 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine speciation in snow during the MOSAiC expedition and its implications for Arctic iodine emissions
Lucy V.
Brown
 a,
Ryan J.
Pound
a,
Matthew R.
Jones†
a,
Matthew J.
Rowlinson
a,
Rosie
Chance
a,
Hans-Werner
Jacobi
c,
Markus M.
Frey
b,
Stephen D.
Archer
d,
Stefanie
Arndt
ef,
Johannes G. M.
Barten
g,
Byron W.
Blomquist
hi,
Ruzica
Dadic
j,
Laurens N.
Ganzeveld
g,
Henna-Reetta
Hannula
k,
Detlev
Helmig
l,
Matthias
Jaggi
j,
Daniela
Krampe
em,
Amy R.
Macfarlane
jno,
Shaun
Miller
b,
Martin
Schneebeli
j and
Lucy J.
Carpenter
a
a,
Ryan J.
Pound
a,
Matthew R.
Jones†
a,
Matthew J.
Rowlinson
a,
Rosie
Chance
a,
Hans-Werner
Jacobi
c,
Markus M.
Frey
b,
Stephen D.
Archer
d,
Stefanie
Arndt
ef,
Johannes G. M.
Barten
g,
Byron W.
Blomquist
hi,
Ruzica
Dadic
j,
Laurens N.
Ganzeveld
g,
Henna-Reetta
Hannula
k,
Detlev
Helmig
l,
Matthias
Jaggi
j,
Daniela
Krampe
em,
Amy R.
Macfarlane
jno,
Shaun
Miller
b,
Martin
Schneebeli
j and
Lucy J.
Carpenter
a
aWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, UK. E-mail: lucy.v.brown@york.ac.uk
bNatural Environment Research Council, British Antarctic Survey, Cambridge, UK
cInstitute for Geosciences and Environmental Research, CNRS/Univ. Grenoble Alpes/INRAE/IRD/G-INP, Grenoble, France
dBigelow Laboratory for Ocean Sciences, East Boothbay, ME, USA
eAlfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung, Bremerhaven, Germany
fInstitute of Oceanography, University of Hamburg, Hamburg, Germany
gMeteorology and Air Quality Section, Wageningen University, Wageningen, the Netherlands
hCooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO, USA
iNational Oceanic and Atmospheric Administration, Physical Sciences Laboratory, Boulder, CO, USA
jWSL Institute for Snow and Avalanche Research SLF, Davos, Switzerland
kFinnish Meteorological Institute, Helsinki, Finland
lBoulder A.I.R., LLC, Boulder, CO, USA
mRegionalverband Ruhr, Essen, Germany
nUiT The Arctic University of Norway, Tromsø, Norway
oNorthumbria University, Newcastle upon Tyne, England, UK
First published on 6th December 2024
Abstract
Photochemical release of iodine from snow has been suggested as a source of reactive iodine to the Arctic atmosphere, however understanding of the underlying mechanism and potential source strength is hindered by a lack of measurements of iodine concentration and speciation in snow. Moreover, the origin of snow iodine is also unknown. Here, we report iodine speciation measurements in Arctic snow on sea ice at a range of snow depths from 177 samples, representing 80 sampling events, from December 2019 to October 2020 collected during the Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition. We demonstrate that while there appears to be a source of iodine, in particular iodate, to the base of the snow over first year ice, this does not influence iodine concentration in the surface snow. There is instead evidence of a top-down source of iodine, potentially from iodine-enriched marine aerosol, as well as some evidence for episodic influx of iodate with dust. The potential for photochemical release of molecular iodine (I2) from iodide in surface snow was investigated, and it was demonstrated that this could provide an iodine emission flux to the Arctic atmosphere comparable to oceanic fluxes. Knowledge of the prevalence and speciation of iodine in Arctic snow will contribute to better understanding of its contribution to observed concentrations of polar iodine oxide (IO), and hence its contribution to the depletion of tropospheric ozone in the Arctic.
Introduction
The influence of halogens on polar atmospheres
Ozone (O3) in the Arctic troposphere can reduce to near-zero concentrations during springtime due to photochemical cycling of reactive halogens. These observations have been called ozone depletion events (ODEs) due to their episodic nature observed at Arctic coastal stations.1 Over the Arctic Ocean, the depletion of tropospheric ozone is widespread and can last for prolonged periods.2,3 The impact of this ozone depletion is hemispheric-wide, as the export of ozone-poor air masses to lower latitudes can reduce background ozone over North America and Europe.4 In addition to their impact on ozone, halogens oxidise atmospheric elemental mercury to more soluble forms, leading to mercury deposition and increased bioavailability.5 Satellite data show that halogen monoxides BrO and IO are widespread in the Antarctic troposphere,6 and BrO in the Arctic troposphere,7 thus halogen chemistry exerts a significant influence on the Arctic and Antarctic regions.ODEs were first observed in the 1980s,8,9 and linked to halogens by Barrie et al. (1988) from a study at Alert, Canada.10 In the transition from winter to spring, depleted ozone concentrations were observed to coincide with increased atmospheric bromine (Br). It is now well established that reactive bromine contributes to the chemical destruction of ozone through a mechanism initiated by sea-salt-enriched snow, which can become airborne in wind-blowing snow events. These events liberate the bromine and aerosol-enriched snow, resulting in the formation and subsequent release of gas phase reactive bromine species.11–15 More recently, the importance of reactive iodine species in the depletion of ozone over the Arctic Ocean has been revealed.16
The mechanism of halogen-driven tropospheric ozone destruction is initiated by photolysis of precursor dihalogen molecules (X2, where X = Br or I) (reaction 1, labelled R1). This leads to production of halogen atoms (X) which rapidly react with O3 (R2) to form bromine or iodine oxide radicals (XO). XO can react with hydroperoxyl (HO2) (R3) or nitrogen dioxide (NO2) (R4) to form HOX and XONO2, which are photolytically broken down17 to reform X atoms (R5 and R6) or enter aerosol where they undergo condensed phase reactions. Aqueous HOX reacts with bromide (Br−), chloride (Cl−) and iodide (I−) (R7) to produce volatile interhalogen (e.g. BrCl) or dihalogen molecules (Br2 and I2) which are re-released to the gas phase to participate in the photochemical cycle. No O3 is produced in this cycle, thus catalytic destruction of ozone occurs. XO cross reactions also occur, to regenerate dihalogens (R8), or halogen radicals (R9 and R10). The combined impact of BrO and IO on ozone loss is greater than the sum of the effects of the individual halogens, because XO cross reactions occur approximately an order of magnitude faster than BrO + BrO.18
In clean air, IO and iodine dioxide (OIO) self-reactions (examples R10 & R11) produce higher iodine oxides (e.g. I2O2, I2O3, and I2O4), which are thought to be subsequently photolysed back to IO or OIO.19 Reactions of iodine oxides with ozone form stable I2O5 (R12), which can polymerise to form iodine oxide particles (IOPs).20,21 This appears to be the dominant mechanism for IOP formation where iodine is present in relatively high concentrations. However, the balance of evidence now suggests that formation of gas phase iodic acid (usually referred to as HIO3) drives IOP formation in multiple environments, including over the central Arctic Ocean.22–25 As well as over the Arctic, gas phase HIO3 has been detected in environments ranging from coastal marine air to the lower free troposphere, and a number of mechanisms for its formation have been proposed.22–24,26,27 IOPs can act as cloud condensation nuclei (CCN), further increasing the impact of iodine emissions on polar environments.22,28–30
| X2 + hv → 2X | (R1) |
| X + O3 → XO + O2 | (R2) |
| XO + HO2 → HOX + O2 | (R3) |
| XO + NO2 → XONO2 | (R4) |
| HOX + hv → X + OH | (R5) |
| XONO2 + hv → X + NO3 | (R6) |
| HOX + X(surface)− → X2 + OH− | (R7) |
| XO + XO → X2 + O2 | (R8) |
| XO + XO → X + X + O2 | (R9) |
| XO + XO → OXO + X | (R10) |
| OXO + OXO → X2O4 | (R11) |
| X2O4 + O3 → X2O5 + O2 | (R12) |
Iodine in the polar troposphere
The presence of iodine in the polar troposphere was first demonstrated in the Arctic by Wittrock et al., (2000), who observed IO using Differential Optical Absorption Spectroscopy (DOAS), in Ny Alesund, Svalbard, from 1995–1998.31 During sunlit periods, stratospheric IO was positively identified for some days each month, with some evidence for tropospheric IO, without further quantification.In the Antarctic troposphere, IO was first measured in 1999 at Neumayer Station by Frieβ (2001) using DOAS. Iodine monoxide was reported to be higher in summer compared to winter, and present at up to 10 pptv.32 At Halley Station, a site 12 km south and 30 km east of the Antarctic ice edge, surface IO was measured by LP-DOAS (Long Path Differential Optical Absorption Spectroscopy) and peaked in Austral spring at around 20 pptv, with a weaker peak in Austral autumn of 6–7 pptv.33 The mixing ratio measured during the springtime peak remains the highest reported mixing ratio of IO in the polar boundary layer. The concentration of IO closely tracked solar radiation, indicating that IO production is photochemically mediated and that it has a short boundary-layer lifetime. Satellite observations retrieved for October 2005 in the Antarctic springtime revealed IO mixing ratios >12 pptv.34 Iodine monoxide peaked over sea ice and around the edge of the Antarctic ice shelf. In the Weddell Sea, IO was measured at a mean mixing ratio of 5.1 pptv, with a maximum of 6.6 pptv.35 Higher IO mixing ratios were again associated with areas with a high ice extent. IO was also later measured over a one-year period by MAX-DOAS in the Western Antarctic at the Bharati station.36 At this station, in summer there can be complete sea-ice melt, while in winter the sea ice can extend several kilometres. Average surface IO mixing ratios were typically below 1 pptv and only a weak increase in IO mixing ratio was observed during spring, with a few autumn days reaching values comparable to the springtime values. The maximum mixing ratio measured was 1.9 ± 0.3 pptv during October.
There are fewer observations of IO in the Arctic and the levels observed are generally lower than those in the Antarctic troposphere.34 At Hudson Bay, Canada, Mahajan et al., (2010) detected IO in spring with peak daytime levels of 3 ± 1 pptv.37 More recently, the Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition in the high Arctic has allowed a much longer time series of IO observations. Between March and October 2020, IO was observed at mixing ratios around 0.2–1.0 pptv throughout the entire sunlit period, with enhanced levels of 2.9 ± 0.3 pptv during spring.16 Model simulations of the halogen-driven chemical destruction of ozone indicated that over the entire sunlit period, IO contributed more to the depletion of tropospheric ozone than BrO.16
In terms of the seasonality of iodine, evidence from aerosol studies at Alert and Mould Bay (two Arctic non-coastal sites) indicate spring and autumn peaks.10 Iodine concentrations measured at a coastal site in the same study were higher, and did not show any seasonal variation. During the Arctic MOSAiC expedition, no autumn peak in IO was recorded, with measurements continuing until late September.16 Evidence for a weak autumn peak in Antarctic IO at Halley Station (western Antarctica, some tens of kilometres from the ice edge) was observed,33 with a similar weak autumn peak reported in East Antarctica at Bharati Station.36
Potential iodine release mechanisms
To understand and predict the concentrations and seasonality of polar iodine and its impact on ozone, its source and release mechanism must be understood. Several potential mechanisms have been postulated. The fact that IO mixing ratios over the Antarctic are higher than in the Arctic has been attributed to Antarctic ice being typically thinner than Arctic ice, supporting under-ice biological processes and allowing faster diffusion of iodine precursors through the ice and snow.19,38 Saiz-Lopez et al., (2008) postulated that HOI and I− on the underside of sea ice, produced by phytoplankton at concentrations highly enriched compared to seawater, could diffuse upwards through brine channels in first year ice, forming an iodine-rich brine layer on the top of sea ice,38 either under the snowpack or directly exposed to the atmosphere. This process was included in a model39 and is commonly used to explain Antarctic IO.36 It has also been invoked for Arctic iodine trends,40 including for the MOSAiC expedition4 and iodine in ice cores.41,42 An additional biological mechanism postulated to explain Arctic IO production is seawater emissions of organo-iodine compounds from open water polynyas.37Alternatively, there is evidence for a snowpack chemical source of IO, as indicated by IO mixing ratios of up to 50 pptv in the interstitial air of the Antarctic snowpack.43 In the Arctic, a photolytic snowpack source of I2 was demonstrated;44 I2 is a photolabile precursor to iodine atoms and hence IO (R1 & R2). From in situ chamber experiments, these authors determined peak I2 levels of 22 pptv within the interstitial air of the snowpack at 40 cm depth. I2 was only observed under sunlit (natural or artificial) conditions. Raso et al., (2017) suggested that the mechanism was photochemical production of I2 from iodide.44 This is known to proceed slowly by O2 oxidation in acidic solution (R13),45 or accelerated in frozen solution by photoinduced oxidation (R14), with the iodine radical recombining with I− to form I2−, and subsequently tri-iodide (I3−, R15 and R16).46 Tri-iodide exists in equilibrium with I2 (R17).47
| 4I− + O2 + 4H+ → 2I2 + 2H2O | (R13) |
I− + O2 + hv → (I−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ˙O2) → I˙ + O2− ˙O2) → I˙ + O2− | (R14) |
| I˙ + I− → I2− | (R15) |
| I2− + I2− → I3− + I− | (R16) |
| I2 + I− ⇌ I3− | (R17) |
| IO3− + hv → → I2, HOI or IO(g) | (R18) |
| IO3− + NO2− → I−, I2(aq), NO3− | (R19) |
| NO2− + 2H+ → H2ONO+ + HONO | (R20) |
| 2H2ONO+ + 2I− → I2 + 2NO + H2 | (R21) |
| HONO + 2I− + O2 + H+ → I3− + NO3− + H2O | (R22) |
Formation of reactive iodine can also occur in dark conditions. It has been demonstrated that a frozen solution of iodate and nitrite formed reactive iodine (R19).50 This reaction was accelerated in frozen conditions compared to room temperature, which was attributed to the freeze-concentration effect increasing concentrations of reactants between ice grains during freezing. Formation of I2 from iodide and nitrite (R20–R22) was also demonstrated from frozen solution,51,52 again with the freeze concentration effect essential for efficient conversion.
Alongside snow and ice sources of IO, oceanic emissions of iodine could provide an important background source for polar iodine. Deposition of ozone to the ocean surface followed by reaction with iodide leads to emission of I2 and HOI, which subsequently form IO during the sunlit period.53 The influence of this mechanism on polar IO levels will depend upon several factors including surface ocean iodide concentrations, tropospheric ozone mixing ratios, wind speeds and the proximity of the location to the ocean.
Aims of this study
Despite the significant impact of reactive halogen chemistry on the polar atmosphere and evidence of a snow/ice source of IO, the mechanism of iodine release and its potential source strength are poorly understood. Consideration of possible release mechanisms suggests that reactive iodine formation is likely to be a function of the chemical speciation of iodine as well as its snow/ice concentration. Adding to the uncertainty in the release mechanism is the fact that there are only very few measurements of the concentration of iodine in snow, and even fewer of its speciation in snow. In the Arctic, snow iodide concentrations have been reported from Utqiagvik, Alaska,44 and in Svalbard, where total iodine (TI)54 and observations of iodide and iodate (IO3−)55 have been reported.In this study, we report iodine speciation measurements from snow on sea ice sampled during the MOSAiC expedition aboard the Research Vessel (RV) Polarstern. Samples were collected on 80 occasions over a 9 months period (December–September) over a range of snow depths. For these samples, full iodine speciation was measured, along with complementary quantification of major ions including sodium (Na+), chloride (Cl−) and calcium (Ca2+). From knowledge of the availability of iodine in snow, potential emissions of reactive iodine to the atmosphere were investigated. This dataset vastly expands the current knowledge of iodine in Arctic snow, allowing analysis of the seasonal changes in snow iodine over 9 months and 3 seasons, as well as consideration of its source and impacts.
Methods
Study site and sampling
The Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition was a year-long (October 2019–September 2020) expedition in the high Arctic aboard the RV Polarstern. The expedition aimed to gain fundamental insights into controls of the coupled Arctic climate system. The drift track of the cruise is displayed in Fig. 1. The expedition was split into five segments called Legs; Legs 1–4 were at the same ice floe, with transit to and from Svalbard for personnel exchange between Legs 3 and 4, and Leg 5 on a different ice floe, following disintegration of the original floe at the end of Leg 4.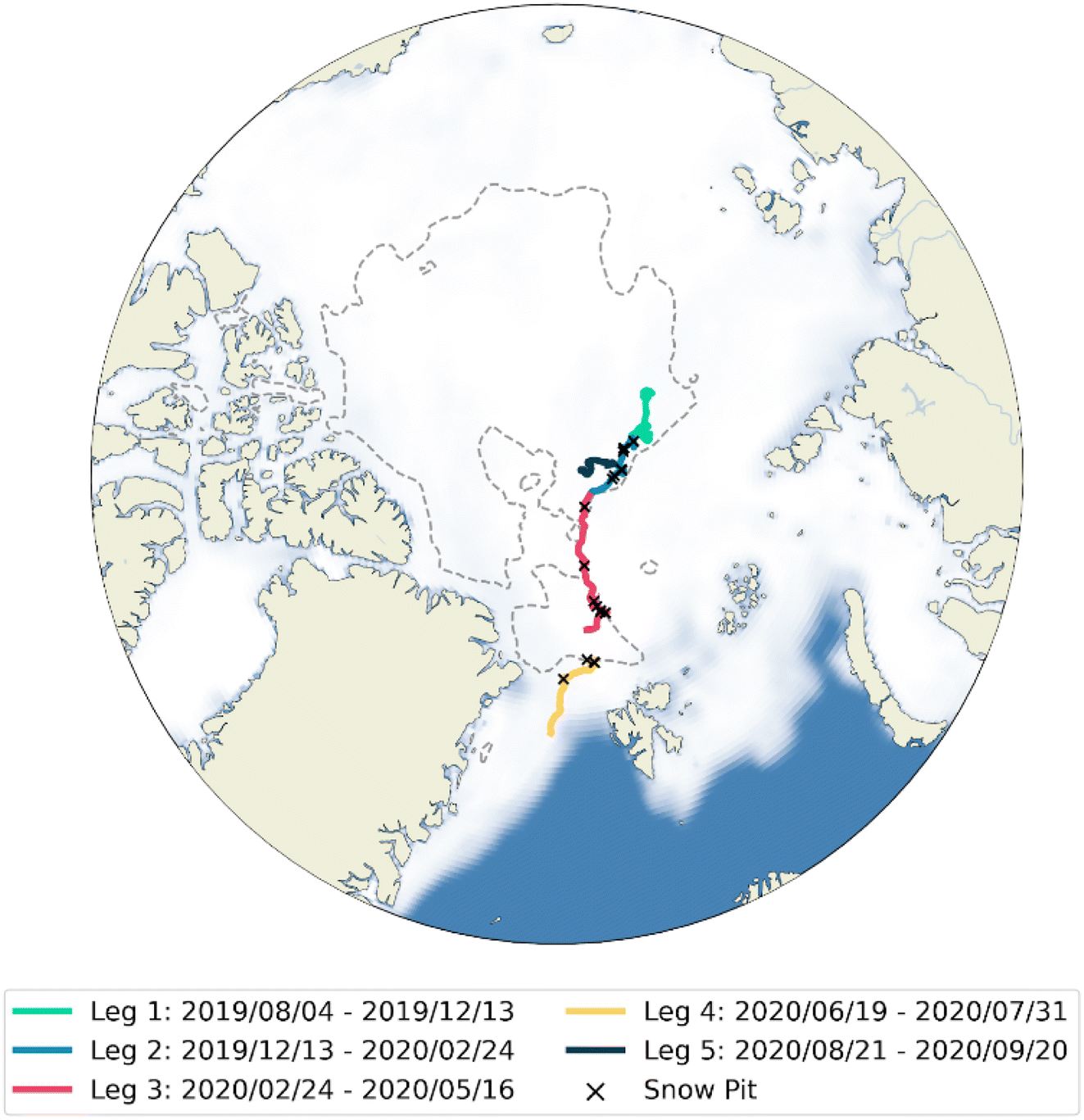 | ||
| Fig. 1 Cruise track of the RV Polarstern during the MOSAiC expedition – each of the 5 Legs is displayed in a different colour. Time in transit is not displayed. The white area is the sea ice coverage in March 2020, representing the maximum sea ice extent, and the summer minimum ice extent (85% sea ice coverage) is shown by the dashed line.56 Locations of snow pits are marked by crosses. | ||
A summary of snow sampling and the complementary ice measurements made are provided in Macfarlane et al., (2023)57 and Nicolaus et al., (2022).59 During this expedition, a comprehensive suite of measurements were carried out to characterise the physical and chemical properties of the snow. The integration of the observed snow parameters obtained during MOSAiC is possible due to the coordinated sampling strategy. This enabled comparison of the physical and chemical properties of the snowpack, whether the measurements were conducted on-site or analysed later in different laboratories after the expedition.
A range of ice types was required due to the change in ice properties with age, both chemical and physical.58 Therefore, snow samples were collected in snow pits, over a range of ice substrates, including first year ice (FYI) and second year ice (SYI), which were determined based on the properties of ice cores drilled in the designated sampling areas.59 Sampling tubes (screw top polypropylene tubes, 50 mL, Corning) were inserted horizontally into the snow pit after excavation. This allowed for vertical profiles of 3 cm vertical resolution of the snowpack.
Iodine concentrations were analysed from 80 sampling events (where a sampling event corresponds to one snow pit) between 30th Dec. 2019 and 18th Sept. 2020. Snow pits were often sampled at multiple depths, therefore, throughout the campaign, 177 measurements of iodine speciation in snow were made. 102 samples, categorised as “surface”, were collected within 5 cm of the snow–air interface, and 53 were collected within 5 cm of the snow–ice interface, categorised as “base” (18 samples fell into both categories, which could happen when the snowpack was <10 cm deep).
For major ion analysis, the sampling tubes were rinsed with UHP water and dried in a class 100 clean laboratory at the British Antarctic Survey (BAS), Cambridge prior to field deployment. All snow samples for iodine and major ions analyses were stored at −20 °C on board RV Polarstern. After the end of the MOSAiC expedition in Oct 2020, the samples were shipped frozen to BAS, Cambridge (for major ion analysis) or York (for iodine speciation analysis) and were only melted overnight (at 4 °C, in the dark) prior to analysis.
For comparison to snow samples, iodine speciation was measured from 16 seawater samples collected by Niskin bottles attached to the CTD (conductivity, temperature and depth) rosette during the cruise. Samples were collected down to a depth of 50 m below the sea ice.
Iodine speciation measurements
Undiluted and unfiltered snow samples were analysed for iodine speciation. Volumes of 400 μL were analysed for iodide by ion chromatography (IC) using an Agilent 1100 HPLC system. The guard and analytical columns used were a Dionex IonPac AS-23 4 × 50 mm and 4 × 250 mm, respectively. The chromatogram was collected for 16.1 minutes following injection, with iodide elution at around 11 minutes. The IC mobile phase was 0.4 M sodium chloride (NaCl, Sigma-Aldrich BioXtra 99.5%, 2 M stock solution) with a flow rate of 0.64 mL min−1. Direct detection of iodide was performed via UV-vis spectrometry using an Agilent 1260 series detector at 226 nm (at this wavelength, iodide has a molar extinction coefficient of ∼2.8 × 104 M−1). The flow cell had a 4 μL volume and 60 mm path length (Agilent G4212-60007).Iodide was calibrated using potassium iodide solutions (KI, Fisher 99%, 0.1 M stock solution) prepared gravimetrically from a stock solution. The reagent stock solutions were 10% weight by volume hydroxylamine hydrochloride (NH2OH–HCl, Sigma-Aldrich Reagent Plus, 99%), 70 mM calcium hypochlorite (Ca(ClO)2, Sigma-Aldrich Technical Grade) and 500 mM sodium sulfite (Na2SO3, Sigma-Aldrich, min 98%, refreshed bi-monthly).
To measure iodate, NH2OH–HCl (final concentration of 7 mM) was added to the thawed snow samples to reduce all iodate to iodide. The iodate fraction was obtained by difference by subsequent measurements of both iodide and total inorganic iodine (TII = iodide + iodate). Total dissolved iodine (TDI) was also measured following digestion of dissolved organic iodine (DOI). To the sample, Ca(ClO)2 was added to a final concentration of 189 μM and left for one hour, followed by addition of Na2SO3 to a final concentration of 380 μM, and NH2OH–HCl to a final concentration of 7 mM. TDI was then quantified as iodide.
The limit of detection (LoD) for iodide (and hence TII and TDI) was 0.12 nM. Further details of the analytical methods for iodide and iodate analysis are described in Jones et al., (2023).60 Iodine in seawater was measured using the same method as for snow samples, however with dilution of seawater (800 μL sample with 850 μL deionised water or reagent).60
Major ion analysis
Snow sampling and analysis are based on the approach by Frey et al., (2020).61 The calibration range of the ion chromatography system required dilution of samples with elevated salinities (>0.5 practical salinity units) with UHP water, typically by a factor of 100 or more for most snow samples.Samples were analysed for major ions using Dionex Integrion ICS-4000 ion chromatography systems with reagent-free eluent generation. A Dionex AS-AP autosampler was used to supply sample water to 250 μL sample loops on each instrument. Cation analyses were performed with a Dionex Ionpac CG16-4 μm (2 × 50 mm) guard column and CS16 (4 μm, 2 × 250 mm) separator column. A 32–45 mM methane sulfonic acid eluent concentration-gradient was used for effective separation of the analytes, before conductivity detection. Anion analyses were performed using a Dionex Ionpac AG17-C (2 μm, 2 × 50 mm) guard column and AS17-C (2 × 250 mm) separator column. A 3.5–27 mM potassium hydroxide eluent concentration-gradient was used for effective separation of the analytes. Calibration was achieved using a range of calibration standards prepared from Sigma-Aldrich standards (1000 ppm) by a series of gravimetric dilutions. Measurement accuracies were evaluated using European reference materials ERM-CA408 (simulated rainwater) and CA616 (groundwater) and were all within 5% and respective LoDs of ∼2 ppbw.
Back trajectories
Back-trajectories were produced using FLEXPART v10.4, a Lagrangian particle dispersion model.62,63 Turbulence is parameterised using the standard Gaussian model,62 while the planetary boundary layer height (PBLH) is calculated using a Richardson number threshold.64 A convection parameterisation is used based on Emanuel and Živković-Rothman (1999).65 Here, FLEXPART is driven by Global Forecast System (GFS) reanalyses at 0.5° × 0.5° resolution, releasing 1000 particles backwards in time over seven days. Trajectories were initialised from the surface every six hours along the path of the MOSAiC expedition. The back trajectories were then binned onto a 0.500° × 0.625° resolution land cover map with sea ice extent from MERRA-2 meteorology.56 Particles below 100 m in altitude were treated as being influenced by surface emissions/interactions from that land cover type.Results and discussion
Summary of iodine and major ions measurements
Of the 177 samples measured for iodine speciation, 24 were below the LoD for iodide, 16 were below the LoD for total inorganic iodine, and 5 were below the LoD for total dissolved iodine. Summary statistics for the measured fractions across the whole campaign are provided in Table 1. Mean, median and standard deviations were calculated assuming that measurements below the LoD = 0.5 × LoD.| Depth | Mean ± 1 sd | Median | Min | Max | n | |
|---|---|---|---|---|---|---|
| Iodide (nM) | Surface | 0.8 ± 0.8 | 0.5 | <0.12 | 3.8 | 102 |
| Bulk | 0.9 ± 0.9 | 0.7 | <0.12 | 4.4 | 69 | |
| Base | 0.9 ± 1.0 | 0.7 | <0.12 | 3.8 | 53 | |
| All | 1.3 ± 5.0 | 0.6 | <0.12 | 65 | 177 | |
| TII (nM) | Surface | 8.1 ± 30 | 1.2 | <0.12 | 205 | 102 |
| Bulk | 52 ± 100 | 1.6 | <0.12 | 510 | 69 | |
| Base | 76 ± 120 | 2.1 | <0.12 | 510 | 53 | |
| All | 32 ± 100 | 1.4 | <0.12 | 862 | 177 | |
| TDI (nM) | Surface | 8.7 ± 30 | 1.9 | <0.12 | 206 | 102 |
| Bulk | 53 ± 110 | 2.5 | <0.12 | 520 | 69 | |
| Base | 77 ± 120 | 2.9 | <0.12 | 520 | 53 | |
| All | 32 ± 98 | 2.1 | <0.12 | 857 | 177 | |
It is important to note that the snowpack is constantly evolving due to erosion and redeposition of snow.66 The stratigraphy of the snow therefore changes with time, while we have denoted samples as “surface”, “base” and “bulk” at the time of sampling, this does not preclude influences from other depths due to reshuffling of the snowpack. It is outside the scope of this work to perform detailed analysis of the evolving snow stratigraphy, instead we have examined general trends across categories, with the understanding that there is uncertainty in the “history” of the snow sample.
A graphical summary of the chemical measurements of the snow samples, along with available literature values of the same variables, are displayed in Fig. 2. The measurements are split into “surface” and “bulk” measurements, where bulk measurements are all non-surface samples (including “base” samples). On four occasions, the total snow height was not available, therefore it was not possible to define the samples collected on these days as either “surface” or “bulk”. One of the samples of these instances (PS122_3_33_66) was extremely high in iodide, iodate and calcium, representing the maximum concentrations of iodide and total inorganic iodine displayed in Table 1. This sampling event was omitted from Fig. 2 as an outlier, to avoid obscuring the other data, but is discussed in the section “Potential top-down inputs of iodine to the snow-pack”.
 | ||
| Fig. 2 Graphical summary of all chemical measurements, including iodide, iodate, total iodine, sodium, chloride and calcium, according to month of the year. Measurements are split into surface (<5 cm from the snow–air interface) and bulk samples (>5 cm from the snow–air interface). Ranges of literature reports of Arctic snow measurements are also displayed, denoted by “F”: Frassati (2024),55 “R”: Raso (2017),44 “S1”: Spolaor (2019)54 measurements from early April 2016, “S2”: Spolaor (2019)54 measurements from late April 2015 and “S3”: Spolaor (2019)54 measurements from January 2017. Limits of detection for iodide and TII are shown by dashed lines. Uncertainties in iodide and total iodine concentrations, shown by error bars, are standard deviations of triplicate measurements. Uncertainties in iodate concentrations are propagated from uncertainties in iodide and total inorganic iodine measurements. Uncertainties in iodide:iodate ratio are propagated from uncertainties in iodide and iodate concentrations. | ||
Fig. 2 (row a) demonstrates the large variability in total iodine concentrations in both the surface and bulk snow samples. Total iodine concentrations in the surface snow ranged from <0.12 to 206 nM (mean = 8.7 nM). In the bulk snow, total iodine was generally higher, ranging from <0.12 to 520.2 nM (mean = 53 nM), excluding the aforementioned outlier. Surface concentrations of total iodine in continental snow at Ny-Alesund, Svalbard (80 °N) were previously reported, measured by ICP-MS.54 Concentrations were measured in light conditions (late April 2015 & early April 2016), ranging from 0.3 to 7 nM and 0.5 to 3 nM and during polar night (January 2017) ranging from 2 to 8 nM. In the current work, surface snow total iodine in January was higher, ranging from 0.6–206 nM (mean 11 nM). In sunlit conditions in April, concentrations from MOSAiC samples ranged from 0.3–6.8 nM (mean 3.5 nM), similar to the results of Spolaor et al., (2019).54
Speciation within total iodine was measured for all samples in the current work. As shown in Fig. 2 (row b), the iodide concentrations in surface and bulk snow were comparable, ranging from below the LoD up to ∼4 nM. In the surface snow, the highest concentrations occurred in January–February, during polar night, and again at the beginning of autumn (September) when light levels decrease in the Arctic. Iodide concentrations were lower during the summer months, not exceeding 2 nM at the surface.
There are only a few previous studies of speciated iodine in Arctic snow samples. A study of continental snow collected at >42 cm depth in Ny-Alesund, Svalbard, reported lower iodide concentrations of 0.5–0.6 nM.55 These samples were collected at the end of the polar night, and are at the lower end of the range of concentrations observed in this work during the unlit period (January/February) in this work. Continental snow samples were also collected in Utqiagvik, Alaska, in January–February 2014 after polar sunrise.44 Surface iodide concentrations were approximately 1–3 nM which are slightly higher than the concentrations described in this work during the sunlit period, which was from mid-March. In the same study, bulk samples were slightly higher in concentration than those reported in the current work, at a range of approximately 1–5 nM (Fig. 2 row b).
As described in Methods, iodate concentrations were obtained by difference between measurements of TII and iodide, which both have LoDs of 0.12 nM. We report iodate concentrations down to a reporting limit of 0.01 nM, however such low concentrations are often within the errors of the measurements they are calculated from (iodide and TII concentration) and so may be considered negligible. For mass balance purposes we have left these negligible iodate concentrations in place. Iodate is displayed in Fig. 2 (row a), and as with total iodine, there is a large variation in iodate concentration throughout the expedition. In surface snow, iodate ranged from <0.01 to 203 nM (mean 7.5 nM), while for bulk snow, the iodate concentration ranged from <0.01 to 509 nM (mean 52 nM). The higher concentrations of total iodine and iodate in bulk snow is examined in the following sections. Bulk iodate was also reported by Frassati et al., (2024) at low concentrations of 0.8–1 nM in the lower layers of continental Arctic snow (February 2022).55
The iodide:iodate ratio in snow samples was analysed (Fig. 2, row c), and it was determined that there was not a consistent ratio in either bulk or surface snow throughout the year. This indicated that there was not a dominant form of inorganic iodine in Arctic snow. In surface snow, the iodide:iodate ratio peaked in spring and autumn, while this trend was not evident for bulk snow. The seasonal trends in iodide:iodate ratio are discussed in section “Potential top-down inputs of iodine to the snow-pack”.
The Na+ and Cl− analyses of the snow allow us to examine potential seawater sources of iodine. Measured in ppbw (equivalent to ng g−1), concentrations were highly variable; between 201 and 6 × 106 ppbw for Na+ and between 388 and 6 × 106 ppbw for Cl− (Fig. 2, row d). However, bulk snow had more instances of very high Na+ and Cl− compared to the surface. Na+ and Cl− were reported by Raso et al., (2017)44 and Na+ by Spolaor et al., (2019);54 in both studies, analysing continental snow, the salt content was at the lower end of the range reported in this work (Fig. 2), attributed to the fact that the MOSAiC samples were collected over sea ice. This indicates the upward transport of sea salt from the saline sea ice surface or the sea water in the current work.
The MOSAiC samples were analysed for calcium ions (Ca2+), displayed in Fig. 2 (row d). Ca2+ is often considered to be the best tracer of continental dust among the major ionic species observed in Arctic snow.67,68 Concentrations were typically below 1000 ppbw, but with episodically high values throughout the snow pit (up to 1.03 × 106 ppbw), which are discussed in the coming sections.
For insight into the source of iodine into the snow, the enrichment factor (EF) of total iodine, iodide and iodate relative to seawater were calculated according to eqn (1), with a seawater Na+ concentration of 0.46 M and the mean (±1 sd) measured under-ice seawater concentrations (see Methods) of total iodine (262 ± 87 nM), iodide (27 ± 11 nM) and iodate (215 ± 76 nM). For calculation of EFs, depth-paired measurements of Na+ and the ion in question must be available. This was not always the case; therefore the EF could not be calculated for every snow sample.
 | (1) |
In the bulk snow pit, total iodine was typically not strongly enriched, except for one sample; EFs ranged from 0.02 to 1475 (the second highest EF was 10), resulting in mean and median EFs of 72 and 3, respectively. Iodide EFs demonstrated a similar trend, whereby EFs were generally low, with episodically high values. Iodide EFs ranged from 0.01 to 1583 (mean = 134, median = 0.9). In the Raso et al., (2017) study,44 iodide EFs were higher in the bulk snow, ranging ∼400–1900; the relatively low sodium content of the bulk continental snow, coupled with the relatively high iodide concentrations leads to these high EFs.
In the current work, two bulk snow samples were highly enriched in iodide and/or total iodine compared to the other samples, occurring on 2nd March 2020 ([iodide] = 3.7 nM) and 23rd April 2020 ([iodide] = 0.8 nM). Both samples were associated with particularly low sodium concentrations: 1540 and 201 ppbw, respectively. In the bulk of the snow pit, iodate enrichment ranged from 0.008 to 585 (mean 26, n = 25). To our knowledge, there are no previously reported iodate bulk EFs from Arctic snow to compare with.
Investigating a bottom-up source of iodine to the snowpack
 | ||
| Fig. 3 Iodide and iodate concentrations, separated into surface (<5 cm from the snow–air interface) and base (<5 cm from the snow–ice interface) values, and the age of the underlying ice (first year ice, FYI, and second year ice, SYI). The boxplots display the median value, and the first and third quantiles. Whiskers extend to the largest and smallest values within 1.5× the interquartile range. Measurements outside this range are displayed by points. Not all measurements displayed in Fig. 2 are visible in this plot, as categorisations of the age of the underlying ice were not always available. | ||
There is a more pronounced difference between FYI and SYI for iodate than iodide. The reason for this difference isn't clear, but some possibilities are specificity in ion exclusion during ice formation (Zhang 2021),73 or higher reactivity of iodide compared to iodate causing a reduction in the concentration. Iodate is generally considered a stable form of iodine, therefore less likely to undergo chemical reaction.
The permeability of FYI compared to SYI is well established, and depends on the rule of fives, which describes how at a brine volume fraction (BVF) of 5%, brine inclusions interconnect, and the ice becomes permeable. This depends on temperature and salinity, and at a bulk salinity of 5 ppt and a temperature of −5 °C, the critical BVF of 5% is reached.58 This permeability difference was demonstrated during the MOSAiC campaign where FYI was shown to reach or exceed the critical 5% BVF throughout, whereas SYI did not.69 Permeation has been suggested as a mechanism for the leaching of iodine-containing salts from seawater upwards through thinner Antarctic ice,38 which typically exists at around 50 cm. During the MOSAiC expedition, the ice thickness of FYI was initially recorded at approximately 30 cm in December, growing to a maximum thickness of up to 2 m in May, and decreasing again through late summer.70 At the time of polar sunrise, when we would expect photolytically driven and biological processes to begin, the ice was present at between approximately 1.25 to 1.70 m thickness. The timescale of liquid diffusion through ice at these thicknesses was approximated following the method described in Saiz-Lopez et al., (2015),38 demonstrating that for ice at the onset of the polar sunrise, for even the fastest potential rate of liquid diffusion, permeation of salts from seawater all the way through ice is not a viable method for transfer of iodine compounds from seawater to the base of the snowpack in the Arctic (Table 2).
| Ice thickness (cm) | Time for diffusion for diffusion coefficients (D, in cm2 s−1) | ||
|---|---|---|---|
| D = 1 × 10−7 | D = 5 × 10−5 | D = 1.3 × 10−4 | |
| 30 | 71 years | 52 days | 20 days |
| 125 | 1240 years | 2.5 years | 350 days |
| 170 | 2300 years | 4.6 years | 1.8 years |
| 200 | 3200 years | 6.3 years | 2.4 years |
The high concentrations of iodate, and to an extent iodide, at the base of the snowpack were therefore attributed to brine formed during ice formation. This brine could be present in brine channels formed in the interior ice (which would therefore not need to permeate up through the whole ice depth), or as a brine skim atop the ice. This brine skim could be expelled upwards during ice formation, or could have been left on top of pancake ice, as a result of wave action.71 The presence of brine in the snowpack is indicated by the elevated concentration of Na+ at the base of FYI, compared to SYI (median 3.3 × 106 compared to 3.1 × 104 ppbw Na+, respectively; a difference of two orders of magnitude). In laboratory and field experiments, it was demonstrated that a dye-spiked brine wicked upwards to a distance of up to 5 cm in snow,72 providing a potential method of transport of seawater salts into snow near the snow–ice interface when snow has sufficiently connected porosity.
The highest concentrations of iodate in the base snow were observed on FYI during the ice growth phase (December until May), supporting brine expulsion during growth. No high iodate concentrations in base snow were observed for the rest of the MOSAiC year, despite there being a higher occurrence of FYI sampled in the later stages of the campaign. A potential reason for this is the flushing of non-saline meltwater through the snow. If surface snow (which was lower in iodate) melted first, this could wash away salts deeper in the snowpack, reducing their concentration. Replenishment of iodate from the seawater would then not be possible, even for sufficiently thin ice, for two reasons: (1) if non-saline meltwater was transported downward through the ice, the salinity of the brine channels would be reduced, and the law of fives would not be satisfied. This would hinder future upward transport of seawater salts, including iodate, through the sea ice, and (2) downwards transport of meltwater through ice and into the underlying seawater could also cause stratification of the seawater below the ice, with a layer of iodate-free fresh water in contact with the bottom of the ice. It is also noteworthy that during the summer it is possible that snow samples were composed of, or included, the melting ice surface. In this case, the concentrations of iodine species reported for summer samples would represent, or contain, the bulk ice (with bulk in this instance meaning both ice grains and channels).
While we do not expect iodate originating from ice growth processes to directly affect snow over SYI, there is a non-zero concentration of iodate in snow over SYI (Fig. 3). In this work, we cannot determine its origin with certainty, however a potential source could be mixing from snow with a higher iodate concentration (whether the source is oceanic or atmospheric), which occurs within and across the snowpack due to erosion and deposition.66
We have indicated that there does not appear to be a strong sea-ice source of salts to the base of the snowpack above SYI, but shown that elevated levels of iodate, and to a lesser extent iodide, are present in the base snow above FYI. We next examine whether the iodine levels in this base snowpack were enriched in concentration with respect to seawater. The total iodine, iodide and iodate EF profiles through the snow above FYI are displayed in Fig. 4. Total iodine and iodate appeared to be slightly enriched at the base of the snowpack (up to EF = 6.6), increasing further towards the snow–air interface.
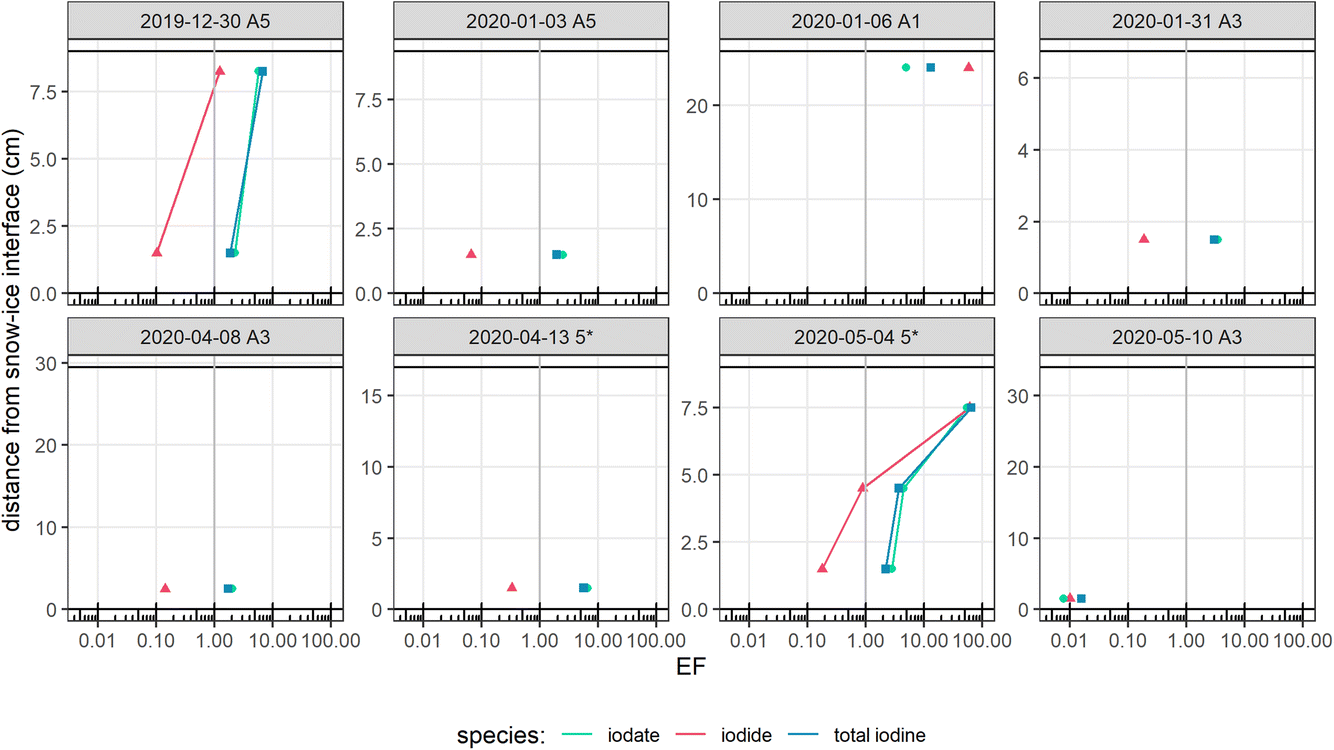 | ||
| Fig. 4 Enrichment factors (EF) for total iodine, iodide and iodate throughout the snowpack, for snow over FYI, with respect to sea water. Each panel represents samples taken from an individual snow pit. The height of the snow pit is indicated by the upper black horizontal lines, with the lower line at the snow–ice interface (h = 0 cm) and the upper line at the snow–air interface (height variable). EFs are displayed on a log scale for total iodine (blue squares), iodate (green circles) and iodide (pink triangles), with a vertical line at EF = 1 as a visual aid. Note the depth scales are variable across snow pits. | ||
For iodide at the base of the snowpack, the EF was consistently below 1, indicating that iodide is depleted relative to seawater. There is therefore no evidence of any significant additional source between the bulk seawater and the base of the snowpack during the whole cruise track. Therefore, the widely used mechanism of a biological source of iodine to the atmosphere appears not to apply here. In this mechanism, phytoplankton on the underside of sea ice produce large quantities of iodine, in equilibrium between I2 and I− + HOI, which are transported upward through the sea ice to the ice–air or ice–snow interface.38 If iodine species were highly enriched under the sea ice and transported upwards through the ice, enriched iodine species at the ice–snow interface would be expected. Instead, the EF of iodide, as for total iodine and iodate, is low towards the ice–snow interface, and appears to increase with proximity to the snow–air interface, indicating that the transfer of iodide inside the snowpack is top-down. We note that the opposite trend was observed in continental snow, in which iodide EFs increased with depth.44 This may be due to the formation of the snowpack in very different environments, however the specific reason for this discrepancy is unclear.
Deviation from an EF of 1 could be due to several factors. There could be ion-specificity in brine rejection; a study of brine rejection of chloride, bromide and fluoride found that fluoride was more efficiently rejected than bromide and chloride, indicating that different halogen species behave differently during ice formation.73 Alternatively, chemical reactions, e.g. between iodide and nitrite during the freeze–thaw process,52 could cause the observed depletion of iodide relative to seawater.
In summary, there appears to be a source of iodine to the base of the snowpack over FYI but not SYI, which is higher in winter and spring during the ice growth phase. This is possibly linked to the presence of brine expelled to the top of the ice, and is slightly enriched in iodate but not in iodide, which was actually depleted compared to seawater.
Potential top-down inputs of iodine to the snowpack
000 ppbw Ca2+), there is not a strong correlation between iodide or iodate with Ca2+, therefore this relationship is only a good predictor when the Ca2+ load is high. Mineral dust as a source of iodate has been identified in several studies, including the AMT21 cruise between the UK and Chile,75 where Saharan dust was associated with the highest iodate measurements during the campaign. Similarly, the highest TII concentrations observed in aerosols during Meteor cruise 55 were observed just off the West coast of Africa, which was subject to the highest levels of input from dust and other aerosol sources.76 Iodide was less strongly correlated to Ca2+, indicating that an additional source other than dust could also be responsible for elevated iodide at the snow surface.
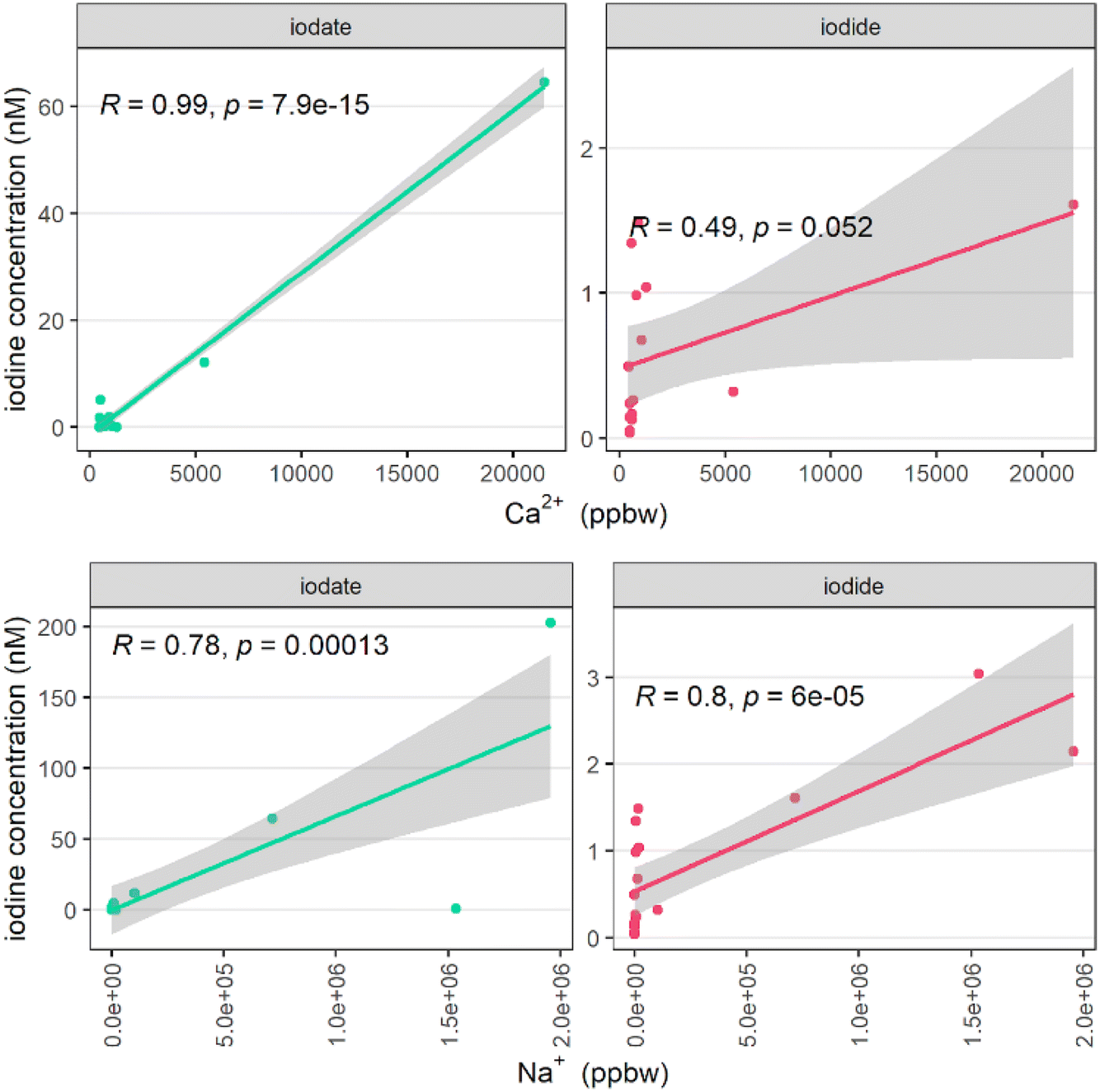 | ||
| Fig. 5 Correlation between iodide and iodate with sodium and calcium (in part per billion by weight, ppbw) in surface snow samples. | ||
For our samples, there was one incident of high dust input, marked by “visible dust” on the snow surface (sampling event PS122_3_33_66, on the 25th March 2020). Samples collected during this event had iodide and iodate concentrations of 65.4 and 797 nM respectively at snow between 5 and 7.5 cm above the snow–ice interface, and 15.6 and 245 nM, respectively, from 7.5 cm up to the snow surface. These levels can be compared with mean surface concentrations of 0.76 ± 0.77 nM iodide and 7.4 ± 30 nM iodate observed in the remaining surface snow samples. Ca2+ was not measured at these particular snow depths, however at 1–4 cm above the snow–ice interface, the calcium concentration was 1.03 × 106 ppbw, the highest measured during the campaign (the mean and standard deviation for the reported snow samples, excluding this sample, was 2685 ± 9457 ppbw). Due to the mismatch in sampling depths, these could not be considered a true matched pair, therefore were not included in Fig. 5, however, they add further evidence to support iodine input from dust to the Arctic snow. Iodate and fine dust load were closely linked in Talos Dome Antarctic ice core samples,77 consistent with the current work.
To investigate a potential sea-salt aerosol (SSA) source of iodine, Na+ was used as a tracer. Both iodide and iodate were correlated with elevated Na+ concentrations, indicating some influence of SSA, with both correlations satisfying statistical significance (p < 0.05 in both cases, Fig. 5). There were however instances of elevated iodide (up to 1.5 nM) without evidence of significant dust or SSA input (i.e. with sodium and calcium concentrations close to zero), the source of which is presumably due to non-SSA input.
In the Arctic, while there are fewer studies available, marine aerosols are also enriched in iodide relative to seawater. In the Canadian Arctic, EFs ranged between 100 and 10000, with a minimum EF in winter, and a peak EF in summer and early autumn.10 A further Arctic study obtained EFs of total iodine in aerosol (relative to Br) of 2676 ± 2180, representing an extreme enrichment of iodine relative to seawater.79 In a cruise between Shanghai and the Arctic ocean, aerosol iodine was again consistently enriched, ranging from EF = 144 to 20675 across the whole cruise track.80 The extreme enrichment of iodine in marine aerosols can be explained by the uptake of gas-phase iodine to existing aerosol.28,81 This is part of the halogen recycling mechanism described by R1–R7.
In the MOSAiC snow samples, the enrichment of iodide in the surface snowpack showed some seasonality, with an increase in the EF through late summer (July to September, Fig. 2). During late summer, the surface snow is largely composed of the surface scattering layer, which consists of partially melted sea ice. The changing composition of the sample substrate from snow to partially melted sea ice could be a potential reason for the change in EF (however iodine speciation in ice was not measured during MOSAiC to confirm this). Another potential reason for the change in EF is changing air masses affecting atmospheric deposition. The back trajectories of the air masses impacting the sampling were compiled to investigate the source air for deposition (Fig. 6). These back-trajectories were calculated for air arriving at the location of the RV Polarstern, at a maximum altitude of 100 m, over the last 7 days. Throughout late summer, it is seen that the air masses had quite different influences, compared to earlier in the year. During July and August, the air masses arriving at the MOSAiC drift track spent a higher percentage of time over the ocean and mixed ice/ocean, compared to earlier in the year. It was previously reported that Antarctic aerosols which had greater exposure to the ice-front in the previous 48 hours had higher concentrations of total dissolved iodine.82 The increasing EF of iodide in snow during this period could therefore reflect an increased EF of iodide in deposited aerosol during the same period.
 | ||
| Fig. 6 Back trajectories for air parcels arriving at the MOSAiC cruise track, covering 7 days and 100 m in altitude. Calculations not performed for periods where the RV Polarstern was in transit. | ||
Iodine speciation in surface snow
The molar ratio of iodide:iodate at the snow surface, shown in Fig. 2 (row c), displayed seasonal trends. During the polar night there were approximately equal instances of iodide and iodate being the dominant form of iodine in the surface snow, presumably reflecting chemical differences in the source of iodine to the snow (potentially from deposited aerosols). There was a sharp decline in the iodide:iodate ratio in March, coincident with the polar sunrise, after which it decreased to values close to the ratio measured for seawater samples collected during MOSAiC (seawater iodide:iodate molar ratio of 0.13). Another study reported concentrations of iodide and iodate in snow collected in Svalbard just after polar sunrise, from which a molar ratio of 0.6 was calculated, however these concentrations were from snow at a depth greater than 42 cm from the snow–air interface.55
The large variation in iodide:iodate molar ratio during the polar night was investigated, assuming the main source of iodine to the surface snow was aerosol deposition. Where speciation of iodine in Arctic aerosols has been reported, there is a similarly large range in ratios. On a cruise between 65 to 85 N, between 31st July and 5th September 2008, an aerosol iodate to iodide ratio of 6.6 ± 4.0 (equating to an iodide to iodate ratio of 0.15) was obtained.80 Conversely, during a cruise in the Arctic ocean between July–September 2003, the mean concentrations of iodide and iodate in aerosol respectively were 13.8 ± 12.7 pmol m−3 and 0.04 ± 0.07 pmol m−3, giving an average iodide:iodate ratio of 345.79 A further study demonstrated that in the ocean between Australia and the Antarctic, and on the Antarctic coast, iodide was present in a higher abundance than iodate in aerosols (mean iodide:iodate ratio of 4.5), however, both were lower in abundance than organic iodine.82 The large variation in iodine speciation in Arctic aerosols was previously attributed to differences in sea-ice extent between sampling campaigns,79 however large variations in iodide:iodate ratios were also observed within campaigns; iodide:iodate ratios in one Arctic expedition ranged from 0.08 to 5.80 The differences in the dominant iodine species between and within campaigns demonstrates the complex cycle of iodine cycling within the Arctic atmosphere and cryosphere. Variation in iodide:iodate ratios in aerosol could contribute to the large variety of iodide:iodate ratios observed in surface snow during polar night during the MOSAiC cruise.
Under the influence of light, the sharp decrease in the iodide:iodate ratio could be caused by photochemical oxidation of iodide in the snow and volatilisation of the products. This has been demonstrated in the lab,46 and in the field.44 Our results indicate that iodide is more readily photo-liberated than iodate. As light begins to decrease from July onwards, there are once more instances of high iodide:iodate ratios. The trend in iodide:iodate ratios could indicate a steady state during early summer whereby iodine (in particular, iodide) is photochemically liberated from snow, accumulated in particles and re-deposited. As light decreases in late summer, less photo-liberation occurs, while deposition continues, causing an increase in iodide and the iodide:iodate ratio. This hypothesis is consistent with the increase in iodide EFs observed during late summer (Fig. 2 row e). Photo-liberation during the day and accumulation in aerosols would result in higher aerosol iodine concentrations during the day and higher snow iodine during the night. We note that observations in the marine atmosphere at Mace Head, Ireland showed significantly higher concentrations of aerosol iodide during the day compared to night-time.83 In snow, total iodine peaked at night-time, and was at its lowest in sunlit hours, again consistent with photo-chemical liberation.54
Atmospheric implications
Having provided evidence that suggests that photo-chemically driven liberation of iodine from the surface snowpack takes place during Arctic summer, we next attempt to broadly estimate the potential magnitude of this emission from the snowpack to the atmosphere. Due to the minimum in the iodide concentration during the sunlit period, as well as the reduction in the iodide:iodate ratio coinciding with polar sunrise, we assume that iodide is photochemically converted to I2, as discussed in the introduction, and that this takes place in the top 5 cm of the snowpack; this corresponds to the depth over which incident UV-vis light is attenuated to 1/e of its incident irradiance in Arctic surface snow.84
In the work of Raso et al., (2017),44 the maximum interstitial air concentration of I2 was calculated from eqn (2). The iodide concentrations used were average concentrations from samples within 5 cm of the snow–air boundary. The volume of liquid was obtained from the snow water equivalent (SWE) of each sample,57 with a 9% assumed freezing expansion applied, and the air volume was 1 − Vliq. The molar volume of air (mvair) was 20.8 L mol−1 (for air at −20 °C), chosen to reflect the conditions of the Raso et al., (2017) experiment.44
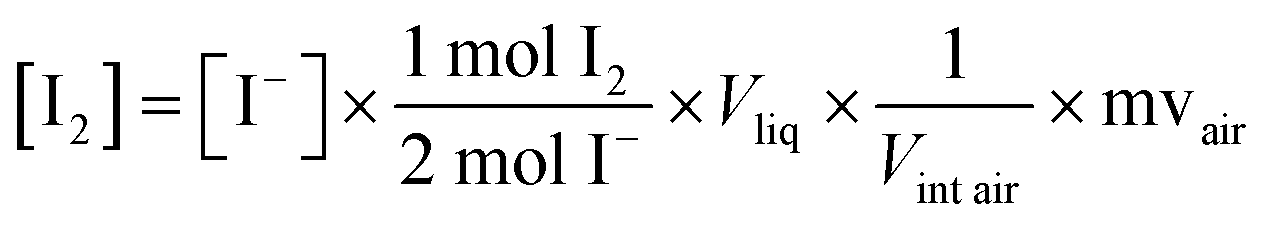 | (2) |
000 pptv were calculated. To account for incomplete conversion of iodide to I2, a scaling factor was calculated based on the work of Raso et al., (2017).44 Using their reported iodide concentration of 2 nM and eqn (2), we obtain a mixing ratio of ∼14000 pptv I2 for complete conversion to I2. Their maximum measured interstitial mixing ratio of 22 pptv I2 results in a scaling factor of 22 pptv/14000 pptv to account for incomplete conversion. The resulting interstitial I2 mixing ratios obtained in this work were between ∼0.09 and 50 pptv (Fig. 7a), reflecting the range in iodide concentrations in different snow samples.
 | ||
| Fig. 7 (a) Interstitial I2 mixing ratios (in parts per trillion by volume, ppbv) predicted from photolysis of iodide, scaled according to interstitial I2 measurements in Raso (2017).44 (b) I2 emission rates, resulting from the calculated interstitial concentrations. | ||
These interstitial air concentrations were converted to an emission of I2 (FI2) from the snowpack following the methodology of Michalowski (2000),85 shown in eqn (3), where the transfer coefficient kt = 1.25 × 10−5 s−1, Vpbl/Vint air is ratio of the volume of the planetary boundary layer to the interstitial air for a unit area, Dsp is the depth of the snow I2 is being produced in, and [I2] is the interstitial I2 concentration. The volume of the boundary layer is calculated from the daily mean planetary boundary layer height (PBLH) from a single-column atmospheric chemistry and meteorology model.86
 | (3) |
Conclusions
In summary, we find that photolysis of iodide in surface snow has the potential to contribute significant emissions of reactive volatile iodine to the Arctic atmosphere. Our data do not support efficient transport of iodine from the underlying sea-ice to the snow surface nor any significant enrichment of iodine at the base of the snow. Thus, it appears that a seawater source of iodine is unlikely to be transferred to the atmosphere in instances where there is ice and/or snow cover. Under-ice biological enrichment of iodine and transport through sea-ice brine has been widely applied as an Antarctic atmospheric iodine source,38 but our results show this is likely not applicable in the Arctic, even over first year ice. Instead, the origin of the iodine in surface snow appears to be linked to the salinity of the snow and the prevalence of air mass origins from the ocean, suggesting a source from iodine-enriched marine aerosol. Tentatively, we also find evidence that dust deposition to snow provides an iodate source, in agreement with previous findings.77 In future field work, it would be valuable to extend the current knowledge base by obtaining concurrent measurements of IO and I2 in the polar atmosphere alongside snow depth profiles of the same measurements. When coupled with complementary measurements including iodide, iodate, light levels and interstitial air volumes, a more sophisticated model of reactive iodine production in the snowpack could be developed. This would reduce uncertainty in I2 emission rates, shed light on the fundamental mechanisms of iodine snow photochemistry, and allow more accurate modelling of atmospheric IO and subsequent impacts on the depletion of tropospheric ozone due to halogen chemistry. Concurrent measurements in both snow and atmospheric aerosol of speciated iodine along with the major ions are also required to confirm the sources of iodine to the polar snow surface.Data availability
The iodine speciation data supporting this research are available for download from the research data repository of the University of York at https://doi.org/10.15124/35dfdf15-42f3-454b-9e61-1d05a6a01e34.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LVB, RJP, MRJ, RC and LJC were supported by the European Research Council (ERC) under the European Union's Horizon 2020 programme (grant agreement no. 833290). This sampling was carried out as part of the international Multidisciplinary drifting Observatory for the Study of the Arctic Climate (MOSAiC) with the tag MOSAiC20192020. We thank all persons involved in the expedition of the research vessel Polarstern during MOSAiC in 2019–2020 (AWI_PS122_00) as listed in Nixdorf et al. (2021).88 We are grateful to Hélène Angot for supporting the field work. Snow sampling on MOSAiC was possible due to the funding received by: the Swiss Polar Institute (SPI reference DIRCR-2018-003) Funder ID: https://doi.org/10.13039/501100015594, European Union's Horizon 2020 research and innovation program projects ARICE (grant 730965) for berth fees associated with the participation of the DEARice project, WSL Institute for Snow and Avalanche Research SLF, WSL 201812N1678, Funder ID: https://doi.org/10.13039/501100015742. ARM received funding from the Swiss National Science Foundation (SNF) project number P500PN_217845. MMF was supported by the UK Natural Environment Research Council (NERC) (NE/S00257X/1), the NERC National Capability International grant SURface FluxEs In AnTarctica (SURFEIT) (NE/X009319/1), and the European Union's Horizon 2020 research and innovation programme under grant agreement no. 101003826 via project CRiceS (Climate Relevant interactions and feedbacks: the key role of sea ice and Snow in the polar and global climate system). SM was supported by core funding from the UK Natural Environment Research Council (NERC) to the British Antarctic Survey's Ice Dynamics and Paleoclimate Program. Funding to support participation in MOSAiC for BWB, SDA, DH, HWJ, JGMB and LNG was supported by the U.S. National Science Foundation, Office of Polar Programs: OPP-1807163, OPP-1807496 and OPP-1914781. SA was supported by the Alfred-Wegener-Institut, Helmholtz-Zentrum für Polar- und Meeresforschung, the University of Hamburg, the German Research Foundation’s (DFG) projects fAntasie (AR1236/3-1) and SnowCast (AR1236/1-1) within its priority program “Antarctic Research with comparative investigations in the Arctic ice areas” (SPP1158), and the DFG Emmy Noether Programme project SNOWflAke (project number 493362232).References
- W. R. Simpson, R. Von Glasow, K. Riedel, P. Anderson, P. Ariya, J. Bottenheim, J. Burrows, L. J. Carpenter, U. Frieß, M. E. Goodsite, D. Heard, M. Hutterli, H.-W. Jacobi, L. Kaleschke, B. Neff, J. Plane, U. Platt, A. Richter, H. Roscoe, R. Sander, P. Shepson, J. Sodeau, A. Steffen, T. Wagner and E. Wolff, Halogens and their role in polar boundary-layer ozone depletion, Atmos. Chem. Phys., 2007, 7, 4375–4418 CrossRef CAS.
- H. Angot, B. Blomquist, D. Howard, S. Archer, L. Bariteau, I. Beck, M. Boyer, M. Crotwell, D. Helmig, J. Hueber, H. W. Jacobi, T. Jokinen, M. Kulmala, X. Lan, T. Laurila, M. Madronich, D. Neff, T. Petäjä, K. Posman, L. Quéléver, M. D. Shupe, I. Vimont and J. Schmale, Year-round trace gas measurements in the central Arctic during the MOSAiC expedition, Sci. Data, 2022, 9, 723 CrossRef CAS PubMed.
- H. W. Jacobi, S. Morin and J. W. Bottenheim, Observation of widespread depletion of ozone in the springtime boundary layer of the central Arctic linked to mesoscale synoptic conditions, J. Geophys. Res.:Atmos., 2010, 115, D17302 CrossRef.
- R. P. Fernandez, L. Berná, O. G. Tomazzeli, A. S. Mahajan, Q. Li, D. E. Kinnison, S. Wang, J. F. Lamarque, S. Tilmes, H. Skov, C. A. Cuevas and A. Saiz-Lopez, Arctic halogens reduce ozone in the northern mid-latitudes, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2401975121 CrossRef CAS PubMed.
- K. Toyota, J. C. McConnell, R. M. Staebler and A. P. Dastoor, Air-snowpack exchange of bromine, ozone and mercury in the springtime Arctic simulated by the 1-D model PHANTAS - Part 1: In-snow bromine activation and its impact on ozone, Atmos. Chem. Phys., 2014, 14, 4101–4133 CrossRef.
- A. Schönhardt, M. Begoin, A. Richter, F. Wittrock, L. Kaleschke, J. C. Gómez Martín and J. P. Burrows, Simultaneous satellite observations of IO and BrO over Antarctica, Atmos. Chem. Phys., 2012, 12, 6565–6580 CrossRef.
- I. Bougoudis, A. M. Blechschmidt, A. Richter, S. Seo and J. P. Burrows, Simulating tropospheric BrO in the Arctic using an artificial neural network, Atmos. Environ., 2022, 276, 119032 CrossRef CAS.
- S. J. Oltmans, Surface ozone measurements in clean air, J. Geophys. Res., 1981, 86, 1174–1180 CrossRef CAS.
- J. W. Bottenheim, A. G. Gallant and K. A. Brice, Measurements of NOY species and O3 at 82° N latitude, Geophys. Res. Lett., 1986, 13, 113–116 CrossRef CAS.
- W. Sturges and L. Barrie, Chlorine, Bromine and Iodine in Arctic Aerosols, Atmos. Environ., 1988, 22, 1179–1194 CrossRef CAS.
- J. C. McConnell, G. S. Hendersont, L. Barrie, J. Bottenheim, H. Niki, C. H. Langfordll and E. M. J. Templeton, Photochemical bromine production implicated in Arctic boundary-layer ozone depletion, Nature, 1992, 355, 150–152 CrossRef CAS.
- G. A. Impey, P. B. Shepson, D. R. Hastie, L. A. Barrie and K. G. Anlauf, J. Geophys. Res.:Atmos., 1997, 102, 16005–16010 CrossRef CAS.
- L. Cao, H. Sihler, U. Platt and E. Gutheil, Numerical analysis of the chemical kinetic mechanisms of ozone depletion and halogen release in the polar troposphere, Atmos. Chem. Phys., 2014, 14, 3771–3787 CrossRef.
- C. R. Thompson, P. B. Shepson, J. Liao, L. G. Huey, C. Cantrell, F. Flocke and J. Orlando, Bromine atom production and chain propagation during springtime Arctic ozone depletion events in Barrow, Alaska, Atmos. Chem. Phys., 2017, 17, 3401–3421 CrossRef CAS.
- M. Hausmann and U. Platt, Spectroscopic measurement of bromine oxide and ozone in the high Arctic during polar sunrise experiment 1992, J. Geophys. Res., 1994, 99, 25399–25413 CrossRef.
- N. Benavent, A. S. Mahajan, Q. Li, C. A. Cuevas, J. Schmale, H. Angot, T. Jokinen, L. L. J. Quéléver, A. M. Blechschmidt, B. Zilker, A. Richter, J. A. Serna, D. Garcia-Nieto, R. P. Fernandez, H. Skov, A. Dumitrascu, P. Simões Pereira, K. Abrahamsson, S. Bucci, M. Duetsch, A. Stohl, I. Beck, T. Laurila, B. Blomquist, D. Howard, S. D. Archer, L. Bariteau, D. Helmig, J. Hueber, H. W. Jacobi, K. Posman, L. Dada, K. R. Daellenbach and A. Saiz-Lopez, Substantial contribution of iodine to Arctic ozone destruction, Nat. Geosci., 2022, 15, 770–773 Search PubMed.
- D. M. Joseph, S. H. Ashworth and J. M. C. Plane, On the photochemistry of IONO2: Absorption cross section (240–370 nm) and photolysis product yields at 248 nm, Phys. Chem. Chem. Phys., 2007, 9, 5599–5607 RSC.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III-gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 2007, 7, 981–1191 Search PubMed.
- A. Saiz-Lopez, J. M. C. Plane, A. S. Mahajan, P. S. Anderson, J.-B. Bauguitte, A. E. Jones, H. K. Roscoe, R. A. Salmon, W. J. Bloss, J. D. Lee and D. E. Heard, On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime, Atmos. Chem. Phys., 2008, 8, 887–900 CrossRef CAS.
- R. W. Saunders and J. M. C. Plane, Fractal growth modelling of I2O5 nanoparticles, J. Aerosol Sci., 2006, 37, 1737–1749 CrossRef CAS.
- O. Gálvez, J. C. Gómez Martín, P. C. Gómez, A. Saiz-Lopez and L. F. Pacios, A theoretical study on the formation of iodine oxide aggregates and monohydrates, Phys. Chem. Chem. Phys., 2013, 15, 15572–15583 RSC.
- A. Baccarini, L. Karlsson, J. Dommen, P. Duplessis, J. Vüllers, I. M. Brooks, A. Saiz-Lopez, M. Salter, M. Tjernström, U. Baltensperger, P. Zieger and J. Schmale, Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions, Nat. Commun., 2020, 11, 4924 CrossRef CAS PubMed.
- M. Sipilä, N. Sarnela, T. Jokinen, H. Henschel, H. Junninen, J. Kontkanen, S. Richters, J. Kangasluoma, A. Franchin, O. Peräkylä, M. P. Rissanen, M. Ehn, H. Vehkamäki, T. Kurten, T. Berndt, T. Petäjä, D. Worsnop, D. Ceburnis, V. M. Kerminen, M. Kulmala and C. O'Dowd, Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 2016, 537, 532–534 CrossRef PubMed.
- X.-C. He, Y. J. Tham, L. Dada, M. Wang, H. Finkenzeller, D. Stolzenburg, S. Iyer, M. Simon, A. Kürten, J. Shen, B. Rörup, M. Rissanen, S. Schobesberger, R. Baalbaki, D. S. Wang, T. K. Koenig, T. Jokinen, N. Sarnela, L. J. Beck, J. Almeida, S. Amanatidis, A. Amorim, F. Ataei, A. Baccarini, B. Bertozzi, F. Bianchi, S. Brilke, L. Caudillo, D. Chen, R. Chiu, B. Chu, A. Dias, A. Ding, J. Dommen, J. Duplissy, I. El Haddad, L. Gonzalez Carracedo, M. Granzin, A. Hansel, M. Heinritzi, V. Hofbauer, H. Junninen, J. Kangasluoma, D. Kemppainen, C. Kim, W. Kong, J. E. Krechmer, A. Kvashin, T. Laitinen, H. Lamkaddam, C. P. Lee, K. Lehtipalo, M. Leiminger, Z. Li, V. Makhmutov, H. E. Manninen, G. Marie, R. Marten, S. Mathot, R. L. Mauldin, B. Mentler, O. Möhler, T. Müller, W. Nie, A. Onnela, T. Petäjä, J. Pfeifer, M. Philippov, A. Ranjithkumar, A. Saiz-Lopez, I. Salma, W. Scholz, S. Schuchmann, B. Schulze, G. Steiner, Y. Stozhkov, C. Tauber, A. Tomé, R. C. Thakur, O. Väisänen, M. Vazquez-Pufleau, A. C. Wagner, Y. Wang, S. K. Weber, P. M. Winkler, Y. Wu, M. Xiao, C. Yan, Q. Ye, A. Ylisirniö, M. Zauner-Wieczorek, Q. Zha, P. Zhou, R. C. Flagan, J. Curtius, U. Baltensperger, M. Kulmala, V.-M. Kerminen, T. Kurtén, N. M. Donahue, R. Volkamer, J. Kirkby, D. R. Worsnop and M. Sipilä, Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 2021, 371, 589–595 CrossRef CAS PubMed.
- H. Finkenzeller, S. Iyer, X. C. He, M. Simon, T. K. Koenig, C. F. Lee, R. Valiev, V. Hofbauer, A. Amorim, R. Baalbaki, A. Baccarini, L. Beck, D. M. Bell, L. Caudillo, D. Chen, R. Chiu, B. Chu, L. Dada, J. Duplissy, M. Heinritzi, D. Kemppainen, C. Kim, J. Krechmer, A. Kürten, A. Kvashnin, H. Lamkaddam, C. P. Lee, K. Lehtipalo, Z. Li, V. Makhmutov, H. E. Manninen, G. Marie, R. Marten, R. L. Mauldin, B. Mentler, T. Müller, T. Petäjä, M. Philippov, A. Ranjithkumar, B. Rörup, J. Shen, D. Stolzenburg, C. Tauber, Y. J. Tham, A. Tomé, M. Vazquez-Pufleau, A. C. Wagner, D. S. Wang, M. Wang, Y. Wang, S. K. Weber, W. Nie, Y. Wu, M. Xiao, Q. Ye, M. Zauner-Wieczorek, A. Hansel, U. Baltensperger, J. Brioude, J. Curtius, N. M. Donahue, I. El Haddad, R. C. Flagan, M. Kulmala, J. Kirkby, M. Sipilä, D. R. Worsnop, T. Kurten, M. Rissanen and R. Volkamer, The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 2023, 15, 129–135 CrossRef CAS PubMed.
- H. Yu, L. Ren, X. Huang, M. Xie, J. He and H. Xiao, Iodine speciation and size distribution in ambient aerosols at a coastal new particle formation hotspot in China, Atmos. Chem. Phys., 2019, 19, 4025–4039 CrossRef CAS.
- T. Jokinen, M. Sipilä, J. Kontkanen, V. Vakkari, P. Tisler, E.-M. Duplissy, H. Junninen, J. Kangasluoma, H. E. Manninen, T. Petäjä, M. Kulmala, D. R. Worsnop, J. Kirkby, A. Virkkula and V.-M. Kerminen, Ion-induced sulfuric acid-ammonia nucleation drives particle formation in coastal Antarctica, Sci. Adv., 2018, 4, eaat9744 CrossRef CAS PubMed.
- A. Saiz-Lopez and R. Von Glasow, Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 2012, 41, 6448–6472 RSC.
- J. D. Allan, P. I. Williams, J. Najera, J. D. Whitehead, M. J. Flynn, J. W. Taylor, D. Liu, E. Darbyshire, L. J. Carpenter, R. Chance, S. J. Andrews, S. C. Hackenberg and G. McFiggans, Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA, Atmos. Chem. Phys., 2015, 15, 5599–5609 CrossRef CAS.
- R. W. Saunders, R. Kumar, J. C. Gomez Martin, A. S. Mahajan, B. J. Murray and J. M. C. Plane, Studies of the Formation and Growth of Aerosol from Molecular Iodine Precursor, Z. Phys. Chem., 2010, 224, 1095–1117 CrossRef CAS.
- F. Wittrock, R. Müller, A. Richter, H. Bovensmann and J. P. Burrows, Measurements of iodine monoxide (IO) above Spitsbergen, Geophys. Res. Lett., 2000, 27, 1471–1474 CrossRef CAS.
- U. Frieß, T. Wagner, I. Pundt, K. Pfeilsticker and U. Platt, Spectroscopic measurements of tropospheric iodine oxide at Neumayer station, Antarctica, Geophys. Res. Lett., 2001, 28, 1941–1944 CrossRef.
- A. Saiz-Lopez, A. S. Mahajan, R. A. Salmon, S. J. B. Bauguitte, A. E. Jones, H. K. Roscoe and J. M. C. Plane, Boundary layer halogens in coastal Antarctica, Science, 2007, 317, 348–351 CrossRef CAS PubMed.
- A. Saiz-Lopez, K. Chance, X. Liu, T. P. Kurosu and S. P. Sander, First observations of iodine oxide from space, Geophys. Res. Lett., 2007, 34, L12812 CrossRef.
- H. M. Atkinson, R. J. Huang, R. Chance, H. K. Roscoe, C. Hughes, B. Davison, A. Schönhardt, A. S. Mahajan, A. Saiz-Lopez, T. Hoffmann and P. S. Liss, Iodine emissions from the sea ice of the Weddell Sea, Atmos. Chem. Phys., 2012, 12, 11229–11244 CrossRef CAS.
- A. S. Mahajan, S. Wagh, R. P. Fernandez, S. Singh, S. Bucci and A. Saiz-Lopez, Differences in iodine chemistry over the Antarctic continent, Polar Sci., 2024, 40, 101014 CrossRef.
- A. S. Mahajan, M. Shaw, H. Oetjen, K. E. Hornsby, L. J. Carpenter, L. Kaleschke, X. Tian-Kunze, J. D. Lee, S. J. Moller, P. Edwards, R. Commane, T. Ingham, D. E. Heard and J. M. C. Plane, Evidence of reactive iodine chemistry in the Arctic boundary layer, J. Geophys. Res.:Atmos., 2010, 115, D20303 Search PubMed.
- A. Saiz-Lopez, C. S. Blaszczak-Boxe and L. J. Carpenter, A mechanism for biologically induced iodine emissions from sea ice, Atmos. Chem. Phys., 2015, 15, 9731–9746 CrossRef CAS.
- R. P. Fernandez, A. Carmona-Balea, C. A. Cuevas, J. A. Barrera, D. E. Kinnison, J. F. Lamarque, C. Blaszczak-Boxe, K. Kim, W. Choi, T. Hay, A. M. Blechschmidt, A. Schönhardt, J. P. Burrows and A. Saiz-Lopez, Modeling the Sources and Chemistry of Polar Tropospheric Halogens (Cl, Br, and I) Using the CAM-Chem Global Chemistry-Climate Model, J. Adv. Model. Earth Syst., 2019, 11, 2259–2289 CrossRef.
- C. A. Cuevas, N. Maffezzoli, J. P. Corella, A. Spolaor, P. Vallelonga, H. A. Kjær, M. Simonsen, M. Winstrup, B. Vinther, C. Horvat, R. P. Fernandez, D. Kinnison, J. F. Lamarque, C. Barbante and A. Saiz-Lopez, Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century, Nat. Commun., 2018, 9, 1452 CrossRef PubMed.
- N. Maffezzoli, B. Risebrobakken, M. W. Miles, P. Vallelonga, S. M. P. Berben, F. Scoto, R. Edwards, H. A. Kjær, H. Sadatzki, A. Saiz-Lopez, C. Turetta, C. Barbante, B. Vinther and A. Spolaor, Sea ice in the northern North Atlantic through the Holocene: Evidence from ice cores and marine sediment records, Quat. Sci. Rev., 2021, 273, 107249 CrossRef.
- J. P. Corella, N. Maffezzoli, A. Spolaor, P. Vallelonga, C. A. Cuevas, F. Scoto, J. Müller, B. Vinther, H. A. Kjær, G. Cozzi, R. Edwards, C. Barbante and A. Saiz-Lopez, Climate changes modulated the history of Arctic iodine during the Last Glacial Cycle, Nat. Commun., 2022, 13, 88 CrossRef CAS PubMed.
- U. Frieß, T. Deutschmann, B. S. Gilfedder, R. Weller and U. Platt, Iodine monoxide in the Antarctic snowpack, Atmos. Chem. Phys., 2010, 10, 2439–2456 CrossRef.
- A. R. W. Raso, K. D. Custard, N. W. May, D. Tanner, M. K. Newburn, L. Walker, R. J. Moore, L. G. Huey, L. Alexander, P. B. Shepson and K. A. Pratt, Active molecular iodine photochemistry in the Arctic, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10053–10058 CrossRef CAS PubMed.
- D. O'Sullivan and J. R. Sodeau, Freeze-induced reactions: Formation of iodine-bromine interhalogen species from aqueous halide ion solutions, J. Phys. Chem. A, 2010, 114, 12208–12215 CrossRef PubMed.
- K. Kim, A. Yabushita, M. Okumura, A. Saiz-Lopez, C. A. Cuevas, C. S. Blaszczak-Boxe, D. W. Min, H.-I. Yoon and W. Choi, Production of Molecular Iodine and Tri-iodide in the Frozen Solution of Iodide: Implication for Polar Atmosphere, Environ. Sci. Technol., 2016, 50, 1280–1287 CrossRef CAS PubMed.
- D. A. Palmer, R. E. Mesmer and R. W. Ramette, Triiodide Ion Formation Equilibrium and Activity Coefficients in Aqueous Solution, J. Solution Chem., 1984, 13, 673–683 CrossRef CAS.
- Ó. Gálvez, M. T. Baeza-Romero, M. Sanz and A. Saiz-Lopez, Photolysis of frozen iodate salts as a source of active iodine in the polar environment, Atmos. Chem. Phys., 2016, 16, 12703–12713 Search PubMed.
- C. A. Cuevas, R. P. Fernandez, D. E. Kinnison, Q. Li, J.-F. Lamarque, T. Trabelsi, J. S. Francisco, S. Solomon and A. Saiz-Lopez, The influence of iodine on the Antarctic stratospheric ozone hole, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2110864119 CrossRef CAS PubMed.
- K. Kim, J. Ju, B. Kim, H. Y. Chung, L. Vetráková, D. Heger, A. Saiz-Lopez, W. Choi and J. Kim, Nitrite-Induced Activation of Iodate into Molecular Iodine in Frozen Solution, Environ. Sci. Technol., 2019, 53, 4892–4900 Search PubMed.
- P. O'Driscoll, K. Lang, N. Minogue and J. Sodeau, Freezing halide ion solutions and the release of interhalogens to the atmosphere, J. Phys. Chem. A, 2006, 110, 4615–4618 CrossRef PubMed.
- P. O'Driscoll, N. Minogue, N. Takenaka and J. Sodeau, Release of nitric oxide and iodine to the atmosphere from the freezing of sea-salt aerosol components, J. Phys. Chem. A, 2008, 112, 1677–1682 CrossRef PubMed.
- L. J. Carpenter, S. M. MacDonald, M. D. Shaw, R. Kumar, R. W. Saunders, R. Parthipan, J. Wilson and J. M. C. Plane, Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 2013, 6, 108–111 CrossRef CAS.
- A. Spolaor, E. Barbaro, D. Cappelletti, C. Turetta, M. Mazzola, F. Giardi, M. P. Björkman, F. Lucchetta, F. Dallo, K. Aspmo Pfaffhuber, H. Angot, A. Dommergue, M. Maturilli, A. Saiz-Lopez, C. Barbante and W. R. L. Cairns, Diurnal cycle of iodine, bromine, and mercury concentrations in Svalbard surface snow, Atmos. Chem. Phys., 2019, 19, 13325–13339 CrossRef CAS.
- S. Frassati, E. Barbaro, C. Rossetti, G. Cozzi, C. Turetta, F. Scoto, M. Roman, M. Feltracco, K. Kim, C. Barbante, A. Gambaro and A. Spolaor, Inorganic iodine and bromine speciation in Arctic snow at picogram-per-grams levels by IC-ICP-MS, J. Anal. Sci. Technol., 2024, 15, 49 CrossRef CAS.
- R. Gelaro, W. McCarty, M. J. Suárez, R. Todling, A. Molod, L. Takacs, C. A. Randles, A. Darmenov, M. G. Bosilovich, R. Reichle, K. Wargan, L. Coy, R. Cullather, C. Draper, S. Akella, V. Buchard, A. Conaty, A. M. da Silva, W. Gu, G. K. Kim, R. Koster, R. Lucchesi, D. Merkova, J. E. Nielsen, G. Partyka, S. Pawson, W. Putman, M. Rienecker, S. D. Schubert, M. Sienkiewicz and B. Zhao, The modern-era retrospective analysis for research and applications, version 2 (MERRA-2), J. Clim., 2017, 30, 5419–5454 CrossRef PubMed.
- A. R. Macfarlane, M. Schneebeli, R. Dadic, A. Tavri, A. Immerz, C. Polashenski, D. Krampe, D. Clemens-Sewall, D. N. Wagner, D. K. Perovich, H. Henna-Reetta, I. A. Raphael, I. Matero, J. Regnery, M. M. Smith, M. Nicolaus, M. Jaggi, M. Oggier, M. A. Webster, M. Lehning, N. Kolabutin, P. Itkin, R. Naderpour, R. Pirazzini, S. Hämmerle, S. Arndt and S. Fons, A Database of Snow on Sea Ice in the Central Arctic Collected during the MOSAiC expedition, Sci. Data, 2023, 10, 398 CrossRef CAS PubMed.
- D. N. Thomas and G. S. Dieckmann, Sea Ice, Wiley-Blackwell, West Sussex, 2ndedn, 2010 Search PubMed.
- M. Nicolaus, D. K. Perovich, G. Spreen, M. A. Granskog, L. von Albedyll, M. Angelopoulos, P. Anhaus, S. Arndt, H. Jakob Belter, V. Bessonov, G. Birnbaum, J. Brauchle, R. Calmer, E. Cardellach, B. Cheng, D. Clemens-Sewall, R. Dadic, E. Damm, G. de Boer, O. Demir, K. Dethloff, D. V. Divine, A. A. Fong, S. Fons, M. M. Frey, N. Fuchs, C. Gabarró, S. Gerland, H. F. Goessling, R. Gradinger, J. Haapala, C. Haas, J. Hamilton, H. R. Hannula, S. Hendricks, A. Herber, C. Heuzé, M. Hoppmann, K. V. Høyland, M. Huntemann, J. K. Hutchings, B. Hwang, P. Itkin, H. W. Jacobi, M. Jaggi, A. Jutila, L. Kaleschke, C. Katlein, N. Kolabutin, D. Krampe, S. S. Kristensen, T. Krumpen, N. Kurtz, A. Lampert, B. A. Lange, R. Lei, B. Light, F. Linhardt, G. E. Liston, B. Loose, A. R. Macfarlane, M. Mahmud, I. O. Matero, S. Maus, A. Morgenstern, R. Naderpour, V. Nandan, A. Niubom, M. Oggier, N. Oppelt, F. Pätzold, C. Perron, T. Petrovsky, R. Pirazzini, C. Polashenski, B. Rabe, I. A. Raphael, J. Regnery, M. Rex, R. Ricker, K. Riemann-Campe, A. Rinke, J. Rohde, E. Salganik, R. K. Scharien, M. Schiller, M. Schneebeli, M. Semmling, E. Shimanchuk, M. D. Shupe, M. M. Smith, V. Smolyanitsky, V. Sokolov, T. Stanton, J. Stroeve, L. Thielke, A. Timofeeva, R. T. Tonboe, A. Tavri, M. Tsamados, D. N. Wagner, D. Watkins, M. Webster and M. Wendisch, Overview of the MOSAiC expedition: Snow and sea ice, Elementa, 2022, 10, 000046 Search PubMed.
- M. R. Jones, R. J. Chance, R. Dadic, H. R. Hannula, R. May, M. Ward and L. J. Carpenter, Environmental iodine speciation quantification in seawater and snow using ion exchange chromatography and UV spectrophotometric detection, Anal. Chim. Acta, 2023, 1239, 340700 CrossRef CAS PubMed.
- M. M. Frey, S. J. Norris, I. M. Brooks, P. S. Anderson, K. Nishimura, X. Yang, A. E. Jones, M. G. Nerentorp Mastromonaco, D. H. Jones and E. W. Wolff, First direct observation of sea salt aerosol production from blowing snow above sea ice, Atmos. Chem. Phys., 2020, 20, 2549–2578 CrossRef CAS.
- I. Pisso, E. Sollum, H. Grythe, N. I. Kristiansen, M. Cassiani, S. Eckhardt, D. Arnold, D. Morton, R. L. Thompson, C. D. Groot Zwaaftink, N. Evangeliou, H. Sodemann, L. Haimberger, S. Henne, D. Brunner, J. F. Burkhart, A. Fouilloux, J. Brioude, A. Philipp, P. Seibert and A. Stohl, The Lagrangian particle dispersion model FLEXPART version 10.4, Geosci. Model Dev., 2019, 12, 4955–4997 CrossRef.
- A. Stohl, M. Hittenberger and G. Wotawa, Validation Of The Lagrangian Particle Dispersion Model Flexpart Against Large-Scale Tracer Experiment Data, Atmos. Environ., 1998, 32, 4245–4264 Search PubMed.
- D. H. P. Vogelezang and A. A. M. Holtslag, Evaluation And Model Impacts Of Alternative Boundary-Layer Height Formulations, Boundary-Layer Meteorol., 1996, 81, 245–269 CrossRef.
- K. A. Emanuel and M. Živković-Rothman, Development and Evaluation of a Convection Scheme for Use in Climate Models, J. Atmos. Sci., 1999, 56, 1766–1782 CrossRef.
- V. Nandan, R. Willatt, R. Mallett, J. Stroeve, T. Geldsetzer, R. Scharien, R. Tonboe, J. Yackel, J. Landy, D. Clemens-Sewall, A. Jutila, D. N. Wagner, D. Krampe, M. Huntemann, M. Mahmud, D. Jensen, T. Newman, S. Hendricks, G. Spreen, A. Macfarlane, M. Schneebeli, J. Mead, R. Ricker, M. Gallagher, C. Duguay, I. Raphael, C. Polashenski, M. Tsamados, I. Matero and M. Hoppmann, Wind redistribution of snow impacts the Ka- and Ku-band radar signatures of Arctic sea ice, Cryosphere, 2023, 17, 2211–2229 CrossRef.
- H. W. Jacobi, D. Voisin, J. L. Jaffrezo, J. Cozic and T. A. Douglas, Chemical composition of the snowpack during the OASIS spring campaign 2009 at Barrow, Alaska, J. Geophys. Res.:Atmos., 2012, 117, D00R13 CrossRef.
- H. W. Jacobi, F. Obleitner, S. Da Costa, P. Ginot, K. Eleftheriadis, W. Aas and M. Zanatta, Deposition of ionic species and black carbon to the Arctic snowpack: Combining snow pit observations with modeling, Atmos. Chem. Phys., 2019, 19, 10361–10377 CrossRef CAS.
- M. Angelopoulos, E. Damm, P. Simões Pereira, K. Abrahamsson, D. Bauch, J. Bowman, G. Castellani, J. Creamean, D. V. Divine, A. Dumitrascu, S. W. Fons, M. A. Granskog, N. Kolabutin, T. Krumpen, C. Marsay, M. Nicolaus, M. Oggier, A. Rinke, T. Sachs, E. Shimanchuk, J. Stefels, M. Stephens, A. Ulfsbo, J. Verdugo, L. Wang, L. Zhan and C. Haas, Deciphering the Properties of Different Arctic Ice Types During the Growth Phase of MOSAiC: Implications for Future Studies on Gas Pathways, Front. Earth Sci., 2022, 10, 864523 CrossRef.
- I. A. Raphael, D. K. Perovich, C. M. Polashenski, D. Clemens-Sewall, P. Itkin, R. Lei, M. Nicolaus, J. Regnery, M. M. Smith, M. Webster and M. Jaggi, Sea ice mass balance during the MOSAiC drift experiment: Results from manual ice and snow thickness gauges, Elementa, 2024, 12, 00040 Search PubMed.
- D. K. Perovich and J. A. Richter-Menge, Surface characteristics of lead ice, J. Geophys. Res., 1994, 99, 16341–16350 CrossRef.
- R. Mallett, V. Nandan, J. Stroeve, R. Willatt, M. Saha, J. Yackel, G. Veysière and J. Wilkinson, Dye tracing of upward brine migration in snow, Ann. Glaciol., 2024, 65, e26 CrossRef.
- S. Zhang, C. Zhang, S. Wu, X. Zhou, Z. He and J. Wang, Ion-Specific Effects on the Growth of Single Ice Crystals, J. Phys. Chem. Lett., 2021, 12, 8726–8731 CrossRef CAS PubMed.
- A. A. Frossard, P. M. Shaw, L. M. Russell, J. H. Kroll, M.R. Canagaratna, D. R. Worsnop, P. K. Quinn and T. S. Bates, Springtime Arctic haze contributions of submicron organic particles from European and Asian combustion sources, J. Geophys. Res.:Atmos., 2011, 116, D05205 CrossRef.
- A. R. Baker and C. Yodle, Measurement report: Indirect evidence for the controlling influence of acidity on the speciation of iodine in Atlantic aerosols, Atmos. Chem. Phys., 2021, 21, 13067–13076 CrossRef CAS.
- A. R. Baker, Inorganic iodine speciation in tropical Atlantic aerosol, Geophys. Res. Lett., 2004, 31, L23S02 Search PubMed.
- A. Spolaor, P. Vallelonga, J. M. C. Plane, N. Kehrwald, J. Gabrieli, C. Varin, C. Turetta, G. Cozzi, R. Kumar, C. Boutron and C. Barbante, Halogen species record Antarctic sea ice extent over glacial-interglacial periods, Atmos. Chem. Phys., 2013, 13, 6623–6635 CrossRef.
- J. C. Gómez Martín, A. Saiz-Lopez, C. A. Cuevas, A. R. Baker and R. P. Fernández, On the Speciation of Iodine in Marine Aerosol, J. Geophys. Res.:Atmos., 2022, 127, e2021JD036081 CrossRef PubMed.
- K. Hui, X. Siqi, Y. Xiawei, L. Bing, L. Wei, Y. Hongxia and X. Zhouqing, Iodine speciation in aerosol particle samples collected over the sea between offshore China and the Arctic Ocean, Adv. Polar Sci., 2015, 26, 215–221 Search PubMed.
- S. Xu, Z. Xie, B. Li, W. Liu, L. Sun, H. Kang, H. Yang and P. Zhang, Iodine speciation in marine aerosols along a 15000-km round-trip cruise path from Shanghai, China, to the Arctic Ocean, Environ. Chem., 2010, 7, 406–412 Search PubMed.
- R. A. Duce, B. J. R. Arimoto, C. K. Unni and P. J. Harder, Atmospheric trace elements at Enewetak Atoll: 1. Concentrations, sources, and temporal variability, J. Geophys. Res.:Oceans, 1983, 88, 5321–5342 CrossRef CAS.
- S. C. Lai, T. Hoffman and Z. Q. Xie, Iodine speciation in marine aerosols along a 30,000 km round-trip cruise path from Shanghai, China to Prydz Bay, Antarctica, Geophys. Res. Lett., 2008, 35, L21803 Search PubMed.
- B. S. Gilfedder, S. C. Lai, M. Petri, H. Biester and T. Hoffmann, Iodine Speciation In Rain, Snow And Aerosols, Atmos. Chem. Phys., 2008, 8, 6069–6084 CrossRef CAS.
- M. D. King and W. R. Simpson, Extinction of UV radiation in Arctic snow at Alert, Canada (82°N), J. Geophys. Res.:Atmos., 2001, 106, 12499–12507 CrossRef CAS.
- B. A. Michalowski, J. S. Francisco, S.-M. Li, L. A. Barrie, J. W. Bottenheim and P. B. Shepson, A computer model study of multiphase chemistry in the Arctic boundary layer during polar sunrise, J. Geophys. Res., 2000, 105, 15131–15145 CrossRef CAS.
- J. G. M. Barten, L. N. Ganzeveld, G. J. Steeneveld, B. W. Blomquist, H. Angot, S. D. Archer, L. Bariteau, I. Beck, M. Boyer, P. von der Gathen, D. Helmig, D. Howard, J. Hueber, H. W. Jacobi, T. Jokinen, T. Laurila, K. M. Posman, L. Quéléver, J. Schmale, M. D. Shupe and M. C. Krol, Low ozone dry deposition rates to sea ice during the MOSAiC field campaign: Implications for the Arctic boundary layer ozone budget, Elementa, 2023, 11, 00086 Search PubMed.
- R. J. Pound, L. V. Brown, M. J. Evans and L. J. Carpenter, An improved estimate of inorganic iodine emissions from the ocean using a coupled surface microlayer box model, Atmos. Chem. Phys., 2024, 24, 9899–9921 CrossRef CAS.
- U. Nixdorf, K. Dethloff, M. Rex, M. Shupe, A. Sommerfeld, D. Perovich, M. Nicolaus, C. Heuzé, B. Rabe, B. Loose, E. Damm, R. Gradinger, A. Fong, W. Maslowski, A. Rinke, R. Kwok, G. Spreen, M. Wendisch, A. Herber, M. Hirsekorn, V. Mohaupt, S. Frickenhaus, A. Immerz, K. Weiss-Tuider, B. König, D. Mengedoht, J. Regnery, P. Gerchow, D. Ransby, T. Krumpen, A. Morgenstern, C. Haas, T. Kanzow, F. R. Rack, V. Saitzev, V. Sokolov, A. Makarov, S. Schwarze, T. Wunderlich, K. Wurr and A. Boetius, MOSAiC Extended Acknowledgement, 2021, DOI:10.5281/zenodo.5179738.
Footnote |
| † Now at: Centre for Ecology and Hydrology, UK. |
| This journal is © The Royal Society of Chemistry 2025 |