
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in covalent organic frameworks as heterogeneous photocatalysts for photochemical conversion
Xiangyu
Zhang†
ac,
Rufan
Chen†
a,
Yongxiang
Zhou
 ac,
Zekun
Chen
ac,
Guo Qin
Xu
ac and
Qing-Hua
Xu
*abc
ac,
Zekun
Chen
ac,
Guo Qin
Xu
ac and
Qing-Hua
Xu
*abc
aNational University of Singapore (Suzhou) Research Institute, Suzhou 215123, China
bCollege of Science, Eastern Institute of Technology, Ningbo 315200, China
cDepartment of Chemistry, National University of Singapore, 117543, Singapore. E-mail: qhxu@eitech.edu.cn/chmxqh@nus.edu.sg
First published on 14th April 2025
Abstract
Covalent organic frameworks (COFs) are a class of porous and crystalline materials constructed from covalently linked building blocks. Over the past decade, dramatic progress has been made in synthesizing new COFs with diverse applications, especially in photocatalysis. In this review, we highlight recent progress in COFs as heterogeneous photocatalysts for their applications toward hydrogen evolution from water splitting, carbon dioxide reduction, organic transformations, and hydrogen peroxide production. We focus on rational design of COF structures as well as our understanding of interplay between COF structures and their optical properties as efficient heterogeneous photocatalysts. Challenges and perspectives are discussed at the end.
Broader contextThe quest for sustainable energy solutions has driven transformative advances in light harvesting and conversion technologies over the past five decades. Solar energy utilization has evolved from simple photon absorption to complicated photocatalytic systems that can efficiently capture, convert, and store light energy. Beginning with fundamental studies of light–matter interactions in semiconductor photocatalysts, the field has progressed through several breakthrough innovations in controlling light absorption, charge separation, and surface reactions. The development of new materials has revolutionized our ability to harness the solar spectrum – from traditional semiconductors that are limited to UV light to advanced covalent organic frameworks (COFs) capable of efficient visible light utilization. Most significantly, advances in the design and synthesis of COFs have enabled unprecedented control over light-harvesting efficiency, charge separation and transport processes, and catalytically active sites. The emergence of precisely engineered COFs has marked a critical convergence of structural order and photophysical functionality, establishing new paradigms for solar energy conversion. These developments have accelerated progress across multiple frontiers, from artificial photosynthesis to solar fuel production, laying the foundation for next-generation technologies that can effectively convert sunlight into chemical energy for practical applications. |
1 Introduction
Fossil fuels have been the main source of energy for economic development since the industrial revolution. The release of greenhouse gases from massive consumption of these natural resources has caused lots of environmental problems and created lots of concerns and demand for alternative energy resources.1–3 Solar energy has been considered as the most promising alternative energy resource as it is sustainable, clean, and low-cost. To be utilized for practical applications, solar energy needs to be coupled with energy collection, conversion and storage processes.4 In nature, green plants convert solar energy into chemical energy by utilization of sunlight as the driving force and enzymes as photocatalysts to transform carbon dioxide and water into carbohydrates and oxygen, offering excellent inspiration for researchers to develop efficient artificial photocatalytic systems.5,6In 1972, Fujishima and Honda reported the pioneering work on titanium dioxide (TiO2) as a photocatalyst to realize water splitting under irradiation with ultraviolet (UV) light in a photoelectrochemical cell.7 Since then, photocatalysis has been applied to various reactions with a focus on the development of low-cost, stable and highly efficient photocatalysts. Many traditional inorganic semiconductors have been explored as photocatalysts for photo-induced production of chemical fuels and value-added compounds. However, their narrow light absorption range resulted in poor harvesting of solar energy.8,9 Organic semiconductors such as carbon nitrides, conjugated microporous polymers (CMPs), linear conjugated polymers and covalent triazine-based frameworks (CTFs) have recently emerged as promising materials for photocatalytic applications.10–13 The light absorption and band structure of these organic semiconductors can be tuned by modular copolymerization strategies. However, their amorphous or semicrystalline structures would still hinder charge transportation and consequently limit their photoactivity.14
The development of novel porous and crystalline materials such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs) has opened up new possibilities for new types of highly efficient photocatalysts.15–20 MOFs and HOFs are constructed via coordination bonds and hydrogen bonding interactions, respectively. Both of them usually exhibit relatively low chemical stability and thus limit their applications.21,22 COFs are a class of polymers that are constructed from molecular building blocks with strong covalent bonds through condensation reactions.23–25 They are particularly interesting as they combine properties of crystallinity, permanent porosity, synthetic versatility and good physical/chemical stability.26–30 The long-range-ordered structure of COFs is beneficial for photo-induced electron transport to the catalyst surface for photocatalytic applications. Furthermore, the diverse selection of chromophores enables tunable absorption and band structures. COFs have integrated the advantages of the abovementioned organic semiconductors while overcoming their limitations at the same time.31,32
Since the first report of a semiconducting COF in 2008,33 numerous efforts have been devoted to the field of COF assisted hydrogen evolution from water splitting,34 photocatalytic carbon dioxide (CO2) reduction35 and organic transformations.36 In this review, we will mainly discuss structural design strategies for the construction of efficient COFs as heterogeneous photocatalysts (Scheme 1). The relationship between structural design and optoelectronic properties will be discussed in detail for different photocatalytic applications. Perspectives of challenges and possible directions for future research will be discussed in the end.
 | ||
| Scheme 1 Schematic illustration of the diverse photocatalytic applications of COF-based heterogeneous photocatalysts. | ||
2 Structural aspects – fundamentals of COFs for photocatalysis
2.1 Structure design of COFs for photocatalysis
Porous and crystalline COFs can be obtained by topological utilization of building blocks through covalent bonds.25 The topology diagram or geometry of monomers determines the dimensionality and spatial orientation of COF skeletons as well as the size and shape of their pores. Monomers with C1, C2, C3, C4 and C6 symmetries have been used for the preparation of two-dimensional (2D) COFs with different topologies. The shapes of monomers and their major combination geometries are summarized in Fig. 1. The design strategy of one knot plus one linker is used most widely for the construction of 2D COFs to produce various backbones and pore channels. Besides that, a multicomponent strategy could allow more than one knot and linker to be integrated into one COF, which brings about more options for the design of different functional COFs.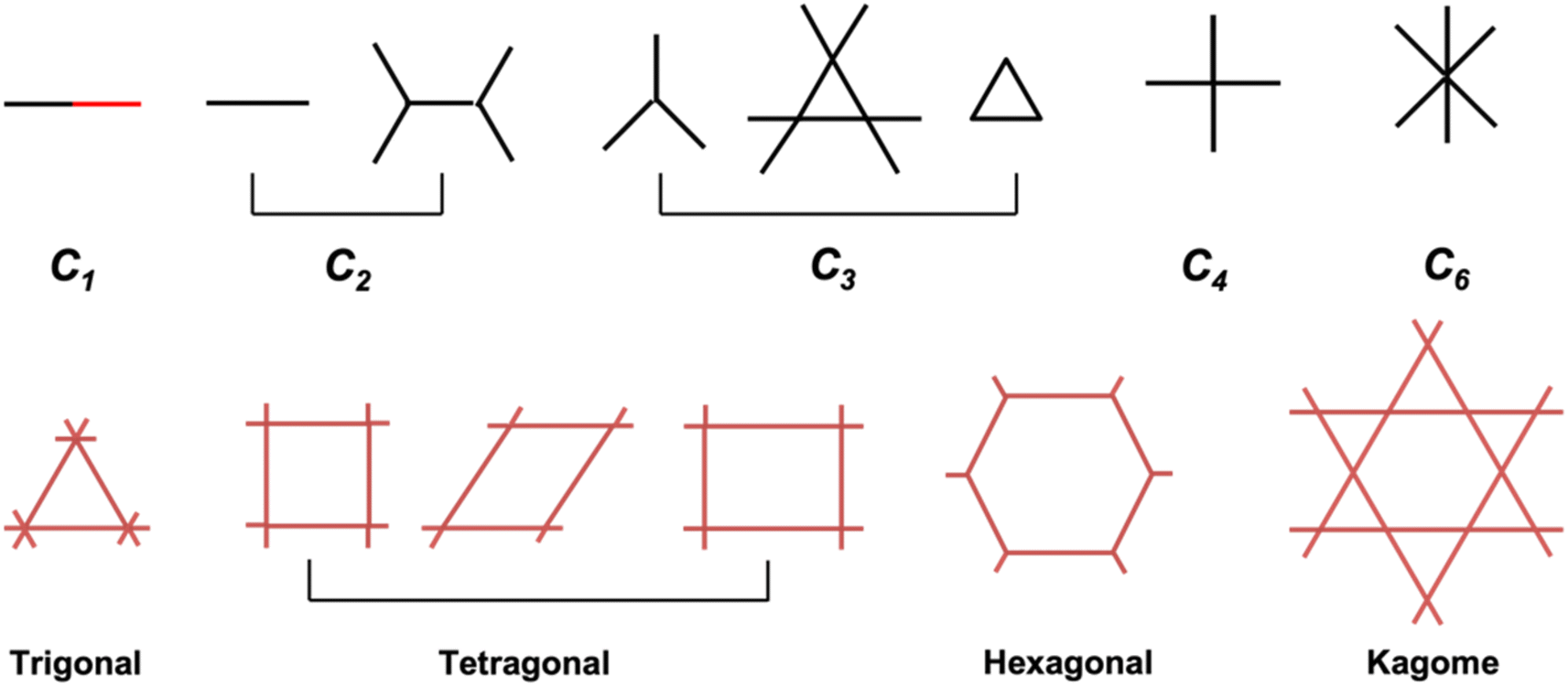 | ||
| Fig. 1 Topological schematics guiding the design of 2D COFs. | ||
Under the guidance of different topology diagrams, precursors of COFs condensed to form covalently linked layered skeletons and aligned pores. For the application of photocatalysis, favorable properties, such as excellent light-harvesting ability, suitable band structure and efficient charge separation, all originate from the structures of COFs. Specifically, skeletons of COFs can be constructed to act as light-harvesters, binding sites, or redox centers by tailored design of numerous building blocks. The high structural porosity gives them high surface areas and permanent pores, enabling enhanced accessibility of sensitizers, sacrificial reagents and cocatalysts. Functional units have been linked via various linkage motifs into stable and conjugated scaffolds, which contribute to increased charge carrier mobility and superior stability to work under harsh conditions. Furthermore, the molecular stacking modes and distances of COFs have been utilized to tune their optical and electronic properties.37–43 Periodically aligned columnar π-arrays can serve as light-harvesting matrices and offer pathways for charge carrier transport. Fully π-conjugated COFs with fully conjugated linkages offer charge transport pathways along with the perpendicular π-column directions. The pores or channels across the materials provide space for reactants to contact reaction centers while the products could be easily released from the catalytic sites.44 Recently, 3D-COFs have attracted significant attention due to their exceptional photocatalytic capabilities. Similar to their 2D counterparts, topology engineering is crucial for the rational design of 3D COFs. Compared to 2D COFs, 3D COFs often demonstrate interconnected channels and large accessible surface areas, which significantly enhance catalytic performance. Moreover, controlling interpenetration and incorporating steric hindrance groups ensure structural robustness, optimizing active site exposure and enhancing catalytic stability.45,46 Various 3D COFs with topologies such as stp, srs, and fcc have been reported as efficient photocatalysts. For instance, Ding et al. developed JUC-640-M COFs based on the stp topology, achieving ultra-large pores, record-low crystal density, and significantly enhanced CO2 photoreduction performance.47 Zhu et al. reported an innovative TMB-COF with srs topology, in which steric hindrance groups effectively convert planar precursors into spatially intricate frameworks, enhancing catalytic site availability.48 Furthermore, Li et al. demonstrated the functionalization of 3D fcc COFs with benzene, pyrazine, and tetrazine groups, significantly tuning reaction microenvironments and boosting photocatalytic urea synthesis via improved electron–hole separation, enhanced light-harvesting, and reactant co-adsorption capabilities.49 These sophisticated structural and functional designs position 3D COFs as promising platforms for high-performance photocatalytic applications.
Linkage selection significantly affects the properties of COFs.50 The boric acid condensation reaction was the earliest reported method for synthesizing COFs. Subsequently, the imine condensation reaction emerged and has become the most widely utilized method, with more than half of all reported COFs synthesized via this route. Traditional imine-linked COFs offer simple synthesis, structural versatility, and good crystallinity, but often require carefully designed monomers or precursors. However, their moderate chemical stability limits practical applications, particularly under harsh photocatalytic conditions. To overcome this limitation, imine-derived linkages—such as β-ketoenamine, triazine, oxazole, and thiazole—have been developed through tandem or post-synthetic modification reactions, transforming imine bonds into more robust structures with improved chemical stability, enhanced structural order, and favorable charge-transfer properties crucial for photocatalysis.51 Meanwhile, sp2 carbon-linked COFs, synthesized via direct C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C condensation, achieve extended π-conjugation, superior electronic conductivity and significantly improved semiconductor properties,52 which greatly enhance charge separation efficiency and boost photocatalytic performance.53 Alternatively, hydrazone-linked COFs, derived from aldehyde and hydrazide precursors, possess exceptional hydrolytic stability, structural flexibility, abundant heteroatomic sites (N and O), and post-synthetic modification capabilities, which enhance intermolecular interactions, facilitate efficient charge-carrier separation, and present diverse catalytic sites, leading to further amplified photocatalytic efficiency.54
C condensation, achieve extended π-conjugation, superior electronic conductivity and significantly improved semiconductor properties,52 which greatly enhance charge separation efficiency and boost photocatalytic performance.53 Alternatively, hydrazone-linked COFs, derived from aldehyde and hydrazide precursors, possess exceptional hydrolytic stability, structural flexibility, abundant heteroatomic sites (N and O), and post-synthetic modification capabilities, which enhance intermolecular interactions, facilitate efficient charge-carrier separation, and present diverse catalytic sites, leading to further amplified photocatalytic efficiency.54
2.2 Advantages of COFs for photocatalysis
COFs are one of the most attractive organic semiconductors pertaining to heterogeneous photocatalysis, owing to several advantages (Fig. 2): (1) the tailorable structure endows COFs with foreseeable semiconducting properties and band structures, and the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of the COF materials could be fine-tuned through molecular design of the well-defined skeletons to tailor their light harvesting ability and semiconducting properties for photocatalytic applications. Also, by utilizing electron deficient or electron donating building blocks, the donor–acceptor structures have been widely used in the construction of COFs to offer enhanced separation efficiencies of electrons and holes. (2) Highly crystalized structure of COFs favors effective charge transfer, as the stacked regularly π-units form an extended framework with close interactions, thereby offering pathways for charge transfer across the material. The well defined structures of COFs also make it clearer to explore the relationship between the structure and photocatalytic activity. (3) The high surface area and porosity of COFs are beneficial not only for the interactions of active sites with the substrate, sacrificial reagent and cocatalyst, but also for effective diffusion during the photocatalytic process. (4) Strong covalent bonds give COFs high stability, especially chemical stability, which makes them resistant to hydrolysis, pH variations, and oxidative or reductive environments and allowing them to work over the long term. The linkage motif in COFs not only provides high stability, but can also be engineered to improve the conjugation in planes to facilitate the charge transfer process. Unlike traditional catalysts that often suffer from fast charge recombination (such as TiO2 and carbon nitride), photocorrosion (such as CdS, In2S3, ZnCdS, and ZnIn2S4), a narrow wavelength range of harvesting light or limited surface area, COFs enhance photocatalytic performance through molecular-level design, allowing for better selectivity, higher quantum efficiency, and metal-free sustainability. In addition, their chemical stability, environmental friendliness, and potential for multi-functional applications (such as H2 production, CO2 reduction, and organic transformations) make them promising next generation photocatalysts. | ||
| Fig. 2 Advantages of COFs for photocatalysis due to their unique properties. | ||
Metal doping and construction of heterojunctions are two very common and effective means to enhance photocatalytic performance. Metal ions and complexes could be bound dispersedly to the backbones of COFs through coordination interaction as new active sites.55,56 The introduction of metals could also facilitate charge transfer and separation, leading to improved photocatalytic activity.57–59 Some metal-modified COFs demonstrate superior photocatalytic performance even without requiring noble-metal co-catalysts, exhibiting excellent catalytic stability and long-term recyclability.57 Metal sites anchored through the backbones of COFs provide exceptional interactions with substrates, thus amplifying the efficiency of photocatalysis. Recent studies have demonstrated that metal clusters integrated within COFs significantly improve local electron density, promote efficient exciton dissociation and thus enhance photoreduction efficiency. Furthermore, precise tuning of the metal environments with distinct coordination geometries reduces energy barriers for critical intermediate formation, resulting in improved catalytic selectivity and activity.58,59 Metal-covalent organic frameworks (MCOFs) serve as a bridge between MOFs and COFs, combining the beneficial properties of both. They maintain the stability, porosity, and tunability of COFs and inherit the rich metal-coordination chemistry of MOFs, yielding frameworks with superior catalytic properties, stability, and multifunctionality. The tunable band structures of COFs are very convenient for them to combine with another semiconductor to construct different types of heterojunctions as the HOMO and LUMO levels of COFs could be easily adjusted for desired heterojunctions. More importantly, strong interactions of COFs with organic or inorganic semiconductors are vital for charge transfer within the heterojunction.
3 Applications in photocatalysis
A typical photocatalytic reaction starts with absorption of light to generate electron–hole pairs, followed by charge separation and transportation to the surface for the oxidation/reduction reactions. Consequently, efforts to enhance the solar energy conversion efficiency of photocatalysts primarily focus on three key areas: improving strength and broadening the spectral range of light absorption, optimizing charge separation and transfer, and accelerating surface reactions. To ensure that photo-excited electrons and holes possess sufficient redox potential to drive surface reactions, the LUMO of covalent organic frameworks must be positioned at a more negative potential than the redox potential of the reduction reaction (Fig. 3). Concurrently, the HOMO should be situated at a more positive potential than the redox potential of the oxidation reaction. COFs provide a unique approach for the application of photocatalysis thanks to their interesting chemical and physical properties such as harvesting of broadband light and efficient charge separation and transfer.60 Recent research progress in COFs in various applications such as photocatalytic hydrogen evolution, carbon dioxide reduction, organic transformations and hydrogen peroxide production will be summarized in this part. | ||
| Fig. 3 Schematic illustration of the processes of photocatalytic water splitting, carbon dioxide reduction (a) and hydrogen peroxide production (b), and corresponding redox potentials of the relevant reactions. | ||
3.1 Photocatalytic H2 evolution
Photocatalytic hydrogen evolution (PHE) from water splitting involves a series of photochemical events and it is key to optimize these events to achieve optimum performance.61 This requires the design of various interfaces to enable continuous flow of electrons and holes to drive the reaction. However, it lacks a general principle for designing organic systems to interlock multiple interfaces to seamlessly integrate light absorption, exciton splitting, charge transfer and transport processes. As COFs enable a systematic design of the skeleton and pores, it is highly possible to engineer various interfaces into one framework. Prior to elucidating specific examples, we have compiled an overview of the PHE performance of various COF-based photocatalysts (Table 1).| Samples | Irradiation conditions | Sacrificial agent | Co-catalyst | HER rate (μmol h−1 g−1) | Ref. |
|---|---|---|---|---|---|
| TFPT-COF | λ > 420 nm | TEOA | Pt | 1970 | 62 |
| N3-COF | λ > 420 nm | TEOA | Pt | 1703 | 63 |
| ZnPor-DETH-COF | λ > 400 nm | TEOA | Pt | 413 | 64 |
| PyTz-COF | AM 1.5 | Ascorbic acid | Pt | 2072.4 | 65 |
| TCDA-COF | 780 nm > λ > 420 nm | Ascorbic acid | Pt | 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
66 |
| TpPa-COF-(CH3)2 | λ > 420 nm | Sodium ascorbate | Pt | 8330 | 67 |
| sp2c-COFERDN | λ > 420 nm | TEOA | Pt | 2120 | 68 |
| PY-DHBD-COF | λ > 420 nm | Ascorbic acid | Pt | 42432 |
69 |
| COF–alkene | λ > 420 nm | TEOA | Pt | 2330 | 70 |
| v-2D-COF-NO1 | λ > 400 nm | TEOA | Pt | 1970 | 71 |
| sp2c-COF-ST | λ > 420 nm | TEOA | Pt | 2150 | 72 |
| sp2c-Py-BT COF | λ > 420 nm | — | NA | 573 | 73 |
| TtaTfa | λ > 420 nm | Ascorbic acid | Pt | 20700 |
74 |
| COF-954 | 780 nm > λ > 420 nm | Ascorbic acid | Pt | 137230 |
75 |
| CYANO-CON | λ > 420 nm | Ascorbic acid | Pt | 134200 |
76 |
| 2D-TP-PB | λ > 420 nm | TEOA | Pt | 24980 |
77 |
| g-C18N3-COF | λ > 420 nm | Ascorbic acid | Pt | 292 | 78 |
| Cu-salphen-HDCOF-NSs | λ > 420 nm | TEA | Cu | 36990 |
79 |
| Co9S8@COF | λ > 420 nm | Ascorbic acid | Co9S8 | 23150 |
80 |
| Zn@H-TpPa | λ > 420 nm | Sodium ascorbate | Pt | 28000 |
81 |
| COF-Cu3TG | λ = 380–800 nm | — | NA | 10470 |
82 |
| TiO2-TpPa-1-COF (1:3) |
λ > 420 nm | Sodium ascorbate | Pt | 11190 |
83 |
| T-COF@CdS-3 | Full spectrum | Ascorbic acid | Pt | 12500 |
84 |
| COF-42 | AM 1.5G | TEOA | Cobaloxime | 163 | 85 |
| NH2-UiO-66/TpPa-1-COF (4:6) |
λ > 420 nm | Sodium ascorbate | Pt | 23410 |
86 |
| ATNT-4 | λ > 420 nm | Ascorbic acid | Pt | 14228.1 |
87 |
| TBTA/g-C3N4 | λ > 420 nm | Ascorbic acid | Pt | 26040 |
88 |
| PEG@BT-COF | λ > 420 nm | Ascorbic acid | Pt | 11140 |
89 |
| MAC-FA1/S-COF | λ > 420 nm | Ascorbic acid | NA | 100000 |
90 |
| COF316/Pt@TpBpy-COF | λ > 420 nm | — | Pt | 220.4 | 91 |
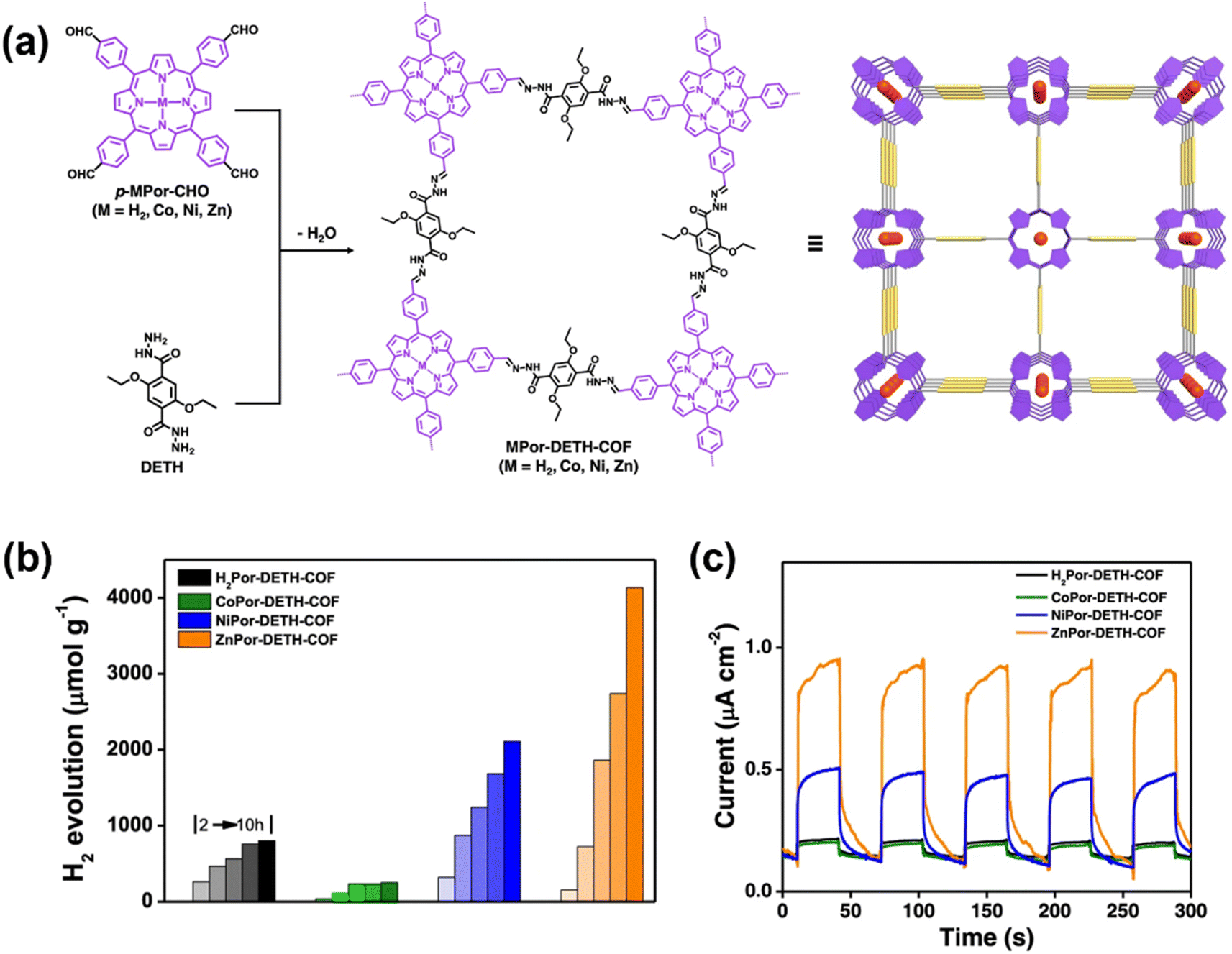 | ||
| Fig. 4 (a) The construction of MPor-DETH-COFs. (b) PHE performance for MPor-DETH-COFs. (c) Photocurrent generation of all four COFs coated on an indium–tin-oxide electrode as a working electrode in a three electrode CV setup upon light on–off switching. Adapted with permission.64 Copyright 2021, Springer Nature. | ||
The donor–acceptor (D–A) system has been demonstrated to be effective in promoting charge transfer and suppressing charge recombination. A 2D D–A COF was constructed with the units of electron-rich pyrene (Py) and electron-deficient thiazolo[5,4-d]thiazole (Tz), which exhibited a PHE rate of 2072.4 μmol h−1 g−1.65 Employing a multicomponent synthesis strategy, a three-component D–π–A structure can be achieved to regulate the photophysical properties of COFs in a more convenient way. The Liu group reported two photoactive 2D COFs with D–π–A structures, in which a three-component donor–π–acceptor COF (TCDA-COF) was constructed from electron-deficient triazine and electron-rich benzotrithiophene units through an sp2 carbon linkage.66 These COFs exhibited a very high PHE rate of 70.8 ± 1.9 mmol h−1 g−1 under visible light irradiation (420–780 nm) with Pt as the co-catalyst.
A fully designable skeleton of COFs makes it very convenient to integrate various functional groups for improved photocatalytic performance. For example, a series of ketoenamine-based COFs, named TpPa-COF-X (X = –H, –(CH3)2 and –NO2), with the same framework were selected as the model system to study the influence of different functional groups on photocatalytic hydrogen evolution (Fig. 5a).67 The obtained TpPa-COF-(CH3)2 with an electron-donating functional group showed the best photocatalytic performance and good stability (Fig. 5b and c), while TpPa-COF-NO2 with an electron-withdrawing functional group exhibited the lowest PHE rate. 3-Ethylrhodanine (ERDN), as an electron deficient functional unit, has been integrated as an end-capping group in the synthetic process of sp2c-COF and produced ERDN-terminated sp2c-COFERDN.68 Because of the push-and-pull effect arising from the ERDN terminating functional group, the PHE rate increased from 1360 μmol h−1 g−1 of sp2c-COF to 2120 μmol h−1 g−1 of sp2c-COFERDN. PY-DHBD-COF with adjacent hydroxyl groups and imine-N throughout the framework allows photogenerated electrons to converge and reduce the adsorbed platinum species into metal clusters, in which uniformly dispersed platinum clusters facilitated electron transfer, resulting in a high PHE rate of 42432 μmol h−1 g−1 at 1 wt% Pt loading.69
 | ||
| Fig. 5 (a) Schematic representation of preparation of TpPa-COF-X. (b) PHE activities and (c) photocatalytic stability of TpPa-COF-X. Reprinted with permission.67 Copyright 2019, Wiley-VCH. | ||
The linkage of COFs is vital for the stability of the materials and affects the photocatalytic performance of COF photocatalysts. Three COFs adopting triphenylbenzene knots and phenyl linkers with different linkages were constructed for PHE.70 Compared to imine- and imide-linked COFs with a low PHE rate of less than 40 μmol h−1 g−1, the cyano-substituted alkene-linked COF showed a relatively high PHE rate of 2.33 mmol h−1 g−1 under the irradiation of visible light. Between two isomeric benzobisoxazole-bridged v-2D-COFs with trans and cis configurations of benzobisoxazole, v-2D-COF-NO1 with the trans configuration linkage exhibited a PHE rate of ∼1.97 mmol h−1 g−1, which is twice that of v-2D-COF-NO2 with the cis configuration linkage.71 Recent investigations by Zhang et al. revealed that sp2-carbon-conjugated COFs (sp2c-COF-ST) constructed through vinylene linkages exhibit reduced effective mass and exciton binding energy, facilitating exciton dissociation and charge separation, thereby enhancing photocatalytic hydrogen evolution performance.72 Subsequently, Xu et al. synthesized a cyanovinylene-linked sp2c-Py-BT COF and imine-linked imine-Py-BT COF, both featuring identical donor–acceptor structures.73 Benefiting from significantly lower exciton binding energy and superior stability conferred by the cyanovinylene linkage, the sp2c-Py-BT COF demonstrated remarkable activity for photocatalytic overall water splitting, achieving an apparent quantum efficiency of 2.53% at 420 nm. In contrast, the imine-Py-BT COF failed to achieve photocatalytic water splitting. The imine linkage has been widely used for the construction of 2D COFs. When the Schiff-base linkage of a D–A COF constructed with triazine and triphenylamine motifs was protonated using ascorbic acid, a PHE rate as high as 20.7 mmol h−1 g−1 was achieved under visible light irradiation.74 The protonated COF showed redshift in the absorption spectrum, improved charge separation and increased hydrophilicity, which are responsible for the dramatically improved photocatalytic performance (Fig. 6). Significant efforts have also been made to understand how protonation enhances the photocatalytic performance of COFs. Protonated COFs were initially employed for applications in sensing.92 In addition to the advantages of improved hydrophilicity and broadened optical absorption range,74 protonation can reverse the direction of charge transfer between different moieties within COFs,93 indicating the potential for synergistic integration with other structural features.94 Li et al. employed highly photoactive oligo(phenylenevinylene) building blocks in the synthesis of imine-linked COFs and implemented the protonation strategy to further enhance the photocatalytic performance. The prepared COF-954 showed an exceptionally high PHE rate of 137.23 mmol h−1 g−1.75 Zhang et al. recently reported two partially protonated COFs with significantly improved photocatalytic efficiency, which was attributed to the formation of homojunctions between pristine and protonated COFs.95 The unique unprotonated/protonated structure generates a strong built-in electric field that effectively facilitates charge separation.
 | ||
| Fig. 6 (a) Synthesis of TtaTfa, TtaTfa AC, TpaTfa, TpaTfa AC, TtaTpa, and TtaTpa AC. AC = ascorbic acid modification. (b) Time course of photocatalytic H2 evolution for TtaTfa, TpaTfa, and TtaTpa using AC as the sacrificial electron donor (SED) (3 mg catalyst, 16 mL 0.1 M AC aqueous solution, 3 μL H2PtCl6 (8 wt%), λ > 420 nm, and 20 °C). (c) Comparison of the photocatalytic HER rates of the above COFs using AC as the SED. Adapted with permission.74 Copyright 2021, Wiley-VCH. | ||
Exfoliation of 2D COFs into nanosheets (NSs) is another promising method to enhance catalytic efficiency as the layer thickness greatly affects the charge separation efficiency of COFs. CYANO-CON NSs were obtained by ball milling a cyano-containing COF (CYANO-COF) under sonication.76 Their atomic force microscopy (AFM) images displayed irregular nanosheet topography with thickness ranging from 4 to 5 nm, corresponding to ∼12–15 COF layers. The CYANO-CON nanosheets showed an impressive apparent quantum efficiency (AQY) of up to 82.6% at 450 nm. Employing a noncovalent functionalization strategy, exfoliation of the bulk crystalline covalent triazine framework (CTF) was greatly facilitated by adding 1-pyrenebutyrate (PB) in water on a large scale.77 PB-modified 2D-TP (2D-TP-PB) was prepared by ball milling homogeneously mixed PB aqueous solution and the bulk CTF. Compared to the as-synthesized CTF, 2D-TP-PB nanosheets demonstrated an optimized band structure, a much higher PHE rate of 24.98 mmol h−1 g−1 and an AQY of up to 27.2% at 420 nm.
Morphological functionalization of COFs has also been utilized to improve their PHE performance. Wei et al. synthesized unsubstituted olefin linked COFs via Knoevenagel condensation and named them g-C18N3-COF.78 It can be organized into microfibrillar structures, facilitating the creation of additive-free, micrometer-thick thin films. The films achieve an AQY of approximately 1.06% at 420 nm, producing H2 at a rate of 14.6 μmol h−1 per 50 mg of COF. The superior photocatalytic performance observed in this thin-film COF can be attributed to several intrinsic advantages of thin-film configurations. COF thin films can efficiently extract and transport charge, reducing electron–hole recombination compared to bulk powders. Their controlled thickness and large surface-to-volume ratio enhance light harvesting and mass transfer, boosting catalytic reaction kinetics. They also offer mechanical strength, easy recovery, and simple device integration.
 | ||
| Fig. 7 (a) Synthesis and structure diagram of COF-Cu3TG, COF-TBTG and JNM-1 (b) The rate of H2 formation of COF-Cu3TG, COF-TBTG and JNM-1 under visible light. (c) Rate of H2 evolution before and after anchoring Ru atoms. (d) XPS spectra for Cu 2p. (e) ELF maps of COF-Cu3TG and COF-Cu3TG-Ru. Reprinted with permission.82 Copyright 2024, Wiley-VCH. | ||
:3) with a weight ratio of 1:3 showed a PHE rate of 11.19 mmol h−1 g−1, which is 5.3, 4.6 and 3.0 times higher than those of TpPa-1-COF, the TiO2/TpPa-1-COF physiosorbed system and their composite without covalent bonds, respectively. A core–shell COF-metal sulfide composite was fabricated by self-polymerization of 1,3,5-benzenetricarboxaldehyde and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine in situ on CdS.84 The T-COF@CdS composites possess well-defined architectures, large contact areas and intimate contact interfaces. T-COF@CdS-3 with a mass ratio of 5:3 (T-COF/CdS) showed the highest PHE rate with an AQY value of 37.8% at 365 nm. Azide-functionalized molecular chloro(pyridine)cobaloxime has been immobilized onto the COF-42 backbone to form a hybrid material, which showed improved and prolonged photocatalytic activity compared to their equivalent physical mixture.85 In this composite, molecular cobaloximes were tethered onto the backbone of propargyl-functionalized COF-42 to replace Pt nanoparticles (NPs) as the cocatalyst for the PHE reaction.
As a similar crystalline material to COFs, MOFs can be easily modified to prepare MOF/COF composites. The Lan group reported a hybrid of NH2-UiO-66 and TpPa-1-COF for PHE.86 NH2-UiO-66 with an exposed –NH2 group was first prepared and added to the reaction system containing Tp and Pa for the synthesis of the NH2-UiO-66/TpPa-1-COF composite. The composite with a weight ratio of 4:6 for NH2-UiO-66:TpPa-1-COF exhibited the best PHE rate of 23.41 mmol h−1 g−1, which is a 20-fold improvement over the pristine TpPa-1-COF. Using a similar strategy, NH2–Ti3C2Tx MXene was synthesized by adding Ti3AlC2, Ti3C2Tx and MXene with exposed –NH2 into a Pyrex tube with 4,6-trihydroxybenzene-1,3,5-tricarbaldehyde (THTA) and benzene-1,4-diamine (BDA).87 The composite ATNT-4 with a mass ratio of 8:4 for NTU–BDA–THTA/NH2–Ti3C2Tx achieved a PHE rate of 14228.1 μmol h−1 g−1, which is about 12.6 times higher than that of the pure NTU–BDA–THTA COF.
Graphitic carbon nitride (g-C3N4) is a type of organic semiconductor with good PHE ability, a simple preparation process, cost-effectiveness, and excellent stability. However, its large band gap limits the utilization of visible light. The hybrid of g-C3N4 and a COF is promising to broaden the light absorption range and facilitate charge separation. A donor–acceptor type COF was used to construct TBTA/g-C3N4 hybrids by in situ condensation of 2,4,6-triformylphloroglucinol (TP) and 4,4′-(benzo-1,2,5-thiadiazole-4,7-diyl)dianiline (BTDA) on g-C3N4.88 The obtained composite showed a PHE rate of 11.73 mmol h−1 g−1 without noble metals as the co-catalyst, which can be further increased to 26.04 mmol h−1 g−1 with Pt as the cocatalyst.
Assembled layers in 2D COFs are not stable under some circumstances for long-term photocatalytic applications in water. The disordered stacking will cause decreased photocatalytic activity. To solve this problem, polyethylene glycol (PEG) was utilized to fill up the mesopore channels of a benzothiadiazole-based COF linked by β-ketoenamine linkages (Fig. 8a).89 This unique procedure prevents the neighboring layers from disordering and retains the columnar π-orbital arrays to facilitate photo-induced charge transport. A significantly improved PHE rate was achieved compared to that of the pristine COF under visible light irradiation (Fig. 8b).
 | ||
| Fig. 8 Illustration of structural transformation of BT-COF and PEG@COF during the deposition of Pt. Reprinted with permission.89 Copyright 2021, Springer Nature. | ||
Heterojunctions were generally constructed through covalent bonding and in situ growth strategies to ensure tight interfacial contact. Li et al. demonstrated an effective approach to constructing Z-scheme heterojunctions by integrating photosensitive metal–organic rings (MAC-FA1) with coral-like S-COF through supramolecular interactions. Compared with covalent binding strategies, this supramolecular approach offers greater synthetic flexibility for combining diverse metal–organic rings with semiconductor materials. In this system, photosensitive MAC-FA1 served as the catalytic site, while S-COF functioned as an efficient light harvesting complex. The optimized 4% MAC-FA1/S-COF heterojunction exhibited remarkable photocatalytic performance with a PHE rate of 100 mmol h−1 g−1 without additional cocatalysts, representing a significant advancement over conventional COF-based composites.90 Luan et al. demonstrated a COF/COF Z-scheme heterojunction by integrating two different COFs through π–π interactions between conjugated aromatic rings. The optimized COF-316/Pt@TpBpy-COF heterojunction with a weight ratio of COF-316:TpBpy-COF = 2:8 exhibited remarkable overall water splitting performance with H2 and O2 evolution rates reaching 220.4 and 110.2 μmol h−1 g−1 respectively under visible light.91 These studies demonstrate that constructing organic heterojunctions through non-covalent interactions offers an effective strategy to achieve efficient charge separation and transfer for photocatalytic applications.
3.2 Photocatalytic CO2 reduction
Carbon dioxide (CO2) is an environmentally harmful greenhouse gas and a potential carbon resource as well. Photo-reduction of CO2 to value-added chemical fuels can not only solve the environmental problem but also provide a new way to develop carbon resources. However, it is very difficult to activate the rigid CO bond in carbon dioxide as the reduction of CO2 involves multiple electrons, which is more complex than water splitting. COFs combine tunable energy diagrams, light-harvesting capability, and catalytic activity at the same time and therefore can act as great candidates for photocatalytic CO2 reduction.96Table 2 summarizes the photocatalytic CO2 reduction performance of different COF-based photocatalysts.
| Samples | Irradiation conditions | CO2 reduction products (μmol h−1 g−1) | Selectivity | Ref. |
|---|---|---|---|---|
| N3-COF | 800 nm ≥ λ ≥ 420 nm | CH3OH (0.57) | — | 97 |
| Re-f-COF | λ > 420 nm | CO (787.5) | — | 98 |
| TpBb-COF | λ ≥ 420 nm | CO (89.9) | — | 99 |
| QL-COF | AM 1.5 | CO (156) | 99.3% | 100 |
| TAPBB-COF | 1000 nm ≥ λ ≥ 200 nm | CO (24.6) | 95.6% | 101 |
| HCOF-2 | λ > 420 nm | CO/CH4 (30.9/9.6) | — | 102 |
| COF-367-CoIII | λ > 380 nm | HCOOH (93) | 97.1% | 103 |
| JUC-640-Co | λ > 380 nm | CO (15100) |
94.4% | 104 |
| Cu4COF-2 | Full spectrum | CO (23.8) | 94.3% | 105 |
| EPCo-COF-AT | λ ≥ 420 nm | CO (17700) |
97.8% | 106 |
| Re-Bpy-sp2c-COF | λ > 420 nm | CO (1040) | 81% | 107 |
| Ni-TpBpy | λ ≥ 420 nm | CO (811.4) | 96% | 108 |
| Fe SAS/Tr-COF | λ > 420 nm | CO (980.3) | 96.4% | 109 |
| DQTP COF-Co | λ ≥ 420 nm | CO (1020) | — | 110 |
| Co-COF | λ ≥ 420 nm | CO (18000) |
95.7% | 111 |
| H-COF-Ni | λ ≥ 420 nm | CO (2847) | 96% | 112 |
| Ni-TP-CON | λ ≥ 420 nm | CO (4360) | 95% | 113 |
| Co/Cu3-TPA-COF | λ ≥ 420 nm | CO (25247.7) |
80.2 | 114 |
| CoNi-COF-3 | λ ≥ 420 nm | CO (2567) | 92.2% | 115 |
| Ru/TpPa-1 | 800 nm ≥ λ ≥ 420 nm | HCOOH (108.8) | — | 116 |
| Ru@TpBpy | 800 nm ≥ λ ≥ 420 nm | HCOOH (172) | — | 117 |
| Cox-COF | λ ≥ 420 nm | CO (4232) | — | 118 |
| TCOF-MnMo6 | 800 nm ≥ λ ≥ 400 nm | CO (37.25) | 100% | 119 |
| BBO-COFBPY-Co | Full spectrum | CO (10552.15) |
91% | 120 |
| COF-318-TiO2 | 800 nm ≥ λ ≥ 380 nm | CO (69.97) | — | 121 |
| TiO2-INA@CuP-Ph COF | AM 1.5 | CO (50.5) | — | 122 |
| NCTS | Full spectrum | CO (7.51) | 100% | 123 |
| T-101/COF | Full spectrum | CO (11.6) | 95% | 124 |
| CdS@COF | λ ≥ 420 nm | CO (507) | 72% | 125 |
| CdS/TpBpy | Full spectrum | CO (5028) | 85% | 126 |
| g-C3N4 (NH)/COF | λ > 400 nm | CO (562.5) | — | 127 |
| TpPa/ZIF-8 | LED | CO (84.87) | > 90% | 129 |
| TMBen-perylene | λ > 420 nm | CO (93) | 96% | 130 |
| NKCOF-113 | λ > 420 nm | HCOOH (360) | — | 131 |
| GO-COF-366-Co | λ > 320 nm | Formate (19750) |
94.4% | 132 |
| CO (6525) | 96.1% | |||
| COF@TI | 800 nm ≥ λ ≥ 400 nm | CO/H2 (470/237) | — | 133 |
The highly flexible structure of COFs makes it easy to decorate functional groups to tune their properties and thus improve photocatalytic performance. COF-366 exhibited high carrier mobility and efficient visible light absorption,134 while its photocatalytic CO production rate was only 8.5 μmol h−1 g−1 in a gas–solid phase reaction under full-spectrum light irradiation.101 Su et al. found that the oxidation reaction was hindered by the negligible difference (0.04 V) between the valence band of COF-366 (VB; +0.86 V) and the redox potential of O2/H2O (+0.82 V).101 A halogenation strategy was proposed to functionalize COF-366 with bromine groups to synthesize a novel COF, namely TAPBB-COF. With a more positive VB value (+1.10 V) than that of COF-366, TAPBB-COF facilitated the oxidation half reaction more effectively. Consequently, TAPBB-COF produced CO at a rate of 24.6 μmol h−1 g−1, which was approximately three-fold higher than that of COF-366 under identical conditions. Jiang et al. prepared three distinct COFs (HCOF-1, HCOF-2 and HCOF-3) by using hydrazine hydrate (HZ) with three different aldehyde precursors, 1,3,5-triformylphenol, 1,3,5-triformylresorcinol, and 1,3,5-triformylphloroglucinol.102 Due to different proportions of hydroxyl groups in the skeleton, HCOF-1 did not exhibit keto–enol tautomerism, while HCOF-2 showed reversible keto–enol tautomerism and HCOF-3 displayed irreversible keto–enol tautomerization. Based on the experimental and density functional theory (DFT) calculation results, reversible keto–enol tautomerism led to an extended light-harvesting range and enhanced charge separation and transportation. Consequently, HCOF-2 showed the highest photocatalytic performance, producing CO and CH4 with a rate of 30.9 and 9.6 μmol h−1 g−1, respectively.
Porphyrin-based COFs with 18 electron π-conjugated macrocycles are promising in photocatalysis due to their efficient visible light absorption and easy incorporation of various metal ions as active sites. Jiang et al. devised a simple strategy to manipulate the cobalt spin state in COF-367-Co by altering the oxidation state of Co in the porphyrin center.103 DFT calculation and experimental results suggest that CoII and CoIII with spin ground states of S = 1/2 and 0 were incorporated into COF-367. Photocatalytic CO2 reduction results reveal that COF-367-CoIII exhibited better activity and remarkably improved selectivity to HCOOH compared with COF-367-CoII. Recently, Fang et al. synthesized a series of 3D stp-topologized porphyrin COFs (JUC-640-M, M = H, Co, or Ni) by polymerizing the 6-connected triptycene-based building unit, 2,3,6,7,14,15-hexa(4′-formylphenyl)triptycene (HFPTP, D3h-symmetrized), with 4-connected 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP-H, D4h-symmetrized) or its metallized derivatives (TAPP-Co and TAPP-Ni), as shown in Fig. 9a.104 Extraordinarily, JUC-640-H has the lowest crystal density (0.106 cm3 g−1) ever reported for any crystalline material and the largest interconnected channels (4.6 nm) among 3D COFs. These features, along with a high surface area (2204 m2 g−1), large and interpenetrated channels and abundant exposed porphyrin moieties (0.845 mmol g−1), made these 3D COFs efficient photocatalysts for CO2 reduction under visible light irradiation. The photocatalytic CO production rate of JUC-640-Co reached as high as 15.1 mmol h−1 g−1, with a high selectivity (94.4%) and stability (Fig. 9b and c). Similarly, the metal phthalocyanines with 18π aromatic macrocyclic structures represent ideal building blocks for COFs, which exhibit exceptional photosensitivity and abundant single-atom sites. Xu et al. prepared highly stable and reactive copper cluster-based COFs (Cu4COF-1 and Cu4COF-2) via a stepwise assembly approach involving epitaxially amino-modified Cu4 clusters (Cu4–NH2), as shown in Fig. 9d–f.105 These Cu4COFs exhibited enhanced stability, narrower band gaps, larger specific surface areas, and superior charge transfer capabilities compared to isolated Cu4 clusters, leading to significantly improved photocatalytic CO2 reduction performance under visible light. Specifically, Cu4COF-2 achieved a CO production rate of 23.8 μmol g−1 h−1 with an impressive selectivity of 94.3% (Fig. 9g). Wang et al. synthesized a novel class of COFs, designated as EPM-COF (M = Co, Ni, Cu), by integrating perfluorinated metallophthalocyanine with ellagic acid. Notably, following alkaline treatment, EPCo-COF demonstrated remarkable photoactivity in CO generation with a production rate of 17.7 mmol h−1 g−1.106
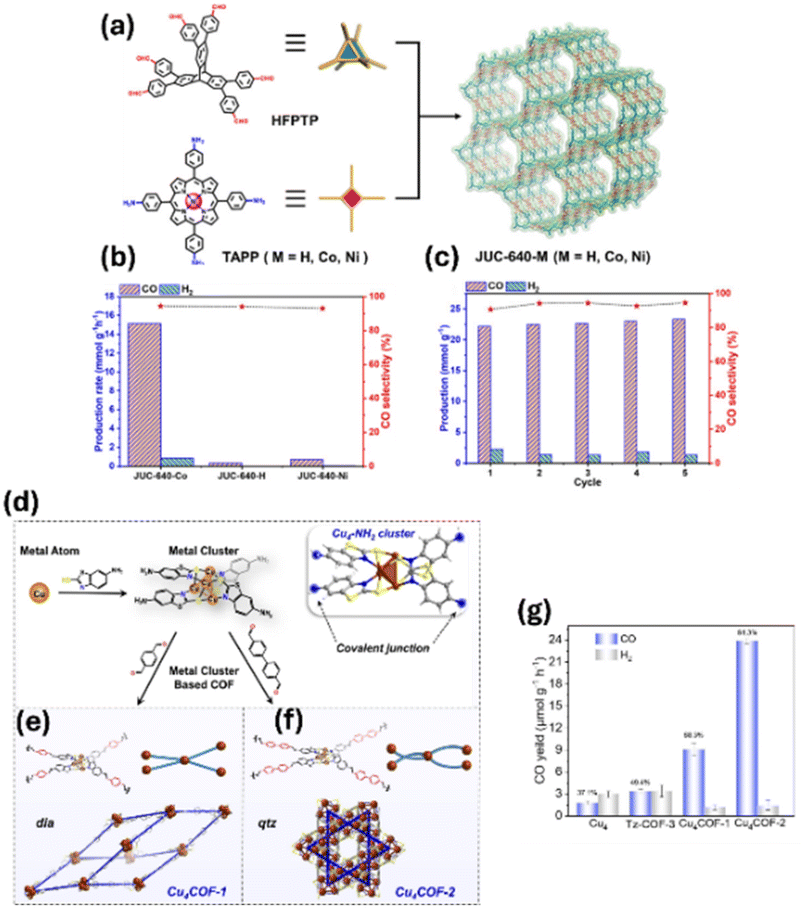 | ||
| Fig. 9 (a) Schematic illustration of the construction of JUC-640-M (M = Co, Ni, or H). (b) Comparison of the photocatalytic activity of JUC-640-M. (c) Stability tests of JUC-640-Co (2 h each cycle). Reprinted with permission.104 Copyright 2023, American Chemical Society. (d) The illustration of step-by-step precise assembly procedures of Cu4 cluster-based COFs; inset: single crystal structure of Cu4. The structural representation of (e) Cu4COF-1 with dia topology and (f) Cu4COF-2 with qtz topology. (g) Photocatalytic CO2-to-CO performance and selectivity of Cu4, Tz-COF-3, Cu4COF-1 and Cu4COF-2. Reprinted with permission. Copyright 2025, Wiley-VCH. | ||
COFs of different morphologies with certain functions have also been prepared to further improve the photocatalytic CO2 reduction performance. Cooper et al. developed fibrous COFs, which exhibit a very high CO production rate of 1.04 mmol h−1 g−1 and a selectivity of 81% during light exposure.107 This excellent performance was primarily ascribed to the unique fiber morphology that can increase the effective light absorption through light scattering, improve the penetration depth, and enhance the photon utilization efficiency. Furthermore, the continuous fiber structure facilitates the efficient transport of photogenerated electrons and holes along a one-dimensional conductive path, greatly hindering the electron–hole recombination. Finally, its high aspect ratio provides a large number of accessible surfaces and facilitates mass transfer, significantly enhancing photocatalytic kinetics.107
 | ||
| Fig. 10 (a) Synthetic of M SAS/Tr-COF. (b) CO generation rate and (c) calculated CO selectivity over Tr-COFs, 0.6 wt% Fe SAS/Tr-COFs, 1.1 wt% Fe SAS/Tr-COFs, and 4.8 wt% Fe SAS/Tr-COFs in 4 h under visible light irradiation. (d) DFT-calculated Gibbs free energy (ΔG, eV) profiles for CO2 photoreduction on Fe SAS/Tr-COFs and Tr-COFs with the corresponding geometry structures and differential charge densities of intermediates over Fe SAS/Tr-COFs. Adapted with permission.109 Copyright 2022, American Chemical Society. | ||
Single metal atoms have also been successfully immobilized onto COFs by metal–O or metal–O/N binding. By exploiting the high affinity of the anthraquinone group toward metal atoms, Lan et al. prepared transition metal modified DQTP (2,6-diaminoanthraquinone-2,4,6-triformylphloroglucinol) COFs (DQTP COF-M, M = Co, Ni, Zn).110 DQTP COF-Co showed an impressive CO production rate of 1.02 × 103 μmol h−1 g−1, whereas DQTP COF-Zn exhibited a high preference for the formation of HCOOH (152.5 μmol h−1 g−1). Wang et al. synthesized two types of COFs with oxygen coordinated Co atoms by using 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT) as a fixed module and varying aldehyde molecules, referred to as Co-2,3-DHTA-COF and Co-TP-COF, containing Co-O4 and Co-O3N sites, respectively (Fig. 11a).111 Based on the experimental and DFT calculation results, the unique Co-O4 coordination environment in Co-2,3-DHTA-COF increased the Co(II) loading amount in the COF, facilitated the charge transfer from the photosensitizer to the catalyst, improved the CO2 adsorption capacity, and lowered the energy barrier of the rate-determining step. Co-2,3-DHTA-COF thus exhibited an outstanding photocatalytic CO production rate of 18000 μmol h−1 g−1 and reached a high selectivity of 95.7% under visible light conditions (Fig. 11b). Moreover, Ni ions were anchored into H-COF by chelating coordination with an N-acylhydrazone linkage.112 Single Cu-O/N sites were formed on the DHTA–TTA 2D COF by binding Cu with the imine and methoxy groups.139 In another study, Ni active sites were immobilized in TP-CON via bis-chelating coordination.113 All the above metallized COFs exhibited excellent photocatalytic CO2 reduction activity. Single cobalt sites have been recently incorporated into the interlayer of COFs (Cu3-TPA-COF, Cu3-TAPB-COF, and Cu3-TAPT-COF) by coordination via imine-N motifs. The Co-imine N moieties were integrated into donor1–acceptor–donor2 architectures to give rise to the formation of a well-separated electron–hole state. This unique configuration efficaciously induces vectorial electron transfer from dual electron-donating domains to the cobalt centers, thereby facilitating CO2 activation and subsequent reduction.114
 | ||
| Fig. 11 (a) Synthesis of 2,3-DHTA-COF and Co-2,3-DHTA-COF. (b) CO and H2 evolution with irradiation time for the CO2RR catalyzed by Co-2,3-DHTA-COF under visible light irradiation. Adapted with permission.111 Copyright 2023, Springer Nature. (c) Schematic of uniformly dispersed POM clusters in the COF by confining them into the pores of the COF through covalent linkages. (d) Time-dependent CO2-to-CO performance for TCOF-MnMo6 and ECOF-MnMo6. Reproduced with permission.119 Copyright 2022, American Chemical Society. | ||
The incorporation of bimetallic sites into COFs to promote photocatalytic CO2 reduction activity by synergistic effects has attracted a lot of attention. Lin et al. introduced a three-step synthesis of FeNi bimetallic COFs. Pure COFs were fabricated from 2,4,6-triformylphloroglucinol and 2,5-diaminobenzenesulfonic acid and NH4-COFs were subsequently obtained by an ammoniating process.140 Metal ions (Fe3+, Ni2+, and Co2+) were attached onto COFs by a cation-exchange method through strong interactions with the abundant –SO3− units. By varying the ratio of Fe/Ni sites on the COFs, the photoreduction of low content CO2 to syngas with a wide range of tunability was realized. In these materials, the Fe and Ni sites play distinct roles in the catalytic process: the Fe sites facilitate H2 production by strongly binding H2O, while the Ni sites favor CO production by preferentially adsorbing CO2. Recently, Lan et al. modified three kinds of benzothiadiazole-based COFs with Co and Ni dual-metal sites by metal–thiadiazole interaction (CoNi-COF-n, n = 0, 1, 2, and 3).115 Benefiting from the synergistic effect of the fully β-ketoenamine-tautomerized COF-3 configuration and dual-metal sites, CoNi-COF-3 showed a high CO production rate of 2567 μmol h−1 g−1 with a selectivity of 92.2%. Besides, metal NPs were decorated onto the COF matrix to accelerate CO2 photoreduction. Fan et al. fixed Ru NPs onto a ketoamine-based COF (Ru/TpPa-1) and achieved a photocatalytic formic acid generation rate of 108.8 μmol h−1 g−1 with a Ru loading of 3.0 wt%.116 The presence of Ru NPs promoted visible light harvesting, improved the charge transfer, and inhibited charge recombination. Ru NPs were also loaded onto a bipyridine-based COF (Ru@TpBpy), which produced HCOOH with a rate of 172 μmol h−1 g−1 under a low Ru loading (0.7 wt%).117 Furthermore, Bi et al. immobilized Co quantum dots (QDs) onto COF-318, realizing a photocatalytic CO generation rate of 4232 μmol h−1 g−1 and a H2 evolution rate of 6611 μmol h−1 g−1.118 Here, Co QDs contribute to the enhanced activity by not only serving as an electron trap to promote charge separation but also boosting the adsorption and activation of CO2 molecules. Moreover, single POM clusters were restricted within the nanopores of COFs. Lan et al. synthesized a subvalent COF, specifically TCOF, via a [4 + 3] Schiff-base condensation of 2,3,6,7-tetrakis(4-formylphenyl)tetrathiafulvalene (TTF-4CHO) and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT) under solvothermal conditions (Fig. 11c).119 TCOF and POM were then covalently bonded to form TCOF-MnMo6 by the Schiff-base reaction between the uncondensed aldehyde functional groups in the pores of TCOF and amine-functionalized POM (MnMo6-2NH2). By integrating the merits of both components, TCOF-MnMo6 demonstrated high performance in yielding CO with a rate of 37.25 μmol h−1 g−1 and a selectivity of approximately 100% (Fig. 11d).
Zhang et al. reported three benzoxazole-based COFs with distinct cobalt coordination geometries (Co–N–O2, Co–N–O3, and Co–N2–O2). Through post-synthetic modification, precise control over the catalytic microenvironment was achieved, where Co was anchored by coordination with imine–nitrogen atoms, imine–nitrogen and adjacent hydroxyl groups, and bipyridine units, respectively. The optimized BBO-COFBPY-Co catalyst demonstrated remarkable performance with a CO production rate of 10.55 mmol h−1 g−1 and a selectivity of 91%, surpassing many previous COF-based systems. Mechanism studies revealed that rational engineering of the Co coordination environment could optimize the local electronic structure of BBO-COFs, promoting efficient exciton dissociation and charge carrier migration while suppressing electron–hole recombination. This work not only achieved high catalytic activity but also provided insights into the influence of metal coordination environments on the photocatalytic CO2 reduction process, complementing other strategies in metal-modified COF design.120
:COF-TiO2, Bi2WO6, and α-Fe2O3), as shown in Fig. 12, by utilizing the interaction of abundant hydroxyl functional groups on the surface of oxide-semiconductors with strong electron-withdrawing substituents on TFPN/TFPC building blocks.121 Among these prepared COF-SC, COF-318-TiO2 demonstrated the maximal CO production rate of 69.67 μmol g−1. In situ XPS spectra were utilized to confirm the Z-scheme charge transfer pathway. The positive shift in Ti 2p binding energy under UV irradiation indicated a decrease in electron density at Ti sites (Fig. 12c), confirming that photogenerated electrons migrated from TiO2 to COF-318. As illustrated in Fig. 12d, electron transfer from TiO2 to COF-318 via covalent bonds results in electron accumulation at the cyano/pyridine units of COF-318 for CO2 reduction, while holes remain in TiO2 to facilitate H2O oxidation. The unsatisfactory intramolecular electron transfer in imine-linked COFs limits its photocatalytic performance. Wang et al. reported a CdS@COF core–shell photocatalyst, where the imine-linked COF was constructed from Zn-porphyrin and Co-bipyridyl units.125 They found that CdS acted as a channel for photo-induced electron transfer from Zn-porphyrin to Co-bipyridyl units to avoid an inefficient pathway via a highly polarized imine linkage. CdS@COF showed an obviously enhanced CO production rate of ∼507 μmol h−1 g−1 compared with pure CdS and the pristine COF. Ye et al. synthesized a van der Waals heterojunction of defective g-C3N4 (NH)/COF with S-scheme charge transfer, exhibiting a CO evolution rate of 11.25 μmol h−1, which is 45 and 15 times higher than that of g-C3N4 and g-C3N4/COF, respectively.128 Su et al. constructed COF/MOF hybrids (TpPa/ZIF-8) by simply grinding a blend of TpPa-1, Zn(OAc)2·2H2O and 2-IM.129 In the absence of photosensitizers and sacrificial agents, the optimized TpPa/ZIF-8 photocatalyst exhibited a CO evolution rate of 84.87 μmol h−1 g−1 when CO2 concentration is only 10%.
 | ||
| Fig. 12 (a) Schematic illustration of the preparation of COF-318-SCs. (b) Photocatalytic performance of various COF-318-SCs as well as bulk COF-318, TiO2, Bi2WO6, and α-Fe2O3. (c) High-resolution in situ XPS for Ti 2p of COF-318-TiO2 in the dark and under 365 nm LED irradiation; (d) schematic illustration of the charge-transfer process under light irradiation with the Z-scheme model. Adapted with permission.121 Copyright 2020, Wiley-VCH. | ||
In addition to traditional semiconductors, dyes and enzymes are also combined with COFs for efficient CO2 photoreduction. Zhou et al. separately combined three polycyclic aromatic hydrocarbons (anthracene, pyrene and perylene) with olefin-linked COFs (TMBen) through the aldol condensation reaction.130 TMBen–perylene establishes a type II band alignment, which significantly restrained charge recombination by accumulation of electrons and holes on TMBen and perylene domains, respectively (Fig. 13a). Consequently, TMBen–perylene showed an 8 times improvement over pristine TMBen in the photocatalytic reduction of CO2 to CO (Fig. 13b). Chen et al. reported the immobilization of formate dehydrogenase in an olefin-linked COF (NKCOF-113) to build a novel photoenzymatic system for photocatalytic HCOOH production.131 In this system, NKCOF-113 served as a photosensitizer to provide photogenerated electrons to the Rh moieties, which react with NAD+ to form a nicotinamide cofactor (NADH). Formate dehydrogenase converted CO2 to formic acid by employing NADH and NAD+ was regenerated simultaneously to participate in the next photocatalysis cycle.
 | ||
| Fig. 13 (a) Schematic illustration of the PAH-functionalized TMBen for photocatalytic CO2 reduction. (b) A comparison of the amount of product obtained with TMBen and TMBen–perylene after 5 hours of reaction. Reproduced with permission.130 Copyright 2022, Wiley-VCH. | ||
Besides dyes and enzymes, graphene-based materials and topological quantum materials have emerged as promising components for COF-based composites. Gong et al. developed a covalently anchored COF-GO composite (GO-COF-366-Co) that exhibited interesting solvent-dependent selectivity for CO2 reduction. In acetonitrile (CH3CN), the composite showed a high selectivity of 94.4% for formate production with a yield of 1.975 mmol h−1 g−1, while switching to an CH3CN/H2O mixture (4:1) led to preferential CO production with 96.1% selectivity and a CO yield of 6.525 mmol h−1 g−1. Photoelectrochemical studies and in situ FTIR revealed that the covalent integration created efficient charge-transfer bridges and stabilized key reaction intermediates through hydrogen bonding interactions, which facilitated efficient charge separation and transfer.132
Very recently Dey et al. demonstrated a novel approach by integrating a 2D COF with a strong topological insulator (TI), PbBi2Te4, to form a unique COF-topological quantum material nano-heterostructure. The robust metallic surface of TI served as an electron reservoir to minimize electron–hole recombination, while the presence of 6s2 lone pairs in Pb2+ and Bi3+ facilitated CO2 binding. This synergistic design enabled tunable syngas production with controllable CO:H2 ratios through adjustment of the acetonitrile/water ratio.133
3.3 Photocatalytic organic transformation
Light-induced organic transformations are regarded as environmentally friendly and sustainable alternatives for the synthesis of fine chemicals. Organic transformation reactions employing traditional catalysts such as metal complexes usually need harsh conditions, and they suffer from poor stability. COFs as photocatalysts for light-induced organic transformations featuring high stability, recyclability and adjustable band structures have been widely utilized in many reactions. Here, we mainly introduce their applications in aerobic oxidation, reductive dehalogenation functionalization and trans–cis transition.C linkage, Por-sp2c-COF possessed a permanent porosity and exhibited high chemical stability in various organic solvents and concentrated HCl or NaOH. The prepared Por-sp2c-COF showed higher photocatalytic activity and better reusability than imine-linked Por-COF for aerobic oxidation of amines to imines under visible light irradiation. Yang et al. reported a stable hydrazone-bridged COF (TFPT-BMTH) with abundant 2-methoxyethoxy groups in the pores.142 Thanks to the introduction of 2-methoxyethoxy groups that enhance the interlayer interactions and increase the hydrophilic ether content, the TFPT-BMTH COF has several advantages, such as excellent stability, reusability, and hydrophilicity, for the application of photocatalytic oxidation of benzylamine in water. Cao et al. constructed three Tp-BTD COF isomers with different stacking modes (AA, AB, and ABC stacking) by reacting 4,7-bis(4-aminophenyl)-2,1,3-benzothiadiazole (BTD) and 2,4,6-triformylphloroglucinol (Tp) at different temperatures and solvent media.143 They found that different layer stacking led to different ratios of enol and keto forms in these three isomers, which can regulate the photocatalytic production efficiency of Type I reactive oxygen species (superoxide radicals, hydroxyl radicals, and hydrogen peroxide) and Type II reactive oxygen species (singlet oxygen). The Tp-BTD COF with ABC stacking mode exhibited the fastest generation rate of Type I reactive oxygen species, thereby achieving a three-fold higher photocatalytic oxidation efficiency of benzylamine than that of AA stacking and AB stacking isomers. A very recent study by Liu and co-workers developed a new donor–acceptor COF with a wide absorption range from 200 to 900 nm exhibiting a photocatalytic conversion of 99% and a selectivity of 98% in 20 min for selective coupling of benzylamine.144 Furthermore, by synthesizing a series of donor–acceptor COFs, named JUC-675 to JUC-677, Fang et al. realized a photocatalytic system capable of simultaneous production of hydrogen peroxide (H2O2) and value-added organic chemical N-benzylbenzdiamine (BBAD), maximizing the use of solar energy (Fig. 14). Among them, JUC-675 showed a high selectivity of 99.9% and a yield of 96% for BBAD in oxidative coupling of benzylamine.145
 | ||
| Fig. 14 Reaction mechanism of the photocatalytic H2O2 production and BBAD co-production system. Reproduced with permission.145 Copyright 2024, Wiley-VCH. | ||
Aerobic oxidation of sulfides is feasible by using reactive oxygen species generated by photoexcited COFs. Chen et al. synthesized an A2B2-Por-COF by a simple acetic acid catalyzed self-condensation reaction.146 Benefiting from the outstanding light-harvesting capability and favorable photoelectric features of porphyrin, the A2B2-Por-COF can effectively oxidize thioanisoles and several methyl phenyl sulfides bearing different substituents with a selectivity of >99% under visible light irradiation. Bai et al. fabricated a noodle-like nanofiber AQ-COF with AB stacking mode, which is different from the previously reported spherical AQ-COFDMF with AA stacking mode.147 The novel micromorphology and stacking module of AQ-COF improved its photocatalytic performance for aerobic oxidation of sulfides to sulfoxides by accelerating electron–hole separation and transfer. Recently, Li et al. developed a benzothiadiazole-based COF (TpBTD-COF) via solvothermal synthesis using 4,7-bis(4-aminophenyl)-2,1,3-benzothiadiazole (BTD) and 2,4,6-triformylphloroglucinol (Tp). Integrating an electron transfer mediator, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), significantly improved the photocatalytic performance for aerobic sulfoxidation. TEMPO facilitated smoother electron transfer and dramatically increased conversions, exhibiting over 2.5 times higher activity compared to systems without the mediator. Moreover, TpBTD-COF exhibited exceptional robustness, sustaining multiple reaction cycles with enhanced photocatalytic activity over time.148 Similarly, Huang et al. reported a benzotrithiophene (BTT)-based sp2 carbon-conjugated COF (BTT-sp2c-COF) that enabled selective oxidation of organic sulfides with the incorporation of TEMPO. TEMPO efficiently mediated hole transfer from BTT-sp2c-COF to sulfide substrates, facilitating oxygen incorporation into sulfoxides through electron transfer.149 Dong et al. also synthesized a pyrene-based azine-linked COF (Py-azine-COF) through aldimine condensation between 1,3,6,8-tetrakis(4-formylphenyl)pyrene and hydrazine hydrate. After incorporating TEMPO as a hole mediator, Py-azine-COF exhibited rapid and highly selective aerobic oxidation of various organic sulfides. Mechanistic studies revealed that TEMPO promoted charge separation and cooperated with superoxide radicals formed by oxygen reduction, thereby enhancing overall catalytic efficiency.150
COFs are also widely employed in photocatalytic oxidative hydroxylation reactions. In a first report on benzoxazole-based COFs as photocatalysts, reversible/irreversible cascade reactions were employed to synthesize three benzoxazole-based COFs (namely LZU-190, LZU-191, and LZU-192).151 The obtained COFs demonstrated impressive acid resistivity, alkali resistivity and photostability and an extended light absorption range resulting from their rigid benzoxazole linkage. Among these COFs, LZU-190 showed admirable photoactivity and reusability, which maintained a phenol yield of 99% for more than 20 cycles in photocatalytic oxidative hydroxylation of arylboronic acids tests. Taking advantage of the excellent stability of COFs with a vinylene linkage, Zhang et al. synthesized three vinylene-bridged COFs (labeled as COF-p-3Ph, COF-p-2Ph, and COF-m-3Ph) by secondary-amine-catalyzed Knoevenagel condensation.152 At low loadings, these COFs can effectively and stably oxidize arylboronic acids to phenols driven by visible light. Jiang et al. introduced a method to obtain different reactive oxygen species by regulating the excitonic effects in the COFs.153 In this work, a series of porphyrinic COFs (DhaTph-M, M = 2H, Zn, Ni) were constructed through Schiff-base condensation. The introduction of metal ions into the porphyrin center was found to disrupt the coexistence of excitons and charge carriers under illumination (Fig. 15a). The presence of Zn2+ in the porphyrin center increased the transformation of singlet excitons to triplet excitons and promoted the energy transfer process, whereas Ni2+ in the porphyrin center facilitated the dissociation of excitons into free carriers and enhanced charge transfer processes. The photocurrent measurements and electrochemical impedance spectroscopy (EIS) results (Fig. 15b and c) show that DhaTph-Ni exhibited the strongest photocurrent response and the lowest charge-transfer resistance among the three COFs, which can be attributed to increased free charge carriers resulting from enhanced exciton dissociation. Conversely, the pronounced electron–hole interaction in DhaTph-Zn resulted in a strong excitonic effect, substantially decreasing free charge carrier generation. Due to the distinct excitonic behavior, DhaTph-Zn and DhaTph-Ni activated oxygen (O2) to form singlet oxygen (1O2) and superoxide radicals (O2˙−), under visible light excitation. Hence, DhaTph-Zn achieved outstanding performance in selective oxidation of organic sulfides, while DhaTph-Ni realized high photoactivity in hydroxylation of boronic acid.
 | ||
| Fig. 15 (a) Schematic displaying DhaTph-M COFs (M = 2H, Zn and Ni) with discriminative oxygen active species selectivity to 1O2 and O2˙− for oxidation of thioanisole and hydroxylation of phenylboronic acid. (b) Photocurrent responses and (c) EIS Nyquist plots for DhaTph-M (M = 2H, Zn, and Ni). Reproduced with permission.153 Copyright 2020, American Chemical Society. | ||
In addition to aerobic oxidation reactions, COFs can also serve as photocatalysts for reductive dehalogenation reactions. Liu et al. prepared a donor–acceptor COF-JLU22 by condensing 1,3,6,8-tetrakis(4-aminophenyl)pyrene and 4,4′-(benzothiadiazole-4,7-diyl)dibenzaldehyde.154 COF-JLU22 exhibited high activity and recyclability as a heterogeneous photocatalyst for reductive dehalogenation of phenacyl bromide derivatives, which were derived from the large BET specific area, good crystallinity and strong light-harvesting ability. Using a series of [3 + 3] 2D COFs with hexagonal structures but distinct compositions as the model material, Yang et al. explored the crucial factors that influence the photocatalytic performance of COFs for the reductive dehalogenation reaction.155 Among them, OH–TFP–TTA showed the best performance for the photocatalytic reductive dehalogenation reaction. The introduction of –OH groups extended the light absorption range of OH–TFP–TTA. The replacement of TAPB units with triazine units introduced donor–acceptor domains that facilitate charge separation. Considering that the construction of a D–A structure in COFs can accelerate the separation of photogenerated charges, Baeyens et al. designed and synthesized three donor–acceptor COFs, BTTZ-por COF, FBQD-por COF and PTBC-por COF, by condensing three electron-deficient aldehydes with electron-donating porphyrin units.156 The PTBC-Por COF showed the best activity in reductive dehalogenation of 2-bromoacetophenone derivatives due to longer carrier lifetimes, better charge separation and lower charge transfer resistance.
Photocatalytic functionalization of organic molecules is an important approach to acquiring various derivatives with diverse properties. By employing three distinct linkers, Liu et al. synthesized three isomorphic pyrene-based COFs to tune their optoelectronic characteristics and band structures.157 They found that visible light absorption and photogenerated charge transfer processes of COFs were improved by the introduction of 4,4′-([1,2,5]thiadiazolo[3,4-c]pyridine-4,7-diyl)-based electron-deficient units. Among the three isomorphic COFs, COF-JLU24 exhibited the highest photocatalytic activity for C-3 functionalization of indoles as well as broad substrate scope and outstanding recyclability as a metal-free photocatalyst. Cai et al. constructed a chemically stable olefin-linked 2D-COF (referred to as TTO-COF) by acid-catalyzed aldol condensation of 2,4,6-tris(4-formyl-phenyl)-1,3,5-triazine and 2,4,6-trimethyl-1,3,5-triazine.158 Owing to high charge carrier conduction efficiency, TTO-COF outperformed imine COFs and g-C3N4 in photocatalytic C–H functionalization of arenes and heteroarenes and showed better stability and recyclability.
Achieving a green and economical trans–cis (E–Z) conversion of alkenes/olefins is of great significance for the synthesis of anticancer drugs, dyes, and scintillators. Banerjee et al. reported visible-light-driven E–Z transformation of olefins by a COF.159 The COF (TpTt) was prepared by reacting melamine/1,3,5-triazine-2,4,6-triamine (Tt) and 2,4,6-triformylphloroglucinol (Tp) aldehyde. In these COFs (TpTt), the triazine core forms strong π–π interactions with the E alkenes to catalyze E–Z photoisomerization and keto functionalities can prolong the lifetime of the excited triplet state. Moreover, the β-ketoenamine linkage gives TpTt high chemical stability. Benefiting from these features, TpTt can effectively and stably convert trans-stilbene to cis-stilbene under blue light irradiation.
 | ||
| Fig. 16 (a) The synthesis process of Ace-COF-Ni. (b) Proposed mechanism of an Ace-COF-Ni catalyzed sulfur–carbon cross-coupling reaction. Adapted with permission.160 Copyright 2021, Wiley-VCH. | ||
In addition, a Pt complex was introduced into COFs for facilitating light driven organic transformations. Alemán et al. used a cis-[PtCl2(DMSO)2] precursor to synthesize a Pt(II) hydroxyquinoline complex, which was then covalently bonded with an imine-based COF.162 The resultant Pt@COF not only brought about high activity for oxidizing sulfides to sulfoxides with a TON of up to 25000, but also exhibited a turnover number (TON) of 7500 for photohydrodebromination of bromo-derivatives. Beyzavi et al. employed 2-(4-formylphenyl)-5-formylpyridine as a COF linker to coordinate with metals for the first time.163 The COF-UARK-49-Pt photocatalyst with 9.1% Pt loading was obtained by reacting cis-[PtCl2(DMSO)2] and COF-UARK-49 in toluene at 50 °C overnight, which exhibited boosted photocatalytic activity of decarboxylative difluoroalkylation and oxidative cyclization reactions compared to pristine COF-UARK-49.
A dual metalized 2D-COF TpBpy for light driven C-N cross-coupling reactions was introduced by Maji et al. A photosensitizing iridium complex and nickel were fixed into the COF pore by chelating with bipyridine sites of the TpBpy COF.164 The Ni-Ir@TpBpy photocatalyst prevented the formation of nickel-black due to strong metal binding sites, demonstrating superior catalytic performance, durability, and versatility. The authors then employed Ni-Ir@TpBpy-catalyzed C–N coupling reactions to enable the functionalization of amide derivatives and the synthesis of some commercially available drug molecules. Duan et al. recently used a bimetallic COF for the oxidative coupling of amines to improve charge separation and catalytic activity. The bimetallic COF-Sr2Fe1 was constructed by coordinating Sr2+ and Fe2+ ions into the porphyrin centers of COF-366. COF-Sr2Fe1 achieved a photocatalytic yield of 97% for the oxidative coupling of benzylamine to N-benzylbenzaldimine under visible light. This performance significantly surpassed that of monometallic COFs (e.g., COF-Sr at 79% and COF-Fe at 6%). DFT calculations showed that Sr2+ enhanced C–N coupling ability, while Fe2+ facilitated dehydrogenation. Photogenerated electrons migrate from Fe2+ to Sr2+, enhancing charge separation and reaction efficiency (Fig. 17a).165 Furthermore, Jiang et al. expanded the topology of 3D COFs by integrating POSS-based frameworks with porphyrin building blocks. The team synthesized four distinct POSS-linked 3D COFs: POSS-MTFPP-COFs-scu (Co, Zn) and POSS-MTFPP-COFs-sqc (Ni, H2). Scu topology exhibited triangular and square channels, while sqc featured interpenetrated frameworks. High surface areas were achieved (up to 1726 m2 g−1 for POSS-ZnTFPP-COF-scu). The frameworks were tested for photocatalytic production of artemisinin, an antimalarial compound. POSS-H2TFPP-COF-sqc achieved the highest yield (63%) by generating singlet oxygen (1O2) effectively, which catalyzed the oxidation of dihydroartemisinic acid (Fig. 17b).166
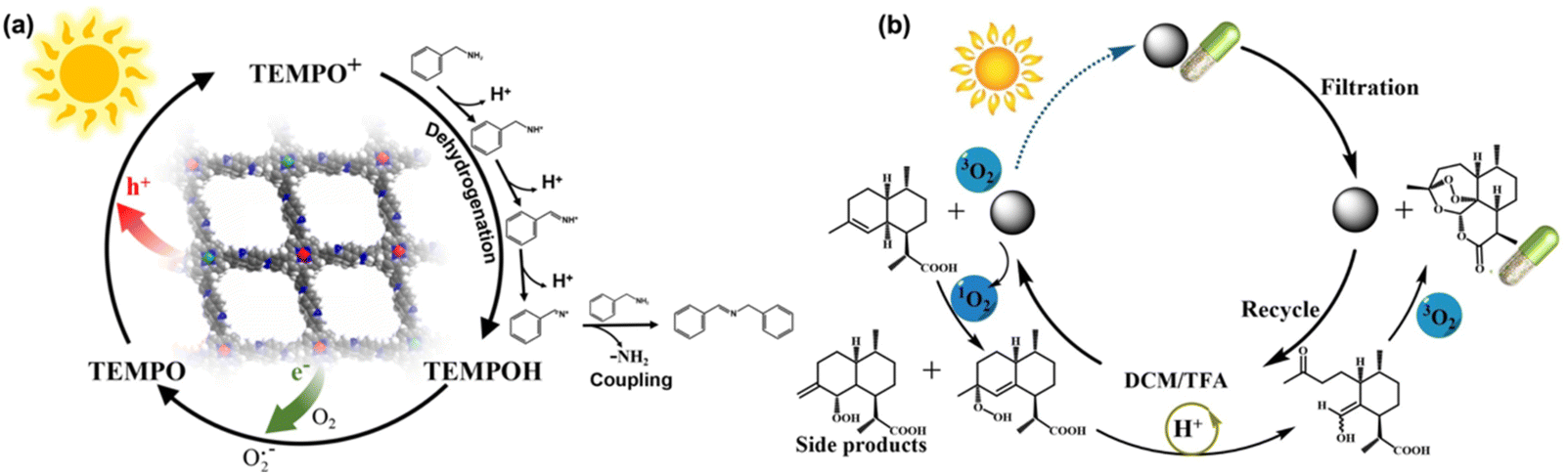 | ||
| Fig. 17 (a) Schematic diagram of the oxidative coupling mechanism of COF-Sr2Fe1 photocatalytic oxidation of benzylamine. Adapted with permission.165 Copyright 2024, Elsevier. (b) Schematic diagram of the reaction mechanism for producing artemisinin by photocatalytic oxidation. Adapted with permission.166 Copyright 2024, Wiley-VCH. | ||
C– double bond linkage confers stability to Por-sp2c-COF under high concentrations of amine. Later, Wang et al. used the same porphyrin molecule to react with p-phenylenediamine to synthesize imine-based Por-COF.168 The reported Por-COF only showed mild performance in selective aerobic oxidation of sulfides under white LED irradiation, while the yield of sulfoxides was obviously enhanced by cooperating with the TEMPO catalyst.
Wang et al. further introduced a seed growth method to synthesize core–shell NH2-MIL-125@TAPB-PDA hybrid materials.169 Specifically, small amounts of terephthaldehyde (PDA) and 1,3,5-tris(4-aminophenyl)benzene (PAPB) precursors were first added into NH2-MIL-125 suspension to generate COF seeds on the NH2-MIL-125 surface. Controlled amounts of PDA and PAPB were subsequently added to obtain NH2-MIL-125@TAPB-PDA photocatalysts with different COF shell thicknesses. The best yield of 94.7% for photo-oxidation of benzyl alcohols to benzaldehydes was achieved by NH2-MIL-125@TAPB-PDA-3 with a COF shell thickness of ∼20 nm, which was a 2.5- and 15.5-fold enhancement compared to NH2-MIL-125 and TAPB-PDA COF, respectively. Huang et al. designed and prepared COF/CdS composites for oxidizing benzyl alcohols to benzaldehydes.170 The COF was prepared by condensation of terephthalaldehyde and 1,3,5-tris(4-aminophenyl)benzene. The synthesis of COF/CdS composites with varying CdS contents was achieved by reaction with cadmium acetate, thioacetamide and COF with different mass ratios. A series of COF/CdS photocatalysts were obtained and denoted as COF/CdS-1, COF/CdS-2, COF/CdS-3 and COF/CdS-4, respectively. COF/CdS-3 gave the highest benzaldehyde yield of 97.1%, which is 2.5 and 15.9 times that of pure CdS and COF, respectively. The boosted photocatalytic oxidation efficiency was attributed to the improved electron–hole separation in the COF/CdS heterojunction. Kuang et al. recently combined Cs2AgBiCl6 (CABC) with a semi-conductive covalent organic framework (C4N) to form a type II heterojunction with staggered band alignment, creating an improved photocatalyst (Fig. 18a).171 In this heterojunction, C4N provided heterogeneous nucleation sites for CABC crystal growth and the oxygen affinity of C4N enhanced aerobic reactivity. The combination enabled better separation of charge carriers. The optimal CABC/C4N heterojunction achieved 100% thioanisole conversion after 6 hours. This was 2.2 times better than that of pure CABC and 7.7 times better than that of pure C4N. The catalyst showed good stability over five cycles (Fig. 18b and c). Tan et al. adopted an in situ encapsulation strategy to develop a novel coralloid W18O49@TpPa-H S-scheme heterojunction for efficient photocatalytic aerobic oxidation.172 In this heterojunction, W18O49 nanobundles acted as “coral bones” wrapped in a branched TpPa-H structure and exhibited broad light absorption across UV-visible-NIR regions. The optimized W18O49@TpPa-H-0.1 achieved a benzylamine conversion rate of 99% in 4 hours with an imide product selectivity of 99%. The photocatalytic performance is 9.9 times better than that of pure W18O49 and 2.8 times better than that of pure TPA-H. W18O49@TpPa-H-0.1 was effective even under 740 nm light and showed good stability over 5 reaction cycles. The research represents an important advancement in developing efficient photocatalysts for organic synthesis, particularly in the selective oxidation of amines to imines under mild conditions solely using light and air as the reagents.
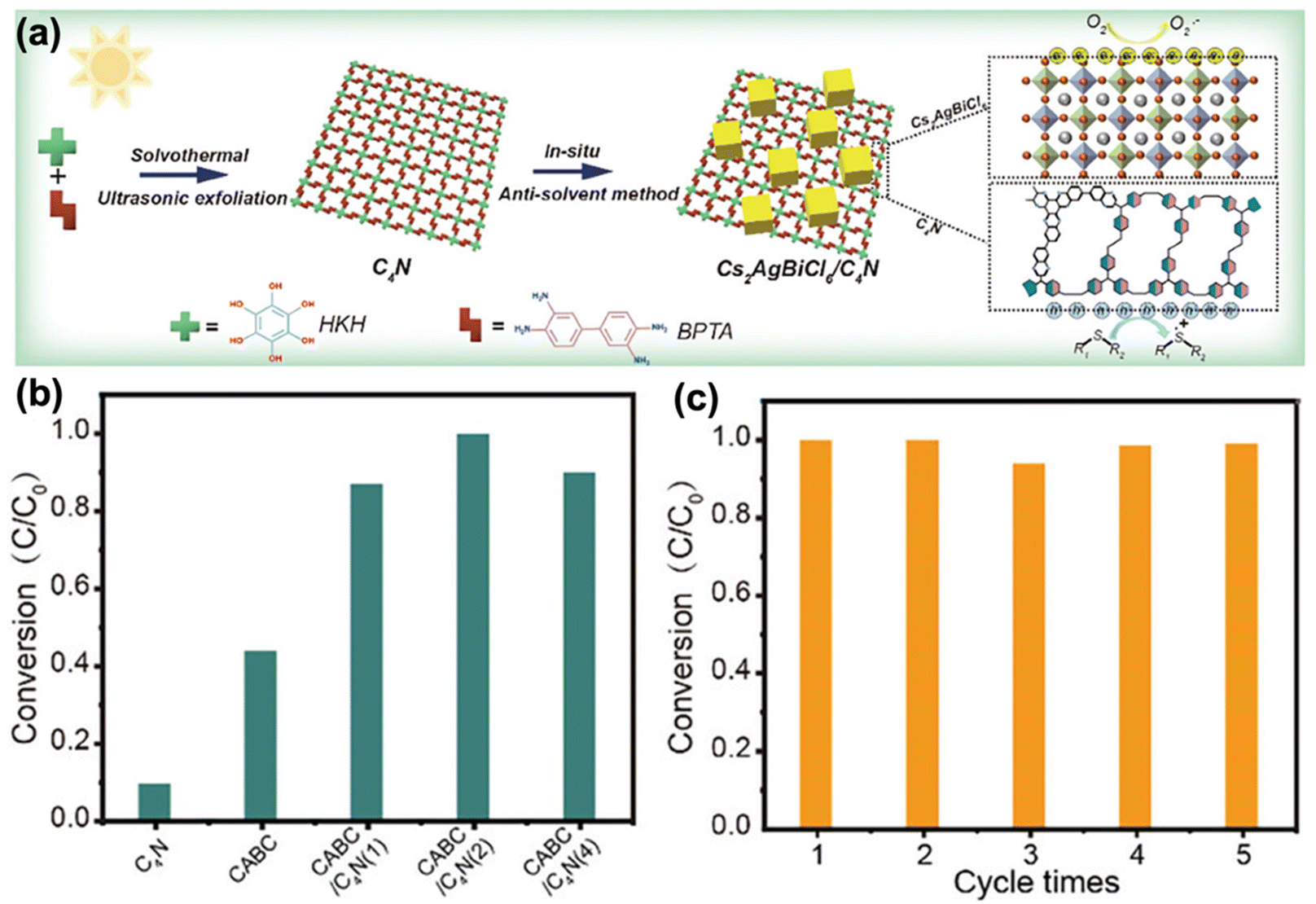 | ||
| Fig. 18 (a) Schematic of synthesis path of the CABC/C4N heterojunction. (b) Comparison of photocatalytic oxidation properties of thioesters. (c) Photocatalytic cycle stability of CABC/C4N. Adapted with permission.171 Copyright 2024, Wiley-VCH. | ||
3.4 Photocatalytic hydrogen peroxide production
H2O2 is a versatile chemical widely used as an antimicrobial disinfectant, bleach, and oxidizing agent in chemical synthesis. Compared with traditional production methods such as the anthraquinone oxidation reaction, photocatalytic synthesis provides an environmentally friendly and cost-effective alternative that uses solar energy and water to reduce pollution. COFs composed of earth-abundant elements like C, H and N show promise as new generation photocatalysts for H2O2 production. In addition to their strong visible light absorption, efficient photo-induced carrier separation, and tunable and diverse structures, COFs allow the formation of intermediates suitable for H2O2 synthesis while hindering its decomposition. Table 3 summarizes the photocatalytic H2O2 production performance of some COF-based photocatalysts.| Samples | Irradiation conditions | Solvent system | H2O2 production rate (μmol h−1 g−1) | Ref. |
|---|---|---|---|---|
| TAPD-(Me)2 COF | 700 nm > λ > 420 nm | H2O:EtOH (1:9, v/v) |
234.52 | 173 |
| TTF-BT-COF | AM 1.5 | H2O | 690 | 174 |
| EBA-COF | λ > 420 nm | H2O:EtOH (9:1, v/v) |
1830 | 175 |
| COF-N32 | λ > 420 nm | H2O | 605 | 176 |
| TaptBtt | λ > 420 nm | H2O | 1407 | 177 |
| TZ-COF | λ > 420 nm | H2O | 268 | 178 |
| COF-2CN | λ > 420 nm | H2O | 1601 | 179 |
| DVA-COF | λ = 420 nm | H2O:BA (9:1, v/v) |
8450 | 180 |
| TF50-COF | λ > 400 nm | H2O:EtOH (9:1, v/v) |
1739 | 181 |
| COF-TfpBpy | λ > 420 nm | H2O | 694 | 182 |
| COF-BPDA-DTP | λ > 420 nm | H2O | 1164 | 183 |
| TiCOF-spn | 780 nm ≥ λ ≥ 420 nm | H2O | 489.94 | 184 |
| COF-NUST-16 | λ > 420 nm | H2O:EtOH (9:1, v/v) |
1081 | 185 |
| CoPcF16 | λ > 400 nm | H2O:EtOH (9:1, v/v) |
2096 | 186 |
| TAPT-TFPA COFs | AM 1.5 | H2O | 2143 | 187 |
| ZnO/TpPa-Cl | AM 1.5 | H2O:EtOH (9:1, v/v) |
2443 | 188 |
| TiO2/BTTA | 780 nm > λ > 350 nm | FAL | 1480 | 189 |
| CDs@CTFs | λ ≥ 420 nm | H2O | 535.41 | 190 |
| Bi4O5Br2/TTD-COF | LED (400–700 nm) | H2O | 5221 | 191 |
| COF-ZCS | λ ≥ 420 nm | H2O | 5171 | 192 |
| CsPbBr3/CTFs | λ = 420 nm | H2O | 1680 | 193 |
| ZnIn2S4/TpPa-1 | 780 nm > λ > 400 nm | H2O:EtOH (9:1, v/v) |
1032 | 194 |
| WO3/Tp-TAPB | λ > 420 nm | H2O | 1488.4 | 195 |
| CTF-1-G/WS2 | λ > 420 nm | H2O | 156 | 196 |
 | ||
| Fig. 19 (a) The TTF-BT-COF with an oxidation–reduction molecular junction produces H2O2via the ORR and WOR simultaneously. (b) Photocatalytic H2O2 production activity of the three COFs. (c) Photocatalytic cycle stability of TTF-BT-COF. Adapted with permission.174 Copyright 2022, Wiley-VCH. (d) Chemical structure of COF-0CN, COF-1CN and COF-2CN. (e) Electrostatic potential distribution of COF-0CN, COF-1CN and COF-2CN. (f) Photocatalytic synthesis of H2O2 from COFs, g-C3N4 and P25. (g) Comparison of the performance of COF-2CN and other photocatalysts. Adapted with permission.179 Copyright 2024, Wiley-VCH. | ||
Modification and doping of COFs constitute pivotal avenues for enhancing the performance of COFs in photocatalytic H2O2 generation. Ni et al. synthesized three COFs, designated as COF-0CN, COF-1CN, and COF-2CN, each featuring a varying degree of cyano group modification (Fig. 19d).179 A positive correlation was observed between the number of cyano groups and the efficiency of charge separation and transport within the COFs. Experimental results and theoretical calculations indicated that cyano group modifications augmented the number of charge transfer pathways between donor and acceptor moieties, thereby facilitating charge separation and transport processes (Fig. 19e). Additionally, the incorporation of dicyano modifications was found to reduce the energy barrier for the two-electron water oxidation reaction. Finally, COF-2CN achieved a high H2O2 production rate of 1601 μmol h−1 g−1 under visible light irradiation using only water and oxygen as reactants (Fig. 19f). This performance was superior to that of most conventional photocatalysts and most recently reported catalysts (Fig. 19g). Chen et al. demonstrated that the anchored vinyl groups in DVA-COF not only broadened the light absorption spectrum but also accelerated charge separation and transfer efficiency, which consequently promoted the generation of H2O2via the 2e− ORR pathway.180 By means of fluorine substitution, plenty of Lewis acid sites were introduced into TF50-COF, which helped fine-tune the electronic structure of proximate carbon atoms, promote O2 adsorption, extend the light-harvesting range and elevate charge separation efficiency.181 The protonation of bipyridine units in COF-TfpBpy was believed to promote the 2e− WOR and further facilitate the formation of endoperoxide intermediate species, which strengthened Yeager-type oxygen adsorption on COF-TfpBpy, accelerating the one-step 2e− ORR process.182 Consequently, COF-TfpBpy exhibited a high solar-to-chemical conversion efficiency of 0.57% at 298 K. Kong and co-workers developed a molecular engineering strategy to fabricate three imine-linked alkyne-containing COFs (COF-BPDA-DTP, -BD, and -PA) that had similar molecular structures but different lengths of linkers.183 They found that longer linkers potentially contribute to a more negative conductive band energy level, increased specific surface area, and enhanced charge separation efficiency. Among the three COFs, COF-BPDA-DTP with the longest linker length exhibited the highest H2O2 production rate of 1164 μmol h−1 gcat−1.
The layered structure of 2D COFs limits the exposure of active sites, which diminishes photocatalytic efficiency, whereas 3D COFs retain intrinsic precursor properties with non-overlapping π-planes. TiCOF-spn integrated a photoactive titanium center and triazine units in a 3D crystalline porous structure, exhibiting a photocatalytic H2O2 production rate of 489.9 μmol h−1 g−1.184 Zhang et al. constructed a 3D COF (COF-NUST-16) with tty topology achieving a photocatalytic H2O2 production rate of 1081 μmol h−1 g−1.185 Notably, its photocatalytic performance was over 4 times greater than that of COF-NUST-17 – a 2D COF with a similar structure, highlighting better active site accessibility and mass transfer of its 3D framework in boosting photocatalytic activity.
 | ||
| Fig. 20 (a) Schematic diagram of the mechanism of photocatalytic H2O2 production in an S-scheme heterojunction. (b) Rate of photocatalytic hydrogen peroxide production by BIT and other catalysts. Adapted with permission.191 Copyright 2024, American Chemical Society. (c) Synthesis diagram of the TT-COF/ZCS composite. Photocatalytic production of H2O2 in (d) oxygen and (e) air environments for TT-COF, ZCS, and TZ-40. (f) Photocatalytic cycle stability of TZ-40. Adapted with permission.192 Copyright 2024, Wiley-VCH. | ||
4 Conclusion and perspective
COFs are novel porous materials constructed from covalently linking organic building blocks and are regarded as one of the most promising materials for heterogeneous photocatalysis because of their molecular backbones that are beneficial for light harvesting and promoting charge separation and transport. On the one hand, molecular chromophores with excellent light harvesting capability can be selected to build the backbone of COFs. On the other hand, conjugated architecture, both in-plane and through stacking, could promote redshift of light absorption, which allows harvesting more portions of solar light. At the same time, the conjugated and crystalline structure of COFs can also facilitate charge transport, prevent charge recombination, and increase charge carrier mobility. Furthermore, covalent bonds of COFs make them stable and robust, which is very important for their applications in photocatalysis. Benefiting from the structural diversity, designability and tunability of COFs, multiple favorable properties can be integrated into one material for photocatalytic applications.In this review, we start with a brief discussion of the structural design concepts of COFs and highlight the merits of COFs as photocatalysts. We then introduce the progress of COFs as photocatalysts in photocatalytic H2 production, CO2 reduction, organic transformations, and H2O2 production in the last five years. Pure COFs exhibit tunable band structures and excitonic behavior and customizable surface properties such as the modification of hydrophilic groups to improve hydrophilicity. Abundant metal anchoring sites such as bipyridine and porphyrin units allow COFs to easily load well-defined metal active sites to realize higher photocatalytic performance. Simultaneously, strong coordination or chelation between the metals and COFs enables long-term stability of metal sites. In addition, higher photocatalytic efficiencies are achieved by combining COFs with other semiconductor materials such as TiO2, CdS, g-C3N4 and MOFs. COF-based composites synergize the benefits of both components and form a type II or Z scheme heterojunction that facilitates the charge separation efficiency.
Despite significant progress in the photocatalytic applications of COFs, some urgent challenges remain to be addressed. Firstly, the solar to hydrogen (STH) and solar to fuel (STF) efficiencies of COFs are not yet satisfactory, especially in the absence of sacrificial agents. Only a few COFs have been reported to achieve high efficiency in photocatalytic H2 production and CO2 reduction using water as the hole scavenger. It is highly desired to construct COFs with suitable band diagrams for simultaneous driving oxidation and reduction half-reactions in one photocatalyst, i.e. overall water splitting. Secondly, conventional synthesis methods of COFs are restricted by harsh reaction conditions. Developing synthetic procedures with mild conditions, low cost, and high yield is imperative for large-scale fabrication of COFs. Even though some COFs with specific linkages can be synthesized on a large scale, there is still a lack of synthetic methods for the fabrication of most COF materials. Lastly, the in-depth mechanism of COF-based photocatalysts is still elusive. Steady and time-resolved spectroscopy can be employed to explore the formation, separation, and recombination of photogenerated carriers. Various in situ techniques such as in situ Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy can be utilized to identify reaction intermediates and study active centers to guide the design of new COFs. Although many problems are yet to be overcome, large-scale applications of COF photocatalysis can still be foreseen through continuous research efforts to overcome these challenges.
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support from the Science and Technology Project of Jiangsu Province (BZ2022056). We also acknowledge the support from the Eastern Institute of Technology, Ningbo, and Shanghai Institute of Organic Chemistry.References
- D. Coumou, G. Di Capua, S. Vavrus, L. Wang and S. Wang, Nat. Commun., 2018, 9, 2959 CrossRef CAS PubMed.
- D. Shindell and C. J. Smith, Nature, 2019, 573, 408–411 CrossRef CAS PubMed.
- J. Zhang, W. Tian, M. P. Chipperfield, F. Xie and J. Huang, Nat. Clim. Change, 2016, 6, 1094–1099 CrossRef.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Q. Guo, Z. Ma, C. Zhou, Z. Ren and X. Yang, Chem. Rev., 2019, 119, 11020–11041 CrossRef CAS PubMed.
- M. D. Regulacio and M.-Y. Han, Acc. Chem. Res., 2016, 49, 511–519 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- S. Tao and D. Jiang, CCS Chem., 2021, 3, 2003–2024 CrossRef CAS.
- Z.-J. Lin, S. A. Mahammed, T.-F. Liu and R. Cao, ACS Cent. Sci., 2022, 8, 1589–1608 CrossRef CAS PubMed.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou and X. Li, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- J. Cao, Z. Yang, W. Xiong, Y. Zhou, Y. Wu, M. Jia, C. Zhou and Z. Xu, Coord. Chem. Rev., 2021, 439, 213924 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eeal1585 CrossRef PubMed.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu and M. Zhang, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- S. Liu, T. Qian, M. Wang, H. Ji, X. Shen, C. Wang and C. Yan, Nat. Catal., 2021, 4, 322–331 CrossRef CAS.
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang and L. Liu, Nature, 2022, 604, 72–79 CrossRef CAS PubMed.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- Y. Li, X. Song, G. Zhang, L. Wang, Y. Liu, W. Chen and L. Chen, ChemSusChem, 2022, 15, e202200901 CrossRef CAS PubMed.
- H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS.
- P. Costa, A. Vega-Peñaloza, L. Cognigni and M. Bonchio, ACS Sustain. Chem. Eng., 2021, 9, 15694–15721 CrossRef CAS.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CAS.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS PubMed.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- N. Keller, M. Calik, D. Sharapa, H. R. Soni, P. M. Zehetmaier, S. Rager, F. Auras, A. C. Jakowetz, A. Görling, T. Clark and T. Bein, J. Am. Chem. Soc., 2018, 140, 16544–16552 CrossRef CAS PubMed.
- N. Keller, D. Bessinger, S. Reuter, M. Calik, L. Ascherl, F. C. Hanusch, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 8194–8199 CrossRef CAS PubMed.
- Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- X. Guan, Q. Fang, Y. Yan and S. Qiu, Acc. Chem. Res., 2022, 55, 1912–1927 CrossRef CAS PubMed.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H. L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- R. M. Zhu, Y. Liu, W. K. Han, J. D. Feng, J. Zhang, H. Pang, J. Zhang and Z. G. Gu, Angew. Chem., Int. Ed., 2025, 64, e202412890 CrossRef CAS PubMed.
- N. Li, J. Zhang, X. Xie, K. Wang, D. Qi, J. Liu, Y. Q. Lan and J. Jiang, Nat. Commun., 2025, 16, 1106 CrossRef CAS PubMed.
- L. Y. Qin, C. D. Ma, J. Zhang and T. H. Zhou, Adv. Funct. Mater., 2024, 34, 2414086 Search PubMed.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Nat. Rev. Chem, 2022, 6, 881–898 CrossRef CAS PubMed.
- W. X. Li, Y. X. Wang, L. Li, X. Y. Huang, M. D. Liu, B. Gui, X. J. Lang and C. Wang, Chin. J. Struct. Chem., 2024, 43, 138196 Search PubMed.
- E. Q. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. H. Chen and D. L. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- H. Zhuang, C. Guo, J. Huang, L. Wang, Z. Zheng, H. N. Wang, Y. Chen and Y. Q. Lan, Angew. Chem., Int. Ed., 2024, 63, e202404941 CrossRef CAS PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- M.-H. Li, C. Xu and Y.-W. Yang, Coord. Chem. Rev., 2024, 512, 215894 CrossRef CAS.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y. Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CrossRef CAS PubMed.
- B. Zhang, H. Li, Y. Kang, K. Yang, H. Liu, Y. Zhao and S. Qiao, Adv. Funct. Mater., 2024, 35, 2416958 CrossRef.
- M. Zhang, P. Huang, J. P. Liao, M. Y. Yang, S. B. Zhang, Y. F. Liu, M. Lu, S. L. Li, Y. P. Cai and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202311999 CrossRef CAS PubMed.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas and S. Ma, Matter, 2020, 2, 416–427 CrossRef CAS.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS PubMed.
- J. L. Sheng, H. Dong, X. B. Meng, H. L. Tang, Y. H. Yao, D. Q. Liu, L. L. Bai, F. M. Zhang, J. Z. Wei and X. J. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- Y. Li, L. Yang, H. He, L. Sun, H. Wang, X. Fang, Y. Zhao, D. Zheng, Y. Qi and Z. Li, Nat. Commun., 2022, 13, 1355 CrossRef CAS PubMed.
- C. Mo, M. Yang, F. Sun, J. Jian, L. Zhong, Z. Fang, J. Feng and D. Yu, Advanced Science, 2020, 7, 1902988 CrossRef CAS PubMed.
- S. Li, R. Ma, S. Xu, T. Zheng, G. Fu, Y. Wu, Z. Liao, Y. Kuang, Y. Hou and D. Wang, J. Am. Chem. Soc., 2022, 144, 13953–13960 CrossRef CAS PubMed.
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z.-A. Lan, X. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 1318–1325 CrossRef CAS PubMed.
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2024, 36, 2305313 CrossRef CAS PubMed.
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg and C. Penschke, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- Y. Zhong, W. Dong, S. Ren and L. Li, Adv. Mater., 2024, 36, 2308251 CrossRef CAS PubMed.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- T. Sun, S. Li, L. Zhang and Y. Xu, Angew. Chem., Int. Ed., 2023, 62, e202306617 CrossRef.
- S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed.
- Y. Zang, R. Wang, P.-P. Shao, X. Feng, S. Wang, S.-Q. Zang and T. C. Mak, J. Mater. Chem. A, 2020, 8, 25094–25100 RSC.
- M. Wang, H. Lv, B. Dong, W. He, D. Yuan, X. Wang and R. Wang, Angew. Chem., Int. Ed., 2024, 63, e202401969 CrossRef CAS PubMed.
- G. Liu, G. Pan, Q. Dang, R. Li, L. Li, C. Yang and Y. Yu, ChemCatChem, 2022, 14, e202101800 CrossRef CAS.
- J. Li, J. Zhou, X. H. Wang, C. Guo, R. H. Li, H. Zhuang, W. Feng, Y. Hua and Y. Q. Lan, Angew. Chem., Int. Ed., 2024, 136, e202411721 CrossRef.
- C.-C. Li, M.-Y. Gao, X.-J. Sun, H.-L. Tang, H. Dong and F.-M. Zhang, Appl. Catal., B, 2020, 266, 118586 CrossRef CAS.
- Y. Wang, Z. Hu, W. Wang, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao, G. Yu and Y.-N. Liu, Chem. Sci., 2021, 12, 16065–16073 RSC.
- K. Gottschling, G. k. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2020, 142, 12146–12156 CrossRef CAS PubMed.
- F. M. Zhang, J. L. Sheng, Z. D. Yang, X. J. Sun, H. L. Tang, M. Lu, H. Dong, F. C. Shen, J. Liu and Y. Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106–12110 CrossRef CAS PubMed.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS PubMed.
- C. Lin, C. Han, L. Gong, X. Chen, J. Deng, D. Qi, Y. Bian, K. Wang and J. Jiang, Catal. Sci. Technol., 2021, 11, 2616–2621 RSC.
- T. Zhou, L. Wang, X. Huang, J. Unruangsri, H. Zhang, R. Wang, Q. Song, Q. Yang, W. Li and C. Wang, Nat. Commun., 2021, 12, 3934 CrossRef CAS PubMed.
- X. A. Li, Z. Z. Liang, Y. C. Zhou, J. F. Huang, X. L. Wang, L. M. Xiao and J. M. Liu, Aggregate, 2024, 5, e442 CrossRef CAS.
- B. B. Luan, X. Chu, Y. Wang, X. Qiao, Y. Jiang and F. M. Zhang, Adv. Mater., 2024, 36, 2412653 CrossRef CAS PubMed.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed.
- Y. Zhong, W. Dong, S. Ren and L. Li, Adv. Mater., 2024, 36, e2308251 CrossRef PubMed.
- X. Zhang, C. Gao, Y. Zhou, R. Chen, X. Guan, Z. Shen, B. Hu and Q.-H. Xu, Sci. China:Chem., 2025, 1–9 Search PubMed.
- H. Dong and F. M. Zhang, Chin. J. Struct. Chem., 2024, 43, 100304 CrossRef.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- D. H. Streater, E. R. Kennehan, D. Wang, C. Fiankor, L. Chen, C. Yang, B. Li, D. Liu, F. Ibrahim and I. Hermans, J. Am. Chem. Soc., 2024, 146, 4489–4499 CrossRef CAS PubMed.
- J.-X. Cui, L.-J. Wang, L. Feng, B. Meng, Z.-Y. Zhou, Z.-M. Su, K. Wang and S. Liu, J. Mater. Chem. A, 2021, 9, 24895–24902 RSC.
- X. Yu, K. Gong, S. Tian, G. Gao, J. Xie and X.-H. Jin, J. Mater. Chem. A, 2023, 11, 5627–5635 RSC.
- L. j. Wang, R. l. Wang, X. Zhang, J. l. Mu, Z. y. Zhou and Z. m. Su, ChemSusChem, 2020, 13, 2973–2980 CrossRef CAS PubMed.
- L. Ai, W. Li, Q. Wang, F. Cui and G. Jiang, ChemCatChem, 2022, 14, e202200935 CrossRef CAS.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H.-L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- Y. Xu, J. P. Dong, L. Wang, R. L. Geng, R. Wang, Y. N. Si, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2025, e202501391 CAS.
- W. Lin, F. Lin, J. Lin, Z. Xiao, D. Yuan and Y. Wang, J. Am. Chem. Soc., 2024, 146, 16229–16236 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- M. Lu, Q. Li, J. Liu, F.-M. Zhang, L. Zhang, J.-L. Wang, Z.-H. Kang and Y.-Q. Lan, Appl. Catal., B, 2019, 254, 624–633 CrossRef CAS.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- S. Yang, R. Sa, H. Zhong, H. Lv, D. Yuan and R. Wang, Adv. Funct. Mater., 2022, 32, 2110694 CrossRef CAS.
- H. Lv, P. Li, X. Li, A. Chen, R. Sa, H. Zhu and R. Wang, Chem. Eng. J., 2023, 451, 138745 CrossRef CAS.
- X. Lan, H. Li, Y. Liu, Y. Zhang, T. Zhang and Y. Chen, Angew. Chem., 2024, e202407092 CAS.
- J. Wang, W. Zhu, F. Meng, G. Bai, Q. Zhang and X. Lan, ACS Catal., 2023, 13, 4316–4329 CrossRef CAS.
- K. Guo, X. Zhu, L. Peng, Y. Fu, R. Ma, X. Lu, F. Zhang, W. Zhu and M. Fan, Chem. Eng. J., 2021, 405, 127011 CrossRef CAS.
- Z. Liu, Y. Huang, S. Chang, X. Zhu, Y. Fu, R. Ma, X. Lu, F. Zhang, W. Zhu and M. Fan, Sustainable Energy Fuels, 2021, 5, 2871–2876 RSC.
- S. Dong, Z. Tan, Q. Chen, G. Huang, L. Wu and J. Bi, J. Colloid Interface Sci., 2022, 628, 573–582 CrossRef CAS PubMed.
- M. Lu, M. Zhang, J. Liu, T.-Y. Yu, J.-N. Chang, L.-J. Shang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- B. Zhang, H. Li, Y. Kang, K. Yang, H. Liu, Y. Zhao and S. Qiao, Adv. Funct. Mater., 2025, 35, 2416958 CrossRef CAS.
- M. Zhang, M. Lu, Z. L. Lang, J. Liu, M. Liu, J. N. Chang, L. Y. Li, L. J. Shang, M. Wang, S. L. Li and Y. Q. Lan, Angew. Chem., 2020, 132, 6562–6568 CrossRef.
- L. Wang, G. Huang, L. Zhang, R. Lian, J. Huang, H. She, C. Liu and Q. Wang, J. Energy Chem., 2022, 64, 85–92 CrossRef CAS.
- Y. Wang, Z. Hu, W. Wang, Y. Li, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao and G. Yu, Appl. Catal., B, 2023, 327, 122419 CrossRef CAS.
- X. An, J. Bian, K. Zhu, R. Liu, H. Liu and J. Qu, Chem. Eng. J., 2022, 442, 135279 CrossRef CAS.
- L. Zou, R. Sa, H. Zhong, H. Lv, X. Wang and R. Wang, ACS Catal., 2022, 12, 3550–3557 CrossRef CAS.
- K. H. Do, D. P. Kumar, A. P. Rangappa, J. Lee, S. Yun and T. K. Kim, J. Mater. Chem. A, 2023, 11, 8392–8403 RSC.
- J. Qiu, Y. Zheng, L. Wang, M. Liu, L. Tian, X. Yu, X. An and G. Lv, J. Mater. Chem. A, 2023, 11, 4572–4578 RSC.
- J. Wang, Y. Yu, J. Cui, X. Li, Y. Zhang, C. Wang, X. Yu and J. Ye, Appl. Catal., B, 2022, 301, 120814 CrossRef CAS.
- R.-G. Yang, Y.-M. Fu, H.-N. Wang, D.-P. Zhang, Z. Zhou, Y.-Z. Cheng, X. Meng, Y.-O. He and Z.-M. Su, Chem. Eng. J., 2022, 450, 138040 CrossRef CAS.
- H. Lin, Y. Liu, Z. Wang, L. Ling, H. Huang, Q. Li, L. Cheng, Y. Li, J. Zhou and K. Wu, Angew. Chem., 2022, 134, e202214142 CrossRef.
- Z. Zhao, D. Zheng, M. Guo, J. Yu, S. Zhang, Z. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200261 CrossRef CAS PubMed.
- Y. N. Gong, J. H. Mei, W. J. Shi, J. W. Liu, D. C. Zhong and T. B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202318735 CrossRef CAS PubMed.
- A. Dey, J. Pradhan, S. Biswas, F. Ahamed Rahimi, K. Biswas and T. K. Maji, Angew. Chem., Int. Ed., 2024, 136, e202315596 CrossRef.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros and X. Duan, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- Y. Xiang, W. Dong, P. Wang, S. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. Jin and Y. Weng, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- J.-X. Cui, Y.-M. Fu, B. Meng, J. Zhou, Z.-Y. Zhou, S.-M. Liu and Z.-M. Su, J. Mater. Chem. A, 2022, 10, 13418–13427 RSC.
- Y. Zhang, L. Cao, G. Bai and X. Lan, Small, 2023, 19, 2300035 CrossRef CAS PubMed.
- M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei and H. Xie, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- W. Tu, Y. Yang, C. Chen, T. Zhou, T. Li, H. Wang, S. Wu, Y. Zhou, D. O'Hare and Z. Zou, Small Struct., 2023, 4, 2200233 CrossRef CAS.
- B. Han, X. Ou, Z. Zhong, S. Liang, H. Deng and Z. Lin, Small, 2020, 16, 2002985 CrossRef CAS PubMed.
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- Z. Liu, Q. Su, P. Ju, X. Li, G. Li, Q. Wu and B. Yang, Chem. Commun., 2020, 56, 766–769 RSC.
- S. Yang, X. Li, Y. Qin, Y. Cheng, W. Fan, X. Lang, L. Zheng and Q. Cao, ACS Appl. Mater. Interfaces, 2021, 13, 29471–29481 CrossRef CAS PubMed.
- Y. Fang, Y. Liu, H. Huang, J. Sun, J. Hong, F. Zhang, X. Wei, W. Gao, M. Shao, Y. Guo, Q. Tang and Y. Liu, Nat. Commun., 2024, 15, 4856 CrossRef CAS PubMed.
- J.-C. Liu, C. Tuo, W.-Y. Xiao, M.-Y. Qi, Y. Yusran, Z.-T. Wang, H. Li, C.-S. Guo, J.-L. Song, S.-L. Qiu, Y.-J. Xu and Q. Fang, Angew. Chem., Int. Ed., 2024, e202416240 Search PubMed.
- W. Hao, D. Chen, Y. Li, Z. Yang, G. Xing, J. Li and L. Chen, Chem. Mater., 2019, 31, 8100–8105 CrossRef CAS.
- Q. Li, X. Lan, G. An, L. Ricardez-Sandoval, Z. Wang and G. Bai, ACS Catal., 2020, 10, 6664–6675 CrossRef CAS.
- X. Li, Y. Wang, F. Zhang and X. Lang, J. Colloid Interface Sci., 2023, 648, 683–692 CrossRef CAS PubMed.
- F. Huang, Y. Wang, X. Dong and X. Lang, Sci. China:Chem., 2023, 66, 3290–3296 CrossRef CAS.
- X. Dong, F. Zhang, Y. Wang, F. Huang and X. Lang, Appl. Catal., B, 2024, 345, 123660 CrossRef CAS.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334–342 CrossRef CAS.
- H. Liu, C. Li, H. Li, Y. Ren, J. Chen, J. Tang and Q. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 20354–20365 CrossRef CAS PubMed.
- H. Shan, D. Cai, X. Zhang, Q. Zhu, P. Qin and J. Baeyens, Chem. Eng. J., 2022, 432, 134288 CrossRef CAS.
- Z. Li, S. Han, C. Li, P. Shao, H. Xia, H. Li, X. Chen, X. Feng and X. Liu, J. Mater. Chem. A, 2020, 8, 8706–8715 RSC.
- Y. Yang, H. Niu, L. Xu, H. Zhang and Y. Cai, Appl. Catal., B, 2020, 269, 118799 CrossRef CAS.
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152–6156 CrossRef CAS PubMed.
- H. Chen, W. Liu, A. Laemont, C. Krishnaraj, X. Feng, F. Rohman, M. Meledina, Q. Zhang, R. Van Deun, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2021, 60, 10820–10827 CrossRef CAS PubMed.
- W. Dong, Y. Yang, Y. Xiang, S. Wang, P. Wang, J. Hu, L. Rao and H. Chen, Green Chem., 2021, 23, 5797–5805 RSC.
- A. López-Magano, A. E. Platero-Prats, S. Cabrera, R. Mas-Ballesté and J. Alemán, Appl. Catal., B, 2020, 272, 119027 CrossRef.
- Z. Almansaf, J. Hu, F. Zanca, H. R. Shahsavari, B. Kampmeyer, M. Tsuji, K. Maity, V. Lomonte, Y. Ha, P. Mastrorilli, S. Todisco, M. Benamara, R. Oktavian, A. Mirjafari, P. Z. Moghadam, A. R. Khosropour and H. Beyzavi, ACS Appl. Mater. Interfaces, 2021, 13, 6349–6358 CrossRef CAS PubMed.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- S. Shi, W. Liu, Y. Li, S. Lu, H. Zhu, M. Du, X. Chen and F. Duan, J. Colloid Interface Sci., 2024, 655, 611–621 CrossRef CAS PubMed.
- Y. Gao, S. Li, L. Gong, J. Li, D. Qi, N. Liu, Y. Bian and J. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202404156 CrossRef CAS PubMed.
- J.-L. Shi, R. Chen, H. Hao, C. Wang and X. Lang, Angew. Chem., Int. Ed., 2020, 59, 9088–9093 CrossRef CAS PubMed.
- K. Feng, H. Hao, F. Huang, X. Lang and C. Wang, Mater. Chem. Front., 2021, 5, 2255–2260 RSC.
- G. Lu, X. Huang, Y. Li, G. Zhao, G. Pang and G. Wang, J. Energy Chem., 2020, 43, 8–15 CrossRef.
- K. Zhang, G. Lu, Z. Xi, Y. Li, Q. Luan and X. Huang, Chin. Chem. Lett., 2021, 32, 2207–2211 CrossRef CAS.
- Q. Qin, Z.-H. Xia, W.-Q. Liu, H.-Y. Chen and D.-B. Kuang, Small, 2024, 20, 2402410 CrossRef CAS PubMed.
- X. Zhao, A. Li, D. Yang, T.-Y. Qiu, Z. Zhao, S.-L. Wang, X. Mu and H.-Q. Tan, J. Colloid Interface Sci., 2024, 653, 67–76 CrossRef CAS PubMed.
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed.
- J.-N. Chang, Q. Li, J.-W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- L. Zhai, Z. Xie, C.-X. Cui, X. Yang, Q. Xu, X. Ke, M. Liu, L.-B. Qu, X. Chen and L. Mi, Chem. Mater., 2022, 34, 5232–5240 CrossRef CAS.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14, 4344 CrossRef CAS PubMed.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. Huixiang Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309480 CrossRef CAS PubMed.
- Y. Hou, P. Zhou, F. Liu, Y. Lu, H. Tan, Z. Li, M. Tong and J. Ni, Angew. Chem., Int. Ed., 2024, 63, e202318562 CrossRef CAS PubMed.
- H. Yu, F. Zhang, Q. Chen, P.-K. Zhou, W. Xing, S. Wang, G. Zhang, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402297 CrossRef CAS PubMed.
- H. Wang, C. Yang, F. Chen, G. Zheng and Q. Han, Angew. Chem., Int. Ed., 2022, 61, e202202328 CrossRef CAS PubMed.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng, J. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H. Huang and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- T. Yang, Y. Wang, Y. Chen, X. Peng, H. Zhang and A. Kong, CrystEngComm, 2023, 25, 4511–4520 RSC.
- W.-K. Han, H.-S. Lu, J.-X. Fu, X. Liu, X. Zhu, X. Yan, J. Zhang, Y. Jiang, H. Dong and Z.-G. Gu, Chem. Eng. J., 2022, 449, 137802 CrossRef CAS.
- M. Wu, Z. Shan, J. Wang, T. Liu and G. Zhang, Chem. Eng. J., 2023, 454, 140121 CrossRef CAS.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- Y. Liu, L. Li, H. Tan, N. Ye, Y. Gu, S. Zhao, S. Zhang, M. Luo and S. Guo, J. Am. Chem. Soc., 2023, 145, 19877–19884 CrossRef CAS PubMed.
- Y. Zhang, J. Qiu, B. Zhu, M. V. Fedin, B. Cheng, J. Yu and L. Zhang, Chem. Eng. J., 2022, 444, 136584 CrossRef CAS.
- Y. Yang, J. Liu, M. Gu, B. Cheng, L. Wang and J. Yu, Appl. Catal., B, 2023, 333, 122780 CrossRef CAS.
- W. Ren, Q. Chang, N. Li, J. Yang and S. Hu, Chem. Eng. J., 2023, 451, 139035 CrossRef CAS.
- J.-Y. Yue, Z.-X. Pan, P. Yang and B. Tang, ACS Mater. Lett., 2024, 6, 3932–3940 CrossRef CAS.
- X. Ma, S. Li, Y. Gao, N. Li, Y. Han, H. Pan, Y. Bian and J. Jiang, Adv. Funct. Mater., 2024, 34, 2409913 CrossRef CAS.
- Y. Zheng, T. Gao, S. Chen, C. T. J. Ferguson, K. A. I. Zhang, F. Fang, Y. Shen, N. A. Khan, L. Wang and L. Ye, Compos. Commun., 2022, 36, 101390 CrossRef.
- G. Xia, J. Qiu, L. Zhang, D. Dai and J. Yao, Colloids Surf., A, 2023, 664, 131124 CrossRef CAS.
- Y. Yang, Y. Li, X. Ma, L. Xie, D. Lv, L. Jiang, J. He, D. Chen and J. Wang, Catal. Sci. Technol., 2023, 13, 5599–5609 RSC.
- Y. Shao, D. You, Y. Wan, Q. Cheng and Z. Pan, Dalton Trans., 2023, 52, 11272–11284 RSC.
Footnote |
| † Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |