
A theoretical study on the environmental oxidation of fenpyrazamine fungicide initiated by hydroxyl radicals in the aqueous phase†
Hisham K.
Al Rawas
*a,
Dinh Hieu
Truong
 bc,
Emma
Schell
d,
Jennifer
Faust
d,
Sonia
Taamalli
a,
Marc
Ribaucour
a,
Abderrahman
El Bakali
a,
Nissrin
Alharzali
e,
Duy Quang
Dao
*bc and
Florent
Louis
a
bc,
Emma
Schell
d,
Jennifer
Faust
d,
Sonia
Taamalli
a,
Marc
Ribaucour
a,
Abderrahman
El Bakali
a,
Nissrin
Alharzali
e,
Duy Quang
Dao
*bc and
Florent
Louis
a
aUniv. Lille, CNRS, UMR 8522, Physico-Chimie des Processus de Combustion et de l'Atmosphère – PC2A, 59000 Lille, France. E-mail: hisham.alrawas@univ-lille.fr
bInstitute of Research and Development, Duy Tan University, Da Nang, 550000, Vietnam. E-mail: daoduyquang@duytan.edu.vn
cSchool of Engineering and Technology, Duy Tan University, Da Nang 550000, Vietnam
dDepartment of Chemistry, College of Wooster, Wooster, OH, USA
eDepartment of Physical and Theoretical Chemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Ilkovičova 6, 84215 Bratislava, Slovakia
First published on 19th December 2024
Abstract
Fenpyrazamine (FPA) is a widely used fungicide in agriculture to control fungal diseases, but its environmental degradation by oxidants and the formation of potential degradation products remain unexplored. This study investigates the oxidation of FPA by hydroxyl radicals (HO˙) using density functional theory (DFT) calculations at the M06-2X/6-311++G(3df,3pd)//M06-2X/6-31+G(d,p) level of theory. Three standard oxidation mechanisms, including formal hydrogen transfer (FHT), radical adduct formation (RAF), and single electron transfer (SET), were evaluated in the aqueous phase, with reaction kinetics analyzed over a temperature range of 283–333 K. As a result, the reactivity order of the mechanisms was determined to be RAF > FHT > SET. At 298 K, the calculated total rate constants for FHT and RAF reactions were competitive, being 6.09 × 109 and 8.21 × 109 M−1 s−1, respectively, while that for SET was slightly lower at 2.35 × 109 M−1 s−1. The overall rate constant was estimated to be 1.67 × 1010 M−1 s−1. The most favourable RAF reaction occurred at the C38![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C39 double bond, while the predominant FHT reactions involved the H15 and H13 hydrogen atoms of the methyl C8 group. The lifetime of FPA in natural water with respect to HO˙ oxidation was predicted to range from 10.84 hours to 2.62 years, depending on environmental conditions. Furthermore, the toxicity assessments revealed that while FPA is neither bioaccumulative nor mutagenic, it poses developmental toxicity and is harmful to aquatic organisms, including fish, daphnia, and green algae.
C39 double bond, while the predominant FHT reactions involved the H15 and H13 hydrogen atoms of the methyl C8 group. The lifetime of FPA in natural water with respect to HO˙ oxidation was predicted to range from 10.84 hours to 2.62 years, depending on environmental conditions. Furthermore, the toxicity assessments revealed that while FPA is neither bioaccumulative nor mutagenic, it poses developmental toxicity and is harmful to aquatic organisms, including fish, daphnia, and green algae.
Environmental significanceFenpyrazamine (FPA) fungicide has been widely approved for use in agricultural activities to control brown rot, stem rot, and gray mold affecting vegetables and fruits in over 30 countries around the globe. This fungicide has low volatility in the atmosphere but presents very high solubility in the aquatic environment. The long-term utilization of FPA may lead to accumulation in the aquatic environment and oxidation to form various degradation products which may be toxic and harmful to aquatic organisms and human health. There is no evidence of the environmental degradation mechanism and possible products of FPA. Thus, this paper sheds light on the hydroxyl radical-initiated oxidation of FPA in the aqueous phase. The thermodynamics and kinetics of the oxidation reactions, and the environmental fate and ecotoxicology of FPA and its degradation products are analyzed in detail. Hopefully, the presented results will provide interesting information for future research in environmental, chemical, physical and analytical chemistry disciplines. |
1 Introduction
Our capacity to produce enough food of adequate quality, essential for the fast-growing and increasingly selective population, is significantly hindered by crop fungi. Pesticides, particularly fungicides, have become an indispensable and effective solution to this pressing issue. Synthetic fungicides have been used to protect crops in most countries worldwide for over a century. Their role in controlling fungal diseases has consistently led to improved crop yields and higher incomes for farmers.1Fungicides, whether natural or manufactured, are important agents that protect plants against fungi and eliminate fungal infections. Alongside insecticides, herbicides, and plant growth regulators, they form a crucial part of the agrochemical store known as pesticides. Their availability ensures the protection of crops and the preservation of their yield, measured in terms of the quantity or the quality of products, providing reassurance to both farmers and consumers.
The term “fungicide” refers to compounds that control a huge scope of organisms, including true fungi (e.g., Ascomycota and Basidiomycota), Oomycota (e.g., Phytophthora and Pythium), and Plasmodiophora. However, fungicides do not contain chemicals that control viruses, bacteria, and nematodes, which are controlled by antibiotics, insecticides, and genetic methods. Since the discovery of different pesticide types, many factors have ensured their continued use and growth in the pesticide industry. These factors encompass a growing global population, rising incomes, and direct advantages for both growers (e.g., reduced labor costs, improved yields, and increased profits) and consumers (e.g., enhanced consistency in food quality, a wider range of produce and lower prices).2 The utilization of fungicides has remarkably grown in the last few decades and is routinely applied throughout the plant growing season. Depending on the crop type, several treatments may be used each season.3
However, the intensive application of fungicides has presented additional issues, such as the development of resistance and potential hazards to farmers and nearby residents during the application process. There are also risks to consumers through residues in food and adverse environmental impacts, including accumulation in soils, contamination of natural waters, and detrimental effects on non-target organisms.4
Within agriculture, diseases like gray mold (Botrytis cinerea) and stem rot (Sclerotinia sclerotiorum) pose significant threats to the yield and productivity of fruit trees and vegetables. Effectively controlling these diseases becomes crucial. Various fungicides, including benomyl, have been formulated to control gray mold. However, since gray mold has a short lifecycle coupled with prolific spore production, it is difficult to control and is classified as a phytopathological fungus prone to developing resistance to fungicides; thus novel fungicides were in demand.5 One such innovation is fenpyrazamine (FPA), a fungicide discovered and developed by Sumitomo Chemical Co., Ltd. Fenpyrazamine (C17H21N3O2S), or S-allyl 5-amino-2,3-dihydro-2-isopropyl-3-oxo-4-(o-tolyl)pyrazole-1-carbothioate, is a fungicidal compound belonging to the pyrazole fungicide family.
Distinguished by its aminopyrazolinone structure, this fungicide stands out as a unique example not found among agricultural chemicals. It has a low vapor pressure of 2.89 × 10−8 Pa at 25 °C, indicating that it is not highly volatile and thus has a limited potential to evaporate into the atmosphere under standard environmental conditions. Moreover, its solubility in water is 20.4 mg L−1 at 20 °C.6
Fenpyrazamine demonstrates notably strong effectiveness against brown rot, stem rot, and gray mold affecting vegetables and fruits. Sumitomo Chemical Co., Ltd registered fenpyrazamine fungicide in Korea and acquired EU registration. The product was introduced as “PROLECTUS®” in October 2012 in Italy and as “Pixio® DF” in January 2014 in Japan. Following that, Sumitomo Chemical Co., Ltd obtained registration for fenpyrazamine and introduced the fungicide in several EU countries and Australia.6 Fenpyrazamine has been registered in about 30 countries all over the world, including the European Union (EU), the USA, Chile, Japan, and Korea. These registrations cover various crops, including almonds, grapes, lettuce, etc.7
The effectiveness of fenpyrazamine against fungi was evaluated, demonstrating potent inhibition of germ tube elongation and inducing morphological alterations in Botrytis cinerea germ tubes. Furthermore, it exhibited significant preventive efficacy, achieving 100% control against gray mold at a low concentration, just 1/16 of the registered concentration (250 mg a.i. per L) for cucumber gray mold in Japan. Fenpyrazamine's target enzyme is 3-keto reductase in the ergosterol biosynthetic pathway. The fungicide also displayed high efficacy in field trials against pathogenic Sclerotiniaceae like Sclerotinia spp., Botrytis spp., and Monilinia spp., demonstrating its practical applicability.6 Fenpyrazamine has been detected in water samples taken from 2 sites of River Arun in the south of England, as reported in ref. 8.
According to the World Health Organization, the acceptable daily intake of fenpyrazamine was established to be 0–0.3 mg per kg body weight, which represents an International Estimated Daily Intake of 0–2% of the upper bound of the ADI. An International Estimated Short-Term Intake was calculated for fenpyrazamine, for which an acute reference dose was established to be 0.8 mg per kg body weight. Moreover, an estimated maximum dietary burden test was conducted, and 2.38 mg kg−1 of fenpyrazamine residue in grape pomace was found.9 In addition, fenpyrazamine was tested and found to be non-carcinogenic in rats with an acute oral LD50 > 2000 mg kg−1 and acute inhalation LD50 > 4.84 mg L−1.10
Like other organic compounds, fenpyrazamine could decompose and react with other species in the environment. Therefore, it is essential to understand the decomposition and oxidation processes of the fenpyrazamine fungicide under environmental conditions to better assess its lifetime, toxicology, and impact on human health and the environment. Facing this task, several research approaches have been investigated, in which quantum chemistry has become one of the indispensable pillars allowing a better understanding of the mechanisms and kinetics of fungicide degradation. Using molecular simulations and density functional theory (DFT) calculations, several theoretical studies have been reported on the reaction mechanisms and the kinetics of HO˙-initiated oxidation of pesticides like dimethoate,11 metazachlor,12 quinmerac,13 phosmet,14 thiram15 and chlorpyrifos.16
No literature data exist on the aquatic degradation of fenpyrazamine (FPA) and its potential degradation products. Hence, this study aims to investigate the degradation mechanisms of FPA induced by HO˙ by considering the different reaction pathways: formal hydrogen transfer (FHT), radical adduct formation (RAF), and single electron transfer (SET). The mechanisms, thermodynamics, and the kinetics of reactions, in addition to the lifetime of FPA, will be examined in the aqueous phase at various temperatures ranging from 283 to 323 K, covering the temperature range of water in environmental settings and water treatment processes. Additionally, the ecotoxicity of FPA and its decomposition products will be assessed in the aquatic environment, highlighting their impact on aquatic species.
2 Computational methods
2.1 Electronic structure and kinetic studies
The computational approaches have highly been recommended in proving effective in accurately calculating radical reactions involved in the degradation or the oxidation of harmful organic molecules.14,16–21Gaussian 16 Rev. C.01 software22 was employed to optimize geometry and perform the calculations of harmonic vibrational frequency using density functional theory (DFT), as well as to find the transition states. The M06-2X method23 in conjunction with the 6-31+G(d,p) Pople-style basis set was used. Additional single-point electronic energy calculations were performed based on the optimized geometries, with the largest Pople basis set, namely 6-311++G(3df,3pd). The computations were conducted in water and utilized the solvation model (SMD).24
The fenpyrazamine structure underwent conformational analysis using MSTor software.25 The most stable conformation was selected for examining the potential energy surfaces (PES) of fenpyrazamine towards HO˙ radicals.
Three standard reaction pathways, including formal hydrogen transfer (FHT), radical adduct formation (RAF), and single electron transfer (SET), were evaluated in the aqueous phase. The Gaussian Post Processor (GPOP) software26 was utilized to compute all rate constants under 1 M standard conditions and over the temperature range of 283–333 K. The kinetic studies of FHT and RAF reactions were established using the pre-reactive complexes scheme introduced by Singleton and Cvetanovic.27
To validate whether the imaginary frequency corresponds to the correct motion along the reaction coordinates, Intrinsic Reaction Coordinate (IRC) calculations were carried out. A spin–orbit coupling (SOC) value of −0.836 kJ mol−1 for HO˙ radicals28 and the zero-point energy (ZPE) corrections were considered in all the reactions. Marcus's theory was applied to estimate the rate constant of the SET reaction.29–31
Thermal rate constants (k) were computed first, followed by considering diffusion rate constants (kD) in the aqueous medium. This was done based on the steady-state Smoluchowski rate coefficient,32 in conjunction with Truhlar33 and the Stokes–Einstein approaches.34,35 The diffusion-corrected apparent rate constants (kapp) were determined through the application of the Collins–Kimball theory,36 and the Eckart method was used for the tunnelling correction.37
The total rate constant (ktotal) of each reaction mechanism was derived by adding up the rate constant (ki) of all individual reactions. The overall rate constant (koverall) was obtained by summing up the total rate constant of the three mechanisms, i.e., koverall = ktotal(FHT) + ktotal(RAF) + ktotal(SET). The branching ratios (Γ) were calculated for each reaction by dividing their respective apparent rate constants (ki) by the overall rate constant (koverall). The detailed calculations are mentioned in the ESI File.†
2.2 Ecotoxicity evaluation
The toxicity of fenpyrazamine and its possible degradation products under environmental conditions and their impact on aquatic organisms were studied using the quantitative structure–activity relationship (QSAR) method. The estimation of the acute and chronic toxicities in fish, daphnia, and green algae, was carried out using the Ecological Structure–Activity Relationship (ECOSAR version 2.2) software.38 Toxicity values for new chemicals can be predicted by inputting the estimated octanol/water partition coefficient (Kow) into a regression equation and adjusting the resulting value based on the compound's molecular weight. ECOSAR currently encompasses over 120 chemical classes, supported by more than 600 QSAR models derived from publicly available experimental data. These regression models predict toxicity by leveraging experimental data from structurally similar compounds. Numerous studies, such as those by Melnikov et al. (2016)39 and Zhou et al. (2021),40 have assessed the performance of various in silico aquatic toxicity prediction models, highlighting their strengths and the limitations imposed by the complexity of certain chemical families. As a result, they highlighted that ECOSAR consists of one of reasonably accurate predictive models for aquatic toxicity.The median lethal concentration (LC50) and the median effect concentration (EC50) values were employed to examine the acute toxicity in fish (96 h of exposure), daphnia (48 h of exposure), and green algae (96 h of exposure), and the chronic toxicity (ChV) values based on the European Union and Chinese criteria (Table S1 in the ESI File†).
The EPI Suite software (EPI Suite 4.1)41 was used to calculate the bioaccumulation factor (BAF) as well as the bioconcentration factor (BCF). They are two related quantities used in environmental toxicology to predict the potential accumulation of a chemical in living organisms. According to REACH Annex XIII (2011) and the Canadian Environmental Protection Act (CEPA), a chemical is considered bioaccumulative if its BAF or BCF values in aquatic organisms exceed 2000 L per kg wet-wt and classified as highly bioaccumulative if the values are higher than 5000 L per kg wet-wt.42
Finally, the Toxicity Estimation Software Tool (T.E.S.T version 5.1)43 was used to assess the developmental toxicity and mutagenicity of fenpyrazamine and its degradation products. The compounds are classified as developmental toxicants (>0.5) and developmental non-toxicants (<0.5), according to whether or not this chemical can cause developmental toxicity effects. In addition, a compound is mutagenicity positive (>0.5) if it can cause mutations in the genetic material of the organisms; otherwise, it is mutagenicity negative (<0.5).43
3 Results and discussion
3.1 Structure and electronic properties of fenpyrazamine
First, a conformational analysis was made using the MSTor software44 to find the most stable conformer of FPA. The ConfGen module of this program allows the generation of Gaussian input files for a set of conformational structures by rotating around specified bonds in an input structure. The 5 torsions combined with 4 initial dihedral angles (0°, 90°, 180°, and 270°) of the FPA molecule led to 372 initial structures. The geometry optimizations at the M06-2X/6-31+G(d,p) level of theory found 15 conformers, where the most stable conformer was selected to investigate the potential energy surfaces of FPA towards HO˙ radicals. The optimized structure of the most stable conformer of FPA in the aqueous phase is shown in Fig. 1. To know more about the electronic properties of FPA molecules, its Fukui indices, highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and the electrostatic potential (ESP) maps were investigated and shown in Fig. S2 and S3 (ESI File).†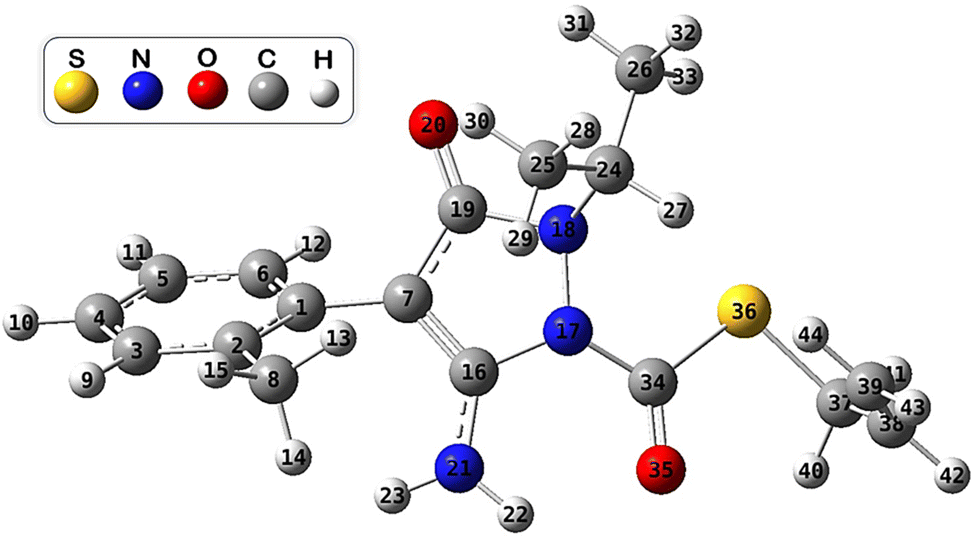 | ||
| Fig. 1 Optimized structure of the most stable conformer of FPA. | ||
From the ESP map, the region of high electron density (in red colour) is located at the CO group and the O atom (O20), while that of the low electron density is located at the C–C bond related to the S atom, and at the N–H bonds. Moreover, it could be seen that the HOMO is primarily situated at the rings, whereas the LUMO is located at the pyrazole ring, and the CO group bonded to the S atom. This information enables the prediction of molecular regions with a high tendency to donate electrons and those that tend to accept them in single electron transfer reactions with external radicals. The results of the calculated Fukui indices for radical attack (f0) show that the NH2, the pyrazole ring, and the CO groups may be favourable sites for the radical reaction.
To identify in which state fenpyrazamine exists in the environment, its pKa and molar fraction were calculated. The pKa value (i.e., 3.42) was obtained based on the calculated Gibbs free energies of both protonated (at N17, N18, N21, O20, and O35 positions) and deprotonated forms of FPA,45 where the protonation was most favourable at the O20 atom.
The results show that FPA predominantly exists in its protonated form in an acidic medium (pH < 3.4), while it predominantly exists in the neutral form when pH > 3.4 (Fig. S4, ESI File).† In the pH range from 6 to 8, which reproduces the environmental conditions of almost all surface water and underground water, FPA exists totally in the neutral form. Thus, according to the primary purpose of this work, which is to evaluate the environmental degradation of the organic contaminant, the neutral form of the FPA molecule in all reactions will be only considered.
3.2 Reaction mechanisms
The degradation process of fenpyrazamine by HO˙ radicals in the environment involves 21 formal H-abstractions (FHT), 12HO˙-additions at the unsaturated atoms (RAF), and a single electron transfer (SET) reaction.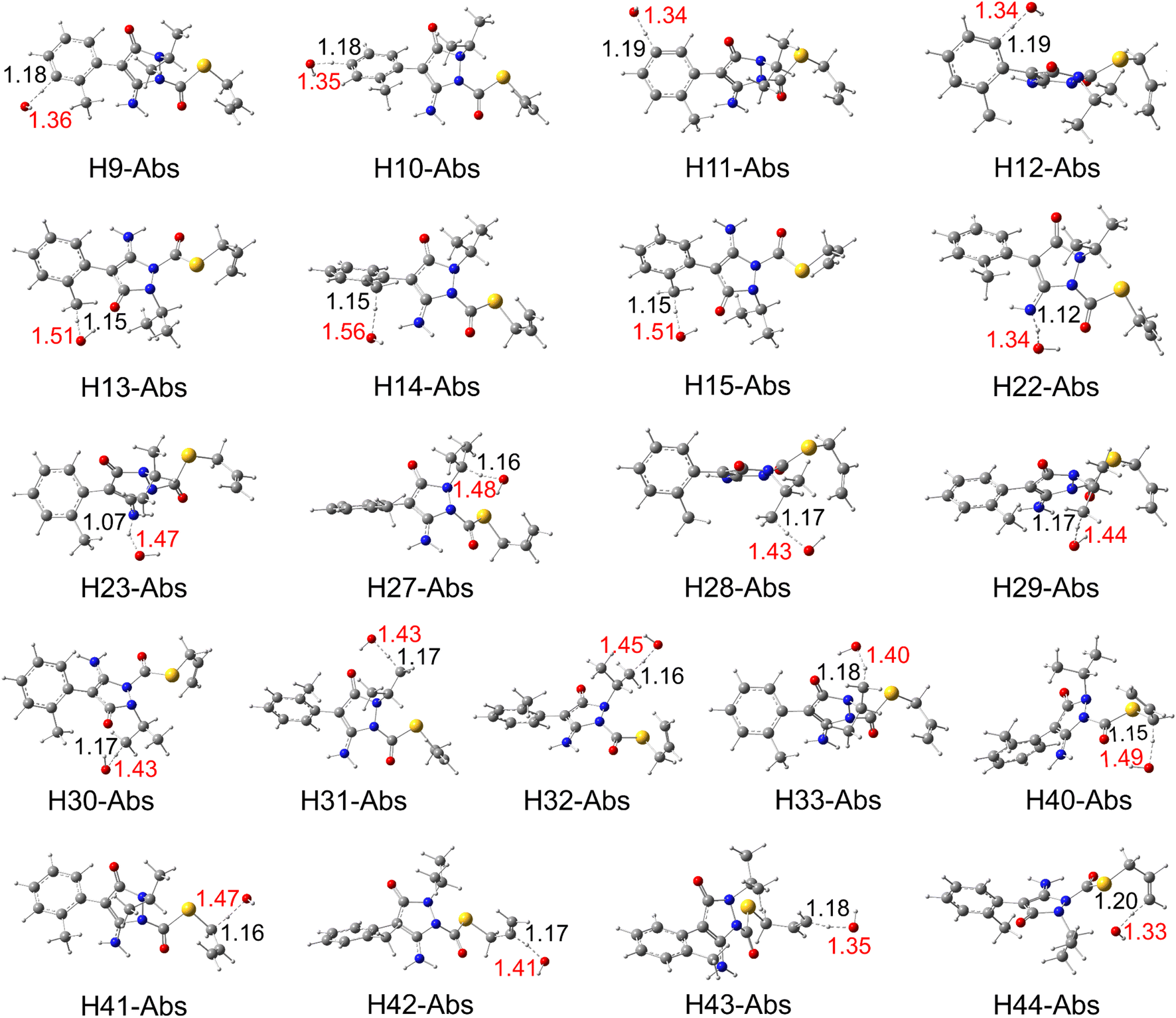 | ||
| Fig. 2 Optimized structures of the transition state (TS) for all formal hydrogen transfer (FHT) reactions in the aqueous phase. | ||
The results of the H-abstraction reaction (Fig. 3) reveal that all the pathways are spontaneous and exergonic, with negative values of their standard Gibbs free reaction energy (ΔrG°) at 298 K. These values range from −144.56 kJ mol−1 (at H41 of the methylene group) to −35.58 kJ mol−1 (at H10 of the benzyl ring). In addition, the Gibbs free activation energy (ΔG°≠) was found to be lowest between 25.65 and 35.55 kJ mol−1, at H13 and H15 of the methyl group (of C8), H40 of the methylene group (of C37 adjacent to the S atom), and H31 of the methyl group (of C26), respectively. It is noteworthy that H31 is stabilised by a hydrogen bond with the oxygen atom O20. However, as expected, the highest ΔG°≠ value of 84.03 kJ mol−1 was seen at H23 of the amino group of N21. This may be due to several factors like steric hindrance and the electron-withdrawing effect of the nitrogen atom rich in electron densities. These factors may make the H-abstraction from this group more challenging than from C–H bonds of the methyl (–CH3) groups or the methylene (–CH2–) ones.
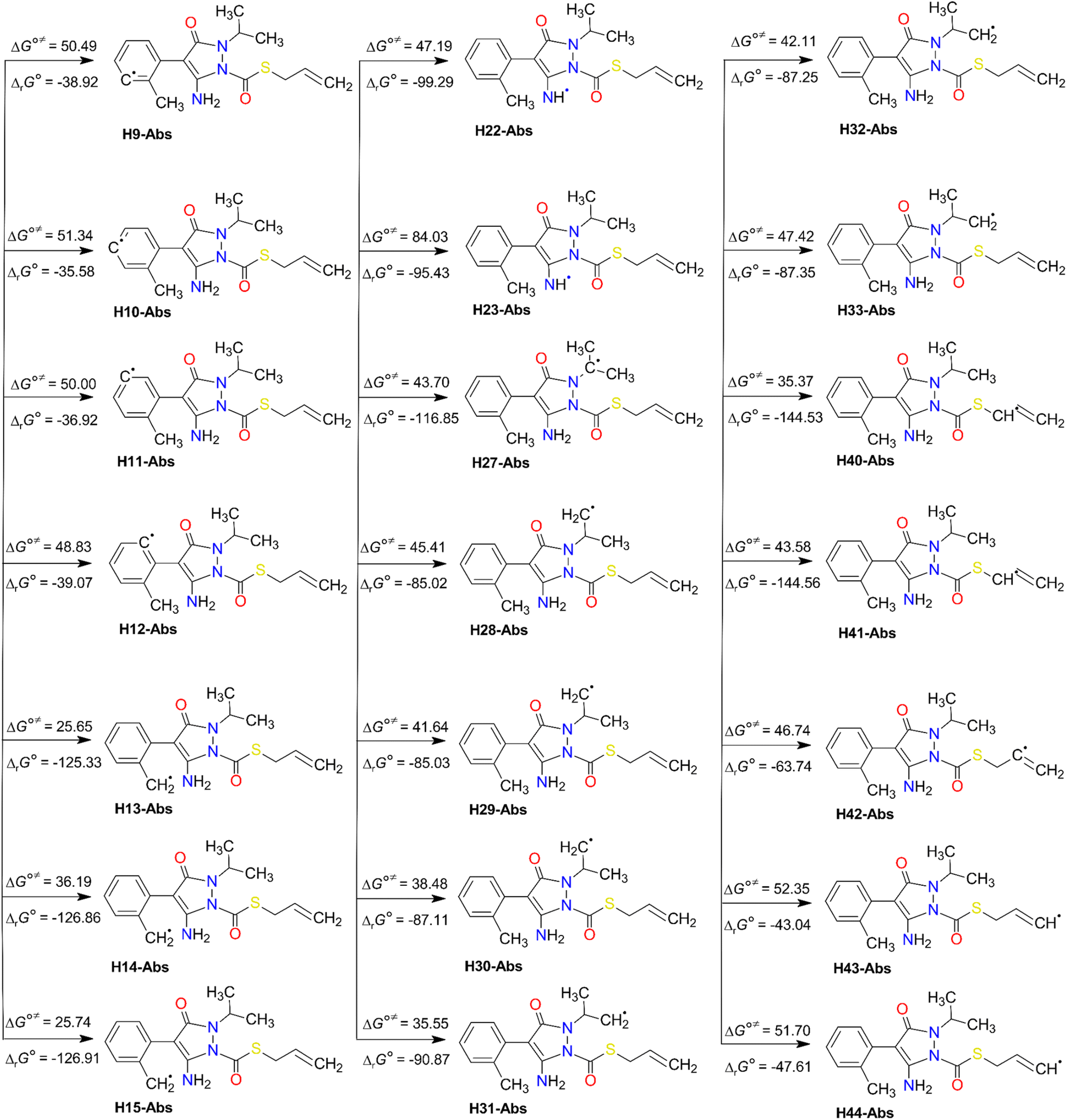 | ||
| Fig. 3 H-abstraction degradation pathways of FPA (with the water product). Standard Gibbs free reaction energies (ΔrG°) and standard Gibbs free activation energies (ΔG°≠) at 298 K. Units are in kJ mol−1. | ||
Furthermore, all the FHT reaction pathways are exothermic, as indicated by their negative standard reaction enthalpy at 0 K  (Table S3, ESI File†). These values vary from −141.59 (at H41) to −34.61 kJ mol−1 (at H11). The lowest energy barrier (E0) at the TS at 0 K was found at H13 (−10.87 kJ mol−1) and H15 (−10.86 kJ mol−1). This indicates that the FHT pathways at the methyl groups (H13/H15 and H30/H31) and the methylene group (H40/H41) are the predominant reactions, with H15 the most favourable. From a thermodynamics point of view, we can conclude that FHT reactions are generally favoured in the order –CH3– > –CH2– > benzyl ring > –NH2. This finding will be validated later in the reaction kinetics section.
(Table S3, ESI File†). These values vary from −141.59 (at H41) to −34.61 kJ mol−1 (at H11). The lowest energy barrier (E0) at the TS at 0 K was found at H13 (−10.87 kJ mol−1) and H15 (−10.86 kJ mol−1). This indicates that the FHT pathways at the methyl groups (H13/H15 and H30/H31) and the methylene group (H40/H41) are the predominant reactions, with H15 the most favourable. From a thermodynamics point of view, we can conclude that FHT reactions are generally favoured in the order –CH3– > –CH2– > benzyl ring > –NH2. This finding will be validated later in the reaction kinetics section.
Additionally, Table S4 (ESI File)† resumes the values of standard enthalpy, entropy, Gibbs free energy of activation 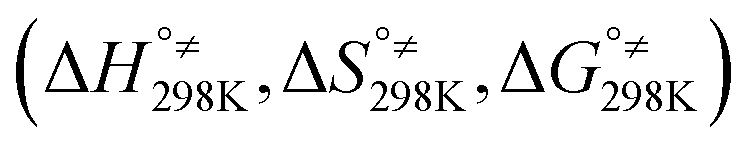 and of reaction
and of reaction 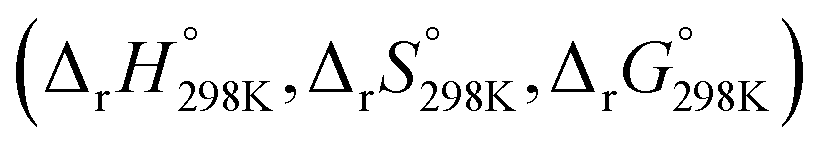 for all the FHT reactions at 298 K temperature calculated at two different DFT levels of theories, i.e., M06-2X/6-31+G(d,p) and M06-2X/6-311++G(3df,3pd), to evaluate the influence of computational levels on the obtained energetic results. As can be seen in Table S4,† differences in standard Gibbs free activation energy (ΔΔG°≠) obtained from the computational M06-2X/6-31+G(d,p) level of theory (level 1) and the M06-2X/6-311++G(3df,3pd) level (level 2) are relatively small, varying from −2.71 kJ mol−1 (at H22 position) to 0.59 kJ mol−1 (at H29). Meanwhile, differences in standard Gibbs free reaction energy (ΔΔrG°) between the two levels are more remarkable, from −7.64 kJ mol−1 (at H22) to −1.60 kJ mol−1 (at H23).
for all the FHT reactions at 298 K temperature calculated at two different DFT levels of theories, i.e., M06-2X/6-31+G(d,p) and M06-2X/6-311++G(3df,3pd), to evaluate the influence of computational levels on the obtained energetic results. As can be seen in Table S4,† differences in standard Gibbs free activation energy (ΔΔG°≠) obtained from the computational M06-2X/6-31+G(d,p) level of theory (level 1) and the M06-2X/6-311++G(3df,3pd) level (level 2) are relatively small, varying from −2.71 kJ mol−1 (at H22 position) to 0.59 kJ mol−1 (at H29). Meanwhile, differences in standard Gibbs free reaction energy (ΔΔrG°) between the two levels are more remarkable, from −7.64 kJ mol−1 (at H22) to −1.60 kJ mol−1 (at H23).
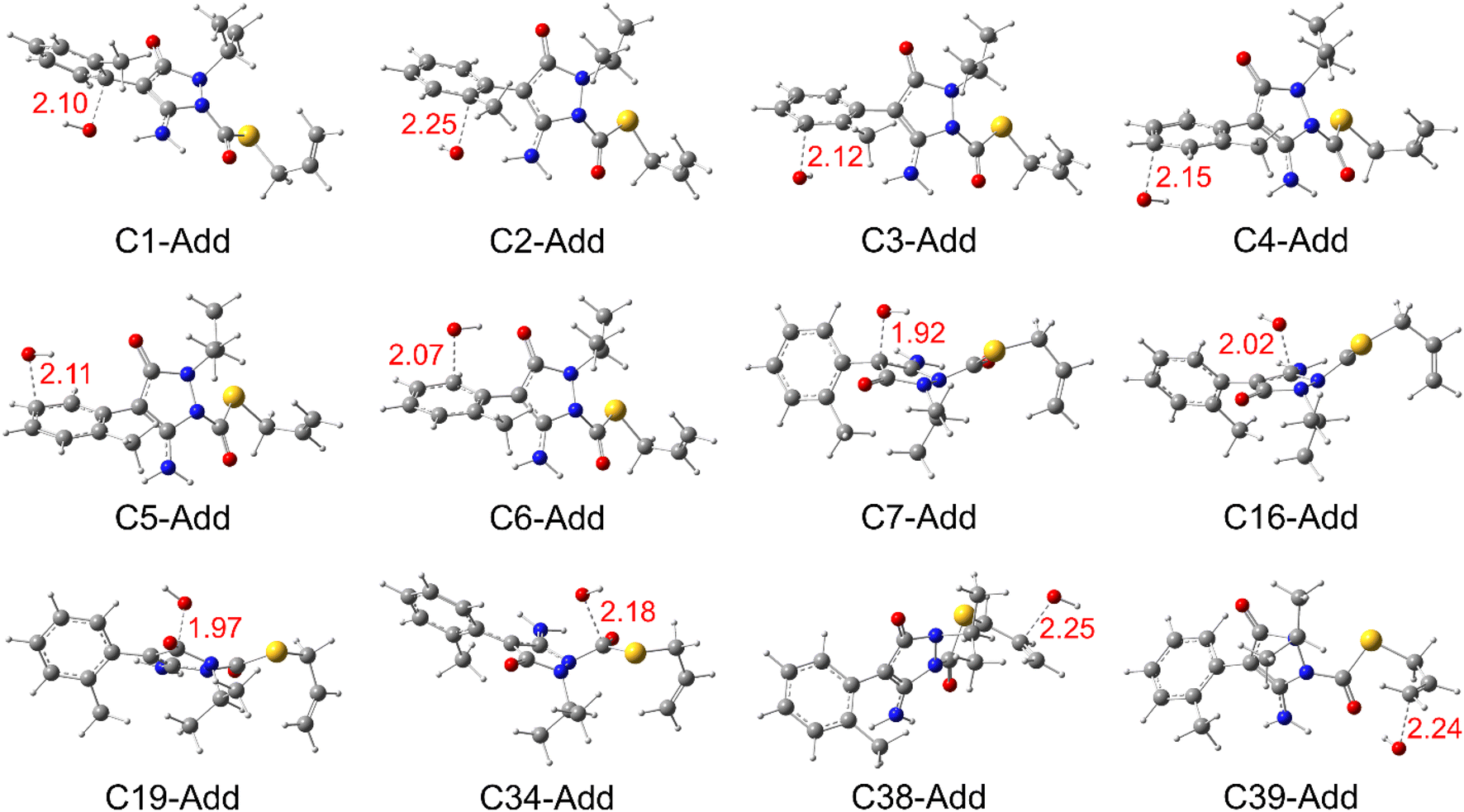 | ||
| Fig. 4 Optimized structures of the transition state (TS) for all the radical adduct formation (RAF) reactions in the aqueous phase. | ||
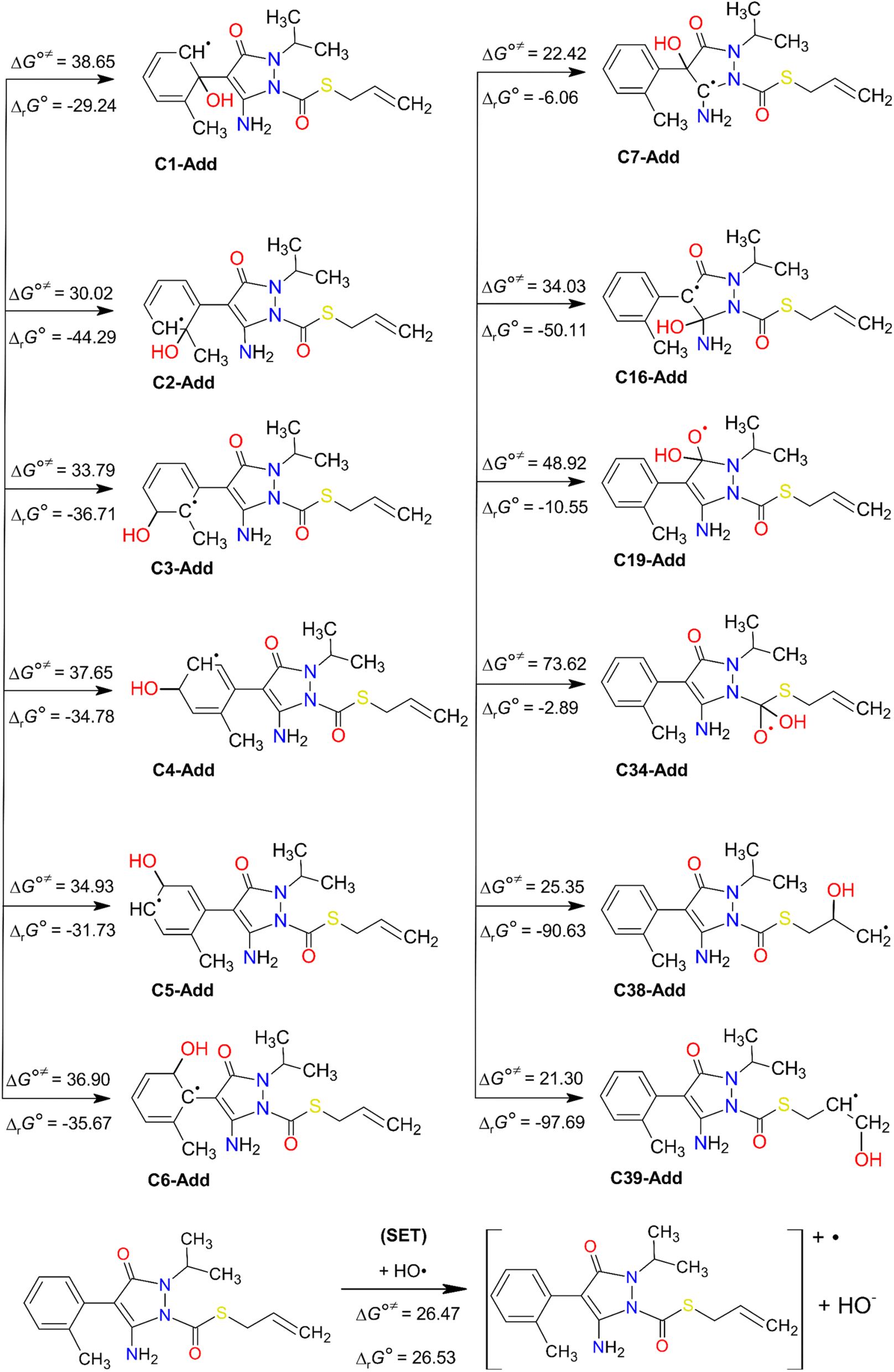 | ||
| Fig. 5 HO˙-addition and SET degradation pathways of FPA. Standard Gibbs free reaction energies (ΔrG°) and standard Gibbs free activation energies (ΔG°≠) at 298 K. Units are in kJ mol−1. | ||
As can be seen in Fig. 5, the HO˙-addition reactions at all the concerned positions were spontaneous and exergonic, having negative values of their standard Gibbs free reaction energy (ΔrG°), ranging from −97.69 (at C39) to −2.89 kJ mol−1 (at C34). The same trend was seen from the value of the standard Gibbs free activation energy (ΔG°≠), which was also lowest at C39 (21.30 kJ mol−1) and highest at C34 (73.62 kJ mol−1). Moreover, all the RAF reaction pathways are exothermic, as indicated by the negative standard reaction enthalpy 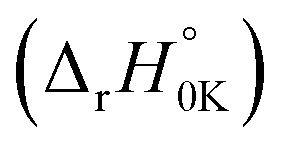 obtained at 0 K (Table S5, ESI File†). The relative values of
obtained at 0 K (Table S5, ESI File†). The relative values of 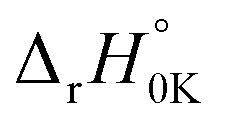 vary from −135.40 (at C39) to −32.21 kJ mol−1 (at C19). These findings indicate that HO˙-addition on C39 is the most favourable thermodynamic pathway due to the lack of steric hindrance. In addition, by comparing the energy barriers (E0) at the TS at 0 K (Table S5†), it is observed that the lowest values are at C2, C7, C38, and C39, which suggest that these pathways are also feasible.
vary from −135.40 (at C39) to −32.21 kJ mol−1 (at C19). These findings indicate that HO˙-addition on C39 is the most favourable thermodynamic pathway due to the lack of steric hindrance. In addition, by comparing the energy barriers (E0) at the TS at 0 K (Table S5†), it is observed that the lowest values are at C2, C7, C38, and C39, which suggest that these pathways are also feasible.
Finally, the differences between the two mentioned computational levels, i.e., M06-2X/6-31+G(d,p) and M06-2X/6-311++G(3df,3pd), are small for the standard Gibbs free activation energies, but they become more pronounced for the energies of reaction. A similar observation can be found for the standard activation and reaction enthalpies. The ΔΔG°≠ values vary in the range from −0.86 kJ mol−1 (at C3) to 3.12 kJ mol−1 (at C34), while the ΔΔrG° values are larger, from −2.73 kJ mol−1 (at C3) to 9.99 kJ mol−1 (at C34) (Table S6, ESI File†).
3.3 Reaction kinetics of fenpyrazamine oxidation
Kinetics of the FHT, RAF, and SET reactions were studied to better understand the oxidation pathways of fenpyrazamine by HO˙ radicals. This was done by calculating the rate constants of each reaction channel over a temperature range of 283–323 K. The results for the rate constant (k) and the branching ratio (Γ, %) for all reactions are presented in Fig. 6, Tables S7 and S8 (ESI File).† Tables S9 and S10† resume tunnelling correction estimated by the Eckart method for FHT and RAF at temperature T from 283 to 333 K.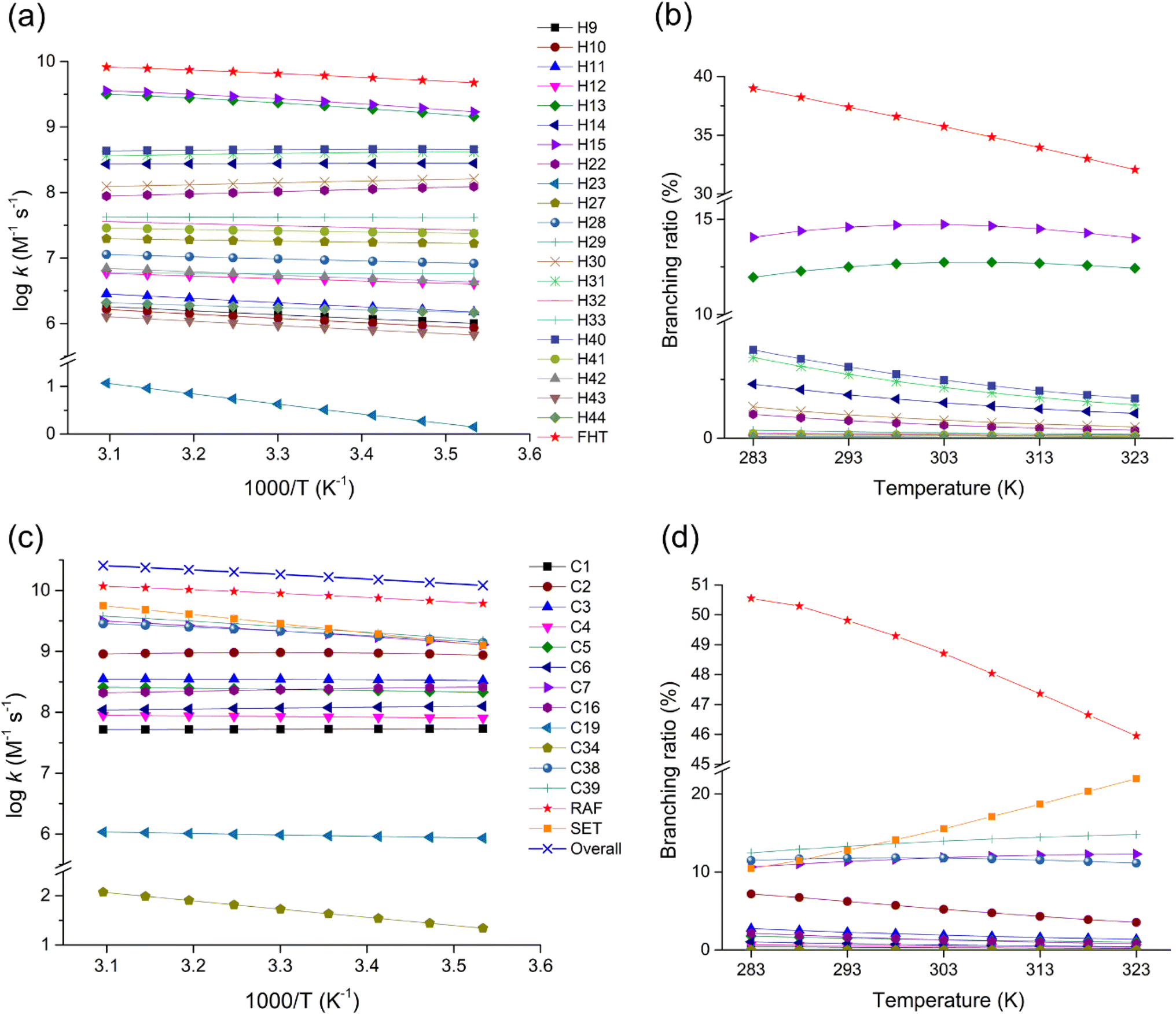 | ||
Fig. 6 Rate constant (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k) and branching ratio (Γ, %) values for the FHT (a and b) and the RAF and SET (c and d) reactions in the temperature range of 283–323 K. k) and branching ratio (Γ, %) values for the FHT (a and b) and the RAF and SET (c and d) reactions in the temperature range of 283–323 K. | ||
The total rate constant of the FHT reactions at 298 K, being 6.09 × 109 M−1 s−1, less than that of the RAF reactions, being 8.21 × 109 M−1 s−1, indicates that the RAF reaction is more predominant. The rate constant of the FHT reaction pathway varies from 3.24 (at H23) and 8.57 × 105 (at H43) to 2.45 × 109 M−1 s−1 (at H15), while those of the RAF reaction range from 4.34 × 101 (at C34) to 2.28 × 109 M−1 s−1 (at C39). The most favourable pathways in the FHT and RAF reactions can be seen from the branching ratio values illustrated in Fig. 6b and d, respectively.
Regarding the effect of temperature, the degradation of FPA by HO˙ has an overall rate constant value ranging from 1.21 × 1010 to 2.56 × 1010 M−1 s−1 as the temperature changes from 283 to 323 K. The total rate constant of the H-abstraction reactions varied from 4.73 × 109 to 8.22 × 109 M−1 s−1, with their total branching ratio values ranging between 39.00 and 32.07%. Meanwhile, the total rate constants of HO˙-addition reactions varied from 6.12 × 109 to 1.18 × 1010 M−1 s−1, representing 50.55 to 45.95%. Moreover, the rate constant values of the SET reaction were observed to be between 1.27 × 109 to 5.63 × 109 M−1 s−1 (2.35 × 109 at 298 K), representing 10.45 to 21.98% of the overall reaction, which is notably high.
As expected, among the H-abstraction reactions, the highest branching ratios were observed at H15 of the methyl group (increasing from 14.06%, reaching 14.73%, then decreasing to 14.01% with T), indicating the predominant pathway, and at H13 of the same group (increasing from 11.96%, reaching 12.74%, then decreasing to 12.43%). These are followed by H40 abstraction of the methylene group (3.76 to 1.69%) and H31 of the methyl group (3.44 to 1.42%).
All the FHT channels show an increase in the rate constant values with increasing temperature, except the channels H14, H22, H30, H31, and H40, which show a slight decline. This phenomenon is probably due to the destabilization of pre-reactive complexes at higher temperatures. Moreover, it could be due to the reduced tunnelling effect at higher temperatures, which decreases the reaction rate.
Concerning the RAF reactions, the highest rate constant value was recognized at the C39 position of the vinyl group, increasing from 1.51 × 109 to 3.80 × 109 M−1 s−1 corresponding to 12.45 to 14.82% of the overall reaction, indicating the most favourable HO˙-addition site. The addition at C38 of the same group follows this channel with the rate constant increasing from 1.39 × 109 to 2.85 × 109 M−1 s−1 and the addition at C7 of the pyrazole ring with the rate constant increasing from 1.29 × 109 to 3.15 × 109 M−1 s−1. In contrast, all the HO˙-addition reaction pathways (except C34) have a decreasing trend in the rate constant values as a function of temperature.
3.4 The lifetime of fenpyrazamine towards HO˙ radicals
To ascertain how long fenpyrazamine persists in the environment when exposed to HO˙ radicals, its lifetime (τ) was computed over the temperature range of 283–323 K at different concentrations of HO˙ in natural waters (i.e., from 10−15 to 10−18 M).46The results presented in Table 1 show that at a low temperature (i.e., 283 K) and low HO˙ concentration (i.e., 10−18 M), the lifetime of FPA was 8.3 × 107 s (2.62 years). As the temperature increases to 323 K, the lifetime decreases to 3.9 × 107 s (1.24 years). Moreover, at 323 K, increasing the [HO˙] from 10−18 to 10−15 M leads to a decrease in the lifetime to 3.9 × 104 s (10.84 hours). In addition, at 298 K, the lifetime varies between 16.67 hours and 1.90 years.
| T (K) | 283 | 288 | 293 | 298 | 303 | 308 | 313 | 318 | 323 |
| k overall, M−1 s−1 | 1.21 × 1010 | 1.35 × 1010 | 1.51 × 1010 | 1.67 × 1010 | 1.83 × 1010 | 2.01 × 1010 | 2.19 × 1010 | 2.38 × 1010 | 2.56 × 1010 |
| [HO˙], M | τ (s) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 × 10−15 | 8.3 × 104 | 7.4 × 104 | 6.6 × 104 | 6 × 104 | 5.5 × 104 | 5 × 104 | 4.6 × 104 | 4.2 × 104 | 3.9 × 104 |
| 1 × 10−16 | 8.3 × 105 | 7.4 × 105 | 6.6 × 105 | 6 × 105 | 5.5 × 105 | 5 × 105 | 4.6 × 105 | 4.2 × 105 | 3.9 × 105 |
| 1 × 10−17 | 8.3 × 106 | 7.4 × 106 | 6.6 × 106 | 6 × 106 | 5.5 × 106 | 5 × 106 | 4.6 × 106 | 4.2 × 106 | 3.9 × 106 |
| 1 × 10−18 | 8.3 × 107 | 7.4 × 107 | 6.6 × 107 | 6 × 107 | 5.5 × 107 | 5 × 107 | 4.6 × 107 | 4.2 × 107 | 3.9 × 107 |
Higher temperatures and concentrations of HO˙ increase the degradation process of FPA, hence decreasing its lifetime.
3.5 Further oxidation reactions of the main-radical product
H15-Abs was the main radical product arising from the abstraction of H15 hydrogen species of fenpyrazamine by HO˙. The further oxidation of this radical product was studied towards different oxidizing agents in aquatic environments like 3O2, HO˙, ˙NO, and ˙NO2 at 298 K, and the results are presented in Fig. 7. Optimized structures and Cartesian coordinates of all the intermediates from the further reactions of the H15-Abs radical are presented in Table S11 (ESI File).†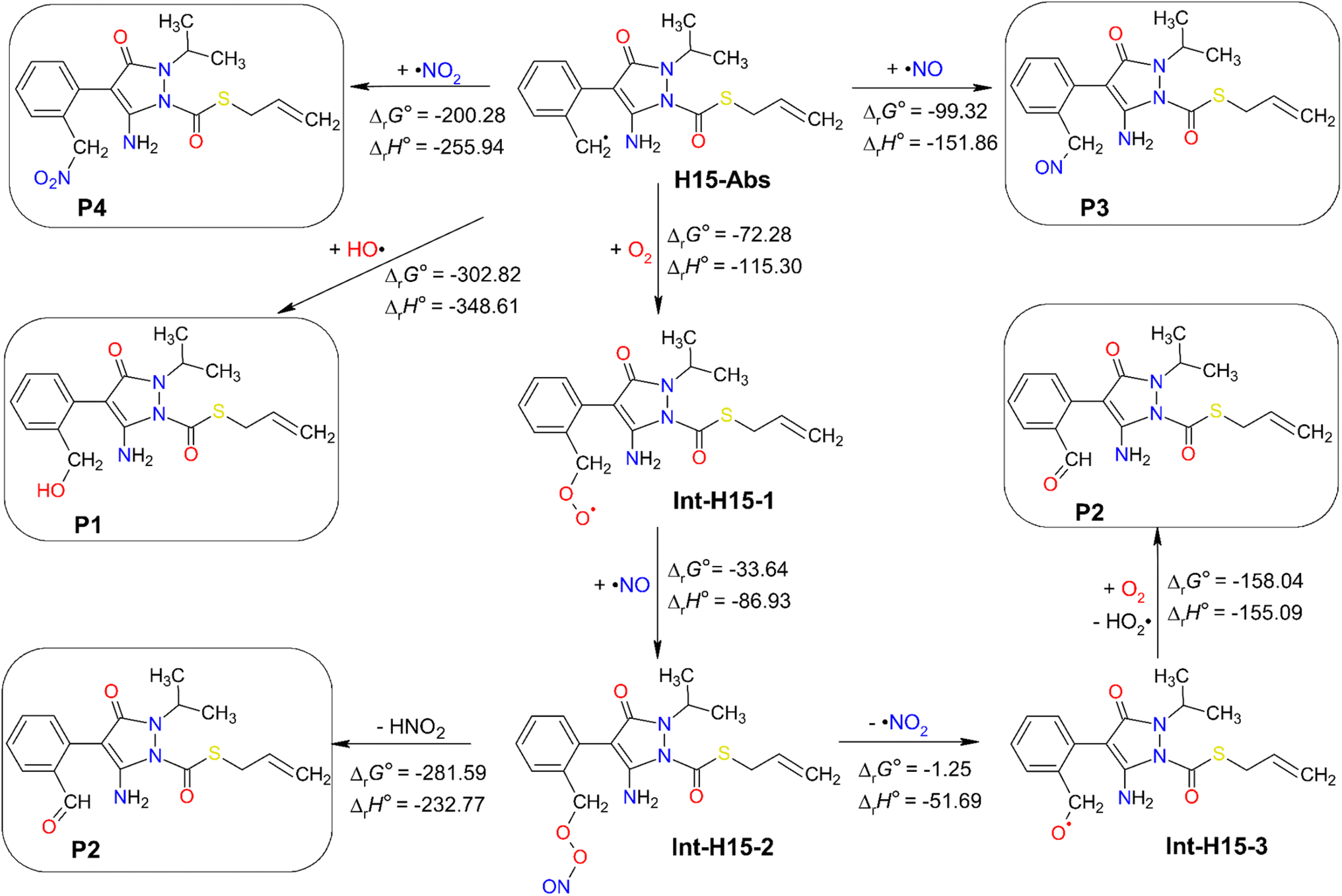 | ||
| Fig. 7 Standard Gibbs free reaction energy (ΔrG°) and standard reaction enthalpy (ΔrH°) calculated for the further reactions of H15-Abs with 3O2, HO˙, ˙NO, and ˙NO2 at 298 K. Units are in kJ mol−1. | ||
All the subsequent reactions are exergonic, spontaneous, and exothermic, indicated by their highly negative ΔrG° and ΔrH° values. The direct addition of HO˙ at C8 of the H15-Abs radical seems to be the most feasible reaction with the lowest ΔrG° value of −302.82 kJ mol−1, generating the neutral product P1. In the same way, 3O2 can attack C8 to form the Int-H15-1 intermediate with a ΔrG° value of −72.28 kJ mol−1. Among various adducts possibly obtained from the O2-addition reactions (at C1–C6, and C7 and C8), the addition to the C8 position exhibits the most predominant reaction (Table S12, ESI).† The Int-H15-1 intermediate can react with ˙NO at the O position to form the Int-H15-2 intermediate, which then undergoes O–O bond cleavage forming ˙NO2 that abstract a hydrogen from C8 producing HNO2 and the neutral product P2. In another pathway, the dissociation of ˙NO2 from Int-H15-2 can form another intermediate Int-H15-3, which in turn reacts with 3O2 to produce HO2˙ and P2. Moreover, the direct reaction of H15-Abs with ˙NO and ˙NO2 can yield the products P3 and P4 with ΔrG° values of −99.32 and −200.28 kJ mol−1, respectively (Fig. 7).
On the other hand, C39-Add was the main radical product obtained from the HO˙-addition at C39 of FPA, forming a radical at C38. Similarly, the essential further oxidation of C39-Add was also studied, and the results are shown in Fig. S5 (ESI File).† Optimized structures and Cartesian coordinates of all the intermediates from the further reactions of C39-Add species are presented in Table S13 (ESI File).†
The addition of HO˙ at C38 of the C39-Add radical produces P5 with the lowest ΔrG° value of −344.92 kJ mol−1, indicating the most feasible reaction. The reaction of the C39-Add radical with 3O2 generates the intermediate Int-C39-1 with ΔrG° value of −100.10 kJ mol−1. Int-C39-1 reacts with ˙NO to form a second intermediate Int-C39-2, which subsequently transforms into HNO2 and the neutral product P6 after the cleavage of the O–O bond and the abstraction of hydrogen from C38 by ˙NO2. In a second pathway, P6 can be produced by the reaction of 3O2 with Int-C39-3 (product of Int-C39-2 after the ˙NO2 dissociation). Furthermore, the neutral products P7 and P8 can be generated from the reaction of C39-Add with ˙NO and ˙NO2, with ΔrG° values of −122.53 and −219.57 kJ mol−1, respectively.
By comparing between H15-Abs and C39-Add radical products, it is observed that the further degradation reactions of C39-Add have lower ΔrG° values than those of H15-Abs. This shows the preference for the radical attack at C38, as previously proved in the kinetics section. Moreover, the highly negative ΔrG° values indicate that these reactions are spontaneous and favourable and can happen at a high rate.
3.6 Ecotoxicity evaluation
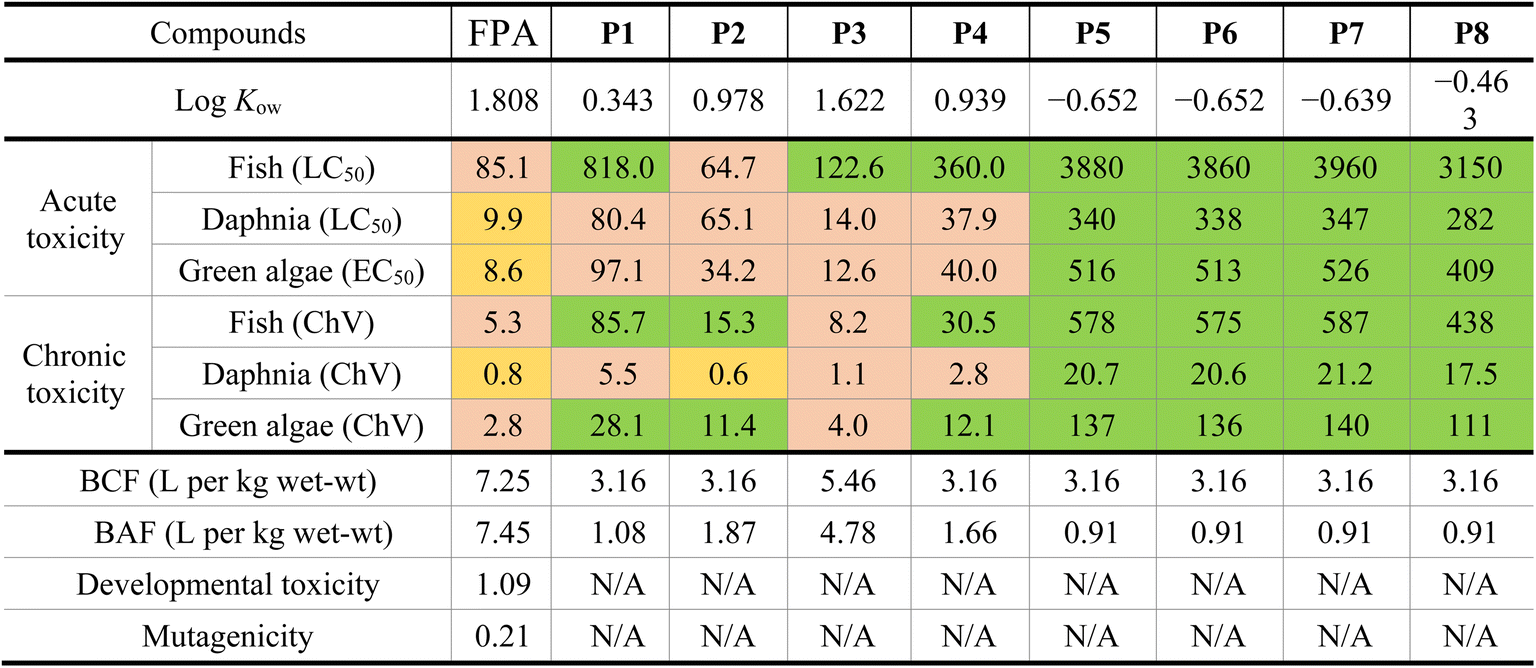
The toxicity estimation of FPA and its essential degradation products was studied for some aquatic species (e.g., fish, daphnia, and green algae) present in the ecosystem, in addition to their BAF, BCF, developmental toxicity, and mutagenicity values. The results are presented in Table 2.
|
The acute toxicity values of FPA were found to be 85.1, 9.9, and 8.6 mg L−1 for fish, daphnia, and green algae, respectively. This means that FPA is harmful to fish and toxic to daphnia and green algae. The values of the chronic toxicity of FPA were 5.3, 0.8, and 2.8 mg L−1, which means it's harmful to fish and green algae but toxic to daphnia.
Concerning the by-products, the data show higher values of LC50 and ChV than those of FPA, which means they are less harmful than the main FPA compound. The products P1, P3, and P4 do not cause acute toxicity to fish; P1, P2, and P4 do not cause chronic toxicity to fish and green algae. Moreover, LC50 and ChV values of the products P5–P7, and P8 show that they are not harmful, and their negative partition coefficient (logKow) values indicate that they are hydrophilic and have high water solubilities.
All the BCF and BAF values of FPA and its degradation products indicate that they are non-bioaccumulative. The BCF and BAF values of FPA were revealed to be 7.25 and 7.45 L per kg wet-wt, respectively, which are higher than those of its by-products, varying from 3.16 to 5.46 L per kg wet-wt for BCF and from 0.91 to 4.78 L per kg wet-wt for BAF.
Fenpyrazamine is considered a developmental toxicant due to its developmental toxicity value of 1.09, whereas it is non-mutagenic with a mutagenicity value of 0.21. On the other hand, no data were found for the developmental toxicity and mutagenicity of the products P1–P8.
4 Conclusions
Fenpyrazamine (FPA) is a new fungicide that has shown strong effectiveness against brown rot, stem rot, and gray mold, which affect vegetables and fruits. The degradation process of FPA induced by HO˙ was studied in the aqueous phase by DFT calculations in the 283–323 K temperature range. Three reaction mechanisms were considered: H-abstraction (FHT), HO˙-addition (RAF), and single electron transfer (SET).Over the temperature range 283–323 K, it is noteworthy that the decreasing order of the three studied reaction mechanisms is as follows: RAF > FHT > SET. At 298 K, very close total rate constants were obtained for the FHT and RAF reactions (i.e., 6.09 × 109 and 8.21 × 109 M−1 s−1, respectively), while that of the SET reaction was 2.35 × 109 M−1 s−1, and 1.67 × 1010 M−1 s−1 for the overall rate constant.
The abstraction of the H15 atom (H15-Abs) of the methyl group (at C8) was observed to be the most favourable abstraction pathway, followed by H13 of the same group (H13-Abs), H40 of the methylene group (H40-Abs), and H31 of the methyl group (H13-Abs). Furthermore, the preferred site for the HO˙-addition was observed at C38/C39 of the vinyl group (C38-Add and C39-Add) and C7 of the pyrazole ring (C7-Add), where C39-Add showed the most favourable pathway.
The lifetime of FPA in natural waters may vary from 10.84 hours to 2.62 years based on the temperature range (283–323 K) and the HO˙ radical concentration (10−15 to 10−18 M).
The further reactions of the main radical product from the FHT reaction (H15-Abs) and that from the RAF reaction (C39-Add) were studied towards different oxidizing agents in aquatic environments like 3O2, HO˙, ˙NO, and ˙NO2 at 298 K, leading to 8 possible products P1–P8.
The toxicity estimation of FPA revealed that it is not bioaccumulative and mutagenic, but it is considered a developmental toxicant, harmful to fish, and toxic to daphnia and green algae. However, the degradation products are less dangerous than FPA itself.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. K. A. R.: data curation, formal analysis, investigation, writing – original draft; D. H. T.: data curation, formal analysis, investigation; E. S.: data curation, formal analysis, investigation; J. F.: formal analysis, writing – review and editing; S. T.: methodology; supervision, validation, writing – review and editing; M. R.: formal analysis, writing – review and editing; A. E. B.: methodology; supervision, validation, writing – review and editing; N. A.: data curation, formal analysis, investigation; D. Q. D.: data curation, formal analysis, supervision, validation, writing – review and editing; F. L.: methodology; project administration, supervision, validation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. Also, SEAGrid (https://www.seagrid.org) is acknowledged for computational resources and services for the selected results used in this publication.47–50 The authors also thank the University of Lille, ERDF, and the HPC facility E-mail: clara@uniba.sk at Comenius University in Bratislava for using their computational facilities. This work is a contribution to the CaPPA project (Chemical and Physical Properties of the Atmosphere), funded by the French National Research Agency (ANR) through the PIA (Programme Investissement d'Avenir) under contract ANR-11-LABX-005-01, to the Institut de Recherches Pluridisciplinaires en Sciences de l'Environnement (IRePSE Fed 4129), and a contribution to the CPER research project ECRIN, with financial support from the French Ministère de l'Enseignement Supérieur et de la Recherche, the Hauts-de-France Region and the European Funds for Regional Economic Development. This work was supported also by the Slovak Research and Development Agency, grant agreement no. APVV-20-0127, as well as the U.S. National Science Foundation (AGS-2045025).References
- A. Gulkowska, I. J. Buerge and T. Poiger, Online solid phase extraction LC-MS/MS method for the analysis of succinate dehydrogenase inhibitor fungicides and its applicability to surface water samples, Anal. Bioanal. Chem., 2014, 406, 6419–6427, DOI:10.1007/s00216-014-8073-4.
- R. P. Oliver and H. G. Hewitt, Fungicides in Crop Protection, CABI, 2nd edn, 2014, p. 201 Search PubMed.
- T. J. Reilly, K. L. Smalling, J. L. Orlando and K. M. Kuivila, Occurrence of boscalid and other selected fungicides in surface water and groundwater in three targeted use areas in the United States, Chemosphere, 2012, 89(3), 228–234 CrossRef CAS.
- H.-W. Dehne, H. B. Deising, U. Gisi, K. H. Kuck, P. E. Russell and H. Lyr, Modern fungicides and antifungal compounds VI. DPG Spectrum Phytomedizin, Proceedings of the 16th International Reinhardsbrunn Symposium, April 25–29, 2010, Friedrichroda, Germany, 2011 Search PubMed.
- P. Leroux, R. Fritz, D. Debieu, C. Albertini, C. Lanen and J. Bach, et al., Mechanisms of resistance to fungicides in field strains of Botrytis cinerea, Pest Manage. Sci., 2002, 58(9), 876–888 CrossRef CAS.
- N. Kimura, M. Hashizume, T. Kusaba and S. Tanaka, Development of the novel fungicide fenpyrazamine, J. Pestic. Sci., 2017, 42(3), 137–143 CrossRef CAS PubMed.
- APVMA, Australian Pesticides and Veterinary Medicines Authority, Data guidelines, 2015, https://www.apvma.gov.au/registrations-and-permits/data-guidelines, accessed 6.25.24 Search PubMed.
- A. C. Taylor, G. R. Fones, A. Gravell and G. A. Mills, Use of Chemcatcher® passive sampler with high-resolution mass spectrometry and multi-variate analysis for targeted screening of emerging pesticides in water, Anal. Methods, 2020, 12, 4015–4027, 10.1039/D0AY01193B.
- FAO, Pesticide residues in food. Joint FAO/WHO meeting on pesticide residues, 2017, https://www.fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Report2017/web_2017_JMPR_Report_Final.pdf, accessed 6.25.24 Search PubMed.
- S. Tanaka, R. Ishikawa and P. Armengaud, Y. Senechal, General characteristics of fenpyrazamine, a novel fungicidal compound for controlling gray mold, 2012, available from: https://www.r4p-inra.fr/wp-content/uploads/2018/01/Tanaka-et-al-2012-AFPP.pdf.
- X. Shi, R. Zhang, Y. Li, Q. Zhang, X. Xu and W. Wang, Mechanism theoretical study on OH-initiated atmospheric oxidation degradation of dimethoate, J. Mol. Struct., 2018, 1163, 61–67 CrossRef CAS.
- D. Q. Dao, S. Taamalli, F. Louis, D. Kdouh, Z. Srour and T. C. Ngo, et al., Hydroxyl radical-initiated decomposition of metazachlor herbicide in the gaseous and aqueous phases: mechanism, kinetics, and toxicity evaluation, Chemosphere, 2023, 312, 137234 CrossRef CAS.
- T. C. Ngo, S. Taamalli, Z. Srour, V. Fèvre-Nollet, A. El Bakali and F. Louis, et al., Theoretical insights into the oxidation of quinmerac herbicide initiated by HO˙ radical in aqueous media: Mechanism, kinetics, and ecotoxicity, J. Environ. Chem. Eng., 2023, 11(3), 109941 CrossRef CAS.
- K. H. Al Rawas, R. A. Mawla, T. Y. N. Pham, D. H. Truong, T. L. A. Nguyen, S. Taamalli, M. Ribaucour, A. E. Bakali, I. Černušák, D. Q. Dao and F. Louis, New insight into environmental oxidation of phosmet insecticide initiated by HO˙ radicals in gas and water – a theoretical study, Environ. Sci., 2023, 25, 2042–2056, 10.1039/D3EM00325F.
- D. H. Truong, T. C. Ngo, T. L. A. Nguyen, T. T. Nguyen, S. Taamalli and A. El Bakali, et al., Oxidation of thiram fungicide by hydroxyl radicals in water: a theoretical study on reaction mechanism and kinetics, Vietnam J. Chem., 2024, 62(4), 547–555 CrossRef CAS.
- D. H. Truong, T. L. A. Nguyen, N. Alharzali, H. K. Al Rawas, S. Taamalli and M. Ribaucour, et al., Theoretical insights into the HO˙-induced oxidation of chlorpyrifos pesticide: Mechanism, kinetics, ecotoxicity, and cholinesterase inhibition of degradants, Chemosphere, 2024, 350, 141085 CrossRef CAS PubMed.
- M. A. Abdel-Rahman, M. F. Shibl, A. M. El-Nahas, S. Abdel-Azeim, S. H. El-demerdash and N. Al-Hashimi, Mechanistic insights of the degradation of an O-anisidine carcinogenic pollutant initiated by OH radical attack: theoretical investigations, New J. Chem., 2021, 45, 5907–5924, 10.1039/D0NJ06248K.
- X. Bo, J. Sun, Q. Mei, B. Wei, Z. An, D. Han, Z. Li, J. Xie, J. Zhan and M. He, Degradation of prosulfocarb by hydroxyl radicals in gas and aqueous phase: Mechanisms, kinetics and toxicity, Ecotoxicol. Environ. Saf., 2020, 191, 110175, DOI:10.1016/j.ecoenv.2020.110175.
- A. Galano and J. R. Alvarez-Idaboy, Kinetics of radical-molecule reactions in aqueous solution: a benchmark study of the performance of density functional methods, J. Comput. Chem., 2014, 35(28), 2019–2026 CrossRef CAS PubMed.
- Y. Huo, M. Li, Z. An, J. Sun, Q. Mei, B. Wei, Z. Qiu, J. Xie and M. He, Ozonolysis of Permethrin in the Atmosphere: Mechanism, Kinetics, and Evaluation of Toxicity, J. Phys. Chem. A, 2021, 125, 7705–7715, DOI:10.1021/acs.jpca.1c04812.
- Z. Qiu, J. Sun, D. Han, F. Wei, Q. Mei, B. Wei, X. Wang, Z. An, X. Bo, M. Li, J. Xie and M. He, Ozonation of diclofenac in the aqueous solution: Mechanism, kinetics and ecotoxicity assessment, Environ. Resour., 2020, 188, 109713, DOI:10.1016/j.envres.2020.109713.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241, DOI:10.1007/s00214-007-0310-x.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396, DOI:10.1021/jp810292n.
- J. L. Bao, L. Xing and D. G. Truhlar, Dual-Level Method for Estimating Multistructural Partition Functions with Torsional Anharmonicity, J. Chem. Theory Comput., 2017, 13, 2511–2522, DOI:10.1021/acs.jctc.7b00232.
- A. Miyoshi, GPOP software. Revision 2022.01.20m1, http://akrmys.com/gpop/ Search PubMed.
- D. L. Singleton and R. J. Cvetanovic, Temperature dependence of the reaction of oxygen atoms with olefins, J. Am. Chem. Soc., 1976, 98, 6812–6819, DOI:10.1021/ja00438a006.
- K. P. Huber and G. Herzberg, Constants of diatomic molecules, in Molecular Spectra and Molecular Structure: IV Constants of Diatomic Molecules, ed. Huber K. P. and Herzberg G., Springer US, Boston, MA, 1979, pp. 8–689, DOI:10.1007/978-1-4757-0961-2_2.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I, J. Chem. Phys., 1956, 24(5), 966–978 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. III. Applications to Data on the Rates of Organic Redox Reactions, J. Chem. Phys., 1957, 26(4), 872–877 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. II. Applications to Data on the Rates of Isotopic Exchange Reactions, J. Chem. Phys., 1957, 26(4), 867–871 CrossRef CAS.
- M. V. Smoluchowski, Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Lösungen, Z. Phys. Chem., 1918, 92U(1), 129–168 CrossRef.
- D. G. Truhlar, Nearly encounter-controlled reactions: The equivalence of the steady-state and diffusional viewpoints, J. Chem. Educ., 1985, 62(2), 104–106 CrossRef CAS.
- A. Einstein, Zur Elektrodynamik bewegter Körper, Ann. Phys., 1905, 322(10), 891–921 CrossRef.
- G. G. Stokes, Mathematical and Physical Papers, Cambridge Core, 2009, DOI:10.1017/CBO9780511702297.
- F. C. Collins and G. E. Kimball, Diffusion-controlled reaction rates, J. Colloid Sci., 1949, 4(4), 425–437 CrossRef CAS.
- B. C. Garrett and D. G. Truhlar, Semiclassical tunneling calculations, J. Phys. Chem., 1979, 83, 2921–2926, DOI:10.1021/j100485a023.
- US EPA, Ecological Structure Activity Relationships (ECOSAR) Predictive Model, 2015, https://www.epa.gov/tsca-screening-tools/ecological-structure-activity-relationships-ecosar-predictive-model Search PubMed.
- F. Melnikov, J. Kostal, A. Voutchkova-Kostal, J. B. Zimmerman and P. T. Anastas, Assessment of predictive models for estimating the acute aquatic toxicity of organic chemicals, Green Chem., 2016, 18(16), 4432–4445 RSC.
- L. Zhou, D. Fan, W. Yin, W. Gu, Z. Wang and J. Liu, et al., Comparison of seven in silico tools for evaluating of daphnia and fish acute toxicity: case study on Chinese Priority Controlled Chemicals and new chemicals, BMC Bioinf., 2021, 22(1), 151 CrossRef CAS.
- US EPA, EPI Suite™-Estimation Program Interface, 2015, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface Search PubMed.
- J. A. Arnot and F. A. Gobas, A review of bioconcentration factor (BCF) and bioaccumulation factor (BAF) assessments for organic chemicals in aquatic organisms, Environ. Rev., 2006, 14, 257–297, DOI:10.1139/a06-005.
- US EPA, Toxicity Estimation Software Tool (TEST), 2015, https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test, accessed 6.9.23 Search PubMed.
- W. Chen, J. Zheng, J. L. Bao, D. G. Truhlar and X. Xu, MSTor 2023: A new version of the computer code for multistructural torsional anharmonicity, now with automatic torsional identification using redundant internal coordinates, Comput. Phys. Commun., 2023, 288, 108740 CrossRef CAS.
- A. M. Rebollar-Zepeda and A. Galano, Quantum mechanical based approaches for predicting pKa values of carboxylic acids: evaluating the performance of different strategies, RSC Adv., 2016, 6, 112057–112064, 10.1039/C6RA16221E.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake, S. A. Rusak, A. L. Rose and T. D. Waite, Methods for reactive oxygen species (ROS) detection in aqueous environments, Aquat. Sci., 2012, 74, 683–734, DOI:10.1007/s00027-012-0251-x.
- R. Dooley, K. Milfeld, C. Guiang, S. Pamidighantam and G. Allen, From Proposal to Production: Lessons Learned Developing the Computational Chemistry Grid Cyberinfrastructure, J. Grid Comput., 2006, 4, 195–208, DOI:10.1007/s10723-006-9043-7.
- K. Milfeld, C. Guiang, S. Pamidighantam and J. Giuliani, in Cluster Computing through an Application-oriented Computational Chemistry Grid, 2005, https://www.semanticscholar.org/paper/Cluster-Computing-through-an-Application-oriented-Milfeld-Guiang/c60eeacc596cf7a0f6c96c1362d6cc318f7dda8a Search PubMed.
- S. Pamidighantam, S. Nakandala, E. Abeysinghe, C. Wimalasena, S. R. Yodage, S. Marru and M. Pierce, Community Science Exemplars in SEAGrid Science Gateway: Apache Airavata Based Implementation of Advanced Infrastructure, Procedia Comput. Sci., 2016, 80, 1927–1939 CrossRef.
- N. Shen, Y. Fan and S. Pamidighantam, E-science infrastructures for molecular modeling and parametrization, J. Comput. Sci., 2014, 5(4), 576–589 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00606b |
| This journal is © The Royal Society of Chemistry 2025 |