studied the dual role of activate carbon in microwave-assisted methane pyrolysis, observing increased methane conversion with higher N2 content in the feed gas, achieving 100% conversion.180 This was assigned to the role of N2 induced energetic micro-plasmas, which are high-energy, short-lived sparks (<1 micron in size) that improve methane conversion.126,181–183
Deng et al. used microwave-sensitive H–(Fe)ZSM-5 catalysts, achieving a significant conversion increase (3% in thermally-heated fixed-bed vs. 40% under microwave conditions), attributing this to micro-plasmas.182 Dominguez et al.183 reported 80% conversion after 10 minutes with activated carbon under microwave heating, compared to 50% with conventional heating at 800 °C, with carbon nanofibers forming only under microwave conditions, suggesting that “micro-plasmas” may play a decisive role in their formation.
Recently, promising results for microwave-assisted methane pyrolysis were reported by Dadsetan et al., using activated carbon as initial seed particles in a fluidized bed reactor.173 The reactor was operated in a bubbling fluidization regime to optimize the residence time and reduce reactor's blockage risk. The most interesting aspect was that pyrolytic carbon formed during the reaction absorbed microwave energy, enabling continuous operation without adding new catalysts. They reported an activation energy of 224 kJ mol−1, 90% methane conversion at 1216 °C, hydrogen selectivity >95%, and over 500 hours of operation without reactor blockage.173 The vertically oriented reactor allowed carbon particles to be collected, ground, and recycled back into the process or stored.
Microwave pyrolysis performance is strongly influenced by operational parameters such as pressure, feed composition, temperature, power, and frequency. Importantly, conversion does not increase linearly with microwave energy input, highlighting the need to optimize power for maximum energy efficiency in hydrogen production. Jiang et al. observed that increasing microwave power by 11% resulted in a 37% increase in methane conversion.174 Microwave methane pyrolysis holds promise due to the low energy consumption (∼8.7 kW h kgH2−1 with 47.6% LHV efficiency), safe, easy, and controllable operation.174 However, the technology has only been conducted in laboratory setups and scaling up remains a significant challenge.173
000 tonnes of carbon black per year.184
Plasma, an ionized state of matter, comprises of reactive components like electrons, ions, radicals, and other excited species. Energy transfer in plasma occurs as accelerated electrons in an electric field transfer energy to heavier particles. Key advantages of the plasma process include direct transfer of energy to the process gas and the ability to achieve high process temperatures unattainable by other methods. These features significantly help accelerate chemical reactions that might otherwise face thermodynamic limitations. Like other non-catalytic pyrolysis methods, plasma pyrolysis can also lead to a wide range of intermediates such as C2H6, C2H4, and C2H2. For a detailed description of plasma chemistry, kinetics, thermodynamics, and electrodynamics, refer to Prof. Alexander Fridman's book, which extensively covers plasma technologies.167
In this review paper, we summarize the development of plasma pyrolysis processes aimed at hydrogen production. For a detailed examination of plasma pyrolysis technologies covering studies aimed at acetylene and higher molecular weight hydrocarbon production, readers are directed to other review papers.185,186 The reported methane decomposition activity via the conversion, hydrogen yield, and energy yield using various plasma technologies is summarized in Table 6.
| Discharge technology | Feed flow (L min−1) | CH4 concentration in feed (%) | Conversion (%) | H2 yield (selectivity) | Energy yield (kW h kgH2−1) | Ref. |
|---|---|---|---|---|---|---|
| a Energy efficiency = HHV of produced H2/(HHV of CH4 + power) | ||||||
| Gliding arc | 50 × 10−3 to 150 × 10−3 | 50 | 21–60 | 19–57 (∼96) | ∼37–100 | 187 |
| Gliding arc | 6 | 9 | 91.8 | 74 (80.7) | ∼100 | 188 |
| Gliding arc | 10 | 50 | 50 | 43 (86) | 36 | 189 |
| Gliding arc | 60 | 1–24 | 20–34 | 15–27 (73–80) | 55–444 | 190 |
| GA with CH4 + Ar, He, CO2 and N2 | 1 | 50–100 | 40–60 | 18–37 (40–62) | 139–322 | 191 |
| Thermal | — | 12.5 | 99.5 | 96 (96) | ∼86 | 184 |
| Thermal with quenching gas | 50–80 (quenching from 100–150) | 100 | 67–96 | 50–72 (71–75) | ∼37–45 | 192 |
| Thermal steam | 100–300 | 100 | 75–88 | — (—) | ∼30–63 (2.5–5.2 kW h m−3 H2) | 193 |
| MW (at 3 kPa) | 98 × 10−3 | 100 | 82 | 57 (70) | 93 | 194 |
| MW (at 1 atm with CH4–H2) | 400 | 20 | 82 | 58 (71) | 657 | 195 |
| MW with H2–CH4 feed at 5.5–11 kPa | 6 | 50–100 | 31–49 | 18–39 (59–79) | 155–211 | 196 |
| MW with H2–CH4 feed at 11 kPa | 60 | 1–34 | 19–64 | 16–47 (74–87) | 83–611 | 190 |
| MW with H2–CH4 feed at 5.5 kPa | 100 × 10−3 | 100 | 19 | 7 (35) | 666 | 197 |
| MW with H2–CH4 feed | 30 | 5 (H2 = 0.0–20%) | 72–95 | 47–50 (52–65) | — | 198 |
| Nanosecond pulse DBD with 5–25 cm outer electrode | 110 × 10−3 | 9 | 23–87 | — (18–80% H2 in output gas) | 0.72–7.3% efficiencya | 199 |
Plasma can be classified based on the gas temperature. In thermal (equilibrium) plasma, the temperature of the excited electrons (Te) is the same as other heavy neutral particles (T0) ranging from 1000 K to 10000 K.186,200 In non-thermal (cold) plasma, gas temperatures (T0) are in the range of 300–500 K (T0 ≪ Te). Finally, warm plasma refers to processes with high gas temperatures (1000–7000 K), but with a significant degree of non-equilibrium (T0 < Te). In addition to gas temperature, the plasma process can be further classified based on the type of discharge (Fig. 17), such as arc plasma, dielectric barrier (DBD), corona, glow, spark, microwave plasma, or gliding arc discharge.185,186
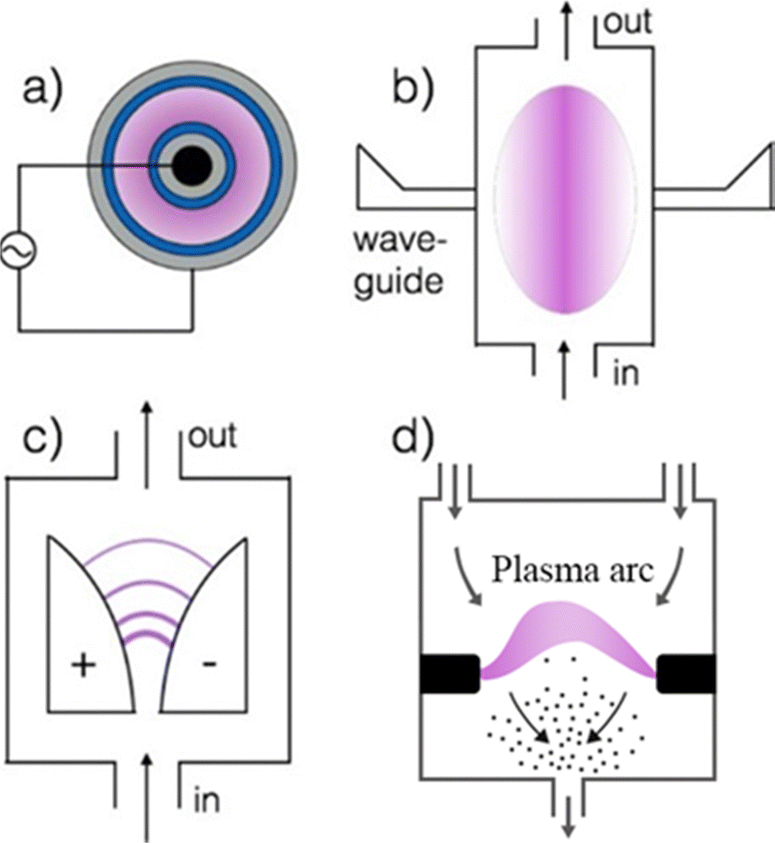 | ||
| Fig. 17 Commonly used plasma reactors for gas conversion: (a) dielectric barrier (grey = inner and outer electrodes, blue = dielectric surfaces), (b) microwave plasma, (c) gliding Arc discharge in standard configuration, and (d) arc plasma, Reproduced from ref. 201 with permission from the American Chemical Society (open access), copyright 2018. | ||
The thermal plasma method for methane pyrolysis was initially developed for conversion of methane to acetylene or ethylene with hydrogen as a by-product. The maximum theoretical yield for acetylene occurs at temperatures between 1427–1727 °C (Fig. 18). To enhance selectivity for acetylene or ethylene, quenching is used to prevent further dehydrogenation and carbon black formation, a kinetically limited process. Huels and Dupont have demonstrated a methane thermal plasma process for higher hydrocarbon production on an industrial scale.202,203 The Huels process uses electrical energy to reform methane-hydrogen mixtures, followed by water spray quenching to cool the product, with reported acetylene yields of 50% and conversion efficiency of 70%.204 The Dupont process, similar to Huels, featuring a magnetically rotated discharge, improved yields to 65–70%.205
 | ||
| Fig. 18 Equilibrium diagram for decomposition of 1 mol of methane versus temperature including carbon, Reproduced from ref. 206 with permission from the American Chemical Society (open access), copyright 2002. | ||
In the last few years, there has been a shift in the use of thermal plasma for methane pyrolysis to focus on hydrogen production.184,192 Research has primarily aimed at optimizing operating temperatures to balance energy efficiency and hydrogen yield. Arc plasma, powered by AC or DC, is commonly used with plasma gases like Ar or N2 for stable operation. Fulcheri and collaborators, in partnership with Monolith Materials, have developed arc plasma reactors.184,207–209 In a recently published study, they demonstrated a plasma reactor with a 3-phase AC power supply, operating at 1727 °C, achieving nearly complete methane conversion (>99%) and hydrogen yields of 96–99%. Monolith Materials Co. utilizes H2 as plasma gas to heat up the CH4 to produce carbon black. The commercial methane pyrolysis units will be discussed in Section 6. Kim et al. optimized a hybrid direct current (DC)-radio frequency (RF) thermal plasma reactor for hydrogen production.210 Their results showed methane decomposition begins above 327 °C, with stable hydrogen and solid carbon production occurring between 1227–2227 °C. At higher temperatures, hydrogen molecules dissociate into atoms, increasing C2H2 selectivity.
As mentioned earlier, since the C2H2 decomposition to hydrogen and carbon is a kinetically limited process, the process is sensitive to the feed flow rate and the use of a quenching gas. The flow rate of CH4 is used as tool to optimize residence time and methane conversion. For instance, Fincke et al. demonstrated this using a combination of longer reactor length (12.7 cm to 61 cm) and a decrease in methane flow rate (121 L min−1 to 90 L min−1), resulting in a 6-fold increase in hydrogen and carbon yields (4.7% to 30%).211 Similarly, Lee et al. demonstrated that lowering methane flow rates from 80 L min−1 to 50 L min−1, led to an increase in methane conversion from 66% to 96%, but with limited impact on hydrogen selectivity.192 In contrast, increasing quenching gas (N2) flow rate from 100 L min−1 to 200 L min−1, had limited impact on either methane conversion or hydrogen selectivity due to the naturally high quenching rate in the triple DC thermal plasma reactor. However, faster quenching rates can enhance acetylene selectivity by stabilizing intermediate radicals. Li et al. demonstrated that increasing the quenching gas flow rate from 0 to 20 L min−1 raised acetylene selectivity from 48% to 75% while reducing hydrogen selectivity from 24% to 12%.212
Different feed compositions impact hydrogen selectivity and conversion in thermal plasma systems. Li et al. showed that pre-mixing methane and argon before fed into a plasma torch is more effective with higher methane conversion rates of ∼95–99%, and a maximum acetylene selectivity of ∼79%, compared to injecting methane after the torch outlet (conversion: 60–75%; maximum acetylene selectivity: 70%).212 This was due to the higher temperatures and longer residence times achieved with the pre-mixing strategy. However, hydrogen selectivity was not reported for the pre-mixed case, suggesting negligible hydrogen production.
Cold plasma has also been used for methane pyrolysis and is commonly demonstrated in a DBD plasma reactor. A DBD reactor consists of two electrodes separated by a dielectric barrier, typically in a cylindrical configuration, operating at low power (watts) and ambient pressure. However, unlike other pyrolysis methods, cold plasma focuses on methane coupling to produce C2+ compounds rather than hydrogen. Xu et al. demonstrated a cylindrical DBD reactor for methane conversion, achieving ∼25% methane conversion, ∼36% hydrogen selectivity, and ∼42% C2H6 selectivity.197 Both methane conversion and hydrogen selectivity increased as a function of residence time and discharge power but came at a cost of reduced energy efficiency. The effectiveness of DBD cold plasma reactors for methane coupling was also demonstrated by Wang et al., reporting 25% methane conversion and 80% total C2 + C3 selectivity.213
To improve low conversion rates in DBD reactors, packing materials and catalysts have been employed. The low gas temperature of the cold plasma in DBD reactors allows the use of catalysts without sintering. Kim et al. used alumina in the packing bed of a cylindrical DBD reactor operating at atmospheric pressure, to increase non-catalytic methane conversion rates from 20% to 60% and increase selectivity of C2 products such as ethylene and acetylene. The enhancement was attributed to enhanced electric fields with the use of dielectric alumina. Alumina has also been used as a support for catalysts such as Pt,214 Cu/Zn,215 Pd,216 and Ni217 for cold plasma pyrolysis in DBD reactors. However, all of these studies primarily focused on increasing selectivity for C2 and C3 products via methane coupling.
In addition to thermal and cold plasma technologies, there have been efforts to conduct methane pyrolysis in a warm plasma with gas temperatures ranging in thousands of Kelvins, but with a significant degree of non-equilibrium. The two common discharges are based on microwave plasma (MP) and gliding-arc plasma (GAP). In an MP reactor, the plasma is generated by supplying energy using waveguides, typically at 2.45 GHz or 915 MHz. Studies by Onoe et al.,218 Heintze et al.219 and Minea et al.220 on MP reactors, have shown that adjusting input power and pressure can control reaction temperature and influence the product distribution in methane pyrolysis. At low pressure and power, conversion rates are lower, with high selectivity for C2 products. Higher pressures and power create a more constricted plasma discharge, resembling thermal plasma, and increase hydrogen and carbon selectivity. Cho et al. demonstrated this effect, achieving high CH4 conversion (>99%) and H2 selectivity (60–85%) with input powers of 1–5 kW compared to previous studies operating at lower wattages.221
Jasiński et al. also demonstrated the application of a microwave plasma at operating powers of 1.5–5 kW for high-efficiency hydrogen production from methane pyrolysis.222,223 Using pure CH4 in a swirl inlet feed, they achieved ∼99% conversion, ∼100% H2 selectivity, and an energy yield of ∼381 gH2 kW h−1. The swirl flow pattern shielded the reactor tube from the plasma's hot core and increased methane residence time in high-temperature regions, enhancing performance compared to CH4 mixed with N2, which resulted in ∼13.2% conversion, ∼96.8% H2 selectivity, and ∼74 gH2 kW h−1. The energetic mass yield of 381 gH2 kW h−1 surpassed other methods, such as gliding arc methane pyrolysis (40 gH2 kW h−1) and water electrolysis (21 gH2 kW h−1).222 In addition to optimizing hydrogen production yields, Tsai et al.224 and Singh et al.225 also demonstrated the use of MP reactors for obtaining high-value carbon with graphite-rhombohedral and graphene structures, respectively.
In addition to the MP reactor, the GAP reactor has also widely been studied for methane pyrolysis in a warm plasma. In the conventional GAP reactor, a plasma discharge is initiated at the narrowest gap between the ‘knife’ shaped electrodes, and it glides up until quenched by the increasing electrode distance. The concept of the GAP reactor is credited to A. Czernichowski, who demonstrated GAP reactors operating from 0.16 atm to 1 atm with input power up to 2 kW.226 Methane conversion of up to 34% was achieved with acetylene being the major product (selectivity: 70–90%). Indarto et al. demonstrated the positive impact of adding Argon or Helium to the methane feed gas in a GAP reactor.191 For pure CH4, a maximum conversion of 45% and H2 selectivity of 40% was achieved. With Ar or He addition at a constant total gas flow rate, methane conversion and H2 selectivity increased to 60% and 70%, respectively. This was attributed to the existence of meta-stable argon or helium in the plasma system which possibly increases the number of energetic unstable species available to react with methane.
Like thermal arc plasma reactors, the feed composition also impacts methane conversion in GAP reactors. Zhou et al. used H2/CH4 mixtures as feed gas and were able to increase methane conversion from 25% to 40% when the H2/CH4 ratio was varied from 1.5 to 7.227 However, the introduction of H2 did not impact product distribution, maintaining C2H2 selectivity >90%. Improved hydrogen selectivity using GAP reactors was demonstrated by Zhang et al. using a mixture of CH4/N2 as feed gas in a rotating gliding arc plasma reactor.188 The highest methane conversion (∼90%) and H2 selectivity (∼80%) were achieved for a CH4/N2 molar ratio of 0.1. At higher ratios, both methane conversion and H2 selectivity decreased, attributed to a reduction in gas temperature.
In summary, plasma reactors for methane pyrolysis yield diverse results depending on process conditions such as gas additions, feed flow rates, input power, and temperature. Interest in plasma pyrolysis has surged due to CO2 concerns and growing hydrogen demand, a trend likely to continue with industrial shifts toward hydrogen. Based on current published research, thermal plasma technologies look most promising for hydrogen production with the best hydrogen yields. However, the energy and costs of hydrogen production via this process will eventually dictate its viability. The reported costs are detailed in Section 5, and industrial advancements are summarized in Section 6.
3.3. Hybrid molten metal/metal-salt methane pyrolysis
Molten metals (Pb, Sb, Bi), molten metal alloys (Ni–Bi, Cu–Bi), and molten salts (KBr, NaBr, NaCl, MnCl2, KCl) are gaining interest for methane pyrolysis.228 The insolubility of carbon particles in molten metal and their lower density allows for easier separation as they float on top. As shown in Fig. 19, in this configuration the natural gas feed is supplied to a reactor operating at 900–1200 °C and containing molten metal media. As the bubbles rise in the reactor, catalytic and non-catalytic methane pyrolysis reactions can occur on the surface and within the bubbles.229,230 These molten metal systems can be designed in either a bubble column or plug reactor configuration with molten metal recirculation pumps to centralize the heat source, improving efficiency.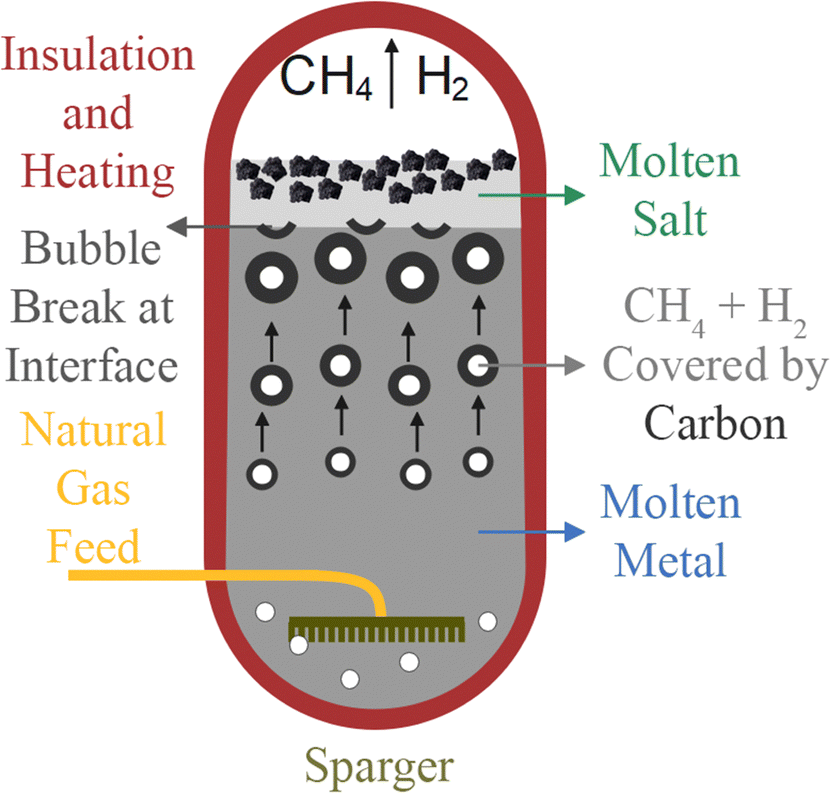 | ||
| Fig. 19 The schematic diagram of the bubble reactor for methane decomposition. | ||
According to several experimental and theoretical studies, the reaction in bubble reactors comprises (1) non-catalytic CH4 conversion due to the high temperature proceeding inside the gas bubbles, and (2) catalytic reaction taking place in the contact area (bubble surface) between the gas and liquid phases.231
The performance of bubble reactors depends on factors like the size of bubbles (Db), gas holdup (α), superficial gas velocity (v), surface tension (σ), viscosity (μ), density (ρ), and reactor design playing pivotal roles.232,233 Based on the literature the orifice size, velocity, and surface tension of liquid have a direct correlation with the initial bubble diameter while the density of molten media has an inverse effect on the initial bubble diameter. A larger bubble diameter will reduce the gas–liquid contact area, lowering catalytic conversion, as shown in Fig. 20.
 | ||
| Fig. 20 The effect of bubbles’ surface area on methane conversion in gas and surface phases reaction, Reproduced from ref. 234 with permission from the Elsevier, copyright 2021. | ||
Studies on bubble size after the orifice and along the reactor length highlight its importance.235–241 Serban et al. found that smaller feed tubes (1/16 vs. 1/4 in) reduced bubble size 10-fold, increasing methane conversion 5-fold due to better heat transfer with molten Sn.242 Mott metal spargers outperform feed tubes, achieving methane conversion near equilibrium limits (87% at 750 °C).
Tate's law (eqn (3)) studied in 1864, states that bubble diameter depends on the orifice diameter and liquid properties.243 Akita and Yoshida (1974) identified orifice size and gas velocity as the key factors influencing bubble size (eqn (4)).238 Mori in 1979 (eqn (5)), expanded on this, incorporating molten medium properties, orifice diameter, and gas velocity, which has been widely used in subsequent studies.244,245 The gas velocity not only impacts the bubble size, but also gas–liquid contact time and exposure to high temperatures. Geißler et al. showed that increasing gas velocity from 50 mL min−1 to 150 mL min−1 reduced methane conversion from 32% to 21% at 1000 °C.246
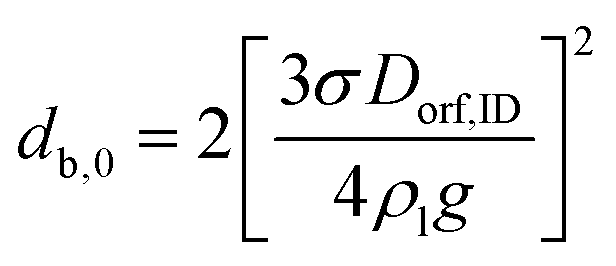 | (3) |
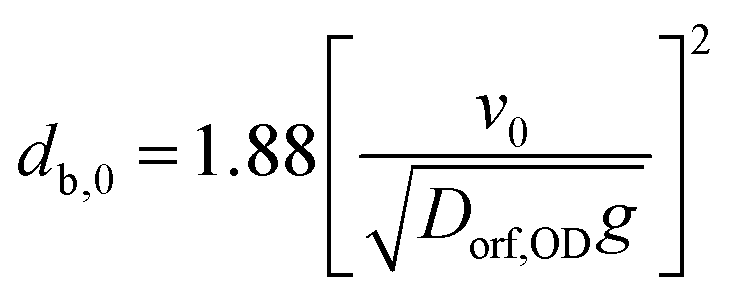 | (4) |
 | (5) |
The variables Dorf, Qg, v0, and g in eqn (3)–(5), represent orifice diameter, gas flow rate, gas velocity, and gravitation acceleration. The OD, ID, and l subscripts correspond to outer diameter, inner diameter, and liquid. Another key factor is gas holdup which can be defined as the volume fraction of gas in the total volume of the gas–liquid phase.247 A smaller gas hold-up requires a larger reactor volume given the same hydrogen production rate, methane conversion, and reaction temperature.
An equation developed by Kataoka and Ishii estimates that gas holdup is directly related to gas velocity, gas density, and liquid viscosity, while indirectly associated with surface tension and liquid density.248 The gas holdup throughout the reactor will be proportionate to the CH4 conversion efficiency due to mole creation. Von Wald et al.57 suggested limiting gas holdup at reactor entrance to 12.5–25.0% of value at the reactor exit, even with 100% CH4-to-H2 conversion.
The catalytic activity of molten metals plays a key role in CH4 decomposition, with performance summarized in Table 7. Upham et al. explored methane pyrolysis by flowing a CH4–Ar mixture (2.25 sccm Ar, 0.25 sccm CH4) over 38.5 mm2 of different molten metals.249 As exhibited in Fig. 21, each pure metal or alloy exhibits different hydrogen production activity.230,249–251
| Molten components | Feed flow rate (sccm), CH4 concentration (%) | Temperature (°C), pressure (bar) | Reactor diameter (mm), length (mm), volume (mL) | Residence time (s) | Conversion (%) | Activation energy (kJ mol−1) (pre-exponential factor) | Ref. |
|---|---|---|---|---|---|---|---|
| Ni0.27–Bi0.73 | 10, 80 | 1000, 2 | 12, 80, 9.04 | — | 15 | 208 (7.88 × 106 cm s−1) | 249 |
| Ni0.27–Bi0.73 | 10, 80 | 1065, 2 | 30, 1.1 (m), 777.15 | — | 95 | 208 (7.88 × 106 cm s−1) | 249 |
| NaKCl | 20, 100 | 1000, 1 | 25, 115, 56.42 | 0.5 | 4.5 | 301 (6.5 × 1010 s−1) | 55 |
| Fe(3%)/NaKCl | 20, 100 | 1000, 1 | 25, 115, 56.42 | 0.5 | 9.5 | 171 (5.7 × 105 s−1) | 55 |
| NaCl–KCl | 100, 100 | 900, — | 27, —, — | — | 12.6 | — | 252 |
| 2 g Ni foam/150 g NaCl–KCl | 100, 100 | 900, — | 27, —, — | — | 14.0 | — | 252 |
| 10 g Ni foam/150 g NaCl–KCl | 100, 100 | 900, — | 27, —, — | — | 18.0 | — | 252 |
| 20 g Ni foam/150 g NaCl–KCl | 100, 100 | 900, — | 27, —, — | — | 16.5 | — | 252 |
| Te | 10, 70 | 980, 1 | 12, 70, 7.91 | 0.5 | 20 | 166 (4.9 × 106 s−1) | 251 |
| Ni0.27–Te0.73 | 10, 70 | 980, 1 | 12, 70, 7.91 | 0.5 | 16 | 193 (1.8 × 107 s−1) | 251 |
| Ni0.5–Te0.5 | 10, 70 | 980, 1 | 12, 70, 7.91 | 0.5 | 10 | 223 (4.7 × 108 s−1) | 251 |
| Ni0.27–Bi0.73 | 10, 70 | 980, 1 | 12, 70, 7.91 | 0.5 | 4 | 208 (6.5 × 105 s−1) | 251 |
| NaCl | 10, 70 | 980, 1 | 12, 70, 7.91 | 0.5 | 1 | 374 (3.9 × 1012 s−1) | 251 |
| Ni0.27–Bi0.73/NaBr (660/260) | 10, 100 | 1000, — | 22, 920, 349.545 | 5.3 | 37.5 | — | 253 |
| Ni0.27–Bi0.73/NaBr (660/260) | 25, 100 | 1000, — | 22, 920, 349.54 | 5.3 | 25 | — | 253 |
| Ni0.27–Bi0.73/KBr (110/240) | 10, 70 | 1000, — | 22, 350, 132.98 | 1.8 | 15 | — | 253 |
| Ni0.27–Bi0.73/KBr | 10, 70 | 1000, — | 22, 350, 132.98 | 2.2 | 27 | — | 253 |
| MnCl2(67)–KCl(33) | 20, 50 | 1000 (1050) | 25, 115, 56.42 | 0.6 | 23 (42) | 161 (1.05 × 105 cm s−1) | 230 |
| MnCl2(50)–KCl(50) | 20, 50 | 1000 (1050) | 25, 115, 56.42 | 0.6 | 18 (35) | 153 (4.00 × 104 cm s−1) | 230 |
| KCl | 20, 50 | 1000 (1050) | 25, 115, 56.42 | 0.6 | 4 (13) | 300 (8.7 × 109 cm s−1) | 230 |
| MnCl2 | 20, 50 | 1000 (1050) | 25, 115, 56.42 | 0.6 | 19 (36) | 177 (6.3 × 105 cm s−1) | 230 |
| NaCl | 15, 100 | 1000 | 16, 180, 36.17 | 0.8 | 5.4 | 231 (2.2 × 108 s−1) | 254 |
| KCl | 15, 100 | 1000 | 16, 180, 36.17 | 0.8 | 5.2 | 236 (3.5 × 108 s−1) | 254 |
| NaBr | 15, 100 | 1000 | 16, 180, 36.17 | 0.8 | 4.3 | 277 (1.4 × 1010 s−1) | 254 |
| KBr | 15, 100 | 1000 | 16, 180, 36.17 | 0.8 | 6.2 | 233 (1.3 × 108 s−1) | 254 |
| GaInSn | 9, 100 | 1300 | — | 1.36 | 80 | — | 255 |
| Cu | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 33.5 | — | 245 |
| Bi | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 67.4 | — | 245 |
| Sn | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 45.4 | — | 245 |
| Cu0.6–Bi0.4 | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 51.3 | — | 245 |
| Cu0.2–Bi0.8 | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 68.4 | — | 245 |
| Cu0.2–Sn0.8 | 500, 100 | 1160, 1 | 65, 65, 215.58 | 26 | 41.2 | — | 245 |
| Cu0.975–Ga0.025 | 500, 100 | 1160, 1 | 65, 65, 215.58 | — | 37.22 | — | 245 |
| Sn | 200, 100 | 1000 (1175) | 45, 1050, 1669.1 | — | 21 (72) | — | 246 |
| Sn | 50, 100 | 1000 (1175) | 45, 1050, 1669.1 | 4.9–3.2 | 32 (77) | — | 246 |
| Sn | 2000, 100 | 1000 (1200) | 110, 140, 1329.8 | — | ∼0 (52) | — | 256 |
| Cu0.5–Bi0.5 | 10, 80 | 1000, 1 | 40.5, 150, | 0.7 | 56 | 222 (3.7 × 108 s−1) | 257 |
| Ga | 450, 50 | 1119, 1 | 36, 150, 150 | 0.5 | 91 | — | 258 |
| Ga | 450, 50 | 950, 1 | 36, 50, 50 | 0.2 | 74 | — | 258 |
| NaBr–KBr | 20, 100 | 850, | —, 120, — | 1 | <1 | 160 | 259 |
| NaBr–KBr/5 wt% MoS2 | 20, 100 | 850, | —, 120, — | 1 | 5.3 | 57 | 259 |
| NaBr–KBr/5 wt% Al2O3 | 20, 100 | 850, | —, 120, — | 1 | <1 | 124 | 259 |
| NaBr–KBr | 15, 100 | 1000, 1 | 26, 570, 302.47 | 2.5 | 18 | 240 (1.07 × 107 s−1) | 234 |
| 5 wt% γ-Al2O3/NaBr–KBr | 15, 100 | 1000, 1 | 26, 180, 95.52 | — | 10.1 | 128 (2.2 × 104 s−1) | 234 |
| NaBr–KBr | 15, 100 | 1000, 1 | 26, 180, 95.52 | — | 4.3 | 246 (4.38 × 108 s−1) | 234 |
| NiMo–Bi (2.3:1.3:96.4 wt%) |
4, 100 | 800, 2 | 8, 10, 0.5 | 7.8 | 9.9 | 81.2 | 260 |
| NiMo–Bi (2.3:1.3:96.4 wt%) |
4, 10 | 800, 2 | 8, 10, 0.5 | 7.8 | 25 | 81.2 | 260 |
| NiMo–Bi (2.3:1.3:96.4 wt%) |
4, 10 | 800, 2 | 8, 30, 1.5 | — | 45 | 81.2 | 260 |
| Non catalytic reaction | — | — | — | — | — | 423 (5.1 × 1014) | 261 |
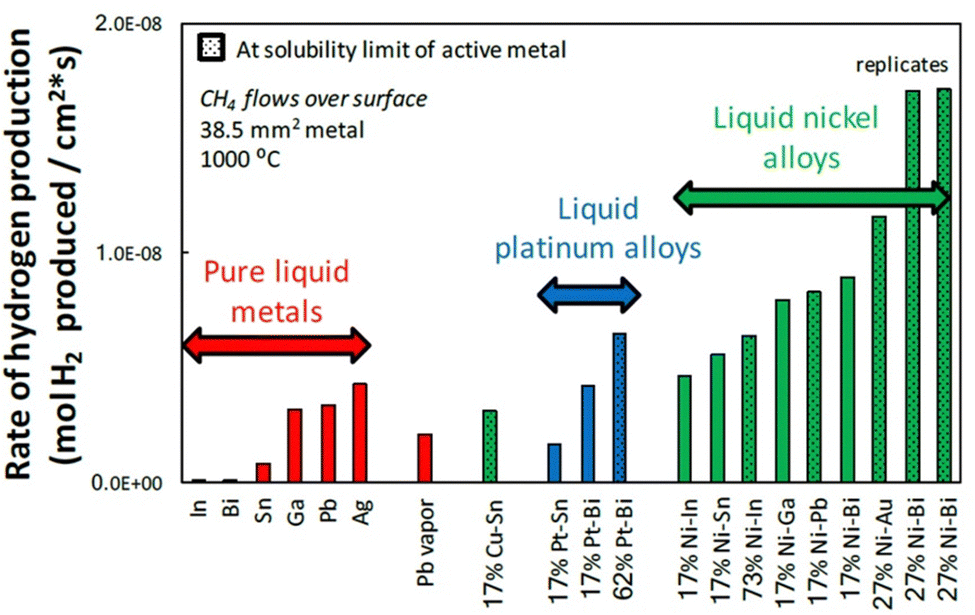 | ||
| Fig. 21 The activity of different pure metals and alloys for the methane pyrolysis process. Adapted from ref. 249 with permission from The American Association for the Advancement of Science, copyright 2017. | ||
Low-melting point (30 °C to 272 °C) metals show some activity, in the order In <Bi < Sn < Ga < Pb with activation energy only slightly lower than the non-catalytic route (422 kJ mol−1).262 Adding active elements such as Ni and Pt significantly improves the activity, proportional to the amount. The alloy composition determines the surface tension, bubble diameters, and catalytic activity. Interestingly, while Ni and Pt have approximately the same intrinsic activity, Ni shows a greater impact on hydrogen production activity in alloys. Cu has also been used for a similar purpose to enhance the activity of Bi, Ni, Sn, and Ga,245 with CuBi achieving the highest conversion (68.4%) at 1160 °C, followed by CuNi, CuSn, and CuGa.245
In a recent study, Chen et al. added Mo to the Ni–Bi alloy with a composition of Ni (2.3 wt%):Mo (1.3 wt%):Bi (96.4 wt%) and achieved a 37 times increase in hydrogen production rate versus Ni–Bi at 800 °C.260 However, the reaction was limited to 9.9% conversion under pure CH4 flow with 10 cm molten alloy height and 50% conversion under 10% CH4 flow with 50 cm reactor height. Different temperatures and molten alloy volumes should be investigated to optimize conversion under a high CH4 flow.
Several researchers have combined solid catalysts with molten media by preparing a slurry in either powder or foam forms. For example, Parkinson et al. demonstrated that the conversion in a 190 mm column of NBr–KBr at 1000 °C more than doubled when 0–5 wt% γ-Al2O3 was introduced and the activation energy decreased from 246 kJ mol−1 to 128 kJ mol−1.234 The decrease in activation energy was linked to the catalytic activity of γ-Al2O3 compared to NBr–KBr. Additionally, the increased slurry density and viscosity extended residence time, further improving conversion.
A promising development is the use of molten salts such as MnCl2, NaBr, KBr, KCl, and NaCl for molten metal pyrolysis.253,263 Their advantages include: (1) lower density and melting point, facilitating circulation, and (2) higher water solubility, aiding carbon separation. However, their high vapor pressure presents challenges, often restricting use to a second layer above molten metal. The elevated vapor pressure increases system operating pressure, and prolonged evaporation and condensation can block downstream equipment.254
Several studies have investigated the impact of molten salts on carbon purity. Using Ni–Bi alloy and molten salts (NaBr, KBr, KCl) for methane decomposition reactions, Rahimi et al. found that employing the second phase reduced metal contamination from 83 wt% to 5 wt%.253 The concentration of contaminants decreased with increasing height of the salt phase layer. In another research, Parkinson et al. assessed CH4 decomposition in molten salts of NaBr, NaCl, KBr, and KCl and applied deionized water washing.254 The CHNS analysis revealed that carbon purity ranged from 55.8 wt% to 92.5 wt% (91.7–97.4 at%), with NaCl yielding the highest purity, followed by NaBr, KCl, (Na, K)Br, and KBr.
Seo et al. investigated a two-layer reactor comprising molten metal (NiBi) and salt (NaCl) with a total length of 10 cm.264 As depicted in Fig. 22, metal/salt combination improved conversion compared to the pure counterpart, reducing liquid costs and highlighting the role of interface in methane conversion. The catalytic activity is primarily attributed to the metal layer, with a 9/1 metal/salt ratio yielding the highest conversion (31% at 1000 °C). The beneficial impact of the second layer is associated with its surface tension, following the order: KBr < KCl < NaBr < NaCl. Furthermore, carbon analysis confirmed higher purity with salt, alone or combined, versus metal.
 | ||
| Fig. 22 The influence of molten media on methane conversion, Reproduced from ref. 264 with permission from Elsevier, copyright 2023. | ||
The process of molten metal methane pyrolysis offers a favorable method for producing hydrogen, mainly because it simplifies the separation of carbon and avoids the problem of catalysts becoming inactive. However molten metals are corrosive and further research is required on reactor design, construction, and to prevent the loss of molten media via carbon product contaminated with liquid.
4. Carbon co-product
The mass ratio of solid carbon to hydrogen in methane pyrolysis is 3:1, making carbon co-product utilization crucial for commercial viability. The common carbon types produced are carbon black (CB),184,193,252 graphite,265–267 carbon nanotubes (CNTs), and carbon fibers, as illustrated in Fig. 23. Keipi et al. classified carbon products by operating temperatures:268 nanotubes at 600–1000 K, graphite-like carbons at 1000–1400 K, and carbon black at 1400–2000 K.
 | ||
| Fig. 23 Carbon products variety based on the methane pyrolysis method and condition, adapted from ref. 261 with permission from the Elsevier, copyright 2016. | ||
The non-catalytic plasma method269 is used for the carbon black (CB) production because of the high temperatures generated by the plasma.184,211 For non-catalytic methane pyrolysis, carbon growth happens through the small (0.1–1 nm) polycyclic aromatic hydrocarbons (PAHs), containing 10 to 18 rings, which grow by incorporating C2H2.270 These PAHs collide via van der Waals forces, forming clusters (1–10 nm) that merge into larger nanodroplets and eventually crystallize into nanoparticles (10–1000 nm). A range of factors, such as the conditions of temperature and pressure, the type of initial carbon sources, and the levels of hydrogen and oxygen, significantly affect the properties of the carbon black produced.
As summarized in Table 8, CB with an average price of $1 per kg is the cheapest industrially produced nanocarbon. In comparison, the price of industrial multi-walled nanotubes is in the range of $400–450 per kg and graphene nanoplatelets range from $450–15000 per kg.271,272 The global consumption of CB in 2019 was around 13.7 Mt, and currently, the major use of CB is as a pigment and reinforcing phase in rubber, plastics, and tire industries.273,274 CB is also used in SiC production, electronic devices, power cable shielding and thermal insulation. Emerging applications include environmental remediation and renewable energy harvesting, particularly for batteries, fuel cells, catalysis, solar cells, and as an adsorbent for purification systems.273 Estimates show an annual growth rate of 4.8% for the global carbon black market from 2023 to 2030, with an anticipated value of USD 31.04 billion by 2030.275
| Carbon types | Price ($ per kg) | Global market size (thousand metric tonnes per year) | Ref. |
|---|---|---|---|
| a Coated spherical graphite for anode in a lithium-ion (Li-ion) battery. b Industrial-grade multi-walled carbon nanotube. c Total graphite market size. | |||
| Carbon black | 0.75–1.10 | 14000–16400 |
276 and 277 |
| Natural graphitea | 2–11a | 1700c | 278–280 |
| Synthetic graphite | 7–20a | 2100 | 278, 280 and 281 |
| Petcoke | 3–25 | 460–3800 | 282 and 283 |
| Activated carbon | 1.25–2.01 | 5.8 | 281 and 284 |
| MWCNTb | 400–450 | <15 | 271 and 285 |
| Single NT | 77000–300000 |
<2 | 285 and 286 |
| Graphene nanoplatelets | 450–15000 |
<1 | 272 |
Graphitic carbon is also experiencing increasing market demand. Natural graphite trades at a price >8 $ per kg while the cost of battery-grade synthetic graphite is approximately 20 $ per kg.278 A two-fold increase in demand is expected by 2028 compared to 2018, driven by graphite demand for Li-ion anode battery applications.281 Currently, it is estimated that graphitic anodes in Li-ion batteries contribute 10–15% of the raw material cost.287 The rise in battery use through energy system electrification will further boost demand for graphitic carbon.
Molten metal pyrolysis is an attractive route for the production of graphitic carbon. Ji et al. evaluated the performance and capacity of various carbons derived from molten salt bubble columns in Li-ion and Na-ion cells.288 Salts containing FeCl3 and MnCl2 produced more graphitic carbons, as confirmed by Raman spectroscopy, and showed the highest capacities of 272 mA h g−1 (FeCl3) and 233 mA h g−1 (MnCl2) in Li-ion half cells. However, the impact of contamination by metals and salts on battery performance needs further investigation.
The microwave-assisted method (at medium temperature) is another way to produce pure graphite.173 Dadsetan et al. investigated this approach using a fluidized bed reactor filled with granular carbon.289 Semi-graphitic carbon was obtained that showed 75% initial coulombic efficiency and ∼200 mA h g−1 specific capacity. Beyond battery applications, these carbons can be used in steelmaking to replace metallurgical coke and enhance slag foaming in electric arc furnaces (EAFs).290 Additionally, further processing can prepare them for graphene production.291
Carbon nanotubes (CNTs) are a valuable by-product of methane pyrolysis with applications in water purification, reinforcement materials, nanoelectronic devices, gas storage, sensors, and biotechnology.292,293 CNTs are typically produced through catalytic routes using metals like Ni, Co, and Fe under conventional or microwave heating.172 Low-temperature conditions are required to produce CNT, so a catalyst is essential to increase conversion. Jiang et al. studied Ni–Pd and Ni–Cu catalysts supported over CNT at 550–600 °C, reporting simultaneous production of H2 and multi-walled carbon nanotubes (MWCNTs).174
Two primary mechanisms have been proposed to describe CNT formation over a catalyst, as depicted in Fig. 24.294–296 The metal–support interaction is the first effective parameter: (a) robust metal–support interaction results in base-growth mechanism, (b) and weaker interaction leads to tip-growth.297 In the base-growth mechanism, carbon is formed over catalyst particles, allowing regeneration.298 In the tip-growth scenario, the carbon formed on the surface diffuses towards the rear side, leading to the leaching of the catalyst particles.295 Lobo et al. linked the initial nucleation process to the growth mechanism,299 with tip growth occurring when CNT nucleation starts between the catalyst and support, and base growth when nucleation begins on unsupported particles.
 | ||
| Fig. 24 Two types of carbon nano tubes deposition mechanisms including (a) tip-growth mechanism and (b) base-growth mechanism, Reproduced from ref. 294 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Catalyst particle size also influences growth mechanism.300 Gohier et al.'s research on Co, Fe, and Ni indicated that particle diameters less than 5 nm resulted in base growth, while diameters >15 nm led to tip-growth.301 Temperature gradients from exothermic methane decomposition and endothermic carbon deposition promote carbon diffusion, and the high surface energy of metals may influence graphene plane deposition and upward lifting as more carbon deposits form.302
Various computational techniques, such as density functional theory, Monte Carlo, classical and quantum molecular dynamics simulations, and thermodynamics, have been employed to investigate these two growth modes.303–305 Despite progress, further research is necessary to gain a comprehensive model is needed to develop a comprehensive model. Describing previous studies in this manner is out of this paper's scope but Chen et al. recently reviewed these simulations and models thoroughly.306
In conclusion, carbon production from methane pyrolysis presents a compelling opportunity. The versatility of carbon products from carbon black for tires and printing inks to graphene for electronics and composites, highlights its potential across diverse industries. However, carbon production from large-scale methane pyrolysis is unlikely to be accommodated by the current market. In contrast, small-scale methane pyrolysis offers a more practical alternative, aligning production with market capacities and regional needs. This decentralized approach enables local distribution of both hydrogen and carbon products, optimizes resource use, and mitigate the risk of excess carbon production, making methane pyrolysis a more sustainable and adaptable technology.
5. Technoeconomic assessment
The economic feasibility of any emerging hydrogen production technology is pivotal to its success. Consequently, techno-economic analyses (TEA) of various methane pyrolysis methods have garnered significant attention in recent literature. These studies delve into several key factors, such as capital expenditure (CAPEX) and operational expenditure (OPEX). These include reactor and equipment costs, natural gas prices, heating sources (e.g., electricity), carbon taxation, and the market value of solid carbon. These evaluations are essential for assessing the viability of methane pyrolysis processes on various scales. Since 2017, TEA for various methods, including catalytic process, molten media, plasma reactors, and thermal decomposition, have been conducted. The TEA results for different routes, adjusted to 2023 US$, are summarized in Table 9.| Process type | Production scale (tH2 per year) | CAPEX (M$) | Annualized CAPEX (M$ per year) | OPEX (M$ per year) | LCOH ($ per kgH2) | Ref. |
|---|---|---|---|---|---|---|
| Ni catalyst | — | 33.6 | 50.6 | 144.9 | — | 307 |
| Solid catalysts | 71000 |
105.9 | 9.4 | 136.1 | 2.4–3.2 | 277 |
| Ni catalyst | 4928–50229 |
36.9–201.3 | 4.3–23.6 | — | — | 308 |
| Carbon catalyst | 32809 |
— | 17.8 | 89.9 | — | 136 |
| Thermal | 140729 |
462.0 | 61.8 | 241 | 1.64 | 309 |
| Thermal | 787 | 5.9 | 0.9 | 1.1 | 3.52 | 310 |
| Thermal | — | 321.7 | 47.9 | 143.6 | — | 307 |
| Ni–Bi MM | 24530 |
142.9 | 12.7 | — | 2.76 | 233 |
| K–Cl MM | 23652 |
421.5 | 32.4 | 53.4 | — | 311 |
| Ni–Bi MM | 10400 |
33.1 | 5.6 | 20.1 | 2.00 | 57 |
| Ni–Bi MM | 100000 |
461.6 | 50.8 | 161.0 | 1.76 | 312 |
| Ni–Bi MM | 200000 |
783.9 | 86.4 | 361.4 | 2.18 | 313 |
| Electron beam plasma | 72000 |
865.4 | 111.8 | 204.3 | 2.83 | 314 |
| Plasma | 699 | 14.1 | — | — | 7.00 | 315 |
| MM-plasma | 215 | 21.8 | 2.2 | 6.0 | 1.30 | 316 |
Recent TEA studies on methane pyrolysis have focused on the catalytic route, emphasizing the challenges of coke formation, which deactivates catalysts, increases pressure drop, and causes reactor blockages. To address this, two approaches are proposed: burning coke to regenerate catalysts and produce heat, as discussed by Muradov,104 though this generates GHG emissions, or continuously feeding fresh catalysts while disposing of deactivated ones.277,307 Lane et al. also proposed a process involving partial oxidation to generate the required reaction heat.308 These studies utilized a fluidized bed reactor to design the catalytic methane pyrolysis process.
The average annualized CAPEX per kilogram of hydrogen produced, calculated using eqn (6) and eqn (7), (where i is discount rate and n is lifetime), ranges from 0.13–0.88 $ per kgH2.136,277,308 The lowest CAPEX is associated with the highest production rate (71 kt per year), benefiting from economies of scale. While the CAPEX is relatively small and comparable to natural gas reforming (0.5–0.7 $ per kgH2), the cost associated with catalyst replacement and feedstock for methane pyrolysis is significant. Fig. 25 shows cost breakdown compiled from different studies, with methane feedstock contribution at 60–66%, catalyst costs at 24–25%, and utilities at only 5–6%. The overall cost of hydrogen is reported to be between 2.3–3.4 $ per kgH2.277,317
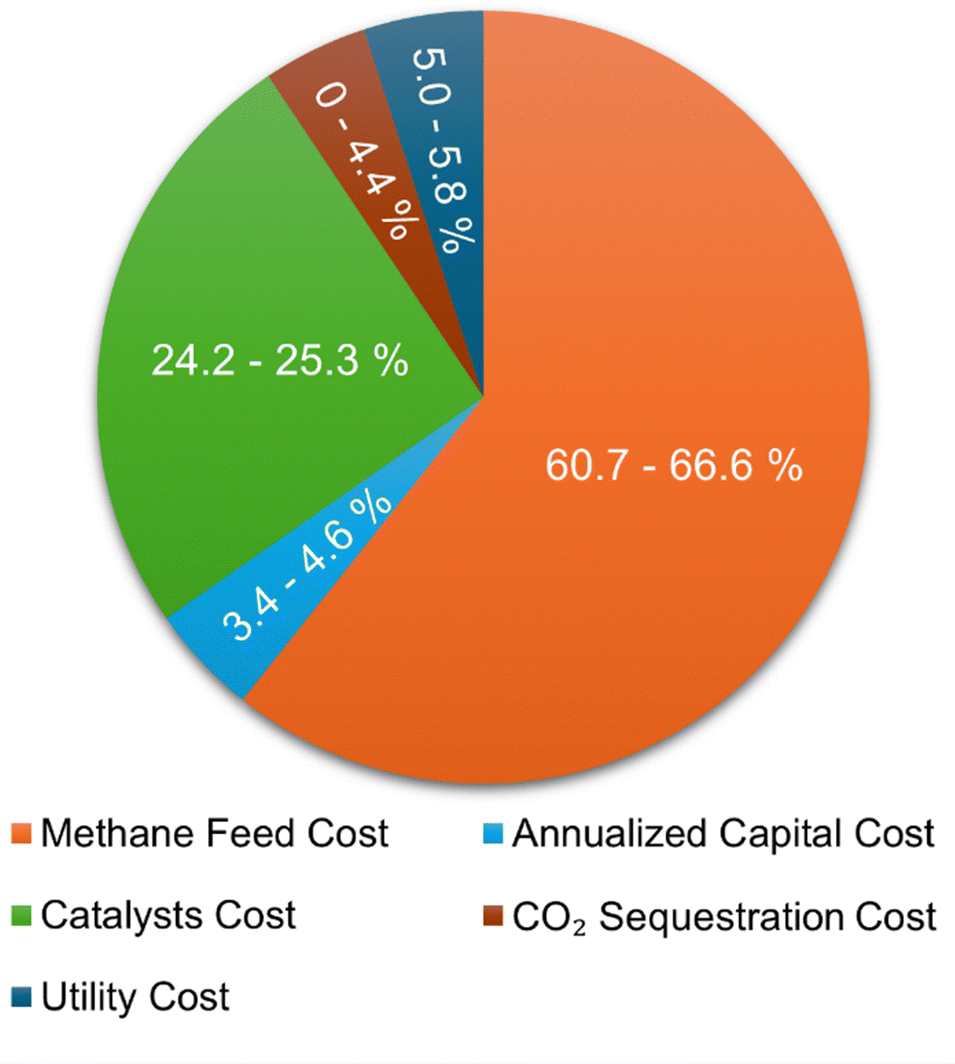 | ||
| Fig. 25 Breakdown of the cost contribution for catalytic methane pyrolysis, reproduced from ref. 277 with permission from the Elsevier, copyright 2021. | ||
The importance of catalyst separation and regeneration was discussed in depth by Zhang et al. for a catalytic methane pyrolysis plant based on a fluidized bed reactor coupled with a hydrogen fuel cell to generate electricity.277 The levelized cost of electricity (LCOE) increases from 177 € per MW h (with catalyst reactivation and carbon byproduct sales) to 356 € per MW h (without catalyst recycling or carbon sales), highlighting importance off catalyst recycling for economic viability. However, a key research gap in published literature is related to detailed assessment of the impact of catalyst performance on the CAPEX and OPEX of the methane pyrolysis process. Finally, future TEA studies could also look into the potential costs of integrating CO2 capture units into catalytic methane pyrolysis plants, particularly if methane is utilized to provide energy.
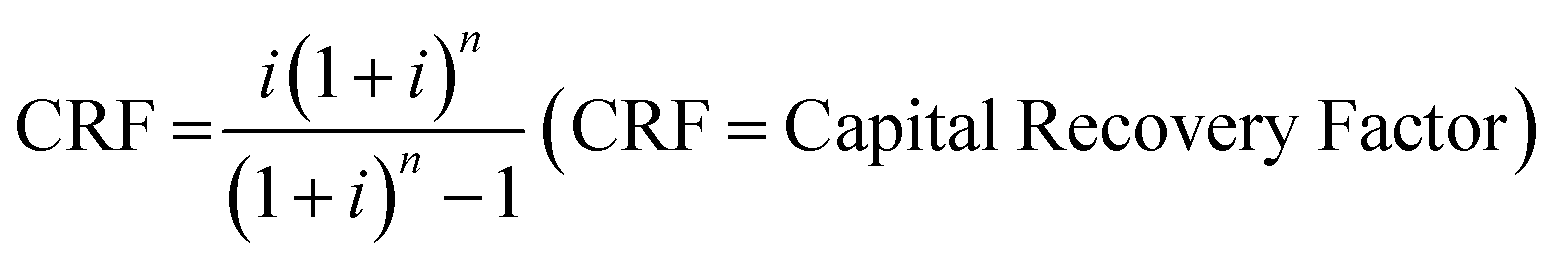 | (6) |
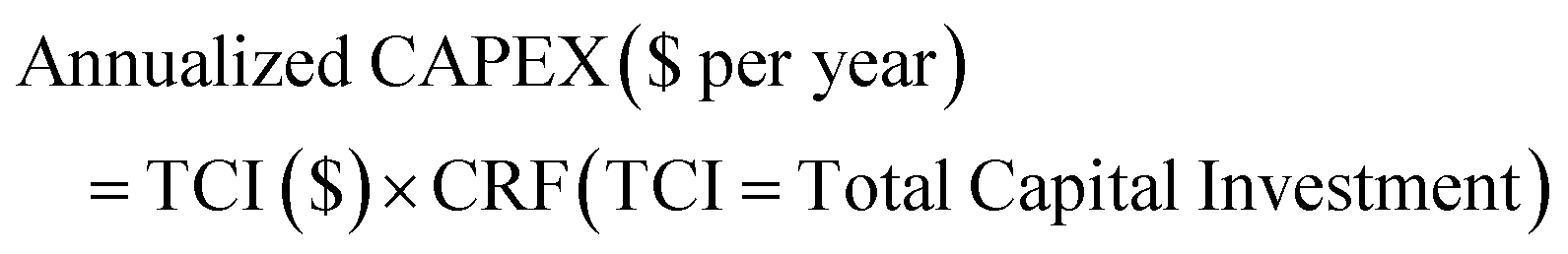 | (7) |
The TEA for thermal pyrolysis of methane at 1400–1500 °C has also been reported in a couple of studies. Keipi et al.310 incorporated a circulating bed reactor for small scale production of 0.8 ktH2 per year while Okeke et al.309 employed a furnace reactor with bottom combustion heat for large-scale production of 140 ktH2 per year. He annualized CAPEX was 0.44 $ per kgH2 for large-scale and 1.22 $ per kgH2 for small-scale production.309,310 While the reported CAPEX for thermal pyrolysis is higher than catalytic routes, the OPEX is lower at 1.45–1.71 $ per kgH2versus 1.92–2.3 $ per kgH2 due to the absence of catalyst regeneration or supply requirement.277,309,310 Okeke et al. also considered various design scenarios, such as air/oxygen-fired methane for a heat source or air/oxygen-fired carbon black as the heat source.309 Since the primary purpose of methane pyrolysis is utilizing NG in a low-emission route, this requires the use of CO2 capture and air separation units because when the carbon is combusted in the absence of a capture unit these methods release significant GHG's (11.2 kgCO2 kgH2−1), higher than those from SMR. The calculated after-tax minimum hydrogen selling price for the four scenarios was between 1.98 $ per kgH2 to 3.15 $ per kgH2, with the lowest cost for O2-fire carbon black with carbon capture.309 In contrast at smaller production scales, Keipi et al.310 estimated 3.52 $ per kgH2, reducible to 2.63 $ per kgH2 with the addition of a gasifier to produce more hydrogen from carbon byproducts.
TEA studies on bubble reactors show promise due to their ability to continuously remove solid carbon. The annualized CAPEX for the molten media method was reported to be 0.43–0.54 $ per kgH2 for production rates of 10–200 ktH2 per year, indicating a linear relationship between CAPEX and production rates.57,233,312,313 This contradicts industrial norms, where CAPEX share decreases with increased capacity, likely due to uncertainties in bubble reactor design and cost estimation.
Ni–Bi, identified as a high-activity molten media for methane pyrolysis,249 was commonly used in these studies, with OPEX ranging from 1.61–2.26 $ per kgH2. It was observed that the source of heat substantially influences the operational cost. For instance, Parkinson et al. demonstrated 2.18 and 1.76 $ per kgH2 for 200 and 100 ktH2 per year production capacity, respectively, since the latter design considered methane combustion instead of electricity for energy supply.312,313 The LCOH ranged from 1.83 to 2.76 $ per kgH2 while the lowest value was reported for the 100 ktH2 per year production rate with a $0.15 per kg carbon sale.57 Molten media type and metal loss rates are critical to process success. Von Wald et al. showed 0.0001 wt% Ni–Bi contamination in solid carbon raised hydrogen costs by 334%. Angikath et al. demonstrated that using Ga and KCl–MnCl2 can reduce reactor costs by 61.3% and 23.8%, respectively, compared to Ni–Bi reactors.233 Future studies should explore alternative media, energy supply scenarios, and the impact of varying conversion rates to optimize the process.
The plasma method has also been explored in TEA studies. Kerscher et al. employed an electron beam plasma reactor requiring multiple electron beam accelerators, as the largest commercial units operate within the range of 250–560 kW,314,318 increasing CAPEX compared to other methods. However, they assumed 100% conversion and selectivity toward H2, resulting in omitting the PSA system in their design. While future advancements may reduce CAPEX for electron beam plasma reactors, the costs associated with PSA systems should still be considered.
In another study, Tabat et al. designed an on-site hydrogen production system using a molten multi-layer reactor (filled with Ni–Br and Mg–Cl2) with a microwave plasma source.316 Their process incorporated a gasification reactor, a WGS reactor, and a bio-methanation reactor to recapture CO2 emissions, achieving a low LCOH of 1.3 $ per kgH2. This was in contrast to another study that reported higher LCOH values even without the bio-methanation unit.310
Regardless of the process type and specific reactor design required, solid carbon sales (Fig. 26) are particularly impactful for driving economic viability since carbon makes up 75% of the product mass. For instance, Keipi et al. concluded that carbon sales of 254–497 $ per t can make thermal pyrolysis competitive versus SMR.310 Parkinson et al. indicated that a CO2 cost penalty of 25.4 $ per t with a carbon sale at 182 $ per t makes molten media pyrolysis cost-effective as SMR. NG price also substantially influences the hydrogen cost due to the fact that about 0.25% of feed gas mass is associated with hydrogen. Von Wald et al. reported a 38% increment in LCOH when the NG price was raised by 50%.57 Similarly, Brandon et al. found that LCOH could rise by 25% and 29% when the feed cost increases by 29% for energy supplied by NG and H2 combustion, respectively.258 Utilizing hydrogen to generate heat requires a higher feed demand (higher sensitivity to NG price) for the process but it also reduces the CO2 emissions.
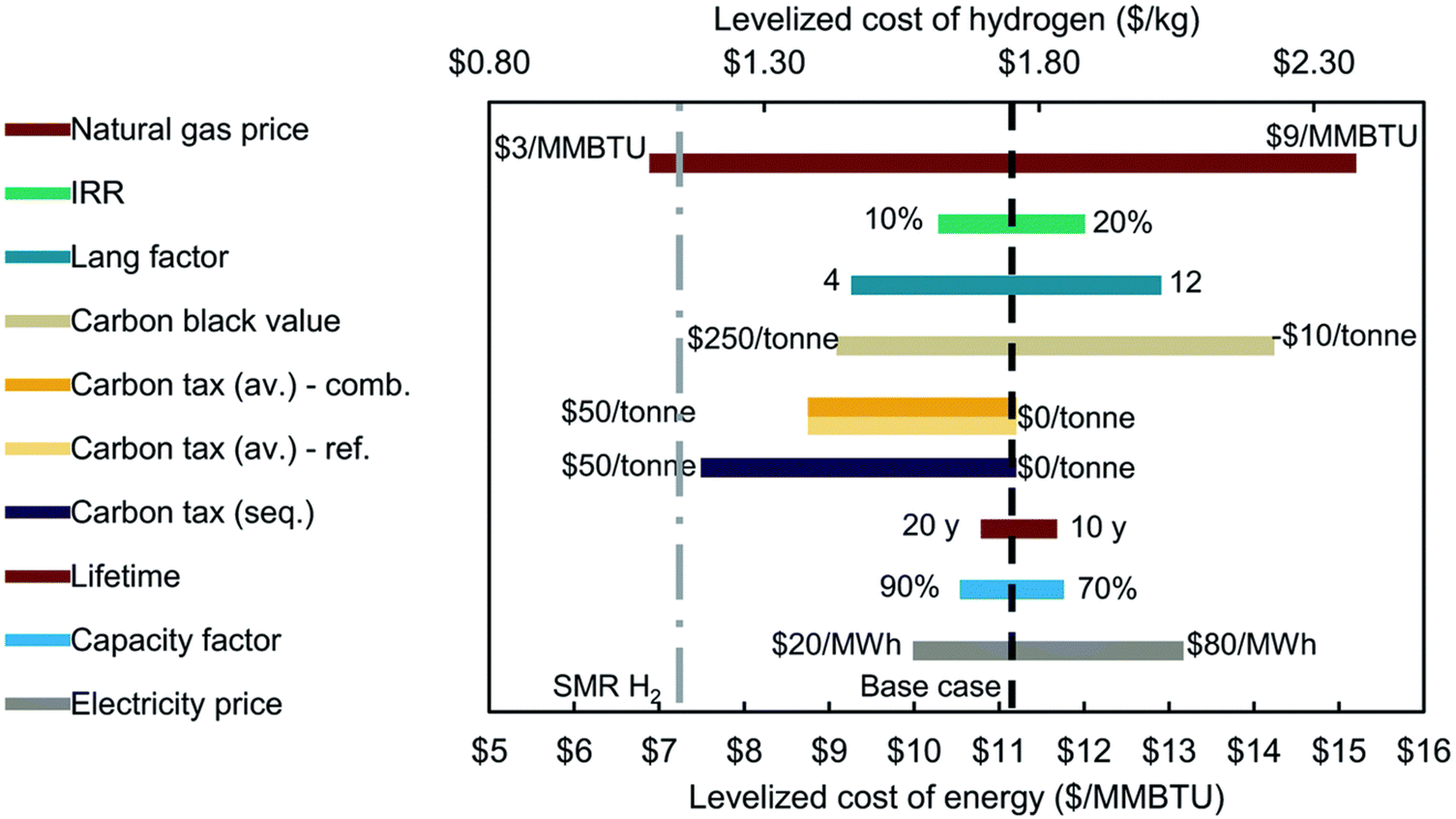 | ||
| Fig. 26 The impact of feed cost, internal rate of return (IRR), lang factor, carbon sale, carbon tax, lifetime, capacity, and electricity cost on the LCOH for small-scale hydrogen production, reproduced from ref. 57 with permission from the Royal Society of Chemistry, copyright 2020. | ||
To summarize, TEA studies indicate that OPEX has a more significant impact than CAPEX for methane pyrolysis. This is largely due to high feedstock costs and energy requirements. Additionally, methane pyrolysis theoretically produces 2 moles of hydrogen per mole of methane, compared to 4 moles in SMR, making feedstock costs per unit of hydrogen higher in pyrolysis. Additionally, handling solid carbon brings additional challenges for methane pyrolysis. Depending on the methods employed, the importance of these parameters varies. Among the different methods, the plasma reactor possesses the highest OPEX at $2.83 per kgH2, highlighting its energy-intensive nature. Future TEA studies should aim to optimize operational conditions for each method and evaluate production scales, including on-site generation and centralized sites with transportation costs.
6. Scale up and commercialization efforts
The growing demand for hydrogen has encouraged several companies and startups to explore different methods of methane pyrolysis for low-emission hydrogen production (Fig. 27). As mentioned earlier, plasma pyrolysis, the most mature methane pyrolysis technology, includes the thermal plasma arc process developed by Kvaerner and SINTEF.319 The process was tested on a pilot scale of 150 kW reactor between 1992 and 1997, recirculated the hydrogen back. In 1999 a commercial plant was commissioned in Canada to produce 20000 tonnes of carbon black per year. However, in 2002 the plant closed down because of technical and financial difficulties.208
 | ||
| Fig. 27 Methane pyrolysis-based companies at various stages of technology development. | ||
Monolith Inc., founded in 2013, adopted technology from Kvaerner and demonstrated it at a pilot-scale (700 tonnes of Cblack per year) in 2014 at their facility in Seaport, California. In 2020, it commissioned its commercial plant in Olive Creek, Nebraska to produce 14000 tonnes of high-quality carbon black per year.184 Recently, the Department of Energy agreed to a $1 billion loan to Monolith to expand its Olive Creek facility,320 enabling production of 180000 tonnes of carbon black and 60000 tH2 per year, primarily for manufacturing 290000 t per year of anhydrous ammonia on-site.320,321 The company had also signed an agreement with Goodyear Tire & Rubber Company to utilize the carbon black to develop tires with a new formulation.322,323 With proven commercial-scale technology, Monolith's process achieves a technology readiness level (TRL) of 9. For cold plasma methane pyrolysis, TOMSK-GAZPROM is the only known active company.324 Collaborating with universities in Tomsk, Siberia, they developed a microwave-heated metallic catalyst bed where micro-discharges between catalyst particles aid natural gas decomposition. However, this technology remains at the laboratory scale.176,325,326
There are a few companies working on non-catalytic thermal methane pyrolysis. BASF and partners are exploring an electrically heated moving-bed reactor where carbon granules flow counter to the gas. Methane pyrolyzes on the carbon granules at 1200–1400 °C, with the granules transferring heat and facilitating solid carbon collection. Funded by the Federal Ministry of Education and Research (BMBF), research on the process was conducted between 2010 and 2020 on a lab-scale, and currently this project has been in the test plant stage since 2021 at BASF's Ludwigshafen site.327
Ekona Power's innovative technology uses pulsed combustion and high-speed gas dynamics for non-catalytic methane pyrolysis.328 This process uses a pulsed ionized stream from intermittent oxy-fired combustion of methane or recycled hydrogen, introduced at high velocities, to mix with methane and produce solid carbon and hydrogen. In 2022, Ekona tested a reactor producing 200 kgH2 per day in British Columbia, Canada, and plans to commission a commercial unit producing 1 tH2 per day in Alberta by 2024–2025.328 The process uses minimal electricity, relying on unconverted natural gas combustion for heat, leading to some GHG emissions.
Among non-catalytic processes, to our knowledge, Aurora Hydrogen is the only start-up that uses microwave technology to obtain CO2-free hydrogen.329 The Aurora process uses carbon as a microwave susceptor, and microwave energy to provide the heat needed to break hydrocarbon chains. This results in an extremely efficient process that uses 80% less electricity than electrolysis. Aurora is currently constructing a 200 kgH2 per day demonstration facility in Edmonton, with plans to scale up future facilities.330
For catalytic pyrolysis, the HAZER process by Hazer Group stands out, producing hydrogen and high-quality graphite with 60% efficiency using iron ore as a catalyst in a fluidized bed at 850 °C.331 The reactor's pressure, temperature, and mass flow can be adjusted to manage the hydrogen production, the quality of the pyrolytic carbon, and the catalyst material's deactivation. In January 2024, Hazer announced the commissioning of their demonstration in Perth, Australia with a capacity to produce 100 tH2 per year.332 Hazer Group has also announced a partnership with FortisBC, and Suncor Energy to construct a methane pyrolysis plant in British Columbia (Canada) to produce 2500 tH2 per year.333 Finally, Hazer Group also has recently signed a memorandum of understanding with Chubu Electric Power and Chiyoda Corporation to prepare a project development plan for a pyrolysis plant in Japan. This project aims to produce 10000 tH2 per year in its first phase and increase to 50000 tH2 per year, and 100000 tH2 per year in the subsequent phase.331
Finland-based Hycamite has also developed proprietary catalysts to produce hydrogen, high-value graphite, and a range of other electrically conductive carbon products suitable for supercapacitors, electronics, and electric cables. Hycamite has been operating a pilot plant since 2021 and has raised over €25 million to scale up the technology. Alberta based Innova Hydrogen Corp. is a start-up company that uses a proprietary catalyst and reactor that breaks down CH4 with the help of high temperatures and turbulence.334 Innova is operating a demo facility in Calgary and is currently designing its first field pilot for deployment in 2024/2025.
Molten media, which allow continuous carbon separation, are being explored by several companies and institutes. C-Zero, a startup from the University of California, Santa Barbara, is focused on commercializing molten salt pyrolysis.335 The company has secured funding from Breakthrough Energy Ventures (led by Bill Gates), SK Gas, Eni, Mitsubishi, and others. The long-term plan is to produce 1000 kgH2 per day at a targeted production cost of 1.5 $ per kgH2, with a pilot plant of 400 kgH2 per day capacity set to be completed in 2024.336,337 Their patent shows that molten salt (any combination of K, Na, Mg, Ca, Mn, Zn, La, or Li, with at least one of F, Cl, Br, I, OH, SO3, or NO3) and molten metal (Ni, Fe, Co, Ru, Ce, MoC, WC, SiC, MgO, CaO, Al2O3, MgF2, CaF2) are the primary media in their reactor to transfer heat and accelerate methane decomposition.338
VulcanX is another start-up focused on the use of molten media at 900 to 1100 °C to decompose natural gas into hydrogen and carbon.339 The plug flow reactor incorporates a recirculating molten metal mechanism which maximizes heat transfer rate and prevents blockage by the solid carbon. It launched its activities in 2017 and completed a 1 kgH2 per day lab prototype in 2020. Having raised funding from Alberta Innovates and other sources, they began designing and building a 55 kgH2 per day pilot plant in 2021–2022. Long-term goals include scaling production to 1–200 tH2 per day at a cost of <$2 per kg H2 with a targeted 70% reduction in GHG emissions compared to SMR.339
7. Government policies and programs to support clean hydrogen
Government policies can create a supportive environment by setting clear targets for hydrogen deployment, funding research, and establishing regulatory frameworks that lower barriers for new technologies along the hydrogen value chain. Incentives such as subsidies, grants, and tax credits reduce the financial risks associated with emerging technologies, making them more competitive.In this section, we examine recent policies, investments, and support mechanisms for hydrogen production and utilization in North America, which could potentially be applicable to the deployment of methane pyrolysis technologies. Canada and the United States have passed legislation to support their low-carbon hydrogen value chains, introducing incentives that position commercial-scale hydrogen as a key to reducing emissions. Both countries aim to expand hydrogen production and transition from traditional SMR methods to cleaner alternatives, such as SMR + CCS, water electrolysis, and methane pyrolysis.340 Although they share these goals, each country employs unique strategies to drive investment in low-carbon hydrogen.
The United States announced its first-ever National Clean Hydrogen Strategy and Roadmap in June 2023. This roadmap was part of key initiatives and investments made under the bipartisan infrastructure law (BIL) announced in 2021, US Department of Energy is investing $9.5 billion in clean hydrogen, including $1 billion for electrolyzer development, $500 million to support the manufacturing of clean hydrogen equipment and $8 billion for regional clean hydrogen hubs.341
Through the Inflation Reduction Act (IRA), which was signed into law in 2022, the United States provides either an investment tax credit (ITC) or hydrogen production tax credit (PTC). The credits can be claimed for initial project capital costs or operating costs based on the carbon intensity of hydrogen produced. The IRA only classifies ‘clean’ hydrogen if the carbon intensity is <4 kgCO2eq. per kgH2 during its lifecycle, and PTC increases as carbon intensity decreases. Clean hydrogen producers could benefit by claiming up to US$ 3.00 per kgH2 if the hydrogen carbon intensity is less than 0.45 kg kgH2−1.341 Alternatively, the IRA offers an ITC, in which up to 30% of the eligible costs are claimable if the hydrogen carbon intensity is less than 0.45 kg kgH2−1 and wage and apprenticeship requirements are met.342 The detailed information about these supports has been summarized in Table 10.340,343
| Carbon intensity tiers (kgCO2eq. per kgH2) | Production tax credit ($ per kgH2) | Maximum investment tax credit (% of project costs) |
|---|---|---|
| <0.45 | $3.00 | 30% |
| 0.45–1.5 | $1.02 | 10% |
| 1.5–2.5 | $0.75 | 7.5% |
| 2.5–4 | $0.60 | 6% |
Canada, on the other hand, outlined their hydrogen strategy in 2020, with a vision of becoming a global leader in producing, using, and exporting low-carbon hydrogen by 2050. The federal government of Canada plans to invest approximately C$11.4 billion in tax credits for clean hydrogen, clean technologies, manufacturing, and carbon capture, utilization, and storage (CCUS) through 2027–28. Like the United States, Canada's tax credits only apply to hydrogen produced with a carbon intensity <4 kgCO2eq. per kgH2 and the rates are at 40%, 25%, or 15% of eligible costs depending on the carbon intensity.340
The PTC in United States significantly larger because it builds up over time-based on production rate over a 10-year period, while Canada's funding is a one-time payment at the start. Allocating tax credits according to production rates would be more effective, as projects vary in their capital and operating cost balance. This approach is also beneficial for new-technology projects that may not reach commercial operation; in these cases, Canada incurs costs, but the U.S. does not.340
Another incentive that the federal government of Canada has considered is the “Low Carbon Economy Challenge” fund, which is a competitive program that aims at reducing GHG emissions. This fund will provide projects with a technology readiness level (TDL) of 1–7 (which includes methane pyrolysis) between C$1 to C$25 million. Eligible projects include but are not limited to carbon capture, low-carbon fuel production for own use, zero-emission vehicle infrastructure and purchase, and fuel transportation infrastructure.344
A key difference between the two countries is that while the United States lacks a federal carbon tax penalty, Canada imposes one of the highest carbon taxes globally. The Canadian carbon tax framework sets a minimum national standard for carbon pricing, allowing provinces to implement their own systems that meet or exceed these standards. This includes a fuel charge and an output-based pricing system (OBPS) that targets large industrial facilities. Facilities covered by the OBPS pay a carbon price only on the portion of their emissions that exceed a defined limit. As of 2024, the tax is C$80 per tonne of equivalent CO2 emissions, increasing by C$15 annually to reach C$170 per tonne in 2030.345
In addition to supporting hydrogen production, the United States Department of Treasury provides the “Alternative Fuel Vehicle Refueling Property Credit,” which is a 30% tax credit for installing infrastructure like hydrogen refueling stations, capped at $1000 for individuals and $100000 per property for businesses.346 In 2024, the US federal government announced $90 million to support hydrogen refueling stations and infrastructure in California, Texas, and Colorado.347
Canada also provides support for hydrogen fueling stations through the “Zero Emission Vehicle Infrastructure Program,” allocating C$400 million in Budget 2022. This program shares the same goal as the Canada Infrastructure Bank (CIB), which created the “Charging and Hydrogen Refueling Infrastructure (CHRI)” initiative and allocated a minimum of C$500 million.348 The goal is to deploy 50000 new zero-emission vehicle charging ports and hydrogen refueling stations by March 2029. Charging and refueling stations installed within the first two years are eligible for CIB financing, covering 65% of capital costs, while installations in years three and four qualify for 50% financing. In underserved areas, the CIB will finance up to 80% of eligible capital costs. This program focuses on large-scale projects with total eligible costs of at least $20 million.349 The total combined funding for the Zero Emission Vehicle Infrastructure Program” and the CIB's initiative is C$1.2 billion.348
In conclusion, both the US and Canada recognize hydrogen as a crucial component in achieving their net-zero goals. Both countries have implemented tax credits and incentives to support hydrogen production, refueling stations, hydrogen fuel cell vehicles, and related infrastructure, alongside carbon capture programs for existing plants.
8. Conclusion
Methane pyrolysis has emerged as a promising technology for hydrogen production, offering a pathway to generate hydrogen with significantly reduced greenhouse gas emissions compared to conventional methods. The integration of methane pyrolysis into existing natural gas infrastructures can facilitate widespread adoption, leveraging current assets and minimizing new capital investments.In this review, we have extensively discussed the various technological approaches to methane pyrolysis, highlighting the distinction between catalytic and non-catalytic processes. Catalytic routes, while efficient in lowering the reaction temperatures and enhancing hydrogen yields, face challenges related to catalyst deactivation due to carbon deposition and contamination of the carbon byproduct. Non-catalytic methods, although requiring higher temperatures, avoid complexities related to catalysts and offer robustness for industrial applications. Throughout the review, the technical feasibility, efficiency, and environmental benefits of different methods have been evaluated. The development of a commercial technology for low-GHG hydrogen production via methane pyrolysis aligns with global CO2 reduction goals. Additionally, the co-production of valuable carbon solids such as carbon black, graphite, and graphene present opportunities for developing new materials and applications.
However, several challenges remain. The scalability of methane pyrolysis technologies needs to be addressed to meet the growing demand for hydrogen in various sectors, including transportation, industry, and power generation. The mismatch between size of hydrogen market relative to carbon market is also a big issue. Developing large volume carbon products would be critical. Additionally, economic barriers due to high operational costs and the need for further technological advancements to optimize process efficiencies and carbon product utilization must be overcome. Furthermore, policy frameworks and market incentives which are currently missing are critical to supporting the commercial viability of methane pyrolysis. Regulatory support for low-GHG technologies, coupled with financial mechanisms such as carbon credits and subsidies for clean hydrogen production, can significantly enhance the attractiveness of methane pyrolysis.
Looking forward, the focus on research and development will be crucial in addressing the technical challenges associated with methane pyrolysis. Innovations in catalyst design are needed to improve resistance to coking and enhance catalyst longevity, reducing operational costs and increasing process efficiency. Exploring novel reactor designs that can effectively handle high throughputs and integrate seamlessly with regeneration and separation processes will also be vital. The potential for integrating renewable energy sources with methane pyrolysis processes presents an exciting avenue for further reducing the carbon footprint of hydrogen production. As the costs of renewable energy continue to decline, the feasibility of such integrations will likely increase, making methane pyrolysis an even more attractive option for sustainable hydrogen production. Moreover, the development of markets for carbon byproducts will play a pivotal role in driving economic viability of methane pyrolysis. High-value applications for carbon solids, such as in battery technologies or advanced composites, could provide significant revenue streams that offset production costs and promote circular economic models.
In conclusion, methane pyrolysis stands at the cusp of significant growth, with the potential to play a key role in the global transition to a NetZero future. Continued technological advancements, supportive policy measures, and robust market development for carbon byproducts will be essential in realizing the full potential of this promising technology.
Author contributions
A. L. M., S. H., M. F., and M. A. K. wrote the manuscript. M. A. K. supervised the project. M. A. K., M. G. K., M. J. T. and R. A. E. revised manuscript and participated in discussions.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Alberta Innovates under the Hydrogen Centre of Excellence Continuous Intake Program (Agreement No. 222301923) and by the Natural Sciences and Engineering Research Council of Canada (NSERC) under the Discovery Grants Program (RGPIN-2024-05032).References
- K. Levin, D. Rich, K. Ross, T. Fransen and C. Elliott, Designing and Communicating Net-Zero Targets, World Resources Institute, 2020, 1–30, https://www.wri.org/research/designing-and-communicating-net-zero-targets Search PubMed.
- Climate Watch Historical Country Greenhouse Gas Emissions Data, (accessed 2022), 2022, https://www.climatewatchdata.org/ghg-emissions.
- J. Whitehead, P. Newman, J. Whitehead and K. L. Lim, Striking the right balance: understanding the strategic applications of hydrogen in transitioning to a net zero emissions economy, Sustainable Earth Rev., 2023, 6, 1, DOI:10.1186/s42055-022-00049-w.
- B. Moon, W. Lee and Y. Lee, The Present Condition and Outlook of Hydrogen Industry in Alberta, Canada, J. Kor. Inst. Gas, 2021, 25, 1–6, DOI:10.7842/kigas.2021.25.1.1.
- Hydrogen, International Renewable Energy Agency, (accessed 2022), 2022, https://www.irena.org/Energy-Transition/Technology/Hydrogen#:~:text=Globalproductionstandsataround,(notanenergysource).
- A. O. Oni, K. Anaya, T. Giwa, G. Di Lullo and A. Kumar, Comparative assessment of blue hydrogen from steam methane reforming, autothermal reforming, and natural gas decomposition technologies for natural gas-producing regions, Energy Convers. Manage., 2022, 254, 115245, DOI:10.1016/j.enconman.2022.115245.
- M. A. Khan, C. MacKinnon, C. Young, D. B. Layzell, M. Adnan Khan, C. MacKinnon, P. Eng Cameron Young and Pe. B. David Layzell, Techno-Economics of a New Hydrogen Value Chain Supporting Heavy Duty Transport, Trans. Accel. Rep., 2022, 4, 1–52 Search PubMed.
- N. Gerloff, Comparative Life-Cycle-Assessment analysis of three major water electrolysis technologies while applying various energy scenarios for a greener hydrogen production, J. Energy Storage, 2021, 43, 102759, DOI:10.1016/j.est.2021.102759.
- S. F. Ahmed, M. Mofijur, S. Nuzhat, N. Rafa, A. Musharrat, S. S. Lam and A. Boretti, Sustainable hydrogen production: Technological advancements and economic analysis, Int. J. Hydrogen Energy, 2022, 47, 37227–37255, DOI:10.1016/j.ijhydene.2021.12.029.
- S. Hasan and R. Shabaneh, The Economics and Resource Potential of Hydrogen Production in Saudi Arabia, King Abdullah Petroleum Studies and Research Center, 2021, 1–39 Search PubMed.
- F. Birol, The Future of Hydrogen, 2019, 1–203, https://www.iea.org/reports/the-future-of-hydrogen Search PubMed.
- E. Lewis, S. McNaul, M. Jamieson, M. S. Henriksen, H. S. Matthews, T. Shultz, T. J. Skone, R. Stevens, J. White, L. Walsh, J. Grove, T. Shultz, P. E. Timothy, J. Skone and R. Stevens, Comparison Of Commercial, State-Of-The-Art, Fossil-Based Hydrogen Production Technologies, National Energy Technology Laboratory (NETL), (accessed 12 April 2022), 2022, 138, https://www.iea-etsap.org/E-TechDS/PDF/I01-ind_boilers-GS-AD-gct.pdf.
- X. Li, X. Hao, A. Abudula and G. Guan, Nanostructured catalysts for electrochemical water splitting: current state and prospects, J. Mater. Chem., 2016, 4, 11973–12000, 10.1039/C6TA02334G.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Future cost and performance of water electrolysis: An expert elicitation study, Int. J. Hydrogen Energy, 2017, 42, 30470–30492, DOI:10.1016/j.ijhydene.2017.10.045.
- K. Zeng and D. Zhang, Recent progress in alkaline water electrolysis for hydrogen production and applications, Prog. Energy Combust. Sci., 2010, 36, 307–326, DOI:10.1016/j.pecs.2009.11.002.
- A. Buttler and H. Spliethoff, Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454, DOI:10.1016/j.rser.2017.09.003.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A comprehensive review on PEM water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 4901–4934, DOI:10.1016/j.ijhydene.2013.01.151.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Seawater electrolysis for hydrogen production: a solution looking for a problem?, Energy Environ. Sci., 2021, 14, 4831–4839, 10.1039/d1ee00870f.
- Next Level Solid Oxide Electrolysis: Upscaling potential and techno-economical evaluation for 3 industrial use cases (accessed 15 May 2023), Institute for Sustainable Process Technology (ISPT), 2023, 1–33, https://ispt.eu/media/20230508-FINAL-SOE-public-report-ISPT.pdf.
- D. Peterson, J. Vickers and D. DeSantis, Hydrogen production cost from PEM electrolysis, Department of Energy (USA), 2019, 1–15, https://www.hydrogen.energy.gov/docs/hydrogenprogramlibraries/pdfs/19009_h2_production_cost_pem_electrolysis_2019.pdf?Status=Master.
- Air Liquide to build 20 MW PEM electrolyser with Hydrogenics tech, Fuel Cells Bulletin, Mark Allen Group, 2019, 9 DOI:10.1016/s1464-2859(19)30108-7.
- M. Naeini, J. S. Cotton and T. A. Adams, An eco-technoeconomic analysis of hydrogen production using solid oxide electrolysis cells that accounts for long-term degradation, Front. Energy Res., 2022, 10, 1015465, DOI:10.3389/fenrg.2022.1015465.
- Y. Yan, V. Manovic, E. J. Anthony and P. T. Clough, Techno-economic analysis of low-carbon hydrogen production by sorption enhanced steam methane reforming (SE-SMR) processes, Energy Convers. Manage., 2020, 226, 113530, DOI:10.1016/j.enconman.2020.113530.
- C. Yang and J. M. Ogden, Analyzing Natural Gas Based Hydrogen Infrastructure – Optimizing Transitions From Distributed To Centralized H2 Production, National Hydrogen Association, 2005, 20.
- Y. Jiang and D. Bhattacharyya, Process modeling of direct coal-biomass to liquids (CBTL) plants with shale gas utilization and CO2 capture and storage (CCS, Appl. Energy, 2016, 183, 1616–1632, DOI:10.1016/j.apenergy.2016.09.098.
- C. N. Hamelinck and A. P. C. Faaij, Future prospects for production of methanol and hydrogen from biomass, J. Power Sources, 2002, 111, 1–22, DOI:10.1016/S0378-7753(02)00220-3.
- A. Elnigoumi, Investigating Cost Effective Pathways for Blue Hydrogen Production in Alberta, Master Thesis, University of Calgary, 2021, https://prism.ucalgary.ca/bitstreams/31e6026d-e1fe-4878-9b8b-4ff0170bbbea/download Search PubMed.
- D. Bellotti, M. Rivarolo and L. Magistri, A comparative techno-economic and sensitivity analysis of Power-to-X processes from different energy sources, Energy Convers. Manage., 2022, 115565, DOI:10.1016/j.enconman.2022.115565.
- A. H. Reksten, M. S. Thomassen, S. Møller-Holst and K. Sundseth, Projecting the future cost of PEM and alkaline water electrolysers; a CAPEX model including electrolyser plant size and technology development, Int. J. Hydrogen Energy, 2022, 47, 38106–38113, DOI:10.1016/j.ijhydene.2022.08.306.
- E. Smith, J. Morris, H. Kheshgi, G. Teletzke, H. Herzog and S. Paltsev, The cost of CO2 transport and storage in global integrated assessment modeling, Int. J. Greenhouse Gas Control, 2021, 109, 103367, DOI:10.1016/j.ijggc.2021.103367.
- N. Ma, W. Zhao, W. Wang, X. Li and H. Zhou, Large scale of green hydrogen storage: Opportunities and challenges, Int. J. Hydrogen Energy, 2024, 50, 379–396, DOI:10.1016/j.ijhydene.2023.09.021.
- P. Muthukumar, A. Kumar, M. Afzal, S. Bhogilla, P. Sharma, A. Parida, S. Jana, E. A. Kumar, R. K. Pai and I. P. Jain, Review on large-scale hydrogen storage systems for better sustainability, Int. J. Hydrogen Energy, 2023, 48, 33223–33259, DOI:10.1016/j.ijhydene.2023.04.304.
- O. Faye, J. Szpunar and U. Eduok, A critical review on the current technologies for the generation, storage, and transportation of hydrogen, Int. J. Hydrogen Energy, 2022, 47, 13771–13802, DOI:10.1016/j.ijhydene.2022.02.112.
- M. R. Usman, Hydrogen storage methods: Review and current status, Renewable Sustainable Energy Rev., 2022, 167, 112743, DOI:10.1016/j.rser.2022.112743.
- J. Andersson and S. Grönkvist, Large-scale storage of hydrogen, Int. J. Hydrogen Energy, 2019, 44, 11901–11919, DOI:10.1016/j.ijhydene.2019.03.063.
- L. Schlapbach and A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 2001, 414, 353–358, DOI:10.1038/35104634.
- A. M. Elberry, J. Thakur, A. Santasalo-Aarnio and M. Larmi, Large-scale compressed hydrogen storage as part of renewable electricity storage systems, Int. J. Hydrogen Energy, 2021, 46, 15671–15690, DOI:10.1016/j.ijhydene.2021.02.080.
- M. A. Khan, M. Powell, M. Tampier, E. Thorn and D. Layzell, Hydrogen and the Decarbonization of Steel Production in Canada: Reaching Economies of Scale, (accessed 2023), 2023, https://transitionaccelerator.ca/reports/hydrogen-and-the-decarbonization-of-steel-production-in-canada/.
- M. A. Khan, D. Layzell, C. MacKinnon and C. Young, The Techno-Economics of Hydrogen Compression, (accessed 2021), 2021, https://transitionaccelerator.ca/reports/technical-brief-the-techno-economics-of-hydrogen-compression/.
- Z. Yanxing, G. Maoqiong, Z. Yuan, D. Xueqiang and S. Jun, Thermodynamics analysis of hydrogen storage based on compressed gaseous hydrogen, liquid hydrogen and cryo-compressed hydrogen, Int. J. Hydrogen Energy, 2019, 44, 16833–16840, DOI:10.1016/j.ijhydene.2019.04.207.
- E. Wolf, Electrochemical Energy Storage for Renewable Sources and Grid Balancing, Elsevier Inc., 2015, 129–142 Search PubMed.
- J. E. Quintos Fuentes and D. M. F. Santos, Technical and Economic Viability of Underground Hydrogen Storage, Hydrogen, 2023, 4, 975–1000, DOI:10.3390/hydrogen4040057.
- J. Hölzen, thesis: Hydrogen-powered aviation – techno-economics of flying with green liquid hydrogen, Gottfried Wilhelm Leibniz Universität, 2024, https://www.repo.uni-hannover.de/handle/123456789/16518.
- A. Ahmad, E. Oko and A. Ibhadon, Comparative energy and exergy analysis of ortho-para hydrogen and non-ortho-para hydrogen conversion in hydrogen liquefaction, Int. J. Hydrogen Energy, 2024, 78, 991–1003, DOI:10.1016/j.ijhydene.2024.06.368.
- M. Gardiner, Energy requirements for hydrogen gas compression and liquefaction as related to vehicle storage needs, (accessed 2009), 2009, https://www.hydrogen.energy.gov/pdfs/9013_energy_requirements_for_hydrogen_gas_compression.pdf.
- J. C. Crivello, B. Dam, R. V. Denys, M. Dornheim, D. M. Grant, J. Huot, T. R. Jensen, P. de Jongh, M. Latroche, C. Milanese, D. Milčius, G. S. Walker, C. J. Webb, C. Zlotea and V. A. Yartys, Review of magnesium hydride-based materials: development and optimisation, Appl. Phys. A:Mater. Sci. Process., 2016, 122, 1–20, DOI:10.1007/s00339-016-9602-0.
- P. C. Rao and M. Yoon, Potential liquid-organic hydrogen carrier (Lohc) systems: A review on recent progress, Energies, 2020, 13, 6040, DOI:10.3390/en13226040.
- C. Chu, K. Wu, B. Luo, Q. Cao and H. Zhang, Hydrogen storage by liquid organic hydrogen carriers: Catalyst, renewable carrier, and technology – A review, Carbon Resour. Convers., 2023, 6, 334–351, DOI:10.1016/j.crcon.2023.03.007.
- H. Huang, Y. Yu and K. H. Chung, Seasonal storage of electricity by hydrogen in benzene-water system, Int. J. Hydrogen Energy, 2012, 37, 12798–12804, DOI:10.1016/j.ijhydene.2012.05.156.
- A. S. Mehr, A. D. Phillips, M. P. Brandon, M. T. Pryce and J. G. Carton, Recent challenges and development of technical and technoeconomic aspects for hydrogen storage, insights at different scales; A state of art review, Int. J. Hydrogen Energy, 2024, 70, 786–815, DOI:10.1016/j.ijhydene.2024.05.182.
- A. Lin and G. Bagnato, Revolutionising energy storage: The Latest Breakthrough in liquid organic hydrogen carriers, Int. J. Hydrogen Energy, 2024, 63, 315–329, DOI:10.1016/j.ijhydene.2024.03.146.
- G. Valenti, Compendium of Hydrogen Energy: Hydrogen Storage, Distribution and Infrastructure, Elsevier, 2015, vol. 2, pp. 27–51 Search PubMed.
- Á. Berenguer-Murcia, J. P. Marco-Lozar and D. Cazorla-Amorós, Hydrogen Storage in Porous Materials: Status, Milestones, and Challenges, Chem. Rec., 2018, 18, 900–912, DOI:10.1002/tcr.201700067.
- H. W. Langmi, J. Ren, B. North, M. Mathe and D. Bessarabov, Hydrogen storage in metal-organic frameworks: A review, Electrochim. Acta, 2014, 128, 368–392, DOI:10.1016/j.electacta.2013.10.190.
- D. Kang, C. Palmer, D. Mannini, N. Rahimi, M. J. Gordon, H. Metiu and E. W. McFarland, Catalytic Methane Pyrolysis in Molten Alkali Chloride Salts Containing Iron, ACS Catal., 2020, 10, 7032–7042, DOI:10.1021/acscatal.0c01262.
- N. Sánchez-Bastardo, R. Schlögl, H. Ruland, M. Keller, N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Methane Pyrolysis for Zero-Emission Hydrogen Production: A Potential Bridge Technology from Fossil Fuels to a Renewable and Sustainable Hydrogen Economy, Ind. Eng. Chem. Res., 2021, 60, 11855–11881, DOI:10.1021/acs.iecr.1c01679.
- G. A. Von Wald, A. M. S. Masnadi, D. C. Upham and A. R. Brandt, Optimization-based technoeconomic analysis of molten-media methane pyrolysis for reducing industrial sector CO2emissions, Sustainable Energy Fuels, 2020, 4, 4598–4613, 10.1039/d0se00427h.
- M. Steinberg, Fossil fuel decarbonization technology for mitigating global warming, Int. J. Hydrogen Energy, 1999, 24, 771–777, DOI:10.1016/S0360-3199(98)00128-1.
- J. M. Smith, H. C. Van Ness, M. M. Abbott and M. T. Swihart, Introduction To Chemical Engineering Thermodynamics, 2018, 8th edn Search PubMed.
- J. Zeng, M. Tarazkar, C. Palmer, M. J. Gordon, H. Metiu and E. W. McFarland, Initial Steps in CH4Pyrolysis on Cu and Ni, J. Phys. Chem. C, 2021, 125, 18665–18672, DOI:10.1021/acs.jpcc.1c03606.
- S. G. Zavarukhin and G. G. Kuvshinov, The kinetic model of formation of nanofibrous carbon from CH4-H2 mixture over a high-loaded nickel catalyst with consideration for the catalyst deactivation, Appl. Catal., A, 2004, 272, 219–227, DOI:10.1016/j.apcata.2004.05.044.
- Q. Chen and A. C. Lua, Kinetic reaction and deactivation studies on thermocatalytic decomposition of methane by electroless nickel plating catalyst, Chem. Eng. J., 2020, 389, 124366, DOI:10.1016/j.cej.2020.124366.
- I. Alstrup and M. T. Tavares, Kinetics of carbon formation from CH4 + H2 on silica-supported nickel and Ni-Cu catalysts, J. Catal., 1993, 139, 513–524, DOI:10.1006/jcat.1993.1045.
- J. W. Snoeck, G. F. Froment and M. Fowles, Filamentous carbon formation and gasification: Thermodynamics, driving force, nucleation, and steady-state growth, J. Catal., 1997, 169, 240–249, DOI:10.1006/jcat.1997.1634.
- M. C. Demicheli, E. N. Ponzi, O. A. Ferretti and A. A. Yeramian, Kinetics of carbon formation from CH4-H2 mixtures on nickel-alumina catalyst, Chem. Eng. J., 1991, 46, 129–136, DOI:10.1016/0300-9467(91)87004-T.
- S. Douven, S. L. Pirard, G. Heyen, D. Toye and J. P. Pirard, Kinetic study of double-walled carbon nanotube synthesis by catalytic chemical vapour deposition over an Fe-Mo/MgO catalyst using methane as the carbon source, Chem. Eng. J., 2011, 175, 396–407, DOI:10.1016/j.cej.2011.08.066.
- U. Narkiewicz, W. Arabczyk and W. Konicki, Fullerenes, Nanotubes Carbon Nanostruct., 2005, 13, 99–105 CrossRef CAS.
- M. Borghei, R. Karimzadeh, A. Rashidi and N. Izadi, Int. J. Hydrogen Energy, 2010, 35, 9479–9488 CrossRef CAS.
- Y. Zhang and K. J. Smith, A kinetic model of CH4 decomposition and filamentous carbon formation on supported Co catalysts, J. Catal., 2005, 231, 354–364, DOI:10.1016/j.jcat.2005.02.010.
- S. K. Saraswat, B. Sinha, K. K. Pant and R. B. Gupta, Kinetic Study and Modeling of Homogeneous Thermocatalytic Decomposition of Methane over a Ni-Cu-Zn/Al2O3 Catalyst for the Production of Hydrogen and Bamboo-Shaped Carbon Nanotubes, Ind. Eng. Chem. Res., 2016, 55, 11672–11680, DOI:10.1021/acs.iecr.6b03145.
- C. J. Chen, M. H. Back and R. A. Back, ACS Symp. Ser., 1976, 1–16 Search PubMed.
- N. Ozalp, K. Ibrik and M. Al-Meer, Kinetics and heat transfer analysis of carbon catalyzed solar cracking process, Energy, 2013, 55, 74–81, DOI:10.1016/j.energy.2013.02.022.
- C. Guéret, M. Daroux and F. Billaud, Methane pyrolysis: Thermodynamics, Chem. Eng. Sci., 1997, 52, 815–827, DOI:10.1016/S0009-2509(96)00444-7.
- K. Salipira, N. J. Coville and M. S. Scurrell, Carbon produced by the catalytic decomposition of methane on nickel: Carbon yields and carbon structure as a function of catalyst properties, J. Nat. Gas Sci. Eng., 2016, 32, 501–511, DOI:10.1016/j.jngse.2016.04.027.
- A. C. Lua and H. Y. Wang, Hydrogen production by catalytic decomposition of methane over Ni-Cu-Co alloy particles, Appl. Catal., B, 2014, 156–157, 84–93, DOI:10.1016/j.apcatb.2014.02.046.
- Y. Shen and A. C. Lua, Sol-gel synthesis of titanium oxide supported nickel catalysts for hydrogen and carbon production by methane decomposition, J. Power Sources, 2015, 280, 467–475, DOI:10.1016/j.jpowsour.2015.01.057.
- G. Wang, Y. Jin, G. Liu and Y. Li, Production of hydrogen and nanocarbon from catalytic decomposition of methane over a Ni-Fe/Al2O3 catalyst, Energy Fuels, 2013, 27, 4448–4456, DOI:10.1021/ef3019707.
- I. W. Wang, D. A. Kutteri, B. Gao, H. Tian and J. Hu, Methane Pyrolysis for Carbon Nanotubes and CO x -Free H 2 over Transition-Metal Catalysts, Energy Fuels, 2019, 33, 197–205, DOI:10.1021/acs.energyfuels.8b03502.
- J. Li, L. Dong, L. Xiong, Y. Yang, Y. Du, L. Zhao, H. Wang and S. Peng, High-loaded Ni-Cu-SiO2catalysts for methane decomposition to prepare hydrogen and carbon filaments, Int. J. Hydrogen Energy, 2016, 41, 12038–12048, DOI:10.1016/j.ijhydene.2016.05.137.
- M. Pudukudy and Z. Yaakob, Methane decomposition over Ni, Co and Fe based monometallic catalysts supported on sol gel derived SiO2 microflakes, Chem. Eng. J., 2015, 262, 1009–1021, DOI:10.1016/j.cej.2014.10.077.
- M. Pudukudy, Z. Yaakob and M. S. Takriff, Methane decomposition into COx free hydrogen and multiwalled carbon nanotubes over ceria, zirconia and lanthana supported nickel catalysts prepared via a facile solid state citrate fusion method, Energy Convers. Manage., 2016, 126, 302–315, DOI:10.1016/j.enconman.2016.08.006.
- G. Sierra Gallego, J. Barrault, C. Batiot-Dupeyrat and F. Mondragón, Production of hydrogen and MWCNTs by methane decomposition over catalysts originated from LaNiO3 perovskite, Catal. Today, 2010, 149, 365–371, DOI:10.1016/j.cattod.2009.06.004.
- M. Pudukudy, Z. Yaakob, Q. Jia and M. S. Takriff, Catalytic decomposition of methane over rare earth metal (Ce and La) oxides supported iron catalysts, Appl. Surf. Sci., 2019, 467–468, 236–248, DOI:10.1016/j.apsusc.2018.10.122.
- M. Pudukudy, Z. Yaakob and Z. S. Akmal, Direct decomposition of methane over SBA-15 supported Ni, Co and Fe based bimetallic catalysts, Appl. Surf. Sci., 2015, 330, 418–430, DOI:10.1016/j.apsusc.2015.01.032.
- D. Kang and J. W. Lee, Enhanced methane decomposition over nickel-carbon-B2O3 core-shell catalysts derived from carbon dioxide, Appl. Catal., B, 2016, 186, 41–55, DOI:10.1016/j.apcatb.2015.12.045.
- N. Bayat, M. Rezaei and F. Meshkani, Hydrogen and carbon nanofibers synthesis by methane decomposition over Ni-Pd/Al2O3 catalyst, Int. J. Hydrogen Energy, 2016, 41, 5494–5503, DOI:10.1016/j.ijhydene.2016.01.134.
- A. M. Amin, E. Croiset and W. Epling, Review of methane catalytic cracking for hydrogen production, Int. J. Hydrogen Energy, 2011, 36, 2904–2935, DOI:10.1016/j.ijhydene.2010.11.035.
- N. Bayat, M. Rezaei and F. Meshkani, Methane decomposition over Ni-Fe/Al2O3 catalysts for production of COx-free hydrogen and carbon nanofiber, Int. J. Hydrogen Energy, 2016, 41, 1574–1584, DOI:10.1016/j.ijhydene.2015.10.053.
- W. Wang, H. Wang, Y. Yang and S. Jiang, Ni-SiO 2 and Ni-Fe-SiO 2 catalysts for methane decomposition to prepare hydrogen and carbon filaments, Int. J. Hydrogen Energy, 2012, 37, 9058–9066, DOI:10.1016/j.ijhydene.2012.03.003.
- E. Tezel, H. E. Figen and S. Z. Baykara, Hydrogen production by methane decomposition using bimetallic Ni–Fe catalysts, Int. J. Hydrogen Energy, 2019, 44, 9930–9940, DOI:10.1016/j.ijhydene.2018.12.151.
- P. Wang, H. Zhu, M. Huang, C. Wan, D. Li and L. Jiang, Catalytic methane decomposition to hydrogen and carbon over hydrotalcite-derivative composition-uniform and sintering-resistant Ni-Fe/Al2O3 alloy catalysts, Int. J. Energy Res., 2022, 46 DOI:10.1002/er.8349.
- I. Suelves, M. J. Lázaro, R. Moliner, B. M. Corbella and J. M. Palacios, Hydrogen production by thermo catalytic decomposition of methane on Ni-based catalysts: Influence of operating conditions on catalyst deactivation and carbon characteristics, Int. J. Hydrogen Energy, 2005, 30, 1555–1567, DOI:10.1016/j.ijhydene.2004.10.006.
- Y. Shen and A. C. Lua, Synthesis of Ni and Ni-Cu supported on carbon nanotubes for hydrogen and carbon production by catalytic decomposition of methane, Appl. Catal., B, 2015, 164, 61–69, DOI:10.1016/j.apcatb.2014.08.038.
- N. Bayat, F. Meshkani and M. Rezaei, Thermocatalytic decomposition of methane to COx-free hydrogen and carbon over Ni–Fe–Cu/Al2O3 catalysts, Int. J. Hydrogen Energy, 2016, 41, 13039–13049, DOI:10.1016/j.ijhydene.2016.05.230.
- P. Li, J. Liu, N. Nag and P. A. Crozier, In situ preparation of Ni-Cu/TiO2 bimetallic catalysts, J. Catal., 2009, 262, 73–82, DOI:10.1016/j.jcat.2008.12.001.
- L. Zhou, L. R. Enakonda, M. Harb, Y. Saih, A. Aguilar-Tapia, S. Ould-Chikh, J. Louis Hazemann, J. Li, N. Wei, D. Gary, P. Del-Gallo and J. M. Basset, Fe catalysts for methane decomposition to produce hydrogen and carbon nano materials, Appl. Catal., B, 2017, 208, 44–59, DOI:10.1016/j.apcatb.2017.02.052.
- X. Weng, J. Zhang, Z. Wu, Y. Liu, H. Wang and J. A. Darr, Continuous syntheses of highly dispersed composite nanocatalysts via simultaneous co-precipitation in supercritical water, Appl. Catal., B, 2011, 103, 453–461, DOI:10.1016/j.apcatb.2011.02.009.
- A. A. Ibrahim, A. H. Fakeeha, A. S. Al-Fatesh, A. E. Abasaeed and W. U. Khan, Methane decomposition over iron catalyst for hydrogen production, Int. J. Hydrogen Energy, 2015, 40, 7593–7600, DOI:10.1016/j.ijhydene.2014.10.058.
- R. Musamali and Y. M. Isa, Decomposition of Methane to Carbon and Hydrogen: A Catalytic Perspective, Energy Technol., 2018, 7, 1800593, DOI:10.1002/ente.201800593.
- S. Takenaka, H. Ogihara, I. Yamanaka and K. Otsuka, Decomposition of methane over supported-Ni catalysts: Effects of the supports on the catalytic lifetime, Appl. Catal., A, 2001, 217, 101–110, DOI:10.1016/S0926-860X(01)00593-2.
- A. E. Awadallah, M. S. Abdel-Mottaleb, A. A. Aboul-Enein, M. M. Yonis and A. K. Aboul-Gheit, Catalytic Decomposition of Natural Gas to CO/CO2-Free Hydrogen Production and Carbon Nanomaterials Using MgO-Supported Monometallic Iron Family Catalysts, Chem. Eng. Commun., 2015, 202, 163–174, DOI:10.1080/00986445.2013.836631.
- Y. Wang, L. Qiu, L. Zhang, D. M. Tang, R. Ma, C. L. Ren, F. Ding, C. Liu and H. M. Cheng, Growth mechanism of carbon nanotubes from Co-W-C alloy catalyst revealed by atmospheric environmental transmission electron microscopy, Sci. Adv., 2022, 8, eabo5686, DOI:10.1126/sciadv.abo5686.
- N. Muradov, F. Smith and A. T-Raissi, Catal. Today, 2005, 102–103, 225–233 CrossRef CAS.
- N. Z. Muradov and T. N. Veziroǧlu, From hydrocarbon to hydrogen-carbon to hydrogen economy, Int. J. Hydrogen Energy, 2005, 30, 225–237, DOI:10.1016/j.ijhydene.2004.03.033.
- M. S. Kim, S. Y. Lee, J. H. Kwak, G. Y. Han and K. J. Yoon, Hydrogen production by decomposition of ethane-containing methane over carbon black catalysts, Korean J. Chem. Eng., 2011, 28, 1833–1838, DOI:10.1007/s11814-011-0064-y.
- S. Patel, S. Kundu, P. Halder, M. H. Marzbali, K. Chiang, A. Surapaneni and K. Shah, Production of hydrogen by catalytic methane decomposition using biochar and activated char produced from biosolids pyrolysis, Int. J. Hydrogen Energy, 2020, 45, 29978–29992, DOI:10.1016/j.ijhydene.2020.08.036.
- S. C. Lee, H. J. Seo and G. Y. Han, Hydrogen production by catalytic decomposition of methane over carbon black catalyst at high temperatures, Korean J. Chem. Eng., 2013, 30, 1716–1721, DOI:10.1007/s11814-013-0107-7.
- S. Y. Lee, B. H. Ryu, G. Y. Han, T. J. Lee and K. J. Yoon, Catalytic characteristics of specialty carbon blacks in decomposition of methane for hydrogen production, Carbon, 2008, 46, 1978–1986, DOI:10.1016/j.carbon.2008.08.008.
- H. Nishii, D. Miyamoto, Y. Umeda, H. Hamaguchi, M. Suzuki, T. Tanimoto, T. Harigai, H. Takikawa and Y. Suda, Catalytic activity of several carbons with different structures for methane decomposition and by-produced carbons, Appl. Surf. Sci., 2019, 473, 291–297, DOI:10.1016/j.apsusc.2018.12.073.
- Y. Shen and A. C. Lua, A trimodal porous carbon as an effective catalyst for hydrogen production by methane decomposition, J. Colloid Interface Sci., 2016, 462, 48–55, DOI:10.1016/j.jcis.2015.09.050.
- Y. Shen and A. C. Lua, Polyol synthesis of nickel-copper based catalysts for hydrogen production by methane decomposition, Int. J. Hydrogen Energy, 2015, 40, 311–321, DOI:10.1016/j.ijhydene.2014.10.071.
- Y. Shen, M. Ge and A. C. Lua, Deactivation of bimetallic nickel-copper alloy catalysts in thermocatalytic decomposition of methane, Catal.:Sci. Technol., 2018, 8, 3853–3862, 10.1039/c8cy00339d.
- N. Muradov, Catalysis of methane decomposition over elemental carbon, Catal. Commun., 2001, 2, 89–94, DOI:10.1016/S1566-7367(01)00013-9.
- M. H. Kim, E. K. Lee, J. H. Jun, S. J. Kong, G. Y. Han, B. K. Lee, T. J. Lee and K. J. Yoon, Hydrogen production by catalytic decomposition of methane over activated carbons: Kinetic study, Int. J. Hydrogen Energy, 2004, 29, 187–193, DOI:10.1016/S0360-3199(03)00111-3.
- S. Krzyzyński and M. Kozłowski, Activated carbons as catalysts for hydrogen production via methane decomposition, Int. J. Hydrogen Energy, 2008, 33, 6172–6177, DOI:10.1016/j.ijhydene.2008.07.091.
- N. Muradov, Z. Chen and F. Smith, Fossil hydrogen with reduced CO2 emission: Modeling thermocatalytic decomposition of methane in a fluidized bed of carbon particles, Int. J. Hydrogen Energy, 2005, 30, 1149–1158, DOI:10.1016/j.ijhydene.2005.04.005.
- F. Cepeda, L. Di Liddo and M. J. Thomson, Enhancing hydrogen production: Modelling the role of activated carbon catalyst in methane pyrolysis, Int. J. Hydrogen Energy, 2024, 83, 410–420, DOI:10.1016/j.ijhydene.2024.08.056.
- D. P. Serrano, J. A. Botas and R. Guil-Lopez, H2 production from methane pyrolysis over commercial carbon catalysts: Kinetic and deactivation study, Int. J. Hydrogen Energy, 2009, 34, 4488–4494, DOI:10.1016/j.ijhydene.2008.07.079.
- S. Y. Lee, J. H. Kwak, G. Y. Han, T. J. Lee and K. J. Yoon, Characterization of active sites for methane decomposition on carbon black through acetylene chemisorption, Carbon, 2008, 46, 342–348, DOI:10.1016/j.carbon.2007.11.049.
- T. Tokunaga, N. Kishi, K. Yamakawa, K. Sasaki and T. Yamamoto, Methane decomposition for hydrogen production by catalytic activity of carbon black under low flow rate conditions, J. Ceram. Soc. Jpn., 2017, 125, 185–189, DOI:10.2109/jcersj2.16246.
- Y. Kameya and K. Hanamura, Kinetic and Raman spectroscopic study on catalytic characteristics of carbon blacks in methane decomposition, Chem. Eng. J., 2011, 173, 627–635, DOI:10.1016/j.cej.2011.08.017.
- Z. Yuan Chen, L. Zhen Bian, L. Jun Wang, Z. You Yu, H. Lei Zhao, F. Shen Li and K. Chih Chou, Topography, structure, and formation kinetic mechanism of carbon deposited onto nickel in the temperature range from 400 to 850 °C, Int. J. Miner., Metall. Mater., 2017, 24, 574–583, DOI:10.1007/s12613-017-1439-9.
- L. Chen, L. E. Noreña, J. A. Wang, R. Limas, U. Arellano and O. A. G. Vargas, Promoting role of amorphous carbon and carbon nanotubes growth modes of methane decomposition in one-pot catalytic approach, Catalysts, 2021, 11, 1217, DOI:10.3390/catal11101217.
- I. R. Hamdani, A. Ahmad, H. M. Chulliyil, C. Srinivasakannan, A. A. Shoaibi and M. M. Hossain, Thermocatalytic Decomposition of Methane: A Review on Carbon-Based Catalysts, ACS Omega, 2023, 8, 28945–28967, DOI:10.1021/acsomega.3c01936.
- D. P. Serrano, J. A. Botas, J. L. G. Fierro, R. Guil-López, P. Pizarro and G. Gómez, Hydrogen production by methane decomposition: Origin of the catalytic activity of carbon materials, Fuel, 2010, 89, 1241–1248, DOI:10.1016/j.fuel.2009.11.030.
- S. R. Patlolla, K. Katsu, A. Sharafian, K. Wei, O. E. Herrera and W. Mérida, A review of methane pyrolysis technologies for hydrogen production, Renewable Sustainable Energy Rev., 2023, 181, 113323, DOI:10.1016/j.rser.2023.113323.
- S. Takenaka, E. Kato, Y. Tomikubo and K. Otsuka, Structural change of Ni species during the methane decomposition and the subsequent gasification of deposited carbon with CO2 over supported Ni catalysts, J. Catal., 2003, 219, 176–185, DOI:10.1016/S0021-9517(03)00152-0.
- J. I. Villacampa, C. Royo, E. Romeo, J. A. Montoya, P. Del Angel and A. Monzón, Catalytic decomposition of methane over Ni-Al2O3 coprecipitated catalysts Reaction and regeneration studies, Appl. Catal., A, 2003, 252, 363–383, DOI:10.1016/S0926-860X(03)00492-7.
- P. Ammendola, R. Chirone, G. Ruoppolo and G. Russo, Combust. Sci. Technol., 2008, 180, 869–882 CrossRef CAS.
- H. F. Abbas and W. M. A. W. Daud, An experimental investigation into the CO2 gasification of deactivated activated-carbon catalyst used for methane decomposition to produce hydrogen, Int. J. Hydrogen Energy, 2010, 35, 141–150, DOI:10.1016/j.ijhydene.2009.10.072.
- J. Zhang, X. Li, H. Chen, M. Qi, G. Zhang, H. Hu and X. Ma, Hydrogen production by catalytic methane decomposition: Carbon materials as catalysts or catalyst supports, Int. J. Hydrogen Energy, 2017, 42, 19755–19775, DOI:10.1016/j.ijhydene.2017.06.197.
- J. L. Pinilla, I. Suelves, R. Utrilla, M. E. Gálvez, M. J. Lázaro and R. Moliner, Hydrogen production by thermo-catalytic decomposition of methane: Regeneration of active carbons using CO2, J. Power Sources, 2007, 169, 103–109, DOI:10.1016/j.jpowsour.2007.01.045.
- Z. Q. Sun, J. H. Wu, M. Haghighi, J. Bromly, E. Ng, H. L. Wee, Y. Wang and D. K. Zhang, Methane cracking over a bituminous coal char, Energy Fuels, 2007, 21, 1601–1605, DOI:10.1021/ef060616v.
- A. Dufour, A. Celzard, V. Fierro, F. Broust, C. Courson, A. Zoulalian and J. N. Rouzaud, Catalytic conversion of methane over a biomass char for hydrogen production: Deactivation and regeneration by steam gasification, Appl. Catal., A, 2015, 490, 170–180, DOI:10.1016/j.apcata.2014.10.038.
- S. Tong, B. Miao, W. Zhang, L. Zhang and S. H. Chan, Optimization of methane catalytic decomposition in a fluidized bed reactor: A computational approach, Energy Convers. Manage., 2023, 297, 117719, DOI:10.1016/j.enconman.2023.117719.
- N. Muradov, Proceedings of the 2000 Hydrogen Program Review, 2000, 1–14.
- T. Marquardt, A. Bode and S. Kabelac, Hydrogen production by methane decomposition: Analysis of thermodynamic carbon properties and process evaluation, Energy Convers. Manage., 2020, 221, 113125, DOI:10.1016/j.enconman.2020.113125.
- J. L. Pinilla, R. Utrilla, R. K. Karn, I. Suelves, M. J. Lázaro, R. Moliner, A. B. García and J. N. Rouzaud, High temperature iron-based catalysts for hydrogen and nanostructured carbon production by methane decomposition, Int. J. Hydrogen Energy, 2011, 36, 7832–7843, DOI:10.1016/j.ijhydene.2011.01.184.
- A. M. Dunker, S. Kumar and P. A. Mulawa, Production of hydrogen by thermal decomposition of methane in a fluidized-bed reactor - Effects of catalyst, temperature, and residence time, Int. J. Hydrogen Energy, 2006, 31, 473–484, DOI:10.1016/j.ijhydene.2005.04.023.
- N. Muradov, Hydrogen via methane decomposition: An application for decarbonization of fossil fuels, Int. J. Hydrogen Energy, 2001, 26, 1165–1175, DOI:10.1016/S0360-3199(01)00073-8.
- M. Hadian, D. P. F. Marrevee, K. A. Buist, B. H. Reesink, A. N. R. Bos, A. P. van Bavel and J. A. M. Kuipers, Kinetic study of thermocatalytic decomposition of methane over nickel supported catalyst in a fluidized bed reactor, Chem. Eng. Sci., 2022, 260, 1179938, DOI:10.1016/j.ces.2022.117938.
- E. E. S. Michaelides, SpringerBriefs in Applied Sciences and Technology, Springer Verlag, 2013, pp. 89–119 Search PubMed.
- A. Helmi, E. Fernandez, J. Melendez, D. A. P. Tanaka, F. Gallucci and M. Van Sint Annaland, Fluidized bed membrane reactors for ultra pure H2 production - A step forward towards commercialization, Molecules, 2016, 21, 376, DOI:10.3390/molecules21030376.
- N. De Nooijer, F. Gallucci and M. Van Sint Annaland, in Fluidization XVI, AIChE, 2019, 119–121.
- J. G. Yates and P. Lettieri, Fluidized-Bed Reactors: Processes and Operating Conditions, 2016, vol. 26 Search PubMed.
- A. Łamacz and G. Łabojko, CNT and H2 production during CH4 decomposition over Ni/CeZrO2. II. catalyst performance and its regeneration in a fluidized bed, Chem. Eng., 2019, 3, 1–18, DOI:10.3390/chemengineering3010025.
- K. K. Lee, G. Y. Han, K. J. Yoon and B. K. Lee, Catal. Today, 2004, 93–95, 81–86 CrossRef CAS.
- A. C. Pivem and M. J. S. De Lemos, Laminar heat transfer in a moving porous bed reactor simulated with a macroscopic two-energy equation model, Int. J. Heat Mass Transfer, 2012, 55, 1922–1930, DOI:10.1016/j.ijheatmasstransfer.2011.11.047.
- Y. Ku, H. C. Wu, P. C. Chiu, Y. H. Tseng and Y. L. Kuo, Methane combustion by moving bed fuel reactor with Fe2O3/Al2O3 oxygen carriers, Appl. Energy, 2014, 113, 1909–1915, DOI:10.1016/j.apenergy.2013.06.014.
- A. Gurgen, Nuclear Power Plant Design and Analysis Codes, 2021, pp. 261–276 Search PubMed.
- B. Bulfin, Thermodynamic limits of countercurrent reactor systems, with examples in membrane reactors and the ceria redox cycle, Phys. Chem. Chem. Phys., 2019, 21, 2186–2195, 10.1039/c8cp07077f.
- A. A. Munera Parra, F. Platte and D. W. Agar, Multiplicity Regions in a Moving-Bed Reactor: Bifurcation Analysis, Model Extension, and Application for the High-Temperature Pyrolysis of Methane, Chem. Ing. Tech., 2016, 88, 1703–1714, DOI:10.1002/cite.201600069.
- M. Shirzad, M. Karimi, J. A. C. Silva and A. E. Rodrigues, Moving Bed Reactors: Challenges and Progress of Experimental and Theoretical Studies in a Century of Research, Ind. Eng. Chem. Res., 2019, 58, 9179–9198, DOI:10.1021/acs.iecr.9b01136.
- WO2013004398A8, 2012.
- S. Schneider, S. Bajohr, F. Graf and T. Kolb, State of the Art of Hydrogen Production via Pyrolysis of Natural Gas, ChemBioEng Rev., 2020, 7, 150–158, DOI:10.1002/cben.202000014.
- A. Bode, D. W. Agar, K. Büker, M. Hensmann, J. Hunfeld, U. Janhsen, D. Klingler and S. Schunck, Methane Pyrolysis and CO2 Activation – Technologies with Application Options for Hydrogen, Carbon and Synthesis Gas Production, Chem. Ing. Tech., 2016, 88, 1342, DOI:10.1002/cite.201650483.
- S. G. Zavarukhin and G. G. Kuvshinov, Mathematical modeling of the continuous process for synthesis of nanofibrous carbon in a moving catalyst bed reactor with recirculating gas flow, Chem. Eng. J., 2008, 137, 681–685, DOI:10.1016/j.cej.2007.06.036.
- J. S. Yun, J. H. Kim, S. C. Kang and J. S. Im, Mechanism of activated carbon-catalyzed methane decomposition process for the production of hydrogen and high-value carbon, Carbon Lett., 2023, 33, 1799–1809, DOI:10.1007/s42823-023-00516-0.
- S. Ahmed, A. Aitani, F. Rahman, A. Al-Dawood and F. Al-Muhaish, Decomposition of hydrocarbons to hydrogen and carbon, Appl. Catal., A, 2009, 359, 1–24, DOI:10.1016/j.apcata.2009.02.038.
- O. Olsvik and F. Billaud, Thermal coupling of methane. A comparison between kinetic model data and experimental data, Thermochim. Acta, 1994, 232, 155–169, DOI:10.1016/0040-6031(94)80055-3.
- A. Holmen, O. Olsvik and O. A. Rokstad, Pyrolysis of natural gas: chemistry and process concepts, Fuel Process. Technol., 1995, 42, 249–267, DOI:10.1016/0378-3820(94)00109-7.
- E. A. Blekkan, R. Myrstad, O. Olsvik and O. A. Rokstad, Characterization of tars and coke formed during the pyrolysis of methane in a tubular reactor, Carbon, 1992, 30, 665–673, DOI:10.1016/0008-6223(92)90186-Z.
- Z. M. Wang, X. Zhang, J. M. Lei, K. R. Jin, D. Wang and Z. Y. Tian, Revisit the PAH and soot formation in high-temperature pyrolysis of methane, J. Anal. Appl. Pyrolysis, 2024, 182, 106668, DOI:10.1016/j.jaap.2024.106668.
- C.-J. Chen, M. H. Back and R. A. Back, The thermal decomposition of methane. II. Secondary reactions, autocatalysis and carbon formation; non-Arrhenius behaviour in the reaction of CH 3 with ethane, Can. J. Chem., 1976, 54, 3175–3184, DOI:10.1139/v76-452.
- G. I. Kozlov and V. G. Knorre, Single-pulse shock tube studies on the kinetics of the thermal decomposition of methane, Combust. Flame, 1962, 6, 253–263, DOI:10.1016/0010-2180(62)90103-7.
- L. S. Kassel, The thermal decomposition of methane1, J. Am. Chem. Soc., 1932, 54, 3949–3961, DOI:10.1021/ja01349a019.
- A. Fridman, Plasma chemistry, Cambridge University Press, 2008, vol. 9780521847 Search PubMed.
- N. Pournoori, H. Delavari and M. Madah, Radio wave/microwave-involved methods for cancer diagnosis, Electromagnetic Waves-Based Cancer Diagnosis and Therapy: Principles and Applications of Nanomaterials, Academic Press, 2023, pp. 1–64 DOI:10.1016/B978-0-323-99628-0.00002-2.
- J. Li, J. Dai, G. Liu, H. Zhang, Z. Gao, J. Fu, Y. He and Y. Huang, Biochar from microwave pyrolysis of biomass: A review, Biomass Bioenergy, 2016, 94, 228–244, DOI:10.1016/J.BIOMBIOE.2016.09.010.
- A. Cappelli, L. Lupori and E. Cini, Baking technology: A systematic review of machines and plants and their effect on final products, including improvement strategies, Trends Food Sci. Technol., 2021, 115, 275–284, DOI:10.1016/j.tifs.2021.06.048.
- X. Zhang, K. Rajagopalan, H. Lei, R. Ruan and B. K. Sharma, An overview of a novel concept in biomass pyrolysis: microwave irradiation, Sustainable Energy Fuels, 2017, 1, 1664–1699, 10.1039/C7SE00254H.
- T. Christiansen, B. Robinson, A. Caiola, C. Jiang and J. Hu, Improved Efficiency of the Microwave-Enhanced Catalytic Pyrolysis of Methane through Supplemental Thermal Heating, Ind. Eng. Chem. Res., 2022, 61, 15832–15841, DOI:10.1021/acs.iecr.2c02093.
- M. Dadsetan, M. F. Khan, M. Salakhi, E. R. Bobicki and M. J. Thomson, CO2-free hydrogen production via microwave-driven methane pyrolysis, Int. J. Hydrogen Energy, 2023, 48, 14565–14576, DOI:10.1016/j.ijhydene.2022.12.353.
- C. Jiang, I. W. Wang, X. Bai, S. Balyan, B. Robinson, J. Hu, W. Li, A. Deibel, X. Liu, F. Li, L. M. Neal, J. Dou, Y. Jiang, R. Dagle, J. A. Lopez-Ruiz and G. Skoptsov, Methane Catalytic Pyrolysis by Microwave and Thermal Heating over Carbon Nanotube-Supported Catalysts: Productivity, Kinetics, and Energy Efficiency, Ind. Eng. Chem. Res., 2022, 61, 5080–5092, DOI:10.1021/acs.iecr.1c05082.
- Y. Y. Tanashev, V. I. Fedoseev, Y. I. Aristov, V. V. Pushkarev, L. B. Avdeeva, V. I. Zaikovskii and V. N. Parmon, Methane processing under microwave radiation: Recent ®ndings and problems.
- S. I. Galanov, A. G. Zherlitsyn, Y. V. Medvedev, O. I. Sidorova and V. P. Shiyan, Production of a highly dispersed carbon material and hydrogen from natural gas in a microwave reactor with metallic catalysts, Russ. J. Appl. Chem., 2011, 84, 997–1002, DOI:10.1134/S1070427211060176.
- B. Fidalgo, Y. Fernández, L. Zubizarreta, A. Arenillas, A. Domínguez, J. J. Pis and J. A. Menéndez, Growth of nanofilaments on carbon-based materials from microwave-assisted decomposition of CH 4, Appl. Surf. Sci., 2008, 254, 3553–3557, DOI:10.1016/j.apsusc.2007.11.037.
- C. Jiang, A. Araia, S. Balyan, B. Robinson, S. Brown, A. Caiola, J. Hu, J. Dou, L. M. Neal and F. Li, Kinetic study of Ni-M/CNT catalyst in methane decomposition under microwave irradiation, Appl. Catal., B, 2024, 340, 123255, DOI:10.1016/J.APCATB.2023.123255.
- K. Conley and A. J. Karttunen, Bridging the Junction: Electrical Conductivity of Carbon Nanotube Networks, J. Phys. Chem. C, 2022, 126, 17266–17274, DOI:10.1021/acs.jpcc.2c03904.
- B. Fidalgo, Y. Fernández, A. Domínguez, J. J. Pis and J. A. Menéndez, Microwave-assisted pyrolysis of CH4/N2 mixtures over activated carbon, J. Anal. Appl. Pyrolysis, 2008, 82, 158–162, DOI:10.1016/j.jaap.2008.03.004.
- J. A. Menéndez, A. Arenillas, B. Fidalgo, Y. Fernández, L. Zubizarreta, E. G. Calvo and J. M. Bermúdez, Microwave heating processes involving carbon materials, Fuel Process. Technol., 2010, 91, 1–8, DOI:10.1016/J.FUPROC.2009.08.021.
- Y. Deng, X. Bai, V. Abdelsayed, D. Shekhawat, P. D. Muley, S. Karpe, C. Mevawala, D. Bhattacharyya, B. Robinson, A. Caiola, J. B. Powell, A. P. van Bavel, J. Hu and G. Veser, Microwave-assisted conversion of methane over H-(Fe)-ZSM-5: Evidence for formation of hot metal sites, Chem. Eng. J., 2021, 420, 129670, DOI:10.1016/J.CEJ.2021.129670.
- A. Domínguez, B. Fidalgo, Y. Fernández, J. J. Pis and J. A. Menéndez, Microwave-assisted catalytic decomposition of methane over activated carbon for CO2-free hydrogen production, Int. J. Hydrogen Energy, 2007, 32, 4792–4799, DOI:10.1016/j.ijhydene.2007.07.041.
- L. Fulcheri, V. J. Rohani, E. Wyse, N. Hardman and E. Dames, An energy-efficient plasma methane pyrolysis process for high yields of carbon black and hydrogen, Int. J. Hydrogen Energy, 2023, 48, 2920–2928, DOI:10.1016/j.ijhydene.2022.10.144.
- J. Feng, X. Sun, Z. Li, X. Hao, M. Fan, P. Ning and K. Li, Plasma-Assisted Reforming of Methane, Adv. Sci., 2022, 9, 2203221, DOI:10.1002/advs.202203221.
- M. Wnukowski, Methane Pyrolysis with the Use of Plasma: Review of Plasma Reactors and Process Products, Energies, 2023, 16, 6441, DOI:10.3390/en16186441.
- M. Kheirollahivash, F. Rashidi and M. M. Moshrefi, Experimental study and kinetic modeling of methane decomposition in a rotating arc plasma reactor with different cross-sectional areas, Int. J. Hydrogen Energy, 2019, 44, 17460–17469, DOI:10.1016/j.ijhydene.2019.05.011.
- H. Zhang, C. Du, A. Wu, Z. Bo, J. Yan and X. Li, Rotating gliding arc assisted methane decomposition in nitrogen for hydrogen production, Int. J. Hydrogen Energy, 2014, 39, 12620–12635, DOI:10.1016/j.ijhydene.2014.06.047.
- S. Heijkers, M. Aghaei and A. Bogaerts, Plasma-Based CH4 Conversion into Higher Hydrocarbons and H2: Modeling to Reveal the Reaction Mechanisms of Different Plasma Sources, J. Phys. Chem. C, 2020, 124, 7016–7030, DOI:10.1021/acs.jpcc.0c00082.
- S. Kreuznacht, M. Purcel, S. Böddeker, P. Awakowicz, W. Xia, M. Muhler, M. Böke and A. von Keudell, Comparison of the performance of a microwave plasma torch and a gliding arc plasma for hydrogen production via methane pyrolysis, Plasma Processes Polym., 2023, 20, 2200132, DOI:10.1002/ppap.202200132.
- A. Indarto, J. W. Choi, H. Lee and H. K. Song, Effect of additive gases on methane conversion using gliding arc discharge, Energy, 2006, 31, 2986–2995, DOI:10.1016/j.energy.2005.10.034.
- Y. H. Lee, J. H. Oh and S. Choi, Evaluation of process conditions for methane pyrolysis applying the triple thermal plasma system, Int. J. Hydrogen Energy, 2023, 48, 27127–27136, DOI:10.1016/j.ijhydene.2023.03.427.
- A. Mašláni, M. Hrabovský, P. Křenek, M. Hlína, S. Raman, V. S. Sikarwar and M. Jeremiáš, Pyrolysis of methane via thermal steam plasma for the production of hydrogen and carbon black, Int. J. Hydrogen Energy, 2021, 46, 1605–1614, DOI:10.1016/j.ijhydene.2020.10.105.
- M. Heintze and M. Magureanu, Methane conversion into acetylene in a microwave plasma: Optimization of the operating parameters, J. Appl. Phys., 2002, 92, 2276–2283, DOI:10.1063/1.1497457.
- C. Shen, D. Sun and H. Yang, Methane coupling in microwave plasma under atmospheric pressure, J. Nat. Gas Chem., 2011, 20, 449–456, DOI:10.1016/S1003-9953(10)60209-5.
- M. Wnukowski, A. W. van de Steeg, B. Hrycak, M. Jasiński and G. J. van Rooij, Influence of hydrogen addition on methane coupling in a moderate pressure microwave plasma, Fuel, 2021, 288, 119674, DOI:10.1016/j.fuel.2020.119674.
- C. Xu and X. Tu, Plasma-assisted methane conversion in an atmospheric pressure dielectric barrier discharge reactor, J. Energy Chem., 2013, 22, 420–425, DOI:10.1016/S2095-4956(13)60055-8.
- M. Wnukowski, J. Gerber and K. Mróz, Shifts in Product Distribution in Microwave Plasma Methane Pyrolysis Due to Hydrogen and Nitrogen Addition, Methane, 2022, 1, 286–299, DOI:10.3390/methane1040022.
- O. Khalifeh, A. Mosallanejad, H. Taghvaei, M. R. Rahimpour and A. Shariati, Decomposition of methane to hydrogen using nanosecond pulsed plasma reactor with different active volumes, voltages and frequencies, Appl. Energy, 2016, 169, 585–596, DOI:10.1016/j.apenergy.2016.02.017.
- A. Fridman, Plasma Chemistry, Cambridge University Press, 2009, pp. 1–11 Search PubMed.
- A. Bogaerts and E. C. Neyts, Plasma Technology: An Emerging Technology for Energy Storage, ACS Energy Lett., 2018, 3, 1013–1027, DOI:10.1021/acsenergylett.8b00184.
- S. PARIMI, thesis: Study of Methane Reforming in Warm Non-Equilibrium Plasma Discharges, Texas A&M University, 2010.
- P. C. Kong, B. a Detering, J. D. Grandy and L. D. Zuck, Plasma Processing of Hydrocarbon, Electric power 2007, 2007, 12.
- H. Gladisch, How Huels makes acetylene by dc arc, Hydrocarbon Processing and Petroleum Refining, 1962, 41, 159–164.
- J. Holmes, Evaluation of Dupont arc process for acetylene and vinyl chloride monomer production, U.S. Department of Energy Office of Scientific and Technical Information DOI:10.2172/1862128.
- S. B. Harrison, Turquoise hydrogen production by methane pyrolysis, 2021, 2–9, https://www.sbh4.de/assets/turquoise-hydrogen-production-by-methane-pyrolysis%2C-petroleum-technology-quarterly-october-2021.pdf.
- L. Fulcheri and Y. Schwob, From methane to hydrogen, carbon black and water, Int. J. Hydrogen Energy, 1995, 20, 197–202, DOI:10.1016/0360-3199(94)E0022-Q.
- M. Gautier, V. Rohani and L. Fulcheri, Direct decarbonization of methane by thermal plasma for the production of hydrogen and high value-added carbon black, Int. J. Hydrogen Energy, 2017, 42, 28140–28156, DOI:10.1016/j.ijhydene.2017.09.021.
- L. Fulcheri, F. Fabry, S. Takali and V. Rohani, Three-Phase AC Arc Plasma Systems: A Review, Plasma Chem. Plasma Process., 2015, 35, 565–585, DOI:10.1007/s11090-015-9619-8.
- K. S. Kim, J. H. Seo, J. S. Nam, W. T. Ju and S. H. Hong, Production of hydrogen and carbon black by methane decomposition using DC-RF hybrid thermal plasmas, IEEE Trans. Plasma Sci., 2005, 33, 813–823, DOI:10.1109/TPS.2005.844526.
- J. R. Fincke, R. P. Anderson, T. A. Hyde and B. A. Detering, Plasma pyrolysis of methane to hydrogen and carbon black, Ind. Eng. Chem. Res., 2002, 41, 1425–1435, DOI:10.1021/ie010722e.
- T. Li, C. Rehmet, Y. Y. Cheng, Y. Jin and Y. Y. Cheng, Experimental Comparison of Methane Pyrolysis in Thermal Plasma, Plasma Chem. Plasma Process., 2017, 37, 1033–1049, DOI:10.1007/s11090-017-9806-x.
- B. Wang, W. Yan, W. Ge and X. Duan, Methane conversion into higher hydrocarbons with dielectric barrier discharge micro-plasma reactor, J. Energy Chem., 2013, 22, 876–882, DOI:10.1016/S2095-4956(14)60267-9.
- L. Liu, S. Das, Z. Zhang and S. Kawi, Nonoxidative Coupling of Methane over Ceria-Supported Single-Atom Pt Catalysts in DBD Plasma, ACS Appl. Mater. Interfaces, 2022, 14, 5363–5375, DOI:10.1021/acsami.1c21550.
- A. Górska, K. Krawczyk, S. Jodzis and K. Schmidt-Szałowski, Non-oxidative methane coupling using Cu/ZnO/Al2O3 catalyst in DBD, Fuel, 2011, 90, 1946–1952, DOI:10.1016/j.fuel.2010.12.023.
- M. Taheraslani and H. Gardeniers, Plasma catalytic conversion of CH4 to Alkanes, olefins and H2 in a packed bed DBD reactor, Processes, 2020, 8, 774, DOI:10.3390/PR8070774.
- P. Chawdhury, S. Bhanudas Rawool, M. Umamaheswara Rao and C. Subrahmanyam, Methane decomposition by plasma-packed bed non-thermal plasma reactor, Chem. Eng. Sci., 2022, 258, 117779, DOI:10.1016/j.ces.2022.117779.
- K. Onoe, A. Fujie, T. Yamaguchi and Y. Hatano, Selective synthesis of acetylene from methane by microwave plasma reactions, Fuel, 1997, 76, 281–282, DOI:10.1016/S0016-2361(96)00228-1.
- M. Heintze, M. Magureanu and M. Kettlitz, Mechanism of C2 hydrocarbon formation from methane in a pulsed microwave plasma, J. Appl. Phys., 2002, 92, 7022–7031, DOI:10.1063/1.1521518.
- T. Minea, D. C. M. van den Bekerom, F. J. J. Peeters, E. Zoethout, M. F. Graswinckel, M. C. M. van de Sanden, T. Cents, L. Lefferts and G. J. van Rooij, Non-oxidative methane coupling to C2 hydrocarbons in a microwave plasma reactor, Plasma Processes Polym., 2018, 15, 1800087, DOI:10.1002/ppap.201800087.
- W. Cho, S. H. Lee, W. S. Ju, Y. Baek and J. K. Lee, Catal. Today, 2004, 98, 633–638 CrossRef CAS.
- M. Jasiński, M. Dors and J. Mizeraczyk, Application of atmospheric pressure microwave plasma source for production of hydrogen via methane reforming, Eur. Phys. J. D, 2009, 54, 179–183, DOI:10.1140/epjd/e2008-00221-1.
- M. Jasiński, M. Dors and J. Mizeraczyk, Production of hydrogen via methane reforming using atmospheric pressure microwave plasma, J. Power Sources, 2008, 181, 41–45, DOI:10.1016/j.jpowsour.2007.10.058.
- C. H. Tsai and K. T. Chen, Production of hydrogen and nano carbon powders from direct plasmalysis of methane, Int. J. Hydrogen Energy, 2009, 34, 833–838, DOI:10.1016/J.IJHYDENE.2008.10.061.
- M. Singh, A. Sengupta, K. Zeller, G. Skoptsov and R. L. van der Waals, Effect of hydrogen concentration on graphene synthesis using microwave-driven plasma-mediated methane cracking, Carbon, 2019, 143, 802–813, DOI:10.1016/j.carbon.2018.11.082.
- A. Czernichowski and P. Czernichowski, Proceedings of the 10th Canadian Hydrogen Conference, Quebec, 2000.
- M. Zhou, Z. Yang, J. Ren, T. Zhang, W. Xu and J. Zhang, Non-oxidative coupling reaction of methane to hydrogen and ethene via plasma-catalysis process, Int. J. Hydrogen Energy, 2023, 48, 78–89, DOI:10.1016/j.ijhydene.2022.09.252.
- K. Wang, W. S. Li and X. P. Zhou, Hydrogen generation by direct decomposition of hydrocarbons over molten magnesium, J. Mol. Catal. A: Chem., 2008, 283, 153–157, DOI:10.1016/j.molcata.2007.12.018.
- M. Msheik, S. Rodat and S. Abanades, Experimental comparison of solar methane pyrolysis in gas-phase and molten-tin bubbling tubular reactors, Energy, 2022, 260, 124943, DOI:10.1016/j.energy.2022.124943.
- D. Kang, N. Rahimi, M. J. Gordon, H. Metiu and E. W. McFarland, Catalytic methane pyrolysis in molten MnCl2-KCl, Appl. Catal., B, 2019, 254, 659–666, DOI:10.1016/j.apcatb.2019.05.026.
- B. T. Le, S. I. Ngo, Y. Il Lim and U. Do Lee, One-dimensional kinetic model with heat transfer and axial dispersion of molten-metal bubble column reactors for hydrogen production via methane pyrolysis, Int. J. Hydrogen Energy, 2023, 48, 35821–35837, DOI:10.1016/j.ijhydene.2023.06.031.
- S. I. Ngo, Y. Il Lim, H. M. Kwon and U. Do Lee, Hydrodynamics of molten-metal bubble columns in the near-bubbling field using volume of fluid computational fluid dynamics, Chem. Eng. J., 2023, 454, 140073, DOI:10.1016/j.cej.2022.140073.
- F. Angikath, F. Abdulrahman, A. Yousry, R. Das, S. Saxena, O. Behar, H. Alhamed, T. Altmann, B. Dally and S. M. Sarathy, Technoeconomic assessment of hydrogen production from natural gas pyrolysis in molten bubble column reactors, Int. J. Hydrogen Energy, 2024, 49, 246–262, DOI:10.1016/j.ijhydene.2023.07.308.
- B. Parkinson, C. F. Patzschke, D. Nikolis, S. Raman and K. Hellgardt, Molten salt bubble columns for low-carbon hydrogen from CH4 pyrolysis: Mass transfer and carbon formation mechanisms, Chem. Eng. J., 2021, 417, 127407, DOI:10.1016/j.cej.2020.127407.
- S. Park, M. Kim, Y. Koo, D. Kang, Y. Kim, J. Park and C. Ryu, Numerical modeling of methane pyrolysis in a bubble column of molten catalysts for clean hydrogen production, Int. J. Hydrogen Energy, 2023, 48, 7385–7399, DOI:10.1016/j.ijhydene.2022.11.068.
- F. Angikath, G. Pezzella and S. M. Sarathy, Bubble-Size Distribution and Hydrogen Evolution from Pyrolysis of Hydrocarbon Fuels in a Simulated Ni0.27Bi0.73Column Reactor, Ind. Eng. Chem. Res., 2022, 61, 12369–12382, DOI:10.1021/acs.iecr.2c01148.
- T. Hibiki, T. Takamasa and M. Ishii, One-dimensional drift-flux model and constitutive equations for relative motion between phases in various two-phase flow regimes at microgravity conditions, Proceedings of the International Conference on Nuclear Engineering (ICONE12), 1977, 3, 377–386, DOI:10.1115/icone12-49037.
- K. Akita and F. Yoshida, Bubble Size, Interfacial Area, and Liquid-Phase Mass Transfer Coefficient in Bubble Columns, Industrial and Engineering Chemistry Process Design and Development, 1974, 13, 84–91, DOI:10.1021/i260049a016.
- D. C. Blanchard and L. D. Syzdek, Production of air bubbles of a specified size, Chem. Eng. Sci., 1977, 32, 1109–1112, DOI:10.1016/0009-2509(77)80150-4.
- A. A. Kulkarni and J. B. Joshi, Bubble formation and bubble rise velocity in gas-liquid systems: A review, Ind. Eng. Chem. Res., 2005, 44, 5873–5931, DOI:10.1021/ie049131p.
- R. J. Andreini, J. S. Foster and R. W. Callen, Characterization of gas bubbles injected into molten metals under laminar flow conditions, Metall. Trans. B, 1977, 8, 625–631, DOI:10.1007/BF02669340.
- M. Serban, M. A. Lewis, C. L. Marshall and R. D. Doctor, Hydrogen production by direct contact pyrolysis of natural gas, Energy Fuels, 2003, 17, 705–713, DOI:10.1021/ef020271q.
- T. Tate, On the magnitude of a drop of liquid formed under different circumstances, London, Edinburgh Dublin Philos. Mag. J. Sci., 1864, 27, 176–180, DOI:10.1080/14786446408643645.
- K. Mori, M. Sano and T. Sato, Size Of Bubbles Formed At Single Nozzle Immersed In Molten Iron, Trans. Iron Steel Inst. Jpn., 1979, 19, 553–558, DOI:10.2355/isijinternational1966.19.553.
- D. Scheiblehner, D. Neuschitzer, S. Wibner, A. Sprung and H. Antrekowitsch, Hydrogen production by methane pyrolysis in molten binary copper alloys, Int. J. Hydrogen Energy, 2023, 48, 6233–6243, DOI:10.1016/j.ijhydene.2022.08.115.
- T. Geißler, A. Abánades, A. Heinzel, K. Mehravaran, G. Müller, R. K. Rathnam, C. Rubbia, D. Salmieri, L. Stoppel, S. Stückrad, A. Weisenburger, H. Wenninger and T. Wetzel, Hydrogen production via methane pyrolysis in a liquid metal bubble column reactor with a packed bed, Chem. Eng. J., 2016, 299, 192–200, DOI:10.1016/j.cej.2016.04.066.
- E. Palo, V. Cosentino, G. Iaquaniello, V. Piemonte and E. Busillo, Thermal Methane Cracking on Molten Metal: Kinetics Modeling for Pilot Reactor Design, Processes, 2023, 11, 1537, DOI:10.3390/pr11051537.
- K. Isao and I. Mamoru, Drift flux model for large diameter pipe and new correlation for pool void fraction, Int. J. Heat Mass Transfer, 1987, 30, 1927–1939, DOI:10.1016/0017-9310(87)90251-1.
- D. C. Upham, V. Agarwal, A. Khechfe, Z. R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Catalytic molten metals for the direct conversion of methane to hydrogen and separable carbon, Science, 2017, 358, 917–921, DOI:10.1126/science.aao5023.
- C. Palmer, M. Tarazkar, M. J. Gordon, H. Metiu and E. W. Mcfarland, Methane pyrolysis in low-cost, alkali-halide molten salts at high temperatures, Sustainable Energy Fuels, 2021, 5, 6107–6123, 10.1039/d1se01408k.
- J. Zeng, M. Tarazkar, T. Pennebaker, M. J. Gordon, H. Metiu and E. W. McFarland, Catalytic Methane Pyrolysis with Liquid and Vapor Phase Tellurium, ACS Catal., 2020, 10, 8223–8230, DOI:10.1021/acscatal.0c00805.
- M. Liu, Z. Huang, Y. Zhou, J. Zhan, K. Zhang, M. Yang and Y. Zhou, Optimized Process for Melt Pyrolysis of Methane to Produce Hydrogen and Carbon Black over Ni Foam/NaCl-KCl Catalyst, Processes, 2023, 11, 1–11, DOI:10.3390/pr11020360.
- N. Rahimi, D. Kang, J. Gelinas, A. Menon, M. J. Gordon, H. Metiu and E. W. McFarland, Solid carbon production and recovery from high temperature methane pyrolysis in bubble columns containing molten metals and molten salts, Carbon, 2019, 151, 181–191, DOI:10.1016/j.carbon.2019.05.041.
- B. Parkinson, C. F. Patzschke, D. Nikolis, S. Raman, D. C. Dankworth and K. Hellgardt, Methane pyrolysis in monovalent alkali halide salts: Kinetics and pyrolytic carbon properties, Int. J. Hydrogen Energy, 2021, 46, 6225–6238, DOI:10.1016/j.ijhydene.2020.11.150.
- A. A. M. Parra and D. W. Agar, Molten metal capillary reactor for the high-temperature pyrolysis of methane, Int. J. Hydrogen Energy, 2016, 42, 13641–13648, DOI:10.1016/j.ijhydene.2016.12.044.
- D. Neuschitzer, D. Scheiblehner, H. Antrekowitsch, S. Wibner and A. Sprung, Methane Pyrolysis in a Liquid Metal Bubble Column Reactor for CO2-Free Production of Hydrogen, Energies, 2023, 16, 7058, DOI:10.3390/en16207058.
- C. Palmer, M. Tarazkar, H. H. Kristoffersen, J. Gelinas, M. J. Gordon, E. W. McFarland and H. Metiu, Methane Pyrolysis with a Molten Cu-Bi Alloy Catalyst, ACS Catal., 2019, 9, 8337–8345, DOI:10.1021/acscatal.9b01833.
- B. Jos and M. Jim, Methane pyrolysis in a molten gallium bubble column reactor for sustainable hydrogen production: Proof of concept & techno-economic assessment, Int. J. Hydrogen Energy, 2021, 46, 4917–4935, DOI:10.1016/j.ijhydene.2020.11.079.
- M. McConnachie, A. Sheil, M. Konarova and S. Smart, Evaluation of heterogeneous metal-sulfide molten salt slurry systems for hydrogen production through methane pyrolysis, Int. J. Hydrogen Energy, 2024, 49, 981–991, DOI:10.1016/j.ijhydene.2023.08.072.
- L. Chen, Z. Song, S. Zhang, C. K. Chang, Y. C. Chuang, X. Peng, C. Dun, J. J. Urban, J. Guo, J. L. Chen, D. Prendergast, M. Salmeron, G. A. Somorjai and J. Su, Ternary NiMo-Bi liquid alloy catalyst for efficient hydrogen production from methane pyrolysis, Science, 2023, 381, 857–861, DOI:10.1126/science.adh8872.
- T. Keipi, K. E. S. Tolvanen, H. Tolvanen and J. Konttinen, Thermo-catalytic decomposition of methane: The effect of reaction parameters on process design and the utilization possibilities of the produced carbon, Energy Convers. Manage., 2016, 126, 923–934, DOI:10.1016/j.enconman.2016.08.060.
- T. G. Wi, Y. J. Park, U. Lee and Y. B. Kang, Methane pyrolysis rate measurement using electromagnetic levitation techniques for turquoise hydrogen production: Liquid In, Ga, Bi, Sn, and Cu as catalysts, Chem. Eng. J., 2023, 460, 141558, DOI:10.1016/j.cej.2023.141558.
- Y. G. Noh, Y. J. Lee, J. Kim, Y. K. Kim, J. S. Ha, S. S. Kalanur and H. Seo, Enhanced efficiency in CO2-free hydrogen production from methane in a molten liquid alloy bubble column reactor with zirconia beads, Chem. Eng. J., 2021, 428, 131095, DOI:10.1016/j.cej.2021.131095.
- J. C. Seo, S. Park, G. Park, Y. Lee, S. J. Han and S. K. Kim, Catalytic CH4 pyrolysis promoted by the interface of a molten metal–salt hybrid system, Gas Sci. Eng., 2023, 115, 205017, DOI:10.1016/j.jgsce.2023.205017.
- M. Abuseada, R. M. Spearrin and T. S. Fisher, Influence of process parameters on direct solar-thermal hydrogen and graphite production via methane pyrolysis, Int. J. Hydrogen Energy, 2023, 48, 30323–30338, DOI:10.1016/j.ijhydene.2023.04.198.
- M. Abuseada and T. S. Fisher, Continuous solar-thermal methane pyrolysis for hydrogen and graphite production by roll-to-roll processing, Appl. Energy, 2023, 352, 121872, DOI:10.1016/j.apenergy.2023.121872.
- Y. Tang, P. Peng, S. Wang, Z. Liu, X. Zu and Q. Yu, Continuous production of graphite nanosheets by bubbling chemical vapor deposition using molten copper, Chem. Mater., 2017, 29, 8404–8411, DOI:10.1021/acs.chemmater.7b02958.
- T. Keipi, thesis: Technology Development and Techno-Economic Analysis of Hydrogen Production by Thermal Decomposition of Methane, 2017, https://tutcris.tut.fi/portal/en/publications/technology-development-and-technoeconomic-analysis-of-hydrogen-production-by-thermal-decomposition-of-methane(ebe2e83b-a19d-4aad-8dd2-4524b7c51cab).html.
- F. Wang, F. Wang, R. Hong, X. Lv and Y. Zheng, High-purity few-layer graphene from plasma pyrolysis of methane as conductive additive for LiFePO4 lithium ion battery, J. Mater. Res. Technol., 2020, 9, 10004–10015, DOI:10.1016/j.jmrt.2020.06.072.
- J. Prabowo, L. Lai, B. Chivers, D. Burke, A. H. Dinh, L. Ye, Y. Wang, Y. Wang, L. Wei and Y. Chen, Solid carbon co-products from hydrogen production by methane pyrolysis: Current understandings and recent progress, Carbon, 2024, 216, 118507, DOI:10.1016/j.carbon.2023.118507.
- Industrial Grade Multi Walled Carbon Nanotubes 10 nm, https://www.cheaptubes.com/product/industrial-grade-multi-walled-carbon-nanotubes-10nm/.
- Graphene Nanoplatelets, https://www.cheaptubes.com/product/graphene-nanoplatelets/.
- S. Khodabakhshi, P. F. Fulvio and E. Andreoli, Carbon black reborn: Structure and chemistry for renewable energy harnessing, Carbon, 2020, 162, 604–649, DOI:10.1016/j.carbon.2020.02.058.
- J. D. Jung, C. G. Jung and J. P. Bouysset, Recovered carbon black: potential markets, ire Waste and Recycling.
- Carbon Black Market Size, Share & Trends Analysis Report By Type, By Application (Tire, Non- Tire Rubber, Plastics, Inks & Coatings, Other), By Grade, By Region, And Segment Forecasts, 2024 - 2030, (accessed 2023), 2023, https://www.grandviewresearch.com/industry-analysis/carbon-black-market.
- Factory Price Carbon Black N220/N330/N550/N660/N990 Powder Carbon Black For Pigment Rubber Tyre Industry, https://www.alibaba.com/product-detail/Factory-Price-Carbon-Black-N220-N330_1600845464072.html?spm=a2700.7724857.0.0.708e985cUpiqUx.
- J. Riley, C. Atallah, R. Siriwardane and R. Stevens, Technoeconomic analysis for hydrogen and carbon Co-Production via catalytic pyrolysis of methane, Int. J. Hydrogen Energy, 2021, 46, 20338–20358, DOI:10.1016/j.ijhydene.2021.03.151.
- GRAPHITE MARKET, https://westwaterresources.net/minerals-portfolio/graphite-market/.
- R. A. Dagle, V. Dagle, M. D. Bearden, J. D. Holladay, T. R. Krause and S. Ahmed, An Overview of Natural Gas Conversion Technologies for Co-Production of Hydrogen and Value-Added Solid Carbon Products, (accessed 2017), 2017, https://www.osti.gov/biblio/1411934.
- Graphite facts, (accessed 2022), 2022, https://natural-resources.canada.ca/our-natural-resources/minerals-mining/mining-data-statistics-and-analysis/minerals-metals-facts/graphite-facts/24027.
- J. Zhang, C. Liang and J. B. Dunn, Graphite Flows in the U.S.: Insights into a Key Ingredient of Energy Transition, Environ. Sci. Technol., 2023, 57, 3402–3414, DOI:10.1021/acs.est.2c08655.
- M. Phagare, North America Calcined Petroleum Coke Market Report 2023 Market Size Split by Type, (accessed 2023), 2023, https://www.cognitivemarketresearch.com/regional-analysis/north-america-calcined-petroleum-coke-market-report.
- Global Calcined Petcoke Market Overview, (accessed 2023), 2023, https://www.marketresearchfuture.com/reports/calcined-petcoke-market-7897?utm_term=&utm_campaign=&utm_source=adwords&utm_medium=ppc&hsa_acc=2893753364&hsa_cam=20373630173&hsa_grp=154349590714&hsa_ad=665813627268&hsa_src=g&hsa_tgt=dsa-2089395973864&hsa_kw=.
- Per Ton Coconut Shell Price Activated Carbon Active Carbon 8 16 Mesh Chemical Auxiliary Agent Norit Activated Carbon Price in Kg, (accessed 1 November 2024), https://www.alibaba.com/product-detail/Per-Ton-Coconut-Shell-Price-Activated_1600729943730.html?spm=a2700.7724857.0.0.4c214d7amvKDSJ.
- H. U. Modekwe, O. O. Ayeleru, M. A. Onu, N. T. Tobias, M. A. Mamo, K. Moothi, M. O. Daramola and P. A. Olubambi, Handbook of Carbon Nanotubes, Springer International Publishing, 2022, pp. 619–633 Search PubMed.
- Single Walled Carbon Nanotubes, https://www.cheaptubes.com/product-category/single-walled-carbon-nanotubes/.
- X. Ma, M. Chen, B. Chen, Z. Meng and Y. Wang, High-Performance Graphite Recovered from Spent Lithium-Ion Batteries, ACS Sustainable Chem. Eng., 2019, 7, 19732–19738, DOI:10.1021/acssuschemeng.9b05003.
- Y. Ji, C. Palmer, E. E. Foley, R. Giovine, E. Yoshida, E. Sebti, A. R. Patterson, E. McFarland and R. J. Clément, Valorizing the carbon byproduct of methane pyrolysis in batteries, Carbon, 2023, 204, 26–35, DOI:10.1016/j.carbon.2022.12.044.
- M. Dadsetan, K. G. Latham, M. F. Khan, M. H. Zaher, S. Manzoor, E. R. Bobicki, M. M. Titirici and M. J. Thomson, Characterization of carbon products from microwave-driven methane pyrolysis, Carbon Trends, 2023, 12, 100277, DOI:10.1016/j.cartre.2023.100277.
- T. Echterhof, Review on the use of alternative carbon sources in EAF steelmaking, Metals, 2021, 11, 1–16, DOI:10.3390/met11020222.
- G. Mishra, A. Warda and S. P. Shah, Carbon sequestration in graphene oxide modified cementitious system, J. Build. Eng., 2022, 62, 105356, DOI:10.1016/j.jobe.2022.105356.
- E. Gonzalez Noya, D. Srivastava and M. Menon, Heat-pulse rectification in carbon nanotube y junctions, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79, 115432, DOI:10.1103/PhysRevB.79.115432.
- X. Zeng, D. Fu, H. Sheng, S. Xie, X. Li, Q. Hu and J. Zou, Growth and morphology of carbon nanostructures by microwave-assisted pyrolysis of methane, Phys. E, 2010, 42, 2103–2108, DOI:10.1016/j.physe.2010.04.002.
- Y. Yan, J. Miao, Z. Yang, F. X. Xiao, H. Bin Yang, B. Liu and Y. Yang, Carbon nanotube catalysts: Recent advances in synthesis, characterization and applications, Chem. Soc. Rev., 2015, 44, 3295–3346, 10.1039/c4cs00492b.
- D. Ayillath Kutteri, I. W. Wang, A. Samanta, L. Li and J. Hu, Methane decomposition to tip and base grown carbon nanotubes and COx-free H2 over mono- and bimetallic 3d transition metal catalysts, Catal.:Sci. Technol., 2018, 8, 858–869, 10.1039/c7cy01927k.
- J. Dijon, P. D. Szkutnik, A. Fournier, T. Goislard De Monsabert, H. Okuno, E. Quesnel, V. Muffato, E. De Vito, N. Bendiab, A. Bogner and N. Bernier, How to switch from a tip to base growth mechanism in carbon nanotube growth by catalytic chemical vapour deposition, Carbon, 2010, 48, 3953–3963, DOI:10.1016/j.carbon.2010.06.064.
- R. T. K. Baker, Catalytic growth of carbon filaments, Carbon, 1989, 27, 315–323, DOI:10.1016/0008-6223(89)90062-6.
- M. He, Y. Magnin, H. Amara, H. Jiang, H. Cui, F. Fossard, A. Castan, E. Kauppinen, A. Loiseau and C. Bichara, Linking growth mode to lengths of single-walled carbon nanotubes, Carbon, 2017, 113, 231–236, DOI:10.1016/j.carbon.2016.11.057.
- L. S. Lobo, Nucleation and growth of carbon nanotubes and nanofibers: Mechanism and catalytic geometry control, Carbon, 2017, 114, 411–417, DOI:10.1016/j.carbon.2016.12.005.
- P. Moodley, J. Loos, J. W. Niemantsverdriet and P. C. Thüne, Is there a correlation between catalyst particle size and CNT diameter, Carbon, 2009, 47, 2002–2013, DOI:10.1016/j.carbon.2009.03.046.
- A. Gohier, C. P. Ewels, T. M. Minea and M. A. Djouadi, Carbon nanotube growth mechanism switches from tip- to base-growth with decreasing catalyst particle size, Carbon, 2008, 46, 1331–1338, DOI:10.1016/j.carbon.2008.05.016.
- H. Dai, A. G. Rinzler, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Single-wall nanotubes produced by metal-catalyzed disproportionation of carbon monoxide, Chem. Phys. Lett., 1996, 260, 471–475, DOI:10.1016/0009-2614(96)00862-7.
- F. Ding, A. Rosén and K. Bolton, Molecular dynamics study of the catalyst particle size dependence on carbon nanotube growth, J. Chem. Phys., 2004, 121, 2775–2779, DOI:10.1063/1.1770424.
- M. He, P. V. Fedotov, A. Chernov, E. D. Obraztsova, H. Jiang, N. Wei, H. Cui, J. Sainio, W. Zhang, H. Jin, M. Karppinen, E. I. Kauppinen and A. Loiseau, Chiral-selective growth of single-walled carbon nanotubes on Fe-based catalysts using CO as carbon source, Carbon, 2016, 108, 521–528, DOI:10.1016/j.carbon.2016.07.048.
- V. L. Kuznetsov, A. N. Usoltseva, A. L. Chuvilin, E. D. Obraztsova and J. M. Bonard, Thermodynamic analysis of nucleation of carbon deposits on metal particles and its implications for the growth of carbon nanotubes, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 64, 2354011–2354017, DOI:10.1103/PhysRevB.64.235401.
- X. Chen, X. Pang and C. Fauteux-Lefebvre, The base versus tip growth mode of carbon nanotubes by catalytic hydrocarbon cracking: Review, challenges and opportunities, Carbon Trends, 2023, 12, 100273, DOI:10.1016/j.cartre.2023.100273.
- X. Zhang, A. Kätelhön, G. Sorda, M. Helmin, M. Rose, A. Bardow, R. Madlener, R. Palkovits and A. Mitsos, CO2 mitigation costs of catalytic methane decomposition, Energy, 2018, 151, 826–838, DOI:10.1016/j.energy.2018.03.132.
- J. M. Lane and P. L. Spath, Technoeconomic Analysis of the Thermocatalytic Decomposition of Natural Gas, U.S. Department of Energy Laboratory (NREL), (accessed 2001), 2001, https://www.nrel.gov/docs/fy02osti/31351.pdf.
- I. J. Okeke, B. A. Saville and H. L. MacLean, Low carbon hydrogen production in Canada via natural gas pyrolysis, Int. J. Hydrogen Energy, 2023, 48, 12581–12599, DOI:10.1016/j.ijhydene.2022.12.169.
- T. Keipi, H. Tolvanen and J. Konttinen, Economic analysis of hydrogen production by methane thermal decomposition: Comparison to competing technologies, Energy Convers. Manage., 2018, 159, 264–273, DOI:10.1016/j.enconman.2017.12.063.
- J. Boo, E. H. Ko, N. K. Park, C. Ryu, Y. H. Kim, J. Park and D. Kang, Methane pyrolysis in molten potassium chloride: An experimental and economic analysis, Energies, 2021, 14, 8182, DOI:10.3390/en14238182.
- B. Parkinson, M. Tabatabaei, D. C. Upham, B. Ballinger, C. Greig, S. Smart and E. McFarland, Hydrogen production using methane: Techno-economics of decarbonizing fuels and chemicals, Int. J. Hydrogen Energy, 2018, 43, 2540–2555, DOI:10.1016/j.ijhydene.2017.12.081.
- B. Parkinson, J. W. Matthews, T. B. McConnaughy, D. C. Upham and E. W. McFarland, Techno-Economic Analysis of Methane Pyrolysis in Molten Metals: Decarbonizing Natural Gas, Chem. Eng. Technol., 2017, 40, 1022–1030, DOI:10.1002/ceat.201600414.
- F. Kerscher, A. Stary, S. Gleis, A. Ulrich, H. Klein and H. Spliethoff, Low-carbon hydrogen production via electron beam plasma methane pyrolysis: Techno-economic analysis and carbon footprint assessment, Int. J. Hydrogen Energy, 2021, 46, 19897–19912, DOI:10.1016/j.ijhydene.2021.03.114.
- R. Dagle, V. Dagle, M. Bearden, J. Holladay, T. Krause and S. Ahmed, R&D opportunities for development of natural gas conversion technologies, Tech. Rep., 2017, 2.1–4.1 Search PubMed.
- M. E. Tabat, F. O. Omoarukhe, F. Güleç, D. E. Adeniyi, A. Mukherjee, P. U. Okoye, C. C. Ogbaga, E. I. Epelle, O. Akande and J. A. Okolie, Process design, exergy, and economic assessment of a conceptual mobile autothermal methane pyrolysis unit for onsite hydrogen production, Energy Convers. Manage., 2023, 278, 116707, DOI:10.1016/j.enconman.2023.116707.
- V. Spallina, D. Pandolfo, A. Battistella, M. C. Romano, M. Van Sint Annaland and F. Gallucci, Techno-economic assessment of membrane assisted fluidized bed reactors for pure H2 production with CO2 capture, Energy Convers. Manage., 2016, 120, 257–273, DOI:10.1016/j.enconman.2016.04.073.
- A. G. Chmielewski, Reviews of Accelerator Science and Technology: Accelerator Applications in Industry and the Environment, World Scientific Publishing Co., 2012, pp. 147–159 Search PubMed.
- B. Gaudernack and S. Lynum, Hydrogen from natural gas without release of CO2 to the atmosphere, Int. J. Hydrogen Energy, 1998, 23, 1087–1093, DOI:10.1016/S0360-3199(98)00004-4.
- J. Shah, Open For Business: LPO Issues New Conditional Commitment for Loan Guarantee, 2021, https://www.energy.gov/lpo/articles/open-business-lpo-issues-new-conditional-commitment-loan-guarantee.
- C. Fromm, Hydrogen Production via Methane Pyrolysis: An Overview of ‘Turquoise’ H2, 2024, https://www.chemengonline.com/fullscreen/hydrogen-production-via-methane-pyrolysis-an-overview-of-turquoise-h2/.
- Monolith raises more 300 million latest funding round led tpg rise climate decarbonization, 2022, https://therisefund.com/news/monolith-raises-more-300-million-latest-funding-round-led-tpg-rise-climate-decarbonization.
- In Industry First, Goodyear Launches Tire With Monolith's Carbon Black, 2023, https://monolith-corp.com/news/in-industry-first-goodyear-launches-tire-with-monolith-s-carbon-black.
- R. Daliah, Technology Landscape: Key Players in Methane Pyrolysis, (accessed 2021), 2021.
- Y. D. Korolev, O. B. Frants, N. V. Landl, V. G. Geyman, A. G. Zerlitsyn, V. P. Shiyan and Y. V. Medvedev, IEEE Trans. Plasma Sci., 2009, 37, 2298–2302 CAS.
- A. G. Zherlitsyn, V. P. Shiyan and P. V. Demchenko, Microwave plasma torch for processing hydrocarbon gases, Resource-Efficient Technologies, National Research Tomsk Polytechnic University, 2016, 2, 11–14, DOI:10.1016/j.reffit.2016.04.001.
- New technologies, https://www.basf.com/us/en/who-we-are/sustainability/we-produce-safely-and-efficiently/energy-and-climate-protection/carbon-management/innovations-for-a-climate-friendly-chemical-production.html#text-1002215085.
- Ekona Technology, https://ekonapower.com/technology/.
- Aurora Hydrog., https://aurorahydrogen.com/technology.
- A. Gillis, Aurora CEO speaks with Edmonton Global at the Canadian H2 Convention, 2023, https://aurorahydrogen.com/news/aurora-speaks-with-edmonton-global-at-the-canadian-h2-convention.
- Hazer Group Process and Updates, https://hazergroup.com.au/investors/#latestupdates.
- Hazer Commercial Demonstration Plant, (accessed 2024), 2024, https://research.csiro.au/hyresource/hazer-commercial-demonstration-plant/.
- Hydrogen partnership to advance new pilot facility with CleanBC support (FortisBC, Suncor, Hazer), 2022, https://www.fortisbc.com/news-events/media-centre-details/2022/07/04/hydrogen-partnership-to-advance-new-pilot-facility-with-cleanbc-support.
- Innova Hydrogen, https://www.innovahydrogen.com/technology.
- M. Mcconnachie, M. Konarova and S. Smart, Literature review of the catalytic pyrolysis of methane for hydrogen and carbon production, Int. J. Hydrogen Energy, 2023, 48, 25660–25682, DOI:10.1016/j.ijhydene.2023.03.123.
- Direct Conversion of Methane to Hydrogen by Pyrolysis: Status and Prospects, Advanced Energy Technology, 2022, 1–17, https://aenert.com/news-events/energy-news-monitoring/n/direct-conversion-of-methane-to-hydrogen-by-pyrolysis-status-and-prospects/.
- S. Coleman, The Future of Energy Looks Turquoise, https://www.ansys.com/blog/future-of-energy-is-turquoise.
- US20210061654A1, 2019.
- VulcanX, https://vulcanx.ca/vulcanx-technology/.
- Analysis Of Canada's 2023 Federal Budget And The United States’ IRA: Hydrogen Incentives (accessed April 14, 2023), (accessed 2023), 2023, https://www.ngif.ca/analysis-of-canadas-2023-federal-budget-and-the-united-states-ira-hydrogen-incentives/.
- U.S. National Clean Hydrogen Strategy and Roadmap (accessed June 2023), 2023, https://www.hydrogen.energy.gov/library/roadmaps-vision/clean-hydrogen-strategy-roadmap.
- Section 45V Credit for Production of Clean Hydrogen, Section 48(a)(15) Election To Treat Clean Hydrogen Production Facilities as Energy Property, 2024, https://www.federalregister.gov/documents/2024/04/11/2024-07644/section-45v-credit-for-production-of-clean-hydrogen-section-48a15-election-to-treat-clean-hydrogen.
- IRA Tax Credits for H2 (accessed 30 Jan 2023), 2023, https://www.curtis.com/our-firm/news/ira-tax-credits-for-h2.
- Low Carbon Economy Challenge (accessed Nov 18, 2024), 2024, https://www.canada.ca/en/environment-climate-change/services/climate-change/low-carbon-economy-fund/challenge.html.
- Fuel Charge Rates for Listed Provinces and Territories for 2023 to 2030 (accessed 13 Dec, 2021), 2021, https://www.canada.ca/en/department-finance/news/2021/12/fuel-charge-rates-for-listed-provinces-and-territories-for-2023-to-2030.html.
- U.S. Department of the Treasury Releases Guidance to Expand Clean Vehicle Recharging and Refueling Infrastructure and Lower Transportation Costs for Americans (accessed September 18, 2024), 2024, https://home.treasury.gov/news/press-releases/jy2586.
- Biden-Harris Administration Announces $623 Million in Grants for EV Charging and Alternative Fueling—Including More Than $90 Million for Hydrogen Infrastructure (accessed January, 30, 2024), https://www.energy.gov/eere/fuelcells/articles/biden-harris-administration-announces-623-million-grants-ev-charging-and#:~:text=Biden-HarrisAdministrationAnnounces%24623,HydrogenInfrastructure%7CDepartmentofEnergy.
- The Zero Emission Vehicle Infrastructure Program—Natural Resources Canada,(accessed2023), 2023, https://www.oag-bvg.gc.ca/internet/docs/parl_cesd_202311_08_e.pdf.
- Charging and Hydrogen Refuelling Infrastructure Initiative, 2024, https://cib-bic.ca/en/charging-and-hydrogen-refuelling-infrastructure-initiative/.
| This journal is © The Royal Society of Chemistry 2025 |