
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-step polymerization for tailored donor–acceptor interactions driving efficient hydrogen evolution in visible-light photocatalysts†
Wooteak
Jung‡
a,
Sanghyeok
An‡
 a,
Gayoung
Ham‡
b,
Chanhyeok
Kim
a,
Soyeon
Lee
c,
Jiwoong
Yang
cd,
Dae Sung
Chung
*a,
Hyojung
Cha
*b and
Taiho
Park
*a
a,
Gayoung
Ham‡
b,
Chanhyeok
Kim
a,
Soyeon
Lee
c,
Jiwoong
Yang
cd,
Dae Sung
Chung
*a,
Hyojung
Cha
*b and
Taiho
Park
*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: dchung@postech.ac.kr
bSchool of Energy Engineering, Kyungpook National University, Daehak-ro 80, Buk-gu, Daegu, 41566, Republic of Korea. E-mail: hcha@knu.ac.kr
cDepartment of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu, 42988, Republic of Korea
dEnergy Science and Engineering Research Center, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu, 42988, Republic of Korea
First published on 28th March 2025
Abstract
The development of materials for organic solar cells has made significant strides through the strategic combination of diverse donor structures with acceptor units in polymer backbones. In contrast, semiconducting polymers for photocatalytic hydrogen evolution have primarily focused on acceptor moieties, with limited exploration of donor contributions, primarily owing to the emphasis on designing active sites for proton reduction in inorganic catalysts. To investigate the impact of highly electron-donating moieties on photocatalytic performance, we designed and synthesized benzothiadiazole (BT)-based polymers with randomly incorporated benzodithiophene (BDT) and fluorene units via a streamlined one-pot Stille–Suzuki two-step polymerization. Comprehensive molecular characterization and optical spectroscopic analyses confirmed the successful synthesis of the target polymers. Photocatalytic hydrogen evolution studies, supported by photophysical and spectroscopic investigations, demonstrated that optimizing the proportion of BDT units in the polymer backbone enhances hydrogen evolution rates significantly. Additionally, comparative analyses further highlighted the distinct differences in the photocatalytic efficiency between the BDT and fluorene donor units, providing critical insights into their functional roles. This work underscores the potential of advancing polymer photocatalysts by fine-tuning donor–acceptor interactions through optimization of donor moiety composition, offering a robust framework for achieving superior photocatalytic performance.
Broader contextPolymer-based photocatalysts emerge as a promising alternative due to their tunable optical properties and visible-light absorption. Despite significant advances in donor–acceptor semiconducting polymers for organic photovoltaics, hydrogen evolution photocatalyst designs have predominantly focused on acceptor moieties, leaving the contributions of donor units largely underexplored. In this study, we precisely integrate varying proportions of benzodithiophene (BDT) donor units into benzothiadiazole (BT)-based polymers using a novel two-step polymerization strategy, combining Stille and Suzuki coupling reactions. The synthesized polymers exhibit extended visible-light absorption, prolonged charge carrier lifetimes, and significantly enhanced photocatalytic hydrogen evolution rates. These findings highlight the critical role of molecular design in optimizing polymer photocatalysts and establish a versatile platform for expanding the structural diversity of materials for next-generation hydrogen evolution systems. |
Introduction
The conversion of solar energy into hydrogen gas has garnered renewed interest, as hydrogen serves as a zero-emission energy carrier with extensive industrial applications.1–4 Since Fujishima and Honda's groundbreaking discovery of photochemical water splitting using titanium dioxide in 1972, research on photocatalysts has predominantly centered on inorganic materials. Although these materials demonstrate high efficiency under ultraviolet (UV) light, they face scalability issues and are restricted by their reliance on UV light.5 Organic conjugated polymers emerged as promising alternatives in 1985 with the introduction of poly(p-phenylene) (PPP).6 This breakthrough catalyzed the development of diverse polymer architectures, such as linear conjugated polymers,7,8 carbon nitrides,9 and covalent organic frameworks (COFs),10–12 which provide tunable optical properties and significant synthetic flexibility. However, many of these structured polymers suffer from limited solubility in conventional organic solvents,13,14 restricting their applicability to visible-light-driven hydrogen production for future application.Linear conjugated polymers provide a promising alternative because of their structural versatility and the ability to finely tune their photochemical properties through donor–acceptor copolymerization.15–17 Early studies in this field highlighted the potential of benzothiadiazole (BT)18 and dibenzothiophene-sulfone-based acceptors,19 with notable systems like F8BT and P10 making significant advancements through strategic modifications to their backbone and side chains.20,21 These design strategies primarily aim to optimize the electronic environment, enhance hydrophilicity, and extend light absorption into the visible spectrum. However, despite these advancements, challenges persist, particularly concerning the solubility and processability of these materials, which restrict their scalability. Recent efforts have increasingly focused on incorporating heteroatoms such as boron and nitrogen into polymer backbones to improve charge separation and transport capabilities.22,23 This trend highlights the critical interplay between molecular architecture and photocatalytic performance and driving innovations in the field.
A significant gap in the field of photocatalytic hydrogen generation lies in the understanding of the influence of donor moieties within the polymer backbone on photocatalytic performance. While “push–pull” mechanisms in organic photovoltaics utilize strong donor–acceptor interactions to achieve efficient charge transfer, hydrogen evolution photocatalysts have traditionally focused on designing acceptor structures not only in linear conjugated polymers but also in conjugated framework systems.24–26 This focus reflects the historical influence of inorganic catalyst research, which prioritized proton reduction sites. Currently, research is being conducted not only on strategies that replace heteroatoms in well-known structures22,27 but also on incorporating various functional groups, such as side chains and discontinuous conjugated polymers,20,28 into established backbone structures. However, compared to the diverse range of photoactive materials used in OPVs, the pool of candidate materials for polymer photocatalysts remains significantly limited. In this context, systematically evaluating donor moieties offers a unique opportunity to redefine the structural principles that drive the development of polymer photocatalysts.
This challenge is further exacerbated by the polymerization methods employed in the synthesis of these photocatalysts. In catalyst-mediated cross-coupling reactions such as the Still reaction and Suzuki–Miyaura polymerization, the choice of donor and acceptor monomers significantly influences the reaction pathways.29–31 These methods involve distinct functional groups tailored to the unique chemical properties of the reactants. Acceptor monomers are typically bromine-substituted at both ends, while donor monomers are functionalized with tin or boron, depending on their specific catalytic reaction pathway. For instance, strong donors used in organic solar cells often require Stille polymerization, in which tin plays a critical role. Conversely, hydrogen evolution photocatalysts, which commonly rely on weaker donor moieties, predominantly employ boron-facilitated Suzuki polymerization. These methodological constraints have slowed the development of polymers tailored for hydrogen evolution, unlike the rapid progress observed in organic solar cells. To address this, new strategies are needed to expand both the range of donor materials and polymerization techniques used for hydrogen evolution photocatalysts.
In this study, we investigated the effects of incorporating a strong donor into a BT-based polymer on its physical and chemical properties. Consequently, we randomly introduced weak and strong donors into a single polymer chain. Unlike conventional random copolymers, which typically use uniform reaction conditions, our approach necessitates the development of a two-step polymerization protocol due to the distinct functional group requirements and reaction conditions associated with varying donor strengths. Specifically, Suzuki copolymerization relies on water for the palladium transmetalation reaction; however, water strongly interacts with tin compounds, causing the decomposition of tin-ended reactants.
To overcome this challenge, we developed a novel synthesis protocol that combines the Stille and Suzuki coupling reactions in a one-pot reaction system, as shown in Scheme 1. The Stille reaction was conducted in toluene to prevent water-induced side reactions. Water was then introduced to initiate the Suzuki reaction and simultaneously degrade any residual tin compounds. This strategy allowed for precise control over the benzodithiophene (BDT) fragment content in the polymer backbone by adjusting the amount of tin compounds. Using this method, we synthesized three new polymers—PBF8BT-1, PBF8BT-2, and PBF8BT-3—with varying BDT contents. This design aims to explore the effect of BDT incorporation on both the optoelectronic properties and hydrogen evolution efficiency of the polymers, facilitating the optimization of the donor–acceptor balance.
 | ||
| Scheme 1 Schematic diagram of polymer photocatalyst and strategy of polymer synthesis. | ||
These conjugated polymers were fabricated into polymer dots (Pdots) using a nanoprecipitation method, with PS-PEG-COOH serving as a surfactant to ensure polymer dispersion in water, as outlined in previous research.32 Dynamic light scattering (DLS) and scanning electron microscopy (SEM) confirmed the morphology of the Pdots. The photocatalytic hydrogen evolution rate (HER) reached up to 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 μmol g−1 h−1 (λ > 420 nm) with the addition of 8 wt% Pt as a cocatalyst and ascorbic acid (AA) as a sacrificial donor, representing nearly a two-fold improvement compared to pristine F8BT Pdots. Furthermore, steady-state and time-resolved photoluminescence (PL and TR-PL) analyses, along with transient absorption spectroscopy (TAS), revealed long-lived charge carriers in the absence of hole scavengers, highlighting the critical role of BDT content in optimizing photocatalytic performance. This study advances the understanding of donor–acceptor dynamics in polymer photocatalysts and establishes a scalable synthetic framework for next-generation materials, paving the way for high-performance solar-driven energy systems.
130 μmol g−1 h−1 (λ > 420 nm) with the addition of 8 wt% Pt as a cocatalyst and ascorbic acid (AA) as a sacrificial donor, representing nearly a two-fold improvement compared to pristine F8BT Pdots. Furthermore, steady-state and time-resolved photoluminescence (PL and TR-PL) analyses, along with transient absorption spectroscopy (TAS), revealed long-lived charge carriers in the absence of hole scavengers, highlighting the critical role of BDT content in optimizing photocatalytic performance. This study advances the understanding of donor–acceptor dynamics in polymer photocatalysts and establishes a scalable synthetic framework for next-generation materials, paving the way for high-performance solar-driven energy systems.
Results and discussion
Synthesis and characterization
The polymers were synthesized according to the reactions shown in Scheme 1. The reference polymer F8BT was synthesized via Suzuki–Miyaura coupling between a dibrominated BT monomer and the corresponding diboronic fluorene derivative, 9,9-dioctyl-9H-fluorene-2,7-diboronic acid bis(pinacol) ester. The PBF8BT series were prepared using a two-step polymerization approach conducted in a one-pot system. Initially, Stille polymerization was performed under anhydrous conditions. After 5 h, water was added to activate the Suzuki reaction because diborylation requires aqueous conditions (detailed synthetic procedures are provided in the ESI†). All the synthesized polymers exhibited solubility in chloroform, indicating their suitability for processing in commercial organic solvents. The successful synthesis of the target polymers was confirmed by 1H-NMR spectroscopy, which displayed characteristic signals corresponding to aromatic and alkyl protons. The ratios of (4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) (dSn-BDT) were 10%, 25%, and 40%, yielding PBF8BT-1, PBF8BT-2, and PBF8BT-3, respectively. The actual molar compositions of BDT were determined to be 11.0%, 33.8%, and 49.0%, as calculated from the integration values of the peaks corresponding to the α-proton in the side chain of BDT (3.2–2.9 ppm, labeled as “a” in Fig. S1–S3, ESI†) and the α-proton of fluorene and the tertiary proton of the ethylhexyl side chain of BDT (2.3–2.0 ppm, labeled as “b,c” in Fig. S1–S3, ESI†). As the proportion of dSn-BDT in the feedstock increased, the corresponding BDT content in the final polymers also increased. This trend is attributed to the highly planar nature of the polymer backbone, which may reduce solubility and limit effective polymer condensation during the second polymerization step. Detailed information about these polymers is presented in Table 1.| Polymers | M n [kg mol−1]a | PDI | T d [°C]b | λ max [nm] | λ edge [nm] | E optg [eV]c | LUMO [eV]d | HOMO [eV]e |
|---|---|---|---|---|---|---|---|---|
| a Determined by gel permeation chromatography (GPC) using tetrahydrofuran as an eluent at 40 °C, relative to a polystyrene standard. b Thermal decomposition temperature (5% weight loss) determined by TGA under N2. c Optical band gap estimated from the absorption edges (λedge) in the solution state. d Lowest unoccupied molecular orbital (LUMO) levels were estimated from the onset of the first reduction potentials with reference to ferrocene at −4.8 eV. e HOMO = LUMO + Eoptg. | ||||||||
| PBF8BT-1 | 13.5 k | 2.27 | 378 | 444 | 583 | 2.13 | −3.68 | −5.81 |
| PBF8BT-2 | 12.7 k | 2.41 | 379 | 451 | 640 | 1.94 | −3.67 | −5.61 |
| PBF8BT-3 | 12.7 k | 2.46 | 389 | 455 | 685 | 1.83 | −3.76 | −5.59 |
To confirm the effective participation of the initial fragments in the subsequent polymerization step, gel permeation chromatography (GPC) analysis was conducted. As shown in Fig. 1(a), the GPC profiles of F8BT and the PBF8BT series exhibited uniform molecular weight distributions, indicating not only the successful integration of the first-stage fragments into the final polymer chains but also the indirect demonstration of the retained reactivity of the intermediate fragments generated in the first step. Additionally, the molecular weight was carefully controlled during the second polymerization step by optimizing the reaction time, thereby minimizing the variability in the polymer properties resulting from molecular weight differences. To directly validate the reactivity of the intermediate fragments synthesized during the first step, these intermediates were isolated, and a separate polymerization reaction was conducted. Reaction intermediates were obtained under conditions identical to those used for PBF8BT-1 synthesis. Polymerization was then carried out by reintroducing the intermediate into the reactor under the standard F8BT synthesis conditions. The resulting polymer was purified using the same procedure as for F8BT. GPC analysis (Fig. S6, ESI†) confirmed the successful synthesis of the target polymer (Mn = 8.9 k, PDI = 1.86), validating the reactivity of the intermediate in the Suzuki reaction. These results collectively demonstrate the effectiveness of the two-step polymerization approach. The modular timing of water addition provides precise control over the reaction sequence, facilitating the synthesis of complex conjugated polymer architectures. This strategy highlights the robustness and scalability of the methodology for producing advanced conjugated polymers tailored for specific functional applications.
 | ||
| Fig. 1 (a) GPC profiles of F8BT and PF8BT polymers. (b) Energy level diagram of F8BT and PBF8BT. (c) UV-vis and (d) PL spectra of solution-state F8BT and PBF8BT polymers dissolved in THF. | ||
Optical and electronic properties
UV-vis spectroscopy and cyclovoltammetry (CV) were conducted to investigate the optical and electronic properties of the polymers and assess the effect of the BDT moiety on the polymer chains. As the BDT ratio in the polymers increased, the highest occupied molecular orbital (HOMO), shown in Fig. 1(b), shifted upward owing to the enhanced p-type properties of the polymers originating from the BDT molecules, consistent with the observed redshift in the λmax of PBF8BT. The exact optical bandgaps (Egap) of the polymers were calculated using Tauc plots (Fig. S7, ESI†), yielding values of 2.13, 1.94, and 1.83 eV, respectively. Furthermore, a higher BDT ratio in the polymers may increase the degree of p-doping, as confirmed by the ultraviolet photoelectron spectroscopy (UPS) of PBF8BT (Fig. S8, ESI†).| (ELUMO = EHOMO + Egap) |
The UV-vis absorption spectra of F8BT and PBF8BT in dilute tetrahydrofuran (THF) are shown in Fig. 1(c), and the corresponding optical properties are summarized in Table 1. All polymers exhibit strong absorption in the 400–550 nm range, characteristic of π–π* transitions in the polymer backbone. However, the PBF8BT polymers display a distinct extension of the absorption edge into the visible region, extending up to 700 nm. This enhanced absorption is directly attributed to the incorporation of BDT units into the polymer backbone, which reduces the optical bandgap and improves the polymers' light-harvesting capability. The broader and redshifted absorption profile of PBF8BT indicates its superior potential for visible-light-driven hydrogen evolution compared to F8BT, as evidenced by the stronger absorption coefficient in the extended spectral region. The PL spectra of F8BT and PBF8BT in solution, shown in Fig. 1(d), reflect trends consistent with their UV-vis absorption properties. The emission peak of F8BT at approximately 540 nm aligns well with previously reported values8 and serves as a reliable reference for comparison. In contrast, the PL maxima of the PBF8BT polymer solution redshifted progressively from 540 to 630 nm as the BDT content in the polymer backbone increased. This redshift is enabled by the higher planarity and stronger donor properties of the BDT moiety compared to those of fluorene. Additionally, a shoulder peak appeared in the PL spectra of PBF8BT, becoming more prominent with higher BDT content. This feature is likely associated with the localized electronic transitions in the BDT moiety and its interaction with neighboring polymer units. Interestingly, while the concentration of the polymer solutions remained constant, the PL intensity of the PBF8BT polymers decreased with increasing BDT ratio. The reduction in PL intensity indicates enhanced non-radiative decay pathways, likely resulting from increased exciton quenching due to intramolecular charge transfer within the polymer chain and efficient separation of electron and exciton states. This separation promotes non-radiative exciton recombination, thereby reducing the overall photoluminescence efficiency.
Density functional theory (DFT) calculations were performed to gain further insight into the structural effects of BDT incorporation, DFT calculations were conducted (Fig. S9, ESI†). These results indicated that introducing BDT significantly reduced the dihedral angle between adjacent polymer units, enhancing the coplanarity of the polymer backbone. This structural feature is crucial for efficient π–π stacking, which enhances exciton and charge-carrier delocalization. However, the same coplanarity that improves delocalization also promotes non-radiative decay pathways, leading to the observed decrease in PL intensity with higher BDT content. Incorporating BDT into the polymer backbone also strengthens the push–pull electronic effect characteristic of donor–acceptor polymers, as shown in Fig. S10 (ESI†). This effect is reflected in the increased bathochromic shift in the emission spectra and enhanced visible-light absorption of the PBF8BT polymers. These changes indicate more effective separation and transfer of charge carriers within the polymer matrix.
Overall, integrating BDT units into the PBF8BT polymer backbone significantly enhanced the visible light absorption and modified the photophysical properties by promoting a planar backbone structure and altering the emission characteristics. The balance between improved absorption and charge recombination dynamics underscores the critical role of molecular architecture in designing high-performance photocatalysts.
Pdot fabrication and photocatalytic hydrogen evolution performance
Pdots were prepared using a nanoprecipitation method with PS-PEG-COOH as a surfactant following procedures from previous research.32 To investigate the hydrodynamic radius and morphology of the Pdots, DLS and liquid phase-transmission electron microscopy (LP-TEM) analyses were performed. The DLS results indicated that the hydrodynamic radii of the Pdots across various conjugated polymers ranged from 70 to 100 nm (Fig. 2), further confirmed by the LP-TEM images, which display spherical Pdots with diameters in the same range. However, no significant differences were observed between the Pdots formed from different conjugated polymers. These findings allow us to focus on the relationship between the hydrogen production performance of each Pdot and the charge carriers generated within the conjugated polymer, independent of any morphological variations. | ||
| Fig. 2 Pdot sizes measured by DLS; insets feature liquid-phase transmission electron microscopy (LP-TEM) images. | ||
Pdots with varying amounts of BDT were evaluated for their photocatalytic HERs under simulated solar illumination (300 W Xe Lamp, 100 mW cm−2, AM 1.5G). Ascorbic acid (AA, pH 4) buffered with NaOH solution was used as the electron donor. In addition, in Fig. S11 (ESI†), the UV-vis absorption spectra of each Pdot did not show significant changes after the deposition of the cocatalysts. As shown in Fig. 3(a) and (b), PBF8BT-2 exhibited an average HER of 7300 μmol h−1 g−1 compared to 3370 μmol h−1 g−1 for PBF8BT-1, both without Pt cocatalysts. When 8 wt% Pt was photodeposited, the average HER increased to 22130 μmol h−1 g−1 for PBF8BT-2 and 5490 μmol h−1 g−1 for PBF8BT-1, showing a consistent trend with cocatalysts conditions. As the amount of Pt cocatalysts increased, the hydrogen evolution rate (HER) of PBF8BT-2 also increased. However, the HER per unit of Pt decreased. We optimized the Pt cocatalyst loading at 8 wt% (Table S2, ESI†). Additionally, in Fig. S12 (ESI†), PBF8BT-2 Pdot exhibit a 2.6-fold higher HER performance compared to F8BT Pdots (without Pt: 1540 μmol g−1 h−1; with Pt: 8300 μmol g−1 h−1). These results indicate that the charge carriers generated in the conjugated polymers significantly influenced hydrogen production, regardless of the presence of the cocatalyst. However, PBF8BT-3, with a high BDT content, exhibited reduced HER performance compared to PBF8BT-2, suggesting that the optimal donor content ratio was achieved in PBF8BT-2, as discussed further in the subsequent photochemical analysis.
 | ||
| Fig. 3 (a) Comparison of photocatalytic hydrogen evolution activities of PBF8BT with and without photo-deposited Pt using ascorbic acid (AA, pH = 4) as a sacrificial agent (λ > 420 nm). (b) HER for each sample (the average of 5 h across five repeated runs). (c) Photocatalytic stability test up to 15 h for optimized PBF8BT-2 nanoparticle. (d) Apparent quantum yield (AQY) of PBF8BT-2 at 400, 420, 450, 500, and 550 nm along with the absorbance spectrum of a 0.2 M (pH = 4) solution of ascorbic acid. The error bars were obtained from the five standards derived from five repeated trials. | ||
For the test of photocatalytic stability test of PBF8BT-2, we conducted a 15-hour stability test Fig. 4(c). After each 5-hour reaction cycle, high-purity argon (>99.99%) was used to purge the reaction solution, and ascorbic acid washing was employed to prevent depletion of the electron donor. The reactivity of the Pdots remained high (≈95%) throughout the first three cycles. This test demonstrates that the photocatalytic stability of PBF8BT-2 was maintained over the 15-hours period. Furthermore, we conducted FT-IR analysis on PBF8BT-2 Pdots before and after the photocatalytic reaction to evaluate potential structural changes (Fig. S14, ESI†). The FT-IR spectra showed no significant differences, confirming that PBF8BT-2 maintains its structural integrity and stability throughout the reaction while sustaining its hydrogen evolution performance. The reactivity of the Pdots remains high (∼95%) throughout the first three cycles. Regarding the apparent quantum yield (AQY) (Fig. 3(d)), PBF8BT-2 with the cocatalysts demonstrated efficiencies of 1.1% at 450 nm and 0.1% at 700 nm. No significant changes were observed in the spectral properties of the PBF8BT-2 Pdots before and after the photocatalytic reaction, indicating their stability under photocatalytic conditions (Fig. S14, ESI†). The slight decrease in the overall absorption is attributed to Pdot aggregation during the photocatalytic process. The residual Pd content of each conjugated polymer is summarized in Table S1 (ESI†).
 | ||
| Fig. 4 Time-resolved photoluminescence (TR-PL) decay kinetics of PBF8BT excited at 450 nm and probed at PL maximum individually, measured via time-resolved photoluminescence (TR-PL). All Pdot solutions were prepared with an absorbance of 0.2 at the excitation wavelength. | ||
Photophysical description of photocatalysts
Time-resolved photoluminescence (TRPL) was used to investigate the radiative decay times of the excited states of the polymers. These TR-PL experiments were conducted under the same conditions as the photocatalytic reaction mixture, maintaining an absorbance of 0.2 at the excitation wavelength. The Pdots were excited at 450 nm and probed at 550 nm for F8BT, as it exhibited the highest emission at 540 nm. For PBF8BT, excitation was performed at 450 nm, and probing was performed at the PL maximum of each polymer—progressively redshifted for PBF8BT-1, PBF8BT-2, and PBF8BT-3. The excited state lifetimes of F8BT were estimated as 0.48 ns, while those of PBF8BT-1, PBF8BT-2, and PBF8BT-3 were measured at 0.71 ns, 0.81 ns, and 0.77 ns, respectively, as shown in Fig. 4. The synthesized F8BT exhibited a lifetime consistent with previously reported values,7,33 whereas all the PBF8BT samples demonstrated a longer carrier lifetime than F8BT. Interestingly, the polymers exhibited similar carrier lifetimes, regardless of the presence of a hole scavenger, possibly due to the charge carriers already being transported to a saturated concentration of Pd in the Pdots.34 Notably PBF8BT-2 exhibited the longest exciton lifetime, suggesting that an optimal concentration of BDT in the polymer backbone enhances exciton lifetime by promoting charge carrier localization and stabilization along the π-conjugated backbone. However, an excessive concentration of BDT may reduce the carrier lifetime due to increased radiative recombination decay. To further investigate the effect of the BDT donor unit, we compared the exciton decay lifetimes of F8BT (λprob = 550 nm) and PBF8BT-2 (λprob = 700 nm) at their respective emission peaks. The overall PL lifetime of the F8BT Pdots is consistently shorter than that of the PBF8BT-2 Pdots. From the TR-PL kinetics, we demonstrated that the BDT donor unit prolonged the exciton lifetime, thereby increasing the charge carrier concentration, reducing recombination, and facilitating proton/hole scavenger activity. The time constants derived from the bi-exponential fits of the TR-PL data are summarized in Table S3 (ESI†).As the charge carrier dynamics in TR-PL are analyzed indirectly, we aimed to confirm the behavior of carriers more directly through TAS. Fig. 5(a) and (b) display the transient absorption spectra of F8BT and PBF8BT in aqueous Pdot solutions, respectively. The experiments, conducted on nanosecond to microsecond (ns–μs) timescales, reveal the emergence of long-lived excited-state absorption, consistent with trends reported for common fluorene-based polymers. For F8BT, ground-state bleaching (GSB) was observed at approximately 500 nm. In contrast, PBF8BT exhibits two distinct GSB features, one around 500 nm from fluorene (F8) and another at 575 nm originating from the BDT moiety. Additionally, positive differences in the optical density corresponding to excited-state absorption (ESA) are observed in Fig. 5(a) and (b). This trend remained unchanged even without the hole scavenger (Fig. S18, ESI†). The kinetics at 550 nm for PBF8BT-2 indicated that the charge carriers originating from BDT exhibited the longest lifetimes, consistent with the lifetimes measured using TR-PL. In contrast, PBF8BT-1 exhibited shorter carrier lifetimes owing to recombination, suggesting that these carriers may not have sufficient time to transfer to the active sites (Fig. 5(c)). In Fig. 5(d), the kinetic profiles at 700 nm, corresponding to the photogenerated polarons on the polymer photocatalysts, revealed a striking result. PBF8BT-3 showed almost no signal, indicating that the photogenerated charge carriers were rapidly lost through non-radiative decay.
 | ||
| Fig. 5 Transient absorption spectra of F8BT and PBF8BT Pdots. (a) F8BT and (b) PBF8BT-2 after photoexcitation with a pump wavelength of 355 nm (2 mJ cm−2). Transient kinetics probed at (c) 550 nm and (d) 700 nm for PBF8BT in aqueous Pdot. All data were obtained at an excitation wavelength of 355 nm and a fluence of 2 mJ cm−2. Suspensions were prepared with an absorbance of 0.42 at 355 nm and carefully purged with argon before the experiment. | ||
After analyzing the kinetics, Fig. 6 illustrates the overall reaction pathway and the proposed mechanism for the polymer. In polymers with a lower BDT content (PBF8BT-1), the carriers generated from BDT do not have a sufficiently long lifetime to facilitate efficient photoredox reactions. In contrast, when the BDT content was too high (PBF8BT-3), most photogenerated carriers were lost through non-radiative decay. This was due to the strong push–pull effect between the donor and acceptor components. These results highlight the need to carefully optimize the BDT content to balance charge generation and stability for efficient photocatalytic activity. To study the effects of the BDT donor units on the photocatalytic activity, we compared the kinetics of F8BT and PBF8BT-2 (Fig. S17, ESI†). The decay of the GSB in F8BT was rapid, with a lifetime of ∼100 ns. In contrast, the GSB lifetime of PBF8BT-2 is significantly extended to over 1000 ns when monitored at 475 nm, corresponding to the fluorene unit. At 585 nm, the GSB kinetics of the PBF8BT-2 Pdot exhibited significant bleaching, whereas that of the F8BT Pdot did not show any signal (Fig. S17(b), ESI†), indicating that the BDT donor moiety generates states, resulting in enhanced charge-carrier stabilization. This trend remained consistent even in the presence of the cocatalysts (Fig. S17(c), ESI†). These findings suggest that the incorporation of BDT units enhances intersystem crossing (ISC) and stabilizes the charge-separated (CS) state by suppressing recombination. In the ESA state, the photo-induced charge carrier states persisted up to 10 μs at a probe wavelength of 700 nm. Considering BDT units are more planar compared to fluorene units, Marcus’ theory predicts a high driving force for ISC due to stable reorganization energy, resulting in low emission and a high ISC quantum yield, which in turn enhances the photocatalytic performance of PBF8BT-2.35,36 From these photophysical analyses, we demonstrate that optimized BDT units suppress unwanted radiative and non-radiative recombination from the excited state to the ground state, thereby prolonging photogenerated charge carrier lifetimes.37 These long-lived charge carriers are more effectively transferred to the cocatalysts (Pt and Pd), enhancing reduction and oxidation with proton and hole scavengers, respectively.
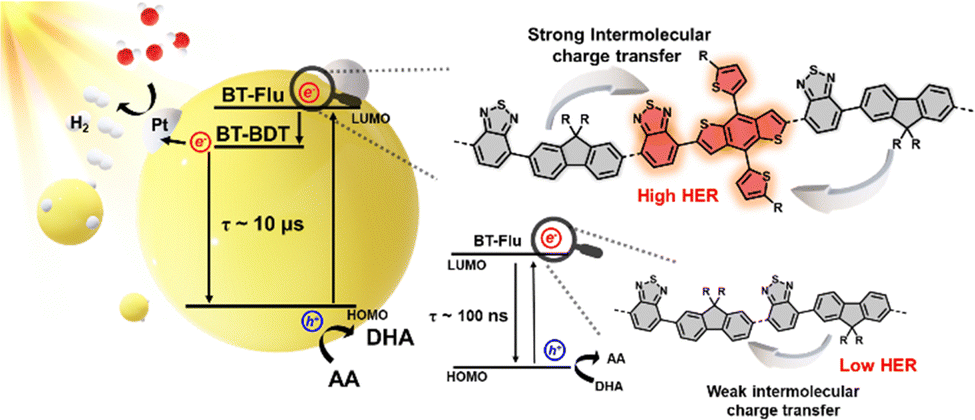 | ||
| Fig. 6 Schematic illustration of photocatalytic processes in PBF8BT Pdots. | ||
Conclusions
This study comprehensively investigated the synthesis, optoelectronic characterization, and photocatalytic performance of PBF8BT polymers with various ratios of BDT donor units. Using a two-step polymerization strategy that combines the Stille and Suzuki coupling reactions, we achieved precise control over the donor–acceptor composition of the polymer backbone. The incorporation of BDT units significantly extended the absorption edge to the visible region (up to 700 nm), enhancing charge carrier separation and lifetime, as demonstrated by steady-state PL, TR-PL, and TAS analyses. Among the synthesized polymers, PBF8BT-2 achieved the highest HER of 22130 μmol h−1 g−1 with an 8 wt% Pt cocatalyst, attributed to its optimized donor–acceptor interactions, which effectively balance charge generation and stability. In contrast, the excessive BDT content in PBF8BT-3 resulted in strong push/pull interactions that increased the non-radiative decay, thereby reducing the photocatalytic efficiency. These findings highlight the critical importance of fine-tuning the molecular architecture to achieve optimal performance.
Mechanistic insights from the TAS analysis revealed that PBF8BT-2 generated long-lived charge-separated states. The planar nature of the BDT units reduced the reorganization energy, enhanced intersystem crossing, and facilitated efficient charge transfer dynamics. This work underscores the importance of optimizing donor moieties to improve visible-light-driven hydrogen evolution. The two-step polymerization method developed in this study offers a scalable and robust framework for synthesizing complex polymer architectures with tailored optoelectronic properties. By combining the design principles of organic photovoltaics and hydrogen evolution photocatalysts, this study lays the groundwork for advancing next-generation polymer-based photocatalysts. The proposed approach offers a sustainable pathway for solar-driven hydrogen production, thus contributing to global efforts to address energy challenges.
Author contributions
W. J. synthesized the materials and wrote the manuscript. S. A. tested the catalyst performance and wrote the manuscript. G. H. measured the photophysics of the materials. C. K. performed the DFT calculations. S. L. and J. Y. measured the morphology of the catalysts. D. C., H. C., and T. P. edited and supervised the manuscript.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work supported by National Research Foundation of Korea (NRF) grants funded by Ministry of Science and ICT (MSIT) (No. 2021R1A5A1084921, No. 2021R1A2C3004420, RS-2023-00213920 and 2022M3H4A1A03067131). The authors appreciate the Pohang Accelerator Laboratory (PAL) for providing the 10A2 and 4D beamlines used in this study.References
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2021, 7, 13–24 CrossRef.
- F. Urbain, V. Smirnov, J.-P. Becker, A. Lambertz, F. Yang, J. Ziegler, B. Kaiser, W. Jaegermann, U. Rau and F. Finger, Energy Environ. Sci., 2016, 9, 145–154 RSC.
- H. Lim, G. H. Han, D. H. Lee, G. Shin, J. Choi, S. H. Ahn and T. Park, Small, 2024, 20, e2400031 Search PubMed.
- H. Lim, I. Jeong, J. Choi, G. Shin, J. Kim, T.-H. Kim and T. Park, Appl. Surf. Sci., 2023, 610, 155601 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- S. An, S. Z. Hassan, J. W. Jung, H. Cha, C. H. Cho and D. S. Chung, Small Methods, 2022, 6, e2200010 CrossRef PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- J. Lee, S. A. Park, S. U. Ryu, D. Chung, T. Park and S. Y. Son, J. Mater. Chem. A, 2020, 8, 21455–21473 RSC.
- J. Lee, G. W. Kim, M. Kim, S. A. Park and T. Park, Adv. Energy Mater., 2020, 10, 1902662 CrossRef CAS.
- Y. Bae, D. Kim, S. Li, Y. Choi, S. Y. Son, T. Park and L. Ye, Prog. Polym. Sci., 2024, 101899 CrossRef CAS.
- J.-W. Ha, J. G. Jung, D. H. Ryu, S. Lee, C. E. Song, B. Lim, Y. J. Jung, J. M. Park and D.-H. Hwang, Macromol. Res., 2023, 31, 25–31 CAS.
- K. Kranthiraja, H. Kim, J. Lee, U. K. Aryal, S. S. Reddy, R. D. Gayathri, T. Gokulnath and S.-H. Jin, Macromol. Res., 2023, 31, 897–905 CAS.
- M. Axelsson, C. F. N. Marchiori, P. Huang, C. M. Araujo and H. Tian, J. Am. Chem. Soc., 2021, 143, 21229–21233 CAS.
- S. A. J. Hillman, R. S. Sprick, D. Pearce, D. J. Woods, W. Y. Sit, X. Shi, A. I. Cooper, J. R. Durrant and J. Nelson, J. Am. Chem. Soc., 2022, 144, 19382–19395 CAS.
- C. L. Chang, T. F. Huang, W. C. Lin, L. Y. Ting, C. H. Shih, Y. H. Chen, J. J. Liu, Y. T. Lin, Y. T. Tseng, Y. H. Wu, Y. E. Sun, M. H. Elsayed, C. W. Chen, C. H. Yu and H. H. Chou, Adv. Energy Mater., 2023, 13, 2300986 CAS.
- C. L. Chang, W. C. Lin, L. Y. Ting, C. H. Shih, S. Y. Chen, T. F. Huang, H. Tateno, J. Jayakumar, W. Y. Jao, C. W. Tai, C. Y. Chu, C. W. Chen, C. H. Yu, Y. J. Lu, C. C. Hu, A. M. Elewa, T. Mochizuki and H. H. Chou, Nat. Commun., 2022, 13, 5460 CAS.
- P. Chen, F. Ji, D. Ma, Y. Xie, X. Wu, M. Zhang, C. Ru, L. Zhou, J. Wu and X. Pan, J. Mater. Chem. A, 2023, 11, 21146–21152 CAS.
- P. Chen, C. Ru, L. Hu, X. Yang, X. Wu, M. Zhang, H. Zhao, J. Wu and X. Pan, Macromolecules, 2023, 56, 858–866 CAS.
- S. U. Ryu, D. H. Ryu, D. H. Lee, Z. U. Rehman, J.-C. Lee, H. Lim, G. Shin, C. E. Song and T. Park, Chem. Eng. J., 2024, 485, 149865 CAS.
- A. F. Saber, A. M. Elewa, H.-H. Chou and A. F. EL-Mahdy, Appl. Catal., B, 2022, 316, 121624 CAS.
- A. F. Saber, A. M. Elewa, H. H. Chou and A. F. EL-Mahdy, ChemCatChem, 2023, 15, e202201287 CAS.
- C. Ru, Y. Wang, P. Chen, Y. Zhang, X. Wu, C. Gong, H. Zhao, J. Wu and X. Pan, Small, 2023, 19, e2302384 Search PubMed.
- T. F. Huang, J. J. Liu, Z. Y. Lai, J. W. Chang, Y. R. Zhuang, Z. C. Jiang, C. L. Chang, W. C. Lin, Y. H. Chen, Y. H. Wu, Y. E. Sun, T. A. Luo, Y. K. Chen, J. C. Yen, H. K. Hsu, B. H. Chen, L. Y. Ting, C. Y. Lu, Y. T. Lin, L. Y. Hsu, T. L. Wu, S. D. Yang, A. C. Su, U. S. Jeng and H. H. Chou, Small, 2024, 20, e2304743 Search PubMed.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 Search PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CAS.
- C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040–3053 CAS.
- M. V. Pavliuk, S. Wrede and H. Tian, Chem. Commun., 2023, 59, 5611–5614 CAS.
- S. An, K. J. Jeong, S. Z. Hassan, G. Ham, S. Kang, J. Lee, H. Ma, J. Kwon, S. Y. Jeong, J. Yang, H. Y. Woo, H. H. Cho, H. Cha, C. Y. Son and D. S. Chung, Adv. Sci., 2024, 11, e2309786 Search PubMed.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francas, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CAS.
- I. Fureraj, J. Wega, E. Balanikas, K. K. Puji Pamungkas, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. Lett., 2024, 15, 7857–7862 CAS.
- C. Schafer, R. Ringstrom, J. Hanrieder, M. Rahm, B. Albinsson and K. Borjesson, Nat. Commun., 2024, 15, 8705 Search PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2022, 34, e2105007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00035a |
| ‡ Wooteak Jung, Sanghyeok An and Gayoung Ham contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2025 |