
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the kinetics enhancement strategies of Si anodes through deconstructing particle–interface–electrode integration
Pingshan
Jia†
a,
Junpo
Guo†
b,
Qing
Li
 a,
Yinan
Liu
a,
Yun
Zheng
a,
Yan
Guo
a,
Yike
Huang
a,
Yingying
Shen
a,
Lifen
Long
a,
Hebin
Zhang
a,
Rong
Chen
a,
Congcong
Zhang
a,
Zhiyuan
Zhang
a,
Jingjun
Shen
a,
Shengyang
Dong
a,
Jiangmin
Jiang
a,
Meinan
Chang
b,
Xupo
Liu
b,
Xiaobing
Wang
b,
Yuxin
Tang
c and
Huaiyu
Shao
*a
a,
Yinan
Liu
a,
Yun
Zheng
a,
Yan
Guo
a,
Yike
Huang
a,
Yingying
Shen
a,
Lifen
Long
a,
Hebin
Zhang
a,
Rong
Chen
a,
Congcong
Zhang
a,
Zhiyuan
Zhang
a,
Jingjun
Shen
a,
Shengyang
Dong
a,
Jiangmin
Jiang
a,
Meinan
Chang
b,
Xupo
Liu
b,
Xiaobing
Wang
b,
Yuxin
Tang
c and
Huaiyu
Shao
*a
aJoint Key Laboratory of the Ministry of Education, Institute of Applied Physics and Materials Engineering, University of Macau, Avenida da Universidade, Taipa, Macao SAR 999078, China. E-mail: hshao@um.edu.mo
bCollaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Chemistry and Chemical Engineering, School of Materials Science and Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
cCollege of Chemical Engineering, Fuzhou University, Fuzhou 350116, China
First published on 6th February 2025
Abstract
The successive introduction of silicon (Si) graphite composite anodes into the global market highlights the tremendous commercial potential of Si anodes. Good kinetic performance related to fast charging capability is the central topic of next-generation Si anodes. However, there is a lack of critical reviews exploring the fundamental limiting factors affecting the kinetics of Si and evaluating the effectiveness of the current strategies. In this review, we deconstruct the particle–interface–electrode integration to analyze key limiting factors of kinetics from a practical application perspective for the first time, including long Li+ diffusion distance and poor conductivity for particles, high Li+ migration impedance at the interface, and insufficient or even interrupted Li+ diffusion paths inside the electrodes. Then, the kinetics enhancement strategies for progressively addressing the above issues are systematically investigated and the quantitative relationships between kinetics and these strategies are deeply discussed. Accordingly, the challenges in quantification and balance for fast-charging Si anodes are identified as the remaining issues, and potential solutions are provided. This review provides valuable guidance on fast-charging Si anodes and suggests promising directions in commercial-oriented Si anode studies.
Broader contextThe pursuit of fast charging is a theme of the contemporary era in the development of high energy density lithium-ion batteries (LIBs). Currently, silicon (Si)–graphite composite anodes are recognized as the most promising choice for high energy density LIBs. However, Si anodes suffer from poor kinetics in multiple dimensions from the perspective of practical applications, resulting in performance that is far behind the targeted fast charging performance. In this context, we deconstruct the particle–interface–electrode integration to analyze key limiting factors of kinetics from a practical application perspective and evaluate effective strategies of progressively enhancing the kinetics of Si anodes, as well as deeply discuss the quantitative relationship between the kinetics and strategies of multiple levels. This review aims to provide valuable insights from an overall perspective of Si-based batteries, and we expect a significant breakthrough in the research and development of high energy density and fast-charging power LIBs based on Si anodes. |
1. Introduction
Promoting electric vehicles (EVs) is an important process toward reaching the carbon neutrality target. Over 26 million electric vehicles (EVs) were on the road in 2022, indicating vigorous development.1 However, EVs still hold a small share of the global market when compared with the dominant traditional fuel vehicles.2 One major bottleneck is their long charging time, which negatively impacts user experience. Consequently, fast charging performance is becoming a critical benchmark for lithium-ion batteries (LIBs) to enhance their competitiveness and cope with more complex driving conditions, thereby expanding the application scenarios.3–11 According to the Goals for Advanced Batteries proposed by the U.S. Advanced Battery Consortium (USABC), power batteries need to obtain a fast charging performance of 80% initial capacity for 15 min charging.12 Currently, the Tesla model S can be recharged up to the 320 km range within 15 min at supercharger locations,13 and BYD can shorten the charging time to 33 minutes,14 which still falls far from the targeted fast charging performance.For the power LIB system, fast charging requires the anode to possess good kinetic performance for fast Li storage.15–19 Based on the properties of the intercalation mechanism, the lithiation of graphite anodes mainly occurs from the sheet edge, which forms the only path for Li+ to intercalate into the graphite layers, resulting in the slow kinetic performance.6,20–24 The limited theoretical capacity of graphite requires the adoption of thicker electrodes to achieve sufficient energy density, which prolongs the Li+ diffusion distance and hampers fast charging performance. Si is regarded as a promising candidate for graphite due to its high capacity (∼3580 mA h g−1, Li15Si4) and low potential (<0.3 V Li+/Li).25 Particularly, notable battery manufacturers such as CATL and Tesla have successfully brought to market the practice of Si–graphite composite anodes to increase the energy density, thereby extending the driving range of EVs.26 Studying the kinetics behavior of Si anodes could facilitate the development of fast-charging Si–graphite composite anodes which are highly anticipated in the market.
Numerous research studies have been conducted on Si anodes from the perspective of alleviating volume expansion.27–30 The volume expansion of Si materials in the composite anode could be mitigated by graphite,31 while the poor kinetic performance of Si materials profoundly hinders the fast charging of these anodes, resulting in them significantly lagging behind the target for fast charging performance. A comprehensive review of the kinetic behavior and performance is crucial to advancing the understanding of practically-oriented Si anode iterations.
On this basis, we deconstruct the particle–interface–electrode integration and propose these limiting factors in three dimensions as follows (Fig. 1): (i) the low intrinsic conductivity and long Li+ diffusion distance cause slow lithiation inside Si materials, resulting in slow kinetics at the particle level;32–35 (ii) the complex solid electrolyte interface (SEI) and the exposed surface of Si materials with poor conductivity result in significant obstruction on kinetics at the interface level;36,37 (iii) the initial electrode porosity and the electrode cracking during operation can affect the Li+ diffusion in the electrode, and the insufficient contact between current collector and electrode also directly impacts the integrity of electrodes, which limits the kinetic performance at the electrode level.38,39
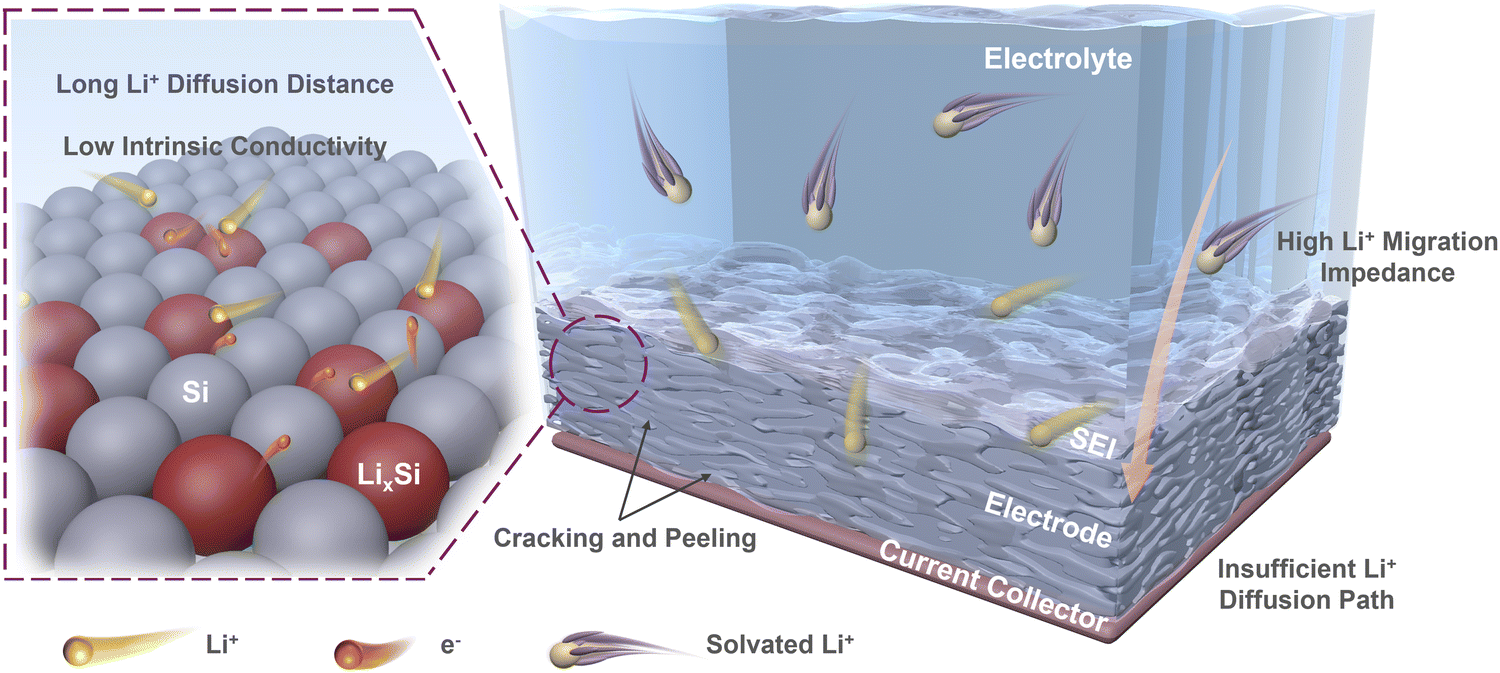 | ||
| Fig. 1 Limiting factors causing poor kinetic performances of Si anodes at particle, interface, and electrode levels. | ||
Herein, the systematic kinetics enhancement strategies of Si anodes from particle, interface, and electrode levels are categorized, as shown in Fig. 2. First, we discuss the adjustments in particle size and porosity of Si materials to balance the shortened Li+ diffusion distance and the increased interface impedance to obtain the optimal Li+ diffusion. Additionally, the element doping and particle compositing strategies are summarized to address poor conductivity at the particle level. Next, the surface coating, SEI optimization strategies, and the quantitative relationship among them are analyzed to reduce the Li+ migration impedance of Si materials at the interface level. Later, the strategies of decreasing electrode porosity and cracks to stabilize the electrical contact of Si materials during operation, as well as the strategy of enhancing the electrode-current collector contact for electrode integrity are reviewed to improve the kinetic performance at the electrode level. In the conclusion, we emphasize quantification and balance as the kinetics enhancement challenges of Si anodes and propose several potential strategies to shed light on future studies.
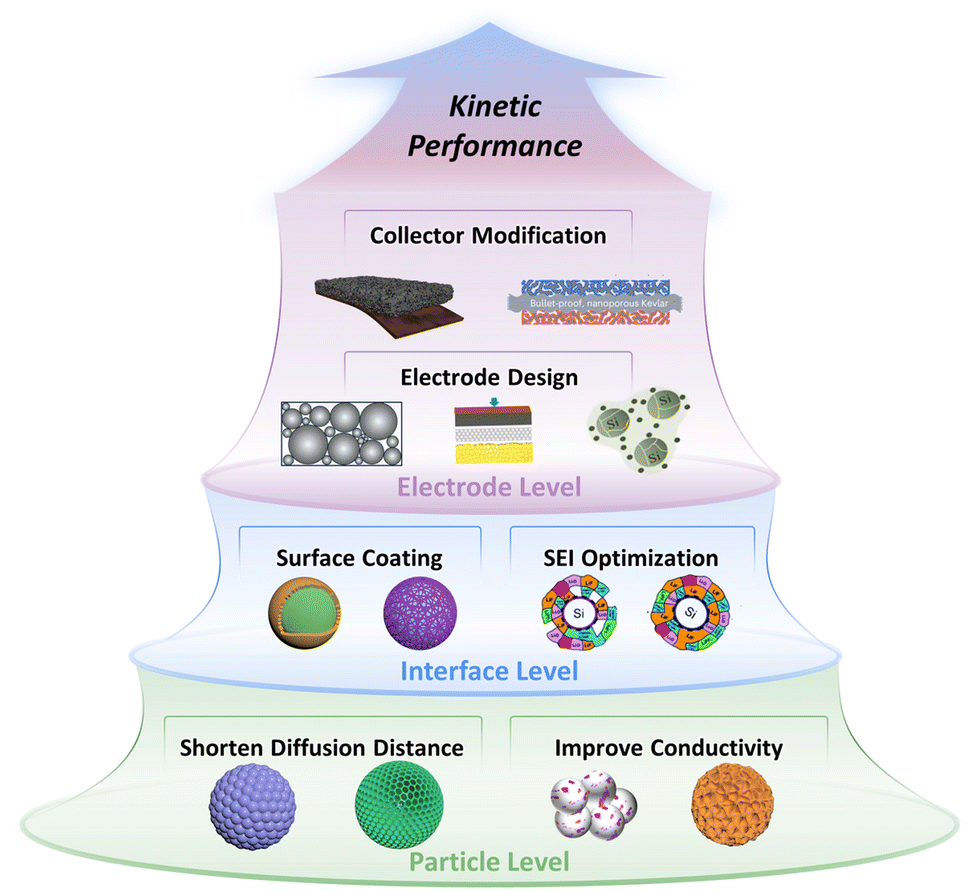 | ||
| Fig. 2 Overview of kinetics enhancement strategies of particle–interface–electrode integration for Si anodes. (Partial images are cited.40–43 Copyright © 2019, American Chemical Society, copyright © 2020 Published by Elsevier Ltd, copyright © 2020 Published by Elsevier Ltd, copyright © 2024, The Author(s), under exclusive license to Springer Nature Limited.) | ||
2. Particle level
Poor Li+ diffusion coefficients (10−14–10−13 cm2 s−1) caused by low intrinsic conductivity of Si leads to their poor kinetic performance.44 Numerous studies are dedicated to revealing the intrinsic mechanism.45 Lee and co-authors investigated the mechanical interaction effects of lithiation Si clusters through in situ TEM.46 As shown in Fig. 3a–c, the unconstrained Si column largely swells along the 〈110〉 orientation. As a comparison, the constrained Si column first swells along the 〈110〉 orientation and gradually touches the two walls. And then, it expands along the 〈100〉 orientation as the compressive stress accumulates. The expansion orientation indicates that the lithiation kinetics of Si along different orientations are different. Yang and co-authors further developed a kinetics model to reveal the lithiation orientation of crystalline Si.47 According to the Li flux profile shown in Fig. 3d, the lithiation orientation dependence in the 〈110〉 orientation is more significant than in other directions. The anisotropic lithiation of Si is controlled by the orientation mobility of crystalline Si and amorphous LixSi. More in-depth study at the atomic scale would be beneficial to further reveal the orientation dependence of Si.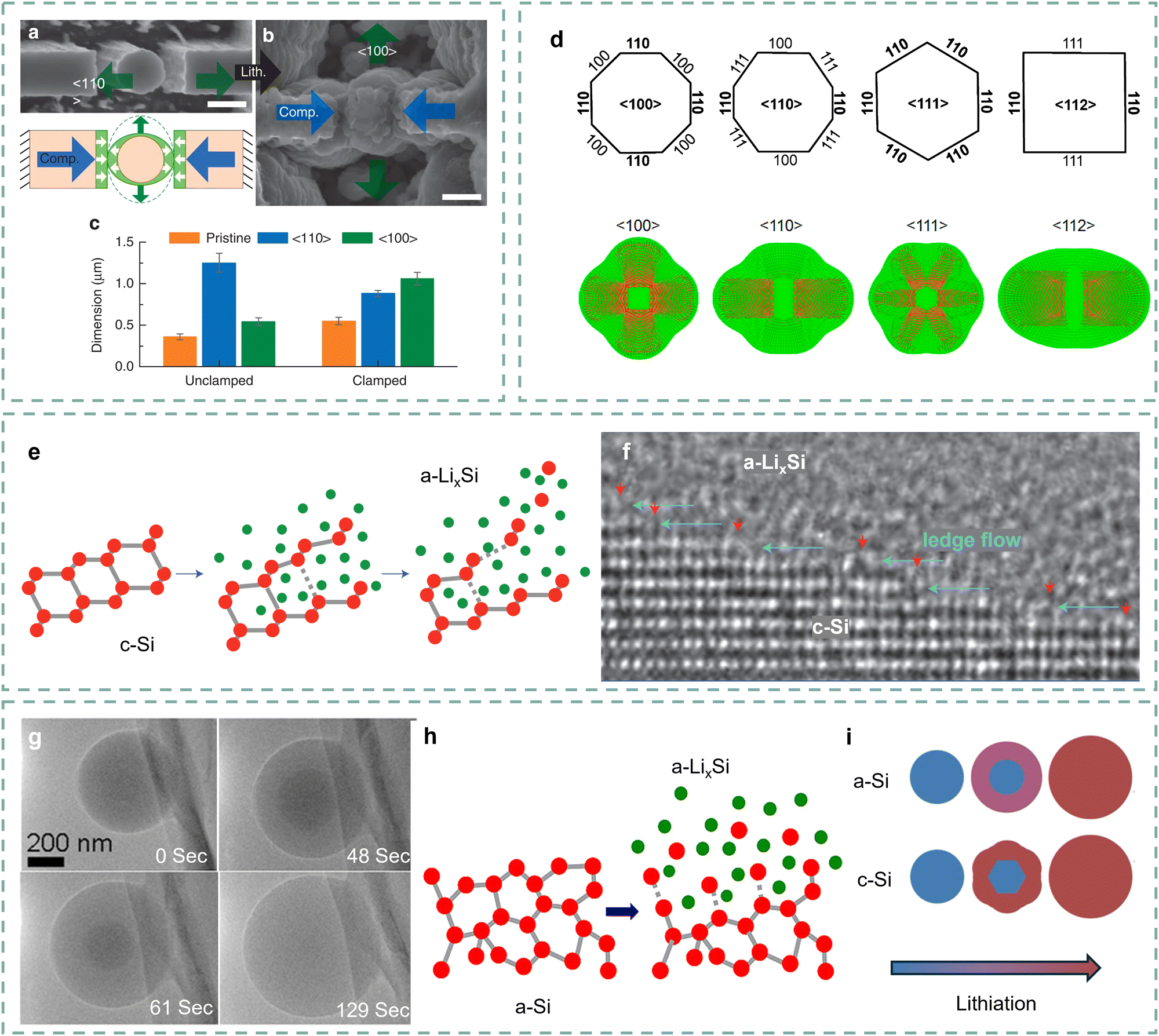 | ||
| Fig. 3 (a) and (b) SEM images of the Si column positioned inside the fixture before (a) and after (b) lithiation, (c) dimension change of the Si column along the 〈110〉 (blue) and 〈100〉 (green) orientations after lithiation.46 Copyright © 2015, The Author(s). (d) The characteristic crystallographic orientations of Si nanowires, and the corresponding Li flux profiles at a representative lithiation snapshot (t = 0.4).47 Copyright © 2012, American Chemical Society. (e) Proposed amorphization mechanism of crystalline Si based on TEM observations, and (f) the ledge-flow dominated lithiation along the (111) plane. Red arrows indicate ledges and cyan arrows ledge-flow directions.48 Copyright © 2012, Springer Nature Limited. (g) Time series of lithiation of the amorphous Si microsphere, and (i) schematic of the lithiation for amorphous Si and crystalline Si.49 Copyright © 2013, American Chemical Society. (h) Mechanism of dissolution for Si atoms from amorphous Si to LixSi.50 Copyright © 2013, American Chemical Society. | ||
Liu and co-authors revealed the causes of orientation mobility by observing the dynamic lithiation interface of single crystalline Si at the atomic scale.48 As shown in Fig. 3e, the Si atom tends to dissociate due to the weakened Si–Si covalent bonds under a high-rate lithiation process.51 They provided a grain boundary ledge flow fact to explain the lithiation kinetics properties of Si anodes. It can be observed that the ledge flow causes the phase transformation from crystalline Si to amorphous LixSi, and the flow rate represents the lithiation kinetics (Fig. 3f). To sum up, this dynamic atomic resolution research on the interface motion and phase transformation reveals the anisotropic lithiation mechanism of Si anodes. It also demonstrates that the high hydrostatic pressure under increasing particle depth will restrict the corresponding lithiation kinetics, indicating the advantages of shortening the Li+ diffusion distance in improving the kinetic performance of Si anodes.52,53
The lithiation of amorphous Si exhibits differences from crystalline Si.45 McDowell and co-authors proposed two-phase lithiation of amorphous Si through in situ observation of the lithiation process.49 As presented in Fig. 3g, an obvious boundary can be found between the Si-rich core in the dark area and the Li-rich shell in the light region, indicating a two-phase lithiation like crystalline Si. Although the amorphous Si lacks long-range ordered structures, as shown in Fig. 3h, its lithiation still involves the breaking of Si–Si bonds.50 Specifically, the Li-rich clusters at the interface between two phases promote the dissociation of Si covalent bonds. In contrast, the amorphous Si presents less hydrostatic pressure and its increased lithiation thickness grows approximately linearly over time, illustrating an isotropic lithiation process (Fig. 3i).54 The kinetics process of amorphous Si is still limited by diffusion distance, as its lithiation velocity is approximately constant.
Hence, the overlong Li+ diffusion distance and intrinsic properties restrict the kinetic performance enhancement of Si anodes. It was reported that the capacity retention of micron-sized bulk Si anodes at a current density of 1C is below 20%,55,56 far below the set fast-charging target of 80% at 4C. Therefore, how to shorten the Li+ diffusion distance and increase the intrinsic conductivity of Si materials at the particle level is significant to improve their kinetic performance, and detailed targeted strategies are introduced in this section.
2.1 Shorten the Li+ diffusion distance
It is well known that reducing Si particle size is the most fundamental way to shorten the Li+ diffusion distance.57 In particular, Si particles with a mean diameter of 150 nm have attracted widespread attention due to an optimized “size effect”.58,59 Therefore, various nanostructures such as nanoparticles, nanowires, hollow nanotubes, and nanofilms/nanosheets have been developed.60–63 It was reported that Si nanoparticles with a particle size smaller than 100 nm, obtained by ball milling, delivered an approximately 30% capacity retention at 2.5 A g−1, an increase compared to micron-sized bulk Si.64 As shown in Fig. 4a, the Si nanowires were initially intended to arrange nanoarrays to buffer volume expansion.65 Importantly, Liu and co-authors found a fast lithiation process of Si nanowires through in situ TEM.66 The Si nanowires possess high lithiation rates of 60–200 nm s−1 in liquid cells (Fig. 4b), which may benefit from the advantage of one-dimensional Li+ transport. Due to the fast kinetics, Si nanowires exhibit good rate performance over 60% capacity retention at 1C.65 Similarly, Si hollow nanotubes also demonstrate the advantage of one-dimensional rapid Li+ diffusion and possess the capability of fast lithiation in the interior of the nanotubes.67,68 Park and co-authors reported a hollow Si nanotube with carbon coatings, delivering over 88% capacity retention at a current density of 5C.68 For the two-dimensional Si materials, Si nanofilms/nanosheets present distinctive lithiation properties.63 As shown in Fig. 4c, Li+ can rapidly diffuse in a two-dimensional orientation parallel to the current collector, which facilitates rapid kinetics compared to Si nanoparticles. The representative Si nanostructures are summarized in Table 1. However, nanostructures imply more contact with electrolytes, which is accompanied by the formation of more SEI, resulting in a potential increase in interface impedance.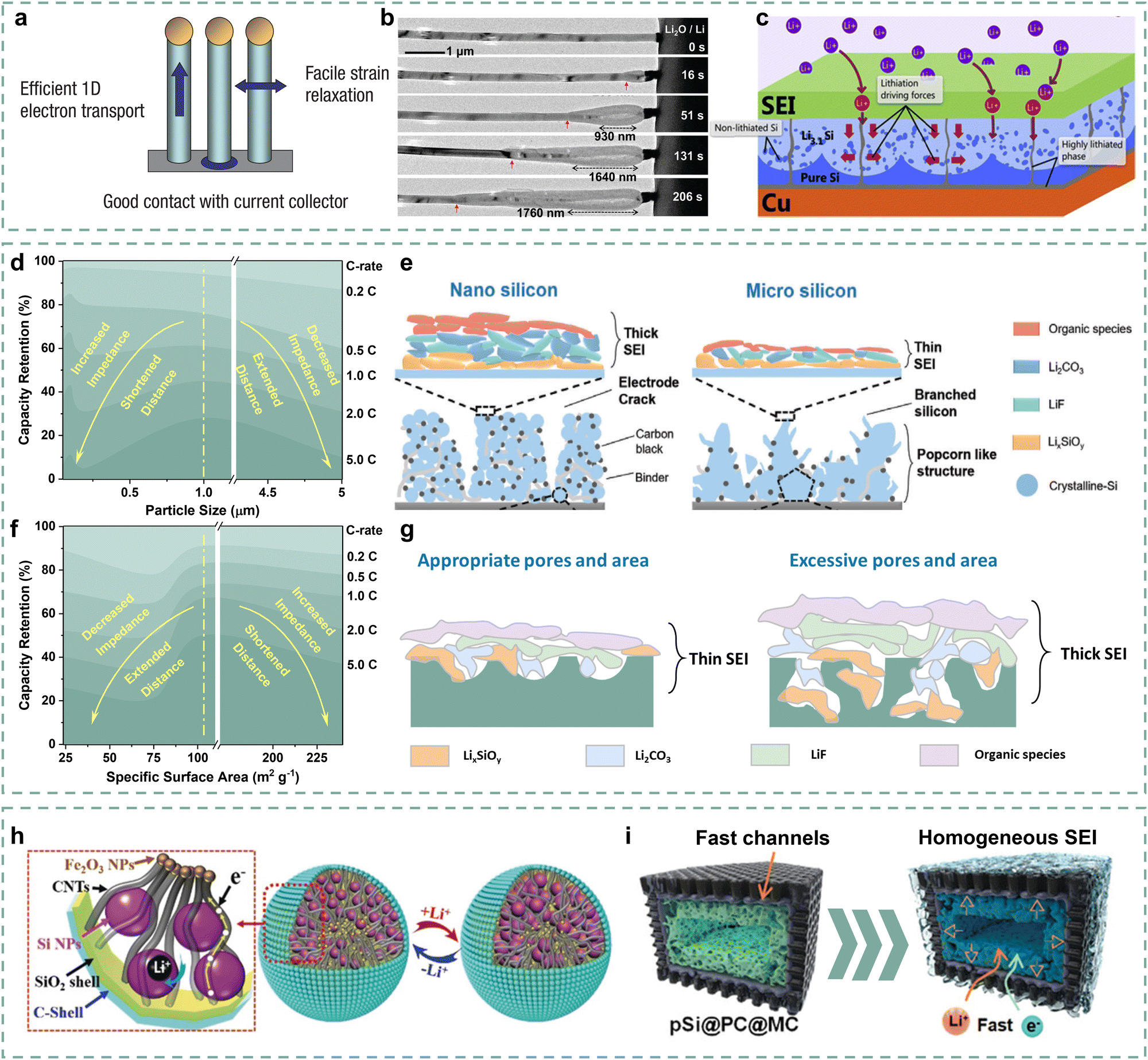 | ||
| Fig. 4 (a) Schematic of lithiation for Si nanowires.65 Copyright © 2007, Springer Nature Limited. (b) Morphology evolution of the Si nanowires during lithiation.66 Copyright © 2011, American Chemical Society. (c) Schematic of the lithiation for Si nanofilms.69 Copyright © 2013, Trans Tech Publications Ltd. (d) The relationship between capacity retention and particle size ranging from 50 nm to 5 μm at different rates and (e) an SEI chemical schematic of nano Si and micron-sized Si anodes.70 Copyright © 2023 Wiley-VCH GmbH. (f) The relationship between capacity retention and surface area at different rates.71 Copyright © 2016 American Chemical Society. (g) SEI chemical schematic of porous Si with different pores and areas. (h) Structural schematic of yolk–shell structured Si/C anodes.72 Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (i) Schematic of the structural properties of pSi@PC@MC anodes.73 Copyright © 2024 Wiley-VCH GmbH. | ||
| Sample | Nanostructure | ICE (%) | Rate performance | Ref. |
|---|---|---|---|---|
| BM-Si | Nanoparticle | 74 | ∼30% at 2.5 A g−1 | 64 |
| Si NP | Nanoparticle | ∼45 | ∼35% at 1 A g−1 | 74 |
| Si NW | Nanowire | 73 | ∼60% at 1C | 65 |
| Si NWs | Nanowire | 69.5 | ∼72% at 0.5C | 75 |
| Si nanotubes | Nanotube | 89 | ∼88% at 5C | 68 |
| DWSiNT | Nanotube | 76 | ∼34% at 12C | 67 |
| Si nanofilm | Nanofilm | 85.1 | 79.7% at 1C | 76 |
| a-C/Si thin film | Nanofilm | 80 | ∼70% at 3.57C | 77 |
| C-SiNS | Nanosheet | 79.4 | ∼50% at 2C | 78 |
Wu and co-authors explored the rate performance differences of Si particles with D50 ranging from 50 nm to 5 μm.70 Si anodes exhibit gradually increased reversible capacity as their particle size decreases from 5 μm to 50 nm, suggesting that a short Li+ diffusion distance can reduce the polarization of Si anodes, thereby increasing their reversible capacity. However, Si anodes with a particle diameter of 1 μm instead of 50 nm present the highest capacity retention at 5C. This confirms that too-small particle sizes promote the electrode–electrolyte contact and SEI formation, which increases the interface impedance, thus obstructing the Li+ migration before reaching particles. Fig. 4d reflects the fact that the kinetic performance of Si anodes has a “volcanic” relationship with particle size, which delivers the highest kinetic performance corresponding to appropriate particle size. The SEI chemical difference of Si anodes with nanometer and micron-sized particle sizes are described in further detail in Fig. 4e. It can be observed that the Si electrode with nanometer particle size forms thicker SEIs than the micron-sized Si electrode due to high surface area and electrode–electrolyte contact. It increases Li+ migration difficulty and distance, as well as polarization during fast lithiation, resulting in poor rate performance. Therefore, balancing the particle size and the interface impedance of the Si anodes is crucial for improving its kinetic performance.
From this perspective, nano-micro-particle design is constantly emerging.72,79–81 Hou and co-authors proposed the 3D plum-pudding-like Si/C anode to balance particle size and interface impedance, ensuring the good fast-charging performance of Si/C electrodes.79 The Si/C material formed by the secondary stacking of nano Si particles delivers a particle size of 2–5 μm. Importantly, the micron-sized Si/C anode delivers a high initial Coulombic efficiency (ICE) of 88%, significantly reducing side reactions such as SEI formation. Hence, this unique structure can shorten the Li+ diffusion distance while reducing SEI formation and interface impedance, thereby accelerating kinetic performance. Furthermore, more functional networks have also been introduced into the constructed micron-sized Si composite particles. Zhang and co-authors developed a yolk–shell Si anode (YS-Si/C) with secondary stacking of Si, CNTs, and Fe2O3 particles and double-layer encapsulation.72 As shown in Fig. 4h, the introduced CNTs and Fe2O3 can serve as the conductive highway among Si particles as well as bridging particles and the shell. Benefiting from the structural design, the YS-Si/C anode delivers a high ICE of 86.9% and 67% capacity retention at a current density of 2 A g−1, indicating that the appropriate balance between diffusion distance and interface impedance has been obtained through this structure.
Porous Si can also greatly shorten the Li+ diffusion distance, thereby ensuring the Si anodes with more active sites and high fast-charging performance.82 Liang and co-authors designed porous Si anodes with different specific surface areas and pore size distributions to investigate the porosity effect on the corresponding Si anode fast-charging performance.71 The synthetic A–Si, C–Si, S–Si, and D–Si materials deliver specific surface areas of 239.3, 23.9, 102.6, and 74.13 m2 g−1 due to their different pore structures, which results in different Li+ diffusion distances. As described in Fig. 4f, it can be observed that the S–Si electrode with mean surface area (102.6 m2 g−1) and appropriate pore size delivers the highest capacity retention compared with other Si anodes, indicating that simply increasing the pore structure/surface area of Si materials cannot be beneficial to improving kinetic performance. As shown in Fig. 4g, the porous structure also tends to promote similarly high interface contact between electrodes and electrolytes, leading to high interface impedance. It can be inferred that increasing active sites and shortening the Li+ diffusion distance of Si materials by optimizing their pore structure, and meanwhile inhibiting the formation of thicker SEIs, contribute to the kinetic performance enhancement of Si anodes.
Furthermore, numerous studies have been conducted to balance the Li+ diffusion distance and interface impedance for porous structures. Coating strategies can effectively inhibit the excessive formation of SEIs and the increase in interface resistance.73,83,84 For example, Ren and co-authors encapsulated porous Si with dense carbon layers, promoting a significant decrease of specific surface area from 350 m2 g−1 to 10 m2 g−1 and an increase of ICE from 68% to 78%.85 Reducing the specific surface area helps to minimize the interface contact with electrolytes, thereby suppressing the formation of SEIs. And the porous Si/C anode also exhibits lower charge transfer impedance than that of porous Si, which further confirms the inhibition of coatings on thicker SEI formation, thus contributing to the kinetics enhancement. Nevertheless, the dense coating can also cover the pore structures, which weakens the active sites. It is important to perform effective strategies to control the appropriate specific surface area and pore structures. Cheng and co-authors proposed a carbon-coated porous Si anode (pSi@PC@MC) to promote appropriate pore structures for kinetics enhancement of typical porous Si anodes.73 The pitch-derived carbon (PC) coating is relatively dense, which helps to avoid direct contact between porous Si and electrolytes. Meanwhile, the dopamine-derived mesoporous carbon (MC) introduces mesopores externally to construct more active sites, thereby promoting the widespread diffusion of Li+. As shown in Fig. 4i, the double coatings of PC and MC largely avoid the contact between porous Si and electrolytes, based on the decrease in specific surface area (222.8 m2 g−1 to 164.1 m2 g−1) and the leap of ICE value (65.92% to 75.6%), which contributes to the thin and homogeneous SEI. Meanwhile, the external mesoporous structures promote electrolyte infiltration and the introduction of more active sites, further providing good kinetic performance. The pSi@PC@MC anode delivers better rate performances of 57.9% and 53.6% capacity retention at current densities of 4 and 6 A g−1 compared with the porous Si anode. Besides, comparison of the pseudocapacitance of porous Si, pSi@MC, and pSi@PC@MC anodes is further persuasive evidence to further demonstrate the importance of controlling appropriate surface areas and pore structures in enhancing the kinetic performance of porous Si anodes.
Therefore, regulating the structure of Si materials through optimizing the particle size of Si particles and pore size distribution of porous Si to balance the Li+ diffusion distance and the interface impedance is necessary to inhibit the excessive formation of SEIs and the increase of interface impedance, thereby significantly improving the kinetic performance of Si anodes.
2.2 Improving the conductivity
The charging process involves Li+ diffusion within the Si anodes and synchronous electron transfer. The imbalance between Li+ diffusion and electron transfer can lead to polarization, which induces over potential and low lithiation depth, resulting in poor rate performance.86–88 Therefore, fast charging requires rapid Li+ diffusion inside Si anodes and the matching electron transfer.89–91 However, the intrinsic properties of Si materials severely hinder the Li+ diffusion and electron transfer within particles, manifesting as locally slow kinetics at the particle level. Therefore, it is necessary to improve the electronic and ionic conductivity of Si materials and balance them.Element doping is a typical and effective strategy to improve the electronic conductivity of Si materials by introducing charge carriers (electrons or holes). For typical III and V groups (boron, phosphorus), as shown in Fig. 5a, the electronic conductivity enhancement mechanism can be simply described as elements incorporated into the lattice structure of Si materials creating p-type Si with three valence electrons or n-type Si with five valence electrons.92 For other potential elements, they can generate multiple ionizations and introduce multiple energy levels, accompanied by the introduction of non-fixed forms of carriers to enhance electronic conductivity.
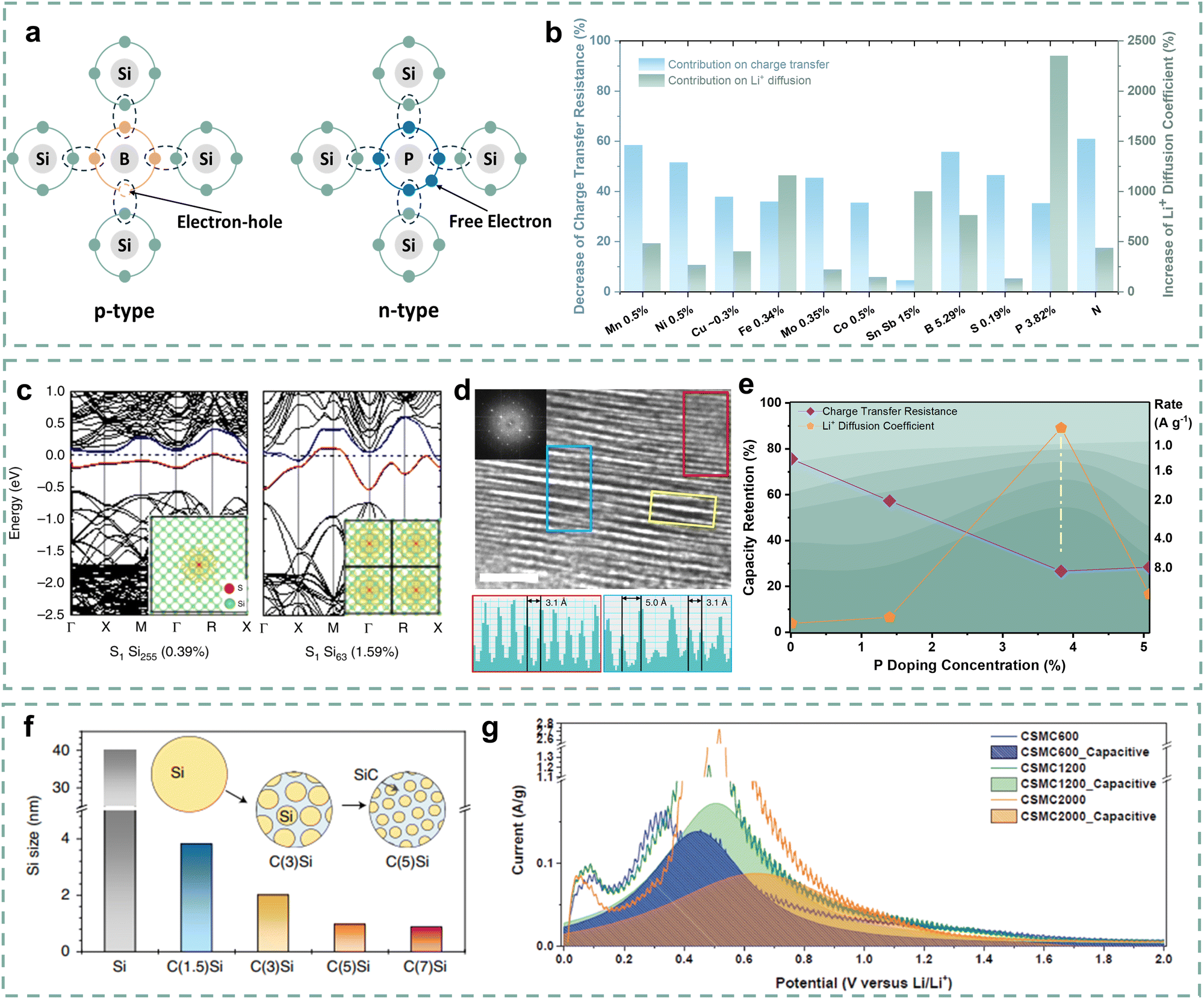 | ||
| Fig. 5 (a) Schematic of the conductivity enhancement mechanism. (b) Doping effects on charge transfer and Li+ diffusion.93–99 (c) Calculated band structures with the S doping in Si. (d) HR-TEM images of S doping Si and the intensity profiles of the selected areas.100 Copyright © 2019, The Author(s). (e) The relationship between capacity retention and P doping concentration at different rates.99 (f) Crystallite size of Si, C(1.5)Si, C(3)Si, C(5)Si, and C(7)Si anodes.101 Copyright © 2021, The Author(s), under exclusive license to Springer Nature Limited. (g) Capacitive contribution of CSMC600, CSMC1200 and CSMC2000 anodes.102 Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Metal alloying and doping are conventional strategies for improving the conductivity of Si materials.103,104 Nulu and co-authors developed Mn doping SiMn05% and Ni doping SiNi05% anode materials to address the poor conductivity of Si materials.93 The SiMn05% and SiNi05% anodes deliver higher Li+ diffusion coefficients and lower charge transfer impedance than the compared Si anode. In particular, the SiMn05% anode delivers a good rate performance with 50% capacity retention at a current density of 3.2 A g−1 due to its good conductivity. Luo and co-authors studied the effect of molybdenum (Mo) doping on the conductivity of Si anodes.95 First-principles calculations show that Mo doping reduces the band gap of Si from 0.61 eV to 0.45 eV and increases the nearby charge density, which means an enhancement of conductivity. Approximately 70% decrease in impedance and an 8.8 times increase in Li+ diffusion coefficient promote the Mo-doped porous nanostructured Si (Mo–PNSi) anode to exhibit an enhanced rate performance of 38.6% capacity retention at a current density of 6.72 A g−1. Various transition metal doping strategies such as Cu, Fe, and Co have also been performed to enhance the conductivity of Si anodes.94,96 Additionally, some electrochemically active metals are also used as dopants to enhance the kinetics of Si anodes while ensuring high reversible capacity. Gao and co-authors proposed to introduce Sn and Sb to improve the electronic conductivity and Li+ diffusion of Si anodes.105 The as-prepared Si8.5Sn0.5Sb anode presents 6000 times the electronic conductivity and 10 times the Li+ diffusion coefficient higher than those of Si particles, respectively. Due to the introduction of electrochemically active Sn and Sb, the composite anode has a high reversible capacity of 1190 mA h g−1 at a current density of 5 A g−1. Fig. 5b presents the contributions of element doping on decreasing charge transfer resistance (RCT) and improving the Li+ diffusion coefficient (DLi+), and detailed information on several element doping strategies is displayed in Table 2. It can be inferred that element doping can reduce the impedance and enhance the Li+ diffusion ability, thereby optimizing the kinetic performance. However, some transition metal ions are easily reduced to metal atoms or dendrites, which tend to cause short circuits.106 Thus, the metal doping strategy of Si materials still needs to be screened and identified for commercial application.
| Element | Concentration (%) | R CT (Ohm) | Original RCT (Ohm) | D Li+ (cm2 s−1) | Original DLi+ (cm2 s−1) | Ref. |
|---|---|---|---|---|---|---|
| Mn | 0.5 | 59 | 101 | 1.90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 10−13 × 10−13 |
3.94 × 10−14 |
93 |
| Ni | 0.5 | 52 | 101 | 1.04 × 10−13 |
3.94 × 10−14 |
|
| Cu | ∼0.3 | 40.3 | 106.5 | 1.41 × 10−11 | 3.53 × 10−12 | 94 |
| Fe | 0.34 | 38.2 | 106.5 | 4.09 × 10−11 | 3.53 × 10−12 | |
| Mn | 0.32 | 40.7 | 106.5 | 1.07 × 10−11 | 3.53 × 10−12 | |
| Mo | 0.35 | 115 | 253.7 | 2.088 × 10−21 | 9.488 × 10−22 | 95 |
| 0.69 | 78.62 | 253.7 | 8.394 × 10−21 | 9.488 × 10−22 | ||
| Co | 0.1 | 8.8 | 15.8 | 4.08 × 10−11 | 6.92 × 10−11 | 96 |
| 0.3 | 6.5 | 15.8 | 7.61 × 10−11 | 6.92 × 10−11 | ||
| 0.5 | 5.6 | 15.8 | 9.99 × 10−11 | 6.92 × 10−11 | ||
| Sn, Sb | 5, 10 | 18.9 | 424.6 | 10 times increase | 105 | |
| B | 5.29 | 7.42 | 13.32 | 8.24 × 10−12 | 1.08 × 10−12 | 97 |
| S | 0.19 | 69.93 | 150.5 | 1.2 × 10−8 | 9.26 × 10−9 | 98 |
| P | 1.39 | 114.4 | 150.9 | 9.637 × 10−14 | 5.68 × 10−14 | 99 |
| 3.82 | 53.1 | 150.9 | 1.335 × 10−12 | 5.68 × 10−14 | ||
| 5.07 | 56.6 | 150.9 | 2.48 × 10−13 | 5.68 × 10−14 | ||
| N | — | 142.7 | 234.3 | 2.82 × 10−12 | 1.22 × 10−12 | 107 |
Non-metal doping (P, S, B, N, etc.) for Si materials has attracted wide attention due to its potential advantages. Since the S atom generally provides two or more electrons as charge carriers,108 Guo and co-authors designed a high-conductivity S-doped porous Si/SiO2 anode for good fast-charging performance.98 After S doping, the S-doped p-Si/SiO2 anode delivers higher reversible capacity with lower loss and rate performance than the p-Si/SiO2 anode. It can be inferred that the capacity and kinetic performance enhancement of the S-doped p-Si/SiO2 anode is largely attributed to the reduced polarization due to high conductivity. Importantly, non-metal doping may alter the crystalline structure of Si, which further affects the Li+ diffusion within the Si particles. Ryu and co-authors proposed that the metallicity of Si will change and transit towards a quasi-metallic state, as the S doping exceeds the equilibrium solid solubility.100 As shown in Fig. 5c, the spatial states of the remaining valence electrons of Si gradually approach and overlap with the increase of S doping, which enhances the band dispersion of the state at the Fermi level and forms the metallic bands. Furthermore, expanded channels (0.5–0.72 nm) have been formed, as the introduction of S-chains expanded the interlayer spacing (Fig. 5d). During the lithiation process, Li2S structures are formed to support the expanded channels, resulting in a lower energy barrier for Li+ diffusion (0.32 eV). Similarly, B, P, or N element doping can also provide additional electrons or holes and impact the crystalline structure. These dopants may cause a decrease in crystal spacing due to the smaller atomic size and affect the Li+ diffusion inside the Si particles.109–111 For example, Wang and co-authors stated that the introduction of P can lead to a decrease in the Li+ diffusion barrier from 0.57 eV to 0.53 eV.112 Je and co-authors discovered a mixed amorphous–crystalline structure with localized atomic distortion in the synthesized B-doped Si (mixed amorphous–crystalline Si, MACS) anodes.113 The localized structural distortion is mainly attributed to the heteroatom-bridge bonds (B–O–Si, B–Si) in crystalline Si, which cause the amorphous phase to wedge into the crystalline phase and reduce the crystalline size, thereby accelerating the Li+ diffusion in the MACS anode. The MACS anode affords ∼36% capacity retention at a current density of 5C due to the effective fast Li+ diffusion. Furthermore, the appropriate doping concentration is important for the high kinetic performance of Si anodes.114 Long and co-authors developed P doping PxSi anode materials through a hydrolysis strategy and further adjusted the P doping concentration through the formation of P–O–Si bonds and the interaction of H/–OH bonds.99 As described in Fig. 5e, as the concentration increases, the PxSi anodes present gradually improving rate performance. And the P0.125Si anode delivers the optimal RCT and DLi+, and the highest rate performance when the P doping concentration increases to 3.82% instead of 5.07%, resulting in a “volcanic” relationship with doping concentration. The P0.125Si anode obtains higher capacity retention of 50% than other PxSi anodes at a current density of 8 A g−1, indicating that the appropriate P doping concentration significantly improves the kinetic performance of the Si anodes.
In addition to element doping, Si composite materials with particle compositing also have obvious structural advantages in enhancing their kinetic performance. Carbon particles have great advantages due to their high conductivity and strong mechanical structure, which mainly contains graphite,115 hard carbon,116 carbon nanotubes,117 and graphene.118 Kim and co-authors designed a Si–graphite composite anode using edge-activated graphite as the substrate.119 They utilized Ni to penetrate the graphite core, forming activated edges and creating numerous active sites. Si is covered on the surface of graphite, which fully fits with the shape of edge-activated graphite, thereby promoting rapid kinetics through these active sites. The Si–graphite composite anode delivers an enhanced rate performance of 20% capacity retention at a current density of 10.5 mA cm−2 (3C). Sung and co-authors further designed a novel C(x)Si anode by embedding nano Si particles into a SiC–C composite matrix (Fig. 5f), which obtains a high capacity retention of 79.6% at a current density of 5C due to good Li+/e− conductivity and short Li+ diffusion distance.101 Besides, adjusting the composite structure is conducive to improving the pseudocapacitive properties of Si anodes, thereby facilitating its kinetic performance. Son and co-authors developed a nanocage-shaped Si/C anode (CSMC) by embedding nano Si particles into a 3D carbon matrix.102Fig. 5g describes the pseudocapacitive contribution to the kinetic performance of these CSMC materials. Benefiting from the synergistic promotion effect of the carbon matrix and Si/C particles, the pseudocapacitive contribution can even reach 88%, which enhances the kinetic performance. As a result, the CSMC600 anode delivers a high capacity retention of 80% at a current density of 20C, which is far superior to the other two compared anodes.
In summary, it can be inferred that both element doping and particle compositing can improve the kinetic performance of Si anodes since they promote good conductivity and Li+ diffusion. The best kinetic performance enhancement can be obtained by regulating the appropriate doping or compositing concentration. Additionally, element doping can bring potential crystal structure changes, which may further improve the Li+ diffusion inside the Si particles. Particle compositing can promote the construction of structure with pseudocapacitive contributions, which is another important factor for developing high kinetics Si anodes at the particle level.
3. Interface level
The interface impedance is the main factor limiting the kinetic performance enhancement of Si anodes, which is also largely attributed to the surface chemical properties of Si anodes, containing material exposed surface properties and the formation of SEIs.120 Bare Si materials generally possess high interface impedance due to poor surface conductivity. SEIs usually contain inorganic layers and organic layers with poor Li+ conductivity, and Li+ migrates cross SEI after desolvation to carry out the lithiation and de-lithiation of Si anodes.121,122 Thus, the chemicals, chemical distribution, and thickness of SEIs affect the Li+ migration and kinetic performance of the Si anodes.123–126 Therefore, how to optimize the surface properties and SEI structure of Si anodes for good kinetic performance is discussed in this section.3.1 Surface coating strategy
Chen and co-authors demonstrated that Si materials naturally have a thin SiOx coating due to the relatively high chemical activity of surface Si atoms, and both bare Si surface and thin SiOx coating have poor Li+/e− conductivity that restricts the corresponding material kinetic performance.127 Several researchers have attempted to adjust the SiOx coating thickness to reduce the interface impedance.128 However, it is a difficult technology with poor effects. Surface coatings have attracted wide attention due to their good Li+/e− conductivity in kinetic performance improvement,129 which mainly contains carbon coatings130 and elastic organic coatings.127A Si anode with carbon coating possesses good kinetic performance due to promising Li+/e− conductivity and strong structural stability.115,131 Xu and co-authors demonstrated the carbon coating contribution of Si anodes on kinetic performance improvement through the in situ TEM method.132Fig. 6a describes the structural differences and dynamic lithiation process of uncoated Si and Si/C anodes, in which the Si/C particle takes only 40 s for full lithiation while the uncoated Si particle takes longer (200 s). This illustrates that carbon coating can accelerate the kinetic performance of Si anodes since the Li+ diffusion of carbon coatings is far superior to Si materials (∼10−7vs. ∼10−12 cm2 s−1),11,133 which can be reconfirmed by the lithiation product thickness comparison between uncoated Si and Si/C particles (Fig. 6b). Numerous studies further focus on the relationship between the carbon coating thickness and the kinetic performance of Si anodes. As described in Fig. 6c, Qi and co-authors investigated the electronic conductivity and rate performance evolution of the Si@C anode with coating thickness ranging from 0 to 9.73 nm.134 The electronic conductivity and capacity retention at high rates exhibit a similar trend, which is a “volcanic” relationship as the coating thickness increases. It can be inferred that the thickness and the accompanied defects of carbon coating can influence the surface area and electronic conductivity of Si@C anodes, thereby affecting their rate performance. The Si@C anode with appropriate carbon coating thickness has an enhanced rate performance due to the good Li+/e− conductivity of the carbon coating, while its rate performance can also be significantly inhibited as the over-thick carbon coating prolongs the Li+ diffusion. Since thinner coating leads to breakage while thicker coating increases Li+ diffusion distance, therefore, an appropriate coating thickness is of great significance in enhancing the kinetic performance of Si anodes at the interface level.
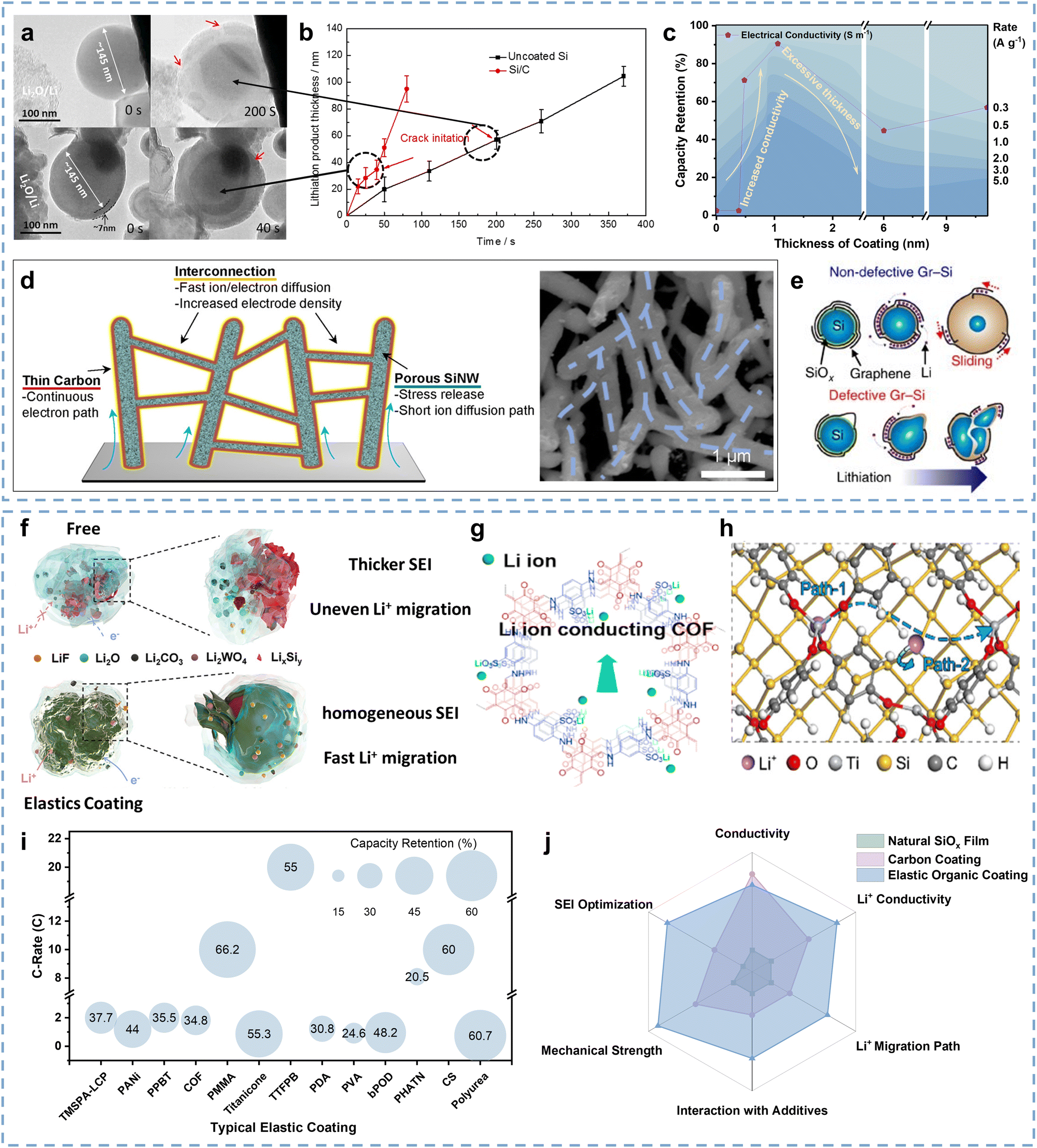 | ||
| Fig. 6 (a) Dynamic lithiation process of uncoated Si and Si/C particles through in situ TEM. (b) Lithiation product thickness comparison of uncoated Si and Si/C anodes.s132 Copyright © 2016 Elsevier B.V. All rights reserved. (c) The relationship between capacity retention and coating thickness at different rates.134 Copyright © 2021 Elsevier Ltd. All rights reserved. (d) Structural schematic of the N-PSi@C anode and the corresponding SEM images.135 Copyright © 2019, American Chemical Society. (e) Schematic of the lithiation of Gr–Si NPs with different graphene encapsulation.136 Copyright © 2015, The Author(s). (f) Schematic of the evolution of interfacial properties and the SEI structure for Si and Si@LCP coating.137 Copyright © 2022 Wiley-VCH GmbH. (g) Structural schematic of the COF coating.138 Copyright © 2020 Published by Elsevier Ltd. (h) Schematic of the Li+ horizontal and vertical diffusion path for Si NPs@titanicone anodes.139 Copyright © 2021 American Chemical Society. (i) Comparison of the rate performance for Si anodes with typical organic coatings.137–147 (j) Normalization for the functionality of natural film, carbon coating, and elastic organic coating. | ||
Furthermore, the structural design of carbon coatings can further improve the kinetics of Si anodes. Wang and co-authors proposed an interconnected carbon coating that encapsulated Si nanowires into a conductive network to facilitate electron transfer.135 As shown in Fig. 6d, the interconnected carbon-coated nanowires accelerate the electron transfer, as well as the space among the nanowires and the pores inside the nanowires provide channels for Li+ diffusion. Conformal coating of thin carbon layers reduces the resistance in the system and ensures a thin SEI outside Si nanostructures. Benefiting from the rapid Li+ diffusion and the matched external electron transfer, the N-PSi@C anode (“N” stands for “network”, “P” stands for “porous”) exhibits an optimized rate performance of ∼48% capacity retention at a current density of 7C. Son and co-authors reported a design to enhance the kinetics of Si anodes by directly growing graphene on Si as a conductive coating.136 As shown in Fig. 6e, the layered stacking of 2D graphene on the surface of Si particles promotes an interconnected percolation network, which increases the conductivity of the Gr–Si materials to 12.8 S cm−1. The Gr–Si composite anode can deliver over 90% capacity retention at a high rate of 10C. Therefore, effective structural design of carbon coatings is beneficial for Li+ diffusion into the Si particles, thereby promoting great kinetics of Si anodes.
Since the huge volume expansion and irreversible structural damage tend to interrupt the Li+ diffusion path of Si anodes, elastic organic coatings have received widespread attention in enhancing the kinetic performance at the interface level.148 Most elastic organic coatings with special chemical groups (C–O and C–F) present good conductivity and are beneficial for structural stability, which is conducive to accelerating the corresponding Li+ migration at the interface.127 For example, some electron-conductive coatings have been developed to facilitate the electron transfer near Si particles. Pan and co-authors proposed a layered conductive polyaniline (LCP) coating for Si anodes.137 The polyaniline chains in the LCP coating allow for electron transfer, resulting in 5 orders of magnitude higher electronic conductivity of the coated Si materials than pure Si, serving as a good electron-conductive coating. And the N-containing in LCP coatings enables dipole–dipole interaction with fluoroethylene carbonate (FEC), which induces uniform LiF formation, thereby forming a homogeneous SEI above the LCP coatings. As shown in Fig. 6f, the designed elastic LCP coating promotes the homogeneous SEI formation and avoids the fragmentation and reconstruction of the SEI, resulting in a uniform and thin SEI that is conducive to Li+ migration. Moreover, the high electronic conductivity of the LCP coating facilitates rapid local electron transfer, which can also enhance the kinetics near the interface. In contrast, the SEI distribution on the pure Si is uneven, and it fractures and reconstructs with volume expansion, resulting in thicker SEI and uneven Li+ migration. Benefiting from the enhanced electronic conductivity and uniform SEI, the composite anode delivers a good rate performance of ∼46% at a current density of 5 A g−1. Wu and co-authors formed a 3D interconnected electron-conductive coating for Si particles by reacting phytic acid with aniline monomers of polyaniline (PANi).149 The PANi coating accelerates the local electron transfer of Si particles and simultaneously extends the electron transfer to the current collector through the interconnection. Similarly, some electron-conductive organic coatings such as poly[3-(potassium-4-butanoate)thiophene] (PPBT) and biphenyl-polyoxadiazole (bPOD) have also been reported to increase the local electronic conductivity to enhance the kinetics of Si anodes.144,150
Moreover, organic coatings with high Li+ conductivity are also conducive to enhancing the kinetics of Si anodes. Ai and co-authors designed a novel Si@COF anode, of which the covalent-organic-framework (COF) coating has good Li+ conductivity (Fig. 6g).138 It can be observed that the Li+ diffusion coefficient of the Si@COF anode is higher than the pure Si anode during lithiation. Additionally, the Si@COF anode delivers better rate performance than the Si anode, which is largely attributed to the high Li+ conductivity, strain, and strength synergy of the COF coating. A thin titanicone coating is another promising choice for Li+ conductivity enhancement at the interface of the Si anodes. Fang and co-authors developed a novel SiNPs@titanicone anode with good fast-charging performance and strong mechanical strength.139 The SiNPs@titanicone-70 anode obtains a high retention capacity of 957 mA h g−1 after 450 cycles due to the low Young's modulus of the titanicone coating, which can release stress and buffer the volume expansion of the Si anodes during lithiation. It can also be inferred that the titanicone coating is beneficial for reducing the SEI thickness of the Si anodes. In contrast, a pure Si anode with thick SEIs will increase the Li+ migration impedance. Benefiting from the long-strand (Ti–O–benzene–O–Ti) group of the titanicone coating, the SiNPs@titanicone anode presents a lower energy barrier, good Li+ conductivity, and more efficient Li+ diffusion path, thereby facilitating the kinetic performance (Fig. 6h). Besides, the titanicone coating is also beneficial for homogenizing the stress distribution of the SiNPs@titanicone anode during lithiation. Benefiting from the lower Li+ migration impedance, the SiNPs@titanicone-70 anode delivers a higher capacity retention (50%) than the other two compared anodes at the current density of 2 A g−1.
Furthermore, the function of coatings has been extended to optimize SEI, thereby further optimizing the surface properties of Si anodes to improve the kinetic performance at the interface. Cao and co-authors developed a chain-like framework (poly-4-trifluoromethylphenylboronic acid, PTFPBA) to form B/F-rich SEIs by polymerizing 2,4,6-tris-4-(trifluoromethylphenyl)boroxine (TTFPB) coating onto the surface of Si anodes.141 The PTFPBA coating exhibits a higher adsorption energy with EC, EMC, DMC, and FEC and lower adsorption energy with LiPF6 than that of bare Si, indicating that the PTFPBA coating can inhibit the growth of SEI and take on several important SEI roles. Benefiting from good Li+/e− conductivity and a high Young's modulus, the Si@TTFPB anode exhibits a higher Li+ diffusion coefficient and better rate performance than the compared Si anode.
As shown in Table 3, elastic coatings typically exhibit one or several functions including the contributions to electronic conductivity, Li+ migration, mechanical strength, and SEI optimization. According to the quantization of the contributions for typical elastic coatings and the comparison of rate performances (Fig. 6i), coatings with higher Li+ conductivity or inducing SEI affinity to Li+ can exhibit better rate performance, illustrating that both coatings’ properties and their impact on SEI should be considered in coating construction to enhance the kinetic performance. As shown in Fig. 6j, elastic organic coatings have a deeper potential to improve the kinetic performance of Si anodes since they possess more comprehensive functions.
| Organic coating | Function | Rate performance | Ref. | |||
|---|---|---|---|---|---|---|
| Electronic conductivity | Li+ migration | Mechanical strength | Optimize SEI | |||
| TMSPA-LCP | ✓ | ✓ | ✓ | 5 A g−1, ∼46% | 137 | |
| PANi | ✓ | ✓ | 3 A g−1, ∼44% | 149 | ||
| PPBT | ✓ | ✓ | 2C, 35.5% | 150 | ||
| COF | ✓ | 5 A g−1, 34.8% | 138 | |||
| PMMA | ✓ | ✓ | 10C, 66.2% | 140 | ||
| Titanicone | ✓ | ✓ | 2 A g−1, 55.3% | 139 | ||
| TTFPB | ✓ | ✓ | 20C, ∼55% | 141 | ||
| PDA | ✓ | 4 A g−1, 30.8% | 142 | |||
| PVA | ✓ | 3 A g−1, 24.6% | 143 | |||
| bPOD | ✓ | ✓ | ✓ | 3 A g−1, 48.2% | 144 | |
| PHATN | ✓ | ✓ | 16.5 A g−1, 20.5% | 145 | ||
| CS | ✓ | ✓ | 10C, ∼ 60% | 146 | ||
| Polyurea | ✓ | ✓ | 2 A g−1, ∼60.7% | 147 | ||
As a result, the surface coating strategy of Si anodes can optimize their surface properties by improving conductivity, facilitating Li+ migration, increasing mechanical strength, and adjusting the distributions or even components of SEIs. Carbon coating can generally improve the conductivity, and promote Li+ migration through potential structural designs. While organic elastic coatings can obtain more functionality through potential functionalization, and better matching with Si particle interfaces and electrolyte interfaces, thereby promoting the kinetics enhancement at the interface more effectively.
3.2 SEI optimization strategy
The chemical distribution of SEIs can affect the Li+ migration barriers. Adopting appropriate SEI optimization strategies is conducive to obtaining superior Li+ migration ability and enhanced kinetic performance of Si anodes from another perspective at the interface level. Typical electrolytes consist of Li salts, solvents, and additives. Many studies have been focused on SEI optimization through the adjustment of electrolyte engineering including Li+ salts, solvents, and additives, thus ensuring the good kinetic performance of Si anodes.151–156Li salt is the Li+ migration carrier in electrolytes, and LiPF6 is a commercial Li salt that easily tends to dissolve in carbonate solvent.157 However, LiPF6 presents a potential corrosion challenge for Si anodes due to its decomposition and HF byproduct.155,158 Lu and co-authors explored the kinetic performance difference among six Li salts on the Si anodes, including LiPF6, LiFSI, LiTFSI, LiBOB, LiDFOB, and LiClO4.155 As shown in Fig. 7a, LiFSI delivers the highest Li+ conductivity of 11.3 mS cm−1 among these individual and composite salts, which is conducive to Li+ migration during desolvation. As a result, the Si anodes based on LiFSI obtain excellent rate performance, including over 70% capacity retention at 5C and over 45% capacity retention at 30C. Besides, the synergy of LiFSI and LiFP6 can also facilitate kinetic performance, which delivers significant improvements in rate performance from 5C to 30C. It is better than Si anodes with pure LiFSI, even though the Li+ conductivity is lower than LiFSI, suggesting potential contributions to SEI optimization. Fig. 7b compares the element distribution around Si particles based on LiFSI and LiPF6. For the Si–LiFSI electrode, Si particles are uniformly surrounded by Li and present uniform Li–O dominated inorganic layers. Since the FSI− possesses good hydrolytic stability and avoids HF formation,159 Li2O in SEI can be preserved while some LiOH phase may be formed.160,161 The Li–O dominant layer is mainly composed of Li2O, Li2CO3, and a small amount of LiF, as well as possibly LiOH, with some Li2CO3 attributed to brief exposure to air. In comparison, in the Si–LiPF6 electrode, the possible oxygen-containing inorganics (Li2O, Li2CO3, etc.) around the Si particles are largely converted into LiF after multiple cycles due to the promotion of HF by LiPF6, which corresponds to the isolated Li–F dominant region in Fig. 7b. Since the Li+ conductivity of LiF is lower than that of Li2O (∼10−9 S cm−1 for LiF and ∼10−7 S cm−1 for Li2O),162–164 the LiF-dominant layer derived from multiple cycles is more unfavorable for Li+ migration. Importantly, the Li–O dominant layer can maintain uniform contact with Si particles after multiple cycles, while the Li–F dominant layer appears as isolated clusters outside the Si particles, with observed gaps between them. It can be inferred that LiFSI is beneficial for the formation of uniform SEI and promotes the smooth migration of Li+, while in the LiPF6 system, the SEI distribution is uneven, which is not conducive to the Li+ migration. Therefore, LiFSI is crucial for constructing uniform SEI to facilitate Li+ migration, which provides more important contributions to accelerate kinetics at the interface than good Li+ conductivity.
 | ||
| Fig. 7 (a) Comparison of rate performance and Li+ conductivity for various Li salts.155 Copyright © 2022 Elsevier Ltd. All rights reserved. (b) Element maps of Si electrodes with LiFSI and LiPF6.165 Copyright © 2022 Royal Society of Chemistry. (c) Schematic of Li+ migration at the SEI.166 Copyright © 2024 Wiley-VCH GmbH. (d) Schematic of SEI formation on Si particles in different solvents.122 Copyright © 2020, The Author(s), under exclusive license to Springer Nature Limited. (e) Schematic of Li+ migration in electrolyte with TFPC.167 Copyright © 2022, American Chemical Society. (f) Schematic of SEI structure with SiCl4.168 Copyright © 2022 Elsevier Ltd. All rights reserved. | ||
It is widely recognized that SEI presents various components, including LixPFy, LiF, Li2O, Li2CO3, and organic Li-OR, LiO2COR, and poly(ethylene-oxide) type polymer/oligomers, stacking on the surface of Si particles.123–125 Ensuring uniform distribution of each component and presenting organic–inorganic stratification to promote Li+ migration is the key to improving kinetics at the interface level (Fig. 7c). Since SEI formation is mainly related to the reduction and decomposition of electrolyte solvents, it is necessary to regulate the solvent composition to construct SEI with optimized components and distributions.169–171 Si anodes largely follow carbonate solvents, which are composed of cyclic alkyl carbonates and one or several linear carbonates.170,172 Ethylene carbonate (EC) has been a widely used solvent component due to its high oxidation stability and high solubility to Li salts.160 Nevertheless, the decomposition of EC through a ring-opening reaction will promote the formation of lithium ethylene dicarbonate (LEDC), which will further react with silicide, causing significant instability of the SEI and rapid thickening with cycling.173,174 Electrolyte additives can promote the SEI formation of Si anodes with thin and uniform distribution.151,169 Schroder and co-authors investigated the positive effect of FEC on SEI structural properties.175 As the in-depth TOF-SIMS analysis described, introducing FEC can increase the inorganic chemical content (LiF and Li–X) while reducing the aliphatic carbon (C sp3, RCO, etc.), thus ensuring good Li+ conductivity and the low Li+ migration impedance of SEI at the interface.176 As a result, the cycled Si-FEC cell delivers slightly higher charge transfer impedance than the compared Si cell at the initial lithiation due to its SEI thickness increase, while an obvious impedance reduction at the de-lithiation because of the stable and uniform SEI formation. It can be inferred that FEC can improve SEI structural strength during lithiation and ensure thin and uniform SEI during cycling to obtain low Li+ migration impedance. Moreover, ether solvents have also received widespread attention due to their high ionic conductivity, low viscosity, and stability in reduction at the anode side. Ji and co-authors designed a mixed electrolyte solvent of tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MTHF) to facilitate the formation of thin and uniform LiF-based SEI.122 The lower thermodynamic reduction sites of ether promote the preferential decomposition of LiPF6, resulting in the preferential growth of fluoride salts on the surface of Si particles. As shown in Fig. 7d, a uniform SEI composed of internal LiF and external few organic species can form on the Si particles in the mixed THF electrolyte, avoiding the excessive thickening of SEI, which is conducive to Li+ migration. In comparison, the SEI formed on Si particles for EC/DMC electrolytes exhibits LiF clusters surrounded by organic components, which may thicken with cycling and hinder the Li+ migration. Benefiting from the regulated thinner SEI, the Si electrode in mixed-THF electrolytes delivers 56.4% capacity retention at a current density of 3C and ∼ 25% capacity retention in EC-DMC electrolytes. However, ether solvents present low oxidation decomposition potential, which limits their practical application. Developing multicomponent mixed solvents to fully utilize advantages is expected to be one of the necessary strategies for optimizing SEI composition and distribution to promote kinetics at the interface.
Limited by the structural defects and challenging regulating technology of natural SEI, researchers have focused on developing artificial SEIs with enhanced uniformity and stability in the kinetic performance of Si anodes. Wen and co-authors proposed a potential trifluoropropylene carbonate (TFPC) additive to promote the formation of SEIs with good Li+ conductivity and structural stability.167 As shown in Fig. 7e, TFPC can promote the uniform formation of SEI on the surface of Si anodes, since its strong electron-withdrawing capability drives ring-opening reactions preferentially on the Si surface. After XPS-etching verification, the SEI induced by TFPC presents an organic–inorganic layered structure, which provides the possibility for the kinetics enhancement of Si anodes, delivering an improved rate performance of 33.6% retention at 5C. Yang and co-authors developed a novel artificial SEI with strong structural strength and chemical stability through the plasticizing reaction between SiCl4 and LEDC.168 As described in Fig. 7f, SiCl4-based electrolytes can facilitate the formation of cross-linked aliphatic carbon in the organic layer and LiCl in the inorganic layer of SEIs, thereby avoiding the decomposition of LEDC during cycling. Benefiting from unique structural advantages, the Si–SiCl4-based cell delivers a lower Warburg factor (σ) and shorter release time (τ0) than the compared Si–SiCl4-free cell, suggesting enhanced Li+ migration through the artificial SEI. As a result, the Si–SiCl4-based cell delivered a significant rate performance enhancement.
Based on uniformity and stability, optimizing the inorganic chemical component and concentration of SEIs is beneficial for improving the kinetic performance of Si anodes. The kinetics of Li+ in different components can be estimated based on the migration barriers and conductivity of Li+.177 The relevant information on potential SEI components is summarized in Table 4, especially Li3N which stands out due to its extremely low migration barrier and high Li+ conductivity, suggesting novel and efficient components urgently need to be introduced smoothly. For the typical components, more attention should be paid to the uniform distribution of components since they present no magnitude of difference among them. However, excessive inorganic chemicals will inevitably increase the density and thickness of SEIs, shielding electron transfer and hindering kinetic performance. Lin and co-authors explored the electron tunneling-thickness effect of Li2CO3, Li3PO4, LiF, and Li layers as artificial SEIs.178 They calculated the Eg, ΔEt, d*, and C of Li2CO3, Li3PO4, LiF, and Li layers of Si anodes. Obviously, the Li2CO3 layer with a critical thickness of 2 nm presents the lowest electron tunneling barrier compared to the Li3PO4 (1.66 nm) and LiF (1.62 nm) layers, illustrating that controlling the chemical distribution and corresponding thickness of SEIs is crucial for enhancing the kinetic performance of Si anodes from the perspective of interface design.
| Component | Li+ migration barrier (eV) | Li+ conductivity (S cm−1) | Ref. |
|---|---|---|---|
| Li2O | 0.28–0.33 | ∼10−7 | 163, 164 and 179 |
| Li2CO3 | 0.28–0.60 | 10−8–10−10 | 180–182 |
| LiOH | 0.46 | — | 183 |
| Li3PO4 | 0.3–0.5 | 8.62 × 10−8 | 184 and 185 |
| LiF | 0.71 | 3 × 10−9 | 183, 186 and 187 |
| 6 × 10−6 (50 °C) | |||
| Li3N | 0.007, 0.038 | 2.085 × 10−4, 5.767 × 10−4 | 188 |
| LiCl | 0.268 | ∼10−8 | 189–191 |
| Li2S | 0.34 | ∼10−9 | 192–194 |
| Li-OR, LiO2COR | — | ∼10−9 | 195–197 |
The components and distributions of SEIs make significant differences in the kinetics at the interface. Although the composition can be effectively adjusted by controlling Li salts, and solvents, and even introducing additives, the transfer ability of Li+ in most SEI components is not outstanding. Therefore, the distribution of SEIs is more important to enhance kinetics at the interface. Constructing thin and uniform SEIs can facilitate the Li+ migration at the interface more effectively and thereby promote interfacial kinetics.
4. Electrode level
During battery operation, the influence of initial structure and subsequent structural evolution of Si electrodes on their kinetic performance is generally overlooked. Although the initial kinetic performance of Si anodes can be significantly improved through particle and interface optimization, the slowly decreasing cycling performance during operation, which involves reversible capacity and cycling stability, has always been a serious challenge.198,199 This is largely attributed to the pore growth, electrode cracking, and material peeling caused by the huge volume expansion of the Si anodes, resulting in Li+ diffusion interruption as another limiting factor that decreases their kinetic performance. Thus, the kinetic performance enhancement of Si anodes from the perspective of the electrode level in this section mainly contains electrode and collector structural design.4.1 Electrode porosity optimization
Electrode pore evolutions can affect the Li+ diffusion paths, especially in thick Si electrodes for high energy density, and the porosity directly determines the diffusion depth of Li+ inside electrodes, greatly affecting the corresponding kinetics (Fig. 8a). Various attempts have been made to optimize the porosity of Si electrodes for their kinetic performance enhancement. Wu and co-authors investigated the porosity effect on the kinetic performance of Si anodes by adjusting their particle size distribution.200 They provided four types of Si materials with D50 distributed at 20.1, 22.7, 15.7 and 13.5 μm. Benefiting from the Li+ diffusion path optimization endowed by moderate particle size distribution, the BSC2 anode delivers the lowest charge transfer impedance. It indicates that the pore structures between large particles can be filled by small particles, thereby reducing the electrode porosity and facilitating Li+ diffusion in the Si electrodes. Sung and co-authors conducted similar attempts on the particle size distribution of Si anodes.201 As shown in Fig. 8b, Si electrodes with small particles generally have low packing densities, while small/large mixture particles contribute to high packing densities. Benefiting from this design, the wide particle-size distribution Si/graphite hybrid (WD-SiG) material achieves a 14% increase in tap density when compared with the SiG material (Fig. 8c). Meanwhile, the WD-SiG anode also delivers low charge transfer impedance and rebounding tendency, indicating good adaptability during stress increases. Therefore, reducing the porosity of Si electrodes to optimize the Li+ diffusion paths and kinetic performance is a consensus. | ||
| Fig. 8 (a) Schematic of Li+ distribution in electrode at high rate.202 Copyright © 2022 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. (b) Particle diameter discrete element modeling simulation. (c) Statistical analysis for SiG and WD-SiG particles.201 Copyright © 2022 Wiley-VCH GmbH. (d) The relationship between capacity retention and electrode porosity at different rates.203 Copyright © 2014 American Chemical Society. (e) Porosity kinetics contribution of Si electrodes in deformation threshold (DFTH) and C-rate threshold (CRTH) conditions.204 Copyright © 2017 Elsevier Ltd. All rights reserved. (f) Schematic of external pressure research and (g) EIS tests of Si electrodes during pressure evolution.41 Copyright © 2020 Published by Elsevier Ltd. (h) Schematic of Li+ diffusion in an electrode with external pressure.205 Copyright © 2023 The Authors. Advanced Energy Materials published by Wiley-VCH GmbH. | ||
In addition, appropriate porosity in thick Si electrodes is also beneficial for their kinetic performance enhancement due to the demand for good electrolyte infiltration and structural buffers. Zhao and co-authors further examined the influence of electrode porosity on the kinetic performance of Si anodes.203 As shown in Fig. 8d, the decrease in electrode porosity can promote the Li+ diffusion in the electrode while excessive reduction of porosity may decrease the Li+ diffusion path, suggesting a “volcanic” relationship between the porosity and kinetic performance. Heubner and co-authors quantified the critical porosity on the kinetic performance of Si anodes depending on the deformation threshold (DFTH) and the C-rate threshold (CRTH) conditions.204 As described in Fig. 8e, the Si electrode with a 20 μm thickness requires 84% porosity to obtain the fast-charging performance at a current density of 5C, while the Si electrode with a 100 μm thickness requires 97% porosity.
Furthermore, appropriate external pressure on the Si electrodes also contributes to kinetic performance enhancement since it provides good Li+/e− contact among active materials during battery operation. As shown in Fig. 8f, Cui and co-authors determined the most appropriate external pressure of Si electrodes by investigating the stress evolution contribution to their kinetic performance.41 During the initial stage of stress increase (<1 MPa), the Si electrodes present few cracks and good Li+/e− conductivity. As the stress further increases (1–2 MPa), the poor electrolyte infiltration and electrode cracks cause a re-increase of charge transfer impedance (Fig. 8g). It can be observed that the Si electrode with 1 MPa external pressure exhibits the lowest impedance, demonstrating kinetic performance enhancement at the electrode level. As described in Fig. 8h, as the appropriate external pressure is applied, the Si particles are in closer contact with each other and adhere more closely to the conductive network, which undoubtedly promotes the Li+ diffusion and electron transfer in the electrode, thereby effectively improving the kinetic performance.
Therefore, appropriate porosity is the prerequisite for enhancing the kinetics at the electrode level, as it affects electrolyte infiltration, Li+ diffusion, and electron transfer. Specifically, loose particle distribution leads to ineffective volume occupation and prolongs the Li+ diffusion inside the electrode, resulting in poor kinetics at the electrode level, while overly dense particle distribution may hinder electrolyte infiltration and obstruct the Li+ diffusion path. Optimizing particle size distribution and applying external pressure are effective means to obtain appropriate gaps among particles and shorten the Li+ diffusion distance while ensuring electrolyte infiltration.
4.2 Electrode integrity maintenance strategy
Significant volume changes of the Si anodes tend to cause electrode cracks during battery operation, which interrupts Li+ diffusion and facilitates the formation of SEIs on the exposed crack surfaces, resulting in increased interface impedance, interrupted Li+ diffusion paths, and kinetic performance decay.67,206 Numerous studies have been conducted to restrict and repair electrode cracks.42,207–209Electrode cracks can seriously affect electrolyte infiltration and Li+ diffusion, and gas generation deteriorates the situation. Vanpeene and co-authors quantified the gas and electrolyte phase variations in electrode cracks through XRCT/XRD analysis.210 The Si standard electrode has an electrolyte phase volume fraction of 13.3% and a gas phase volume fraction of 35% (Fig. 9a), indicating that the infiltration of electrolyte into the electrode is incomplete, which is largely attributed to the obstruction of gas production to the electrolyte deep into the electrode. They further proposed a Si matured electrode which is obtained by placing the Si standard electrode in a humid environment to improve the microstructural environment inside the electrode for gas phase decrease.211 For the Si standard electrode, cracks start to form at 30% SOC and gradually grow with the charging depth increases, eventually connecting with each other to form the interrupted Li+ diffusion paths. In contrast, the growth of cracks in matured electrodes is slower, which avoids the formation of interconnected cracks that penetrate the electrodes. As a result, the Si matured electrode presents a much lower crack volume fraction of 3.5% than the Si standard electrode of 11.6% and reduces the gas phase volume fraction by ∼75% (Fig. 9b–d), indicating that cracks and gas generation can be effectively suppressed in the matured electrode. Therefore, addressing the crack and gas generation challenge to ensure electrode integrity is important for stabilizing the Li+ diffusion paths of Si electrodes and enhancing the kinetic performance.
 | ||
| Fig. 9 (a) Electrolyte phase and gas phase distribution profiles of Si standard electrodes.210 Copyright © 2018 Elsevier Ltd. All rights reserved. (b) Crack distribution 3D views of Si standard and matured electrodes, (c) crack volume fraction, and (d) electrolyte phase and gas phase volume fraction of Si standard and matured electrodes.211 Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) Schematic showing a comparison of electrical connections between a Si/PF-COONa electrode and conventional Si electrode.212 Copyright © 2017 Elsevier Ltd. All rights reserved. (f) Schematic of the self-healing and Li+ transfer of SHP-PEG binder.213 Copyright © 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (g) Schematic of the π⋯π stacking, hydrogen bonds, and the Li+ diffusion through electron-rich coordination sites for PSEA binder.214 Copyright © 2022 Wiley-VCH GmbH. (h) Mechanical mechanism and schematic of PAA-P(HEA-co-DMA) binder.209 Copyright © 2018 Elsevier Inc. (i) Element mapping of the initial cycled Si and Ga/Si anodes.215 Copyright © 2018 Elsevier Ltd. All rights reserved. | ||
Binders play a crucial role in maintaining electrode integrity by fixing Si particles, conductive agents, and current collectors in the electrode, which can effectively suppress the electrode cracking, thereby ensuring stable kinetics during cycling. Importantly, binders can also be extended with good conductivity by grafting various effective e−/Li+-conductive polymers to further enhance the kinetics at the electrode level, with the premise of ensuring electrode integrity.216–219 Endowing effective electronic conductivity to the binders can assist or replace the conductive agents to construct a complete conductive network throughout the Si electrodes, thereby accelerating electron transfer in the electrode. For example, Song and co-authors proposed a polyfluorene-type cross-linked conductive binder (CCB) serving as the secondary conductive network inside the Si electrode.220 The polyfluorene-type CCB exhibits good electronic conductivity of 5.5 × 10−2 S cm−1, promoting uniform current distribution inside the electrode and electronic depolarization effect.221 The Si-CCB electrode delivers a capacity retention of ∼71% at 1 A g−1, higher than that of conventional CMC (56%) and PAA (62.5%) binders. Liu and co-authors developed a conductive polymer binder (sodium poly[9,9-bis(3-propanoate)fluorine], PF-COONa) with high conductivity provided by the main chains (n-type doped polyfluorene) and strong adhesion of the carboxyl side chains, to construct conductive agent free Si electrodes.212 As shown in Fig. 9e, the designed PF-COONa can maintain constant electrical connections even as the Si particles break into pieces, while the conventional conductive agents are pushed away and disconnected from Si particles due to the volume expansion of Si particles. Benefiting from good electronic conductivity and electrical connections, the Si/PF-COONa electrode delivers 41.5% capacity retention at 8.4 A g−1, presenting superior kinetics at the electrode level. Moreover, the influence of ionic conductivity on kinetics is more significant, as superior ion conductive binders can provide potential Li+ diffusion paths.217,219 Munaoka and co-authors designed a functional binder (self-healing polymer incorporated with polyethylene glycol, SHP-PEG) with high Li+ conductivity.213 As shown in Fig. 9f, self-healing polymer (SHP) is rich in dynamic hydrogen bonds that endow the binders with significant mechanical strength to ensure electrode integrity (Fig. 9h-i), while polyethylene glycol (PEG) units assist in the Li+ transfer in the oxygen-rich channels of the binders (Fig. 9h-ii). Introducing 40% PEG2000 can promote an enhancement of Li+ conductivity by 6 times. The Si/SHP-PEG electrode demonstrates 36% capacity retention at a current density of 2C. Li and co-authors proposed that oxygen heteroatoms can provide complexation sites to serve as Li+ diffusion paths.222 They further developed a high ionic conductivity binder (GG-g-PAM) by grafting polyacrylamide (PAM) onto the guar gum (GG) backbone, where the grafting chains buffer stress and oxygen heteroatoms promote Li+ diffusion. Similarly, Hu and co-authors introduced side chains with oxygen atoms onto the binders to provide electron-rich coordination sites,214 developing an interface-adaptive triblock binder (PSEA), including segments of hydrophobic polystyrene (S), elastic poly(2-(2-methoxyethoxy)ethyl acrylate) (E), and hydrophilic poly(acrylic acid) (A) (Fig. 9g). Meanwhile, the electrode integrity is double guaranteed by π⋯π stacking and abundant hydrogen bonds at the hydrophilic side. The Si/PSEA electrode presents an enhanced capacity retention of ∼50% at a current density of 2C, while the Si/CMC-SBR electrode exhibits only ∼42% capacity retention. Therefore, optimizing the Li+ conductivity of the binder usually involves constructing electron-rich coordination sites to promote the Li+ transfer, which can provide potential Li+ diffusion paths, thereby enhancing the kinetics at the electrode level. Table 5 enumerates typical conductive binders for Si anodes, in which the increases in Li+ conductivity often promote more significant kinetics enhancement, demonstrating the urgency of optimizing the ionic conductivity inside the Si electrodes.
| Binder | Conductive function | Rate performance | Cycling stability | Ref. |
|---|---|---|---|---|
| Polyfluorene-type CCB | Electron conductive | 1 A g−1, ∼71% | 0.8 A g−1, 250 cycles, 88% | 220 |
| PPyE | Electron conductive | 2C, ∼45% | 2C, 1000 cycles, ∼1500 mA h g−1 | 223 |
| PFPQ-COONa | Electron conductive | 1C, ∼36% | 0.5C, 400 cycles, 901 mA h g−1 | 224 |
| Polyimine | Electron conductive | 2.5C, 28.2% | 1C, 1000 cycles, 82.4% | 225 |
| PF-COONa | Electron conductive | 8.4 A g−1, 41.5% | 4.2 A g−1, 1000 cycles, 999 mA h g−1 | 212 |
| APA/CNT | Electron conductive | 0.5 A g−1, 84.5% | 0.2 A g−1, 240 cycles, 87.7% | 226 |
| PP@CA | Electron conductive | 3.2 A g−1, 64.3% | 2 A g−1, 2000 cycles, 588 mA h g−1 | 227 |
| b-POB | Electron/ion conductive | 1C, 48.5% | 1C, 100 cycles, ∼1800 mA h g−1 | 228 |
| SHP-PEG | Ion conductive | 2C, ∼36% | 0.5C, 150 cycles, 80% | 213 |
| GG-g-PAM | Ion conductive | 8 A g−1, ∼43% | 1 A g−1, 100 cycles, 83.9% | 222 |
| SSIP | Ion conductive | 1C, ∼65% | 0.5C, 400 cycles, 1620 mA h g−1 | 229 |
| c-PEO-PEDOT:PSS/PEI | Ion conductive | 8 A g−1, 50.4% | 1 A g−1, 500 cycles, 2027 mA h g−1 | 230 |
| PSEA | Ion conductive | 2C, ∼50% | 0.5C, 400 cycles, 82.1% | 214 |
| SHA | Ion conductive | 1C, ∼44% | 0.2C, 100 cycles, 1770.1 mA h g−1 | 231 |
Furthermore, various self-healing strategies have emerged to further maintain the integrity of Li+ diffusion paths.232–234 Xu and co-authors restricted the formation of Si electrode cracks and repaired them through a novel polymer binder with high mechanical strength and good self-healing ability.209 The PAA-P(HEA-co-DMA) binder is synthesized through a thermal condensation reaction between PAA and P(HEA-co-DMA). Benefiting from the rigid and soft chain networks which provide good stretching and shrinking ability (Fig. 9h), the Si/PAA-P(HEA-co-DMA) electrode presents fewer cracks and better Li+ diffusion path integrity than the Si electrode. As a result, the Si/PAA-P(HEA-co-DMA) electrode delivers a significantly higher rate performance than the compared Si/PAA electrode. Moreover, Han and co-authors proposed a novel electrode crack repair strategy through liquid metal with high Li+/e− conductivity, which could fill these cracks and preserve electrode structural integrity.215 As shown in Fig. 9i, Ga fills the Ga/Si electrode cracks, repairing the conductive network and Li+ diffusion paths, which contributes to lower impedance. And the Si/Ga electrode delivers a higher kinetic performance of 16% capacity retention at 10C. According to Table 6, self-healing strategies can effectively promote the repair of electrode cracks, ensuring the integrity of the Li+ diffusion path. After comparison, liquid metal can effectively maintain the Li+ diffusion path and enhance electron transfer in the electrode due to its excellent conductivity and fluidity, thus improving the kinetics more significantly.
| Healing material | Rate performance | Cycling stability | Ref. |
|---|---|---|---|
| Ga/Si | 10C, ∼16% | 4C, 1500 cycles, 81.3% | 215 |
| Ga12.6Sn1.0 | 3C, 54.6% | 2C, 900 cycles, 95.7% | 235 |
| EGaIn-Mxene | 2C, 30.8% | 2C, 100 cycles | 236 |
| GaGeSiP3 | 20 mA cm−2, 57.1% | 6 mA cm−2, 2000 cycles, 90% | 237 |
| PAA-P(HEA-co-DMA) | 1.96C, 72.7% | 5 A g−1, 200 cycles, 90% | 209 |
| XPAA-DABBF | 5C, 57.2% | 0.5C, 500 cycles, 47.65% | 238 |
| PAA-DA/PVA | 4 A g−1, ∼63% | 4 A g−1, 500 cycles, 50.8% | 239 |
| PAA-DA | 1C, 66.8% | 0.5C, 300 cycles, 1834.1 mA h g−1 | 240 |
| TCB | 3C, 49.8% | 0.5C, 200 cycles, 621.2 mA h g−1 | 241 |
| DNB | 4C, 18.2% | 1C, 300 cycles, 1115 mA h g−1 | 242 |
| yCDp/Py-PAA | 2C, 54.1% | 0.5C, 300 cycles, 86.4% | 243 |
As a result, maintaining the integrity of Si electrodes is crucial for ensuring good kinetics during cycling. Binders should receive more attention since their potential for functionalization can satisfy multiple requirements on conductivity and self-healing. In particular, the development of binders with high conductivity helps to obtain matching rapid Li+ diffusion and electron transfer, which can effectively improve the kinetic performance at the electrode level.
4.3 Current collector modification strategy
It is well known that the current collector plays an important role in electron collection and electrode support to maintain Li+ diffusion path integrity and efficiency at the electrode level.244 Cu foil is recognized as an important commercial anode current collector due to its good electronic conductivity. However, Si materials tend to fall off the typical Cu foil since the smooth surface cannot provide enough adhesion for the electrode during battery operation. Thus, optimizing the surface characteristics of the current collector through a targeted modification strategy can also enhance and maintain the kinetic performance of the Si anodes.245 Xue and co-authors increased the surface roughness of Cu foil to provide an enhanced adhesion strength with Si materials, thereby ensuring the Si/m-Cu electrodes deliver higher kinetic performance than the compared Si/Cu electrode.246 Shen and co-authors further developed carbon-coated Cu foil through an in situ chemical vapor deposition strategy.247 The Si/coated Cu foil pouch cell delivers lower initial impedance and operation impedance after 300 cycles than the compared Si/Cu foil pouch cell, suggesting that Cu foil with carbon coating is beneficial for maintaining the Li+ diffusion path integrity. As a result, the Si/coated Cu foil pouch cell has a higher rate performance than the Si/Cu foil pouch cell (Fig. 10a). Furthermore, Zhang and co-authors developed multi-shelled Si@Cu microparticles supported on a 3D Cu collector to further improve the adhesion and electron transfer between Si materials and Cu collector.248 As shown in Fig. 10b, Si materials are confined within rigid Cu meshes, ensuring the integrity of Li+ diffusion paths and promoting kinetic performance at the electrode level. Benefiting from the interconnected 3D Cu network with rich pore structures, the multi-shelled Cu@Si@Cu electrode has a good rate performance of 76% capacity retention at a current density of 10C. | ||
| Fig. 10 (a) Rate performance of Si/bare Cu and Si/coated Cu electrodes.247 Copyright © 2020 The Authors. Published by ESG. Published by Elsevier B.V. (b) Schematic of kinetics enhancement for multishelled Cu@Si@Cu microparticles coated on 3D Cu current collectors.248 Copyright © 2018 American Chemical Society. (c) Stress distribution of standard and porous Cu collectors.249 Copyright © 2018 Elsevier B.V. All rights reserved. (d) Li+ concentration distribution simulation of TCC and PCC electrode on a full charge, (e) ionic conductivity comparison of PE, Celgard 2325, PCC matrix and PCC, and (f) rate performance of Si/PCC and Si/TCC electrodes.43 Copyright © 2024, The Author(s), under exclusive license to Springer Nature Limited. | ||
Additionally, Moon and co-authors proposed using porous Cu collectors to release the stress of the Si anode during cycling, thereby ensuring the integrity of the electrode.249 As shown in Fig. 10c, the pores of the porous current collector can release the stress generated by volume expansion, thus keeping the internal stress of the electrode at a lower level compared to the standard current collector, which can improve the kinetics at the electrode level. Li+ usually bypasses the traditional current collector (TCC) rather than passing through due to high compactness, which increases the Li+ diffusion distance, thus restricting the kinetic performance of the Si anodes.250 Ye and co-authors designed a porous current collector (PCC) by modifying nanoporous Kevlar film to resolve this challenge.43 As described in Fig. 10d, an even Li+ concentration ranging from 1.0 to 0.8 along the distance from separator to PCC can be observed. As a comparison, the TCC exhibits an uneven and wider Li+ concentration range (1.0–0.3) along the distance from the separator to the TCC. The rich porosity and high Li+ conductivity can effectively shorten Li+ diffusion distance and accelerate Li+ migration, thereby ensuring the high kinetic performance of Si/PCC electrodes (Fig. 10e and f). As a result, the Si/PCC pouch cell delivers a better rate capacity retention of 78.3% at 4C (62.3% vs. Si/TCC) and 70.5% at 6C (33.4% vs. Si/TCC), respectively. Therefore, the adhesion strength and Li+/e− conductivity of the current collector foil in the kinetic performance enhancement of Si anodes cannot be neglected.
The ability of the current collector to collect and transfer electrons can also limit the kinetic performance at the electrode level.244 Although Cu foil has superior conductivity, the contact between the electrode and the current collector remains the limiting factor. For Si electrodes, volume changes make active materials tend to peel from the current collector, further exacerbating the unstable contact. Improving the adhesion between the electrode and the current collector is the most effective method to enhance the kinetic performance of Si anodes at the electrode–collector interface.
5. Summary and perspective
To conclude, the particle–interface–electrode integration of Si anodes is deconstructed in this review to identify the main issues that limit the kinetic performance: long Li+ diffusion distance and poor intrinsic conductivity for particles, high Li+ migration impedance at the interface due to defective material surface and limited migration barrier, and insufficient or even interrupted Li+ diffusion paths for initial and cycled electrodes. Through vertical combining of particle, interface, and electrode levels and fully considering their mutual influences, we have evaluated effective strategies for progressively enhancing the kinetics of Si anodes. Specifically, as follows:(i) Si materials with appropriate particle size or pore size distribution largely possess enhanced kinetic performance due to the balance between Li+ diffusion distance and Li+ migration impedance.
(ii) Element doping and particle compositing strategies could effectively enhance the conductivity or promote the Li+ diffusion inside particles. And further adjusting the doping or compositing concentration can obtain the correspondingly optimal kinetic performance.
(iii) Coating and SEI optimization strategies could improve the interface properties of Si anodes, which is conducive to facilitating Li+ migration at the interface, thereby significantly reducing the Li+ migration impedance of Si anodes.
(iv) Appropriate particle size distribution and external pressure, effective binder strategies, as well as modification of the roughness and pore structure of the current collector can adjust electrode porosity and suppress the occurrence of cracks and peeling for initial or cycling electrodes, thus ensuring the integrity of Si electrodes and sufficient Li+ diffusion paths.
Although the emerging strategies promote significant academic and commercial achievements for Si anodes, quantifying the quantitative relationship between kinetics and the strategies of multiple dimensions, as well as balancing their mutual influence, remains a serious challenge of fast charging Si anodes for power batteries. For this reason, several potential solutions require further exploration.
(i) Quantifying the optimal particle size and pore size. The particle size and pore size distribution of Si materials are closely related to their Li+ diffusion distance and involve mutual influence among particle, interface, and even electrode multiple levels. Establishing two theoretical models of “particle size – Li+ diffusion distance” and “pore size distribution – Li+ diffusion distance” with the optimal values is a challenging but necessary topic.
(ii) Establishing the Li+ diffusion barrier comparison table of Si anodes. Although doping element types and the corresponding concentration of Si materials affect the conductivity, most attempts are undirected and inefficient. Therefore, it is necessary to establish the above standard comparison table based on doping elements and their concentration to overcome this challenge.
(iii) Evaluating the contributions of coatings to kinetics for Si anodes. Numerous coatings have been established for Si anodes, endowed with various functions including conductivity, Li+ migration, mechanical strength, and SEI formation. However, their contributions to kinetic behavior and the performance of Si anodes lack quantification. Establishing a database to summarize the enhancement percentage on rate performance through different coatings and their thickness is a significant quantitative method to select the optimal coating suitable for fast-charging Si anodes.
(iv) Constructing targeted SEI precisely. Although in-depth research has been conducted on various components of the SEI, a single component is not sufficient to support the revelation of the complex and disordered SEI integration, resulting in difficulty in controlling the uniformity, composition, thickness, etc. More attention should be paid to the development and introduction of SEIs dominated by components with excellent Li+ conductivity (e.g. Li3N). For typical SEIs, the optimization should be holistically discussed from multiple perspectives of composition, spatial distribution, and overall thickness, avoiding focusing solely on the effect of a single component in a single perspective.
(v) Maintaining Li+ diffusion path integrity during cycling. The formation of porosity, cracks, and peeling of Si electrodes due to the huge volume change and uneven stress distribution tend to interrupt the Li+ diffusion path. How to quantify their impact on the kinetic performance degradation of Si electrodes remains a challenge. The theoretical model “local stress–strain distribution” should be applied to the Si anodes that have undergone enhancement at particle and interface levels. And then calculate the critical stress of crack generation, which is beneficial for further optimizing the Si electrodes and fully exerting the enhancement strategies from multiple levels.
(vi) Promoting dry electrode technology on Si electrodes. Dry electrode technology can avoid the uncontrollable migration of non-active materials stemming from the drying process in the wet process, thereby ensuring a uniform ion/electron transfer network, which is beneficial for improving the fast-charging performance of Si anodes theoretically. However, severe volume expansion can hinder the application of the current dry process on Si anodes. Specifically, Si particles, conductive agents, and binders all suffer from agglomeration stemming from the lack of solvent dispersion, resulting in poor conductivity and electrode instability. Futhermore, volume expansion can exacerbate this instability. It is crucial to develop materials (e.g. binders and conductive agents) and optimize dry processes adaptable to Si anodes. In particular, the development of dry electrode technologies suitable for Si anodes should focus on the adaptation of surface energy among binders, conductive agents, and Si particles to promote the balance between binder cohesion and the adhesion to current collector and materials, thus enhancing the kinetics at the electrode level.
In summary, the kinetic performance enhancement of Si anodes requires design strategies from multiple dimensions. Benefiting from the viewpoints of particle, interface, and electrode levels, we expect this review would contribute to the research and development of high energy density and fast-charging power LIBs based on Si anodes, leaping towards the 4C fast charging era (Fig. 11).
 | ||
| Fig. 11 Prospects for improving the kinetic performance of Si anodes. | ||
Author contributions
P. Jia: conceptualization, visualization, writing – original draft, J. Guo: conceptualization, writing – review & editing, Q. Li: writing – review & editing, Y. Liu: investigation, formal analysis, Y. Zheng and Y. Guo: formal analysis, validation, Y. Huang, Y. Shen, L. Long, S. Dong, and J. Jiang: validation, H. Zhang, R. Chen, C. Zhang, Z. Zhang, and J. Shen: formal analysis, M. Chang, X. Liu, and X. Wang: visualization, Y. Tang: writing – review & editing, H. Shao: writing – review & editing, supervision, funding acquisition, project administration.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicting interests regarding the content of this article.Acknowledgements
This work was financially supported by the Science and Technology Planning Project of Shenzhen of China (Shenzhen-Hong Kong-Macao Category C) (No. SGDX20220530111004028), Macau Science and Technology Development Fund (FDCT) for funding of the Macao Centre for Research and Development in Advanced Materials (2022–2024) (No. 0026/2022/AMJ and 006/2022/ALC), the Natural Science Foundation of Guangdong Province (No. 2023A1515010765), the Science and Technology Planning Project of Guangdong Province of China (No. 2023A0505030001), and a Multi-Year Research Grant (MYRG) from University of Macau (No. MYRG-GRG2023-00140-IAPME-UMDF).References
- I. E. A. (2023), Global EV Outlook 2023, https://www.iea.org/reports/global-ev-outlook-2023, (accessed April, 2023).
- Z. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- J. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef.
- D. Meng, Z. Xue, G. Chen, D. Zhou, Y. He, Z. Ma, Y. Liu and L. Li, Energy Environ. Sci., 2024, 17, 4658–4669 RSC.
- M. M. Yuan, H. J. Liu and F. Ran, Mater. Today, 2023, 63, 360–379 CrossRef CAS.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- A. Zhang, Z. Bi, G. Wang, S. Liao, P. Das, H. Lin, M. Li, Y. Yu, X. Feng, X. Bao and Z. Wu, Energy Environ. Sci., 2024, 17, 3021–3031 RSC.
- C. Y. Wang, T. Liu, X. G. Yang, S. H. Ge, N. V. Stanley, E. S. Rountree, Y. J. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- S. H. Park, P. J. King, R. Y. Tian, C. S. Boland, J. Coelho, C. F. Zhang, P. McBean, N. McEvoy, M. P. Kremer, D. Daly, J. N. Coleman and V. Nicolosi, Nat. Energy, 2019, 4, 560–567 CrossRef CAS.
- C. F. Zhang, S. N. Park, A. Seral-Ascaso, S. Barwich, N. McEyoy, C. S. Boland, J. N. Coleman, Y. Gogotsi and V. Nicolosi, Nat. Commun., 2019, 10, 849 CrossRef PubMed.
- X. Su, Q. L. Wu, J. C. Li, X. C. Xiao, A. Lott, W. Q. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- U. S. A. B. Consortium, Low-Cost/Fast-Charge EV Goals, https://uscar.org/usabc/.
- Tesla Model S, https://www.tesla.com/models.
- B.Y.D, https://www.bydauto.com.cn/pc/brandRelated?id=3.
- M. Armand and J. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- W. X. Huang, Y. S. Ye, H. Chen, R. A. Vilá, A. Xiang, H. X. Wang, F. Liu, Z. A. Yu, J. W. Xu, Z. W. Zhang, R. Xu, Y. C. Wu, L. Y. Chou, H. S. Wang, D. T. Boyle, Y. Z. Li and Y. Cui, Nat. Commun., 2022, 13, 7091 CrossRef PubMed.
- D. P. Finegan, A. Quinn, D. S. Wragg, A. M. Colclasure, X. K. Lu, C. Tan, T. M. M. Heenan, R. Jervis, D. J. L. Brett, S. Das, T. Gao, D. A. Cogswell, M. Z. Bazant, M. Di Michiel, S. Checchia, P. R. Shearing and K. Smith, Energy Environ. Sci., 2020, 13, 2570–2584 RSC.
- M. Salah, P. Murphy, C. Hall, C. Francis, R. Kerr and M. Fabretto, J. Power Sources, 2019, 414, 48–67 CrossRef CAS.
- H. Jin, Y. Huang, C. Wang and H. Ji, Small Sci., 2022, 2, 2200015 CrossRef CAS.
- Y. Liu, H. Shi and Z. Wu, Energy Environ. Sci., 2023, 16, 4834–4871 RSC.
- H. Zhang, L. Wang, H. Li and X. M. He, ACS Energy Lett., 2021, 6, 3719–3724 CrossRef CAS.
- A. Zulke, Y. Li, P. Keil, R. Burrell, S. Belaisch, M. Nagarathinam, M. P. Mercer and H. E. Hoster, Batteries Supercaps, 2021, 4, 934–947 CrossRef.
- Y. F. Li, Q. M. Li, J. L. Chai, Y. T. Wang, J. K. Du, Z. Y. Chen, Y. C. Rui, L. Jiang and B. H. J. Tang, ACS Mater. Lett., 2023, 5, 2948–2970 CrossRef CAS.
- X. Xia, X. Y. Qian, C. Chen, W. Y. Li, D. F. He, G. Y. He and H. Q. Chen, J. Energy Storage, 2023, 72, 108715 CrossRef.
- M. Jiang, J. Chen, Y. Zhang, N. Song, W. Jiang and J. Yang, Adv. Sci., 2022, 9, 2203162 CrossRef CAS PubMed.
- M. Peng, K. Shin, L. Jiang, Y. Jin, K. Zeng, X. Zhou and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, e202206770 CrossRef CAS PubMed.
- S. Chae, S. Choi, N. Kim, J. Sung and J. Cho, Angew. Chem., Int. Ed., 2020, 59, 110–135 CrossRef CAS PubMed.
- C. Y. Chou and G. S. Hwang, Surf. Sci., 2013, 612, 16–23 CrossRef CAS.
- B. Peng, F. Y. Cheng, Z. L. Tao and J. Chen, J. Chem. Phys., 2010, 133, 034701 CrossRef PubMed.
- W. H. Wan, Q. F. Zhang, Y. Cui and E. G. Wang, J. Phys.:Condens. Matter, 2010, 22, 415501 CrossRef PubMed.
- Q. F. Zhang, Y. Cui and E. G. Wang, J. Phys. Chem. C, 2011, 115, 9376–9381 CrossRef CAS.
- L. Wang, J. Yu, S. Y. Li, F. S. Xi, W. H. Ma, K. X. Wei, J. J. Lu, Z. Q. Tong, B. Liu and B. Luo, Energy Storage Mater., 2024, 66, 103243 CrossRef.
- Q. Y. Man, Y. L. An, C. K. Liu, H. T. Shen, S. L. Xiong and J. K. Feng, J. Energy Chem., 2023, 76, 576–600 CrossRef CAS.
- H. H. Zheng, L. Tan, G. Liu, X. Y. Song and V. S. Battaglia, J. Power Sources, 2012, 208, 52–57 CrossRef CAS.
- I. V. Thorat, D. E. Stephenson, N. A. Zacharias, K. Zaghib, J. N. Harb and D. R. Wheeler, J. Power Sources, 2009, 188, 592–600 CrossRef CAS.
- Q. Li, X. S. Liu, X. Han, Y. X. Xiang, G. M. Zhong, J. Wang, B. Z. Zheng, J. G. Zhou and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 14066–14075 CrossRef CAS PubMed.
- J. Cui, X. Chen, Z. Zhou, M. Zuo, Y. Xiao, N. Zhao, C. Shi and X. Guo, Mater. Today Energy, 2021, 20, 100632 CrossRef CAS.
- H. Chen, Z. Z. Wu, Z. Su, S. Chen, C. Yan, M. Al-Mamun, Y. B. Tang and S. Q. Zhang, Nano Energy, 2021, 81, 105654 CrossRef CAS.
- Y. S. Ye, R. Xu, W. X. Huang, H. Y. Ai, W. B. Zhang, J. O. Affeld, A. Cui, F. Liu, X. Gao, Z. Y. Chen, T. Y. Li, X. Xiao, Z. W. Zhang, Y. C. Peng, R. A. Vila, Y. C. Wu, S. T. Oyakhire, H. Kuwajima, Y. Suzuki, R. Matsumoto, Y. Masuda, T. Yuuki, Y. Nakayama and Y. Cui, Nat. Energy, 2024, 9, 11 Search PubMed.
- F. H. Du, K. X. Wang and J. S. Chen, J. Mater. Chem. A, 2016, 4, 32–50 RSC.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4984 CrossRef CAS PubMed.
- S. W. Lee, H. W. Lee, I. Ryu, W. D. Nix, H. J. Gao and Y. Cui, Nat. Commun., 2015, 6, 7533 CrossRef CAS PubMed.
- H. Yang, S. Huang, X. Huang, F. F. Fan, W. T. Liang, X. H. Liu, L. Q. Chen, J. Y. Huang, J. Li, T. Zhu and S. L. Zhang, Nano Lett., 2012, 12, 1953–1958 CrossRef CAS PubMed.
- X. H. Liu, J. W. Wang, S. Huang, F. F. Fan, X. Huang, Y. Liu, S. Krylyuk, J. Yoo, S. A. Dayeh, A. V. Davydov, S. X. Mao, S. T. Picraux, S. L. Zhang, J. Li, T. Zhu and J. Y. Huang, Nat. Nanotechnol., 2012, 7, 749–756 CrossRef CAS PubMed.
- M. McDowell, S. Lee, J. Harris, B. Korgel, C. Wang, W. Nix and Y. Cui, Nano Lett., 2013, 13, 758–764 CrossRef CAS PubMed.
- J. Wang, Y. He, F. Fan, X. Liu, S. Xia, Y. Liu, C. Harris, H. Li, J. Huang, S. Mao and T. Zhu, Nano Lett., 2013, 13, 709–715 CrossRef CAS PubMed.
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J. M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed.
- M. McDowell, I. Ryu, S. Lee, C. Wang, W. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed.
- Z. Jia and T. Li, J. Mech. Phys. Solids, 2016, 91, 278–290 CrossRef CAS.
- M. T. McDowell, S. W. Lee, J. T. Harris, B. A. Korgel, C. M. Wang, W. D. Nix and Y. Cui, Nano Lett., 2013, 13, 758–764 CrossRef CAS PubMed.
- Q. Y. Liu, C. L. Pang, W. L. Chen, Z. X. Rao, H. Q. Lu, L. H. Xue and W. X. Zhang, Energy Technol., 2019, 7, 1900487 CrossRef.
- F. F. Zhao, M. Zhao, Y. R. Dong, L. Ma, Y. Zhang, S. L. Niu and L. M. Wei, Powder Technol., 2022, 404, 117455 CrossRef CAS.
- J. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- Z. Bitew, M. Tesemma, Y. Beyene and M. Amare, Sustainable Energy Fuels, 2022, 6, 1014–1050 RSC.
- H. Li, H. Li, Y. Lai, Z. Yang, Q. Yang, Y. Liu, Z. Zheng, Y. Liu, Y. Sun, B. Zhong, Z. Wu and X. Guo, Adv. Energy Mater., 2022, 12, 2102181 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- A. Casimir, H. Zhang, O. Ogoke, J. Amine, J. Lu and G. Wu, Nano Energy, 2016, 27, 359–376 CrossRef CAS.
- A. Mukanova, A. Jetybayeva, S. T. Myung, S. S. Kim and Z. Bakenov, Mater. Today Energy, 2018, 9, 49–66 CrossRef.
- T. Kasukabe, H. Nishihara, S. Iwamura and T. Kyotani, J. Power Sources, 2016, 319, 99–103 CrossRef CAS.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- X. H. Liu, H. Zheng, L. Zhong, S. Huan, K. Karki, L. Q. Zhang, Y. Liu, A. Kushima, W. T. Liang, J. W. Wang, J. H. Cho, E. Epstein, S. A. Dayeh, S. T. Picraux, T. Zhu, J. Li, J. P. Sullivan, J. Cumings, C. S. Wang, S. X. Mao, Z. Z. Ye, S. L. Zhang and J. Y. Huang, Nano Lett., 2011, 11, 3312–3318 CrossRef CAS PubMed.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. B. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 309–314 Search PubMed.
- M. Park, M. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- A. Arie and J. Lee, Nanotechnol. Appl. Energy Environ., 2013, 737, 80–84 Search PubMed.
- F. Wu, Y. Dong, Y. F. Su, C. X. Wei, T. R. Chen, W. A. Yan, S. Y. Ma, L. Ma, B. Wang, L. Chen, Q. Huang, D. Y. Cao, Y. Lu, M. Wang, L. Wang, G. Q. Tan, J. H. Wang and N. Li, Small, 2023, 19, 2301301 CrossRef CAS PubMed.
- J. Liang, X. N. Li, Z. G. Hou, W. Q. Zhang, Y. C. Zhu and Y. T. Qian, ACS Nano, 2016, 10, 2295–2304 CrossRef CAS PubMed.
- L. Zhang, C. Wang, Y. Dou, N. Cheng, D. Cui, Y. Du, P. Liu, M. Al-Mamun, S. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 8824–8828 CrossRef CAS PubMed.
- Z. Cheng, H. Lin, Y. Liu, J. Li, H. Jiang and H. Zhang, Small, 2024, 20, 2407560 CrossRef CAS PubMed.
- Y. Xu, E. Swaans, S. Chen, S. Basak, P. Harks, B. Peng, H. Zandbergen, D. Borsa and F. Mulder, Nano Energy, 2017, 38, 477–485 CrossRef CAS.
- H. Chen, Y. Xiao, L. Wang and Y. Yang, J. Power Sources, 2011, 196, 6657–6662 CrossRef CAS.
- Y. Cheng, C. Chen, S. Wang, Y. Li, B. Peng, J. Huang and C. Liu, Nano Energy, 2022, 102, 107688 CrossRef CAS.
- M. Datta, J. Maranchi, S. Chung, R. Epur, K. Kadakia, P. Jampani and P. Kumta, Electrochim. Acta, 2011, 56, 4717–4723 CrossRef CAS.
- J. Ryu, D. Hong, S. Choi and S. Park, ACS Nano, 2016, 10, 2843–2851 CrossRef CAS PubMed.
- G. L. Hou, B. L. Cheng, Y. B. Cao, M. S. Yao, B. Q. Li, C. Zhang, Q. H. Weng, X. Wang, Y. Bando, D. Golberg and F. L. Yuan, Nano Energy, 2016, 24, 111–120 CrossRef CAS.
- N. Liu, Z. Lu, J. Zhao, M. McDowell, H. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- S. Chae, Y. Xu, R. Yi, H. Lim, D. Velickovic, X. Li, Q. Li, C. Wang and J. Zhang, Adv. Mater., 2021, 33, 2103095 CrossRef CAS PubMed.
- J. Entwistle, A. Rennie and S. Patwardhan, J. Mater. Chem. A, 2018, 6, 18344–18356 RSC.
- W. Liu, J. Z. Wang, J. T. Wang, X. Z. Guo and H. Yang, J. Alloys Compd., 2021, 874, 159921 CrossRef CAS.
- Y. C. Zhao, C. G. Liu, Y. Sun, R. W. Yi, Y. T. Cai, Y. Q. Li, I. Mitrovic, S. Taylor, P. Chalker, L. Yang and C. Z. Zhao, J. Alloys Compd., 2019, 803, 505–513 CrossRef CAS.
- W. Ren, Y. Wang, Z. Zhang, Q. Tan, Z. Zhong and F. Su, J. Mater. Chem. A, 2016, 4, 552–560 RSC.
- D. Kim, S. Hwang, J. Cho, S. Yu, S. Kim, J. Jeon, K. Ahn, C. Lee, H. Song and H. Lee, ACS Energy Lett., 2019, 4, 1265–1270 CrossRef CAS.
- M. Liu, W. Xu, S. Liu, B. Liu, Y. Gao and B. Wang, Adv. Sci., 2024, 11, 2402915 CrossRef CAS PubMed.
- Y. Chen, F. Guo, L. Yang, J. Lu, D. Liu, H. Wang, J. Zheng, X. Yu and H. Li, Chin. Phys. B, 2022, 31, 078201 CrossRef CAS.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 RSC.
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS PubMed.
- H. R. Xia, W. Zhang, S. K. Cao and X. D. Chen, ACS Nano, 2022, 16, 8525–8530 CrossRef CAS PubMed.
- G. L. Hornyak, J. Dutta, H. F. Tibbals and A. Rao, Introduction to nanoscience, CRC press, 2008 Search PubMed.
- A. Nulu, V. Nulu and K. Y. Sohn, J. Alloys Compd., 2022, 911, 164976 CrossRef CAS.
- A. Nulu, Y. G. Hwang, V. Nulu and K. Y. Sohn, Nanomaterials, 2022, 12, 3004 CrossRef CAS PubMed.
- C. Luo, X. Zhou, J. Ding, H. Xu, X. Wang, H. Yao, J. Tang and J. Yang, Compos. Commun., 2021, 28, 100941 CrossRef.
- A. Nulu, V. Nulu and K. Sohn, ChemElectroChem, 2021, 8, 1259–1269 CrossRef CAS.
- X. Han, Z. Zhang, H. Chen, L. Luo, Q. Zhang, J. Chen, S. Chen and Y. Yang, J. Mater. Chem. A, 2021, 9, 3628–3636 RSC.
- D. Guo, J. Wang, Y. Mai, P. Yang, J. W. Zhou, X. J. Xu, Y. Cheng, X. Y. Dai, Y. J. Gu and F. Z. Wu, Ceram. Int., 2023, 49, 5799–5807 CrossRef CAS.
- B. Long, Y. L. Zou, Z. Y. Li, Z. S. Ma, W. J. Jiang, H. Y. Zou and H. Y. Chen, ACS Appl. Energy Mater., 2020, 3, 5572–5580 CrossRef CAS.
- J. Ryu, J. Seo, G. Song, K. Choi, D. Hong, C. Wang, H. Lee, J. Lee and S. Park, Nat. Commun., 2019, 10, 2351 CrossRef PubMed.
- J. Sung, N. Kim, J. Ma, J. H. Lee, S. H. Joo, T. Lee, S. Chae, M. Yoon, Y. Lee, J. Hwang, S. K. Kwak and J. Cho, Nat. Energy, 2021, 6, 1164–1175 CrossRef.
- Y. Son, J. Ma, N. Kim, T. Lee, Y. Lee, J. Sung, S. H. Choi, G. Nam, H. Cho, Y. Yoo and J. Cho, Adv. Energy Mater., 2019, 9, 1803480 CrossRef.
- N. Mahmood, J. H. Zhu, S. Rehman, Q. Li and Y. L. Hou, Nano Energy, 2015, 15, 755–765 CrossRef CAS.
- A. Nulu, V. Nulu and K. Y. Sohn, ChemElectroChem, 2021, 8, 1214 CrossRef CAS.
- Y. Gao, L. Fan, R. Zhou, X. Du, Z. Jiao and B. Zhang, Nano-Micro Lett., 2023, 15, 222 CrossRef CAS PubMed.
- Y. Y. Zhu, J. Xie, A. Pei, B. F. Liu, Y. C. Wu, D. C. Lin, J. Li, H. S. Wang, H. Chen, J. W. Xu, A. K. Yang, C. L. Wu, H. X. Wang, W. Chen and Y. Cui, Nat. Commun., 2019, 10, 2067 CrossRef PubMed.
- Y. Dong, C. Liu, F. Li, H. Jin, B. Li, F. Ding, Y. Yang, Z. Yang and F. Yuan, ACS Appl. Electron. Mater., 2023, 6, 816–827 CrossRef.
- S. Y. Li, Z. M. Wu, L. Y. Du, Y. L. Shi, F. Tang, R. Li and Y. D. Jiang, J. Mater. Sci.:Mater. Electron., 2018, 29, 288–293 CrossRef CAS.
- M. Chen, Q. Zhou, J. Zai, A. Iqbal, T. Tsega, B. Dong, X. Liu, Y. Zhang, C. Yan, L. Zhao, A. Nazakat, E. SharelPeisan, C. Low and X. Qian, Nano Res., 2021, 14, 1004–1011 CrossRef CAS.
- C. Hou, Y. Qu, H. Xie, H. Tian, X. Wang, H. Lu, J. Wu, Y. Ma and J. Hou, J. Mater. Sci.:Mater. Electron., 2024, 35, 132 CrossRef CAS.
- X. Qu, X. Zhang, Y. Wu, J. Hu, M. Gao, H. Pan and Y. Liu, J. Power Sources, 2019, 443, 227265 CrossRef CAS.
- Z. Wang, Q. Su, H. Deng, W. He, J. Lin and Y. Fu, J. Mater. Chem. A, 2014, 2, 13976–13982 RSC.
- M. Je, G. Song, S. Lee, H. Park, J. Kim and S. Park, J. Mater. Chem. A, 2023, 11, 1694–1703 RSC.
- F. Z. Zhang, W. Luo and J. P. Yang, Chem. – Asian J., 2020, 15, 1394–1404 CrossRef CAS PubMed.
- M. Ko, S. Chae, J. Ma, N. Kim, H. W. Lee, Y. Cui and J. Cho, Nat. Energy, 2016, 1, 16113 CrossRef CAS.
- S. Yi, Z. L. Yan, X. D. Li, Z. Wang, P. P. Ning, J. W. Zhang, J. L. Huang, D. R. Yang and N. Du, Chem. Eng. J., 2023, 473, 145161 CrossRef CAS.
- C. M. Wang, X. L. Li, Z. G. Wang, W. Xu, J. Liu, F. Gao, L. Kovarik, J. G. Zhang, J. Howe, D. J. Burton, Z. Y. Liu, X. C. Xiao, S. Thevuthasan and D. R. Baer, Nano Lett., 2012, 12, 1624–1632 CrossRef CAS PubMed.
- M. Ko, S. Chae, S. Jeong, P. Oh and J. Cho, ACS Nano, 2014, 8, 8591–8599 CrossRef CAS PubMed.
- N. Kim, S. Chae, J. Ma, M. Ko and J. Cho, Nat. Commun., 2017, 8, 812 CrossRef PubMed.
- Q. Fang, S. Xu, X. Sha, D. Liu, X. Zhang, W. Li, S. Weng, X. Li, L. Chen, H. Li, B. Wang, Z. Wang and X. Wang, Energy Environ. Sci., 2024, 17, 6368–6376 RSC.
- K. Cheng, S. Tu, B. Zhang, W. Wang, X. Wang, Y. Tan, X. Chen, C. Li, C. Li, L. Wang and Y. Sun, Energy Environ. Sci., 2024, 17, 2631–2641 RSC.
- J. Chen, X. Fan, Q. Li, H. Yang, M. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- C. Xu, F. Lindgren, B. Philippe, M. Gorgoi, F. Björefors, K. Edström and T. Gustafsson, Chem. Mater., 2015, 27, 2591–2599 CrossRef CAS.
- Z. Y. He, C. X. Zhang, Y. K. Zhu and F. Wei, Energy Environ. Sci., 2024, 17, 7 Search PubMed.
- Z. Chen, A. Soltani, Y. Chen, Q. Zhang, A. Davoodi, S. Hosseinpour, W. Peukert and W. Liu, Adv. Energy Mater., 2022, 12, 2200924 CrossRef CAS.
- G. R. Zheng, Y. X. Xiang, L. F. Xu, H. Luo, B. L. Wang, Y. Liu, X. Han, W. M. Zhao, S. J. Chen, H. L. Chen, Q. B. Zhang, T. Zhu and Y. Yang, Adv. Energy Mater., 2018, 8, 1801718 CrossRef.
- H. Dang, Y. Y. Peng, L. Wang, X. Y. Li and F. Ran, J. Energy Storage, 2023, 74, 109526 CrossRef.
- F. L. Wang, G. Chen, N. Zhang, X. H. Liu and R. Z. Ma, Carbon Energy, 2019, 1, 219–245 CrossRef CAS.
- W. L. An, B. A. Gao, S. X. Mei, B. Xiang, J. J. Fu, L. Wang, Q. B. Zhang, P. K. Chu and K. F. Huo, Nat. Commun., 2019, 10, 1447 CrossRef PubMed.
- Z. L. Xu, K. Cao, S. Abouali, M. A. Garakani, J. Q. Huang, E. K. Heidari, H. T. Wang and J. K. Kim, Energy Storage Mater., 2016, 3, 45–54 CrossRef.
- H. Tachikawa and A. Shimizu, J. Phys. Chem. B, 2006, 110, 20445–20450 CrossRef CAS PubMed.
- C. Qi, S. Li, Z. Yang, Z. Xiao, L. Zhao, F. Yang, G. Ning, X. Ma, C. Wang, J. Xu and J. Gao, Carbon, 2022, 186, 530–538 CrossRef CAS.
- B. Wang, J. Ryu, S. Choi, X. Zhang, D. Pribat, X. Li, L. Zhi, S. Park and R. Ruoff, ACS Nano, 2019, 13, 2307–2315 CAS.
- I. Son, J. Park, S. Kwon, S. Park, M. Rümmeli, A. Bachmatiuk, H. Song, J. Ku, J. Choi, J. Choi, S. Doo and H. Chang, Nat. Commun., 2015, 6, 7393 CrossRef CAS PubMed.
- S. Pan, J. Han, Y. Wang, Z. Li, F. Chen, Y. Guo, Z. Han, K. Xiao, Z. Yu, M. Yu, S. Wu, D. Wang and Q. Yang, Adv. Mater., 2022, 34, 2203617 CrossRef CAS PubMed.
- Q. Ai, Q. Y. Fang, J. Liang, X. Y. Xu, T. S. Zhai, G. H. Gao, H. Guo, G. F. Han, L. J. Ci and J. Lou, Nano Energy, 2020, 72, 104657 CrossRef CAS.
- J. B. Fang, S. Z. Chang, Q. Ren, T. Q. Zi, D. Wu and A. D. Li, ACS Appl. Mater. Interfaces, 2021, 13, 32520–32530 CrossRef CAS PubMed.
- W. Wang, Y. Wang, W. B. Huang, M. Zhou, L. Z. Lv, M. Shen and H. H. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 6919–6929 CrossRef CAS PubMed.
- Z. Cao, X. Y. Zheng, Y. Wang, W. B. Huang, Y. C. Li, Y. H. Huang and H. H. Zheng, Nano Energy, 2022, 93, 106811 CrossRef CAS.
- Y. Bie, J. Yang, X. Liu, J. Wang, Y. Nuli and W. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 2899–2904 CrossRef CAS PubMed.
- W. Shi, H. Wu, J. Baucom, X. Li, S. Ma, G. Chen and Y. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 39127–39134 CrossRef CAS PubMed.
- Y. Yu, C. Yang, Y. Jiang, J. Zhu, Y. Zhao, S. Liang, K. Wang, Y. Zhou, Y. Liu, J. Zhang and M. Jiang, Small, 2023, 19, 2303779 CrossRef CAS PubMed.
- Q. Wang, M. Zhu, G. Chen, N. Dudko, Y. Li, H. Liu, L. Shi, G. Wu and D. Zhang, Adv. Mater., 2022, 34, 2109658 CrossRef CAS PubMed.
- J. Sun, Y. Li, L. Lv, L. Wang, W. Xiong, L. Huang, Q. Qu, Y. Wang, M. Shen and H. Zheng, Adv. Funct. Mater., 2024, 34, 2410693 CrossRef CAS.
- T. Mu, Y. Sun, C. Wang, Y. Zhao, K. Doyle-Davis, J. Liang, X. Sui, R. Li, C. Du, P. Zuo, G. Yin and X. Sun, Nano Energy, 2022, 103, 107829 CrossRef CAS.
- T. W. Kwon, J. W. Choi and A. Coskun, Chem. Soc. Rev., 2018, 47, 2145–2164 RSC.
- H. Wu, G. H. Yu, L. J. Pan, N. A. Liu, M. T. McDowell, Z. A. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 CrossRef PubMed.
- R. Na, K. Minnici, G. Zhang, N. Lu, M. González, G. Wang and E. Reichmanis, ACS Appl. Mater. Interfaces, 2019, 11, 40034–40042 CrossRef CAS PubMed.
- Y. P. Wang, A. Attam, H. G. Fan, W. S. Zheng and W. Liu, Small, 2023, 19, 2303804 CrossRef CAS PubMed.
- A. M. Haregewoin, A. S. Wotango and B. J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- G. G. Eshetu and E. Figgemeier, ChemSusChem, 2019, 12, 2515–2539 CrossRef CAS PubMed.
- J. M. M. de la Hoz, F. A. Soto and P. B. Balbuena, J. Phys. Chem. C, 2015, 119, 7060–7068 CrossRef.
- L. Z. Lv, Y. Wang, W. B. Huang, Y. Y. Wang, G. B. Zhu and H. H. Zheng, Electrochim. Acta, 2022, 413, 140159 CrossRef CAS.
- Y. Yu, H. Koh, Z. Zhang, Z. Yang, A. Alexandrova, M. Agarwal, E. Stach and J. Xie, Energy Environ. Sci., 2023, 16, 5904–5915 RSC.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2013, 25, 394–404 CrossRef CAS.
- A. V. Plakhotnyk, L. Ernst and R. Schmutzler, J. Fluorine Chem., 2005, 126, 27–31 CrossRef CAS.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS PubMed.
- J. Shin, T. Kim, Y. Lee and E. Cho, Energy Storage Mater., 2020, 25, 764–781 CrossRef.
- K. Tasaki, A. Goldberg, J. Lian, M. Walker, A. Timmons and S. Harris, J. Electrochem. Soc., 2009, 156, A1019–A1027 CrossRef CAS.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- J. Sun, J. Yan, F. Li, J. Li, J. Ma, G. Xu, P. Han, G. Hou, Y. Tang, S. Dong, J. Huang and G. Cui, Adv. Mater., 2024, 36, 2405384 CrossRef CAS PubMed.
- S. Lorger, K. Narita, R. Usiskin and J. Maier, Chem. Commun., 2021, 57, 6503–6506 RSC.
- K. Asheim, P. E. Vullum, N. P. Wagner, H. F. Andersen, J. P. Maehlen and A. M. Svensson, RSC Adv., 2022, 12, 12517–12530 RSC.
- X. Li, Z. Chen, X. Liu, L. Guo, A. Li, X. Chen and H. Song, Adv. Funct. Mater., 2024, 34, 2401686 CrossRef CAS.
- Z. Y. Wen, F. Wu, L. Li, N. Chen, G. Q. Luo, J. G. Du, L. Y. Zhao, Y. Ma, Y. J. Li and R. J. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 38807–38814 CrossRef CAS PubMed.
- Z. Yang, M. X. Jiang, C. Cui, Y. X. Wang, J. W. Qin, J. Wang, Y. X. J. Wang, B. G. Mao and M. H. Cao, Nano Energy, 2023, 105, 107993 CrossRef CAS.
- Y. G. Zhang, N. Du and D. R. Yang, Nanoscale, 2019, 11, 19086–19104 RSC.
- Z. Xu, J. Yang, H. Li, Y. Nuli and J. Wang, J. Mater. Chem. A, 2019, 7, 9432–9446 RSC.
- W. Cai, Y. Yao, G. Zhu, C. Yan, L. Jiang, C. He, J. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- Y. Tian, Y. An and B. Zhang, Adv. Energy Mater., 2023, 13, 2300123 CrossRef CAS.
- K. Leung, F. Soto, K. Hankins, P. Balbuena and K. Harrison, J. Phys. Chem. C, 2016, 120, 6302–6313 CrossRef CAS.
- F. Soto, Y. Ma, J. de la Hoz, J. Seminario and P. Balbuena, Chem. Mater., 2015, 27, 7990–8000 CrossRef CAS.
- K. Schroder, J. Avarado, T. A. Yersak, J. C. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS.
- P. Lu, C. Li, E. W. Schneider and S. J. Harris, J. Phys. Chem. C, 2014, 118, 896–903 CrossRef CAS.
- Q. Zhao, S. Stalin and L. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- Y. X. Lin, Z. Liu, K. Leung, L. Q. Chen, P. Lu and Y. Qi, J. Power Sources, 2016, 309, 221–230 CrossRef CAS.
- M. Islam and T. Bredow, J. Phys. Chem. C, 2009, 113, 672–676 CrossRef CAS.
- X. Yi, Y. Guo, S. Pan, Y. Wang, S. Chi, S. Wu and Q. Yang, Trans. Tianjin Univ., 2023, 29, 73–87 CrossRef CAS.
- H. Iddir and L. Curtiss, J. Phys. Chem. C, 2010, 114, 20903–20906 CrossRef CAS.
- S. Shi, Y. Qi, H. Li and L. Hector, J. Phys. Chem. C, 2013, 117, 8579–8593 CrossRef CAS.
- X. Ma, X. Shen, X. Chen, Z. Fu, N. Yao, R. Zhang and Q. Zhang, Small Struct., 2022, 3, 2200071 CrossRef CAS.
- Y. Du and N. Holzwarth, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 76, 174302 CrossRef.
- H. Liu and S. Yen, J. Power Sources, 2006, 159, 245–248 CrossRef CAS.
- C. Li, L. Gu and J. Maier, Adv. Funct. Mater., 2012, 22, 1145–1149 CrossRef CAS.
- D. Lin, Y. Liu, W. Chen, G. Zhou, K. Liu, B. Dunn and Y. Cui, Nano Lett., 2017, 17, 3731–3737 CrossRef CAS PubMed.
- W. Li, G. Wu, C. Araújo, R. Scheicher, A. Blomqvist, R. Ahuja, Z. Xiong, Y. Feng and P. Chen, Energy Environ. Sci., 2010, 3, 1524–1530 RSC.
- Y. Shi, L. Hu, Q. Li, Y. Sun, Q. Duan, Y. Jiang, Y. Xu, J. Yi, B. Zhao and J. Zhang, Energy Storage Mater., 2023, 63, 103009 CrossRef.
- H. Duan, C. Wang, X. Zhang, J. Fu, W. Li, J. Wan, R. Yu, M. Fan, F. Ren, S. Wang, M. Zheng, X. Li, J. Liang, R. Wen, S. Xin, Y. Guo and X. Sun, J. Am. Chem. Soc., 2024, 146, 29335–29343 CrossRef CAS PubMed.
- R. Courtcastagnet, C. Kaps, C. Cros and P. Hagenmuller, Solid State Ionics, 1993, 61, 327–334 CrossRef CAS.
- T. Wi, S. Park, S. Yeom, M. Kim, I. Kristanto, H. Wang, S. Kwak and H. Lee, ACS Energy Lett., 2023, 8, 2193–2200 CrossRef CAS.
- F. Han, J. Yue, X. Fan, T. Gao, C. Luo, Z. Ma, L. Suo and C. Wang, Nano Lett., 2016, 16, 4521–4527 CrossRef CAS PubMed.
- F. Altorfer, W. Buhrer, I. Anderson, O. Scharpf, H. Bill, P. Carron and H. Smith, Physica B, 1992, 180, 795–797 CrossRef.
- L. Ünal, V. Maccio-Figgemeier, L. Haneke, G. Eshetu, J. Kasnatscheew, M. Winter and E. Figgemeier, Adv. Mater. Interfaces, 2024, 11, 2400024 CrossRef.
- M. Zhang, H. Wang, A. Shao, Z. Wang, X. Tang, S. Li, J. Liu and Y. Ma, Adv. Energy Mater., 2024, 14, 2303932 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- N. Kim, Y. Kim, J. Sung and J. Cho, Nat. Energy, 2023, 8, 921–933 CrossRef CAS.
- J. Xiao, F. Shi, T. Glossmann, C. Burnett and Z. Liu, Nat. Energy, 2023, 8, 329–339 CrossRef.
- S. J. Wu, B. Yu, Z. H. Wu, S. Fang, B. M. Shi and J. Y. Yang, RSC Adv., 2018, 8, 19894 RSC.
- J. Sung, N. Kim, S. P. Kim, T. Lee, M. Yoon and J. Cho, Batteries Supercaps, 2022, 5, e202200136 CrossRef CAS.
- L. Lu, Y. Lu, Z. Zhu, J. Shao, H. Yao, S. Wang, T. Zhang, Y. Ni, X. Wang and S. Yu, Sci. Adv., 2022, 8, eabm6624 CrossRef CAS PubMed.
- H. Zhao, N. Yuca, Z. Zheng, Y. Fu, V. Battaglia, G. Abdelbast, K. Zaghib and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 862–866 CrossRef CAS PubMed.
- C. Heubner, U. Langklotz and A. Michaelis, J. Energy Storage, 2018, 15, 181–190 CrossRef.
- H. Yu, L. Wang, Z. Zhang, Y. Li, S. Yang and X. He, Adv. Funct. Mater., 2024, 34, 2406966 CrossRef CAS.
- B. Lu, C. Q. Ning, D. X. Shi, Y. F. Zhao and J. Q. Zhang, Chin. Phys. B, 2020, 29, 026201 CrossRef CAS.
- N. Yuca, I. Kalafat, E. Guney, B. Cetin and O. S. Taskin, Materials, 2022, 15, 2392 CrossRef CAS PubMed.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. A. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- Z. X. Xu, J. Yang, T. Zhang, Y. N. Nuli, J. L. Wang and S. I. Hirano, Joule, 2018, 2, 950–961 CrossRef CAS.
- V. Vanpeene, A. King, E. Maire and L. Roué, Nano Energy, 2019, 56, 799–812 CrossRef CAS.
- V. Vanpeene, J. Villanova, A. King, B. Lestriez, E. Maire and L. Roué, Adv. Energy Mater., 2019, 9, 1803947 CrossRef.
- D. Liu, Y. Zhao, R. Tan, L. Tian, Y. Liu, H. Chen and F. Pan, Nano Energy, 2017, 36, 206–212 CrossRef CAS.
- T. Munaoka, X. Yan, J. Lopez, J. To, J. Park, J. Tok, Y. Cui and Z. Bao, Adv. Energy Mater., 2018, 8, 1703138 CrossRef.
- L. Hu, M. Jin, Z. Zhang, H. Chen, F. Ajdari and J. Song, Adv. Funct. Mater., 2022, 32, 2111560 CrossRef CAS.
- B. Han, Y. Yang, X. B. Shi, G. Z. Zhang, L. Gong, D. W. Xu, H. B. Zeng, C. Y. Wang, M. Gu and Y. H. Deng, Nano Energy, 2018, 50, 359–366 CrossRef CAS.
- Y. M. Zhao, F. S. Yue, S. C. Li, Y. Zhang, Z. R. Tian, Q. Xu, S. Xin and Y. G. Guo, InfoMat, 2021, 3, 460–501 CrossRef CAS.
- H. Chen, M. Ling, L. Hencz, H. Ling, G. Li, Z. Lin, G. Liu and S. Zhang, Chem. Rev., 2018, 118, 8936–8982 CrossRef CAS PubMed.
- H. Zhao, J. Li, Q. Zhao, X. Huang, S. Jia, J. Ma and Y. Ren, Electrochem. Energy Rev., 2024, 7, 11 CrossRef CAS.
- Q. He, J. Ning, H. Chen, Z. Jiang, J. Wang, D. Chen, C. Zhao, Z. Liu, I. Perepichka, H. Meng and W. Huang, Chem. Soc. Rev., 2024, 53, 7091–7157 RSC.
- Z. Song, S. Chen, Y. Zhao, S. Xue, G. Qian, J. Fang, T. Zhang, C. Long, L. Yang and F. Pan, Small, 2021, 17, 2102256 CrossRef CAS PubMed.
- W. Ren, K. Wang, J. Yang, R. Tan, J. Hu, H. Guo, Y. Duan, J. Zheng, Y. Lin and F. Pan, J. Power Sources, 2016, 331, 232–239 CrossRef CAS.
- Z. Li, G. Wu, Y. Yang, Z. Wan, X. Zeng, L. Yan, S. Wu, M. Ling, C. Liang, K. Hui and Z. Lin, Adv. Energy Mater., 2022, 12, 2201197 CrossRef CAS.
- S. Park, H. Zhao, G. Ai, C. Wang, X. Song, N. Yuca, V. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2015, 137, 2565–2571 CrossRef CAS PubMed.
- Y. Zhao, L. Yang, Y. Zuo, Z. Song, F. Liu, K. Li and F. Pan, ACS Appl. Mater. Interfaces, 2018, 10, 27795–27800 CrossRef CAS PubMed.
- S. Gao, F. Sun, A. Brady, Y. Pan, A. Erwin, D. Yang, V. Tsukruk, A. Stack, T. Saito, H. Yang and P. Cao, Nano Energy, 2020, 73, 104804 CrossRef CAS.
- F. Zhang, H. Xia, T. Wei, H. Li, M. Yang and A. Cao, Energy Environ. Sci., 2024, 17, 238–248 RSC.
- B. Chen, D. Xu, S. Chai, Z. Chang and A. Pan, Adv. Funct. Mater., 2024, 34, 2401794 CrossRef CAS.
- Y. Yu, J. Zhu, K. Zeng and M. Jiang, J. Mater. Chem. A, 2021, 9, 3472–3481 RSC.
- Y. Cai, C. Liu, Z. Yu, W. Ma, Q. Jin, R. Du, B. Qian, X. Jin, H. Wu, Q. Zhang and X. Jia, Adv. Sci., 2023, 10, 2205590 CrossRef CAS PubMed.
- W. Zeng, L. Wang, X. Peng, T. Liu, Y. Jiang, F. Qin, L. Hu, P. Chu, K. Huo and Y. Zhou, Adv. Energy Mater., 2018, 8, 1702314 CrossRef.
- Z. Sun, J. Zhu, C. Yang, Q. Xie, Y. Jiang, K. Wang and M. Jiang, ACS Appl. Mater. Interfaces, 2023, 15, 12946–12956 CrossRef CAS PubMed.
- W. Ma, S. Wan, X. Cui, G. Hou, Y. Xiao, J. Rong and S. Chen, Adv. Funct. Mater., 2023, 33, 2212821 CrossRef CAS.
- S. Wang and M. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- R. Narayan, C. Laberty-Robert, J. Pelta, J. Tarascon and R. Dominko, Adv. Energy Mater., 2022, 12, 2102652 CrossRef CAS.
- J. Yang, J. Li, Z. Yang, J. Liu, Y. Xiang and F. Wu, ACS Appl. Energy Mater., 2022, 5, 12945–12952 CrossRef CAS.
- Z. Ju, B. Zhang, T. Zheng, A. Marschilok, E. Takeuchi, K. Takeuchi and G. Yu, Nano Lett., 2024, 24, 6610–6616 CrossRef CAS PubMed.
- W. Li, Y. Li, J. Wang, S. Huang, A. Chen, L. Yang, J. Chen, L. He, W. Pang, L. Thomsen, B. Cowie, P. Xiong, Y. Zhou, G. Jang, D. Min, J. Byun, L. Xu, J. Huang, K. Roh, S. Kang, M. Liu, X. Duan and H. Park, Energy Environ. Sci., 2024, 17, 5387–5398 RSC.
- W. Jang, S. Kim, Y. Kang, T. Yim and T. Kim, Chem. Eng. J., 2023, 469, 143949 CrossRef CAS.
- X. Wan, C. Kang, T. Mu, J. Zhu, P. Zuo, C. Du and G. Yin, ACS Energy Lett., 2022, 7, 3572–3580 CrossRef CAS.
- X. Wan, T. Mu, B. Shen, Q. Meng, G. Lu, S. Lou, P. Zuo, Y. Ma, C. Du and G. Yin, Nano Energy, 2022, 99, 107334 CrossRef CAS.
- X. Lin, Y. Wen, J. Wang, S. Wang, X. Sun, H. Liu and X. Xu, Green Chem., 2024, 26, 2078–2086 RSC.
- M. Jiang, P. Mu, H. Zhang, T. Dong, B. Tang, H. Qiu, Z. Chen and G. Cui, Nano-Micro Lett., 2022, 14, 87 CrossRef CAS PubMed.
- J. Kim, J. Choi, K. Park, S. Kim, K. Nam, K. Char and J. Choi, Adv. Energy Mater., 2022, 12, 2103718 CrossRef CAS.
- D. Rehnlund, F. Lindgren, S. Böhme, T. Nordh, Y. M. Zou, J. Pettersson, U. Bexell, M. Boman, K. Edström and L. Nyholm, Energy Environ. Sci., 2017, 10, 1350–1357 RSC.
- Y. Yang, W. Yuan, X. Q. Zhang, Y. Z. Ke, Z. Q. Qiu, J. Luo, Y. Tang, C. Wang, Y. H. Yuan and Y. Huang, Appl. Energy, 2020, 276, 115464 CrossRef CAS.
- W. D. Xue, X. Tian, Y. W. Xue, T. Peng, Y. Li and J. L. Sun, ChemElectroChem, 2019, 6, 3039–3042 CrossRef CAS.
- X. H. Shen, L. Shao, Z. Y. Tian, Z. W. Hu and G. L. Cao, Int. J. Electrochem. Sci., 2020, 15, 9013–9023 CrossRef.
- Z. Zhang, Z. Wang and X. Lu, ACS Nano, 2018, 12, 3587–3599 CrossRef CAS PubMed.
- S. H. Moon, S. J. Kim, M. C. Kim, J. Y. So, J. E. Lee, Y. K. Shin, W. G. Bae and K. W. Park, Mater. Chem. Phys., 2019, 223, 152–156 CrossRef CAS.
- P. C. Zhu, D. Gastol, J. Marshall, R. Sommerville, V. Goodship and E. Kendrick, J. Power Sources, 2021, 485, 229321 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |