
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concurrent electrode–electrolyte interfaces engineering via nano-Si3N4 additive for high-rate, high-voltage lithium metal batteries†
Jinuk
Kim‡
 a,
Dong Gyu
Lee‡
b,
Ju Hyun
Lee‡
a,
Saehun
Kim‡
a,
Cheol-Young
Park
a,
Jiyoon
Lee
b,
Hyeokjin
Kwon
a,
Hannah
Cho
a,
Jungyoon
Lee
a,
Donghyeok
Son
a,
Hee-Tak
Kim
a,
Nam-Soon
Choi
*a,
Tae Kyung
Lee
*b and
Jinwoo
Lee
*a
a,
Dong Gyu
Lee‡
b,
Ju Hyun
Lee‡
a,
Saehun
Kim‡
a,
Cheol-Young
Park
a,
Jiyoon
Lee
b,
Hyeokjin
Kwon
a,
Hannah
Cho
a,
Jungyoon
Lee
a,
Donghyeok
Son
a,
Hee-Tak
Kim
a,
Nam-Soon
Choi
*a,
Tae Kyung
Lee
*b and
Jinwoo
Lee
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea. E-mail: nschoi@kaist.ac.kr; jwlee1@kaist.ac.kr
bDepartment of Materials Engineering and Convergence Technology, Gyeongsang National University (GNU), 501 Jinju-daero, Jinju 52828, Republic of Korea. E-mail: tklee8865@gnu.ac.kr
First published on 13th February 2025
Abstract
Electrolyte engineering is emerging as a key strategy for enhancing the cycle life of lithium metal batteries (LMBs). Fluorinated electrolytes have dramatically extended cycle life; however, intractable challenges in terms of rate capability and fluorine overuse persist. Here, we introduce a lithiophilic, solvent-interactive, and fluorine-free nano-Si3N4 additive that facilitates the fine-tuning of weak Li+ solvation to form inorganic-rich solid–electrolyte interphase (SEI) layers. Additionally, the alloying and conversion reactions between nano-Si3N4 and Li generated a fast Li+-conductive SEI, overcoming the poor rate performance of weakly solvating electrolytes. Simultaneously, nano-Si3N4 interacts with ethylene carbonate (EC), minimizing hydrogen (H)-transfer reactions and scavenging HF, thus increasing the high-voltage tolerance. Consequently, nano-Si3N4 extends the cyclability of the commercial carbonate-based electrolyte in 360 W h kg−1-level Li||LiNi0.8Co0.1Mn0.1O2 (NCM811) pouch-cells, resulting in 74% capacity retention after 100 cycles, whereas failure occurred without it. Our study provides an in-depth understanding of the working mechanisms of suspension electrolytes through comprehensive analysis.
Broader contextElectrolyte engineering has emerged as a pivotal strategy for enhancing the cycle life of LMBs. In particular, weakly Li+-solvating electrolytes (primarily fluorinated electrolytes) induce the formation of an anion-derived, inorganic-rich SEI layer, significantly extending the cycle life of LMBs. However, these electrolytes present three challenging trade-offs: (1) inducing clustering of Li+ ions and anions, leading to sluggish Li+ transport and reduced rate performance; (2) elevating the fluorine content in the electrolyte, prompting economic and environmental concerns, thereby constraining sustainability; and (3) readily decomposing to form corrosive hydrofluoric acid (HF) under elevated temperatures and high voltage. In this study, we introduced a lithiophilic, solvent-interactive, and fluorine-free nano-Si3N4 additive to address the above challenges. Nano-Si3N4 fine-tunes the Li+ solvation environment to a weaker state, while facilitating the formation of Li3N- and LixSiyNz-based fast Li+-conductive SEIs through alloying and conversion reactions with Li, thereby overcoming trade-offs. Concurrently, nano-Si3N4 strongly interacts with the electrolyte solvent, reducing its oxidative decomposition. Furthermore, nano-Si3N4 scavenges corrosive HF, significantly reducing cathode degradation. Consequently, our designed electrolyte achieves an extended cycle life under practical conditions (N/P = 2 and E/C = 2.5 g Ah−1), including high voltage (4.5 V vs. Li/Li+), high rate (1C), and high temperature (50 °C). |
1. Introduction
As the threat of global warming intensifies, a growing movement continues to push for replacing conventional internal combustion engine vehicles with electric vehicles,1 leading to an increase in societal demand for next-generation batteries with energy densities higher than those of commercial lithium-ion batteries (LIBs, ∼250 W h kg−1).2,3 LMBs have emerged as the most promising candidates, primarily owing to the low reduction potential (−3.04 V vs. standard hydrogen electrode) and high theoretical capacity (3860 mA h g−1) of Li. However, significant challenges still exist in extending the poor cycle life of LMBs, especially under practical conditions (negative to positive ratio, i.e., N/P ratio <2, and electrolyte to capacity ratio, i.e., E/C ratio <3 g Ah−1) because the highly reducing nature of Li leads to uncontrolled (electro)chemical reactions with the electrolytes.4,5 Consequently, an inhomogeneous and thick SEI layer forms, resulting in electrolyte depletion, increased interfacial resistance, and the development of Li dendrites and dead Li.6–9Electrolyte engineering is a pivotal approach to address these challenges. Notably, a higher anion/solvent ratio (i.e., weaker Li+ solvation) in the Li+ solvation shell leads to the formation of anion-derived, inorganic-rich (i.e., LiF-rich) SEI layers as Li+ undergoes desolvation during electrodeposition near the surface of Li.10–12 The resulting LiF-rich SEI layer inhibits the growth of Li dendrites13–15 owing to its high Young's modulus (64.97 GPa)16 and enhances the cycle life through its high band gap (8.9 eV)17 and low electrolyte swelling ratio.18,19 Moreover, a weaker Li+ solvation environment has been found to enhance high-voltage tolerance by mitigating Al dissolution20,21 and tuning the cathode–electrolyte interface (CEI) layer.22,23 Several strategies have been proposed to modulate the Li+ solvation environment, including (1) high-concentration electrolytes (HCEs),21,23 (2) localized high-concentration electrolytes (LHCEs),24–30 (3) solvent fluorination,10,31,32 and (4) weakly Li+ solvating solvents.21,33–36
Nonetheless, intractable challenges persist in electrolytes and must be addressed to realize the practical implementation of LMBs. The primary challenge revolves around the trade-off between long-term cyclability and rate capability, which is influenced by the Li+ solvation environment. Weak Li+ solvation prolongs the cyclability of LMBs yet induces the clustering of Li+ ions and anions, consequently diminishing the rate capability. Conversely, strong Li+ solvation predominantly enhances rate capability owing to fast ion conduction.10,37 However, this leads to the formation of an organic-rich SEI layer, which compromises the cyclability of Li. Another challenge is the correlation between the cyclability and the fluorine content of the electrolyte. Most electrolyte engineering approaches aim to improve the cyclability of LMBs by increasing the fluorine content of salts,21,38 solvents,10,31,32 and diluents.24–30 However, excessive use of fluorine can raise economic and environmental concerns, along with potential risks to human health, thus limiting its sustainability.39 The other challenge is ensuring high-voltage tolerance. Continuous oxidation of the electrolyte not only leads to an unstable CEI but also leads to structural degradation of the cathode.40 Furthermore, fluorinated electrolytes tend to form hydrofluoric acid (HF),41 which causes severe corrosion to both the CEI layer and the cathode.42 Therefore, a novel approach to addressing these challenges is crucial for realizing practical LMBs.
Herein, we introduce a lithiophilic, solvent-interactive, and fluorine-free nano-Si3N4 additive (Fig. S1, ESI†) to a carbonate-based electrolyte (Blank: 1 M lithium hexafluorophosphate (LiPF6) in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl carbonate (DEC) (1:1 vol%) + 10 wt% fluoroethylene carbonate (FEC); x wt% nano-Si3N4 in the Blank is denoted as x-SN) to address the above challenges (Fig. 1). Nano-Si3N4 fine-tuned the Li+ solvation environment, leading to the formation of a more anion-derived, inorganic-rich SEI layer compared to the Blank. Furthermore, nano-Si3N4 directly participated in the formation of Li3N- and LixSiyNz-based SEI layers via alloying and conversion reactions with Li. The high Li+ conductivities of the Li3N (up to 10−3 S cm−1)43 and LixSiyNz (up to 10−4 S cm−1)44 SEI layers enhance the rate performance despite the lower ionic conductivity of 3-SN (see the detailed discussion in Fig. S2, ESI†), which overcomes the trade-off. Moreover, the fast Li+-conductive SEI layer promoted spherical Li growth,45,46 thus enhancing the cycle life of 3-SN. Simultaneously, nano-Si3N4 enhanced the oxidation stability of the Blank. In particular, EC strongly interacts with nano-Si3N4, resulting in a reduction in its H-transfer reaction. Additionally, as confirmed by 19F nuclear magnetic resonance (NMR), nano-Si3N4 scavenged corrosive HF, significantly reducing the structural degradation of the NCM811 and CEI layers. As a result, our designed electrolyte has achieved extended cycle life under various practical (N/P = 2 and E/C = 2.5 g Ah−1) conditions, including high-voltage (4.5 V vs. Li/Li+), high-rate (1C), and high-temperature (50 °C), not only compared to the Blank but also to previously reported nano-silica additives.47 Moreover, 3-SN delivered 74% capacity retention over 100 cycles in a 360 W h kg−1-level (N/P = 2 and E/C = 2.5 g Ah−1) pouch cell, whereas the Blank caused cell failure. This marks the first confirmation of the feasibility of applying suspension electrolytes in practical pouch cells (Table S1, ESI†).
:diethyl carbonate (DEC) (1:1 vol%) + 10 wt% fluoroethylene carbonate (FEC); x wt% nano-Si3N4 in the Blank is denoted as x-SN) to address the above challenges (Fig. 1). Nano-Si3N4 fine-tuned the Li+ solvation environment, leading to the formation of a more anion-derived, inorganic-rich SEI layer compared to the Blank. Furthermore, nano-Si3N4 directly participated in the formation of Li3N- and LixSiyNz-based SEI layers via alloying and conversion reactions with Li. The high Li+ conductivities of the Li3N (up to 10−3 S cm−1)43 and LixSiyNz (up to 10−4 S cm−1)44 SEI layers enhance the rate performance despite the lower ionic conductivity of 3-SN (see the detailed discussion in Fig. S2, ESI†), which overcomes the trade-off. Moreover, the fast Li+-conductive SEI layer promoted spherical Li growth,45,46 thus enhancing the cycle life of 3-SN. Simultaneously, nano-Si3N4 enhanced the oxidation stability of the Blank. In particular, EC strongly interacts with nano-Si3N4, resulting in a reduction in its H-transfer reaction. Additionally, as confirmed by 19F nuclear magnetic resonance (NMR), nano-Si3N4 scavenged corrosive HF, significantly reducing the structural degradation of the NCM811 and CEI layers. As a result, our designed electrolyte has achieved extended cycle life under various practical (N/P = 2 and E/C = 2.5 g Ah−1) conditions, including high-voltage (4.5 V vs. Li/Li+), high-rate (1C), and high-temperature (50 °C), not only compared to the Blank but also to previously reported nano-silica additives.47 Moreover, 3-SN delivered 74% capacity retention over 100 cycles in a 360 W h kg−1-level (N/P = 2 and E/C = 2.5 g Ah−1) pouch cell, whereas the Blank caused cell failure. This marks the first confirmation of the feasibility of applying suspension electrolytes in practical pouch cells (Table S1, ESI†).
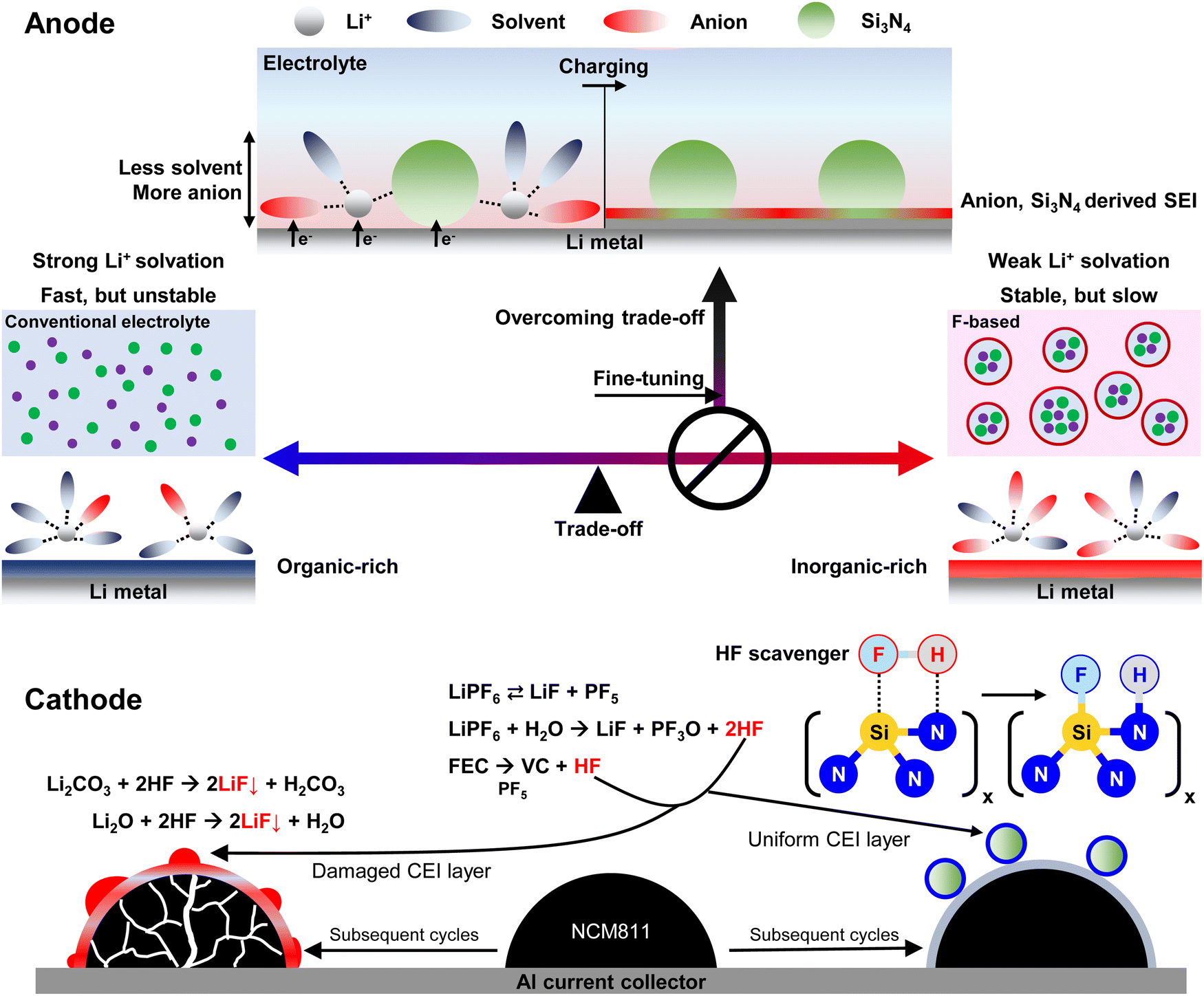 | ||
| Fig. 1 Design logic for introducing a nano-Si3N4 additive for concurrent electrode–electrolyte interface engineering. Conventional fluorinated electrolytes, including HCEs, LHCEs, and fluorinated solvents, exhibit a highly weak Li+ solvation environment. On the other hand, the nano-Si3N4 additive facilitates slightly weaker Li+ solvation compared to the Blank, resulting in a minor ionic conductivity decrease. It will be mitigated by SEI layer engineering, which involves the alloying and conversion reaction of nano-Si3N4 with Li metal. As a result, our electrolyte design effectively overcomes two key trade-offs: those between cycle life and rate performance, and the excessive use of fluorine. Moreover, nano-Si3N4 acts as an HF scavenger. The initial structure of CEI and the cathode can be maintained by nano-Si3N4 even under high-voltage and high-temperature operation. | ||
2. Results and discussion
2.1. Impact of nano-Si3N4 on the Li+ solvation environment
EC is a commonly used solvent in LIBs due to its strong solvating power, attributed to its high dielectric constant (89.78) and donor number (16.4 kcal mol−1).11 However, in LMBs, EC tends to produce an inadequate organic and Li2CO3-rich SEI layer because of severe (electro)chemical decomposition by Li.48,49 Therefore, regulating the reactivity of EC with Li and controlling the composition of the SEI layer to ensure an extended cycle life are imperative.
Fig. 2 illustrates the impact of nano-Si3N4 on the Li+ solvation environment and the resulting improvements in the electrochemical performance. As the nano-Si3N4 content increased from 0 to 5 wt%, a noticeable upfield shift was observed in the 7Li NMR (Fig. 2a), indicating an increase of electron density of the Li+, likely due to the increased Li+-anion coordination or Li+-nano-Si3N4 interactions. Consequently, this led to weaker Li+ solvation, and the effect gradually diminished as the concentration increased (Fig. 2b). Moreover, we quantitatively measured the relative solvation energy in a H-cell.50 The open-circuit voltage (OCV) of 3-SN is −41 mV, while that of the Blank is −24 mV. This implies that 3-SN has a weaker Li+ solvation environment, with a higher ΔGsolv of 1.6 kJ mol−1 than the Blank (Fig. 2c and d). Fig. 2e illustrates the interaction between nano-Si3N4 and EC/DEC (Blank solvent) using Raman spectroscopy. In the Blank solvent, both EC and DEC exhibited doublet peaks of νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1796, 1807 cm−1, and 1740, 1770 cm−1, respectively.51 The EC peaks of 3-SN showed a positive shift at 1798 and 1810 cm−1, which represents a more pronounced shift compared to DEC (less than 1 cm−1). This suggests that the oxygen within the CO bond of carbonates, particularly EC, interacts with nano-Si3N4.52
O at 1796, 1807 cm−1, and 1740, 1770 cm−1, respectively.51 The EC peaks of 3-SN showed a positive shift at 1798 and 1810 cm−1, which represents a more pronounced shift compared to DEC (less than 1 cm−1). This suggests that the oxygen within the CO bond of carbonates, particularly EC, interacts with nano-Si3N4.52
 | ||
| Fig. 2 Characterization of the Li+ solvation environment and its impact on electrochemical performances. (a) and (b) The 7Li NMR results of the Blank and 1, 3, and 5-SN. The 7Li NMR results were obtained by using NMR coaxial inserts with an inner tube containing a 1 M LiCl D2O reference solution. (c) The potentiometric measurements of OCVs in an H-cell comparing two different electrolytes (reference electrolyte: 1 M LiFSI DMC (dimethyl carbonate)). (d) Calculated solvation free energies from (c). (e) Raman shifts between the Blank solvent and x-SN. The doublet peaks correspond to the vibrational modes of the CO bonds (νCO) present in EC and DEC, due to Fermi resonance. (f) Modified Aurbach's coulombic efficiency tests under high current density (2 mA cm−2, detailed in supplementary methods, ESI†). (g) Tafel plots of the Blank and 3-SN in Li||Li symmetric cells. (h) Calculated exchange current densities from (g). | ||
To evaluate the impact of modifying the Li+ solvation environment on the cyclability of Li, we conducted modified Aurbach's coulombic efficiency (C.E.) tests53 (Fig. 2f and Table S2, see the detailed discussion in Fig. S3, ESI†). As shown in Fig. 2f, 3-SN exhibited a significantly higher C.E. under high current density (2 mA cm−2) compared to the Blank (Blank: 95.79% and 3-SN: 98.62%), along with less than half of the nucleation overpotential (Blank: 98.9 mV, 3-SN: 47.1 mV). Moreover, through a series of electrochemical tests, we verified that 3-SN shows a lower nucleation and growth overpotential, along with enhanced cyclability of Li, specifically at high current densities (Fig. S4–S9, Tables S3 and S4, see the detailed discussion in Fig. S9, ESI†). In contrast, the absence of improvements in the C.E. with the application of 3 wt% of other metal nitrides and boron nitride indicates that the enhanced cyclability of 3-SN results from the alloying and conversion reactions of nano-Si3N4 (see the detailed discussion in Fig. S10, ESI†). Fig. 2g and h illustrate the Tafel plot and its exchange current density. The exchange current density of 3-SN was 32% higher than that of the Blank. This suggests that the interfacial reaction kinetics of Li are improved45 due to the fast Li+-conductive Si,N-based SEI layer.44 Consequently, the faster Li+-conductive SEI layer and lower nucleation overpotential facilitate the growth of larger and more spherical Li nuclei (Fig. S11, ESI†).45,54,55
Furthermore, we theoretically analyzed the solvation environment using nano-Si3N4 as the suspension electrolyte via molecular dynamics (MD) simulations. The solvation environment of Li+ was investigated using the Blank and 3-SN. For 3-SN, a significant decrease in the ratio of EC participating in Li+ solvation was observed compared with the Blank, along with the substantial involvement of nano-Si3N4 in the Li+ solvation environment (Fig. 3a and b and Fig. S12, and Table S7, ESI†). This indicates that nano-Si3N4 influences and regulates the solvation environment around Li+ by decreasing the solvation ratio of EC to DEC, which is consistent with experimental observations. Based on the MD simulation results, we additionally performed density functional theory (DFT) calculations to investigate the interaction characteristics of nano-Si3N4 and EC molecules through the calculations of binding energies between the α-Si3N4 (101) surface (Fig. S13, ESI†) and electrolyte molecules (i.e., LiPF6, EC, DEC, and FEC). We found that the EC had strong chemical binding with the Si dangling atom of the α-Si3N4 (101) surface (Fig. 3c and Fig. S14–S17, ESI†), as experimentally demonstrated in Fig. 2e. Its involvement in chemical bonding with the double bond O of EC and its strong binding strength suggest that strong chemical binding with nano-Si3N4 can critically inhibit the oxidation decomposition of EC. Although the DEC molecule also showed covalent bonding with the α-Si3N4 (101) surface, the binding strength was much weaker than that of the EC molecule. Moreover, Li+ showed ionic interactions with the N dangling atom of nano-Si3N4, which supports the results of the NMR measurements (Fig. 2a) and MD simulations.
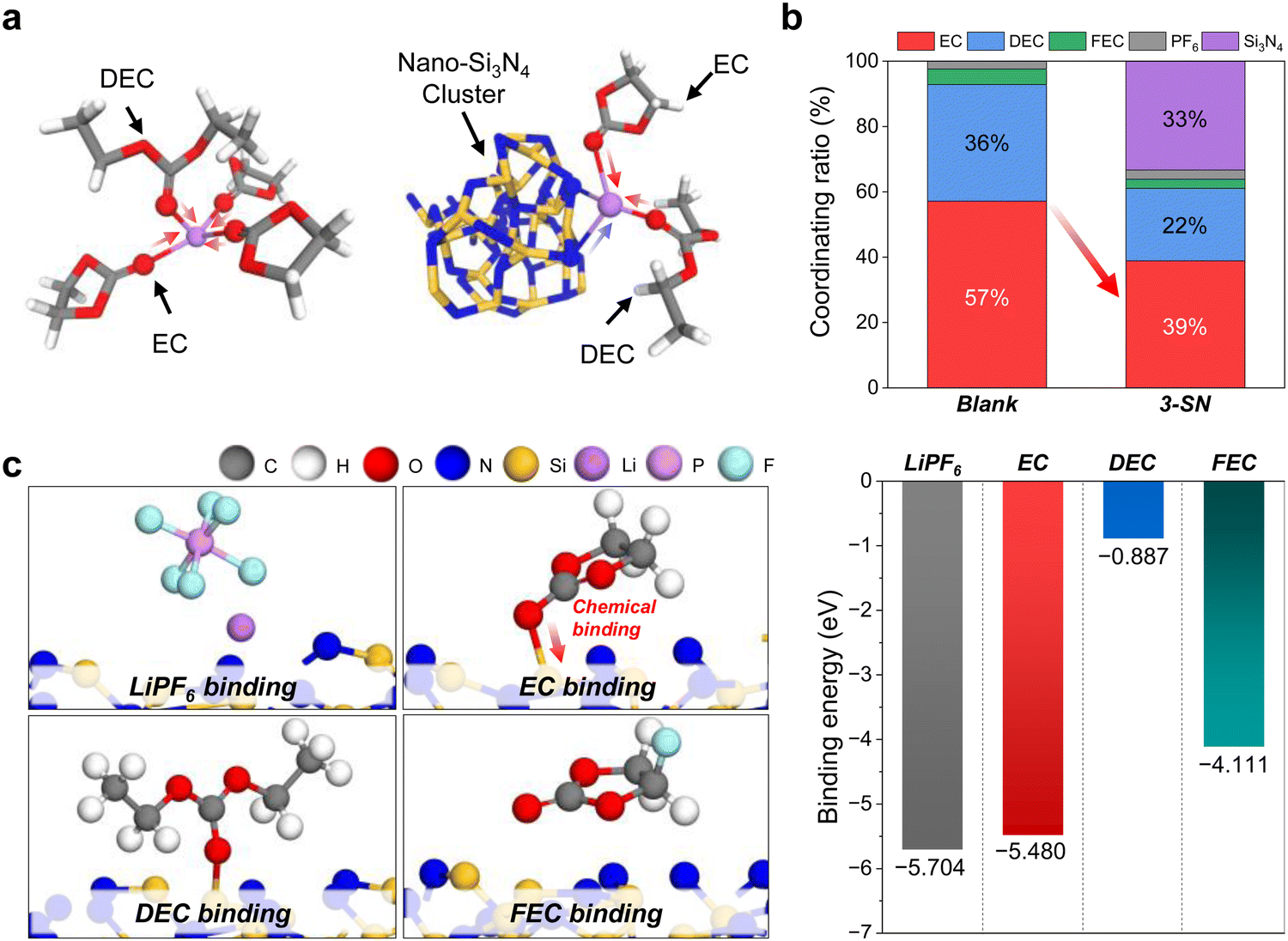 | ||
| Fig. 3 Theoretical investigation of influence of the nano-Si3N4 on Li+ solvation environment. (a)–(c) Solvation and interaction characteristics of the electrolyte and nano-Si3N4. (a) Coordinated structures around Li+ in the Blank and 3-SN. (b) The ratio of electrolytes coordinated with Li+ according to the Blank and 3-SN. (c) Interaction characteristics of LiPF6, EC, DEC, and FEC on the α-Si3N4 (101) surface. | ||
2.2. Bi-layered SEI formed by nano-Si3N4
As confirmed by molecular simulations (Fig. 3), nano-Si3N4 reduced the number of solvent molecules in the Li+ solvation shell, presumably forming an inorganic-rich SEI layer. Furthermore, we hypothesized that the alloying and conversion reactions of nano-Si3N4 contributed to the formation of Si,N-based SEI layers, enhancing the interfacial kinetics (Fig. 2f–h). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) provided clear evidence that 3-SN formed a less-organic-based SEI layer (Fig. 4a). Conversely, 3-SN predominantly formed an inorganic SEI layer originating from the reductive decomposition of LiPF6 (Fig. 4b). These findings were further corroborated by the X-ray photoelectron spectroscopy (XPS) results (Fig. S18, ESI†). In addition, we confirmed the presence of the SEI layer associated with nano-Si3N4. SiN− encompasses both nano-Si3N4, which has not yet contributed to the SEI layer, and LixSiyNz, arising from the conversion reaction of nano-Si3N4 into the SEI layer (Fig. 4c). In contrast, LiSi− includes LixSiyNz and the LixSiy, resulting from the reaction between Li and nano-Si3N4. Unlike SiN−, LiSi− is barely located in the topmost region of the SEI layer, indicating that alloying and conversion reactions commence in close proximity to the Li. The XPS results further reinforced these observations, providing a plausible mechanism for the formation of the SEI layer facilitated by nano-Si3N4 (Fig. S19–S22, see the detailed discussion in Fig. S19 and S22, ESI†). Furthermore, this bi-layered structure of the SEI layer, which is composed of the Si,N-based inner SEI layer, and LiF-rich outer SEI layer, synergistically enhances the cycle life of Li and delivers tolerance to chemical corrosion (see the detailed discussion in Fig. S23 and S24, ESI†).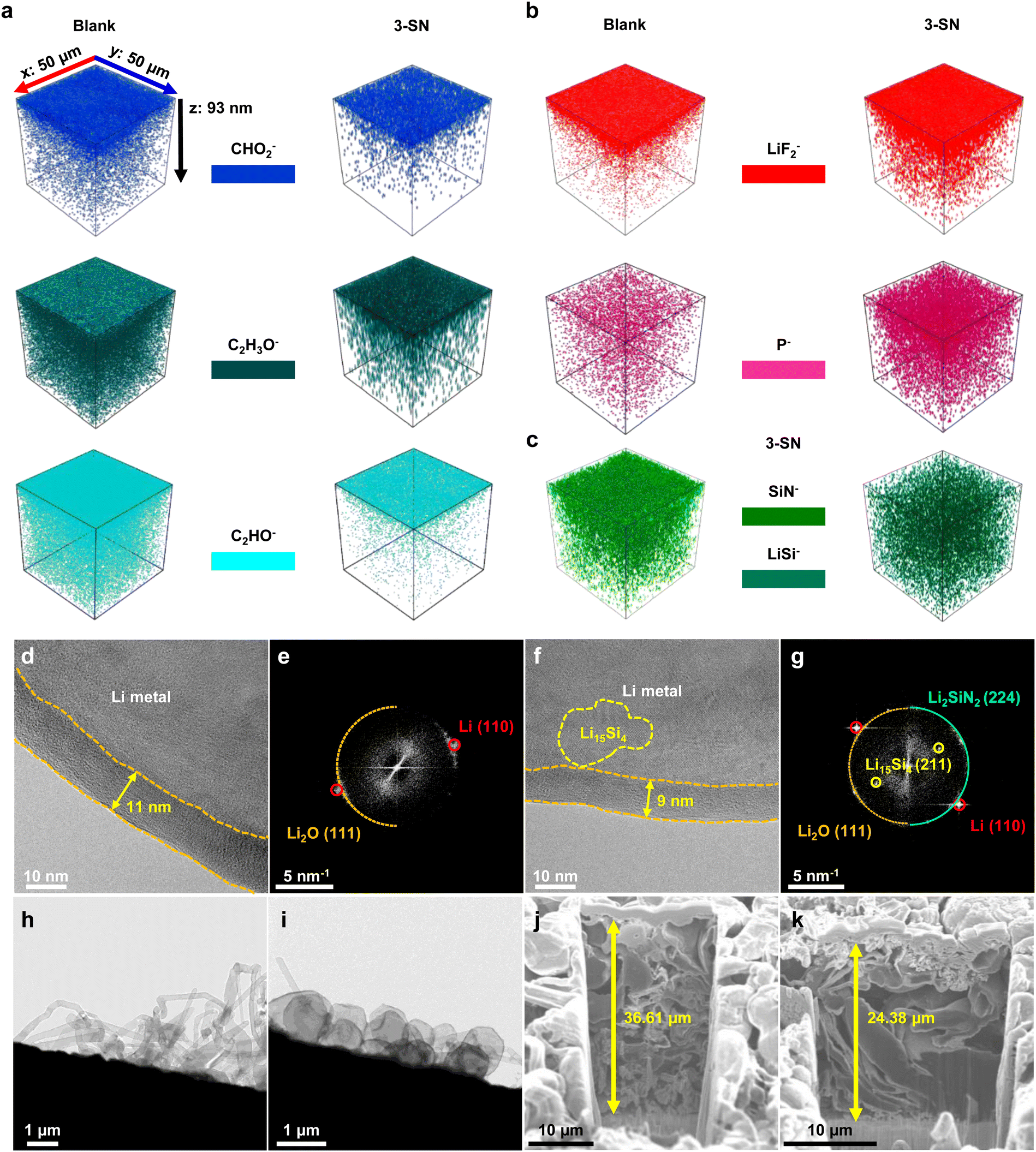 | ||
| Fig. 4 Observations of the SEI layer and Li metal growth morphology. (a)–(c) TOF-SIMS analysis of Li metal anodes retrieved from Li||Li symmetric cells after deposition in the second cycle. (a) 3D reconstruction of the TOF-SIMS signal originating from the organic SEI layer. (b) 3D reconstruction of the TOF-SIMS signal originating from the inorganic SEI layer. (c) 3D reconstructions of the TOF-SIMS signal originating from Si3N4 in 3-SN. The z-axis depth is a theoretical value obtained under the same sputtering conditions applied to the lithium metal. (d)–(g) Cryo-TEM image and its fast Fourier transform (FFT) pattern of the SEI layer after deposition under 0.5 mA cm−2, 0.17 mA h cm−2. Information on crystal structures were obtained from the Materials Project.56 (d) and (e) Blank. (f) and (g) 3-SN. (h) and (i) Cryo-TEM images of the Li growth morphology. (h) Blank, (i) 3-SN. (j) and (k) Cross-sectional focused ion beam-scanning electron microscopy (FIB-SEM) images after Li deposition under 0.4 mA cm−2, 4 mA h cm−2 in Li||Cu half-cells. (j) Blank, (k) 3-SN. The theoretical thickness of 4 mA h cm−2 Li is 20 μm. | ||
Cryo-transmission electron microscopy (Cryo-TEM) images revealed noticeable differences in the SEI layers between the Blank and 3-SN. In the case of the Blank, an 11 nm-thick SEI layer was formed, with the only identified crystalline SEI layer being Li2O, as observed in the fast Fourier transform (FFT) patterns56 (Fig. 4d and e). Conversely, the SEI layer formed by 3-SN was comparatively thinner (9 nm), and displayed FFT patterns of Li2O, Li2SiN2 and Li15Si4 (Fig. 4f and g and Fig. S25 and Table. S8, ESI†). Additionally, Li2SiN2 and Li15Si4 were detected between the Li and SEI layers, indicating that the alloying and conversion reactions occur in close proximity to the Li, as corroborated by TOF-SIMS and XPS (Fig. 4c and Fig. S19–S22, ESI†). The SEI layer generated by nano-Si3N4 enhances Li+ conductivity, thus significantly influencing the morphology of Li deposits (Fig. 4h–k and Fig. S26–S28, ESI†).45
2.3. High-voltage tolerance enabled by nano-Si3N4
Carbonate-based electrolytes generally possess moderate oxidation stabilities; however, they are unsuitable for high-voltage (above 4.3V vs. Li/Li+).57,58 Specifically, EC is highly susceptible to H-transfer reactions,22,59 which are thermodynamically more favorable than other oxidation decomposition pathways.60,61 However, our chronoamperometry results at 4.5V vs. Li/Li+ of Li||NCM811 indicated that the oxidative decomposition of 3-SN was significantly less than that of the Blank (Fig. S29, ESI†). Moreover, we discovered that the dissociation energy of the C–H bond of bound-EC (i.e., EC interacting with nano-Si3N4) was higher than that of free-EC in both 1H and 2H-transfer by performing the DFT calculations (Fig. 5a and b). Given the significance of the bond dissociation energy in the oxidation stability of electrolyte solvents,22 these findings imply that the H-transfer reaction in bound-EC on the surface of nano-Si3N4 is thermodynamically less favorable than that in free-EC (details of the H-transfer reaction are in Note S1, ESI†). Therefore, the moderate oxidation stability of the Blank can be enhanced by introducing nano-Si3N4, which can regulate the Li+ solvation environment and hinder the H-transfer reaction because of its strong chemical binding to EC. | ||
| Fig. 5 Dissociation energy of C–H bond of EC and characterization results of the cycled NCM811 cathode and CEI layer. (a) and (b) Dissociation energies of the C–H bond of EC and bound-EC. (a) Dissociation of the C–H bond from EC to EC (–H) and bound-EC to bound-EC (–H). (b) Dissociation of the C–H bond from EC (–H) to EC (–2H) and bound-EC (–H) to bound-EC (–2H). Orange highlighted hydrogen atoms indicate the dissociating atoms. (c) and (d) HADDF-STEM images of NCM811 retrieved from Li||NCM811 after 50 cycles under 4.5–3.0 V and 0.5C/1D. (c) Blank, (d) 3-SN. High resolution TEM images and FFT patterns of retrieved NCM811 from Li||NCM811 after 50 cycles under 4.5–3.0 V and 0.5C/1D. (e) and (f) Blank, (g) and (h) 3-SN. | ||
The oxidation decomposition of the electrolyte not only leads to the formation of the CEI layer but also induces a phase transition of NCM811 from a layered (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) to a rock salt (Fmm) structure due to the reduction of Ni4+ on the NCM811 surface to Ni2+.40 In Fig. 5c and d, 3-SN produced a much thinner reconstruction layer (1.2–3.5 nm) than the Blank (9.1 nm). The peak shift in the X-ray diffraction (XRD) pattern was more pronounced in the Blank, indicating a more severe phase transition (Fig. S30, ESI†). Additionally, the cross-sectional images of the cycled NCM811 with the Blank display prominent cracks, whereas 3-SN maintains its initial morphology (Fig. S31, ESI†).
m) to a rock salt (Fmm) structure due to the reduction of Ni4+ on the NCM811 surface to Ni2+.40 In Fig. 5c and d, 3-SN produced a much thinner reconstruction layer (1.2–3.5 nm) than the Blank (9.1 nm). The peak shift in the X-ray diffraction (XRD) pattern was more pronounced in the Blank, indicating a more severe phase transition (Fig. S30, ESI†). Additionally, the cross-sectional images of the cycled NCM811 with the Blank display prominent cracks, whereas 3-SN maintains its initial morphology (Fig. S31, ESI†).
Fig. 5e–h illustrate the structure of the CEI layer on the cycled NCM811. The Blank exhibited the formation of a thick and uneven CEI layer ranging from 4–15 nm with excessive LiF and NiF2. The excessive fluorination originates from the conversion of CEI components, such as Li2CO3, to LiF due to HF generated within LiPF6-based electrolytes16,62 (see the detailed discussion in Fig. S32, ESI†). Notably, NiF2 is also formed by HF attack after the undesirable reduction of Ni4+ to Ni2+ by taking electrons from the electrolyte at the deep charged (delithiated) state of the NCM811. Conversely, the CEI layer formed by 3-SN was thin and uniform with an amorphous structure (3.4 nm, Fig. 5h). Two explanations support these results. (1) The H-transfer reaction not only degrades the cathode but also generates HF59 (Note S1, ESI†). 3-SN is less vulnerable to H-transfer reactions than the Blank (Fig. 5a and b), thus it effectively minimizes the formation of HF and cathode degradation. (2) Similar to SiO2, Si3N4 can act as an HF scavenger47,63–65 to mitigate corrosive damage of the cathode. Previous studies have demonstrated that Si–N bonds scavenge HF, resulting in the formation of Si–F and N–H bonds.66–68 After storing the 3-SN at a high temperature of 60 °C for 7 days, the 19F NMR reveals the absence of HF in 3-SN, demonstrating the beneficial role of nano-Si3N4 as a HF scavenger (Fig. S33, details will be discussed in Fig. S38, ESI†). Our DFT results also reveal that the reaction of HF with the α-Si3N4 (101) surface is thermodynamically favorable (Fig. S34, ESI†). Excessive formation of HF can lead to the degradation of the NCM811, Al current collector and the CEI layer, thereby increasing the dissolution of both Ni2+ and Al3+ (Fig. S35, ESI†).69,70 In summary, the high-voltage tolerance of 3-SN originates from its ability to reduce H-transfer reactions and corrosive HF attacks at the cathodes, thus mitigating the degradations of the electrolyte, cathode, and CEI layer.
2.4. Superior electrochemical performances of 3-SN
We found that nano-Si3N4 led to several beneficial changes. Subsequently, we conducted electrochemical tests under various conditions to assess its impact on the electrochemical performance within the practical Li||NCM811 full cell (Fig. 6). Under mild cycling conditions, 3-SN achieved 85% capacity retention after 150 cycles, whereas the Blank retained only 29% (Fig. 6a and Fig. S36, ESI†). Moreover, a more pronounced gap was observed in the electrochemical performance under higher cut-off voltages (Fig. 6b and Fig. S37 and S38, ESI†). While the Blank exhibited a rapid capacity decay around the 20th cycle, 3-SN maintained 80% retention even after 100 cycles. These results were attributed to the nano-Si3N4 mitigating the H-transfer reaction and scavenging HF. The use of nano-Si3N4 as a protective layer for Li metal has been previously reported;71,72 however, these studies did not address high-voltage tolerance. Unlike suspension electrolyte, this approach is inadequate for improving high-voltage stability, as it does not directly prevent the degradation of NCM811. This limitation was experimentally confirmed using a mechanochemical coating method73–75 (Fig. S39, ESI†). Fig. 6c and Fig. S40 (ESI†) illustrate the rate performance of 3-SN. At all discharge C-rates, 3-SN consistently demonstrated a superior discharge capacity, and the gap became more pronounced as the C-rate increased (at 2C, Blank: 128 mA h g−1, 3-SN: 167 mA h g−1). Similarly, rate tests conducted under various conditions highlighted the exceptional rate performance of 3-SN, indicating its potential for fast charging (Fig. S41–S43, ESI†). Moreover, 3-SN exhibited superior electrochemical performance at high rates compared to the Blank at both 4.3 V and 4.5 V vs. Li/Li+ (Fig. S44, ESI†). These results were attributed to the decrease in the interfacial resistance within the electrode–electrolyte interface at both the anode and cathode (Fig. 4 and 5). Additionally, 3-SN also delivers outstanding electrochemical performance at a high-temperature of 50 °C (Fig. 6d). The hydrolysis of LiPF6 accelerates with increasing temperature, resulting in the vulnerability of the Blank to high temperatures. Furthermore, electrochemical tests confirmed that the high-voltage tolerance and rate performance of 3-SN surpassed those of previously reported nano-silica additives (Fig. 6e and Fig. S44–S46, see the detailed discussion in Fig. S45, ESI†).47,64 3-SN exhibited slower sedimentation compared to other electrolyte additives, resulting in a negligible effect on electrochemical performance (see the detailed discussion in Fig. S47, ESI†). We further verified the effect of the nano-Si3N4 additive on a practical pouch cell (Fig. 6f and Fig. S48, ESI†). In a 360 W h kg−1-level pouch cell, 3-SN demonstrated 74% capacity retention over 100 cycles, whereas the Blank exhibited cell failure. This result suggests that even under lean electrolyte conditions (2.5 g Ah−1), 3-SN sufficiently wets the electrode and separator, enabling the diffusion of the suspensions. Furthermore, we demonstrated that the incorporation of nano-Si3N4 in lithium–sulfur batteries can lead to long-term cyclability, even under practical conditions (Fig. S49, ESI†).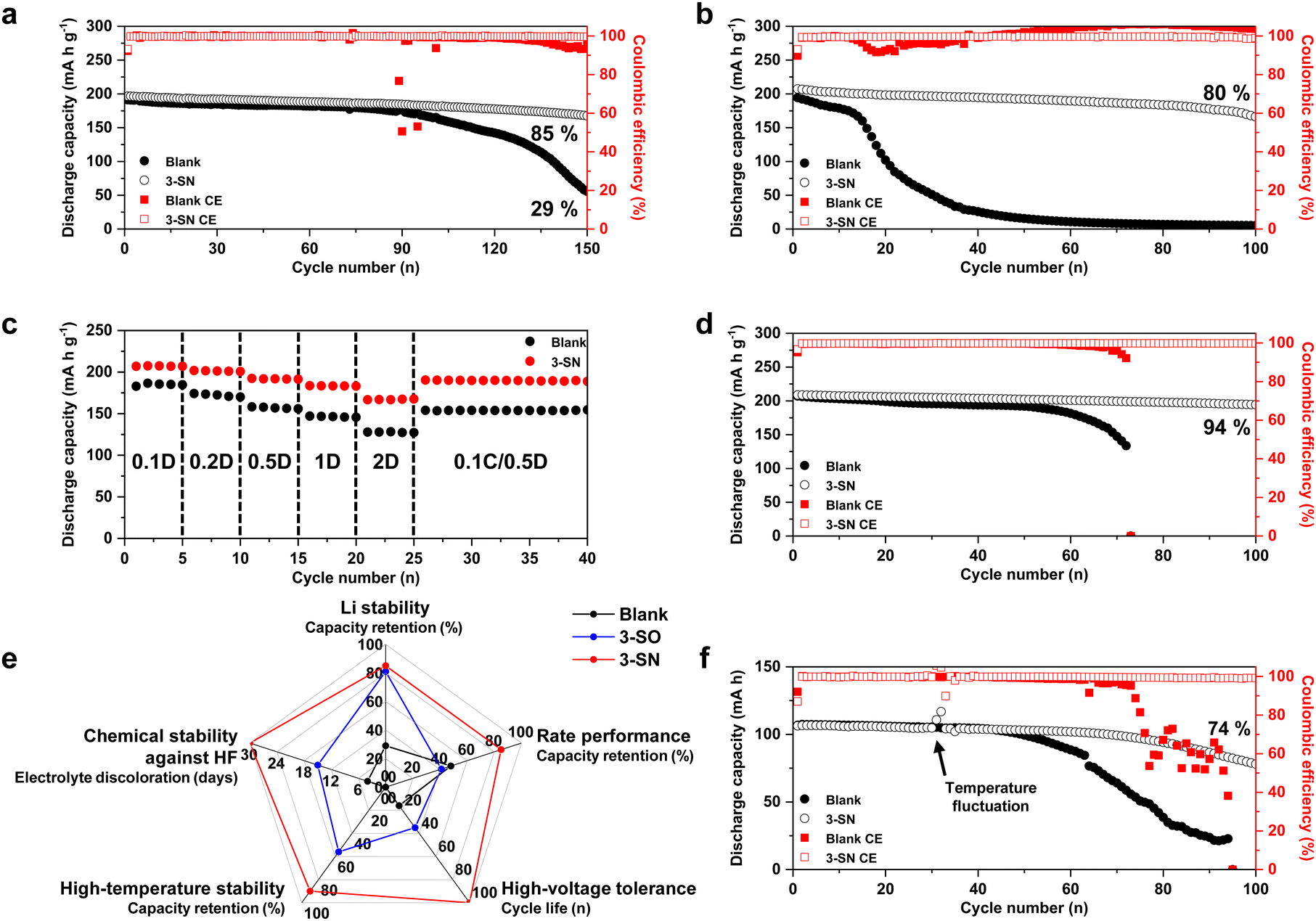 | ||
| Fig. 6 Electrochemical performances of Li||NCM811 full-cells under practical conditions. (a)–(d) N/P = 2.5 (50 μm Li || 3.8 mA h cm−2 NCM811), E/C = 4 μL mA h−1 in coin-cells. (a) 4.3–3.0 V, 0.2C(CC/CV: 0.05C)/0.5D. (b) 4.5–3.0 V, 0.2C/0.5D. (c) Rate test with constant charging C-rate (0.1C). (d) 4.3–3.0 V, 0.2C(CC/CV: 0.05C)/0.5D, under 50 °C. (e) Comprehensive evaluation of the electrolytes. 3-SO stands for 3 wt% of nano-silica within the Blank. Li stability represents the capacity retention in Fig. 6a and Fig. S44a (ESI†) at 150 cycles. Rate performance represents the capacity retention in Fig. S44b (ESI†), at 70 cycles. High-voltage tolerance represents the cycle life (cycle number at 80% capacity retention) in Fig. 6b and Fig. S44c (ESI†). High-temperature stability represents the capacity retention in Fig. 6d and Fig. S44e (ESI†), at 130 cycles. Chemical stability against HF represents the period during which electrolyte discoloration occurs in Fig. S45 (ESI†). (f) 360 W h kg−1-level pouch-cell, N/P = 2 (40 μm Li || 3.8 mA h cm−2 NCM811) E/C = 2.5 g Ah−1, 0.2C(CC/CV: 0.05C)/0.5D. | ||
3. Conclusion
In this study, we propose a multifunctional nano-suspension electrolyte to address the trade-offs and challenges persisting in LMB electrolytes. Nano-Si3N4 enhances the electrochemical performance of LMBs in multiple ways (Fig. 7): (1) modifying the Li+ solvation environment to form an inorganic-rich SEI layer, (2) forming a Si,N-based fast Li+-conductive SEI layer through alloying and conversion reactions, (3) mitigating the H-transfer reaction through EC-Si3N4 interactions, and (4) scavenging HF to minimize corrosive side reactions. (1) and (2) enhanced the cyclability of the Li metal, which is a crucial weakness of carbonate-based electrolytes. Moreover, (2) provides fast interfacial kinetics of the SEI layer, overcoming the trade-off originating from the Li+ solvation strength. (3) and (4) increase the moderate oxidation stability of the carbonate-based electrolytes to a higher level. Consequently, nano-Si3N4 in the carbonate-based electrolyte delivered outstanding electrochemical performance (74% retention at 100 cycles) in a 360 W h kg−1-level pouch cell. We expect that this study will provide a comprehensive understanding of the mechanisms involved in suspension electrolytes in LMBs, ultimately paving the way for the use of suspensions in large-scale LMBs. | ||
| Fig. 7 Overall working mechanisms of the nano-Si3N4 additive. | ||
Author contributions
J. K. and J. L. (Jinwoo Lee) conceived the idea and designed the logic and experiments. N-S. C., T. K. L., and J. L. (Jinwoo Lee) directed the project. J. K. designed and conducted a series of characterization methods of the Li+ solvation environment, electrochemical tests, and overall post-mortem analysis. D. G. L. conducted molecular simulations, including DFT and MD. J. H. L. conducted 7Li and 19F NMR measurements, post-mortem XPS analysis, and electrochemical tests. S. K. designed and conducted post-mortem analysis for cathode materials. C-Y. P. conducted SEM observations. J. L. (Jiyoon Lee) assisted with molecular simulations. H. K., H. C., and H.-T. K. designed and fabricated pouch cells. J. L. (Jungyoon Lee) assisted with cryo-TEM and SEM observations. D. S. designed and fabricated lithium–sulfur batteries. All authors discussed and analyzed the results. J. K., D. G. L., T. K. L., and J. L. (Jinwoo Lee) wrote the manuscript. J. H. L., S. K., and N-S. C. commented on and revised the manuscript. All authors commented on the manuscript and J. L. (Jinwoo Lee) supervised the work.Data availability
The data that support the findings of this study are included in the article and its ESI.† These data are also available from the corresponding authors upon request.Conflicts of interest
J. K., J. H. L., and J. L. (Jinwoo Lee) declare that this work has been filed for a KR Provisional Patent Application (number 10-2023-0069509). The other authors declare no competing interests.Acknowledgements
This work was supported by LG energy solution-KAIST Frontier Research Laboratory (2024), the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (grant number: RS-2023-00235596), and Learning & Academic research institution for Master’s∙PhD students, and Postdocs (LAMP) Program of the NRF grand funded by the Ministry of Education (grant number: RS-2023-00301974).References
- J. Tollefson, Nature, 2023, 616, 424 CrossRef CAS PubMed.
- X. Zeng, M. Li, D. Abd El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- H. Liu, X. Sun, X.-B. Cheng, C. Guo, F. Yu, W. Bao, T. Wang, J. Li and Q. Zhang, Adv. Energy Mater., 2022, 12, 2202518 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- G. Lu, J. Nai, D. Luan, X. Tao and X. W. D. Lou, Sci. Adv., 2023, 9, eadf1550 CrossRef CAS PubMed.
- Y. An, Y. Zeng, D. Luan and X. W. D. Lou, Matter, 2024, 7, 1466–1502 CrossRef CAS.
- H. Li, D. Chao, B. Chen, X. Chen, C. Chuah, Y. Tang, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2020, 142, 2012–2022 CrossRef CAS PubMed.
- M. Li, C. Wang, K. Davey, J. Li, G. Li, S. Zhang, J. Mao and Z. Guo, SmartMat, 2023, 4, e1185 CrossRef CAS.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- P. Xiao, X. Yun, Y. Chen, X. Guo, P. Gao, G. Zhou and C. Zheng, Chem. Soc. Rev., 2023, 52, 5255–5316 RSC.
- G.-X. Li, P. Lennartz, V. Koverga, R. Kou, A. Nguyen, H. Jiang, M. Liao, D. Wang, N. Dandu, M. Zepeda, H. Wang, K. Wang, A. T. Ngo, G. Brunklaus and W. Donghai, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2311732121 CrossRef CAS PubMed.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. D. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- Y. Zhai, Z. Zhong, N. Kuang, Q. Li, T. Xu, J. He, H. Li, X. Yin, Y. Jia, Q. He, S. Wu and Q.-H. Yang, J. Am. Chem. Soc., 2024, 146, 15209–15218 CrossRef CAS PubMed.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- Y. Wang, F. Liu, G. Fan, X. Qiu, J. Liu, Z. Yan, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2021, 143, 2829–2837 CrossRef CAS PubMed.
- S. Park, S. Kim, J.-A. Lee, M. Ue and N.-S. Choi, Chem. Sci., 2023, 14, 9996–10024 RSC.
- Z. Zhang, Y. Li, R. Xu, W. Zhou, Y. Li, S. T. Oyakhire, Y. Wu, J. Xu, H. Wang, Z. Yu, D. T. Boyle, W. Huang, Y. Ye, H. Chen, J. Wan, Z. Bao, W. Chiu and Y. Cui, Science, 2022, 375, 66–70 CrossRef CAS PubMed.
- C. Jin, Y. Huang, L. Li, G. Wei, H. Li, Q. Shang, Z. Ju, G. Lu, J. Zheng, O. Sheng and X. Tao, Nat. Commun., 2023, 14, 8269 CrossRef CAS PubMed.
- S. Kim, J.-A. Lee, T. K. Lee, K. Baek, J. Kim, B. Kim, J. H. Byun, H.-W. Lee, S. J. Kang, J.-A. Choi, S.-Y. Lee, M.-H. Choi, J.-H. Lee and N.-S. Choi, Energy Environ. Sci., 2023, 16, 5108–5122 RSC.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J. C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
- Y. Huang, R. Li, S. Weng, H. Zhang, C. Zhu, D. Lu, C. Sun, X. Huang, T. Deng, L. Fan, L. Chen, X. Wang and X. Fan, Energy Environ. Sci., 2022, 15, 4349–4361 RSC.
- E. Park, J. Park, K. Lee, Y. Zhao, T. Zhou, G. Park, M.-G. Jeong, M. Choi, D.-J. Yoo, H.-G. Jung, A. Coskun and J. W. Choi, ACS Energy Lett., 2022, 8, 179–188 CrossRef.
- G.-X. Li, V. Koverga, A. Nguyen, R. Kou, M. Ncube, H. Jiang, K. Wang, M. Liao, H. Guo, J. Chen, N. Dandu, A. T. Ngo and D. Wang, Nat. Energy, 2024, 9, 817–827 CrossRef CAS.
- L. Le, M. Liao, A. Nguyen and D. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 37497–37503 CrossRef CAS PubMed.
- Q.-K. Zhang, S.-Y. Sun, M.-Y. Zhou, L.-P. Hou, J.-L. Liang, S.-J. Yang, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306889 CrossRef CAS PubMed.
- Q.-K. Zhang, X.-Q. Zhang, L.-P. Hou, S.-Y. Sun, Y.-X. Zhan, J.-L. Liang, F.-S. Zhang, X.-N. Feng, B.-Q. Li and J.-Q. Huang, Adv. Energy Mater., 2022, 12, 2200139 CrossRef CAS.
- W. Cai, Y. Deng, Z. Deng, Y. Jia, Z. Li, X. Zhang, C. Xu, X. Q. Zhang, Y. Zhang and Q. Zhang, Adv. Energy Mater., 2023, 13, 2301396 CrossRef CAS.
- M. Kim, J. An, S.-J. Shin, I. Hwang, J. Lee, Y. Park, J. Kim, E. Park, J. Kim, G. Park, S. Kim, A. Coskun and J. W. Choi, Energy Environ. Sci., 2024, 17, 6079–6090 RSC.
- K. Lee, S.-H. Kwon, J. Kim, E. Park, I. Kim, H. C. Ahn, A. Coskun and J. W. Choi, ACS Energy Lett., 2024, 9, 2201–2211 CrossRef CAS.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- M. Fang, X. Yue, Y. Dong, Y. Chen and Z. Liang, Joule, 2024, 8, 91–103 CrossRef CAS.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- S. Kim and V. G. Pol, ChemSusChem, 2023, 16, e202202143 CrossRef CAS PubMed.
- R. Xu, J.-F. Ding, X.-X. Ma, C. Yan, Y.-X. Yao and J.-Q. Huang, Adv. Mater., 2021, 33, 2105962 CrossRef CAS PubMed.
- C.-B. Jin, N. Yao, Y. Xiao, J. Xie, Z. Li, X. Chen, B.-Q. Li, X.-Q. Zhang, J.-Q. Huang and Q. Zhang, Adv. Mater., 2023, 35, 2208340 CrossRef CAS PubMed.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- W. Liu, C. Yi, L. Li, S. Liu, Q. Gui, D. Ba, Y. Li, D. Peng and J. Liu, Angew. Chem., Int. Ed., 2021, 133, 13041–13050 CrossRef.
- J. Kim, J. Kim, J. Jeong, J. Park, C.-Y. Park, S. Park, S. G. Lim, K. T. Lee, N.-S. Choi, H. R. Byon, C. Jo and J. Lee, Energy Environ. Sci., 2022, 15, 4109–4118 RSC.
- T. Li, X.-Z. Yuan, L. Zhang, D. Song, K. Shi and C. Bock, Electrochem. Energy Rev., 2019, 3, 43–80 CrossRef.
- H. Zhang, Z. Zeng, F. Ma, X. Wang, Y. Wu, M. Liu, R. He, S. Cheng and J. Xie, Adv. Funct. Mater., 2022, 33, 2212000 CrossRef.
- J. G. Han, K. Kim, Y. Lee and N. S. Choi, Adv. Mater., 2019, 31, e1804822 CrossRef PubMed.
- Z. Wang, Y. Wang, C. Wu, W. K. Pang, J. Mao and Z. Guo, Chem. Sci., 2021, 12, 8945–8966 RSC.
- S. Chae, S. Park, K. Ahn, G. Nam, T. Lee, J. Sung, N. Kim and J. Cho, Energy Environ. Sci., 2020, 13, 1212–1221 RSC.
- X.-R. Chen, Y.-X. Yao, C. Yan, R. Zhang, X.-B. Cheng and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 7743–7747 CrossRef CAS PubMed.
- Y.-H. Tan, Z. Liu, J.-H. Zheng, Z.-J. Ju, X.-Y. He, W. Hao, Y.-C. Wu, W.-S. Xu, H.-J. Zhang, G.-Q. Li, L.-S. Zhou, F. Zhou, X. Tao, H.-B. Yao and Z. Liang, Adv. Mater., 2024, 36, 2404815 CrossRef CAS PubMed.
- J. Lee, H.-S. Lim, X. Cao, X. Ren, W.-J. Kwak, I. A. Rodriguez-Perez, J.-G. Zhang, H. Lee and H.-T. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 37188–37196 CrossRef CAS PubMed.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Adv. Mater., 2021, 33, 2100404 CrossRef CAS PubMed.
- Y. Guo, S. Pan, X. Yi, S. Chi, X. Yin, C. Geng, Q. Yin, Q. Zhan, Z. Zhao, F.-M. Jin, Y.-B. He, F. Kang, S. Wu and Q.-H. Yang, Adv. Mater., 2024, 36, 2308493 CrossRef CAS PubMed.
- S. C. Kim, X. Kong, R. A. Vila, W. Huang, Y. Chen, D. T. Boyle, Z. Yu, H. Wang, Z. Bao, J. Qin and Y. Cui, J. Am. Chem. Soc., 2021, 143, 10301–10308 CrossRef CAS PubMed.
- G. Yang, I. N. Ivanov, R. E. Ruther, R. L. Sacci, V. Subjakova, D. T. Hallinan and J. Nanda, ACS Nano, 2018, 12, 10159–10170 CrossRef CAS PubMed.
- Y.-H. Tan, G.-X. Lu, J.-H. Zheng, F. Zhou, M. Chen, T. Ma, L.-L. Lu, Y.-H. Song, Y. Guan, J. Wang, Z. Liang, W.-S. Xu, Y. Zhang, X. Tao and H.-B. Yao, Adv. Mater., 2021, 33, 2102134 CrossRef CAS PubMed.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J.-G. Zhang, Adv. Energy Mater., 2017, 8, 1702097 CrossRef.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- Y. Fang, S. L. Zhang, Z.-P. Wu, D. Luan and X. W. D. Lou, Sci. Adv., 2021, 7, eabg3626 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- P. Xiao, Y. Zhao, Z. Piao, B. Li, G. Zhou and H.-M. Cheng, Energy Environ. Sci., 2022, 15, 2435–2444 RSC.
- X. Wang, D. Ren, H. Liang, Y. Song, H. Huo, A. Wang, Y. Gao, J. Liu, Y. Gao, L. Wang and X. He, Energy Environ. Sci., 2023, 16, 1200–1209 RSC.
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Energy Environ. Sci., 2020, 13, 183–199 RSC.
- L. Giordano, P. Karayaylali, Y. Yu, Y. Katayama, F. Maglia, S. Lux and Y. Shao-Horn, J. Phys. Chem. Lett., 2017, 8, 3881–3887 CrossRef CAS PubMed.
- J. Wu, Y. Wu, L. Wang, H. Ye, J. Lu and Y. Li, Adv. Mater., 2024, 36, 2308193 CrossRef CAS PubMed.
- D. M. Knotter and T. J. J. Denteneer, J. Electrochem. Soc., 2001, 148, F43 CrossRef CAS.
- M. Lim, H. An, J. Seo, M. Lee, H. Lee, H. Kwon, H.-T. Kim, D. Esken, R. Takata, H. A. Song and H. Lee, Small, 2023, 19, 2302722 CrossRef CAS PubMed.
- H.-J. Kwon and J.-G. Park, J. Korean Phys. Soc., 2022, 81, 903–909 CrossRef CAS.
- J. G. Han, K. Kim, Y. Lee and N.-S. Choi, Adv. Mater., 2019, 31, 1804822 CrossRef PubMed.
- K. Kim, D. Hwang, S. Kim, S. O. Park, H. Cha, Y.-S. Lee, J. Cho, S. K. Kwak and N.-S. Choi, Adv. Energy Mater., 2020, 10, 2000012 CrossRef CAS.
- J.-G. Han, M.-Y. Jeong, K. Kim, C. Park, C. H. Sung, D. W. Bak, K. H. Kim, K.-M. Jeong and N.-S. Choi, J. Power Sources, 2020, 446, 227366 CrossRef CAS.
- E. Yoon, J. Lee, S. Byun, D. Kim and T. Yoon, Adv. Funct. Mater., 2022, 32, 2200026 CrossRef CAS.
- M. Fang, B. Du, X. Zhang, X. Dong, X. Yue and Z. Liang, Angew. Chem., Int. Ed., 2024, 136, e202316839 CrossRef.
- H. Cheng, D. Li, B. Xu, Y. Wei, H. Wang, B. Jiang, X. Liu, H. Xu and Y. Huang, Energy Storage Mater., 2022, 53, 305–314 CrossRef.
- Z. Liu, X. Wu, P. Hu and C. Shang, J. Colloid Interface Sci., 2023, 652, 50–56 CrossRef CAS PubMed.
- K. Long, S. Huang, H. Wang, A. Wang, Y. Chen, Z. Liu, Y. Zhang, Z. Wu, W. Wang and L. Chen, Energy Environ. Sci., 2024, 17, 260–273 RSC.
- W. Pan, S. Huang, K. Long, X. Liu, P. Qing, H. Liu, Y. Jin, Y. Chen, H. Li, L. Mei, Z. Wu and L. Chen, Mater. Today Energy, 2024, 45, 101675 CrossRef CAS.
- X. Liu, K. Long, S. Huang, P. Xiao, C. Ling, Y. Chen, Z. Wu, Y. Zhang and L. Chen, J. Power Sources, 2024, 612, 234843 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03862b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |