
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Failure mode diagnosis and stabilization of an efficient reverse-bias bipolar membrane CO2 to CO electrolyzer†
Sven
Brückner‡
 a,
Oleksandr
Bondarchuk‡
bcd,
Ana
Araújo
be,
Wen
Ju
af,
Rosalía
Cid
g,
Elvira
Paz
b,
Florian
Krebs
a,
Olívia Salomé G. P.
Soares
e,
Isilda
Amorim
b,
Zhipeng
Yu
b,
Pierre
Schröer
a,
Philipp
Hauke
a,
Manuel Fernando R.
Pereira
e,
Lifeng
Liu
bh and
Peter
Strasser
*a
a,
Oleksandr
Bondarchuk‡
bcd,
Ana
Araújo
be,
Wen
Ju
af,
Rosalía
Cid
g,
Elvira
Paz
b,
Florian
Krebs
a,
Olívia Salomé G. P.
Soares
e,
Isilda
Amorim
b,
Zhipeng
Yu
b,
Pierre
Schröer
a,
Philipp
Hauke
a,
Manuel Fernando R.
Pereira
e,
Lifeng
Liu
bh and
Peter
Strasser
*a
aThe Electrochemical Energy, Catalysis, and Materials Science Laboratory, Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bInternational Iberian Nanotechnology Laboratory (INL), 4715-330 Braga, Portugal
cSPIN-Lab Centre for Microscopic Research on Matter, University of Silesia in Katowice, 75 Pułku Piechoty Str. 1A, Chorzów 41-500, Poland
dInstitute of Chemistry, University of Silesia in Katowice, 9 Szkolna Str., 40-006 Katowice, Poland
eLSRE-LCM – Laboratory of Separation and Reaction Engineering-Laboratory of Catalysis and Materials, Faculty of Engineering and ALiCE – Associate Laboratory in Chemical Engineering, Faculty of Engineering, University of Porto, 4200-465 Porto, Portugal
fDepartment of Electrochemistry and Catalysis, Leibniz Institute for Catalysis, 18059, Rostock, Germany
gCentre for Cooperative Research on Alternative Energies (CIC energiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, 01510 Vitoria-Gasteiz, Spain
hSongshan Lake Materials Laboratory (SALB), Dongguan, Guangdong 523808, People's Republic of China
First published on 29th May 2025
Abstract
Efficient and stable CO2-to-CO electrolyzers are key process components for the generation of green synthesis gas and its downstream conversion and valorization to carbonaceous e-chemicals and e-fuels. While alkaline CO2 electrolyzers suffer from low CO2 utilization due to cathodic carbonate formation and crossover, acidic CO2 electrolyzers suffer from low CO faradaic efficiency. Reverse-bias bipolar membrane (BPM) cell architectures have been proposed to promote cathodic proton transport, yet resulted in limited cell lifetimes due to complex degradation and failure regimes. A thorough diagnosis of BPM cell dynamics is missing to date. Here, we build and diagnose an efficient zero-gap reverse-bias BPM CO2-to-CO electrolyzer cell deploying CO-selective single Ni atom cathode catalysts. We analyzed its key cell performance parameters and diagnosed the cell stability and failure regimes over 100 hours. The electrolyzer cell showed excellent performance up to 500 mA cm−2 with CO faradaic efficiency near 100%. The proton-controlled ion transport in the cathode was directly confirmed by an experimental carbon cross-over coefficient (CCC) of zero, suggesting minimal carbon loss due to carbonate formation. This was coupled to a high single pass conversion of ∼70% at the largest current densities and 60 vol% CO in the cell outlet, ideally suited for process cascade involving electro- or thermal catalytic steps. While use of a N2 bleed for internal reference has been known to be critical for accurate evaluation of cell performance, we now propose the experimental N2 vol% in the combined cell outlet and bleed flow to be also a valuable diagnostic tool to recognize and analyze cell failure regimes.
Broader contextElectrochemical CO2 reduction is an emerging and promising technology for converting CO2 into value-added products, thereby helping to close the anthropogenic carbon cycle. However, neutral-alkaline CO2 electrolysis suffers from parasitic acid–base CO2 consumption by hydroxide ions, leading to prohibitively low carbon utilization. Owing to their acidic cathode environments, bipolar membrane-based CO2 electrolysis offers improved carbon efficiencies, yet brings along its own characteristic failure modes. Diagnostic tools that monitor, identify, and help mitigate failure modes are missing. This contribution reports experimental strategies to track and mitigate BPM cell failure to extend the lifetime of CO2 electrolyzers. |
Introduction
Direct CO2 electrolysis has emerged as a focal point of scientific exploration and technological advancements in the field of power-to-hydrocarbon e-fuels. Especially, the CO2 to CO cascade has witnessed significant breakthroughs for its inherent high catalytic selectivity and process energy efficiency, and proper catalysts for this conversion can be broadly categorized into two classes: coin metals Ag and Au, where silver has garnered considerable attention,1–6 and the more recent emergence of the family of metal–nitrogen–carbon (MNC) single site catalyst materials.7–12Currently, our understanding indicates that both catalyst classes exhibit good selectivity and demonstrate the ability to achieve high current densities, while there is a considerable cost difference. However, there also remains a knowledge gap regarding the stability and degradation of these two classes of catalysts. On one hand, it is known that silver suffers from cathodic corrosion, akin to other noble metals such as gold and platinum.13,14 On the other hand, despite their proven application in acidic proton exchange membrane environments, the stability of metal–nitrogen–carbon (MNC) catalysts against metal atom leaching is not fully understood. Computational calculations indicate that CO can stabilize lower coordinated Ni species,15 which may also apply to NiNC catalysts. Moreover, MNC catalysts offer unique advantages in terms of relative intermediate binding energies to their coordinative single-metal active sites. This distinctive feature enhances the intrinsic selectivity of the single metal atom active sites toward CO2 reduction compared to that toward hydrogen evolution, and it also allows for efficient metal utilization.16 These characteristics create exciting opportunities for exploring and developing more efficient catalysts for sustainable CO2 conversion at industrial scales.
To fully uncover the true potential of the catalyst, it is essential to evaluate its performance in a full two-electrode electrolyzer cell device. The zero-gap membrane electrode assembly (MEA) cell configuration provides closer proximity between the catalyst and the electrode, enabling efficient charge transfer and low potential losses. However, technical challenges such as flooding and salt precipitation must be addressed and overcome for optimal electrolyzer performance and stability. This work will address these issues.
In the zero-gap cell, salt precipitation caused by exceeding bicarbonate solubility limits has remained a key challenge that could be addressed by cathode water management or by choice of a suitable cation offering larger solubility limits.17 The key advantage of a zero-gap cell is the significantly lower cell potential and energy efficiency compared to liquid cathode gap cells.6,12,18,19 The choice of the type of ion transported across the solid electrolyte membrane offers options including the cation-exchange membrane (CEM), the anion-exchange membrane (AEM) and the bipolar membrane (BPM). BPM based electrolyzers have been operated in both reverse-bias and forward-bias modes. In forward-bias BPM electrolyzer designs, cathodic hydroxide anions are neutralized by acid-based recombination inside the BPM minimizing CO2 depletion by carbonate formation. The spontaneous recombination lowers required cell voltage. By contrast, the reverse bias mode generates a pH gradient by water dissociation. Protons channeled toward the cathode neutralize carbonate at the expense of cell voltage.
State-of-the-art efficient CO2 electrolyzers have been designed using AEMs, thanks to the pH dependent Nernst potential of the competing proton discharge process. However, AEM based cells suffer from significant cathodic flow depletion: if every generated OH− is converted to CO32−, one loses around 7 ml A−1 min−1 CO2.20,21 This drop in flow makes an accurate measurement of the cathodic gas outflow rate from the electrolysis cell critically important. Careless overestimation of cathode outlet flow rates can result in overestimated CO faradaic efficiencies and overestimated cell performance.11,12,22,23 In addition to the gas flow depletion challenge in AEM cell designs, the use of non-noble Ni-based zero-gap anodes in direct contact with the AEM, even in presence of high-pH anolytes, is precluded by anodic corrosion due to low local pH caused by (bi)carbonate crossover.24,25
In this contribution, we design, operate, and diagnose an efficient reverse-bias, zero-gap BPM CO2-to-CO electrolyzer using an acid-stable NiNC single metal atom catalyst. The BPM dissociates water in the reverse bias mode and channels protons to the cathode, which recombine with catalytically generated OH−, thereby eliminating undesired cathodic (bi)carbonate accumulation and gas flow depletion.26–31 At the same time, the acid-stable NiNC single metal atom catalyst with its low atomic H chemisorption32,33 ensures high faradaic efficiencies toward CO. While we diagnose and identify the proton as the dominant transport ion at the cathode using the recently introduced kinetic carbon crossover coefficient (CCC),16 we demonstrate, distinguish, and analyze common electrolyzer failure modes such as salt blocking at cell inlet and outlet. Our BPM electrolyzer could be continuously operated for over 100 h. It showed near 100% CO faradaic efficiencies at over 400 mA cm−2 CO partial current density, coupled to 70% single pass conversion (SP) and a previously unachieved 60% CO-rich outlet gas stream associated with a low CO2 stoichiometric ratio λ.
CO2 reduction catalyst synthesis and characterization
We synthesized a number of distinct NiNC catalysts (NiNC-600C, NiNC-800C, NiNC-1000C) from a Ni-imidazole framework precursor using a 2-step pyrolysis process at varying temperatures. The final pyrolysis step temperature is thereby eponymous. The catalysts obtained were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), N2 adsorption/desorption isotherms, X-ray photoelectron spectroscopy (XPS), Fig. 1, elemental analysis, inductively-coupled plasma optical emission spectroscopy (ICP-OES, Table S2, ESI†) and X-ray diffractometry (XRD, Fig. S1, ESI†). SEM revealed (Fig. 1a, c and e) a slight morphology change with increasing pyrolysis temperature, with more flakes and particles rather than continuous carbon structures. At a smaller length scale, TEM images (Fig. 1b, d and f) suggested no significant differences from earlier comparable synthesis results.16 The morphology change is mirrored in the Brunauer–Emmett–Teller (BET) surface area values (Fig. 1g, h and Table S3, ESI†), where an increase in surface area, mesopores, and micropores was observed from NiNC-600C to NiNC-800C. At even higher temperatures, the mesopores started to collapse coupled with lower BET values. During the pyrolysis, carbon-embedded metallic Ni nanoparticles formed, while the stacking order of the carbon increased with higher temperatures, seen by more prominent (002) carbon reflection in the XRD (Fig. S1, ESI†). Thanks to the final acid washing of all catalysts, accessible metallic Ni particles were removed to ensure no/low HER activity.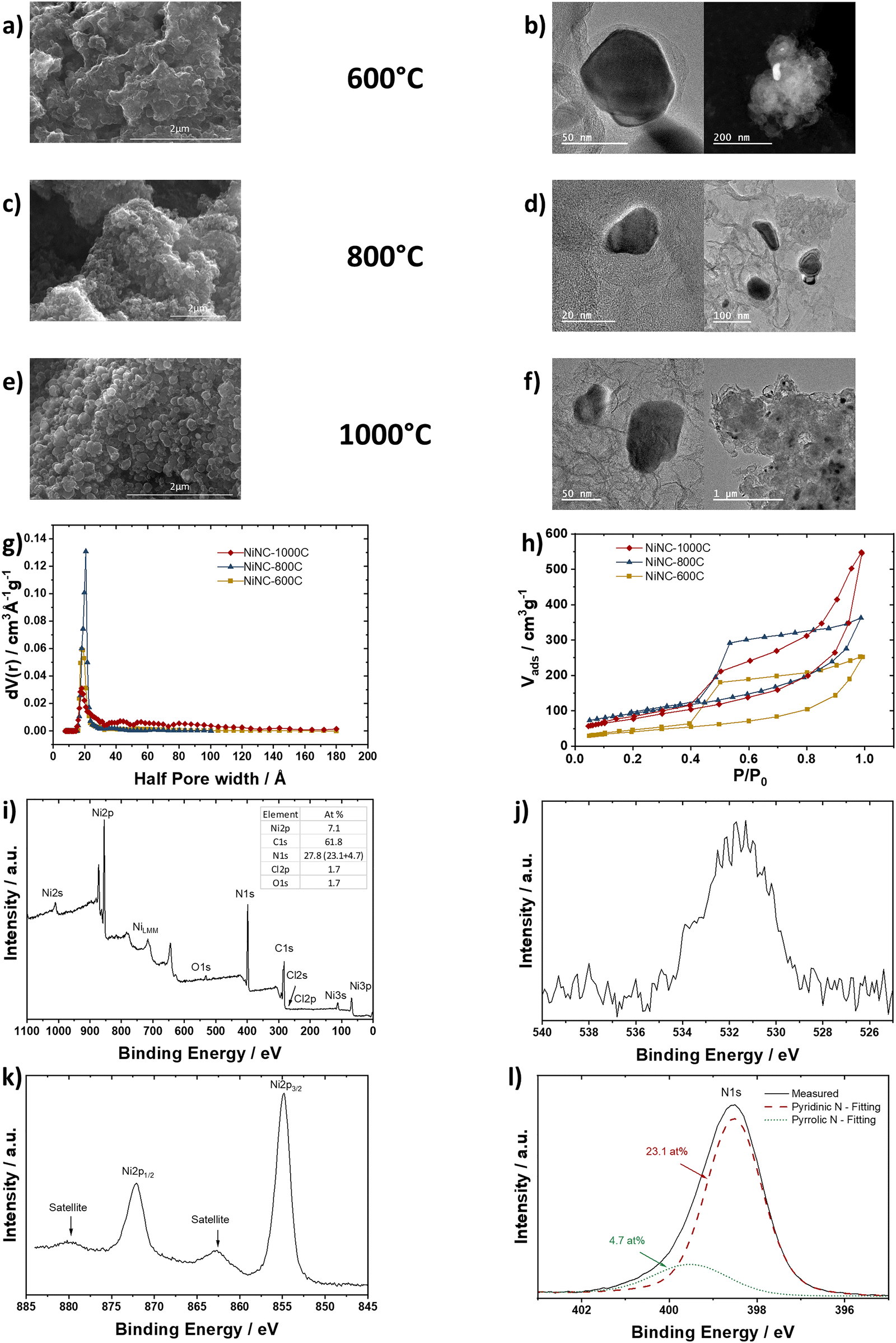 | ||
| Fig. 1 Characterization of the NiNC catalyst. (a) SEM and (b) TEM images of the NiNC-600C. (c) SEM and (d) TEM images of the NiNC-800C. (e) SEM and (f) TEM images of the NiNC-1000C. (g) Pore distribution and (h) adsorption/desorption isotherm plots. (i) survey XPS of NiNC-1000C, (j) insights of satellite and main peaks of nickel, (k) oxygen and (l) nitrogen, respectively. | ||
Fig. 1i presents the survey XPS spectrum of the NiNC-1000C sample. The strong C 1s signal is mainly due to the carbon framework, where the single atom nickel is embedded into. A small oxygen peak in Fig. 1j at ∼532 eV is due to typical surface contamination, like COOH, CO and COH. The shape and peak position of the O 1s signal apparently indicate no significant amount of metal oxides. The estimated atomic concentration Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio is 1:3.8 ≈ 4, very different from that in the “Ni3N” (Note S1, ESI†). The Ni 2p3/2 main peak position is at 854.9 eV (Fig. 1k), meaning the chemical shift of the Ni 2p level is larger by 1.3 eV than that of NiO (853.6 eV). The satellite peak is shifted by 7.9 eV to the higher binding energies from the main peak. The intensity of the satellite is 12% of the intensity of the main peak. This is a much smaller relative satellite intensity compared to the 77% for NiO. The Ni 2p3/2 XPS peak of NiO is dominated by the 2p5d8 core hole configuration34 – the high spin configuration of Ni cation. According to Grosvenor et al. the relatively weak satellite, observed for NiNC-1000C, is indicative of low-spin configuration of Ni ions.35 Given this and taking into account the substantial chemical shift of the main peak to higher binding energy together with the observed Ni:N concentration ratio, one could speculate that the dominating oxidation state of Ni in the NiNC-1000C sample is higher than +2 and the correspondent final state is 2p5d7. In the same time this conclusion is not in accord with the reported Ni 2p spectra for Ni(OH)2, where Ni is in +2 oxidation state, and Ni 2p3/2 peak appears at 855.8 eV.36 As an alternative explanation one can consider the square-planar structure where 4 nitrogen atoms are around Ni which is in low spin configuration and its oxidation state is +2 with the fully occupied 3dxy, 3dxz, 3dyz, 3dz2 orbitals and empty 3dx2−y2 orbital – similar to what can be found in nickel porphyrin.37 To check this explanation out we performed XPS characterization of oxide free Ni porphyrin and Ni phthalocyanine films in situ grown via thermal evaporation. The results are presented in the ESI.†
:N ratio is 1:3.8 ≈ 4, very different from that in the “Ni3N” (Note S1, ESI†). The Ni 2p3/2 main peak position is at 854.9 eV (Fig. 1k), meaning the chemical shift of the Ni 2p level is larger by 1.3 eV than that of NiO (853.6 eV). The satellite peak is shifted by 7.9 eV to the higher binding energies from the main peak. The intensity of the satellite is 12% of the intensity of the main peak. This is a much smaller relative satellite intensity compared to the 77% for NiO. The Ni 2p3/2 XPS peak of NiO is dominated by the 2p5d8 core hole configuration34 – the high spin configuration of Ni cation. According to Grosvenor et al. the relatively weak satellite, observed for NiNC-1000C, is indicative of low-spin configuration of Ni ions.35 Given this and taking into account the substantial chemical shift of the main peak to higher binding energy together with the observed Ni:N concentration ratio, one could speculate that the dominating oxidation state of Ni in the NiNC-1000C sample is higher than +2 and the correspondent final state is 2p5d7. In the same time this conclusion is not in accord with the reported Ni 2p spectra for Ni(OH)2, where Ni is in +2 oxidation state, and Ni 2p3/2 peak appears at 855.8 eV.36 As an alternative explanation one can consider the square-planar structure where 4 nitrogen atoms are around Ni which is in low spin configuration and its oxidation state is +2 with the fully occupied 3dxy, 3dxz, 3dyz, 3dz2 orbitals and empty 3dx2−y2 orbital – similar to what can be found in nickel porphyrin.37 To check this explanation out we performed XPS characterization of oxide free Ni porphyrin and Ni phthalocyanine films in situ grown via thermal evaporation. The results are presented in the ESI.†
The N 1s spectrum (Fig. 1l) exhibits a single peak with the maximum at 398.5 eV, shifted by 0.5 eV with respect to the “Ni3N” sample. The observed singlet shape of N 1s is in contrast to the nickel nitrides prepared in a similar way. Pyridinic and pyrrolic functional groups were reported based on the multiple component fitting of the N 1s spectra.10,38,39 Fig. S7 (ESI†) compares N 1s spectra of the “Ni3N” with the N 1s spectrum from oxygen-free VN prepared via thermal nitridation of pure vanadium.40 The “Ni3N” and VN spectra reveal quite similar asymmetric shapes, characteristic of conductive materials, while the NiNC-1000C spectrum has a clear minimum on the higher binding energy side of the N 1s peak. The minimum is followed by the onset of the inelastic losses. The distance from the onset to the N 1s core-level peak can be used to determine the band gap (∼4.5 eV) or, rather, the HOMO–LUMO gap according to Nichols et al.41 and references therein. A further comparison between NiNC-100C and reference materials like Ni-TPP, Ni PC and Ni3N can be found in the Note S1 (ESI†).
Design and performance of a zero-gap BPM electrode assembly
Mass transport and solubility limitations of gaseous CO2 in liquid aqueous electrolytes preclude the use of fully immersed electrodes, where industrial current densities on the order of several hundreds of mA cm−2 are of the essence. Instead, gas diffusion electrodes (GDEs) composed of a porous transport layer (PTL) equipped with a catalyst-coated micro porous layer (MPL) separate the CO2 gas channel from the liquid catholyte compartment (one-gap design) or from the ion exchange membrane (zero-gap design). Fig. 2 displays the zero-gap CO2 electrolyzer cell design where the anodic and cathodic flow fields are integrated into the current collectors. In between the two current collectors is the BPM assembly consisting of the anode, the BPM and the NiNC-catalyst on the carbon cloth PTL (see box in Fig. 3). | ||
| Fig. 2 Schematic of the zero gap ccs cell with insight in the BPM and cathode processes. The cell contains a flow field integrated current collector, a NiFe-LDH@NF anode, a bipolar membrane and a NiNC catalyst coated GDL. The insight shows the direction of the membrane, CEM to cathode, and the assumed neutralization of the carbonate by protons to recover CO2. | ||
 | ||
| Fig. 3 Electrochemical zero-gap cell performance. (a) Polarization curves of catalyst with different pyrolysis temperatures and (b) FE at different current densities. (c) The CCC proves that no carbonate formation is noticeable and (d) N2 vol% of the GC over different current densities to prove no flow depletion, expected 20 vol% (5 ml min−1 N2 and 20 ml min−1 CO2). (e) Single pass conversion and CO2 to CO ratio at different current densities and (f) the CO ratio in the outlet stream of the electrolyzer cell. All test represent the mean value of at least 2 individual tests, the standard deviation can be found in the ESI.† | ||
The cell components in the enlarged box in Fig. 3 highlight the working principle of a BPM assembly where water is dissociated at the interface of a cationic (CEM) and an anionic (AEM) exchange membrane, before H+ and OH− move in opposite directions. Unlike for AEM design, in the BPM design, OH− move exclusively across the AEM to the anode. There, they help maintain the pH enabling the use of platinum-group-metal (PGM)-free anodes such as NiFe-LDH grown on nickel foam working as anode.42–44 On the other hand, the protons move across the CEM to the cell cathode and buffer OH− or CO32− to water and CO2, respectively. The OH− are generated from the catalytic conversion of water to hydrogen or the conversion of CO2 to CO and form CO32−. The H+ at the cathode will thus regenerate CO2. For AEM electrolysis, techno-economic analysis showed that the CO2 membrane crossover in AEM systems coupled to subsequent anodic CO2 separation and regeneration is quite energy- and cost intensive.29,45
Fig. 3 shows six key cell performance parameters vs. the applied current density of the reverse-bias zero gap BPM electrolyzer cell, in particular cell voltage, CO faradaic efficiency (FECO), carbon cross over coefficient (CCC), N2 vol% in cell outlet flow rate, single pass conversion (SP), and the CO concentration in the outlet flow. Each performance parameter was evaluated for each of the three NiNC cathode catalysts at a CO2 inlet flow rate of 20 ml min−1 or 4 ml min−1 cm−2 in a current density range from 0–500 mA cm−2. The polarization curves (Fig. 3a) of all three cathodes were very similar up to 200 mA cm−2. Beyond that, the rise in cell voltage varied for each catalyst, likely as a result of increasing H2 faradaic efficiency (FEH2) for the NiNC-600C and the NiNC-1000C. Drawing 500 mA cm−2 proved problematic for NiNC-800C likely due to slow water dissociation rates. While trends in FECO (Fig. 3b) did not correlate with the pyrolysis temperatures, they followed trends in BET surface areas, suggesting a relation to catalyst porosity. Notably, despite the reverse-bias low-pH conditions at the cathode, the BPM cell reached favorable FECO values of 80–100% at current densities up to 500 mA cm−2.
In order to characterize the ion transport in the cathodes of CO2 electrolyzers, we recently introduced the concept of the carbon cross over coefficient, CCC.16 Illustrated in the context of a AEM CO2 electrolyzer,16 the CCC proved a diagnostic tool to distinguish ion transport regimes dominated by protons (CCC ∼ 0), carbonate (CCC ∼ 1), or bicarbonate ions (CCC ∼ 2). Equivalently, the CCC provided insight in the non-electrocatalytic consumption of CO2 by OH−.16Fig. 3c reports experimental CCC values in the present BPM CO2 electrolyzer environment. Unlike for AEM CO2 electrolyzers, the experimental CCC values are near 0 over the entire current density range, suggesting protons to be the dominant ion at the cathode and implying no significant loss of CO2 by carbonate formation. This directly confirms the hypothesized functional concept of a reverse-bias BPM-based cell cathode. Here, we further introduce the experimental volume fraction of N2 in the cell outlet (Fig. 3d) as a new diagnostic tool to characterize failure modes and degradation regimes of CO2 electrolyzers. A small N2 “bleed” is introduced after the cell and prior to the gas chromatographic analysis to provide an internal calibration for accurate determination of the outlet flow. For a CO2 to CO electrolyzer operating at FECO ∼ 100% without any non-electrocatalytic loss of CO2, a constant N2 bleed of 5 ml min−1 against the CO2 inlet feed of 20 ml min−1 should result in an experimental N2 fraction of about 20 vol%, as evidenced in Fig. 3d. By contrast, in case of (bi)carbonate formation from OH− and CO2, the volumetric outlet flow rate decreases, while the experimental N2 vol% increases. In absence of carbonate formation, yet significant competitive H2 evolution, the outlet flow rate increases, while the N2 vol% decreases, as observed for NiNC-800C (blue line in Fig. 3d) above 200 mA cm−2 where the FECO gradually dropped. These considerations underline the diagnostic value of the N2 vol%, which will be elaborated further below. Next, the single pass conversions of the BPM electrolyzer were evaluated for each NiNC catalyst (Fig. 3e). Thanks to the low carbonate formation in the proton-rich cathode suggested by CCC ∼ 0, the present BPM CO2 electrolyzer design displayed a very high ∼70% single pass conversion (SP) that significantly surpassed the theoretical limit of 50% of AEM CO2 electrolyzer systems that are characterized by CCC ∼ 1. These favorable SP values resulted in CO-enriched outlet flows of up to 60 vol% at 400–500 mA cm−2 for NiNC-1000C (Fig. 3f) that are relevant for cascaded CO2 conversion using tandem electrolyzer cell designs46 or other subsequent CO-based, e.g. synthesis gas related, thermal catalytic processes.
One of the challenges of BPM cell designs remained the limiting water dissociation rates at higher current densities coupled with high overpotentials. The Boettcher group showed how self-made BPMs overcome this challenge; we prepared CEM-AEM combinations47–49 and succeeded in significantly decreasing cell potentials to below 4 V even at high current densities of 600 mA cm−2 (Fig. S9, ESI†). The significantly lowered cell voltage penalty using TiOx water dissociation catalysts is testament to the technological viability of reversed-bias BPM electrolyzer schemes. It voids the commonly held view that excessive cell voltages will preclude the wider use of reverse-bias BPM cells. In Table S4 (ESI†) we can show that we could improve the achievable current density, faradaic efficiency and even lower drastically the cell potential.
We note that minor, though finite, K+ migration from anode to cathode across the BPM does impact the electroneutrality balance, as well. K+ transport rates lower the required water dissociation rate (H2O → H+ + OH−) in the BPM. While this may benefit overpotentials, the resulting potassium carbonate formation may affect stability (Fig. S8a, ESI†) and promotes CO2 consumption. To lower K+ cross-over-rates, we carried out control experiments using 0.1 M KOH instead of 1 M KOH, which lowered the FECO (Fig. S8b, ESI†). There appears to exist a trade-off between product selectivity and K+ cross-over in BPM systems, in line with a previous report on Ag catalysts.30
Failure mode diagnosis and stabilization of zero-gap BPM CO2 electrolyzers
In order to learn more about the nature, the diagnosis, and the mitigation of failure modes of zero-gap BPM CO2 electrolyzers, we investigated the stability of the BPM cell at an initial constant current density of 100 mA cm−2 for 100 h. Fig. 4a and b report the temporal evolution of the cell voltage Ecell, FECO, FEH2, and N2 vol% over the test time. We observed an initial stable performance (FECO = 100%, N2 bleed = 20 vol%) below 3.25 V cell potential for around 10 h. This stable regime was followed by an unstable regime, characterized by increased, fluctuating Ecell and scattered FECO and FEH2. During the unstable regime, the N2 vol% values rose to 100% (yellow period in Fig. 4b), followed by a period of fluctuating N2 vol% values (grey period in Fig. 4b). Correlating the dynamic performance parameters in Fig. 4a and b with an inspection of the cathodic CO2 gas channels, we realized that the N2 bleed vol% values provided a surprisingly useful diagnostic test for the detailed salt blocking locations (Fig. 4b and c): N2 vol% values of 100% correlated with complete salt blocking near the cell outlet (marked “outlet” in Fig. 4b). This is because during this particular type of salt blocking failure, the cell outlet gas flow ceased completely, while the N2 bleed flow rose to 100 vol%, being the only gas detected. Regimes with strongly fluctuating values of N2 vol%, by contrast, represented a failure mode characterized by dynamic partial salt blocking (marked “partial” in Fig. 4b). Incomplete dynamic salt blocks in the cathode flow channel led to reduced CO2 feeds. This causes the FEH2 to rise in order to satisfy the applied current density. As a result of this, FECO and FEH2 fluctuated irregularly. Occasional cathode flushing helped normalize the N2 vol% values, suggesting the removal of salt blocks.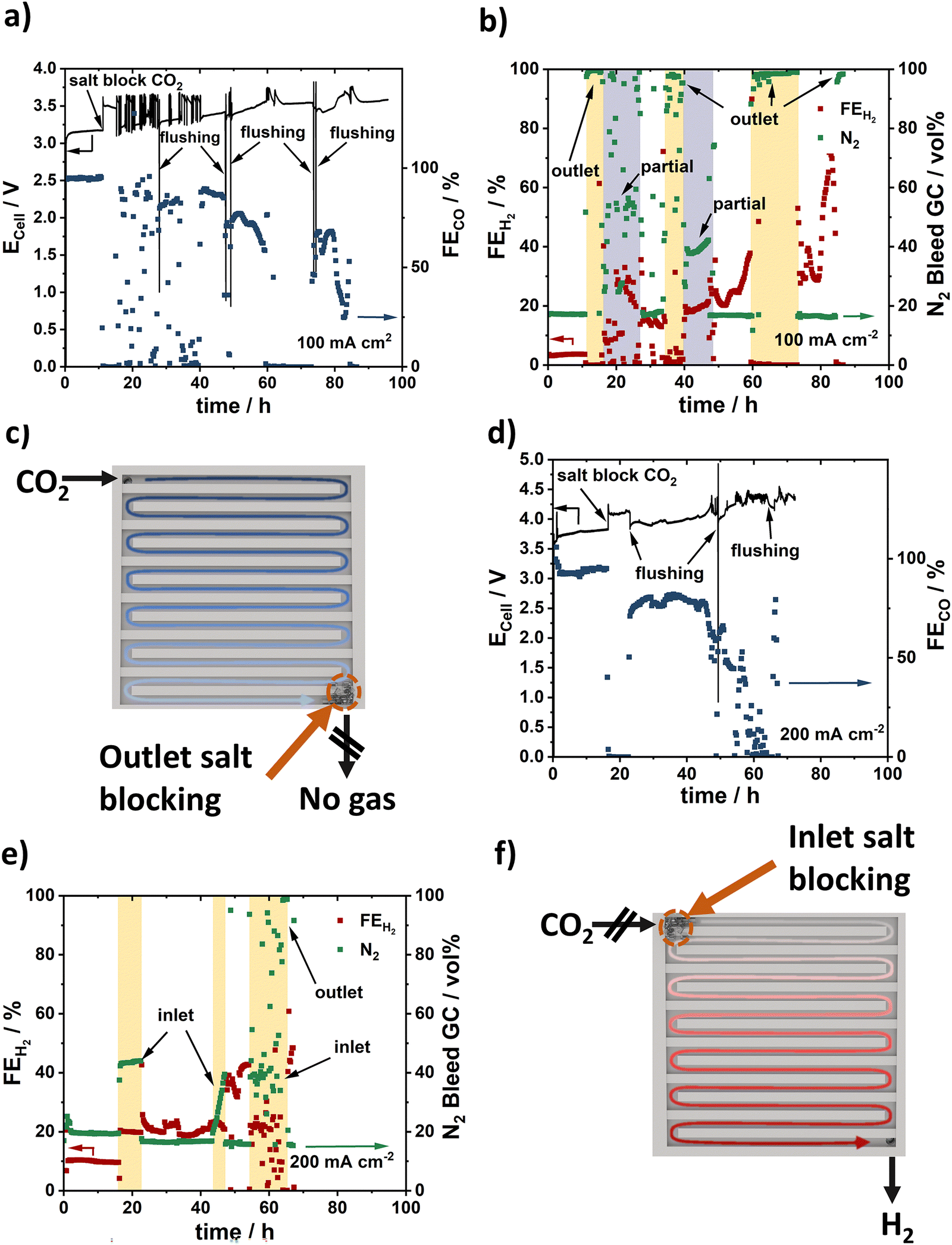 | ||
| Fig. 4 Electrochemical zero gap cell stability. (a) Cell voltage (solid line) and FECO (blue squares) during a 100 h stability test at constant current density of 100 mA cm−2. Salt blocking failure events (“salt block CO2”) are indicated. “flushing” marks salt removal by water flushing of cathode channel. (b) FEH2 and actual N2 vol% in the exit flow. Expected N2 vol% from N2-bleed is 20%. The N2 vol% is used as diagnostic tool to determine where and what type of cell blocking occurs. “outlet” (yellow periods) marks full-blocking failure regimes characterized by N2 vol% ∼ 100% coupled to FEH2 ∼ 0% associated with complete flow blockage near cell outlet. “partial” (grey periods) marks partial blocking failure regimes characterized by very scattered N2 vol% values between 20% and 100%. (c) Schematic illustration of salt blocking at the cell exit derived from data in (b). (d) Cell voltage (solid line) and FECO (blue squares) during a 100 h stability test at constant current density of 200 mA cm−2. (e) FEH2 and actual N2 vol% in the exit flow. “inlet” (yellow periods) marks failure regimes characterized by elevated N2 vol% > 20% and elevated FEH2 associated with salt blockage near cell inlet. (f) Schematic illustration of salt blocking at the inlet of cell derived from data in (e). N2 bleed: 5 sccm, CO2 feed: 20 sccm. The data shown in the figure correspond to single tests. | ||
We then tested the system stability at a constant current density of 200 mA cm−2 (Fig. 4d and e). After an initial stable regime at 3.7 V and FECO > 90%, the cell entered its first failure mode regime after ca. 18 h. This failure regime appeared distinct from the one observed in Fig. 5a and b: while Ecell rose, and FECO dropped, N2 vol% and FEH2 values stabilized at 40% and 20%, respectively, without fluctuations (yellow period in Fig. 5e). This failure regime involves salt blocking near the cell inlet (marked “inlet”), the location of which we were able to confirm experimentally (Fig. 4f). Inlet salt blocking is characterized by time-stable faradaic H2 evolution from water coupled to exclusive detection of H2 and N2 in the outlet flow, because CO2 input feed is blocked. Again, cathode flushing was able to alter or partially mitigate salt blocking (see >65 h), corroborating the importance of cathodic water management in zero-gap CO2 electrolyzers. Our stability studies hence suggested that our BPM zero-gap CO2 electrolyzer performance was limited by the system stability and not by any single component stability.
 | ||
| Fig. 5 Optimized stability test. (a) Cell voltage and FE of a stability test at 100 mA cm−2 using a 1 M CsOH. (b) FEH2 and N2 vol% in the GC to prove no salt blocking. The data shown in the figure correspond to single tests. | ||
In AEM CO2 electrolyzer systems, salt block degradation can be addressed by (i) water management (ii) cell temperature adjustments or (iii) salt solubility increases, e.g., replacing K+ with Cs+. We investigated whether these mitigation strategies would also apply to BPM CO2 electrolyzer and opted for strategy (iii). In a control experiment, we used a 200 ml 1 M CsOH anolyte at 100 mA cm−2.
Use of Cs at the anode clearly solved failure modes associated with salt precipitation and allowed stable operation over at least 100 h at close to 100% FE, 3.25 V (Fig. 5a) and only a small pH change (from pH 14 to pH 13 after 100 h) at the anode. In order to verify that N2 vol% continues to act as diagnostic tool, we monitored N2 vol% values and confirmed their time-dependent stability at nearly 20% over 100 h. These results corroborate that the favorable solubility of Cs-(bi)-carbonate suppressed undesired salt blocking (Fig. 5b). We note in passing that, in our view, cell stability tests at current densities of about 100 mA cm−2 reveal system stability rather than catalyst component stability. This makes component stability tests in a liquid electrolyte environment of a commonly employed H-cell operating at even smaller current densities very doubtful. This is why we investigated the BPM electrolyzer stability at higher current densities of 200 mA cm−2 at extended test times (Fig. S10, ESI†). For the initial 24 h, the cell performance maintained favorable 90% FECO with a slight rise in Ecell from 3.9 to 4.3 V. Then, we noticed a pH change on the anode to pH 8–9 after 140 h (100 h at 100 mA cm−2 and 40 h at 200 mA cm−2). This was associated with a drastic black anolyte color suggesting corrosion of the Ni-based anode catalyst. Post-testing analysis of the anode revealed a clear morphology change at the front side of the anode (Fig. S11, ESI†) and we saw individual particles in the completely blackened anode (Fig. S12, ESI†) with the anode back side still resembling metallic Ni foam (Fig. S13, ESI†). Finally, after a test time of 170 h, the zero gap CO2 electrolyzer ceased operation with complete loss of CO selectivity. After disassembly, pinholes were observed all over the membrane, indicating possible impact of the anode restructuring. Clearly, when PGM-free anodes are used, tracking the evolution of anodic pH is critical.
Conclusion
We have presented the first zero-gap reverse-bias BPM CO2-to-CO electrolyzer cell deploying CO-selective single Ni atom cathode catalysts. Furthermore, we have reported the usefulness of the novel concept of the carbon crossover coefficient, CCC, and the N2 bleed as diagnostic tools to predict and understand the dominant transport ion and the type of failure regimes. We have analyzed key cell performance parameters and diagnosed its cell stability over 100+ hours. While use of a N2 bleed for internal reference has been known to be critical for accurate evaluation of cell performance, we now propose the experimental N2 vol% in the combined cell outlet and bleed flow to be a valuable diagnostic tool to recognize and analyze cell failure regimes due to cathodic salt blocking.The zero-gap BPM CO2-to-CO electrolyzer cell showed excellent performance over a wide current density range of 0 to 500 mA cm−2 with FECO near 100%. The proton-controlled ion transport in the cathode was confirmed by a carbon cross-over coefficient (CCC) near zero, suggesting minimal carbon loss due to carbonate formation. This was coupled with a high single pass conversion of ∼70% at the largest current densities.
The experimental N2 vol% values proved a diagnostic tool for the localization of system instabilities due to salt blocking. Complete salt blocking near the cell outlet resulted in experimental N2 volume fractions of close to 100%, as the gas outflow from the cell ceased. Salt blacks near the cell inlet resulted in time stable increases of the experimental N2 vol% to around 40%, with FEH2 nearing 20%. Partial dynamic salt blocking, however, resulted in highly fluctuating values of N2, FECO, and FEH2.
Short term mitigation of system failure modes was achieved by cathodic water management. Increasing salt solubility, by contrast, proved a superior stabilization strategy for 100+ hours. Only at larger current densities (here 200 mA cm−2) and further extended test times (100–200 h), individual component stability failures became noticeable as anolytes approached near neutral pH. This should be considered when CO2 electrolysis component stability is evaluated in low-current and/or short-term H-Cell environments.
Author contributions
Conceptualization: Sven Brückner and Ana Araújo. Investigation and formal analysis: Sven Brückner, Ana Araújo, Oleksandr Bondarchuk, Elvira Paz, Rosalía Cid, Isilda Amorim, Yu Zhipeng, Pierre Schröer, Florian Krebs, Philipp Hauke. Writing – original draft: Sven Brückner, Oleksandr Bondarchuk, Wen Ju. Writing – review & editing: all authors. Supervision: Peter Strasser, Oleksandr Bondarchuk, Manuel Fernando R. Pereira, Olívia Salomé G. P. Soares, Lifeng Liu. Funding acquisition: Peter Strasser, Oleksandr Bondarchuk, Manuel Fernando R. Pereira, Olívia Salomé G. P. Soares, Lifeng Liu, Ana Araújo.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
Funding: Technical University Berlin. The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 851441, SELECTCO2. This work partially supported by national funds through FCT/MCTES (PIDDAC): LSRE-LCM, UIDB/50020/2020 (DOI: https://doi.org/10.54499/UIDB/50020/2020) and UIDP/50020/2020 (DOI: https://doi.org/10.54499/UIDP/50020/2020); and ALiCE, LA/P/0045/2020 (DOI: https://doi.org/10.54499/LA/P/0045/2020). Ana Araújo, acknowledges PhD Scholarship grant from FCT (SFRH/BD/143490/2019). OSGPS acknowledges FCT funding under the Scientific Employment Stimulus – Institutional Call CEECINST/00049/2018.References
- N. Hoshi, M. Kato and Y. Hori, Electrochemical reduction of CO on single crystal electrodes of silver Ag(111), Ag(100) and Ag(110), J. Electroanal. Chem., 1997, 440(1–2), 283–286 CrossRef CAS.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Insights into the electrocatalytic reduction of CO(2) on metallic silver surfaces, Phys. Chem. Chem. Phys., 2014, 16(27), 13814–13819 RSC.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim and C. M. Friend, et al., Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles, J. Am. Chem. Soc., 2015, 137(43), 13844–13850 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu and Q. Liu, et al., Shape-Dependent Electrocatalytic Reduction of CO(2) to CO on Triangular Silver Nanoplates, J. Am. Chem. Soc., 2017, 139(6), 2160–2163 CrossRef CAS PubMed.
- X. Yuan, Y. Wu, B. Jiang, Z. Wu, Z. Tao and X. Lu, et al., Interface Engineering of Silver-Based Heterostructures for CO(2) Reduction Reaction, ACS Appl. Mater. Interfaces, 2020, 12(50), 56642–56649 CrossRef CAS PubMed.
- A. Ozden, Y. J. Liu, C. T. Dinh, J. Li, P. F. Ou and F. P. G. de Arquer, et al., Gold Adparticles on Silver Combine Low Overpotential and High Selectivity in Electrochemical CO Conversion, Acs Appl. Energy Mater., 2021, 4(8), 7504–7512 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Metal-Doped Nitrogenated Carbon as an Efficient Catalyst for Direct CO2 Electroreduction to CO and Hydrocarbons, Angew. Chem., 2015, 127(37), 10908–10912 CrossRef.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev and V. Bon, et al., Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO(2), Nat. Commun., 2017, 8(1), 944 CrossRef PubMed.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, Electrochemical Reduction of CO on Metal-Nitrogen-Doped Carbon Catalysts, ACS Catal., 2019, 9(8), 7270–7284 CrossRef CAS.
- C. Li, W. Ju, S. Vijay, J. Timoshenko, K. Mou and D. A. Cullen, et al., Covalent Organic Framework (COF) Derived Ni-N-C Catalysts for Electrochemical CO(2) Reduction: Unraveling Fundamental Kinetic and Structural Parameters of the Active Sites, Angew. Chem., Int. Ed., 2022, 61(15), e202114707 CrossRef CAS PubMed.
- H. Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E. S. Jeong, V. Subramanian and U. Sim, et al., Achieving highly efficient CO to CO electroreduction exceeding 300 mA cm with single-atom nickel electrocatalysts, J. Mater. Chem. A, 2019, 7(17), 10651–10661 RSC.
- T. T. Zheng, K. Jiang, N. Ta, Y. F. Hu, J. Zeng and J. Y. Liu, et al., Large-Scale and Highly Selective CO Electrocatalytic Reduction on Nickel Single-Atom Catalyst, Joule, 2019, 3(1), 265–278 CrossRef CAS.
- S. J. Raaijman, N. Arulmozhi and M. T. M. Koper, Anisotropic Cathodic Corrosion of Gold Electrodes in the Absence and Presence of Carbon Monoxide, J. Phys. Chem. C, 2020, 124(52), 28539–28554 CrossRef CAS.
- T. J. P. Hersbach and M. T. M. Koper, Cathodic corrosion: 21st century insights into a 19th century phenomenon, Curr. Opinion Electrochem., 2021, 26 Search PubMed.
- P. Prslja and N. López, Stability and Redispersion of Ni Nanoparticles Supported on N-Doped Carbons for the CO2 Electrochemical Reduction, ACS Catal., 2020, 11(1), 88–94 CrossRef.
- S. Brückner, Q. Feng, W. Ju, D. Galliani, A. Testolin and M. Klingenhof, et al., Design and diagnosis of high-performance CO2-to-CO electrolyzer cells, Nat. Chem. Eng., 2024, 1(3), 229–239 CrossRef.
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, Investigation of Electrolyte-Dependent Carbonate Formation on Gas Diffusion Electrodes for CO(2) Electrolysis, ACS Appl. Mater. Interfaces, 2021, 13(13), 15132–15142 CrossRef CAS PubMed.
- Y. Y. Gu, J. C. Wei, J. D. Li, L. Y. Wang and X. Wu, Long-term-stability continuous flow CO reduction electrolysers with high current efficiency, Sustainable Energy Fuels, 2021, 5(3), 758–766 RSC.
- W. Ren, X. Tan, C. Jia, A. Krammer, Q. Sun and J. Qu, et al., Electronic Regulation of Nickel Single Atoms by Confined Nickel Nanoparticles for Energy-Efficient CO(2) Electroreduction, Angew. Chem., Int. Ed., 2022, 61(26), e202203335 CrossRef CAS PubMed.
- G. O. Larrazabal, P. Strom-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen and I. Chorkendorff, et al., Analysis of Mass Flows and Membrane Cross-over in CO(2) Reduction at High Current Densities in an MEA-Type Electrolyzer, ACS Appl. Mater. Interfaces, 2019, 11(44), 41281–41288 CrossRef CAS PubMed.
- G. O. Larrazábal, M. Ma and B. Seger, A Comprehensive Approach to Investigate CO2 Reduction Electrocatalysts at High Current Densities, Acc. Mater. Res., 2021, 2(4), 220–229 CrossRef.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng and G. Li, et al., Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities, Nat. Commun., 2020, 11(1), 593 CrossRef CAS PubMed.
- X. Lv, Q. Liu, H. Yang, J. Wang, X. Wu and X. Li, et al., Nanoconfined Molecular Catalysts in Integrated Gas Diffusion Electrodes for High-Current-Density CO2 Electroreduction, Adv. Funct. Mater., 2023, 33(32), 2301334 CrossRef CAS.
- A. Vass, B. Endrodi, G. F. Samu, A. Balog, A. Kormanyos and S. Cherevko, et al., Local Chemical Environment Governs Anode Processes in CO(2) Electrolyzers, ACS Energy Lett., 2021, 6(11), 3801–3808 CrossRef CAS PubMed.
- A. Vass, A. Kormanyos, Z. Koszo, B. Endrodi and C. Janaky, Anode Catalysts in CO(2) Electrolysis: Challenges and Untapped Opportunities, ACS Catal., 2022, 12(2), 1037–1051 CrossRef CAS PubMed.
- D. A. Vermaas and W. A. Smith, Synergistic Electrochemical CO Reduction and Water Oxidation with a Bipolar Membrane, ACS Energy Lett., 2016, 1(6), 1143–1148 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Electrolytic CO(2) Reduction in a Flow Cell, Acc. Chem. Res., 2018, 51(4), 910–918 CrossRef CAS PubMed.
- M. A. Blommaert, D. Aili, R. A. Tufa, Q. Li, W. A. Smith and D. A. Vermaas, Insights and Challenges for Applying Bipolar Membranes in Advanced Electrochemical Energy Systems, ACS Energy Lett., 2021, 6(7), 2539–2548 CrossRef CAS PubMed.
- M. A. Blommaert, S. Subramanian, K. Yang, W. A. Smith and D. A. Vermaas, High Indirect Energy Consumption in AEM-Based CO(2) Electrolyzers Demonstrates the Potential of Bipolar Membranes, ACS Appl. Mater. Interfaces, 2022, 14(1), 557–563 CrossRef CAS PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, Cation-Driven Increases of CO(2) Utilization in a Bipolar Membrane Electrode Assembly for CO(2) Electrolysis, ACS Energy Lett., 2021, 6(12), 4291–4298 CrossRef CAS PubMed.
- K. Xie, R. K. Miao, A. Ozden, S. Liu, Z. Chen and C. T. Dinh, et al., Bipolar membrane electrolyzers enable high single-pass CO(2) electroreduction to multicarbon products, Nat. Commun., 2022, 13(1), 3609 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Single site porphyrine-like structures advantages over metals for selective electrochemical CO reduction, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- S. Vijay, W. Ju, S. Bruckner, S. C. Tsang, P. Strasser and K. R. Chan, Unified mechanistic understanding of CO reduction to CO on transition metal and single atom catalysts, Nat. Catal., 2021, 4(12), 1024–1031 CrossRef CAS.
- P. S. Bagus, C. J. Nelin, C. R. Brundle, B. V. Crist, E. S. Ilton and N. Lahiri, et al., Main and Satellite Features in the Ni 2p XPS of NiO, Inorg. Chem., 2022, 61(45), 18077–18094 CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds, Surf. Interface Anal., 2004, 36(12), 1564–1574 CAS.
- M. C. Biesinger, L. W. Lau, A. R. Gerson and R. S. Smart, The role of the Auger parameter in XPS studies of nickel metal, halides and oxides, Phys. Chem. Chem. Phys., 2012, 14(7), 2434–2442 CAS.
- G. I. Svirskiy, N. N. Sergeeva, S. A. Krasnikov, N. A. Vinogradov, Y. N. Sergeeva and A. A. Cafolla, et al., Electronic structure of nickel porphyrin NiP: Study by X-ray photoelectron and absorption spectroscopy, Phys. Solid State, 2017, 59(2), 368–377 CAS.
- J. Li, S. Y. Hu, Y. Li, X. B. Fan, F. B. Zhang and G. L. Zhang, et al., Pyrrolic N anchored atomic Ni-N3-C catalyst for highly effective electroreduction of CO2 into CO, Carbon, 2023, 206, 62–71 CAS.
- P. Hou, X. Wang, Z. Wang and P. Kang, Gas Phase Electrolysis of Carbon Dioxide to Carbon Monoxide Using Nickel Nitride as the Carbon Enrichment Catalyst, ACS Appl. Mater. Interfaces, 2018, 10(44), 38024–38031 CAS.
- O. Bondarchuk, A. Morel, D. Bélanger, E. Goikolea, T. Brousse and R. Mysyk, Thin films of pure vanadium nitride: Evidence for anomalous non-faradaic capacitance, J. Power Sources, 2016, 324, 439–446 CAS.
- M. T. Nichols, W. Li, D. Pei, G. A. Antonelli, Q. Lin and S. Banna, et al., Measurement of bandgap energies in low-k organosilicates, J. Appl. Phys., 2014, 115(9), 094105 Search PubMed.
- P. Hauke, M. Klingenhof, X. L. Wang, J. F. de Arau and P. Strasser, Article Efficient electrolysis of 5-hydroxymethylfurfural to the biopolymer-precursor furandicarboxylic acid in a zero-gap MEA-type electrolyzer, Cell Rep. Phys. Sci., 2021, 2(12), 100650 CrossRef CAS.
- P. Hauke, S. Brückner and P. Strasser, Paired Electrocatalytic Valorization of CO and Hydroxymethylfurfural in a Noble Metal-free Bipolar Membrane Electrolyzer, ACS Sustainable Chem. Eng., 2023, 11(37), 13628–13635 CrossRef CAS.
- M. Klingenhof, P. Hauke, M. Kroschel, X. L. Wang, T. Merzdorf and C. Binninger, et al., Anion-Tuned Layered Double Hydroxide Anodes for Anion Exchange Membrane Water Electrolyzers: From Catalyst Screening to Single-Cell Performance, ACS Energy Lett., 2022, 7(10), 3415–3422 CrossRef CAS.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O’Brien, A. Gaona and J. Wicks, et al., Downstream of the CO2 Electrolyzer: Assessing the Energy Intensity of Product Separation, ACS Energy Lett., 2021, 6(12), 4405–4412 CrossRef CAS.
- T. Moller, M. Filippi, S. Bruckner, W. Ju and P. A. Strasser, CO(2) electrolyzer tandem cell system for CO(2)-CO co-feed valorization in a Ni-N-C/Cu-catalyzed reaction cascade, Nat. Commun., 2023, 14(1), 5680 CrossRef PubMed.
- Y.-L. Kao, L. Chen, S. W. Boettcher and D. Aili, Divergent Synthesis of Bipolar Membranes Combining Strong Interfacial Adhesion and High-Rate Capability, ACS Energy Lett., 2024, 9(6), 2953–2959 CrossRef CAS.
- L. Chen, Q. Xu, S. Z. Oener, K. Fabrizio and S. W. Boettcher, Design principles for water dissociation catalysts in high-performance bipolar membranes, Nat. Commun., 2022, 13(1), 3846 CrossRef CAS PubMed.
- J. C. Bui, E. W. Lees, D. H. Marin, T. N. Stovall, L. Chen and A. Kusoglu, et al., Multi-scale physics of bipolar membranes in electrochemical processes, Nat. Chem. Eng., 2024, 1(1), 45–60 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01817j |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |