
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpeciating volatile organic compounds in indoor air: using in situ GC to interpret real-time PTR-MS signals†
Jenna C.
Ditto
 *a,
Han N.
Huynh
bc,
Jie
Yu
d,
Michael F.
Link
e,
Dustin
Poppendieck
e,
Megan S.
Claflin
f,
Marina E.
Vance
g,
Delphine K.
Farmer
h,
Arthur W. H.
Chan
di and
Jonathan P. D.
Abbatt
d
*a,
Han N.
Huynh
bc,
Jie
Yu
d,
Michael F.
Link
e,
Dustin
Poppendieck
e,
Megan S.
Claflin
f,
Marina E.
Vance
g,
Delphine K.
Farmer
h,
Arthur W. H.
Chan
di and
Jonathan P. D.
Abbatt
d
aDepartment of Energy, Environmental, and Chemical Engineering, Washington University in St. Louis, USA. E-mail: dittoj@wustl.edu
bCooperative Institute for Research in Environmental Sciences (CIRES), University of Colorado Boulder, USA
cNOAA Chemical Sciences Laboratory, USA
dDepartment of Chemistry, University of Toronto, Canada
eEngineering Laboratory, National Institute of Standards and Technology, USA
fAerodyne Research, Inc, USA
gDepartment of Mechanical Engineering, University of Colorado Boulder, USA
hDepartment of Chemistry, Colorado State University, USA
iDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Canada
First published on 18th December 2024
Abstract
Proton transfer reaction mass spectrometry (PTR-MS) is often employed to characterize gas-phase compounds in both indoor and outdoor environments. PTR-MS measurements are usually made without upstream chromatographic separation, so it can be challenging to differentiate between an ion of interest, its isomers, and fragmentation products from other species all detected at the same mass-to-charge ratio. These isomeric contributions and fragmentation interferences can confound the determination of accurate compound mixing ratios, the assignment of accurate chemical properties, and corresponding analyses of chemical fate. In this study, we deployed a gas chromatograph upstream of a PTR-MS to investigate contributions of isomers and fragmentation products for select indoor air-relevant chemicals. Measurements were made in a test house across a variety of indoor chemical sources, oxidants, and environmental conditions during the Chemical Assessment of Surfaces and Air (CASA) study. Observed confounding signals at each extracted ion chromatogram ranged from 0% (C2H6OH+, C8H24O4Si4H+, and C10H30O5Si5H+) to 98% (at C5H9+). For many ions, confounding signals varied between indoor conditions, and there were also differences between confounding signals across indoor vs. outdoor measurements. The relative contribution of sets of key structural isomers (e.g., C6–C8 carbonyls, xylenes, trimethylbenzenes, and monoterpenes) remained consistent throughout the measurement period despite changing indoor conditions. These relatively stable isomer distributions yielded stable chemical property assignments for these isomer sets. Taken together, these observations can inform future interpretations of PTR-MS signals measured in different indoor conditions without upstream chromatography.
Environmental significanceProton transfer reaction mass spectrometry (PTR-MS) is an important tool for characterizing the chemical complexity and dynamics of indoor air. However, without upstream chromatography, PTR-MS detects many ions at the same mass-to-charge ratio and cannot distinguish between the identities of the ions contributing to the total observed signal. Other ions co-occurring alongside a target compound may include its isomers as well as fragmentation products of higher molecular weight species. Here, we investigate the relative contributions of select indoor relevant compounds, contributions of their isomers, and contributions of fragmentation interferences under different indoor environmental conditions. These findings will inform and improve indoor PTR-MS measurement interpretation, including for the calculation of compound mixing ratios, assignment of chemical properties, and analyses of chemical fate. |
1 Introduction
Proton transfer reaction mass spectrometry (PTR-MS) is a powerful tool to measure the composition and dynamics of gas-phase compounds in outdoor and indoor environments. For example, within the built environment, recent PTR-MS measurements have been made in occupied homes,1–3 unoccupied buildings,4–7 workplace settings,8–11 and other public indoor spaces like museums and athletic facilities.12–15 These real-time measurements enable researchers to capture rapid compositional changes in the complex mixture of indoor gases.The analysis of PTR-MS data can be challenging, as numerous chemical structures may co-occur at the same mass-to-charge (m/z) ratio. For example, in the PTR-MS data sets, many structural isomers are often present for any particular chemical of interest. In addition, while proton transfer is usually considered a soft ionization method, it is known to fragment some parent ions due to the relatively high collision energy of reagent ions with analytes as well as due to reaction exothermicity. While these fragmentation patterns are sometimes tabulated in libraries,16 it is challenging to parse out the role of parent ions vs. fragmentation products of higher molecular weight compounds in a compositionally complex and variable mixture. Instrument operating conditions (e.g., ionization region parameters, big segmented quadrupole (BSQ) voltages (in the case of Vocus PTR-MS), and inlet capillary configuration) can also impact product ion distributions, i.e., the relative contributions of ions resulting from proton transfer, water clustering, and other ionization pathways.17
To address the speciation of isomers and to better understand the role of parent ion fragmentation, gas chromatography (GC) can be deployed upstream of a PTR-MS to sample and chromatographically separate chemicals in indoor air. This is important for accurate identification and quantitation of volatile organic compounds (VOCs), both of which can have impacts on molecular property assignment and interpretation of real-world measurements. Here, we give three example benefits of deploying GC upstream of PTR-MS instruments.
First, isomer speciation with GC enables an assessment of relative isomer contributions and a better understanding of their ultimate chemical fate indoors. For instance, monoterpenes are all detected at the parent ion C10H17+ in PTR-MS and without GC separation, the contribution of individual monoterpenes to the isobaric mixture is unknown. Monoterpene isomers have different properties like vapor pressure, aqueous solubility, and reactivity with ozone or hydroxyl radicals, that together contribute to their different atmospheric lifetime and secondary organic aerosol yields.18 Characterizing the monoterpene isomer distribution with GC is needed for a robust analysis of their persistence and impacts indoors (and outdoors). Similarly, in the case of isomers like carbonyls that may have different sensitivities in the PTR-MS, due to either differences in proton transfer rate constant and/or differences in their fragmentation patterns, implementing GC upstream of PTR-MS allows users to identify contributing isomer peaks and accurately assess their contributions to the overall signal and overall conversion from detected ions per second to mixing ratio.
Second, GC separation allows for a more complete assessment of important contributors to a single PTR-MS m/z signal. It is now well-known that the C5H9+ signal, commonly attributed to isoprene's parent ion in PTR-MS, has important contributions from the fragmentation products of medium chain aldehydes. In the indoor environment where aldehyde concentrations may be elevated (for instance, due to emissions from cooking19 or due to formation from ozonolysis of unsaturated precursors20), ignoring these contributions could yield overestimates of indoor isoprene mixing ratios indoors.
Last, m/z speciation by GC aids in the interpretation of real-time PTR-MS observations. For example, at the HOMEChem (House Observations of Microbial and Environmental Chemistry) campaign,21m/z 69.069 was assigned the ion formula C5H9+ and interpreted as isoprene.22 Based on this identification and isoprene's octanol–air and water–air partitioning coefficients, it was estimated that this compound would exist exclusively in the gas phase (vs. in surface reservoirs) in the test house. As expected for a gas phase chemical, m/z 69.069 showed a significant drop in signal intensity during house ventilation experiments. However, m/z 69.069 then showed a rapid signal recovery after the house was closed again despite no obvious source of isoprene. This rapid recovery after the ventilation event suggests that the m/z 69.069 chemical also resided within surfaces reservoirs and experienced changes to surface emission rates as a function of house ventilation rate. Thus, this m/z could have had contributions from both true isoprene that persisted in the gas phase (e.g., from human breath), and from fragments of higher molecular weight aldehydes, where the aldehydes themselves likely persisted within surface reservoirs.
GC-PTR-MS approaches, in various forms, have been used for many years in outdoor and laboratory environments. Early applications of GC-PTR-MS measurements include those by Karl et al.23 and Fall et al.,24 who sampled biogenic VOCs at a remote outdoor site and in laboratory experiments. Shortly after, de Gouw et al. used a similar GC-PTR-MS system to investigate the chemical composition and common PTR-MS confounding signals for ion m/z ratios typically measured in urban air and in remote air.25 Recently, Coggon et al. collected and compiled PTR-MS and GC-PTR-MS measurements from several U.S. cities and aircraft campaigns and focused on quantifying interferences for commonly measured species in outdoor air: isoprene, benzene, and acetaldehyde.26
Similarly, a few past studies have deployed GC-PTR-MS systems in indoor environments to probe both isomer and fragmentation contributions. Schripp et al. described early applications of PTR-MS in the indoor environment, exploring a range of different indoor processes including a study of VOC emissions during paint drying, VOC emissions from a laser printer, VOC diffusion through building materials, and more.27 They specifically called for deployment of GC-MS systems alongside PTR-MS measurements for improved VOC identification. Claflin et al. deployed a GC-PTR-MS system with in situ adsorbent tube sampling and desorption in an athletic facility.28 They tracked the mixing ratios of a variety of prominent compounds in indoor air, and performed a quantitative assessment of monoterpene isomer distributions throughout the measurement period. In addition, Ernle et al. deployed a PTR-MS in parallel with a custom fast GC-MS and combined data from both techniques to measure volatile emissions from humans and elucidate interferences from fragments of C5–C10 aldehydes on m/z 69, where m/z 69 is commonly attributed to isoprene.29
Despite these advancements, a comprehensive assessment of isomer contributions and fragment interferences for commonly measured ions in typical indoor environmental conditions has not yet been performed. Evaluating the contribution of these potentially confounding signals is needed for a more robust interpretation of PTR-MS signals in indoor air. In the indoor environment, VOC sources, identities, concentrations, and chemistry differ from the outdoors.30 For example, indoor sources like cooking, cleaning, building materials, and humans dominate VOC emissions. Indoor VOC concentrations can be higher than outdoors due to source proximity and limited ventilation. Chemical transformations indoors are driven by different oxidant and light availability. Indoor surfaces play an important role as chemical sources, sinks, and media for multiphase chemistry due to the large surface area to volume ratio indoors relative to outdoors. Environmental conditions (e.g., temperature, humidity, and ventilation rate) may also impact indoor chemistry and surface fluxes. As a result, a specific focus on PTR-MS confounding signals for indoor VOCs is needed, as what is known about outdoor VOC isomer and fragment roles may not be applicable in the indoor environment.
Here, we applied the in situ Aerodyne Research Inc. GC system integrated with Vocus 2R proton transfer reaction time-of-flight mass spectrometer28 to automatically switch between real-time and GC sampling in a test house. Here on, these techniques will be called “PTR-MS” or “GC-PTR-MS” respectively. In this study, we identified isomer contributions and fragmentation interferences for key ions of indoor relevance using GC. Sampling was performed during the Chemical Assessment of Surfaces and Air (CASA) study.31 During this field campaign, a range of primary pollutant sources (e.g., cooking, wildfire smoke), oxidant conditions (e.g., variable ozone mixing ratios), and environmental conditions (e.g., changes to ventilation, relative humidity) were introduced to the test house. The goal of this work was to assess the variability of these isomer contributions and fragment-derived interferences in the PTR-MS measurements with changing indoor air composition.
2 Materials and Methods
CASA took place at the NIST Net-Zero Energy Residential Test Facility (NZERTF) in Gaithersburg, Maryland, from March 2 to April 11, 2022. The house's total volume was 1100 m3 and consisted of a basement, first floor (where most experiments were performed and measurements were collected), second floor, and attic. The outdoor average air change rate in the house during GC-PTR-MS measurement periods was 0.21 h−1, mostly controlled by a mechanical heat recovery ventilation system.During CASA, a range of indoor perturbation experiments were performed. The following experiments were captured by GC-PTR-MS measurements: cooking, injections of ammonia and carbon dioxide (to investigate indoor acid–base chemistry, henceforth referred to as “acid-base experiments”), fresh wildfire smoke addition, fresh wildfire smoke addition at high relative humidity, fresh wildfire smoke addition followed by introduction of ozone, aged wildfire smoke addition, aged wildfire smoke addition followed by introduction of ozone, no mechanical ventilation, background measurements, and background measurements at high relative humidity. Periodic outdoor air measurements were also collected for comparison. Experimental details for perturbations captured by GC-PTR-MS measurements are summarized in Table S1.† The number of samples collected under each indoor condition is summarized in Table S2.†
2.1 In situ gas chromatography
The in situ GC was installed upstream of the PTR-MS and sampled VOCs onto a multi-bed adsorbent trap. The adsorbent trap contained Tenax TA, Graphitized Carbon, and Carboxen 1000. The trap was held at 15 °C by a Peltier cooler during sampling. The instrument pulled 100 standard cubic centimeters per minute (sccm) air flow through the adsorbent trap for 10 minutes, resulting in a 1 L sample. These adsorbent tubes were evaluated by the manufacturer with sample volumes to 2 L of air at 150 sccm for a range of VOCs and showed no significant breakthrough. This is consistent with publicly available breakthrough data for a range of VOCs from product manufacturers and past testing of similar adsorbent materials under similar sampling conditions.32The trap was ramped to 300 °C to desorb analytes and transfer them to a packed focusing trap also held at 15 °C. The focusing trap was then quickly heated to 300 °C to desorb analytes and transfer them to the GC column. The oven was equipped with a mid-polarity MXT-624 GC column (30 m × 0.25 mm × 1.4 μm). From testing with authentic standards, we determined that this column was capable of separating species with Kovats non-polar retention indices in the range of 400–1100. Helium was used as carrier gas and flowed through the column at 2 sccm. The column oven temperature ramp started at 35 °C and increased to 225 °C over 10 minutes according to the following temperature ramp: hold at 35 °C for 55 s, increase from 35 °C to 100 °C from 55 s to 150 s, increase from 100 °C to 150 °C from 150 s to 350 s, increase from 150 °C to 225 °C from 350 s to 500 s, hold at 225 °C for 120 s. At the end of the column, analytes passed through a 0.6 m heated passivated stainless steel transfer line and were combined with 145 sccm zero air flow prior to entering the Vocus inlet. Sample collection on the adsorbent trap lasted 10 minutes and GC analysis lasted 10 minutes, for a total runtime of 20 minutes.
2.2 Proton transfer reaction mass spectrometry
GC eluents were ionized and detected by a Vocus PTR-MS as mentioned above.33 The ion source was operated with the following parameters: 100 °C reactor temperature, 2.2 mbar focusing ion molecule reactor pressure, 430 V discharge voltage, Vocus ion molecule reactor voltage difference (front-back) of 570 V (resulting in a reduced electric field (E/N) ratio of approximately 133 Td, similar to the range of E/N values in the compilation of measurements described in Coggon et al., which spanned 120–140 Td; many observations from Coggon et al. will be discussed further here for comparison with our observations26), voltage difference between skimmer and IMR back of −39 V, voltage difference between BSQ front and skimmer of −4.2 V, and BSQ RF voltage of 325 V.Instrument resolution (M/ΔM) at the time of the campaign was approximately 9000 at m/z 200. Data were collected from m/z 3–499 every 0.2 seconds to achieve enough data points over each chromatographic peak for optimal peak shape and statistics. The GC-PTR-MS ran at select times during the campaign (Table S1†) and real-time PTR-MS ran for the rest of the field campaign.
2.3 Instrument inlet
The GC-PTR-MS system was housed in the NZERTF garage. Using a 30.5 m, 1.27 cm (½′′) OD, 0.95 cm (3/8′′) ID piece of perfluoroalkoxy (PFA) tubing held at 50 °C, the instrument was connected to the main living/dining area on the first floor of the test house. Throughout the campaign, 4 L min−1 of room air was pulled through the inlet tubing. The inlet was protected from particle infiltration by a Teflon filter housed in a PFA filter holder.2.4 GC-PTR-MS zeroing and calibration
Zero air samples were collected both in-field and post-campaign with zero air. During the campaign, calibrations were performed with the following VOCs in a calibration cylinder: ethanol, acetonitrile, acrolein, furan, isoprene, 1,4-dioxane, toluene, 2-hexanone, hexanal, octamethylcyclotetrasiloxane (D4 siloxane), and limonene. During CASA, 1 L zero air samples and 1 L calibration gas samples were collected on the GC every 3–5 hours.After the campaign, we performed the following additional calibrations from another calibration cylinder: 2-butanone, acrylonitrile, acetone, benzene, o-xylene, chlorobenzene, 1,3,5-trimethylbenzene, α-pinene, and phenol. Acetonitrile, toluene, and isoprene were also present in this mixture and re-calibrated to track during vs. post-campaign signal differences. Finally, the following additional calibrations were performed by evaporating liquid authentic standards into zero air flow,34 and collecting a 1 L sample of that flow onto the GC sample trap as above: pyrrole, β-pinene, nonanal, furfural, 2-heptanone, camphene, 3-carene, and butanal. For calibrated VOCs, we express their mixing ratios in parts-per-billion (ppb), which is defined as the mole fraction of a nmol of VOC per mole of air.
2.5 Data post-processing
All data processing was performed in Tofware v3.2.5 and in TERN 2.2.19b. First, in Tofware, mass calibrations were performed (mean residual for each ion was <10 ppm, where ppm is defined as μmol in 1 mole of air), then peak shape and peak width were defined. Calibrant ions were specifically targeted and their high-resolution m/z peaks were fit. Data were then imported to TERN, and peak areas for each high resolution m/z were determined by mathematically fitting peak functions to the acquired chromatographic data, while minimizing residuals between the mathematically fitted peak and the acquired signal.352.6 Considerations when interpreting these results
We note that the analyses presented below are inherently limited by the trapping and desorption efficiency of the thermal desorption (TD) pre-concentration unit and the specifications of the GC column type and temperature ramp method; compounds that do not effectively desorb from the adsorbent trap, that are thermally labile and fragment in the TD system, or that are not transferred through the GC column will not be detectable by the downstream PTR-MS (for example, peroxides, and high or low volatility analytes below or above the Kovats non-polar retention index range of 400–1100 mentioned above). As such, only ions that are both TD-GC amenable are included in these analyses and careful attention should be paid to the operating conditions of the TD-GC-PTR-MS setup used here when extrapolating these findings to other systems.All chemicals are tracked in this work at their protonated m/z. For most of the compounds analyzed (i.e., those in Table 1), we do not expect any interferences from water cluster adducts, but water clustering could create artifacts for ions with two or more oxygen atoms in their molecular formula. The one exception here is 1,4-dioxane, which was observed to have a small water cluster contribution. Also, we did not observe significant contribution from charge transfer or hydride transfer products in the extracted ion chromatograms (EICs) for these selected chemicals. A detailed analysis of product ion distribution is outside the scope of this paper, but we refer readers to Link et al.'s product ion distribution investigation.17 We note that different PTR-MS instruments from other manufacturers have different ionization region configurations, which may result in different product ion distributions (e.g., ratios of charge transfer products to proton transfer products).
| Compound | Indoor average % of EIC | Indoor standard deviation | % range across indoor conditions | Difference (average % of EIC indoor–outdoor) | EIC figure example |
|---|---|---|---|---|---|
| a EIC not integrated due to lack of visible confounding EIC peaks. | |||||
| Dominant peak as parent ion (>75% of EIC) | |||||
| D4 siloxanea | 100 | — | — | 13 | S5 |
| D5 siloxanea | 100 | — | — | 45 | S6 |
| Ethanola | 100 | — | — | 0 | S7 |
| Furfural | 79 | 1 | 77–81 | 11 | S8 |
| Nonanal | 86 | 2 | 84–89 | 1 | S9 |
| Acetone | 79 | 3 | 74–83 | −4 | S10 |
| Pyrrole | 91 | 5 | 84–98 | 21 | S11 |
| Acetonitrile | 79 | 6 | 71–92 | 24 | S12 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Significant contribution from peaks outside 100 s of parent ion (>50% of EIC) | |||||
| Isoprene | 2 | 2 | 0–6 | −18 | S13 |
| Benzene | 24 | 8 | 13–41 | −19 | S14 |
|
|||||
| Significant contribution from peaks within 100 s of parent ion (>25% of EIC) | |||||
| 2-Hexanone | 9 | 2 | 6–13 | −8 | S15 |
| Limonene | 8 | 2 | 4–10 | 1 | S16 |
| Butanal | 10 | 3 | 5–16 | −8 | S17 |
| 1,4-Dioxane | 8 | 3 | 4–14 | 4 | S18 |
| Hexanal | 70 | 3 | 66–75 | 37 | S15 |
| 1,3,5-Trimethylbenzene | 13 | 3 | 9–20 | 2 | S19 |
| 2-Butanone | 57 | 5 | 50–65 | −34 | S17 |
| o-Xylene | 33 | 6 | 29–49 | 10 | S20 |
| 2-Heptanone | 49 | 6 | 41–60 | 13 | S21 |
| α-Pinene | 58 | 6 | 53–75 | −20 | S16 |
|
|||||
| Increased sensitivity to indoor conditions (highest standard deviations) | |||||
| Acrolein | 56 | 8 | 46–71 | 18 | S22 |
| Furan | 42 | 11 | 26–56 | −14 | S23 |
| Toluene | 55 | 12 | 33–72 | −18 | S24 |
| Acrylonitrile | 70 | 14 | 46–87 | −28 | S25 |
| Chlorobenzene | 72 | 18 | 36–97 | 59 | S26 |
Peak areas from the nearest zero air sample were subtracted from all sample peaks for each target chemical. A discussion of possible positive and negative artifacts and agreement between GC-PTR-MS and real-time PTR-MS is presented in Section S2 and Fig. S2–S4.† In general, we observed good agreement between GC-PTR-MS and real-time PTR-MS signals, indicating no significant losses in the GC sampling and analysis system, and no significant positive or negative artifacts.
2.7 Searching for isomers and fragments based on peak retention time
To investigate the distribution of isomers and fragments for each compound of interest, we identified the retention time of each compound and set a range of 100 s around that compound's retention time to estimate possible isomer contributions. This range of 100 s was determined based on inspecting EICs for all calibrated compounds which contained a series of isomers, including the C6 carbonyls, xylenes, trimethylbenzenes, and terpenes. The C6 carbonyls (at C6H12OH+, including 3-hexanone, 2-hexanone, and hexanal, identified by authentic standards and Kovats numbers) eluted within <10 s of each other. The xylenes (at C8H11+, including m-xylene, p-xylene, o-xylene) eluted within <50 s of each other. The trimethylbenzenes (at C9H13+, including 1,2,3-trimethylbenzene, 1,3,4-trimethylbenzene, and 1,3,5-trimethylbenzene) eluted within <60 s of each other. The terpenes (at C10H17+, including α-pinene, camphene, β-pinene, carene, and limonene) eluted within <80 s of each other. Thus, a conservative window of 100 s was set as the expected time window during which isomers of a particular compound may elute given the GC column and oven temperature program used here. After 100 s, most EIC peaks likely consisted of fragments of higher molecular weight species formed in the ionization region. This window is subject to some uncertainty and should be treated as an estimated rather than absolute range for isomer elution and fragmentation product elution. We note that this 100 s window is also specific to the chromatographic separation method used here, and similar analysis of isomer elution would need to be re-performed for other chromatographic methods and conditions. Peaks falling within this 100 s range may include isomers as well as other closely eluting peaks corresponding to fragments or other unidentified species, and are labeled below as “Peaks within 100 s”. Peaks falling outside this range are labeled as “Peaks outside of 100 seconds”, and possibly include a greater contribution from fragments as well as other unidentified species.2.8 Computing relative contribution of compounds to their EIC
For GC-PTR-MS measurements, when reporting distributions of parent ion proton transfer products and other confounding EIC peaks, peak areas in units of ion counts are used. However, most data here are presented as the peak area of a particular compound of interest relative to the area under its full EIC, and are thus shown in units of percent contribution. A baseline was set for each EIC by averaging the first and last 10 s of each EIC (inspecting values to ensure limited signal change across chromatogram start and end periods), and this baseline value was subsequently subtracted from the EIC. If, upon inspection, an EIC exhibited significant increasing baseline, then EIC integration was limited to the region with steady baseline signal. For example, for acrylonitrile, EIC integration was consistently limited to only the first 400 s of elution time due to a reliably and substantially increasing baseline from 400 to 600 s in all chromatograms. For three chemicals with no visible additional EIC peaks where the parent ion contributed >85% of the EIC signal but less than 100% when the baseline was integrated (C2H6OH+ (ethanol), C8H24O4Si4H+ (D4 siloxane), and C10H30O5Si5H+ (D5 siloxane)), the integral of the baseline was not included and parent ion peaks were assigned a contribution of 100% to their EICs to limit the relative over-importance of baseline noise in these three specific cases. The role of impurities in contributing to the observed ionization region fragmentation: In the Vocus PTR-MS, the BSQ severely reduces the transmission of low molecular weight ions below approximately m/z 50.33 This makes it difficult to accurately quantify the roles of other impurities that may be contributing to unintended fragmentation in the system including O2+ and NO+. While the fragmentation reactions occurring in our instrument's ionization region may be occurring by multiple mechanisms, including via collision with impurity ions or via dehydration and subsequent fragmentation (e.g., for protonated acids and alcohols), a precise identification of these mechanisms is outside the scope of this work. Through our study, we intend to highlight that under typical Vocus PTR-MS operating conditions (e.g., conditions listed in the Materials and Methods section), we observe interferences from unintended fragmentation products, and we aim to bring attention to the relative role of these fragmentation interferences under these typical Vocus PTR-MS settings.3 Results
VOC mixing ratios in the test house varied with each experiment. We focus on select gases calibrated in-field via a calibration gas cylinder and campaign-wide time series for those gases as in Fig. 1. We note that other VOCs indoors are often present at high levels and could also have confounding additional signals at their m/z ratios (for instance, formic acid, acetic acid, acetaldehyde, and methanol4 among others), but we focus on directly calibrated chemicals for this analysis because we could definitively identify the ion in question amongst other additional confounding peaks in the sometimes complex EICs. Mixing ratios ranged from ppt-level (where ppt is defined as pmol in 1 mole of air) to ppb-level and varied based on the experiment performed. Outdoor mixing ratios, while not the focus of this work, are presented in Fig. 1 for contrast. As mentioned in the Materials and Methods section, we only consider ions that are TD-GC amenable in all subsequent analyses.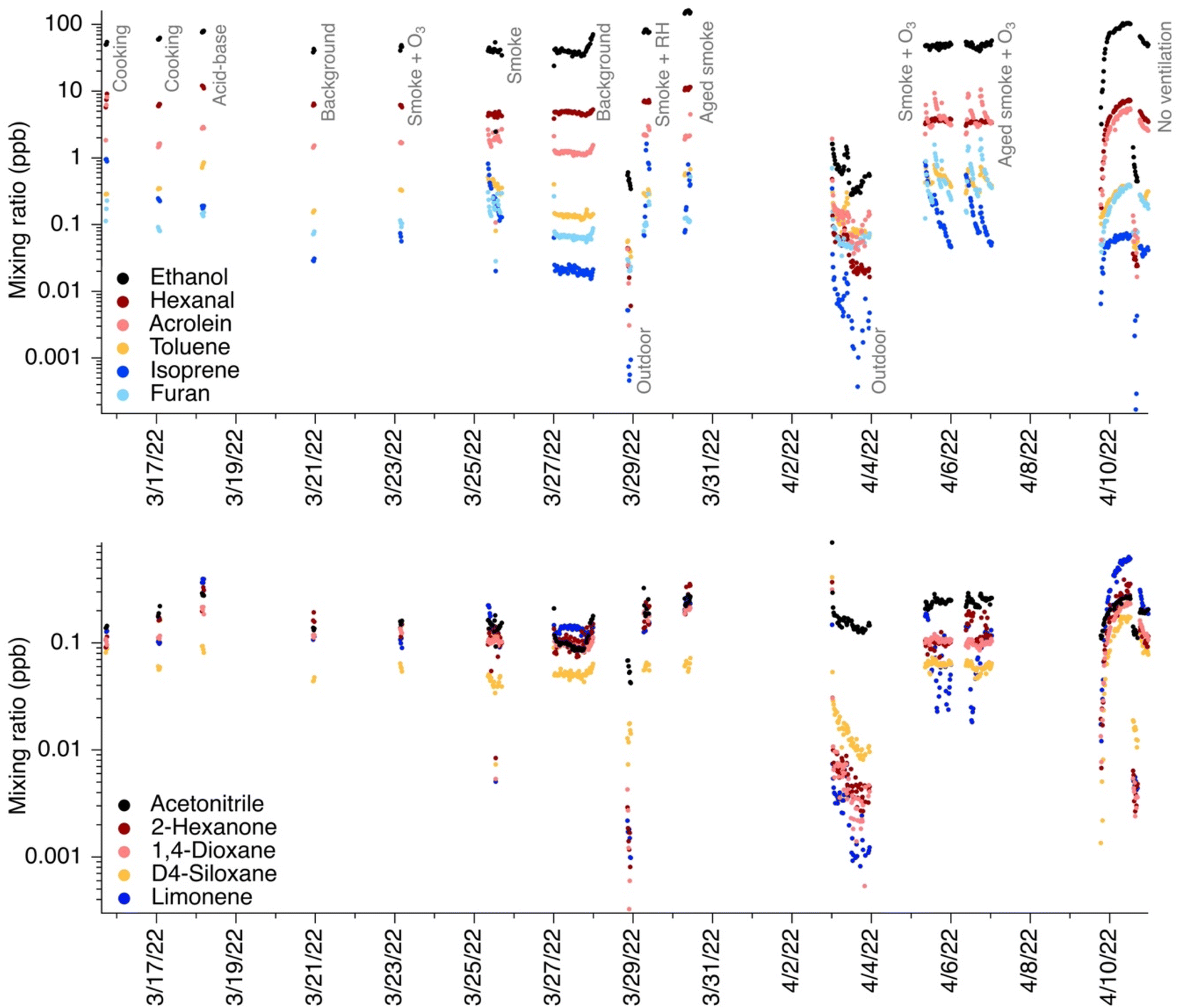 | ||
| Fig. 1 Campaign-wide mixing ratios of GC-PTR-MS in-field calibrated species. Each marker represents the mixing ratio of a target chemical from a GC chromatogram collected either indoors or outdoors. Key experiments performed closest to or during GC-PTR-MS sampling are labeled in grey text in the top panel. | ||
For compounds commonly reported in indoor air, we calculated the peak area of each compound of interest and compared it to the area under the total EIC at that m/z. Other peaks in an EIC include isomers of the species of interest, fragments of protonated higher molecular weight compounds, or unidentified species. Table 1 summarizes the average contribution of each ion of interest to its EIC. All data are displayed in Fig. S1.† Fig. S5–S26† show example EICs for each chemical; note that these EICs show average background signals and average outdoor signals, but do not reflect chromatograms during each different experiment type.
In subsequent descriptions of results, 325 ambient chromatograms are considered. In this section, key sources of target chemicals are mentioned, focusing on those present in the NZERTF during or close to GC-PTR-MS measurements (cooking, acid–base (i.e., injections of ammonia and carbon dioxide), biomass burning smoke, building material emissions, outdoor air infiltration, humans (not directly captured by measurements, but humans entered/exited the house periodically throughout the campaign), and cleaning (not directly captured by measurements, but surface cleaning days occurred between GC-PTR-MS measurement periods)).
3.1 The chemicals targeted here have varying levels of contributions to their EICs
We grouped compounds together in three categories: (1) compounds with significant contribution from their parent ion, defined here for comparison purposes as >75% of their EIC; (2) compounds with significant contributions from peaks outside of 100 s from the parent ion, defined here for comparison as >50% of their EIC (i.e., other peaks in the EIC do not elute close to the parent ion peak); and (3) compounds with significant contributions from other peaks within 100 s from the parent ion, defined here for comparison purposes as >25% of their EIC, and with only <25% of their EIC being from peaks outside of 100 s (i.e., other peaks in the EIC elute close to the parent ion peak).Some observed chemicals showed significant contribution from the parent ion. Fig. 2A highlights two examples of species which showed limited interference from fragments, limited contributions from isomers, and limited contributions from other unidentified ions: acetone and pyrrole. These two ions are elaborated on below. Other species falling into this category include furfural, nonanal, acetonitrile, ethanol, D4 siloxane, and D5 siloxane. This set of compounds tended to have consistent contributions from the parent ion across indoor conditions (standard deviation ≤6%, Table 1).
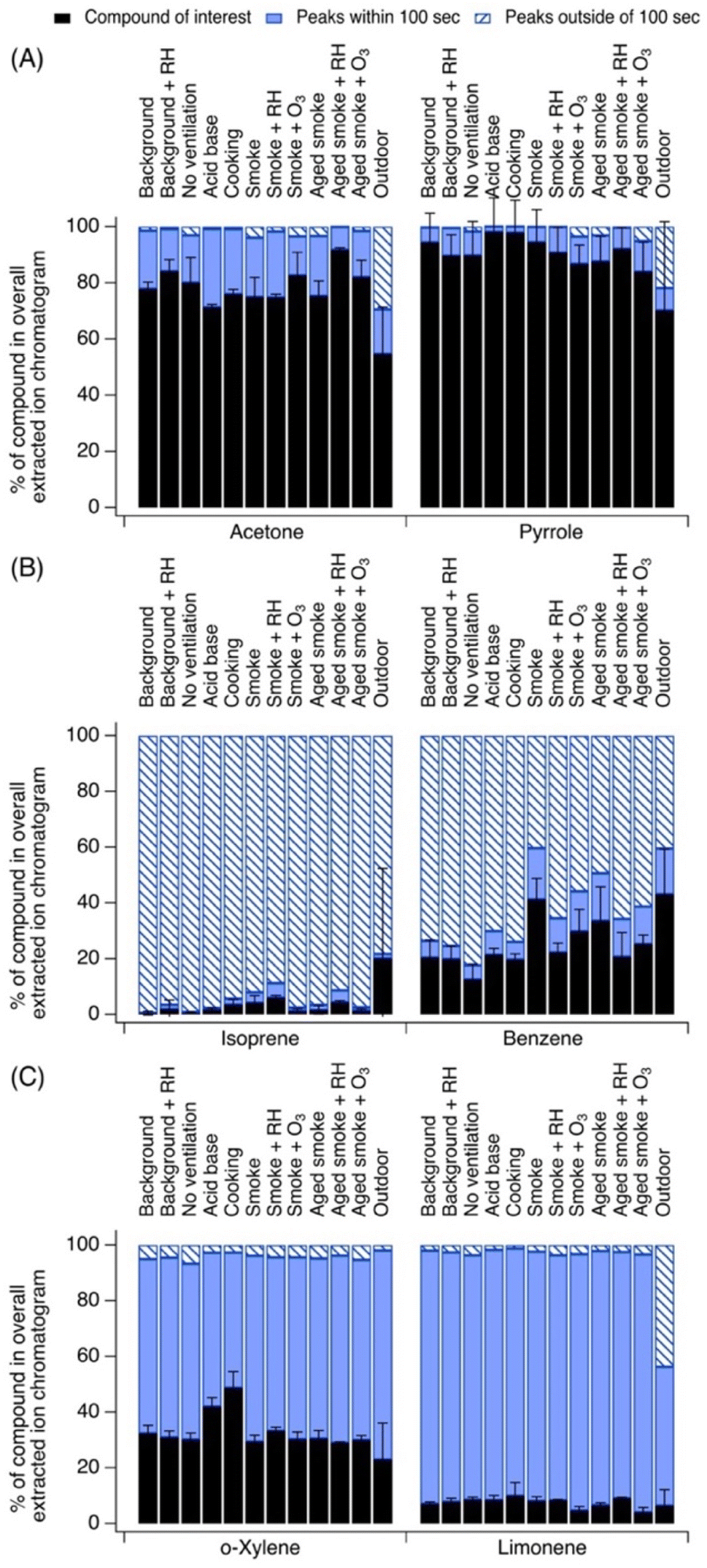 | ||
| Fig. 2 Select ions showing (A) dominant contribution from parent ion in EIC, (B) dominant EIC contributions from peaks eluting within of 100 s of the parent ion's retention time, and (C) dominant EIC contributions from peaks eluting outside of 100 s of parent ion's retention time. Error bars represent standard deviation from the mean. Other examples of species in these categories are discussed in Section S1.† | ||
Fig. 2B highlights two examples of species which showed significant interference from peaks eluting greater than 100 s from the parent ion's retention time: isoprene and benzene. Both of these ions are well-known to have significant fragment interference from other deployments of PTR-MS in outdoor settings, and exhibited these strong fragment interferences consistently throughout the campaign (standard deviation ≤8%, Table 1).
Fig. 2C highlights two examples of species which showed significant confounding contributions from closely eluting peaks, appearing within 100 s of the parent ion's retention time: o-xylene and limonene. Other species falling into this category included 1,3,5-trimethylbenzene, α-pinene, 2-butanone, butanal, 1,4-dioxane, 2-hexanone, hexanal, and 2-heptanone. These species also exhibited consistent behavior throughout the campaign (standard deviation ≤ 6%, Table 1).
3.1.1.1 Acetone. Acetone contributed on average 79 ± 3% to the EIC at C3H6OH+. A minor peak for propanal was present in some chromatograms, as well as small contributions from fragments of other ions such as C4H8OH+, C3H6O2H+, C3H8O3H+, and C5H10OH+. The contributions of these fragments were slightly elevated during cooking experiments (3% instead of 1% to 2%), suggesting that these could be aldehydes emitted from heated cooking oils19 that fragment and contribute to the EIC at C3H6OH+. Both Coggon et al. and DeGouw et al. observed acetone and propanal at this ion m/z outdoors, with the predominant signal coming from acetone,25,26 consistent with our observations here. Key sources of acetone indoors include human breath and a solvent in some personal care products,36 as well as a solvent in a cleaning product possibly applied in the home prior to the campaign.37 Acetone's relatively consistent contribution to its EIC reflects its consistent higher levels in the house (due to its prominent human and building sources) relative to the levels of its other confounding EIC peaks.
3.1.1.2 Pyrrole. Pyrrole contributed 91 ± 4% of its indoor EIC at C4H5NH+. Other contributors included small contributions from possible isomers eluting immediately before and after the pyrrole peak. Indoors, pyrrole may arise from cooking4,38 and from wood smoke.39 Despite its more episodic sources, pyrrole's relatively consistent contribution to its EIC also reflects its higher concentrations in the house relative to the levels of its other confounding EIC peaks, which had low intensity and did not exhibit significant temporal variability throughout the campaign.
3.1.2.1 Isoprene. Here, we observed that isoprene contributed on average only 2 ± 2% to its EIC at C5H9+ indoors. Fewer contributions from other species were observed outdoors due to a greater natural isoprene source. Indoors, interferences were caused almost exclusively by fragmentation products of higher molecular weight aldehydes which were prominent in the house including major contributions from dehydration and fragmentation products of pentanal (C5H10OH+), octanal (C8H16OH+), and nonanal (C9H18OH+). This is consistent with past outdoor and indoor measurements, though the contribution of fragments was generally higher in our observations than other past mostly outdoor studies. For example, Coggon et al. observed that interferences at C5H9+ in outdoor Los Angeles measurements represented 90% of the C5H9+ signal at nighttime. In Pasadena during the day, closer to isoprene-emitting vegetation, interferences were reduced to 10%. Similarly low interferences were measured at a site on Long Island.26 Vermeuel et al. collected comparable measurements in a forest in northern Wisconsin,40 where they observed interferences at C5H9+ reaching nearly 90%, with large contributions from octanal and nonanal fragments. They attributed the interferences to ozone reactions with adsorbent material in the GC's thermal desorption preconcentration unit rather than a true aldehyde source, given the improbability of these aldehydes occurring naturally in the forest. Notably, Coggon et al. discuss recommendations for corrections to the C5H9+ signal using real-time data and the signal of interfering species, leveraging diurnal patterns in outdoor isoprene levels in urban outdoor air. However, since interferences are sensitive to the environment being sampled, these recommendations may or may not be applicable to indoor locations.
In our indoor measurements, the minor contribution of isoprene from outdoor infiltration is exacerbated by the higher mixing ratios of aldehydes from cooking or from ozonolysis reactions. One key source of isoprene indoors is human breath,41 but the CASA study occurred in an unoccupied test house that had little human presence overall, and no human presence during the measurements themselves, thus limiting the importance of this source relative to others. Sources of aldehydes (and thus their interfering fragmentation products at C5H9+) – including cooking (e.g., from the autoxidation of heated oils,19 which could have arisen during pan frying experiments) and ozonolysis of unsaturated hydrocarbons (e.g., squalene and unsaturated triglycerides1,42) – were likely present at higher levels than isoprene itself in this environment.
3.1.2.2 Benzene. Benzene contributed on average 24 ± 8% to the EIC at C6H7+, with notably lower interferences during fresh smoke experiments indoors, as well as during outdoor sampling. Interferences in this case were driven largely by fragments of higher molecular weight aromatics including species like ethylbenzenes, propyl benzenes, butyl benzenes, diethylbenzenes, and benzaldehyde, and unidentified ions. de Gouw et al. reported that under optimized E/N conditions, benzene might have limited interference, though they reported some contribution from ethyl benzene at the C6H7+ ion.25 Coggon et al. reported contributions form ethyl benzene and benzaldehyde at C6H7+as well, consisting of 3% of signal measured in Pasadena but 38% of signal measured at a background site near Las Vegas and 44% measured in Detroit.26 As noted above, Coggon et al. also presented recommendations for correcting benzene signals using interfering time series in outdoor urban air. One suggested method is to use the benzene charge transfer product (C6H6+), as Coggon et al. reported it to have no interferences.26 In our CASA observations, the benzene charge transfer product represented 50–75% of the signal at the C6H6+ EIC but the EIC was not entirely free from confounding peaks. The presence of the charge transfer product may also be subject to the levels of impurities (O2+, NO+) in the system thus it should be used cautiously, but it may serve as an additional point of comparison if users need to simplify the benzene EIC for identification and quantification.
While monoterpenes and monoterpenoid fragments may also contribute at C6H7+, we observed very limited fragmentation interference from these precursors in our sampling conditions.
In this environment, indoor sources of benzene, toluene, ethylbenzene, and xylene (BTEX) include cooking43 and infiltrated wildfire smoke.39 Other aromatics like trimethylbenzenes (discussed below) have similar indoor sources.44 In occupied environments, one possible contributor to benzene signals is benzalkonium chloride from the application of disinfectants.45
3.1.3.1 o-Xylene. o-Xylene contributed on average 33 ± 6% to the C8H11+ EIC indoors. This was consistent across all indoor experiments, with slightly less contribution from confounding peaks in the EIC during cooking and acid–base experiments. Major ions at C8H11+ included isobaric species m-xylene, p-xylene, and ethylbenzene. The contributions of interfering fragments of higher molecular weight species were also consistent across experiments, arising from higher molecular weight aromatics.
3.1.3.2 Limonene and α-pinene. Limonene contributed on average 8 ± 2% to the EIC at C10H17+ indoors, while α-pinene contributed on average 58 ± 6%. α-Pinene's contribution was the greatest cooking and when sampling outdoors, while limonene's contributions were more similar across all measured chromatograms. Other terpenes detected at C10H17+ included camphene, β-pinene, carene.
While limonene is expected to dominate the terpene distribution indoors due to its prominence in many fragranced consumer products (e.g., as observed by Claflin et al. in an athletic center28) this was a test house with no such sources. Thus, α-pinene dominated the terpene distribution, likely arising from building material emissions. For example, upon sampling in the NZERTF following initial construction, Poppendieck et al. measured α-pinene emission rates at 9–11 times limonene's emission rates.37 Some α-pinene could have also infiltrated from outdoor air, but as shown in Fig. S16,† the α-pinene signal was stronger indoors than outdoors, suggesting that infiltration was not the dominant source in the test house during these measurements.
Monoterpenes have many indoor sources including the building materials of a home (for example, from wood products),37 cooking with herbs and spices,46 and cleaning products and other fragranced consumer goods and personal care products.47 Wildfire smoke infiltration experiments also likely contributed monoterpenes to the house's air.39
3.2 The chemicals observed have different indoor and outdoor confounding peaks in their EICs
The role of confounding peaks in the EICs collected indoors vs. outdoors was sometimes variable. Some species showed similar behavior across the two environments, while others differed greatly. For instance, Fig. 3A shows two examples of species with similar indoor vs. outdoor contributions of the parent ion peak (<5% difference): 1,3,5-trimethylbenzene and nonanal. Other chemicals that exhibited very little difference indoors vs. outdoors in terms of their EIC contribution include limonene, 1,4-dioxane, and acetone. In contrast, Fig. 3B shows two examples of species with differing indoor vs. outdoor contributions of the parent ion and confounding EIC peaks (>25% difference): chlorobenzene and 2-butanone. Other chemicals in this category included acetonitrile, hexanal, and acrylonitrile.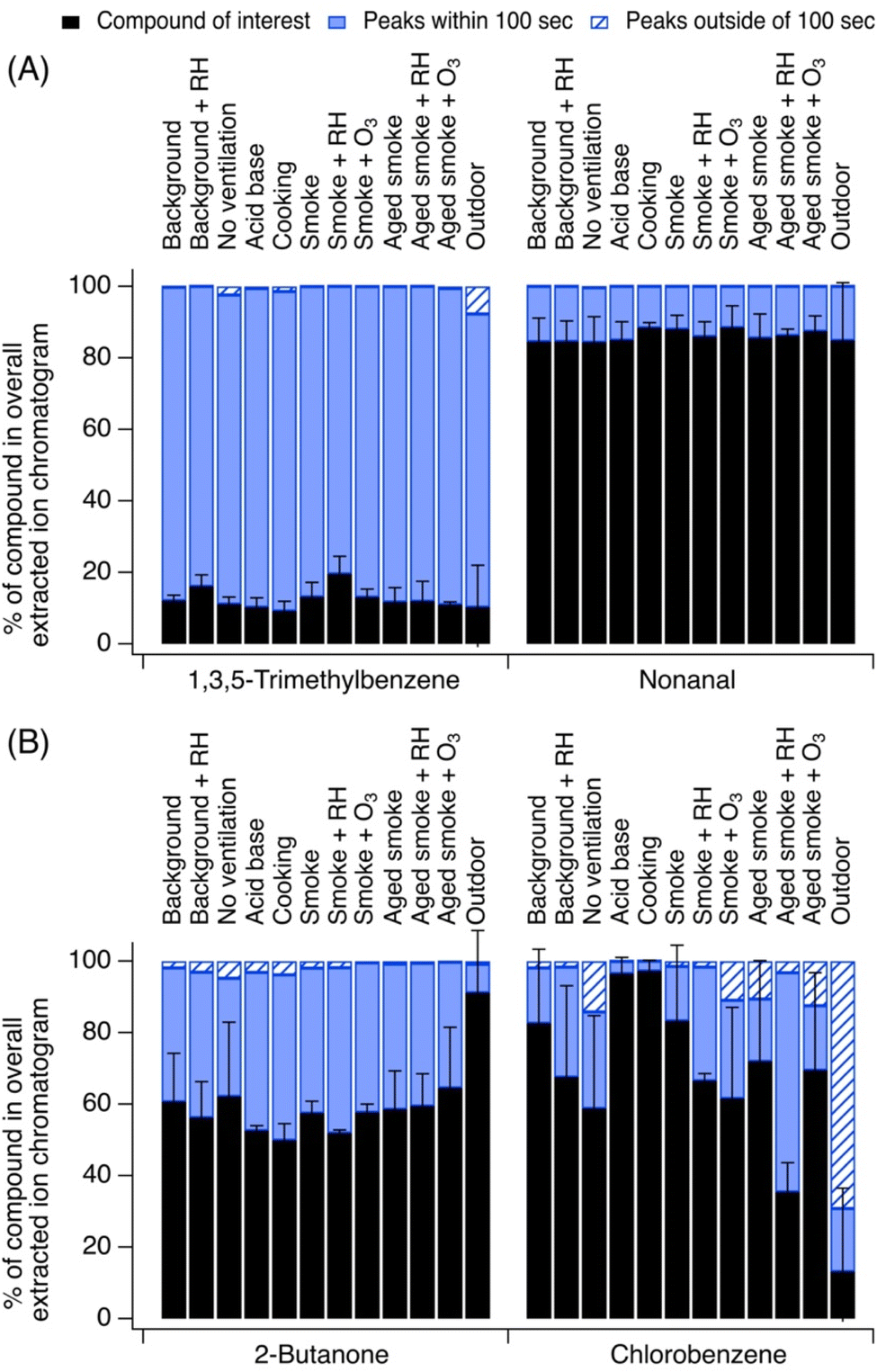 | ||
| Fig. 3 Select ions showing (A) species with similar indoor vs. outdoor parent ion and confounding peak contributions and (B) species with differing indoor vs. outdoor contributions. Error bars represent standard deviation from the mean. | ||
3.2.1.1 1,3,5-Trimethylbenzene. 1,3,5-Trimethylbenzene contributed on average 13 ± 3% to the EIC at C9H13+ indoors, consistently across indoor experiments (relative to 10 ± 11% outdoors).
Contributions of fragmentation products at this EIC were negligible, and nearly all other signals at C9H13+ came from expected isomers including 1,2,3-trimethylbenzene and 1,2,4-trimethylbenzene. Two other unidentified but likely isomer peaks were also observed in the EIC, which could be propylbenzene or ethyl toluene structures.
3.2.1.2 Nonanal. Nonanal contributed on average 86 ± 2% to its EIC at C9H18OH+, consistently across indoor experiments (relative to 85 ± 16% outdoors). Interference from fragments of higher molecular weight species and unknown ions was negligible. Some signal at this EIC was caused by a minor 9-carbon ketone contribution. The use of heated cooking oils is an important source of indoor nonanal, and nonanal is also formed as an ozonolysis product of unsaturated fatty acids and hydrocarbons likely present on building material surfaces and furnishings.1,48,49
3.2.2.1 2-Butanone and n-butanal. These 4-carbon carbonyls had very minimal EIC contributions from fragments or unknown ions but showed a distribution of isomer peaks at C4H8OH+. 2-Butanone (methyl ethyl ketone) contributed on average 57 ± 5% to this EIC indoors with the lowest contributions from other confounding EIC peaks outside (2-butanone had a contribution of 91 ± 17% of the EIC outdoors) which is consistent with it being a major isoprene oxidation product. Indoors, butanal contributed 10 ± 3% to this EIC (relative to 17 ± 15% outdoors). These contributions were consistent across indoor experiment types. We observed three other closely eluting peaks (i.e., well within 100 s of the carbonyl set) which were likely structural isomers that may include isobutyraldehyde as well as unsaturated alcohol-containing structures, ether-containing structures, or heterocyclic oxygen-containing structures (e.g., tetrahydrofuran). 2-Butanone may have arisen from the house materials themselves,37 while butanal could have arisen from heated cooking oil use,50 and both may have been present in wood smoke.39
3.2.2.2 Chlorobenzene. Chlorobenzene contributed 72 ± 18% of its indoor EIC at C6H5ClH+. Contributions varied with source; confounding EIC peaks were the lowest during acid–base experiments (chlorobenzene was 97 ± 3% of the EIC) and cooking (chlorobenzene was 97% ± 3 of the EIC), while they were higher in smoke experiments (chlorobenzene was 35 to 83% of the EIC) and the highest outdoors (chlorobenzene was 13 ± 23% of EIC). The outdoor chlorobenzene signal was one order of magnitude lower than indoors, and a noisy baseline contributed to higher uncertainty in peak integration, yielding more “apparent” non-chlorobenzene signal. The major interfering peak, observed both indoors and outdoors, eluted 3 minutes earlier than chlorobenzene itself. Its identity is uncertain as it is unlikely to be caused by a fragment of a higher molecular weight chlorinated compound (e.g., a dichlorobenzene) due to its early elution time. Structural isomers are also unlikely. Chlorobenzene is sometimes used as a solvent in pesticides (along with dichlorobenzenes51) or as a neat chemical in moth repellents52 and thus could be present in homes that periodically apply these products. It can also be used as a solvent for paints. None of these sources of chlorobenzene were likely to be strong in the NZERTF.
3.3 The chemicals observed have confounding peak signals with variable responses to changing indoor conditions
Last, we emphasize chemicals that showed sensitivity towards specific indoor activities, and chemicals that did not. Fig. 4A highlights acrolein and furan, which exhibited variability in their contributions to their EICs across indoor activities (>5% standard deviation in Table 1). Other chemicals in this grouping include acetonitrile, benzene, o-xylene, 2-heptanone, α-pinene, toluene, acrylonitrile, and chlorobenzene. Fig. 4B contrasts this by highlighting furfural and hexanal, which exhibited similar contributions throughout the campaign (<5% standard deviation). Many other chemicals fall into this grouping including nonanal, acetone, isoprene, 2-hexanone, limonene, butanal, 1,4-dioxane, hexanal, and 1,3,5-trimethylbenzene. | ||
| Fig. 4 Select ions showing (A) species that showed variable contribution to their EIC across the indoor conditions sampled and (B) with consistent contribution throughout the campaign. Error bars represent standard deviation from the mean. | ||
3.3.1.1 Acrolein. Acrolein contributed on average 56 ± 8% to the EIC at C3H4OH+ with the lowest level of confounding EIC peaks measured during aged smoke experiments (acrolein was 50 to 70% of its EIC). Interferences came from signals such as C5H10OH+ (pentanal), C6H12OH+ (hexanal), C7H14OH+ (heptanal), C8H16OH+ (octanal), and C9H18OH+ (nonanal). The contribution of these fragments outdoors was greater than in any indoor condition (acrolein was only 31 ± 36% of its EIC), but indoors there were several strong sources of acrolein introduced to the house which served to increase the contribution of acrolein itself to the C3H4OH+ signal (e.g., heated oils used in cooking experiments,50 smoke infiltration experiments39). Acrolein may also come from wood and wood-based products.53 A recent HOMEChem study showed that acrolein had a high hazard quotient,54 so it is important to accurately quantify this chemical in indoor air, accounting for both the role of fragments and isomers.
3.3.1.2 Furan. Furan contributed on average 42 ± 11% to the EIC at C4H4OH+. Interferences were strong across indoor experiments. Interferences were slightly reduced during smoke experiments (furan was 36 to 56% of its EIC), which is consistent with wildfire smoke being an important source of furan (and a range of alkyl-substituted and oxygenated furan derivatives).39 Indoors, the contribution of fragments and unknown ions to the EIC signal was generally greater, due to a major interference from a fragmentation product of furfural at C5H4O2H+.
3.3.2.1 Furfural. Furfural contributed on average 79 ± 1% to its EIC at C5H4O2H+ and this was consistent across indoor experiments. It had little indoor interference due to fragments or other unknown ions. Outdoors, contributions at this EIC from other interfering species were more important, including C6H12O2H+, C6H6O2H+, and C8H16O2H+. Indoors, wood decomposition is an important furfural source.2 Furfural could have also entered the home during wood smoke infiltration experiments.39
3.3.2.2 n-Hexanal and 2-hexanone. Hexanal contributed 70 ± 3% to the EIC at C6H12OH+, and 2-hexanone contributed on average 9 ± 2%. Both remained consistent across indoor experiments but hexanal showed increased presence of other confounding EIC peaks outdoors (hexanal was only 34 ± 19% of its EIC). For both C6 carbonyls, the presence of other EIC peaks was almost exclusively due to closely eluting carbonyl isomers observed (3-hexanone, 2-hexanone). Hexanal dominated the C6 carbonyl signal, and it is a commonly measured compound in indoor air. Hexanal was one of the major species measured from the house's building materials,37 likely derived from wood products. One past study examined volatile emissions from building materials including flooring, wall coverings, and adhesives, and classified hexanal as the dominant odiferous building material-derived compound.55 Hexanal is also an aldehyde emitted from heated cooking oils.50 Hexanal can be formed from the ozonolysis of linoleic acid, which may be present on indoor surfaces in the house from indoor cooking oil use.20 2-Hexanone is not commonly measured indoors, though it may sometimes be used as a solvent and has been detected in low levels in residential environments.56
4 Discussion
4.1 Importance of accounting for instrument operating conditions and sampling environment conditions
The presence of confounding EIC peaks, beyond the parent ion, at each ion of interest can be instrument operating condition-specific (varying by ionization region pressure, temperature, and voltage parameters, often summarized by the quantity of reduced electric field or E/N), and environment-specific. For instance, an indoor environment containing high quantities of outdoor air might contain more true isoprene (C5H9+) than an indoor environment with high quantities of C5–C10 aldehydes (known contributors of fragments at the C5H9+ ion).29 The impact of these aldehyde fragments might be enhanced for instruments running at higher E/N conditions or reduced in instruments operating at lower E/N conditions. Importantly, other voltage settings in the front end of the instrument such as the voltage settings of the instrument's skimmers and the voltage at the front end of the BSQ can also influence fragmentation patterns for the same E/N settings.26In addition, as the concentration of the target species increases in indoor air, perhaps due to an emission event, this could increase its prominence over any other species contributing to the EIC. An emission event of an interfering species could also cause confounding peaks for a particular chemical to change with time.
Therefore, both instrument operating conditions and sampling environment must be factored into any analyses of PTR-MS ion contributions. As such, while it can be difficult to generalize the findings for the compounds discussed here, observations from this unoccupied test house can be used to understand how different mixtures of gases under different indoor conditions may yield distinct contributions of different chemical species within each PTR-MS signal.
4.2 General trends in the contributions of isomers and interferences of fragments in the GC-Vocus setup at CASA
In this study, we observed some parent ions that were the primary contributor to their EICs, including species that often showed elevated levels indoors, that did not have prominent isomers, and that did not overlap with fragmentation products of higher molecular weight species that eluted from the column with this chromatographic method (e.g., furfural from building materials, nonanal from cooking and ozonolysis of unsaturated hydrocarbons on surfaces, siloxanes from personal care products, acetone from human breath). Of the species classified as having their parent ion as their dominant EIC peak in Table 1, furfural, nonanal, the siloxanes, and pyrrole eluted from the GC column more than halfway through the elution time. The later these compounds elute, the lower the chance of fragmentation product interference from higher molecular weight chemicals, as those chemicals must also be TD-GC amenable with this chromatographic method. They each also had strong indoor signals and unique chemical structures without important isomers in this particular indoor environment. Acetone, ethanol, and acetonitrile all contain 2–3 carbon atoms; due to their small size, they are unlikely to have many prevalent isomers but could have their signals confounded by higher molecular weight fragmentation products eluting later during the chromatographic method. In this case, fragmentation interference was not present, allowing these parent ions to dominate their EICs as well.Some chemicals, like isoprene and benzene, had significant contributions from fragments of higher molecular weight species and from unknown ions. For instance, isoprene had notable aldehyde fragment interference, as described above. For benzene, its EIC showed contributions from other aromatics like ethylbenzenes and benzaldehyde. These interference patterns have been observed in outdoor air as well, as discussed above.
Additionally, some chemicals showed notable contributions from isobaric species, reflecting the chemical complexity of VOCs indoors (e.g., terpenes, xylenes, trimethylbenzenes, saturated short chain carbonyls). The dominant isomer in these cases may be sensitive to the other sources present in the indoor space. Instrument sensitivity towards different isomers may also vary. Sensitivity may vary due to differences in proton transfer rate constants or fragmentation patterns. For example, in the case of the carbonyls investigated here, the aldehyde and ketone functional groups have fairly similar proton transfer rate constants,57 but it is well-known that aldehydes fragment extensively and as a result have lower signal at the parent ion.29 Thus, while an aldehyde may have a low apparent signal at its parent ion, accounting for the role of its fragmentation may mean that its overall contribution to the distribution of isomers is greater than it initially appears relative to a corresponding ketone. Both indoor environmental sources and sensitivity differences are important factors to consider when assigning a particular identity or combination of identities to a single PTR-MS m/z signal.
Indoor vs. outdoor patterns of confounding ions in each EIC may also vary. This similarly emphasizes the importance of understanding the sampling environment and key sources in order to understand the contributions different chemical species for particular ions of interest. For example, some species like 1,3,5-trimethylbenzene and nonanal had very similar parent ion contributions relative to confounding EIC peaks indoors and outdoors, suggesting that the relative composition and abundance of additional EIC peaks in each of these examples is similar across both indoor and outdoor environments studied here. In contrast, 2-butanone and chlorobenzene differed more significantly between the two environments. Thus, an overarching conclusion from this work is that while outdoor evaluations of confounding species in each EIC may sometimes apply to the indoor environment, studies focused specifically on indoor conditions are needed to robustly determine where overlaps exist and where indoor conditions merit unique treatment of PTR-MS data.
4.3 Changes to the levels of confounding peaks in each EIC as a function of indoor activities
Last, different chemicals had different sensitivities towards indoor environmental conditions or activities. Some species were insensitive to indoor conditions, such as furfural, hexanal, nonanal, D4 and D5 siloxane, ethanol, acetone, isoprene, 2-hexanone, limonene, butanal, 1,4-dioxane, and 1,3,5-trimethylbenzene. Some of these species had building material emissions sources (e.g., furfural, hexanal, D4 siloxane) or strong signals in the background air from past human presence or activities (e.g., D5 siloxane, acetone, limonene) but without another important source from the perturbation experiments performed here. We note that when sampling over a short time period (weeks-months) without any humans present in the house, we did not observe much change to these signals, but building material emissions may change over the course of many years and may therefore yield different emissions as time passes,58 and the active presence of humans in the space would also cause significant variability to these signals.Some chemicals were more sensitive to indoor activities, such as acrolein, furan, acetonitrile, benzene, o-xylene, 2-heptanone, α-pinene, toluene, acrylonitrile, and chlorobenzene. Again, this emphasizes the importance of understanding the sampling environment when interpreting indoor PTR-MS measurements, as the indoor activities play a role not only in shaping the dynamics and composition of the indoor VOCs, but also in determining the dynamically changing instrument interferences.
For species sensitive to variations in indoor activities, we investigated whether changes to the contribution of the peak of interest were caused by changes to the contribution of a particular source of that chemical of interest, or changes to the contributions from isomers, fragments, or other ions in the chromatogram. We performed this analysis for acrolein, furan, toluene, and benzene (Fig. S27†), selected because of their high signal strength and minimal known isomer contributions.
Relative to background conditions, acrolein shows a pronounced parent ion peak in acid–base, cooking, and smoke addition experiments under high humidity. In these conditions, the presence of additional peaks in the EICs is also prominent, especially during cooking when the acrolein peak itself is a factor of 3 larger than under average background conditions. However, confounding peaks in the EICs are also about a factor of 3 larger (indicative of the presence of higher molecular weight aldehydes emitted during the thermal degradation of the heated cooking oils, as discussed above). During fresh and aged smoke experiments, the contribution of acrolein itself is a factor of 1.5 larger than the average background conditions. The levels of confounding EIC peaks are similar in size for background and smoke experiments on average, suggesting that smoke is a strong source of acrolein and a less important source of the interfering aldehyde species.
Furan peaks across background, acid–base, and cooking conditions are overall similar, indicating no strong source during those times. Furan peak contributions increase by a factor of 2.5 in fresh and aged smoke experiments relative to background, suggesting a strong smoke source. However, at the same time, the presence of other peaks in the EICs also generally increase, by a factor of approximately 1.5, indicating that the smoke additions were also likely a source of furan derivatives interfering at the same m/z.
For toluene, the roles of other contributing EIC peaks were greatest in the background samples and generally smallest in the fresh and aged smoke experiments. While the peak area of toluene itself varies from experiment to experiment, the peak of toluene itself is about a factor of 2 stronger in the smoke experiments (as toluene may be part of the smoke emission mixture) relative to the house background, and the signal of confounding EIC peaks decreased during those experiments to 0.7 times the background levels, indicating fewer relative contributions from higher molecular weight aromatics at that time. In contrast, during acid–base and cooking experiments, the toluene signal is strong (3 to 5 times greater than background) but the signal of confounding EIC peaks is prominent relative to the house background (2 times greater than the background). Similar behavior is observed for benzene in these conditions, though with overall larger interferences due to fragments of higher molecular weight species and unknown ions.
Together, this emphasizes that the relative contribution of chemicals of interest may vary in their overall EICs as a function of source strength of the target chemical and source strength of the other confounding isomer or fragment species. In this study, we observed both phenomena occurring for the ions discussed.
4.4 Implications for chemical properties and fate
In this section, we demonstrate how variable contributions of each compound of interest to its EIC might influence the properties assigned to that ion if the ion is mis-attributed to a particular target species. Efforts to understand differences in the physicochemical parameters of isomers relative to the difference in estimation method (e.g., for Henry's Law, considering differences between estimates when applying HWINb (a bond contribution method used in Estimation Programs Interface (EPI) Suite), vs. GROMHE (a group contribution method)) show that often the difference in the values of the parameters themselves for two isomers in question can be greater than differences arising from applying a different estimation method.59 While molecular-formula based methods for estimating physicochemical parameters are convenient and work well to represent an approximate average of isomers present at a particular formula, including more specific structural information greatly improves the estimate.For this analysis, we selected species for which we could fully identify all isomer peaks within an EIC. We obtained estimated values for pure component octanol–air partitioning coefficient (log Koa), Henry's law constant (H), and ozone reaction rate constant (kO3) from EPI Suite. We converted peak areas to mixing ratios for each compound in the isomer set using isomer-specific sensitivity values. Then, we used the relative contribution of each isomer to estimate an isomer-weighted average of the pure component property value for log Koa, H, and kO3. This analysis was performed to investigate how much an isomer-weighted property estimate might vary from the pure-component property value, i.e., to study a scenario in which a particular m/z is assigned one particular molecular identity instead of considering its contributing isomers. Data are further discussed in Section S3 and Fig. S28.†
Differences between isomer-weighted mixture estimates for log Koa and Henry's law constants and pure component values are summarized in Table S3.† Overall, the consistent isomer distribution led to consistent property estimates for each of these species that didn't deviate much from the pure component value corresponding to the dominant isomer (e.g., log Koa values for 3-hexanone: 3.5 × 100, for 2-hexanone: 3.8 × 100, for hexanal: 3.8 × 100, and for the isomer-weighted mean: 3.8 × 100 – in this environment, hexanal was the dominant isomer present). This suggests that a simple approximation of composition for this isomer set, that considers the dominant species without detailed isomer speciation, would on average estimate chemical fate in this test house with reasonable accuracy. We observed similar behavior across the carbonyl sets examined in Table S3.† However, this assumes the instrument's sensitivities of the isomers are known – if the isomer sensitivities are similar or are quantified and accounted for, this simplification holds true. If the isomer sensitivities are not similar or these differences are not accounted for, this assertion may fail.
Across the duration of the campaign, the mean isomer-weighted kO3 for the monoterpene mixture ranged from 3.2 × 10−16 to 4.2 × 10−16 cm3 per molecules per s. This range is smaller than the range of kO3 values for each pure monoterpene (1.2 × 10−17 to 4.4 × 10−16 cm3 per molecules per s), meaning that the distribution of monoterpene isomers did not vary much over the course of the measurement period. Differences between mixture estimates and pure component estimates are summarized in Table S4.† Thus, due to the consistent terpene isomer distribution, this also suggests that a simple approximation of composition for monoterpenes in this test house that considers a dominant species without detailed isomer speciation would, on average, estimate chemical fate in this specific environment with reasonable accuracy. However, this evaluation should be performed in other indoor environments to assess whether these assumptions are applicable in other spaces with different sources (including the presence of humans), oxidant, light level, and environmental parameters.
In this work, we used GC-PTR-MS to identify the contributions of isomers and fragmentation interferences for commonly measured PTR-MS ions when sampling indoors. The type and degree of confounding EIC peaks at each ion was compound-dependent, environment dependent, as well as instrument-condition dependent, and thus all these factors must be considered when interpreting future indoor PTR-MS measurements. We highlighted examples of compounds whose parent ion dominated their EIC, whose EIC was confounded significantly by fragmentation products and other unknown ions products, whose EIC was confounded by isobaric species, whose contribution patterns changed between indoor and outdoor environments (or remained constant), and whose contribution patterns were sensitive to indoor activities (or remained constant).
Given the standard operating procedures of the Vocus PTR-MS where the BSQ limits observation of the reagent ions, and critically, the observation and ability to calculate ratios of H3O+ to NO+ and O2+ impurities, it is not straightforward in studies such as ours to assess the roles played by such impurity ions in ion fragmentation in our study. In the future, an expanded Vocus PTR-MS sampling protocol involving routine changes to the BSQ settings to monitor those ions could be valuable if an in-depth analysis of fragmentation products and fragmentation mechanisms is desired. Additionally, the Vocus PTR-MS measurement community could begin to routinely report ratios of charge transfer products to protonated adducts for key ions, like benzene (i.e., C6H6+/C6H7+), as a way to provide a baseline assessment of how much charge transfer from impurity ions may be contributing to observed signals.
This analysis focused on an unoccupied test house, built with low-VOC emitting building materials but without any furnishings present and with minimal influence from humans and consumer product use.37 Occupied homes – which may show increased contributions from many of these sources – might exhibit different emissions profiles for some chemicals and may therefore have different PTR-MS contributions from isomers and fragments at these chemicals' parent ion m/z. This study aimed to understand the role of these confounding EIC peaks in an unoccupied test house, but future studies are needed in occupied settings to build a more robust characterization of how occupancy – including increased prominence of human emissions, use of consumer products, and presence of furniture – changes these observed trends.
Disclaimer
Certain equipment, instruments, software, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement of any product or service by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose. The policy of NIST is to use the International System of Units in all publications. In this document, however, some units are presented in the system prevalent in the relevant discipline.Data availability
Tabulated data are available as part of the ESI and additional data are available on request to the corresponding author.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the entire CASA and NIST (Brian Dougherty, Andrew Mundy, Stephen Zimmerman, and Dan Greb) teams who contributed to this field campaign. J. C. D. and A. W. H. C. acknowledge funding from the Canada Research Chairs program (CRC-2019-00028). J. C. D., H. N. H., J. Y., and J. P. D. A. acknowledge funding from the Alfred P. Sloan Foundation (G-2019-11404). D. K. F. and M. E. V. acknowledge funding from the Alfred P. Sloan Foundation (G-2020-13929).References
- Y. Liu, P. K. Misztal, C. Arata, C. J. Weschler, W. W. Nazaroff and A. H. Goldstein, Observing ozone chemistry in an occupied residence, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(6), e2018140118 CrossRef CAS PubMed.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements, Indoor Air, 2019, 29(4), 630–644 CAS.
- B. Molinier, C. Arata, E. F. Katz, D. M. Lunderberg, Y. Liu, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Volatile Methyl Siloxanes and Other Organosilicon Compounds in Residential Air, Environ. Sci. Technol., 2022, 56(22), 15427–15436 CrossRef CAS PubMed.
- C. Arata, P. K. Misztal, Y. Tian, D. M. Lunderberg, K. Kristensen, A. Novoselac, M. E. Vance, D. K. Farmer, W. W. Nazaroff and A. H. Goldstein, Volatile organic compound emissions during HOMEChem, Indoor Air, 2021, 31(6), 2099–2117 CrossRef CAS PubMed.
- X. Ding, J. Jiang, A. Tasoglou, H. Huber, A. D. Shah and N. Jung, Evaluation of Workplace Exposures to Volatile Chemicals During COVID-19 Building Disinfection Activities with Proton Transfer Reaction Mass Spectrometry, Ann. Work Exposures Health, 2023, 67(4), 546–551 CrossRef CAS PubMed.
- J. M. Mattila, C. Arata, A. Abeleira, Y. Zhou, C. Wang, E. F. Katz, A. H. Goldstein, J. P. D. Abbatt, P. F. DeCarlo, M. E. Vance and D. K. Farmer, Contrasting Chemical Complexity and the Reactive Organic Carbon Budget of Indoor and Outdoor Air, Environ. Sci. Technol., 2022, 56(1), 109–118 CrossRef CAS PubMed.
- M. F. Link, J. Li, J. C. Ditto, H. Huynh, J. Yu, S. M. Zimmerman, K. L. Rediger, A. Shore, J. P. D. Abbatt, L. A. Garofalo, D. K. Farmer and D. Poppendieck, Ventilation in a Residential Building Brings Outdoor NOx Indoors with Limited Implications for VOC Oxidation from NO3 Radicals, Environ. Sci. Technol., 2023, 57(43), 16446–16455 CrossRef CAS PubMed.
- S. Liu, R. Li, R. J. Wild, C. Warneke, J. A. de Gouw, S. S. Brown, S. L. Miller, J. C. Luongo, J. L. Jimenez and P. J. Ziemann, Contribution of human-related sources to indoor volatile organic compounds in a university classroom, Indoor Air, 2016, 26(6), 925–938 CrossRef CAS PubMed.
- X. Tang, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Volatile Organic Compound Emissions from Humans Indoors, Environ. Sci. Technol., 2016, 50(23), 12686–12694 CrossRef CAS PubMed.
- X. Tang, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Siloxanes Are the Most Abundant Volatile Organic Compound Emitted from Engineering Students in a Classroom, Environ. Sci. Technol. Lett., 2015, 2(11), 303–307 CrossRef CAS.
- J. C. Ditto, L. R. Crilley, M. Lao, T. C. VandenBoer, J. P. D. Abbatt and A. W. H. Chan, Indoor and outdoor air quality impacts of cooking and cleaning emissions from a commercial kitchen, Environ. Sci.: Processes Impacts, 2023, 25(5), 964–979 RSC.
- J. Williams, C. Stönner, J. Wicker, N. Krauter, B. Derstroff, E. Bourtsoukidis, T. Klüpfel and S. Kramer, Cinema audiences reproducibly vary the chemical composition of air during films, by broadcasting scene specific emissions on breath, Sci. Rep., 2016, 6(1), 25464 CrossRef CAS PubMed.
- T. Wu, T. Földes, L. T. Lee, D. N. Wagner, J. Jiang, A. Tasoglou, B. E. Boor and E. R. I. Blatchley, Real-Time Measurements of Gas-Phase Trichloramine (NCl3) in an Indoor Aquatic Center, Environ. Sci. Technol., 2021, 55(12), 8097–8107 CrossRef CAS PubMed.
- D. Pagonis, D. J. Price, L. B. Algrim, D. A. Day, A. V. Handschy, H. Stark, S. L. Miller, J. de Gouw, J. L. Jimenez and P. J. Ziemann, Time-Resolved Measurements of Indoor Chemical Emissions, Deposition, and Reactions in a University Art Museum, Environ. Sci. Technol., 2019, 53(9), 4794–4802 CrossRef CAS PubMed.
- Z. Finewax, D. Pagonis, M. S. Claflin, A. V. Handschy, W. L. Brown, O. Jenks, B. A. Nault, D. A. Day, B. M. Lerner, J. L. Jimenez, P. J. Ziemann and J. A. Gouw, Quantification and source characterization of volatile organic compounds from exercising and application of chlorine-based cleaning products in a university athletic center, Indoor Air, 2021, 31(5), 1323–1339 CrossRef CAS PubMed.
- D. Pagonis, K. Sekimoto and J. de Gouw, A Library of Proton-Transfer Reactions of H3O+ Ions Used for Trace Gas Detection, J. Am. Soc. Mass Spectrom., 2019, 30(7), 1330–1335 CrossRef CAS PubMed.
- M. F. Link, M. S. Claflin, C. E. Cecelski, A. A. Akande, D. Kilgour, P. A. Heine, M. Coggon, C. E. Stockwell, A. Jensen, J. Yu, H. N. Huynh, J. C. Ditto, C. Warneke, W. Dresser, K. Gemmell, S. Jorga, R. L. Robertson, J. de Gouw, T. Bertram, J. P. D. Abbatt, N. Borduas-Dedekind and D. Poppendieck, Product Ion Distributions using H3O+ PTR-ToF-MS: Mechanisms, Transmission Effects, and Instrument-to-Instrument Variability, EGUsphere, 2024, 1–37 Search PubMed.
- D. K. Farmer and M. Riches, Measuring Biosphere–Atmosphere Exchange of Short-Lived Climate Forcers and Their Precursors, Acc. Chem. Res., 2020, 53(8), 1427–1435 CrossRef CAS PubMed.
- M. Takhar, Y. Li, J. C. Ditto and A. W. H. Chan, Formation pathways of aldehydes from heated cooking oils, Environ. Sci.: Processes Impacts, 2023, 25(2), 165–175 RSC.
- A. Wisthaler and C. J. Weschler, Reactions of ozone with human skin lipids: Sources of carbonyls, dicarbonyls, and hydroxycarbonyls in indoor air, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(15), 6568–6575 CrossRef CAS PubMed.
- D. K. Farmer, M. E. Vance, J. P. D. Abbatt, A. Abeleira, M. R. Alves, C. Arata, E. Boedicker, S. Bourne, F. Cardoso-Saldaña, R. Corsi, P. F. DeCarlo, A. H. Goldstein, V. H. Grassian, L. H. Ruiz, J. L. Jimenez, T. F. Kahan, E. F. Katz, J. M. Mattila, W. W. Nazaroff, A. Novoselac, R. E. O'Brien, V. W. Or, S. Patel, S. Sankhyan, P. S. Stevens, Y. Tian, M. Wade, C. Wang, S. Zhou and Y. Zhou, Overview of HOMEChem: House Observations of Microbial and Environmental Chemistry, Environ. Sci.: Processes Impacts, 2019, 21(8), 1280–1300 RSC.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac, M. E. Vance, W. W. Nazaroff and J. P. D. Abbatt, Surface reservoirs dominate dynamic gas-surface partitioning of many indoor air constituents, Sci. Adv., 2020, 6(8), eaay8973 CrossRef CAS PubMed.
- T. Karl, R. Fall, P. J. Crutzen, A. Jordan and W. Lindinger, High concentrations of reactive biogenic VOCs at a high altitude site in late autumn, Geophys. Res. Lett., 2001, 28(3), 507–510 CrossRef CAS.
- R. Fall, T. Karl, A. Jordan and W. Lindinger, Biogenic C5 VOCs: release from leaves after freeze–thaw wounding and occurrence in air at a high mountain observatory, Atmos. Environ., 2001, 35(22), 3905–3916 CrossRef CAS.
- J. de Gouw, C. Warneke, T. Karl, G. Eerdekens, C. van der Veen and R. Fall, Sensitivity and specificity of atmospheric trace gas detection by proton-transfer-reaction mass spectrometry, Int. J. Mass Spectrom., 2003, 223–224, 365–382 CrossRef CAS.
- M. M. Coggon, C. E. Stockwell, M. S. Claflin, E. Y. Pfannerstill, X. Lu, J. B. Gilman, J. Marcantonio, C. Cao, K. Bates, G. I. Gkatzelis, A. Lamplugh, E. F. Katz, C. Arata, E. C. Apel, R. S. Hornbrook, F. Piel, F. Majluf, D. R. Blake, A. Wisthaler, M. Canagaratna, B. M. Lerner, A. H. Goldstein, J. E. Mak and C. Warneke, Identifying and correcting interferences to PTR-ToF-MS measurements of isoprene and other urban volatile organic compounds, 2023 [cited 2023 Dec 28], https://www.egusphere.copernicus.org/preprints/2023/egusphere-2023-1497/.
- T. Schripp, S. Etienne, C. Fauck, F. Fuhrmann, L. Märk and T. Salthammer, Application of proton-transfer-reaction-mass-spectrometry for Indoor Air Quality research, Indoor Air, 2014, 24(2), 178–189 CrossRef CAS PubMed.
- M. S. Claflin, D. Pagonis, Z. Finewax, A. V. Handschy, D. A. Day, W. L. Brown, J. T. Jayne, D. R. Worsnop, J. L. Jimenez, P. J. Ziemann, J. de Gouw and B. M. Lerner, An in situ gas chromatograph with automatic detector switching between PTR- and EI-TOF-MS: isomer-resolved measurements of indoor air, Atmos. Meas. Tech., 2021, 14(1), 133–152 CrossRef CAS.
- L. Ernle, N. Wang, G. Bekö, G. Morrison, P. Wargocki, C. J. Weschler and J. Williams, Assessment of aldehyde contributions to PTR-MS m/z 69.07 in indoor air measurements, Environ. Sci.: Atmos., 2023, 3(9), 1286–1295 CAS.
- J. P. D. Abbatt and C. Wang, The atmospheric chemistry of indoor environments, Environ. Sci.: Processes Impacts, 2020, 22(1), 25–48 RSC.
- D. K. Farmer, M. E. Vance, D. Poppendieck, J. P. D. Abbatt, M. R. Alves, K. C. Dannemiller, C. Deeleepojananan, J. C. Ditto, B. Dougherty, O. R. Farinas, A. H. Goldstein, V. H. Grassian, H. N. Huynh, D. Kim, J. C. King, J. Kroll, J. Li, M. F. Link, L. Mael, K. Mayer, A. B. Martin, G. Morrison, R. O’Brien, S. Pandit, B. J. Turpin, M. Webb, J. Yu and S. M. Zimmerman, The Chemical Assessment of Surfaces and Air (CASA) Study: Using chemical and physical perturbations in a test house to investigate indoor processes, Environ. Sci.: Processes Impacts, 2024 10.1039/D4EM00209A.
- R. Sheu, A. Marcotte, P. Khare, S. Charan, J. C. Ditto and D. R. Gentner, Advances in offline approaches for chemically speciated measurements of trace gas-phase organic compounds via adsorbent tubes in an integrated sampling-to-analysis system, J. Chromatogr. A, 2018, 1575, 80–90 CrossRef CAS PubMed.
- J. Krechmer, F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, D. Worsnop, K. Fuhrer, M. Gonin and J. De Gouw, Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 2018, 90(20), 12011–12018 CrossRef CAS PubMed.
- Q. Liu and J. P. D. Abbatt, Liquid crystal display screens as a source for indoor volatile organic compounds, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(23), e2105067118 CrossRef CAS PubMed.
- G. Isaacman-VanWertz, D. T. Sueper, K. C. Aikin, B. M. Lerner, J. B. Gilman, J. A. de Gouw, D. R. Worsnop and A. H. Goldstein, Automated single-ion peak fitting as an efficient approach for analyzing complex chromatographic data, J. Chromatogr. A, 2017, 1529, 81–92 CrossRef CAS PubMed.
- C. Heeley-Hill A, K. Grange S, W. Ward M, C. Lewis A, N. Owen, C. Jordan, G. Hodgson and G. Adamson, Frequency of use of household products containing VOCs and indoor atmospheric concentrations in homes, Environ. Sci.: Processes Impacts, 2021, 23(5), 699–713 RSC.
- D. G. Poppendieck, L. C. Ng, A. K. Persily and A. T. Hodgson, Long term air quality monitoring in a net-zero energy residence designed with low emitting interior products, Build. Sci., 2015, 94, 33–42 Search PubMed.
- C. Wang, J. M. Mattila, D. K. Farmer, C. Arata, A. H. Goldstein and J. P. D. Abbatt, Behavior of Isocyanic Acid and Other Nitrogen-Containing Volatile Organic Compounds in The Indoor Environment, Environ. Sci. Technol., 2022, 56(12), 7598–7607 CrossRef CAS PubMed.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 2018, 18(5), 3299–3319 CrossRef CAS.
- M. P. Vermeuel, G. A. Novak, D. B. Kilgour, M. S. Claflin, B. M. Lerner, A. M. Trowbridge, J. Thom, P. A. Cleary, A. R. Desai and T. H. Bertram, Observations of biogenic volatile organic compounds over a mixed temperate forest during the summer to autumn transition, Atmos. Chem. Phys., 2023, 23(7), 4123–4148 CrossRef CAS.
- B. You, W. Zhou, J. Li, Z. Li and Y. Sun, A review of indoor Gaseous organic compounds and human chemical Exposure: Insights from Real-time measurements, Environ. Int., 2022, 170, 107611 CrossRef CAS PubMed.
- Z. Zhou, S. Zhou and J. P. D. Abbatt, Kinetics and Condensed-Phase Products in Multiphase Ozonolysis of an Unsaturated Triglyceride, Environ. Sci. Technol., 2019, 53(21), 12467–12475 CrossRef CAS PubMed.
- Y. S. Kashtan, M. Nicholson, C. Finnegan, Z. Ouyang, E. D. Lebel, D. R. Michanowicz, S. B. C. Shonkoff and R. B. Jackson, Gas and Propane Combustion from Stoves Emits Benzene and Increases Indoor Air Pollution, Environ. Sci. Technol., 2023, 57(26), 9653–9663 CrossRef CAS PubMed.
- C. P. Weisel, S. Alimokhtari and P. F. Sanders, Indoor Air VOC Concentrations in Suburban and Rural New Jersey, Environ. Sci. Technol., 2008, 42(22), 8231–8238 CrossRef CAS PubMed.
- L. G. Jahn, M. Tang, D. Blomdahl, N. Bhattacharyya, P. Abue, A. Novoselac, L. H. Ruiz and P. K. Misztal, Volatile organic compound (VOC) emissions from the usage of benzalkonium chloride and other disinfectants based on quaternary ammonium compounds, Environ. Sci.: Atmos., 2023, 3(2), 363–373 CAS.
- F. Klein, N. J. Farren, C. Bozzetti, K. R. Daellenbach, D. Kilic, N. K. Kumar, S. M. Pieber, J. G. Slowik, R. N. Tuthill, J. F. Hamilton, U. Baltensperger, A. S. H. Prévôt and I. El Haddad, Indoor terpene emissions from cooking with herbs and pepper and their secondary organic aerosol production potential, Sci. Rep., 2016, 6(1), 36623 CrossRef CAS PubMed.
- C. M. F. Rosales, J. Jiang, A. Lahib, B. P. Bottorff, E. K. Reidy, V. Kumar, A. Tasoglou, H. Huber, S. Dusanter, A. Tomas, B. E. Boor and P. S. Stevens, Chemistry and human exposure implications of secondary organic aerosol production from indoor terpene ozonolysis, Sci. Adv., 2022, 8(8), eabj9156 CrossRef CAS PubMed.
- G. C. Morrison and W. W. Nazaroff, Ozone Interactions with Carpet: Secondary Emissions of Aldehydes, Environ. Sci. Technol., 2002, 36(10), 2185–2192 CrossRef CAS PubMed.
- M. Nicolas, O. Ramalho and F. Maupetit, Reactions between ozone and building products: Impact on primary and secondary emissions, Atmos. Environ., 2007, 41(15), 3129–3138 CrossRef CAS.
- F. Klein, S. M. Platt, N. J. Farren, A. Detournay, E. A. Bruns, C. Bozzetti, K. R. Daellenbach, D. Kilic, N. K. Kumar, S. M. Pieber, J. G. Slowik, B. Temime-Roussel, N. Marchand, J. F. Hamilton, U. Baltensperger, A. S. H. Prévôt and I. El Haddad, Characterization of Gas-Phase Organics Using Proton Transfer Reaction Time-of-Flight Mass Spectrometry: Cooking Emissions, Environ. Sci. Technol., 2016, 50(3), 1243–1250 CrossRef CAS PubMed.
- J. Y. Chin, C. Godwin, C. Jia, T. Robins, T. Lewis, E. Parker, P. Max and S. Batterman, Concentrations and Risks of p-Dichlorobenzene in Indoor and Outdoor Air, Indoor Air, 2013, 23(1), 40–49 CrossRef CAS PubMed.
- T. Yoshida, M. Mimura and N. Sakon, Estimating household exposure to moth repellents p-dichlorobenzene and naphthalene and the relative contribution of inhalation pathway in a sample of Japanese children, Sci. Total Environ., 2021, 783, 146988 CrossRef CAS PubMed.
- A. Schieweck, E. Uhde and T. Salthammer, Determination of acrolein in ambient air and in the atmosphere of environmental test chambers, Environ. Sci.: Processes Impacts, 2021, 23(11), 1729–1746 RSC.
- A. L. Hodshire, E. Carter, J. M. Mattila, V. Ilacqua, J. Zambrana, J. P. D. Abbatt, A. Abeleira, C. Arata, P. F. DeCarlo, A. H. Goldstein, L. H. Ruiz, M. E. Vance, C. Wang and D. K. Farmer, Detailed Investigation of the Contribution of Gas-Phase Air Contaminants to Exposure Risk during Indoor Activities, Environ. Sci. Technol., 2022, 56(17), 12148–12157 CrossRef CAS PubMed.
- L. Xu, Y. Hu and W. Liang, Composition and correlation of volatile organic compounds and odor emissions from typical indoor building materials based on headspace analysis, Build. Sci., 2022, 221, 109321 Search PubMed.
- J. A. Jurvelin, R. D. Edwards, M. Vartiainen, P. Pasanen and M. J. Jantunen, Residential Indoor, Outdoor, and Workplace Concentrations of Carbonyl Compounds: Relationships with Personal Exposure Concentrations and Correlation with Sources, J. Air Waste Manage. Assoc., 2003, 53(5), 560–573 CrossRef CAS PubMed.
- T. Salthammer, U. Hohm, M. Stahn and S. Grimme, Proton-transfer rate constants for the determination of organic indoor air pollutants by online mass spectrometry, RSC Adv., 2023, 13(26), 17856–17868 RSC.
- H. N. Huynh, J. C. Ditto, J. Yu, M. F. Link, D. Poppendieck, D. K. Farmer, M. E. Vance and J. P. D. Abbatt, VOC emission rates from an indoor surface using a flux chamber and PTR-MS, Atmos. Environ., 2024, 338, 120817 CrossRef CAS.
- G. Isaacman-VanWertz and B. Aumont, Impact of organic molecular structure on the estimation of atmospherically relevant physicochemical parameters, Atmos. Chem. Phys., 2021, 21(8), 6541–6563 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00602j |
| This journal is © The Royal Society of Chemistry 2025 |