
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid-heating-assisted metal–support interaction formation: Pd nanoparticles on boron nitride nanotubes as electrocatalysts for high N2-to-ammonia yields†
Ji-Yoon
Song‡
ab,
Jaehyun
Park‡
c,
Minsung
Kang‡
 b,
Wooseok
Lee
d,
Jee Ho
Ha
c,
Jinpil
Ha
a,
Won Bo
Lee
d,
Aqil
Jamal
e,
Issam
Gereige
e,
YongJoo
Kim
*f,
Hyeonsu
Jeong
*b,
Seokhoon
Ahn
*b,
Seok Ju
Kang
*c and
Hee-Tae
Jung
*a
b,
Wooseok
Lee
d,
Jee Ho
Ha
c,
Jinpil
Ha
a,
Won Bo
Lee
d,
Aqil
Jamal
e,
Issam
Gereige
e,
YongJoo
Kim
*f,
Hyeonsu
Jeong
*b,
Seokhoon
Ahn
*b,
Seok Ju
Kang
*c and
Hee-Tae
Jung
*a
aDepartment of Chemical & Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST) 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: heetae@kaist.ac.kr
bInstitute of Advanced Composite Materials, Korea Institute of Science and Technology, Jeonbuk 55324, Republic of Korea. E-mail: ahn75@kist.re.kr; jeonghs98@kist.re.kr
cDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: sjkang@unist.ac.kr
dSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul 08826, Republic of Korea
eSaudi Aramco, Research and Development Center, Dhahran 31311, Saudi Arabia
fDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: cjyjee@korea.ac.kr
First published on 12th March 2025
Abstract
The electrochemical nitrogen (N2) reduction reaction (eNRR) is pivotal for synthesizing green ammonia (NH3) under ambient conditions. However, challenges such as mitigating the detrimental hydrogen evolution reaction (HER) and overcoming the sluggish proton-coupled electron transfer (PCET) step limit the efficiency of the eNRR process. Here, we present a metal–support heterostructure catalyst comprising uniform and high-density palladium nanoparticles (Pd NPs) on defective boron nitride nanotubes (D-BNNTs) via the rapid radiative Joule-heating method. Notably, the strong electronic metal–support interaction (EMSI) between the BNNT defects and Pd NPs creates an electron-deficient state in the Pd NPs, significantly reducing the PCET step and suppressing the HER. This unique configuration of the Pd NPs supported on the D-BNNT catalyst exhibits outstanding NH3 selectivity, achieving 68.0% in neutral aqueous electrolytes and 58.9% in acidic media with a yield rate of 8.69 × 10−10 mol s−1 cm−2. This approach offers a strategic pathway for catalyst engineering in electrochemical reactions, presenting significant potential for practical applications.
Broader contextThe electrochemical nitrogen reduction reaction (eNRR) for ammonia synthesis is regarded as a potential alternative against the energy-intensive Haber–Bosch process. However, the efficiency and selectivity of nitrogen-to-ammonia conversion have been hindered by the competing hydrogen evolution reaction (HER) and the slow proton-coupled electron transfer (PCET) step. Herein, a novel catalyst design using palladium nanoparticles (Pd NPs) on defect-rich boron nitride nanotubes (BNNTs) is presented. The key innovation lies in exploiting the strong electronic metal–support interaction (EMSI) between Pd and BNNT defect sites, creating an electron-deficient state of Pd NPs by altering the electronic structure with high density. This lowers the activation barrier of the PCET step of the eNRR process and also suppresses the HER, achieving high faradaic efficiency and ammonia yields. These findings underscore the importance of exploring EMSI effects in metal–support heterostructure catalysts to make ambient eNRR a practical solution for sustainable and green ammonia production. |
Introduction
Electrochemical nitrogen (N2) fixation toward green ammonia (NH3) production has been proposed to overcome the intensive energy consumption and enormous carbon footprint associated with the Habor–Bosch process.1–5 In particular, the electrocatalytic N2 reduction reaction (eNRR) allows the synthesis of NH3 from water using N2 gas as a reactant under ambient conditions. It becomes more alluring because its driving forces rely upon renewable sources such as solar, wind, etc.6–12 However, a major challenge is the competing hydrogen evolution reaction (HER), which remains an insurmountable bottleneck because both reactions involve the proton-coupled electron transfer (PCET) step under the potential range of below 0 V versus the reversible hydrogen electrode (RHE) scale. From a thermodynamic perspective, the activation energy required to break the triple bond in N2 demands a higher overpotential than that for the HER, making N2 adsorption generally less favorable than that of hydrogenic ions (H+ or H3O+).10–12 As a consequence of these fatal issues, a novel way for developing efficient electrocatalysts is of great interest in both suppressing the HER and boosting the PCET step.Among various approaches to limit the HER and accelerate the PCET step to improve eNRR performance, modulating the electronic structure of the electrocatalyst through the metal–support heterostructure, known as the electronic metal–support interaction (EMSI) effect, has emerged as a potent strategy for regulating the PCET step during the eNRR.13–15 This EMSI effect profoundly influences catalytic activity by altering the energy of the d-band center of metals, thereby redistributing the charge density.16 Various metal–support heterostructures such as Mo/VO2, Ru/TiO2, Fe/TiO2, Cu/CeOx, FeCu/g-CN, Au/TiO2, PdCu/TiO2, Cu/TiO2, and Cu/polyetherimide have shown potential in enhancing eNRR performance and suppressing the HER by creating Lewis-acidic sites and promoting asymmetric electron distributions.13–15,17–23 Nevertheless, prior attempts have struggled to achieve significant electron transfer to activate the PCET step towards the exothermic state, which remained limited in performance improvement. Boron nitride (BN), with its high hydrophobicity and a stronger proton adsorption energy compared to the carbon surface, has received interest as a support material for HER suppression.24,25 Additionally, defective BN can significantly alter the electronic structure of the metal catalysts, enhancing their electrochemical reaction performance through the EMSI effect.26–28 Nevertheless, applying BN analogs as a supporting material in the eNRR poses challenges due to their insulating nature and the difficulty of uniformly depositing metal nanoparticles on their surface.
In this context, we present a catalyst engineering approach utilizing defect-rich boron nitride nanotubes (BNNTs) as a support for metal nanoparticles. Before using BNNTs as a support material for metal nanoparticles, it was observed that raw BNNTs contain amorphous boron and various hBN-like impurities of differing sizes (unreacted hBN, crystalline hBN, hBN shells, etc.).29,30 These impurities were removed by applying previously reported methods, and BNNTs with large impurities removed were used.31 Using a rapid radiative Joule-heating (RRJH) process, high-density Pd nanoparticles are uniformly decorated on defective BNNT (D-BNNT) catalysts. The resulting catalysts impressively suppress the HER, enhancing the selectivity of the eNRR. The improvement of electrochemical performance is carefully confirmed through a combination of computational studies and experimental evidence, which indicates that the defects in BNNT support induce a significant change in the electronic structure of metal catalysts, leveraging the EMSI effect of BNNT support for high-performance eNRR. This catalyst achieves a faradaic efficiency (FE) of 58.9% and a yield rate (YR) of 8.69 × 10−10 mol s−1 cmgeo−2 in 0.25 M H2SO4 at −0.1 VRHE, marking the highest areal NH3 production rate among catalyst engineering approaches.
Results and discussion
Computational studies of eNRR electrocatalysis
To elucidate the crucial role of defects in the BNNT support in the proton-coupled electron transfer (PCET) step and hydrogen evolution reaction (HER), we conducted density functional theory (DFT) calculations of charge modulations and their eNRR pathways (Fig. 1). Boron nitride, serving as a support material, not only exhibits high hydrophobicity but also exhibits a stronger proton adsorption energy compared to carbon surfaces, enabling the successful suppression of the HER.24,25 We utilized a well-known eNRR metal catalyst of palladium (Pd) to enhance eNRR performance due to its low overpotential towards the eNRR.32 Therefore, we postulate that the defects in the BNNT support might alter the electronic structure of the Pd metal catalyst, which results in substantial improvement in activity due to the highly localized electrons increasing the electron transfer between the Pd metal–BNNT support interface and maximizing the electronic metal-to-support interaction (EMSI) effect.25,33–37 | ||
| Fig. 1 Computational studies of electronically tuned catalysts for eNRR electrocatalysis. (a) Bader charges for each Pd atom on the crystalline plane of BN (left) and defective BN (right) supports. (b) Average electron density of Pd on supports. (c) Calculated absorption configurations of the Gibbs free energy reaction path for the PCET step on bare Pd, Pd on the plane, and defects (enzymatic). (d) Gibbs free energy change of the PCET step in alternative, distal, enzymatic pathways, and *N2 selectivity on the catalyst surface. (e) Schematic illustration of the expected eNRR performance of Pd/D-BNNTs. | ||
A single layer of boron nitride and six Pd (111) clusters were used for the calculation as illustrated in Fig. 1a (more details in the Experimental and Fig. S1–S3 for the optimization process of DFT, ESI†). The Bader charge results show that the defect site in BN attracts more electrons from the Pd cluster (0.064 for each Pd atom) than that of the plane (crystalline structure) of BN support (0.012 for each Pd atom), indicating that the active electronic effect occurs between Pd and the defect site than the plane of BN (Fig. 1b).
To explore the influence of the highly electron-deficient state of Pd on eNRR performance, we further carried out DFT calculations of Pd slabs on the plane and defect site. Given that the initial hydrogenation step of adsorbed nitrogen is the PCET step for the NRR process, we first investigated the Gibbs free energy change for converting *N2 to *N2H (ΔGPCET). As mentioned earlier, Bader charge analysis revealed that the strong EMSI effect between the BN and Pd NPs results in an electron-deficient state for the Pd NPs. Thus, an electron-deficient Pd slab was modeled to represent Pd NPs on BN using calculated Bader charges. Compared to the neutral Pd slab, electron-deficient Pd slabs significantly stabilized N2 adsorption and stabilized N2H adsorption, reducing ΔGPCET from 0.808 eV to 0.682 eV and 0.005 eV at Pd on the plane and Pd on the defect, respectively, in the enzymatic pathway (Fig. 1c). In addition to the tremendous decrease of ΔGPCET in the enzymatic pathway, the Pd slab on the defect can effectively drop ΔGPCET during the alternative/distal pathway and suppress the adsorption of *H, which is the reactant of the competing HER (Fig. 1d). H adsorption (*H) is thermodynamically more favored than N2 adsorption (*N2), and most surfaces of metal catalysts are covered by *H.38 These adsorbed *H sites not only promote unwanted side reactions but, more seriously, also obstruct the active sites for initiating NRR by blocking the N2 adsorption sites (*N2), which caused a considerable decreasing the FE and yield rate of NH3. For that reason, *H/*N2 selectivity has been considered an important factor in enhancing the FE and yield rate of NH3. *H/*N2 selectivity (ΔΔG(*H/*N2)) can be confirmed by comparing ΔG(*N2) and ΔG(*H), ΔG(*N2) − ΔG(*H).39,40 The H adsorption energies of the Pd, Pd on the plane, and Pd on the defect are similar with values of −0.738, −0.727, and −0.711 eV, respectively (Table S1, ESI†). Contrary to this, the ΔG(*N2) sites in the enzymatic pathway were significantly different at −0.157, −0.18, and −0.551 eV, respectively. Each *H/*N2 selectivity was 0.581 (Pd), 0.547 (Pd on the plane), and 0.16 (Pd of the defect) eV, indicating that the catalyst surface of Pd on the defect is significantly suppressing H adsorption. These simulation results account for Pd slabs on the defect sites of BN are highly impactive to eNRR activity, suggesting the realization of dense formation of metal nanoparticles is crucial to carry out high eNRR performance.
Catalyst preparation and characterization
Fig. 2a illustrates the overall scheme for the fabricating procedure of the Pd/D-BNNT heterostructure. Synthesizing dense Pd NPs on the BN surface is challenging due to the limited binding affinity between the metal and BN support. Specifically, using a thermal synthetic method with a furnace often leads to the formation of agglomerated metal nanoparticles by Ostwald ripening, migration, and coalescence, mainly due to its long thermal exposure time.41–43 Therefore, we adopted the rapid radiative Joule-heating (RRJH) process to facilitate the formation of high-density metal nanoparticles without significant aggregation (Fig. S4 and S5, ESI†). This short thermal exposure time of less than 0.5 seconds allows the dense Pd NPs to form on boron nitride (Pd/BNNT) and enables precise control over the structural validation and electronic properties of Pd nanoparticles. | ||
| Fig. 2 Morphological and electronic structural characterization. (a) Schematic illustration of the fabrication process of the defective BNNT (D-BNNT) and Pd/BNNT/CP electrode. The temperature versus time plot shows the RRJH operation profile. The inset shows the digital photograph image of the sample during the RRJH process. (b) Cross-sectional SEM image of the Pd/D-BNNT/CP electrode and corresponding EDS mapping images in the red dotted square region. The blue, green, and magenta maps represent C, N, and Pd, respectively. (c) Top-view SEM image of the Pd/BNNT layer. (d) HRTEM bright-field image of the Pd/BNNT layer. (right) FFT patterns indicate the Pd (111) and BNNT (002) planes. (e) Plot of Pd NP size distribution. (f) XPS B 1s spectra of D- and C-BNNTs. (g) XPS Pd 3d spectra of Pd/D-BNNTs, Pd/C-BNNTs, and Pd/CP. (h) Plot of the quantitative relationship between the defect amount in BNNT and binding energy shift. (i) Plot of the bandgap structures of C- and D-BNNT layers before and after Pd NP formation. | ||
The cross-sectional scanning electron microscopy (SEM) image of Pd/BNNT/CP reveals that the Pd NPs were selectively formed on the BNNT layer. This is further confirmed by corresponding X-ray energy-dispersive spectroscopy (EDS) mapping images (Fig. 2b). The magenta color, representing Pd atoms, was predominantly observed on the green-colored layer, indicating the presence of N atoms within BNNTs, while Pd showed marginal contrast in the blue-colored area representing C atoms. One plausible explanation for this observation is the high polarity of the BNNT surface due to the formation of B–N bonds, which are the group III–V compounds in the periodic table leading to electron localization due to its larger electronegativity difference, which provides anchoring sites for Pd metal NPs under high-temperature conditions during a rapid Joule-heating process.44
The top view of the Pd/BNNT layer, as displayed in Fig. 2c and Fig. S6 (ESI†), shows that Pd NPs are well dispersed on the BNNT layer over a large area with a uniform particle size of approximately 17 nm (as shown in the inset histogram in Fig. 2e). The results reveal that the ultra-short heat exposure time (≈0.5 s) and rapid cooling rate (650 K s−1) successfully prevent the aggregation typically caused by Ostwald ripening and coalescence which is in contrast to the conventional furnace and hydrothermal synthesis protocol.39,41,45
The formation of the metallic Pd state was further confirmed by X-ray diffraction (XRD) spectroscopy and X-ray photoelectron spectroscopy (XPS) of the surface of the Pd/BNNT layer (Fig. S7, ESI†). In the resulting XRD pattern, BNNTs have been reported to exhibit a broad and weak peak at 25.6°.46 CP has been reported to exhibit a very sharp diffraction peak at 26.5°.47,48 This strong peak from CP obscured the broad and weak diffraction pattern of BNNTs, making it indistinguishable (Fig. S7, ESI†). On the other hand, the distinct peaks at 40.1°, 46.7°, and 63.2° correspond to Pd (111), (200) and (220) planes, respectively.44,49 Additionally, the presence of Pd 3d peaks at 341.0 and 335.9 eV and Pd 3p peaks at 560.6 eV in the XPS survey spectrum supports the formation of Pd NPs on the BNNT layer following the RRJH process. Moreover, the high-resolution transmittance electron microscopy (HRTEM) bright-field image of the Pd/BNNT layer in Fig. 2d visualizes apparent BNNT bundles in a dotted square and Pd NPs in yellow dotted square regions. The corresponding fast Fourier transformation (FFT) patterns extracted from these dotted square regions in the HRTEM image reveal lattice spacing of 0.34 nm and 0.22 nm, attributed to the (002) plane of BNNTs and (111) plane of Pd, respectively (Fig. S6 and S7, ESI†). The measured lattice distance extracted from the FFT patterns is well-matched with XRD patterns (Fig. S8, ESI†). Furthermore, Scherrer ring patterns corresponding to the (111), (200), and (220) planes of Pd and the (100) and (002) planes of BNNTs clearly indicate the coexistence of both materials in selected area electron diffraction (SAED) patterns (Fig. S9, ESI†). Thus, it is reasonable to conclude that our synthetic approach combined with the polar chemical bonds of BN and a rapid Joule-heating synthesis allowed us to fabricate an electrocatalyst electrode that potentially caused the strong EMSI effect.
The formation of the BNNT defects generated by the jet plasma process was verified by XPS investigation. In the high-resolution XPS B 1s spectra (Fig. 2f and Fig. S10, ESI†), the deconvolution of the D-BNNTs and C-BNNTs shows peaks arising from B–N at ∼190.5 eV and N–B–O at ∼191.5 eV, respectively.44,50,51 Among these, the presence of N–B–O has been reported to increase as the defects in BNNTs increase.52 The D-BNNTs showed a significantly higher B–N–O bond concentration (filled with pink gradation in the deconvoluted plot) compared to the C-BNNTs. This result indicates that the crystalline structure and N–B–O bond orientate from dangling B–O bonds where N-vacant regions are considered defect sites in the BNNT structure.44,53,54 The N–B–O to B–N bond ratios are 0.36 for D-BNNTs and 0.12 for C-BNNTs, indicating that D-BNNTs have three times higher defect sites than C-BNNTs (Fig. S10, ESI†). The strong interaction between Pd and D-BNNTs is further characterized by XPS Pd 3d spectra in Fig. 2g. The binding energy shift of Pd on D-BNNTs (Pd/D-BNNTs), C-BNNTs (Pd/C-BNNTs) and carbon paper (Pd/CP) indicates the interaction between Pd metal and support. For instance, the binding energy in Pd/D-BNNTs (335.49 eV) and Pd/C-BNNTs (335.33 eV) negatively shifts relative to that in a control sample of Pd/CP (335.22 eV). Significantly, the binding energy of Pd/BNNTs is more negatively shifted ∼0.27 eV than that of Pd/C-BNNTs (∼0.11 eV). This larger shift in Pd on D-BNNTs indicates a more significant charge transfer between Pd metal and D-BNNT support, resulting in a substantial change in the electronic structure of the heterogeneous catalyst.44,55 Furthermore, the amount of defects in BNNTs is quantitatively related to corresponding binding energy shifts of Pd 3d on each BNNT (Fig. 2h), indicating electrons in Pd become significantly deprived by defective sites on BNNTs, and this correlation corresponds with Fig. 1a.
The bandgap of electrodes is also characterized by reflective electron energy loss spectroscopy (REELS) (Fig. 2i and Fig. S11, ESI†). Pristine C-BNNTs showed a band gap of 5.90 eV, while the bandgap of pristine D-BNNTs was slightly narrower, with a value of 5.78 eV. The 0.12 eV bandgap difference between D-BNNTs and C-BNNTs strongly signifies a higher concentration of defects from the valence band, such as N vacancies and N–B–O bonds, leading to a higher localization state.54 After the formation of Pd NPs through RRJH, the altered band structures of BNNT substrates were observed. A narrowing of the band gap and the appearance of mid-gap after combination with Pd NPs (Fig. S11, ESI†) reflect the strong electronic interaction between Pd and BNNT supports.53,56 Notably, the difference in the bandgap of D-BNNTs before and after Pd decoration is higher than that of C-BNNTs, which supports that the higher polarity from rich defects on D-BNNTs supplies the higher charge transfer. Thus, the observed electronic effect through theoretical simulation and experimental analyses suggests that the strong interaction between the Pd metal and D-BNNT (Pd/D-BNNT) heterostructures positively enhances eNRR performance.
Electrochemical N2 reduction performance of Pd/D-BNNTs
Fig. 3 presents the electrochemical eNRR performances of Pd/D-BNNTs. In Fig. 3a, the N2 gas contact angle of Pd/D-BNNTs is shown alongside the BNNT sample, illustrating that Pd/D-BNNTs maintain a similar N2 contact angle to the BNNT sample. This observation suggests the preservation of the hydrophobic nature of the BNNT supports even after decorating the Pd NPs, which is advantageous for facilitating efficient three-phase interfaces, thereby contributing to the improvement of N2 adsorption kinetics during the eNRR process.57,58 The advantages arising from the strong interaction between the Pd and defective BNNTs are further supported by measuring the eNRR and H2 evolution activity using a conventional H-type three-electrode beaker cell alongside comparisons with Pd/C-BNNTs and Pd/CP (Fig. S12, ESI†). To ensure a supply of pure N2 gas to the electrolytic cell, the N2 gas was passed through a 0.5 M KMnO4 gas trap and subsequent moisture trap before being introduced into the electrolysis system (Fig. S13 and S14, ESI†). Upon analyzing the linear sweep voltammetry (LSV) trajectories and corresponding Tafel plots obtained under N2-saturated 0.25 M H2SO4 aqueous electrolyte, a noticeable decrease in current response with an increased Tafel slope of 131 mV dec−1 was observed for the Pd/D-BNNT sample. Compared to those of Pd/C-BNNTs (101 mV dec−1) or Pd/CP (96 mV dec−1), the high Tafel slope can be attributed to the reduced H2 evolution activities (Fig. S15, ESI†). Real-time differential electrochemical mass spectroscopy (DEMS) measurement was further conducted under a gas-tight three-electrode cell system with LSV measurement to corroborate the HER activities (Fig. 3b).59 The resulting H2 gas (m/z = 2) evolution graph reveals the delayed H2 evolution potential of the Pd/D-BNNT sample compared to Pd on CP. These results further imply an efficient HER blocking by the BNNT substrate through the redistributing electronic structure, driven by the interaction between the Pd metal and defect-rich BN analogs, thus enhancing the catalytic activity towards the high-efficiency eNRR.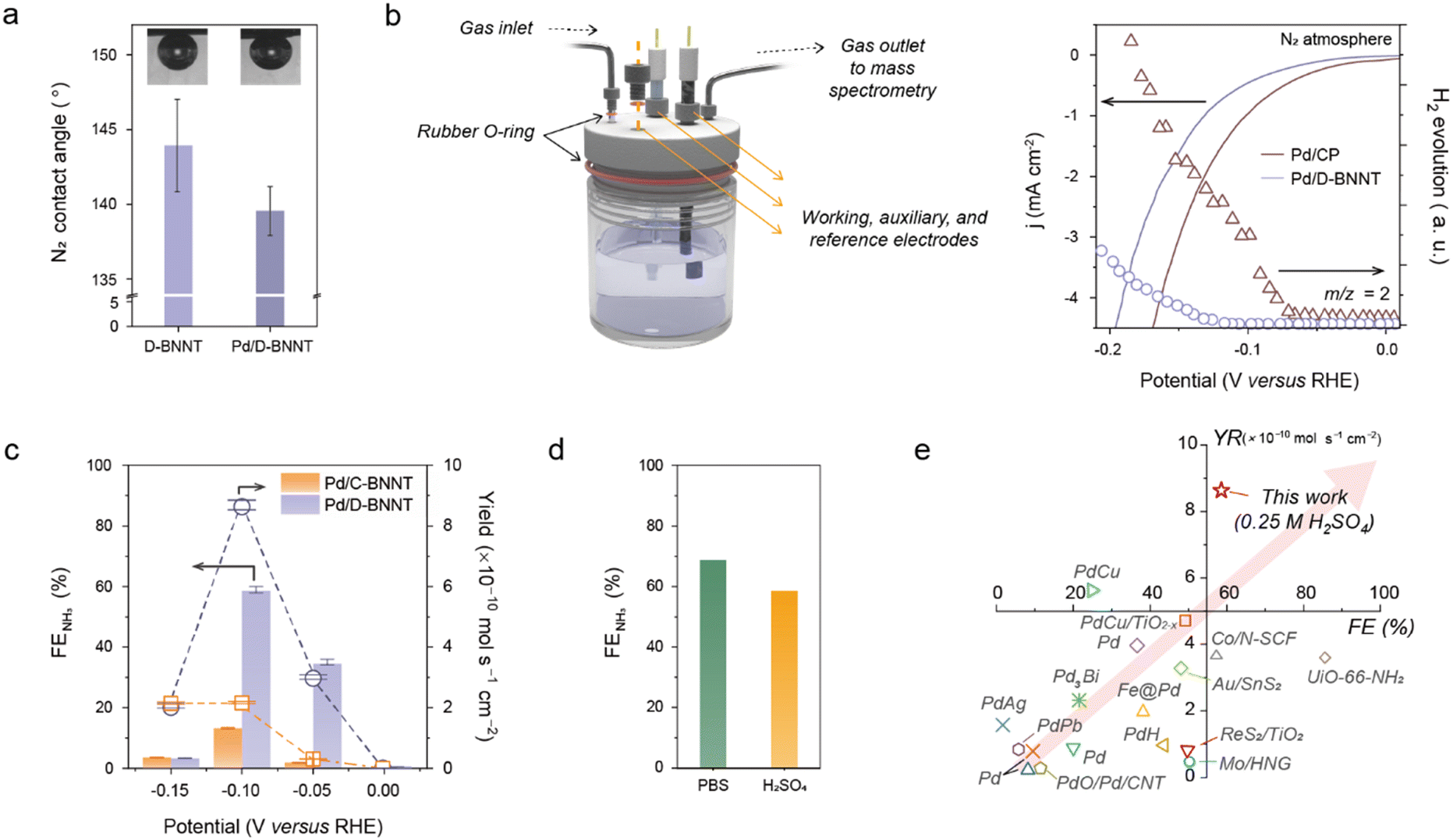 | ||
| Fig. 3 Electrochemical performance of the Pd–D-BNNT catalyst under ambient conditions. (a) N2 gas contact angle measurement of D-BNNT and Pd/D-BNNT electrode materials. (b) A schematic illustration of the gas-tight three-electrode cell (left) and plot of real-time H2 gas (m/z = 2) evolution measurement results of D-BNNT and Pd/D-BNNT electrocatalysts under LSV measurement at a scan rate of 2 mV s−1 (right). (c) Calculated faradaic efficiency (FENH3) and yield of NH3 as a result of time-dependent chronoamperometry measurement with Pd/C-BNNT and Pd/D-BNNT catalysts, respectively. (d) Plot of pH-dependent FE evaluation under PBS (pH 7) and H2SO4 (pH 1) solutions. (e) Literature comparison of FENH3 and yield of NH3 with state-of-the-art electrocatalysts for eNRR. The detailed values for previous reports, including this work, are noted in Table S2 (ESI†). | ||
Following the time-dependent chronoamperometry (CA) measurement at various potentials, the eNRR activities were evaluated by measuring the yield and faradaic efficiency (FE) of the NH3 product (Fig. S16, ESI†). The produced NH3 was measured with colorimetric analysis and 1H NMR spectra (Fig. S17–S19, ESI†).60,61 As the H2 evolution was monitored through the DEMS measurement (Fig. S20, ESI†), the optimal values of FE and yield for Pd/D-BNNTs were determined to be 58.9 (±1.07)% and 8.69 (±0.158) × 10−10 mol s−1 cmgeo−2, respectively, at a potential of −0.10 V versus RHE with a partial NH3 current density response (jNH3) of −0.245 mA cmgeo−2 (Fig. 3c and Fig. S21, ESI†). These values were achieved by optimizing the Pd content and thickness of the BNNT layer (Fig. S22 and S23, ESI†). We also evaluated the eNRR performance of Pd/C-BNNTs and Pd/CP (5.96%) which shows negligible yield and unsatisfied FE, indicating the strong interaction between the metal and defect-rich BNNTs plays a critical role in suppressing the HER and accelerates the eNRR (Fig. S24, ESI†). Interestingly, when electrolysis is conducted using the D-BNNT-coated CP (BNNT/CP) under N2 or Ar bubbling, marginal NH3 or HER activity was observed under the potential range of 0.0 to −0.2 V versus RHE, suggesting that neither the BNNT itself nor the nitrogen atoms in the BNNT layer participate in the electrochemical reduction of N2 (Fig. S25, ESI†). Remarkedly, using the 0.1 M phosphate-buffered saline (PBS) solution (pH 7.4) with Pd/D-BNNT achieved an exceptionally high FE towards NH3, approximately 68.0% at the potential of −0.05 V versus RHE, despite reduced effective current densities for NH3. This underscores the importance of adequately regulating proton and charge transport in the electrocatalyst for N2 electrolysis as promising approaches toward high eNRR performances (Fig. 3d and Fig. S26–S28, ESI†).10,12,62 The comparison of overall eNRR performance concerning the FE and yield of NH3 with the previously reported state-of-art electrocatalysts is summarized in Fig. 3e and Table S2 (ESI†), revealing the RRJH-induced Pd/D-BNNT electrocatalysts can be a viable approach to achieve higher active and selective towards eNRR by regulating the charge transport kinetics of metals on the BNNT layer.
Fig. 4 shows the qualification of the product from eNRR and its stability. We note that eliminating the possibility of a false-positive eNRR process is important to accurately assess the true catalytic performances of metal-based electrocatalysts for the eNRR. Rigorous control experiments and purity checks are essential to ensure reliability and advance the eNRR credibly. Thereby, the presence of byproducts and contaminants during NH3 production is further confirmed by investigating the hydrazine (N2H4) and NOx contaminants. The method was validated using a colorimetric assay (Watt and Chrisp reaction and Griess reaction).63,64 The results indicate that there is negligible N2H4 (FE < 1%) throughout the entire potential range (Fig. S29, ESI†). Moreover, to rule out the possibility of nitrate or nitrite (NO3− or NO2−) contamination from sources such as the fed N2 gas, Nafion membrane, or any instruments, the concentration of NOx was measured in a controlled solution, where the N2 gas was bubbled with OCV held for one hour (Fig. 4a and Fig. S30, ESI†). To ascertain the origin of the produced ammonia, the eNRR was conducted using isotope 15N2 gas. The data reveals a distinct peak split for 15NH3 with a coupling constant (1J, 15N–1H) of ≈73.4 Hz in 1H NMR measurement, whereas a triplet for 14NH3 was obtained with 1J (14N–1H) = 54.2 Hz from 14N2 experiments (Fig. 4b). According to the time-dependent electrolysis test, the calculated FE value obtained from the spectrophotometric analysis and 1H NMR calibration for one- and two-hours 15N2 experiments also agree well with those of the 14N2 experiments (Fig. 4c and Fig. S31 and S32, ESI†). This confirms that NH3 produced in our experiments originated solely from the N2 gas fed during the electrochemical reactions.
 | ||
| Fig. 4 Qualification of products from N2 electrolysis under ambient conditions. (a) UV-Vis spectroscopic investigations of possible contamination of NO2− and hydrazine byproducts. (b) 1H NMR spectra and (c) comparison of the calculated product FE at −0.1 V versus RHE from normal and isotope-labeled N2 (15N2) gas-fed. The full 1H NMR spectra can be found in Fig. S36–S42 (ESI†), raw NMR. (d) Full-pathway DFT calculation of the eNRR process of the Pd/D-BNNT electrocatalyst. (e) Comparison of FE and the yield of NH3 before and after the accelerated durability test (ADT). | ||
Regarding alternative and distal pathways in Fig. 4d, the PCET step was thermodynamically uphill compared to the enzymatic step, but the N2 adsorption step was considerably more stabilized. The first NH3 production step (*N2H3 → *N + NH3) in the distal pathway is hard to proceed with due to a high energy barrier of approximately 1.89 eV, which can be speculated that the enzymatic pathway is the dominant pathway to produce NH3 of the Pd/D-BNNTs. Based on these results, the electron-deficient state of the Pd slab significantly promotes the PCET step of the NRR process over the HER process. Moreover, the theoretical results align with the experimental NRR results, providing strong support for the successful implementation of catalytic engineering of NRR electrocatalysts based on the EMSI effect. Additionally, the eNRR performance measurement following the accelerated durability test (ADT) in an Ar atmosphere shows the robust electrocatalytic NRR performance of Pd/D-BNNTs. Following 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles of CV and consecutively performed CA results demonstrated negligible FE and morphological degradation, indicating the feasibility of Pd/D-BNNTs as an efficient eNRR electrocatalyst (Fig. 4e and Fig. S33 and S34, ESI†).
000 cycles of CV and consecutively performed CA results demonstrated negligible FE and morphological degradation, indicating the feasibility of Pd/D-BNNTs as an efficient eNRR electrocatalyst (Fig. 4e and Fig. S33 and S34, ESI†).
Conclusions
We demonstrated the strong EMSI effect between the defect sites of BNNTs and Pd NPs for the efficient catalyst for the eNRR. Employing the robust and straightforward RRJH method, we successfully fabricated uniform and dense Pd NPs on the BNNT support and confirmed through careful theoretical studies and various experimental results. The resulting Pd/D-BNNT catalyst achieved a high NH3 selectivity of approximately 68% in neutral aqueous electrolytes and a formation rate of 8.69 × 10−10 mol s−1 cmgeo−2 in acidic media in a low potential range (−0.05 to −0.1 VRHE) under ambient conditions. This performance is primarily attributed to the proposed EMSI effect between Pd NPs and defective BNNTs. Moreover, we examined the real-time HER suppression phenomenon by in situ DEMS and the reduction of Gibbs free energy for the PCET step using DFT calculations, further supporting the superior performance of the Pd/D-BNNT catalyst. These findings provide a viable strategy for designing eNRR catalysts and highlight the potential applications of metal and BN heterostructures using the RRJH method. Looking forward, our interests would move forward to the high-entropy alloy electrocatalyst for additional electronic structure modifications.Experimental
Materials
Palladium chloride (PdCl2, 99.9%) and ethanol (anhydrous, ≥99.5%) were purchased from Sigma-Aldrich and used without further purification. A proton exchange membrane (Nafion® 117, Dupont) and all electrode instruments were purchased from Wizmac (South Korea). All chemicals for color reagents were purchased from Sigma-Aldrich and used without further purification.DFT calculations
All DFT calculations were performed using the Vienna ab initio simulation package (VASP).65,66 We used the projector augmented wave (PAW) method to describe the effective potential between ionic cores and electrons and adopted the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) for simulating the exchange–correlation energy.67 The plane-wave energy cutoff was set to 520 eV, and the Brillouin zone integrations were performed using the Monkhorst–Pack scheme with a (4 × 4 × 1) k-point grid. For calculating adsorption energies, we used a Pd slab containing three (111) planes (27 atoms total), and a 40 Å vacuum layer along the z-axis was set to prevent the interaction between two adjacent periodic images. The Pd atoms in the bottom two layers of the slab were fixed at their bulk positions, while the atoms in the top layer and the absorbate molecules were allowed to relax. In geometry optimizations, the total energy was converged to 10−6 eV, and forces acting on ions were required to be less than 0.01 eV Å−1.The Gibbs free energy change (ΔG) of the eNRR intermediates absorbed Pd was calculated using the following equation:
| ΔG = ΔEDFT + ΔEZPE − TΔS |
Synthesis of Pd/BNNTs
Defective BNNTs (D-BNNT) were synthesized using thermal jet-plasma systems.29,30 To remove the amorphous boron source in the as-prepared D-BNNTs, the washing process reported in previous studies was adopted.68 A prepared sample was treated with thermal oxidation in a muffle furnace at 670 °C for over thirteen hours to oxidize amorphous boron into B2O3. Subsequently, B2O3 was removed through the washing process with hot distilled water/methanol over three times. The pre-treated D-BNNTs were dispersed in a 60 wt% tert-butanol/distilled water at a concentration of 0.15 mg ml−1 and allowed to settle for twelve hours to remove h-BN-like impurities. The purified BNNTs in the supernatant were used to synthesize Pd/BNNTs.31 Then, the obtained supernatant was poured onto carbon paper (CP, Sigracet 39 AA, Fuel Cell Store) and slowly vacuum-filtered. Washing with ethanol and drying at 60 °C for over twenty-four hours gives a D-BNNT/CP sample.The D-BNNT on CP was cut into a length of 25.0 mm and a width of 5.00 mm. To this, 76.0 μL of precursor solution (0.1 M PdCl2 in ethanol) was drop-cast on the D-BNNT side and dried overnight in a 60 °C oven. For RRJH, the PdCl2/D-BNNT electrode was connected to a copper tape (3M™) with a length of 20.0 mm (this synthetic method is based on the carbothermal shock (RRJH) method).69,70 A current of 2.80 A is applied to the PdCl2/D-BNNT electrode for 0.50 seconds by using a power supply (Keithley 2461) in an Ar-filled glovebox. RRJH temperature was measured using an infrared thermometer (Optis). An as-synthesized Pd/D-BNNT electrode was used without further washing.
Crystalline BNNTs (C-BNNTs, BNNT SP10-R Refined Puffball, BNNT LLC., USA) were synthesized by the laser ablation method and were used without further purification.71 Pd/C-BNNT and Pd/CP samples were prepared using the same method as Pd/D-BNNTs, using C-BNNTs or without the BNNT layer.
Physical characterization
SEM images were taken using an FEI Magellan 400. XRD was conducted with a Rigaku Smartlab system using a Cu-Kα source. TEM bright field images and EELS were conducted using a Titan G2 Cube at 80 kV and a Tecnai G2 F20 X-Twin (FEI, USA), and cross-sectional TEM specimens were prepared using an FIB-SEM (Helios 650). XPS analyses were performed using a K-Alpha spectrometer (Thermo VG Scientific). The binding energy was corrected to 284.8 eV for the C–C bond. REELS for bandgap analysis was conducted using an Axis-Supra (Kratos). The N2 gas contact angle was measured by a captive N2 gas bubble using a homemade container and DSA100S (KRÜSS, Germany). Gas products were analyzed using a lab-built gas-tight three-electrode cell and differential electrochemical mass spectrometry (DEMS) system.72 A DEMS system was constructed using a series connection of Ar gas (99.999%) or Ar/N2 gas (10:90 in volume; purity of N2: 99.999%), a multiport valve (VICI Valco, USA), a lab-built gas-tight electrochemical cell (Fig. 3b), and a quadrupole mass spectrometer (RGA200, Stanford Research System, USA). An RGA200 mass spectrometer was preliminarily calibrated using an Ar/H2 mixed gas with various mixing ratios. Throughout the CA measurement, the respective carrier gas periodically sweeps the evolved H2 gases in the headspace to deliver them to the mass spectrometer. The recorded mass spectrum of m/z = 2 was used as an H2 gas signal.
Setup for eNRR measurement
All electrochemical measurements were recorded using a multichannel electrochemical workstation (VMP3, Bio-Logic Corp., France). A typical two-compartment H-type cell was fabricated using respective catalyst electrodes, a graphite rod, and a saturated calomel electrode (SCE; saturated KCl filling) as working, counter, and reference electrodes. The graphite rod electrode was separated using a Nafion® 117 membrane. A Nafion 117 membrane was used after pretreatment.73 Initially, a dry Nafion membrane of size 1.5 × 1.5 cm underwent a sequential treatment step involving 5% H2O2, distilled water (MiliQ®), and 0.5 M H2SO4, and further rinsed under ambient conditions with ultrasonication for twenty minutes per step. Subsequently, the membrane was thoroughly washed with distilled water, immersed in distilled water, heated at 80 °C for a minimum of twelve hours, and stored at room temperature until use. The membrane underwent a final treatment with 0.1 M H2SO4 and distilled water in sequence before use. The reference electrode was calibrated to the reversible hydrogen electrode (RHE) scale. The calibration was performed by recording the open-circuit potential (OCP) in a saturated H2 electrolyte using the platinum rod as both working and counter electrodes: V(RHE) = V(SCE) + 0.248 V for 0.25 M H2SO4 and V(RHE) = V(SCE) + 0.610 V for 0.1 M PBS solution, respectively (Fig. S35, ESI†). Each chamber was filled with 30.0 mL of electrolyte. The electrolytes were pre-purged by bubbling N2 (99.999% purity, 10 ml min−1) for one hour and continuously purged with N2 at the same flow rate of 10 ml min−1 during electrolysis. Every measurement was performed with a newly synthesized catalyst electrode. A controlled experiment with an Ar gas was performed with the same setting. All polarization curves were presented after iR-compensation with the series resistance of catalysts, which was determined by electrochemical impedance spectroscopy (EIS) measurements.Product analysis
The produced NH3 was quantified by using the modified indophenol blue method.60 The colorimetric reagent was prepared by dissolving NaOH (4.00 g, 1.00 M), salicylic acid (5.00 g, 5.00 wt%), and sodium citrate (5.00 g, 5.00 wt%) in 100 mL of distilled water. To a 2.00 mL aliquot of the respective electrolyte after electrolysis were added 2.00 mL of the above colorimetric reagent, 1.00 mL of 0.05 M sodium hypochlorite (NaClO) in distilled water, and 0.20 mL of 1.00 wt% sodium nitroferricyanide (Na[Fe(NO)(CN)5]) in distilled water. After standing for two hours in the dark, the UV-vis absorption spectrum (V-750, JASCO, Japan) was recorded. The absorbance at a wavelength of 660 nm was used to determine the NH3 concentration. The standard solution was prepared using ammonium chloride (NH4Cl). 1H NMR spectroscopy (VNMRS 600, Agilent Technologies Inc, USA) was measured by mixing the solution with DMSO-d6 as an internal reference. The calibration was performed by calculating the relative intensity of NH3 with the DMSO-d6 peak after baseline correction. 14NH4Cl and 15NH4Cl were used as standard solutions. The hydrazine was determined by the Watt and Chrisp test.63 A mixed solution of hydrochloric acid (HCl, conc., 10.0 mL), ethanol (100 mL), and p-dimethylaminobenzaldehyde (2.00 g) was used as a color reagent. Hydrazine monohydrate (N2H4·H2O) was used as a standard reagent. After the eNRR measurement, 1.50 mL of electrolyte was taken and mixed with the same volume of color reagent solution as the above. The mixed solution was allowed to stand in the dark for half an hour before recording UV-vis absorption spectra. An absorbance peak at a wavelength of 460 nm was used for hydrazine detection (Fig. S29, ESI†). The nitrite ion concentration was determined using the Griess test.64 Two color reagents (reagents A and B) were respectively prepared. Reagent A: to a 10.0 mL of 37 wt% HCl was dissolved in 35.0 mg of sulfanilamide, and the mixture was diluted to 200 mL with distilled water. Reagent B: to 10 mL of 37 wt% of HCl was added 29.0 mg of 1-naphthyl amine and diluted to 200 mL with distilled water. These color reagents were stored in the dark and used within a week. 10.0 mL of the target sample was mixed with 1.00 mL of reagents A and B. The reaction mixture was shaken well and stood for ten minutes. The absorbance at a wavelength of 541 nm was used for the concentration of NO2− determination. NaNO2 was used for standard solution preparation (Fig. S30, ESI†).The faradaic efficiencies of NH3 and H2 and the NH3 yield were calculated using the following equations:
| FE(NH3) = eNH3 × MNH3 × F ÷ Qtot × 100 (%) |
| FE(H2) = eH2 × MH2 × F ÷ Qtot × 100(%), |

| Yield(NH3) = (CNH3 × Velectrolyte) ÷ (t × A) |
485 C mol−1), the measured amount of NH3 and H2 evolution from the indophenol protocol and DEMS measurement, the number of electrons (3 for the NRR and 2 for the HER), the total charge passed under CA measurement, the concentration of the measured NH3, the volume of the electrolyte, the time passed during CA measurement, and the area of the electrode, respectively.
15N2 isotope-labeling experiments
Isotope labeling experiments were carried out using 15N2 as the feeding gas (Sigma-Aldrich, 98%). Due to the high cost of 15N2, the electrolyte was first purged with Ar gas for one hour, and Ar gas purging was maintained in the headspace to prevent the N2 gas from entering the air. The 15N2 gas was bubbled at a fixed flow rate of 10.0 ml min−1 for ten minutes before electrolysis and continuously purged at a flow rate of 5 ml min−1 during electrolysis. During the electrolysis at a fixed potential of −0.10 V, 2.00 mL of electrolyte was obtained after one hour to investigate the concentration of 15NH3 as a function of electrolysis time. The 1H NMR calibration curve for 15NH3 was determined by using various concentrations of 15NH4Cl as the standard solution (Fig. S32, ESI†).Author contributions
J.-Y. S., J. P., M. K. and S. A. proposed the project and conceptualized it. J.-Y. S. designed and conducted the synthesis and characterization of electrocatalysts. M. K. purified BNNTs and formed the BNNT layer. J. P. designed the experiments and performed all electrochemical studies. J. H. H. helped with the electrochemical measurement. W. L., W. B. L., and Y. K. conducted computational studies. Y. K., H. J., S. A., S. J. K., and H.-T. J. supervised this project. A. J. and I. G. supported the design and concept of electrocatalysts. The manuscript was written by J.-Y. S., J. P., S. J. K., and H.-T. J. with comments and inputs from all authors. All the authors revised and approved the paper.Data availability
All starting materials used in this work are readily available from formal vendors. The data that support the findings of this study are included in the published article and its ESI.† The raw data are also available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00211808 and 2022R1A2C200652111), and the Saudi Aramco-KAIST CO2 Management Center, Korea Institute of Science and Technology (KIST) Open Research Program and KAIST-UC Berkeley-VNU Climate Change Research Center (No. 2021K1A4A8A01079356).Notes and references
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074, DOI:10.1016/j.joule.2018.04.017.
- B. H. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296, DOI:10.1038/s41929-019-0252-4.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639, DOI:10.1038/ngeo325.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303, DOI:10.1038/nature11475.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik and A. K. Jones, Science, 2018, 360, eaar6611, DOI:10.1126/science.aar6611.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68, DOI:10.1016/j.cattod.2016.05.008.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900, DOI:10.1126/science.aaq1684.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56, 10.1039/C7EE02220D.
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727, DOI:10.1038/s41586-022-05108-y.
- S. Liu, Z. Wang, H. Zhang, S. Wang, P. Wang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2020, 8, 14228–14233, DOI:10.1021/acssuschemeng.0c05375.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193, 10.1039/D0EE03596C.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795, DOI:10.1038/s41467-018-04213-9.
- M. Xie, F. Dai, H. Guo, P. Du, X. Xu, J. Liu, Z. Zhang and X. Lu, Adv. Energy Mater., 2023, 13, 2203032, DOI:10.1002/aenm.202203032.
- W. P. Utomo, H. Wu and Y. H. Ng, Small, 2022, 18, 2200996, DOI:10.1002/smll.202200996.
- G. Ren, M. Shi, S. Liu, Z. Li, Z. Zhang and X. Meng, Chem. Eng. J., 2023, 454, 140158, DOI:10.1016/j.cej.2022.140158.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300, DOI:10.1002/adma.202003300.
- X. Wang, S. Qiu, J. Feng, Y. Tong, F. Zhou, Q. Li, L. Song, S. Chen, K. H. Wu and P. Su, Adv. Mater., 2020, 32, 2004382, DOI:10.1002/adma.202004382.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550, DOI:10.1002/adma.201606550.
- C. Liu, X. Guo, Z.-F. Huang, J. Li, L. Gan, L. Pan, C. Shi, X. Zhang, G. Yang and J.-J. Zou, Mater. Chem. Front., 2022, 6, 2190–2200, 10.1039/D2QM00453D.
- Y.-X. Lin, S.-N. Zhang, Z.-H. Xue, J.-J. Zhang, H. Su, T.-J. Zhao, G.-Y. Zhai, X.-H. Li, M. Antonietti and J.-S. Chen, Nat. Commun., 2019, 10, 4380, DOI:10.1038/s41467-019-12312-4.
- P. Jing, P. Liu, M. Hu, X. Xu, B. Liu and J. Zhang, Small, 2022, 18, 2201200, DOI:10.1002/smll.202201200.
- M. Guo, L. Fang, L. Zhang, M. Li, M. Cong, X. Guan, C. Shi, C. Gu, X. Liu and Y. Wang, Angew. Chem., 2023, 135, e202217635, DOI:10.1002/anie.202217635.
- X. Feng, J. Liu, L. Chen, Y. Kong, Z. Zhang, Z. Zhang, D. Wang, W. Liu, S. Li and L. Tong, J. Am. Chem. Soc., 2023, 145, 10259–10267, DOI:10.1021/jacs.3c01319.
- H. Hu and J.-H. Choi, RSC Adv., 2020, 10, 38484–38489, 10.1039/D0RA08138H.
- V. S. K. Choutipalli, K. Esackraj, E. Varathan and V. Subramanian, Appl. Surf. Sci., 2022, 602, 154406, DOI:10.1016/j.apsusc.2022.154406.
- H. Zhang, D. Li, Q. Li, K. Guo, C. Yu, J. Lin, C. Tang and Y. Huang, Colloids Surf., A, 2022, 646, 128947, DOI:10.1016/j.colsurfa.2022.128947.
- J. Wu, L. Wang, X. Yang, B. Lv and J. Chen, Ind. Eng. Chem. Res., 2018, 57, 2805–2810, DOI:10.1021/acs.iecr.7b04864.
- L. Huang, X.-F. Liu, J. Zou, X. Duan, Z.-C. Chen, Z.-H. Zhou, L. Ye, X. Liang, S.-Y. Xie and Y. Yuan, J. Catal., 2022, 406, 115–125, DOI:10.1016/j.jcat.2022.01.004.
- K. S. Kim, C. T. Kingston, A. Hrdina, M. B. Jakubinek, J. Guan, M. Plunkett and B. Simard, ACS Nano, 2014, 8, 6211–6220, DOI:10.1021/nn501661p.
- K. S. Kim, G. Sigouin, H. Cho, M. Couillard, M. Gallerneault, S. Y. Moon, H. S. Lee, M. J. Kim, S. G. Jang and H. Shin, ACS Omega, 2021, 6, 27418–27429, DOI:10.1021/acsomega.1c04361.
- M. Kang, J. Kim, H. Lim, J. Ko, H.-S. Kim, Y. Joo, S. Y. Moon, S. G. Jang, E. Lee and S. Ahn, Nanomaterials, 2023, 13, 2593, DOI:10.3390/nano13182593.
- S. Assad, T. Tariq, M. Z. Idrees, A. M. Butt, K. Bakhat and U. Shamraiz, J. Electroanal. Chem., 2023, 931, 117174, DOI:10.1016/j.jelechem.2023.117174.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487, DOI:10.1021/jacs.7b05213.
- X. Mao, S. Zhou, C. Yan, Z. Zhu and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1110–1116, 10.1039/C8CP07064D.
- C. Liu, Q. Bao, X. Zhao and Y. Li, ACS Appl. Mater. Interfaces, 2022, 14, 30626–30638, DOI:10.1021/acsami.2c00501.
- Y. Huang, T. Yang, L. Yang, R. Liu, G. Zhang, J. Jiang, Y. Luo, P. Lian and S. Tang, J. Mater. Chem. A, 2019, 7, 15173–15180, 10.1039/C9TA02947H.
- S. Gao, Z. Ma, C. Xiao, W. Du, X. Sun, Q. Li, R. Sa and C. Sun, Appl. Surf. Sci., 2022, 591, 153130, DOI:10.1016/j.apsusc.2022.153130.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709, DOI:10.1021/acscatal.6b03035.
- C. Kim, J. Y. Song, C. Choi, J. P. Ha, W. Lee, Y. T. Nam, D. M. Lee, G. Kim, I. Gereige and W. B. Jung, Adv. Mater., 2022, 34, 2205270, DOI:10.1002/adma.202205270.
- H. Tao, C. Choi, L.-X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang and A. W. Robertson, Chem, 2019, 5, 204–214, DOI:10.1016/j.chempr.2018.10.007.
- Y.-C. Han, M.-L. Liu, L. Sun, X.-C. Li, Y. Yao, C. Zhang, S.-Y. Ding, H.-G. Liao, L. Zhang and F. R. Fan, Nano Energy, 2022, 97, 107125, DOI:10.1016/j.nanoen.2022.107125.
- S. Venkateswarlu, S. Kim, M. Balamurugan, Y. Son, M. Yoon, K. T. Nam, S. S. Han and M. J. Kim, Appl. Catal., B, 2024, 345, 123609, DOI:10.1016/j.apcatb.2023.123609.
- A. Abdelhafiz, B. Wang, A. R. Harutyunyan and J. Li, Adv. Energy Mater., 2022, 12, 2200742, DOI:10.1002/aenm.202200742.
- Q. Li, X. Wang, Z. Xie, X. Peng, L. Guo, X. Yu, X. Yang, Z. Lu, X. Zhang and L. Li, Appl. Catal., B, 2022, 305, 121020, DOI:10.1016/j.apcatb.2021.121020.
- Y. Li, H. Wu, J. Zhang, Q. Lu, X. Han, X. Zheng, Y. Deng and W. Hu, J. Mater. Chem. A, 2023, 11, 10267–10276, 10.1039/D2TA09161E.
- J. Ko, D. Kim, G. Sim, S. Y. Moon, S. S. Lee, S. G. Jang, S. Ahn, S. G. Im and Y. Joo, Small Methods, 2023, 7, 2201341, DOI:10.1002/smtd.202201341.
- C. G. Hawkins, A. Verma, W. Horbinski, R. Weeks, P. P. Mukherjee and L. Whittaker-Brooks, ACS Appl. Mater. Interfaces, 2020, 12, 21788–21798, DOI:10.1021/acsami.9b21702.
- Y. Xie and Y. Zhou, J. Mater. Res., 2019, 34, 2472–2481, DOI:10.1557/jmr.2019.224.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677, 10.1039/C9TA06523G.
- G. Han, D. Zhang, C. Kong, B. Zhou, Y. Shi, Y. Feng, C. Liu and D.-Y. Wang, Chem. Eng. J., 2022, 437, 135482, DOI:10.1016/j.cej.2022.135482.
- D. M. Marincel, M. Adnan, J. Ma, E. A. Bengio, M. A. Trafford, O. Kleinerman, D. V. Kosynkin, S.-H. Chu, C. Park and S. J. Hocker, Chem. Mater., 2019, 31, 1520–1527, DOI:10.1021/acs.chemmater.8b03785.
- S.-W. Jeon, S. C. Kang, H. Choi, H. I. Yoo, S. Y. Moon and T.-H. Kim, ACS Appl. Nano Mater., 2023, 7, 394–403, DOI:10.1021/acsanm.3c04575.
- Z.-Q. Xu, C. Elbadawi, T. T. Tran, M. Kianinia, X. Li, D. Liu, T. B. Hoffman, M. Nguyen, S. Kim and J. H. Edgar, Nanoscale, 2018, 10, 7957–7965, 10.1039/C7NR08222C.
- Q. Weng, D. G. Kvashnin, X. Wang, O. Cretu, Y. Yang, M. Zhou, C. Zhang, D. M. Tang, P. B. Sorokin and Y. Bando, Adv. Mater., 2017, 29, 1700695, DOI:10.1002/adma.201700695.
- J. Pu, K. Zhang, Z. Wang, C. Li, K. Zhu, Y. Yao and G. Hong, Adv. Funct. Mater., 2021, 31, 2106315, DOI:10.1002/adfm.202106315.
- S. Chahal, T. K. Sahu, S. Kar, S. J. Ray, V. Biju and P. Kumar, J. Phys. Chem. C, 2022, 126, 21084–21093, DOI:10.1021/acs.jpcc.2c06693.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663, DOI:10.1002/aenm.202103663.
- Y. Chen, J. Cai, P. Li, G. Zhao, G. Wang, Y. Jiang, J. Chen, S. X. Dou, H. Pan and W. Sun, Nano Lett., 2020, 20, 6807–6814, DOI:10.1021/acs.nanolett.0c02782.
- P. Sharma, J. Han, J. Park, D. Y. Kim, J. Lee, D. Oh, N. Kim, D.-H. Seo, Y. Kim and S. J. Kang, JACS Au, 2021, 1, 2339–2348, DOI:10.1021/jacsau.1c00444.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841, DOI:10.1038/nmat3696.
- A. C. Nielander, J. M. McEnaney, J. A. Schwalbe, J. G. Baker, S. J. Blair, L. Wang, J. G. Pelton, S. Z. Andersen, K. Enemark-Rasmussen and V. Colic, ACS Catal., 2019, 9, 5797–5802, DOI:10.1021/acscatal.9b00358.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703, DOI:10.1002/anie.201609533.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008, DOI:10.1021/ac60072a044.
- H.-Y. Liu, H. M. Lant, J. L. Troiano, G. Hu, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2022, 144, 8449–8453, DOI:10.1021/jacs.2c01788.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758, DOI:10.1103/PhysRevB.59.1758.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169, DOI:10.1103/physrevb.54.11169.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865, DOI:10.1103/PhysRevLett.77.3865.
- H. Harrison, J. T. Lamb, K. S. Nowlin, A. J. Guenthner, K. B. Ghiassi, A. D. Kelkar and J. R. Alston, Nanoscale Adv., 2019, 1, 1693–1701, 10.1039/C8NA00251G.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494, DOI:10.1126/science.aan5412.
- J.-Y. Song, C. Kim, M. Kim, K. M. Cho, I. Gereige, W.-B. Jung, H. Jeong and H.-T. Jung, Sci. Adv., 2021, 7, eabk2984, DOI:10.1126/sciadv.abk2984.
- M. W. Smith, K. C. Jordan, C. Park, J.-W. Kim, P. T. Lillehei, R. Crooks and J. S. Harrison, Nanotechnology, 2009, 20, 505604, DOI:10.1088/0957-4484/20/50/505604.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161–1166, DOI:10.1021/jz200352v.
- F. Hanifpour, A. Sveinbjörnsson, C. P. Canales, E. Skulason and H. D. Flosadottir, Angew. Chem., 2020, 132, 23138–23142, DOI:10.1002/ange.202007998.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00210e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |