
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-plating aqueous Zn–iodine batteries enabled via halogen-complexation chemistry for large-scale energy storage†
Hong
Li‡
 ac,
Bosi
Huang‡
a,
Mingyan
Chuai‡
de,
Zhiyang
Zheng
a,
Hao
Chen
b,
Zhihong
Piao
a,
Guangmin
Zhou
*a and
Hong Jin
Fan
*bc
ac,
Bosi
Huang‡
a,
Mingyan
Chuai‡
de,
Zhiyang
Zheng
a,
Hao
Chen
b,
Zhihong
Piao
a,
Guangmin
Zhou
*a and
Hong Jin
Fan
*bc
aTsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China. E-mail: guangminzhou@sz.tsinghua.edu.cn
bSchool of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: fanhj@ntu.edu.sg
cEnergy Research Institute@NTU (ERI@N), Nanyang Technological University, Singapore 637553, Singapore
dState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, Fujian, China
eMindu Innovation Laboratory, Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou 350108, Fujian, China
First published on 13th February 2025
Abstract
Aqueous Zn–I2 batteries are promising candidates for grid-scale energy storage due to their low cost, high voltage output and high safety. However, Ah-level Zn–I2 batteries have been rarely realized due to formidable issues including polyiodide shuttling and zinc dendrites. Here, we develop 10 Ah dual-plating Zn–I2 batteries (DPZIB) by employing ZnIxG4(tetraglyme) complex chemistry, in which zinc and iodine are iteratively dissolved and deposited in the aqueous electrolyte. The battery contains no membrane or high-cost electrolytes. The G4 strengthens the Zn–I bond by acting as an electron donor, and meanwhile, it enhances the reductivity of electrolyte by its complexation with Zn2+. Such halogen-complexation chemistry endows static DPZIB with shuttle-free properties, negligible self-discharge, and minimal zinc dendrites. The battery delivers a capacity of 301.5 mA h over 1800 h at 5 mA cm−2, a low capacity decay (0.028% drop per cycle for 800 cycles at 25 mA cm−2), and a scalable capacity of up to 10.8 A h. As a proof of concept, we demonstrate an integrated system encompassing a membrane-free Zn–I2 flow battery to store solar electricity in the daytime and power electronics at night.
Broader contextAqueous Zn–I2 batteries are promising for large-scale energy storage because of the high theoretical capacity and superior safety. However, the short cycle life has been the bottleneck that is caused by both Zn anode instability and polyiodide shuttling. Instead of depositing surface protection layers, it is more technologically viable to engineer the aqueous electrolytes by introducing additives to fundamentally resolve the dissolution of polyiodide from the cathodes and enhance the reversibility of the Zn anode. In this work, we demonstrate a facile dual-plating strategy to construct aqueous Zn–I2 batteries that can run longer and realize Ah-level capacity. In this design, the active materials of zinc and iodine are iteratively dissolved and deposited, eliminating the requirement of any membrane or electrode framework for either the cathode or anode. Specifically, we utilize ZnIxG4 complexes, in which three additives (ZnI2, KI, and G4) work together to inherently diminish the polyiodide ions and prevent the growth of Zn dendrites, leading to enhanced conversion efficiency on both sides. The prototype of flow batteries integrated with photovoltaic panels is demonstrated. |
Introduction
Large-scale electrical energy storage (EES) devices are crucial in the extensive deployment of renewable energy, to buffer the impact of intermittent supplies of solar and wind electricity. For this purpose, batteries with the properties of low-cost, high-safety, long-lifespan, and high energy density are required.1–5 Many rechargeable batteries, such as lithium-ion batteries, zinc–air batteries, lead acid batteries and flow batteries, have been applied for EES systems.2,6 However, none of them fulfills the above requirements and further improvement remains critically necessary. The focus of energy storage development has shifted from solely maximizing energy density to optimizing the levelized cost of energy, which accounts for energy density, initial cost, and cycling life over the system's lifespan.7 In this context, battery systems that leverage non-flammable and cost-efficient aqueous electrolytes present a promising avenue to meet the demands of gigawatt-scale energy storage applications.8–10 Aqueous Zn–I2 batteries have emerged as promising candidates for this purpose.11–15 However, a significant challenge lies in the dissolution of polyiodide species (such as I3−/I5−), which leads to the loss of active materials and rapid capacity decay. Additionally, the direct interaction between the zinc anode and these shuttling polyiodide intermediates exacerbates zinc corrosion, resulting in diminished coulombic efficiency (CE) and eventually battery failure.11,13,14,16 This issue becomes even more pronounced in large-scale systems, where the issues of polyiodide shuttling and zinc degradation are more severe.17–19 To address these challenges, it is crucial to prevent polyiodide shuttling and zinc corrosion for improving the long-term cyclability and scalability of Zn–I2 batteries.One prevailing strategy is to employ porous materials as the I2 host to trap the polyiodide, such as micro-mesoporous carbon, graphene, metal organic frameworks, and covalent organic frameworks.20–22 However, due to the weak adsorption with polyiodide species, these host materials cannot effectively block the shuttle effect, especially for high-capacity and long-term cycling. Additionally, catalysts were deployed to facilitate the conversion of iodine species to suppress the generation of polyiodide intermediates.23,24 However, catalysts cannot fundamentally prevent the dissolution of polyiodide from the cathodes. Moreover, the discharge capacities of these ‘host–guest’ systems are often constrained by the mass of the redox-inactive host materials. Hence, to realize scalable energy storage with Ah-level capacity, it remains highly desirable to explore alternative energy storage mechanisms that maximize the utilization of active materials while ensuring high reversibility for the Zn–I2 batteries.
Herein, we propose a dual-plating strategy to construct aqueous Zn–I2 batteries potentially possible for grid-scale energy storage. The dual-plating Zn–I2 batteries (DPZIB) have a thermodynamically stable working process, in which the active materials of zinc and iodine are iteratively dissolved and deposited, eliminating the requirement of membrane or electrode frameworks for either the cathode or anode. It intrinsically avoids electrode deterioration and failure that happen often to intercalation type electrodes. This dual-plating architecture does not require complicated and time-consuming manufacturing procedures from conventional metal-ion batteries. Fig. 1(a) and (b) illustrate the key difference in the electrolyte structure. We show that the DPZIB operates through a redox-active ZnIxG4(tetraglyme) complex, which increases the binding between Zn and I, and reconstructs the Zn2+ solvated structure. With such halogen-complexation chemistry in the DPZIB, issues of polyiodide shuttling and zinc dendrites are effectively addressed. The constructed static DPZIB delivers a high capacity of 301.5 mA h at 5 mA cm−2 after 1800 h of cycling and shows a low capacity decay rate of 0.028% over 800 cycles. As a demonstration, we also constructed membrane-free Zn–I2 flow batteries powered by silicon photovoltaics. Owing to their inherent safety, high cycling stability, and simple manufacturing process, the DPZIB presented in this study might be a feasible alternative to Li-ion batteries in large-scale energy storage.
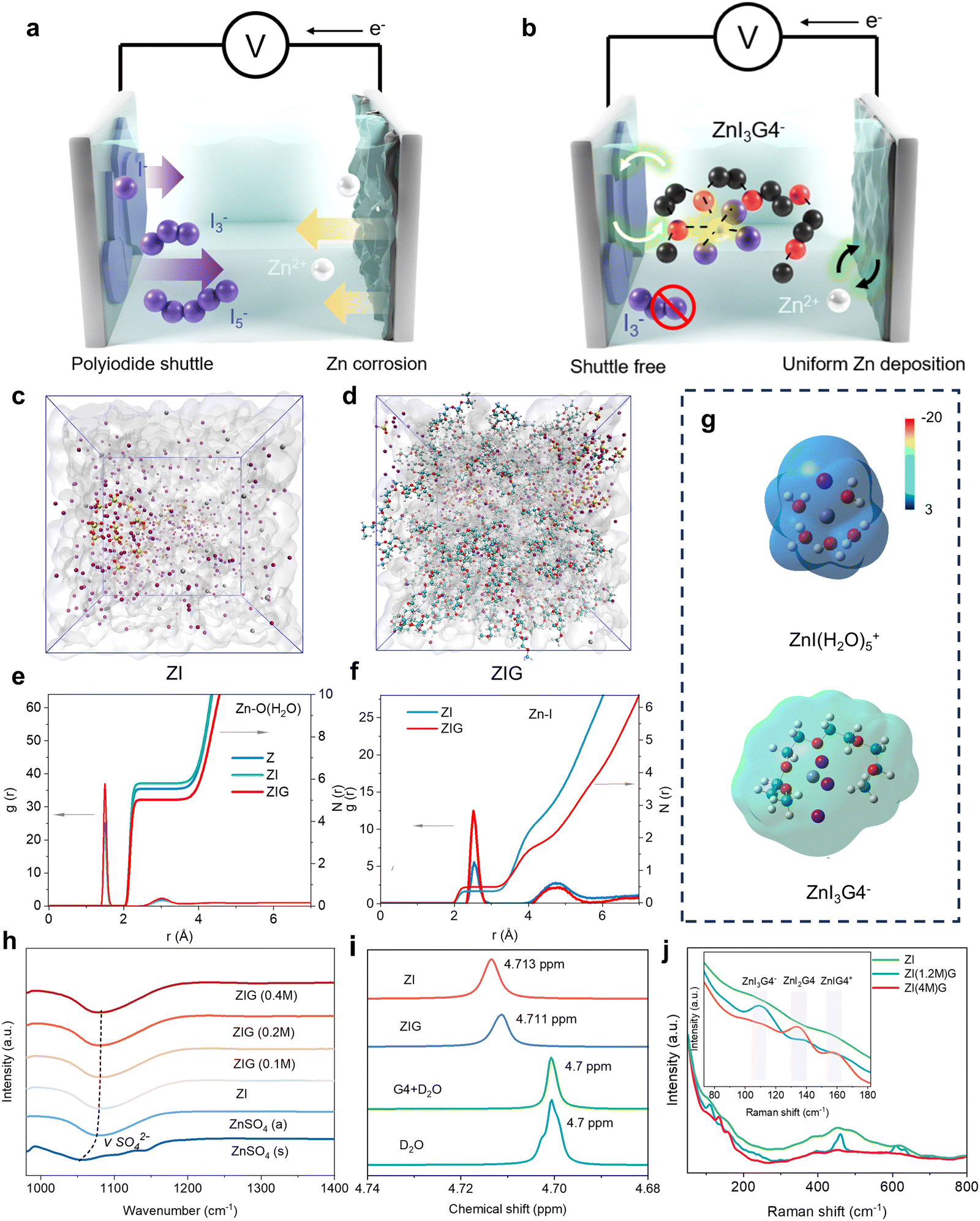 | ||
| Fig. 1 The concept of DPZIB and the electrolyte structure evolution. (a) Schematic illustration of the traditional Zn–I2 batteries and (b) our dual-plating Zn–I2 batteries. The MD snapshots of (c) ZI and (d) ZIG electrolytes. Radial distribution functions and the coordination number for (e) Zn–O (H2O) and (f) Zn–I. (g) Electrostatic potential distributions of ZnI(H2O)5+ (upper) and ZnI3G4− (bottom). (h) FTIR spectra for ZnSO4(s), ZnSO4(a) and ZIG with different amounts of G4; (i) 2H NMR spectra of H2O in pure D2O, G4 + D2O, ZI and ZIG. (j) Raman spectra of ZI and ZIG electrolytes with different concentrations of iodine ion. | ||
Results and discussion
Design concept of dual-plating aqueous Zn–I2 batteries
The zinc anode is highly reversible as a plating electrode in aqueous electrolytes. However, for large-scale systems, the issue of zinc dendrites becomes pronounced during repeated Zn2+ plating and stripping. While iodine presents itself as a promising candidate for a reversible halogen plating electrode, the key issues associated with the shuttling of polyiodide ions and aggregation of solid I2 within the electrolyte must be addressed. In a 1 M zinc sulfate electrolyte, Zn2+ forms a solvation structure, Zn(H2O)62+, due to its strong interaction with water molecules. This solvation structure facilitates electron transfer from the water molecules to the cation via the metal–OH bond, which in turn weakens the O–H bond. Consequently, this promotes zinc dendrites and the hydrogen evolution reaction (HER) at the zinc metal anode.To mitigate these issues, in our design, we introduced ZnI2 and KI into the zinc sulfate electrolyte, in which halogen ions can partially participate in the solvation structure of Zn(H2O)62+ to form ZnI(H2O)5+ owing to its robust coordination capability with transition metal ions. But this is still insufficient for achieving highly reversible plating as the additives cannot bind with intermediate product I3− nor prevent the formation of free iodine ions. To strengthen the binding between Zn2+ and polyiodide ions, we introduce a G4 additive as a strong electron donor into the electrolytes. The lone pair of oxygen atoms in G4 allows it to coordinate with Zn2+,25 and the redistribution of charges may enhance the interaction between Zn and I, thus preventing the generation of free iodine ions, which will be proved in the later section. Notably, the G4 donates electrons to the entire solvation structure and weakens the electron transfer from H2O to Zn2+, enabling the suppression of HER and Zn anode corrosion. In a nutshell, this approach may address key challenges in aqueous Zn–I2 batteries by ensuring both the stability of the zinc anode and the efficiency of iodine plating, paving the way for long cycle life and scalable energy storage systems.
Electrolyte structure evolution
Molecular dynamics simulation (MD) was conducted to quantify and elucidate the evolution of the Zn2+ solvation structure of three electrolytes: ZnSO4 (Z), ZnSO4 + ZnI2 + KI (ZI), and ZnSO4 + ZnI2 + KI + G4 (ZIG). The ZIG electrolytes were prepared by dispersing a certain amount KI and ZnI2 in 0.7 M ZnSO4 aqueous electrolytes with 0.4 M G4 as an additive (denoted as ZIG(0.4 M)). In the pure ZnSO4 electrolyte, Zn(H2O)62+ is confirmed as the dominant solvation structure, with a coordination number (CN) of Zn–O(H2O) reaching 5.80 and an average coordination distance of ∼2.15 Å (Fig. 1(e) and Fig. S1, ESI†). Upon the addition of KI and ZnI2, the I− occupies the Zn2+ solvation shell to form ZnI(H2O)5+ evidenced by a sharp Zn–I peak at 2.53 Å and a CN of 0.38, while the CN of Zn–O(H2O) decreases to 5.5 (Fig. 1(c)–(f)). After G4 was further added, the solution immediately became strikingly yellow (Fig. S2, ESI†), indicating the formation of complex compounds between G4 and the original species in the ZI electrolyte. The added G4 molecules replace the solvated H2O from the primary Zn2+ solvation shell and weaken the CN of Zn–O(H2O) from 5.5 to 5.0, demonstrating the superior capability of G4 to remove coordinated water (Fig. 1(d) and (f)). This is also confirmed by the Zn–O(G4) coordination structure with a pair peak of 2.1 Å and the CN of 2.9 (Fig. S3, ESI†). Moreover, the CN of the Zn–I coordination structure increases from 0.38 to 0.48 to form ZnI3G4− with the addition of G4. Owing to the strong electron-donating ability of the –COC– group in G4 molecules, the electrostatic potential in the solvation structure ZnI3G4− is lower compared with ZnI(H2O)5+ and Zn(H2O)62+ (Fig. 1(g) and Fig. S4, ESI†). The low electrostatic potential can effectively shield the repulsion between Zn2+, providing the ZIG with the highest ionic conductivity (47.6 mS cm−2) to make the Zn2+ diffusion more kinetically favored (Fig. S5, ESI†). Therefore, MD results suggest that G4 partially replaces the original H2O molecules to adjust the Zn2+ solvation structure and diminishes the free iodine ions by constructing ZnI3G4(yH2O)−.More evidence to the solvation structure evolution is provided by spectroscopy results. The Fourier transform infrared (FTIR) spectra presents a negative shift from 1055 to 1076 cm−1 after the ZnSO4 powders dissolve in the aqueous solution (Fig. 1(h)). This noticeable blue shift suggests the generation of the primary Zn2+ solvation shell with weakened interaction between SO42− and Zn2+. The vibration stretching of SO42− (v(SO42−)) shifts to higher wavenumbers after the addition of ZnI + KI or ZnI + KI + G4. As the amount of G4 additive increases to 0.4 M, the v(SO42−) gradually increases from 1080 to 1082 cm−1, implying a modification in the solvation structure of Zn2+ with the addition of G4. These results suggest the G4 molecules participate in the primary Zn2+ solvation shell formation and weaken the solvation interaction with H2O and Zn2+. The influence of the G4 molecules on solvated H2O molecules was investigated via liquid-phase nuclear magnetic resonance (2H NMR) analyses conducted in various electrolyte solutions utilizing D2O as the solvent (Fig. 1(i)). In comparison to pure D2O and D2O/G4, the 2H resonance peak in ZI electrolytes exhibits a shift from 4.7 ppm to 4.713 ppm. This shift can be attributed to a decrease in the electron density of neighboring atoms, resulting in a reduced shielding effect on the protons in H2O molecules. The result suggests the robust coordination between H2O and Zn2+ that decreases the number of free H2O molecules. However, after the introduction of G4 molecules into ZI electrolytes, the peak moves back to 4.711 ppm, indicating that the G4 molecules participate in the coordination environment of Zn2+, thereby releasing the partially confined H2O molecules. Raman spectroscopy was performed to analyze the potential solvation structure of Zn2+ with different concentrations of I− (1.2 M I− and 4 M I− were denoted as ZI(1.2 M)G and ZI(4 M)G, respectively). New peaks located at 100–160 cm−1, corresponding to the zinc–iodine complexes, are observed only when both I− and G4 are present (Fig. S6, ESI†). The observed Raman peaks at about 110 cm−1 are assigned to ZnI3G4− species in the ZI(1.2 M)G4 while peaks appearing at 133, and 159 cm−1 are attributed to ZnI2G4, and ZnIG4+ species in the ZI(4 M)G, respectively. Raman results suggest that once G4 and iodide co-salts (KI + ZnI2) are added into the ZnSO4 electrolyte, I− could be detained by the complex superhalide ions (e.g., ZnI3G4−), demonstrating the synergistic halogen-complexation process and effective diminishing of free iodide via adding G4 and I− (Fig. 1(j)).7,16
Theoretical insights into halogen-complexation chemistry
The halogenation-complex mechanism is further analyzed via density functional theory (DFT) calculations. The Gibbs free energy for the formation of ZnIxG4 (x = 1, 2, 3) superhalides was calculated (Fig. 2(a)). Both G4 and I− exhibit robust coordination capability with Zn2+ to partially replace H2O with energetically favorable structures (Fig. S7 and S8, ESI†). These formed superhalides (ZnIG4+, ZnI2G4, and ZnI3G4−) are exothermic with negative Gibbs free energy, suggesting that the process is thermodynamically feasible. The maximum coordination of three iodide ions in the ZnIxG4 superhalides is attributed to two key factors: the steric hindrance from the functional group of G4 and the reduced Lewis acidity of Zn2+ upon binding with I−, which limits further ligand coordination (Fig. S9, ESI†). The electrolyte structure influences battery performances stemming from the intricate balance between free iodide species, hydrated Zn2+ and Zn2+-complex ions. DFT calculations were conducted to depict the dominant Raman modes of G4, zinc iodide and potassium iodide species in an aqueous medium. As more iodide ions coordinate in the ZnIxG4 superhalides, the Raman mode frequency decreases from 167 to 111 cm−1 (Fig. 2(b)). This trend is consistent with the experimental results (Fig. 1(j)), further validating the presence of ZnIxG4 superhalides in the ZIG electrolytes. Differential charge density and atomic charge analyses provide insights into the stability of various solvation configurations. The ZnIxG4 superhalides display higher charge densities than ZnI(H2O)5+, suggesting stronger electronic coupling after the G4 complexation (Fig. 2(c) and Fig. S10, ESI†). Due to the stronger electron-donating capability of the –COC– group of G4 compared with water, O with strong electronegativity can donate electrons to both Zn2+ and I−. And G4 blocks the electron transfer from water to Zn2+ and enables higher charges of Zn2+ and I− (Fig. 2(d) and Fig. S11, ESI†). As more iodide ions are coordinated in the Zn2+-complex ions, more charge is transferred. This is verified by the changes of the atomic charges. In addition, the bond lengths of Zn–I in ZnIG4+, ZnI2G4, and ZnI3G4− are all shorter than that in the ZnI(H2O)5+, suggesting that Zn–I bonds are strengthened after its complexation. The lowest unoccupied molecular orbital (LUMO) energy levels of the various solvation structures are ultimately investigated to assess their reducibility. The ZnIxG4 superhalides display a higher LUMO energy level compared with both ZnI(H2O)5+ and Zn(H2O)62+, implying its high reductive stability to resist the HER (Fig. 2(e)). Hence, the identification of ZnIxG4 species underscores their crucial role as iodide carriers within the electrolyte systems. They efficiently reduce the number of free iodide ions, and suppress the water-induced parasitic reactions. This effect will benefit the cyclic stability of the I2-based zinc metal anode during deep cycling (to be discussed below). | ||
| Fig. 2 Theoretical calculations for the electrolyte structures. (a) Free energy change in the Zn2+ solvation and halogen-complex transformation processes (C, H, O, Zn, and I atoms are represented by gray, white, red, pink, blue, and violet balls, respectively). (b) DFT calculated major Raman modes of ZnI2, KI, ZnSO4, and G4 in water. (c) Electron density differences (blue and light-yellow regions denote charge depletion and accumulation, respectively), (d) atomic charge, and (e) LUMO energy of different Zn2+ solvation structure. | ||
Energy storage mechanism of DPZIB
To clarify the storage mechanism of the dual-plating Zn–I2 batteries, the charge/discharge processes were in situ inspected under optical microscopy (Fig. 3(a)). In the beginning, only the carbon fibers of the graphite felt, which served as the substrate for iodine deposition, are visible (Fig. 3(b1) and Fig. S12, ESI†). When a charge current is applied for 210 s, yellow iodine films immediately cover the carbon fiber. As the charge depth increases, more iodine is deposited on the carbon fiber, corresponding to darker color. After charging, the electrode was washed with water, and the active material was extracted for UV-visible analysis, which revealed a broad peak at ∼200 cm−1 corresponding to iodine (Fig. 3(c)). Upon discharging, the iodine that has grown on the carbon fiber gradually fades away. A few I2 particles are observed on the carbon fiber at the discharge time of 420 s (Fig. 3(b2)). These observations indicate the reversible I2 stripping and deposition processes during cycling.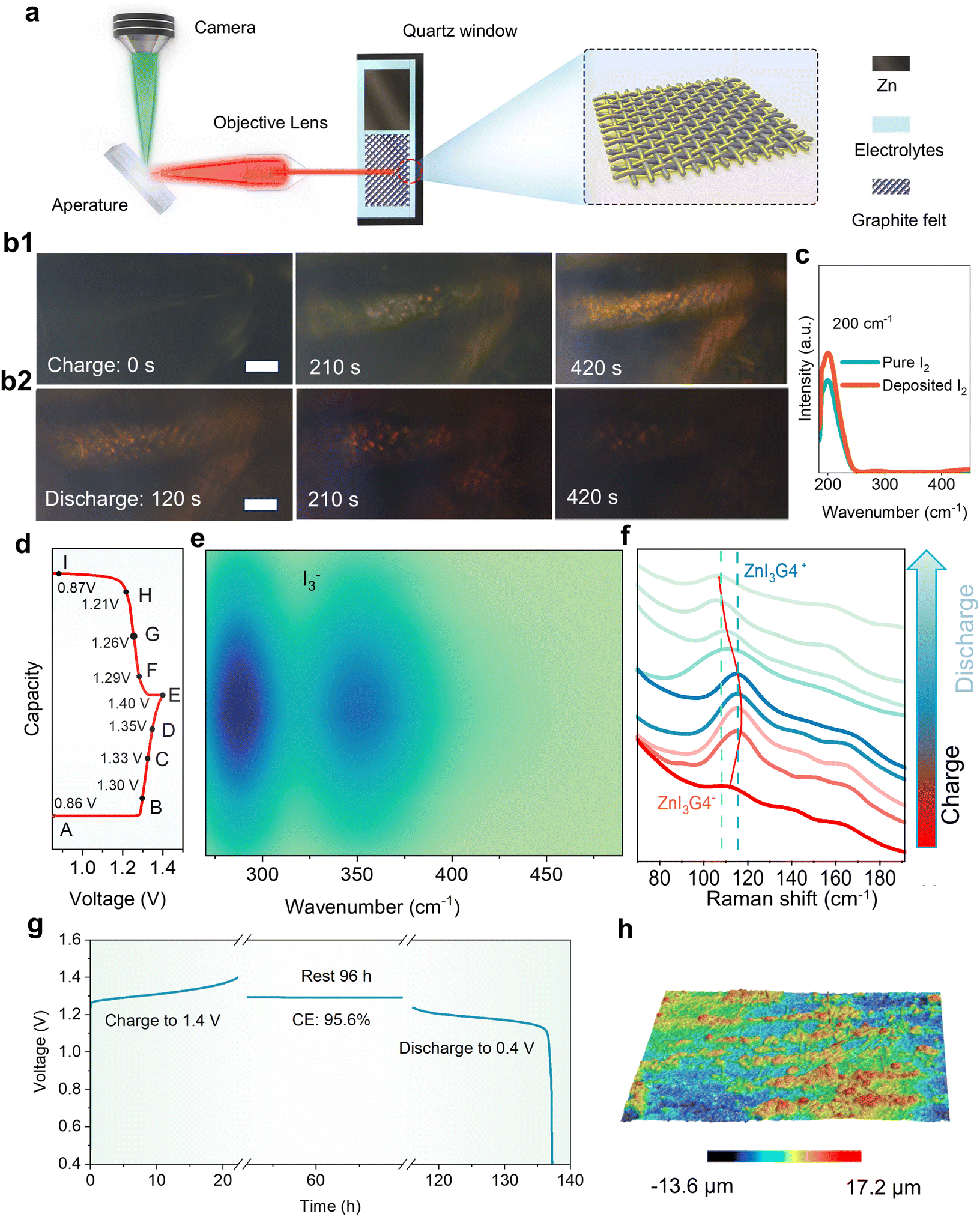 | ||
| Fig. 3 Energy storage mechanism of the DPZIB. (a) Schematic of the in situ microscope experimental setup. (b) Optical microscope images of electrode surface morphology at different charge/discharge time. The scale bars are 10 μm. (c) UV-vis spectra of pure I2 and the deposited I2. (d and e) Charge/discharge curves of DPZIB with selective voltages for in situ UV-vis tests. (f) The corresponding ex situ Raman test. (g) Self-discharge time–potential curves of the DPZIB, and (h) CLSM 3D image of Zn anode after cycling. | ||
It is commonly agreed that the undesirable polyiodide shuttling is the main problem of the Zn–I2 batteries. The oxidation of I− to I2 during charging produces I3− as an intermediate which can easily diffuse to the zinc anode, resulting in serious self-discharging and zinc anode consumption.
| I3− + Zn → Zn2+ + 3I− | (1) |
| I− + I2 → I3− | (2) |
Battery performance and potential for scalable energy storage
The aqueous Zn–I2 dual-plating battery can be assembled by immersing a piece of zinc metal and graphite felt in a homogeneous solution (Fig. 4(a)). The easy structure and low cost of the materials ensures high competency of this battery for scalable implementation. When a normal ZI electrolyte is used, the battery voltage gradually decreases as the charging time increases, even with the increasing current densities (Fig. S17, ESI†). This could be related to polyiodide shuttling and the absence of solid iodine deposition on the carbon felt. For the DPZIB with ZnIxG4 superhalides, the battery delivers a high initial capacity of 304.1 mA h at the current of 5 mA cm−2 with a high energy efficiency of 89.1% (Fig. 4(b)). Here the energy efficiency is defined as the ratio of measured capacity to the theoretical one. After continuous charge/discharge cycles for over 1800 hours, the battery maintains an average coulombic efficiency of 98.6%, with energy efficiency ranging from 85.9% to 93.7% (Fig. 4(c)). The battery delivers capacities of 313.0, 267.5, 211.2, 163.1, 120.0, and 61.3 mA h at the currents of 5, 10, 15, 18, 20, and 25 mA cm−2, respectively (Fig. S18, ESI†). When the current reduces back to 5 mA cm−2, the capacity recovers back to 310.0 mA h. At a high current of 25 mA cm−2, the battery displays a small capacity decay of 0.028% per cycle with a coulombic efficiency of 97.6% after 800 cycles (Fig. 4(d)). The reversibility is also supported by the highly overlapping of the charge/discharge profiles which consistently exhibit distinct plateaus (inset of Fig. 4(d)). The high reversibility and long-term cycling stability of the static DPZIB are attributed to the iterative dissolution and deposition of cathodes and anodes that intrinsically eliminate electrode deterioration during cycling. To scale up the dual-plating battery, we fabricated large cells and obtained capacities up to 10.8 A h at a cell volume of 3176 cm3 (electrolyte volume is 1443 cm3) (Fig. 4(e) and Fig. S19, ESI†). This capacity outperforms most previously reported aqueous batteries that rely on ion intercalation or deposition/dissolution mechanisms (Fig. 4(f)).6,26–34 In our dual-plating system, the capacity is influenced by the ion concentration, volume of the electrolyte, and the energy efficiency. To scale up the capacity under a constant ion concentration, it is also necessary to improve the energy efficiency by, for example, exploring catalysts to boost the I−/I2 redox reaction kinetics. | ||
| Fig. 4 Electrochemical performance of the DPZIB. (a) Schematic illustration of the configuration of the DPZIB. (b) Cycling performance and (c) time–voltage curves of DPZIB at a current of 5 mA cm−2. (d) Long cycling performance of the DPZIB. (e) Capacity of a large-sized static DPZIB and (f) comparison to various aqueous batteries. | ||
The capacity of our designed DPZIBs primarily derives from the carrier ions instead of number of ions intercalated into host lattices or involved in solid–liquid conversion reactions. This allows an independent engineering of power and energy in a flow battery by adjusting electrolyte conditions and electrode surface area. As a proof of concept, we assembled a flow Zn–I2 battery without using an ion exchange membrane to separate the catholytes and anolytes (Fig. S20, ESI†). This membrane-free configuration renders rapid charge transfer and lowers the manufacturing cost, making it suitable for scalable energy storage. In this setup, a relatively high concentration of iodine ions (4 M) was utilized to enhance the energy density. At a constant charging capacity of 2.65 A h at the current of 10 mA cm−2, no significant capacity decay or energy efficiency drop was observed over 30 cycles (Fig. 5(a) and (b)). This suggests minimal side reactions during cycling. Moreover, the coulombic efficiencies under a flow mode at a wide range of currents (5–40 mA cm−2) are relatively stable, although the values are all lower than 100% in the large-sized cells (Fig. 5(c)). The low efficiencies could be related to the temperature fluctuations from day to night and variations at the flow reaction interfaces. Furthermore, these Zn–I2 flow battery modules can be integrated with green energy harvesting devices to buffer the intermittent nature of solar and wind energy, offering a continuous power supply to household electronics such as LED light chains from the day to the night (Fig. 5(d) and (e)). Considering the high cycling stability, intrinsic safety, and low cost of the membrane-free DPZIBs, we envision its potential as a safe alternative to Li-ion batteries in grid-scale energy storage. The next step is to further improve the working voltage and rate performance.
 | ||
| Fig. 5 Proof-of-concept membrane-free flow battery based on the DPZIB. (a) GCD curves, (b) cycling performance and (c) rate capability. (d) Schematic illustration of the integration of membrane-free DPZIB flow battery in grid-scale energy storage system, and (e) a prototype that continuously powers a LED light chain (5 W). | ||
Conclusions
We have demonstrated a low-cost, highly reversible dual-plating Zn–I2 battery via the redox-active ZnIxG4 complexes in an aqueous electrolyte. This system differs from the conventional I− ↔ I2 reactions of the iodine cathode by eliminating the active cathode materials or complex electrode frameworks. Hence, it simplifies the manufacturing process for active materials and electrodes, an essential step in traditional metal-ion batteries. The energy storage is realized based on the principles of the synergistic halogen-complexation chemistry, where G4 serves as an electron donor to increase the interaction between Zn and I. Meanwhile, the reconstructed Zn2+ solvated structure corresponds to the reduction of free iodine and improvement in the conversion efficiency. Moreover, this designed reaction redox also bypasses the challenges of polyiodide shuttling, self-discharge and zinc corrosion, all being crucial to the implementation of aqueous batteries for large-scale application. A prototype energy harvesting system is demonstrated by integrating the DPZIB, Si photovoltaics, and LEDs.Nonetheless, the present challenges for its application in grid-scale energy storage include the relatively low energy density and rate performance compared to the mainstream lithium ion batteries and vanadium redox flow batteries. Possible approaches include anode-free configuration, further optimization of electrolytes, and exploration of catalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors appreciate the financial support from the National Research Foundation, Singapore, under its Singapore–China Joint Flagship Project (Clean Energy) and the Singapore Ministry of Education, under its AcRF Tier 1 project (RT8/22), the Joint Funds of the National Key Research and Development Program of China (2019YFA0705703), Guangdong Innovative and Entrepreneurial Research Team Program (2021ZT09L197), Guangdong Basic and Applied Basic Research Foundation (2023B1515120099). M. Y. Chuai thanks the financial support from the Postdoctoral Fellowship Program (GZC20232668).Notes and references
- J. Meng, Q. Tang, L. Zhou, C. Zhao, M. Chen, Y. Shen, J. Zhou, G. Feng, Y. Shen and Y. Huang, Joule, 2020, 4, 953 CrossRef CAS.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610 CrossRef CAS PubMed.
- G. Choi, P. Sullivan, X. L. Lv, W. Li, K. Lee, H. Kong, S. Gessler, J. R. Schmidt and D. Feng, Nature, 2024, 635, 89 CrossRef CAS PubMed.
- W. Fan, H. Wang and J. Wu, eScience, 2024, 4, 100248 CrossRef.
- H. Li, M. Zhang, Z. Zheng, X. Wu, X. Xiao, Z. Piao, C. Li, Y. Jia, J. Yang and G. Zhou, Adv. Mater., 2024, e2409771 Search PubMed.
- Z. Zheng, X. Zhong, Q. Zhang, M. Zhang, L. Dai, X. Xiao, J. Xu, M. Jiao, B. Wang, H. Li, Y. Jia, R. Mao and G. Zhou, Nat. Commun., 2024, 15, 753 CrossRef CAS PubMed.
- J. J. Hong, L. Zhu, C. Chen, L. Tang, H. Jiang, B. Jin, T. C. Gallagher, Q. Guo, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 15910 CrossRef CAS PubMed.
- L. Yuan, J. Hao, B. Johannessen, C. Ye, F. Yang, C. Wu, S.-X. Dou, H.-K. Liu and S.-Z. Qiao, eScience, 2023, 3, 100096 CrossRef.
- D. E. Ciurduc, C. de la Cruz, N. Patil, A. Mavrandonakis and R. Marcilla, Mater. Today Energy, 2023, 36, 101339 CrossRef CAS.
- K. Xu, X. Zheng, R. Luo, J. Sun, Y. Ma, N. Chen, M. Wang, L. Song, Q. Zhao and W. Chen, Mater. Today Energy, 2023, 34, 101284 CrossRef CAS.
- P. F. Zhang, J. H. Li, S. J. Zhang, D. C. Li, S. Y. Zeng, S. L. Xu, Q. X. Yao, L. Y. Liu, L. Ding, H. X. Li, Y. Y. Hu, J. T. Li and Y. Zhou, Adv. Funct. Mater., 2023, 34, 2306359 CrossRef.
- Q. Zhang, Y. Ma, Y. Lu, Y. Ni, L. Lin, Z. Hao, Z. Yan, Q. Zhao and J. Chen, J. Am. Chem. Soc., 2022, 144, 18435 CrossRef CAS PubMed.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, e2201716 CrossRef PubMed.
- H. Zhao, D. Yin, Y. Qin, X. Cui, J. Feng, Y. Zhang, L. Zhao, N. Gao, M. Cui, C. Xiao, G. Feng, Y. Su, K. Xi and S. Ding, J. Am. Chem. Soc., 2024, 146, 6744 CrossRef CAS PubMed.
- W. Li, H. Xu, H. Zhang, F. Wei, T. Zhang, Y. Wu, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Energy Environ. Sci., 2023, 16, 4502 RSC.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS PubMed.
- Q. Zhang, Y. Ma, Y. Lu, X. Zhou, L. Lin, L. Li, Z. Yan, Q. Zhao, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 23357 CrossRef CAS PubMed.
- X. Zheng, Z. Liu, J. Sun, R. Luo, K. Xu, M. Si, J. Kang, Y. Yuan, S. Liu, T. Ahmad, T. Jiang, N. Chen, M. Wang, Y. Xu, M. Chuai, Z. Zhu, Q. Peng, Y. Meng, K. Zhang, W. Wang and W. Chen, Nat. Commun., 2023, 14, 76 CrossRef CAS PubMed.
- Y. Gao, M. Wang, H. Wang, X. Li, Y. Chu, Z. Tang, Y. Feng, J. Wang, Y. Pan, Z. Ma, Z. Yang, D. Zhou and X. Li, J. Energy Chem., 2023, 84, 62 CrossRef CAS.
- W. Li, L. Huang, H. Zhang, Y. Wu, F. Wei, T. Zhang, J. Fu, C. Jing, J. Cheng and S. Liu, Matter, 2023, 6, 2312 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, e2004240 CrossRef PubMed.
- H. Gan, H. Li, M. Xu, C. Han and H.-M. Cheng, Joule, 2024, 8, 3054 CrossRef CAS.
- P. Hei, Y. Sai, W. Li, J. Meng, Y. Lin, X. Sun, J. Wang, Y. Song and X. X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202410848 CrossRef PubMed.
- F. Yang, J. Long, J. A. Yuwono, H. Fei, Y. Fan, P. Li, J. Zou, J. Hao, S. Liu, G. Liang, Y. Lyu, X. Zheng, S. Zhao, K. Davey and Z. Guo, Energy Environ. Sci., 2023, 16, 4630 RSC.
- A. Zhou, H. Wang, X. Hu, F. Zhang, Y. Zhao, Z. Hu, Q. Zhang, Z. Song, Y. Huang, L. Li, F. Wu and R. Chen, Sci. Bull., 2023, 68, 2170 CrossRef CAS PubMed.
- N. Hu, W. Lv, W. Chen, H. Tang, X. Zhang, H. Qin, D. Huang, J. Zhu, Z. Chen, J. Xu and H. He, Adv. Funct. Mater., 2023, 34, 2311773 CrossRef.
- J. Huang, Y. Zhong, H. Fu, Y. Zhao, S. Li, Y. Xie, H. Zhang, B. Lu, L. Chen, S. Liang and J. Zhou, Adv. Mater., 2024, 36, e2406257 CrossRef PubMed.
- L. Ma, Y. Ying, S. Chen, Z. Chen, H. Li, H. Huang, L. Zhao and C. Zhi, Adv. Energy Mater., 2022, 12, 2201322 CrossRef CAS.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, S. U. Lee and J.-H. Lee, Nat. Energy, 2021, 6, 592 CrossRef CAS.
- Y. Wang, L. e Mo, X. Zhang, Y. Ren, T. Wei, Y. He, Y. Huang, H. Zhang, P. Tan, Z. Li, J. Zhou and L. Hu, Adv. Energy Mater., 2024, 14, 2402041 CrossRef CAS.
- W. Wu, C. Li, Z. Wang, H.-Y. Shi, Y. Song, X.-X. Liu and X. Sun, Chem. Eng. J., 2022, 428, 131283 CrossRef CAS.
- Y. Xu, M. Zhang, R. Tang, S. Li, C. Sun, Z. Lv, W. Yang, Z. Wen, C. C. Li, X. Li and Y. Yang, Energy Environ. Sci., 2024, 17, 6656 RSC.
- L. Hu, C. Dai, Y. Zhu, X. Hou, Z. Liu, X. Geng, H. Wang, J. Chen, N. Sun, Q. Rong, Y. Zhu, X. He and Y. Lin, Energy Environ. Sci., 2024, 17, 5552 RSC.
- Z. He, J. Guo, F. Xiong, S. Tan, Y. Yang, R. Cao, G. Thompson, Q. An, M. De Volder and L. Mai, Energy Environ. Sci., 2023, 16, 5832 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00027k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |