
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanoenzymatic hydrolysis of cotton to cellulose nanocrystals†
Sandra
Kaabel
 *ab,
Inge
Schlapp-Hackl
b,
Eero
Kontturi
b and
Mauri A.
Kostiainen
b
*ab,
Inge
Schlapp-Hackl
b,
Eero
Kontturi
b and
Mauri A.
Kostiainen
b
aDepartment of Chemistry and Material Science, Aalto University, 02150 Espoo, Finland. E-mail: sandra.kaabel@aalto.fi
bDepartment of Bioproducts and Biosystems, Aalto University, 02150 Espoo, Finland
First published on 12th November 2024
Abstract
Solid-state mechanoenzymatic approaches for the production of cellulose nanocrystals from cotton were examined using three commercially available cellulase systems from Trichoderma reesei, Aspergillus niger and a Cellic CTec2 cellulase blend. A rapid and sharp drop in the degree of polymerization, together with the proportional increase in cellulose crystallinity and generation of nanoscale particles, indicates that cotton is extensively transformed to cellulose nanocrystals with just 15 minutes of ball milling of cotton in the presence of the cellulase enzymes. Subsequent aging of the solid reaction mixture at 55 °C did not significantly affect the degree of polymerization, but resulted in higher material losses due to the increased production of glucose. These results reveal that the endo-activity of the commercial cellulase preparations on cellulose is particularly efficient in solid reaction mixtures (solid loading 50 wt%), allowing for a rapid acid-free generation of cellulose nanocrystals with as low as 0.085 wt% enzyme loading.
Introduction
Cellulose yields valuable renewable nanostructured materials, cellulose nanofibers (CNFs) and cellulose nanocrystals (CNCs), through mechanical, chemical and/or enzymatic processing.1 Because of the high tensile strength, high aspect ratio and tuneable surface chemistry, cellulose nanomaterials have a strong impact on the properties of composites, films, foams, fibres and emulsions, which has quickly led to an increased demand across industries.2 Food and beverage, textile and personal care industries are, for example, driving the demand due to the wide applicability of nanocelluloses in sustainable packaging solutions.3,4 The nanocellulose market is projected to grow from USD 0.34 billion in 2022 to USD 3.38 billion by 2032 with a forecast compound annual growth rate (CAGR) of 23.9%, based on the market analysis report carried out by Global Market Insights Inc (2023). This increased demand is, however, met with some persistent challenges in nanocellulose production. Mechanical CNF production processes are energy intensive,5 while industrial production of CNCs generally relies on a large amount of hazardous concentrated acids,6,7 which need to be recycled or disposed of.8 Furthermore, acid hydrolysis, particularly with sulphuric acid, can lower the thermostability of the obtained CNCs through chemical modification.8,9 Enzyme-mediated extraction of nanocelluloses, on the other hand, proceeds under milder reaction conditions, retains the native surface chemistry of cellulose and does not generate harmful waste.10,11 Cellulase enzymes, consisting of endoglucanases (EG, EC 3.2.1.4), cellobiohydrolases (CBH, EC 3.2.1.91), and β-glycosidases (BG, EC 3.2.1.21), which synergistically hydrolyse the β-1,4-glucosidic bonds of cellulose, have been applied to facilitate chemical or mechanical processing of cellulose11 or as the main step in nanocellulose production.12–28 For the former, the precise role of enzymes can be difficult to pick apart, for example in a common case when the enzymatic hydrolysis step is carried out on microcrystalline cellulose, which consists of crystalline cellulose with a low degree of polymerization (DP) generally obtained through acid hydrolysis. In such systems, the morphology of the resulting nanomaterials obtained from enzymatic treatment is presumably strongly influenced by the hydrolytic extent of the starting microcrystalline cellulose. Studies reporting enzymatic hydrolysis as the main step for converting high DP cellulosic materials (e.g. wood-sourced cellulose or cotton linters) to nanocrystals, however, are generally carried out in dilute aqueous solutions (1–2 wt% solid loading) and report long reaction times (24–72 h) and modest CNC yields (ca. 10%). Meanwhile, mechanochemical and related solid-state approaches have delivered efficient methods to depolymerize cellulose to glucose and water-soluble oligomers, through enzymatic hydrolysis29–32 or acid hydrolysis,33–42 by grinding solid reactants in the absence of bulk solvents. Such approaches can accelerate the enzymatic and chemical reactions, which take place in the highly concentrated and mechanically agitated solid reaction mixtures.43,44 Mechanochemical methods can be especially valuable for the production and refining of cellulose nanomaterials, by minimizing the waste generated and the energy consumed by heating the aqueous media in dilute reaction mixtures.24,45,46The objective of the present work was to investigate the feasibility of obtaining cellulose nanocrystals from cotton linters by employing mechanoenzymatic approaches in the presence of commercially available enzymes, focusing on the interplay of cellulase enzymes in the highly concentrated reaction settings (50% w/v) under different ball milling and aging conditions. Following the release of glucose and the recovery of hydrolysed cellulose, the analysis of cellulose molecular mass and morphology after milling and during aging provides a systematic picture of the action of three different commercial cellulase mixtures on cotton cellulose, culminating in a rapid low-waste and acid-free method to generate CNCs.
Results and discussion
Cellulose substrates and enzymatic hydrolysis
Raw cotton balls (CB), without pre-treatment or further purification steps, were powdered to <600 μm particles using a Wiley knife mill. Cellulose fibres remained intact upon pre-milling, providing a minimally processed substrate (DPn = 924 ± 19, DPw = 1518 ± 22, Đ = 1.64 ± 0.01) in comparison to commercially available microcrystalline cellulose, which is often used as a starting material for enzymatic hydrolysis,47–50 but represents an acid-processed low DP cellulose (DPn = 102, DPw = 282, Đ = 2.8) containing micron-scale particles (Fig. S1†). To facilitate comparisons with benchmarked CNCs,51,52 powdered Whatman 1 filter paper (FP) was also studied as a substrate here (DPn = 656 ± 8, DPw = 1089 ± 23, Đ = 1.66 ± 0.06), as it is commonly used in lab-scale sulphuric acid and HCl-gas CNC production protocols. The hydrolytic reactions were carried out using a mechano-enzymatic approach,44,53,54 in which cellulose powder is ball milled in the presence of the enzyme and a small amount of buffer (100 mM NaPB, pH 6) at a liquid-to-solid ratio η = 1.0 μL mg−1, i.e., a solid loading of 50 wt%. The buffer provides the hydrolysis substrate (≈11 molar equivalents of water to anhydrous glucose unit), enzyme buffering media, and reaction lubricant assisting molecular collisions. Mechanochemical liquid-assisted grinding (LAG) reactions, by ball milling or extrusion, are often faster than conventional solution reactions due to enhanced mixing at high concentrations.55–57 The amount of the buffer used here fully absorbs the cotton substrate, resulting in moist solid reaction mixtures. The action of commercially available cellulase systems from Trichoderma reesei (Tr), Aspergillus niger (An) and a Cellic CTec2 blend was compared after a brief period of ball milling (15 or 30 minutes) and after subsequent static incubation (24 or 72 hours of aging at 55 °C), with the extent of enzymatic hydrolysis monitored by quantifying the released glucose and by characterization of the recovered hydrolysed cellulose.Glucose yield as a measure of the hydrolysis extent of cotton
Influential mechanoenzymatic reaction parameters with respect to the type of cellulase preparation – the type of buffer, enzyme loading, liquid-to-solid ratio, and reaction duration – were screened based on glucose assays, which represents the hydrolysis end-product, after hydrolysis of cellulose by endoglucanases and cellobiohydrolases followed by cellobiose hydrolysis to glucose by β-glucosidase.58 Regardless of the cotton material or the type of cellulase used, the amount of glucose released was negligible after a short 15 minute milling step (Fig. 1b, at 0 h of aging) and gradually increased during the aging period. At a concentration of 15 U g−1 cellulose, the Tr cellulases proved to be the most active, producing 20–25% yield of glucose from both cellulose materials within 72 hours. When applied at a comparable concentration (U g−1), the glucose production with An was below 10% regardless of the cotton source, while CTec2 cellulases appeared sensitive to the cellulose source, resulting in ca. 20% yield of glucose from CBs and 12% from FP (Fig. 1b, data in Tables S1 and S3†). Therefore, further reaction parameters were screened using Tr cellulases. This revealed that varying the buffer type (100 mM NaPB at pH 6, 50 mM citrate at pH 6, 50 mM NaOAc at pH 5 or H2O), the liquid-to-solid ratio (1.0–2.0 μL mg−1) and the initial milling duration (5–30 min) had a small effect on the amount of glucose obtained in 72 hours (Fig. 1d, e and f, respectively, Table S2†), while increasing the enzyme loading (1.6 to 8.2 wt%) led to much higher glucose yields (Fig. 1c and Table S1†). | ||
| Fig. 1 (a) General reaction scheme for the mechanoenzymatic hydrolysis of cotton (CB – cotton balls, FP – Whatman 1 filter paper), combining a brief period of ball milling with subsequent aging. (b) Glucose yields from studied cotton sources: right after milling (0 h), and after subsequent aging for 24 or 72 h at 55 °C, by comparing Trichoderma reesei (Tr), Aspergillus niger (An) and Cellic CTec2 cellulase enzymes to a blank reaction. Enzyme loading for Tr, An and CTec2 was adjusted based on respective activity (see the ESI†), to 1.56 wt% for Tr, 0.085 wt% for An and 0.305 wt% for CTec2. Note that the enzyme loading (wt%) is based on all proteins contained in the cellulase preparation (see the ESI†). (c–f) Glucose yield from CB after ball milling + 72 h of aging, by varying the enzyme loading (c), the type of buffer (d), the amount of buffer (e) and the initial milling duration (f). Unless the specific parameter was varied, the base conditions for (c–f) were 400 mg of CB, 1.56 wt% of Tr, NaPB (100 mM, pH 6, η = 1.0 μL mg−1), 15 min of ball milling and 72 h of aging at 55 °C. | ||
Hydrolysis extent based on recovered hydrolysed cellulose
Given the likely variation in the relative activity of the cellulase enzymes (EG, CBH and BG) within the commercial Tr, An and CTec2 enzyme preparations, the correlation between the glucose yield and the yield of cellulose nanocrystals (CNCs) is not straightforward. Higher glucose yields, which are associated with higher weight losses, are not optimal for reaching a high yield of CNCs, as ideally CNCs would be obtained by controlled partial hydrolysis of cotton fibres with minimal material loss. Therefore, to identify the reaction conditions favouring partial hydrolysis of cellulose to CNCs, the bulk hydrolysed cellulose from the reactions with identified influential variables (cellulase mixture type and loading) was characterized at different timepoints by gel-permeation chromatography (GPC), powder X-ray diffraction (PXRD) and atomic force microscopy (AFM), to determine the hydrolysis degree (DPn, DPw and ĐM), the changes in cellulose crystallinity (CrI) and the morphology of the hydrolysed cellulose, respectively.Isolation of hydrolysed cellulose
Isolation of hydrolysed cellulose from water-soluble hydrolysis products could be easily achieved by washing the solids with either water or ethanol. However, these conditions were not successful in removing the enzymes, which remained adsorbed to the solid material. Therefore, alternative cellulose washing conditions were tested based on a reported pH-triggered cellulase desorption protocol (see the ESI† for details, recovered cellulose amounts given in Table 1).59 Gently stirring the crude reaction mixture at 1 wt% for 1 hour in a pH adjusted 50 mM citrate buffer (pH 10), followed by filtration on a 0.22 μM nylon filter removed up to 40% of the proteins depending on the cellulase preparation type and aging duration. A clear correlation could be observed between decreased removal efficiency and duration of the aging step, with a bigger proportion of cellulases removed right after milling (7% Tr, 40% An and 15% CTec2), compared to that after 24 h (≤10% Tr and CTec2, 25% An) and 72 h (≤5% Tr and CTec2, 20% An) of aging. This shows that cellulases proceeding furthest in the hydrolysis of cellulose to glucose are tightly bound, resulting in lower desorption of the enzymes (confirmed by elemental analysis, see Table S5†). This is consistent with earlier observations that some cellulases adsorb to cellulose surfaces nearly irreversibly,60 which implies that a longer hydrolysis duration leads to lower enzyme recyclability and increased protein impurities in the recovered hydrolysed cellulose.| Entry | Cotton & enzyme | Enzyme loading | Hydrolysis conditions | Recovered cellulose % | DPn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
DPwa |
Đ (Mw/Mn)b |
CrI (CI) % |
|---|---|---|---|---|---|---|---|---|
| a The degree of polymerization is given as the average of two measurements. Measurement deviation, which was up to 20%, is included in ESI Tables S1 and S4.† b Dispersity, given as the average of two measurements. | ||||||||
| 1 | CB start | N/A | None | N/A | 920 | 1520 | 1.6 | 66 (62) |
| 2 | CB + blank | 0 wt% | 15 min + 0 h | 91 | 1300 | 1720 | 1.3 | ND |
| 3 | CB + blank | 0 wt% | 15 min + 24 h | 90 | 1070 | 1565 | 1.5 | ND |
| 4 | CB + blank | 0 wt% | 15 min + 72 h | 94 | 1130 | 1620 | 1.4 | ND |
| 5 | CB + Tr | 1.6 wt% | 15 min + 0 h | 91 | 290 | 1100 | 4.0 | 71 (69) |
| 6 | CB + Tr | 1.6 wt% | 15 min + 24 h | 77 | 280 | 950 | 3.5 | 76 (73) |
| 7 | CB + Tr | 1.6 wt% | 15 min + 72 h | 68 | 260 | 1050 | 4.0 | 77 (76) |
| 8 | CB + An | 0.085 wt% | 15 min + 0 h | 100 | 290 | 1220 | 4.2 | 72 (72) |
| 9 | CB + An | 0.085 wt% | 15 min + 24 h | 86 | 240 | 1040 | 4.4 | 74 (73) |
| 10 | CB + An | 0.085 wt% | 15 min + 72 h | 93 | 300 | 1010 | 3.4 | 75 (74) |
| 11 | CB + CTec2 | 0.3 wt% | 15 min + 0 h | 94 | 450 | 1290 | 2.9 | 72 (69) |
| 12 | CB + CTec2 | 0.3 wt% | 15 min + 24 h | 80 | 250 | 820 | 3.3 | 74 (76) |
| 13 | CB + CTec2 | 0.3 wt% | 15 min + 72 h | 76 | 290 | 1150 | 4.0 | 78 (77) |
| 14 | CB + Tr (4.1 wt%) | 4.1 wt% | 15 min + 72 h | 61 | 360 | 1280 | 3.6 | ND |
| 15 | CB + Tr (8.2 wt%) | 8.2 wt% | 15 min + 72 h | 49 | 510 | 1410 | 2.8 | ND |
| 16 | CB + Tr | 1.6 wt% | 30 min + 0 h | 100 | 340 | 980 | 2.9 | ND |
| 17 | CB + Tr | 1.6 wt% | 30 min + 24 h | 80 | 185 | 645 | 3.5 | ND |
| 18 | CB + Tr | 1.6 wt% | 30 min + 72 h | 68 | 205 | 620 | 3.0 | ND |
| 19 | FP start | N/A | none | N/A | 655 | 1090 | 1.7 | ND |
| 20 | FP + blank | 0 wt% | 15 min + 0 h | 92 | 580 | 1000 | 1.7 | ND |
| 21 | FP + blank | 0 wt% | 15 min + 24 h | 92 | 500 | 970 | 1.9 | ND |
| 22 | FP + Tr | 1.6 wt% | 15 min + 0 h | 88 | 250 | 720 | 2.9 | ND |
| 23 | FP + Tr | 1.6 wt% | 15 min + 24 h | 74 | 360 | 1040 | 2.9 | ND |
Degree of polymerization of hydrolysed cellulose
Reactions in the absence of cellulases did not lead to any significant changes in the degree of polymerization of cotton (Fig. 2b and c), showing that cellulose is not hydrolysed solely by the reaction conditions applied. A rapid decrease in cellulose molecular mass was observed, however, when the cotton materials were exposed to cellulases under these conditions (Fig. 2b and d–f). Within the initial 15 minutes of ball-milling, the number average degree of polymerization (DPn) of cellulose drops by 50–75% (Fig. 2b and Table 1, entries 5, 8 and 11), indicating that endoglucanases, which cleave glycosidic bonds at disordered points mid-chain,61 are particularly active under mechanically agitated moist-solid reaction conditions, rapidly producing shorter polymers. Similar DPn values obtained after the milling step indicate that endo-activity on cellulose is comparable in the three cellulases tested. A limited decrease in DPn during subsequent aging (Table 1 entries 6, 7, 9, 10, 12 and 13) allows us to conclude that easily accessible parts of cellulose fibres (less-ordered and exposed) are enzymatically hydrolysed already with the initial short milling step. Previously, mixing24,62 or extrusion63 of dissolving pulp at a solid loading of 20–30 wt% in the presence of endoglucanases was shown to strongly enhance the hydrolytic action of cellulase enzymes, owing to fibre–fibre friction at a low water content, leading to a 54% drop in the pulp molar mass and the disruption of the microfibril structure during 2 hours of treatment. This provided access to low-DP pulp for improved dissolution in NaOH/ZnO in the Biocelsol process,63 and showcased the potential of enzymes for the sustainable production of microfibrillated and nanoscale cellulose materials from the pulp in the HefCel process.24 Our results, on one hand, clearly support this finding and furthermore show that enzymatic treatments at an even higher solid loading (50 wt%) can be achieved by brief ball-milling, yielding a dramatic 75% drop in DPn within 15 minutes, using a commercial cellulase preparation (Tr or An cellulases). An cellulases appear to have particularly high endo-activity, leading to a low DPn at a significantly lower enzyme loading (0.085 wt%) compared to Tr cellulases (1.56 wt%), and compared to the cellulase preparation applied in the HefCel process (0.6 wt%).24 For all three cellulase preparations tested here, the decrease in the weight average degree of polymerization DPw was smaller (15–45%) compared to the drop in DPn, leading to an increase in the dispersity of cellulose to Đ(Mw/Mn) = 3–4 when compared to the cotton starting materials (Fig. 2b). A similar increase in dispersity is observed in the acid-catalyzed production of CNCs from cotton.7,64 A shift in the molecular mass distribution (Fig. 2c–f) of cellulose to a lower Mw by mechanoenzymatic reactions here follows the activity trends observed in glucose assays – with the most pronounced shift observed for Tr upon aging, followed by CTec2 and the least pronounced shift observed for An. Notably, increasing the loading of the Tr enzyme, to either 4.2 or 8.2 wt%, did not bring a larger decrease of the DPn or DPw of cellulose (Table 1, entries 11 and 12). The resulting bimodal molecular mass distributions (Fig. 2g, 72 h data shown) rather indicate that a larger amount of long polymer chains is retained, compared to when less enzyme is used. This may be explained by the presence of excess cellobiohydrolase enzymes at high enzyme loading. Cellobiohydrolase enzymes are responsible for the processive hydrolysis of cellulose from chain ends, and strong non-productive binding of these enzymes along the cellulose fibre can interrupt the action of endo-active hydrolases,65,66 leading to retention of longer cellulose chains. Mechanoenzymatic reactions have been shown to benefit from longer or intermittent milling of the reaction mixture, which improves the mixing of the solids and can aid in the exposure of new polymer surfaces for productive enzyme adsorption.29–31,67–69 Increasing the initial milling duration from 15 to 30 minutes led to a comparable ca. 75% drop in DPn (Table 1, entries 5 and 16), showing that increasing the milling time does not directly impact the DPn obtained. However, following the DP over the course of the aging step showed that longer initial milling led to lower DPn and DPw after aging (Table 1, entries 17 and 18), indicating that the activity of both cellobiohydrolases and endoglucanases benefit from the improved mixing before the aging step. A rapid drop in the DPn and DPw was also observed when FP was exposed to Tr cellulases during 15 minutes of ball milling (Fig. 2h), implying rapid and extensive mid-chain hydrolysis of cellulose from either source which allows for the isolation of CNCs.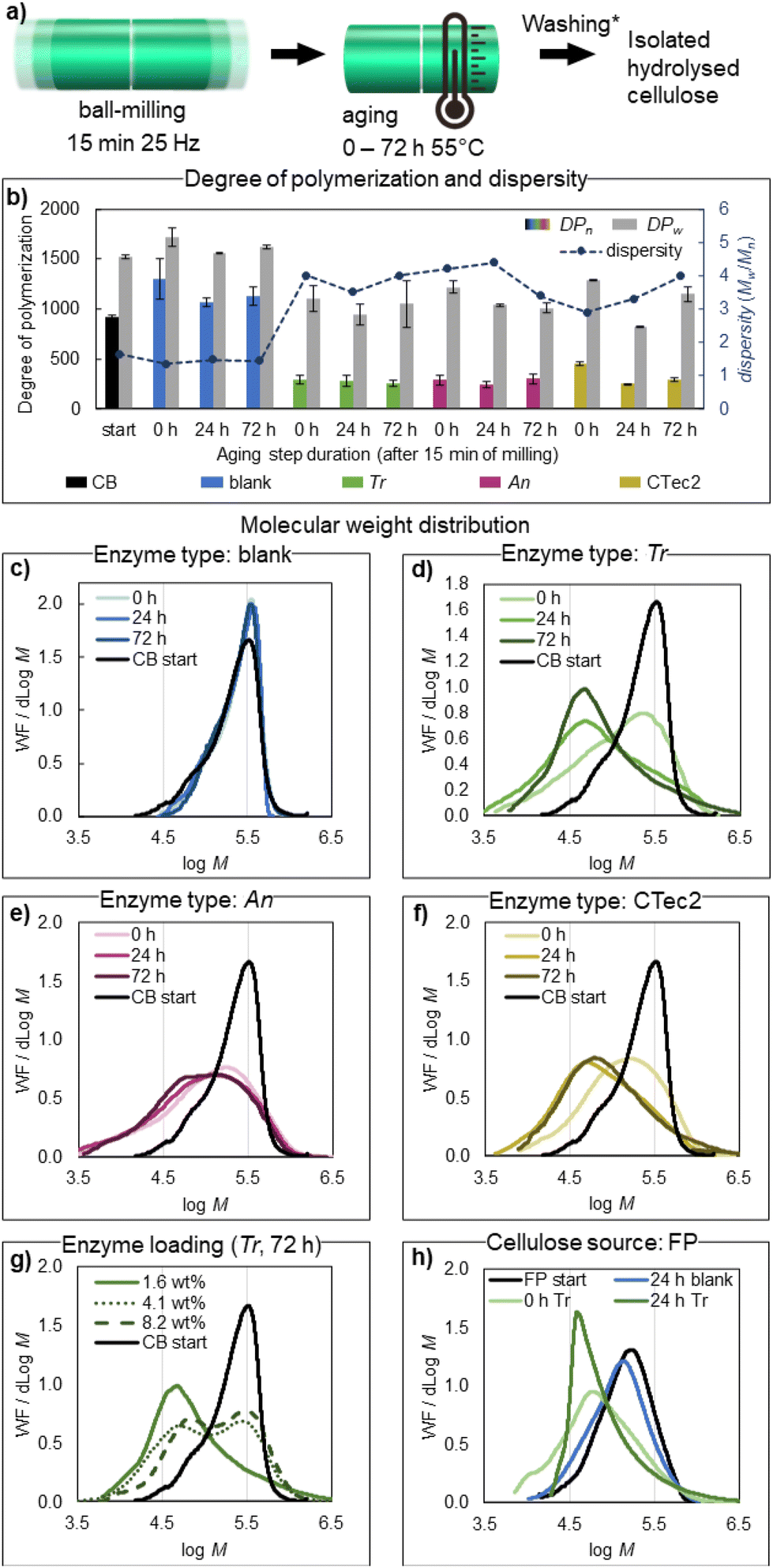 | ||
| Fig. 2 (a) Scheme of the isolation of hydrolysed cellulose, where washing (*) denotes the removal of the soluble hydrolysis products, and pH-triggered enzyme desorption carried out by stirring the crude reaction mixture for 1 hour at 1 wt% in a pH-adjusted 50 mM citrate buffer (pH 10), followed by washing with a 0.22 μM nylon filter, see the ESI† for details. (b) Number average (DPn) and weight average (DPw) degrees of polymerization and dispersity Đ (Mw/Mn) of the isolated hydrolysed cellulose, right after milling (0 h), and after subsequent aging for 24 or 72 h at 55 °C, by comparing Trichoderma reesei (Tr), Aspergillus niger (An) and Cellic CTec2 enzymes to a blank reaction. A line is drawn connecting the independent dispersity data points to guide the eye. Each sample was measured twice, and error bars represent the measurement variation. Reactions with the Tr enzyme were carried out in triplicate, and the respective error bars represent the experimental standard deviation. (c–h) Molecular mass Mw distribution of the isolated hydrolysed cellulose from reactions with the identified influential variables: enzyme type (c–f), enzyme loading (f) and cellulose source (h), compared to the respective starting material. Full experimental details of the reactions are given in Table S1.† | ||
Structure, properties and particle size of isolated hydrolysed cellulose
X-ray diffraction analysis of bulk isolated enzymatically treated materials after milling (15 minutes of milling, denoted on the graphs as 0 h of aging) and after milling + aging (24 and 72 h) shows that the crystalline structure of cellulose Iβ is well preserved (Fig. S2†). In fact, the crystallinity of cellulose increased by ca. 10% relative to the starting material (Fig. 3b) already with the brief enzymatic treatment during ball milling, with a further slower increase observed upon subsequent aging for 24 or 72 h. Crystallinity indices based on the Segal peak height method (CrI, Fig. 3a)70 and the peak fitting method (CI, Fig. 3a) are reported here to give an estimate of the relative changes in crystallinity compared to the starting material (CBs) under different reaction conditions. Neither should be taken as the absolute quantity of crystalline fractions in the materials.71,72 An equally sharp increase in crystallinity (CrI) from 66% to 71–72% for all three tested enzymes, observed right after milling, coincides with the sharp levelling off in DPn, as observed by GPC, showing the action of endoglucanases preferentially hydrolysing the disordered parts of cellulose fibres. A further increase in sample crystallinity by aging for 24 or 72 h is likely due to the generation and removal of soluble hydrolysis products from cellulose, as the magnitude of the CrI change for Tr (77%), An (75%) and CTec2 (78%) correlates well with the respective glucose production. Similar to previous reports on enzymatic CNC production,17 the hydrolysed materials isolated retain the native surface chemistry of cellulose (a zeta potential of up to −26 mV, Fig. 3c), which does not provide enough charge repulsion to obtain good dispersion of the particles in water. This contrasts with CNCs obtained from e.g. sulfuric and phosphoric acid hydrolysis or by involving TEMPO-oxidation post-treatment, where negatively charged functional groups are installed to CNC surfaces (resulting in zeta potentials of −35 mV and lower), which enables good dispersion of the particles through electrostatic repulsion.6,7,73 Cellulose treated with An and CTec2 enzymes retain similar zeta potentials of −26.3 ± 0.87 mV (An) and −22.2 ± 0.6 mV (CTec2) to the starting material with a value of −24.9 ± 0.7 mV (pre-milled CB); however, the Tr treated sample has a distinctly different zeta potential of −16.6 ± 0.2 mV (Tr). A similar variation in the resulting zeta potential arising from the enzymatic mixture used was observed in the hydrolysis of eucalyptus cellulose pulp.74 Here, the variation likely reflects the inefficient removal of the Tr enzymes upon product isolation (washing buffer, pH 10), which remain on the surface of the product to a larger extent compared to the An and CTec2 cellulases, due to the higher enzyme loading. An earlier onset of thermal degradation (276 °C) and reduced mass loss (71%) of the Tr-treated material upon heating, compared to the materials treated with An (onset 334 °C, mass loss 90%), CTec2 (onset 335 °C, mass loss 92%), and a pure buffer without enzymes (onset 323 °C, mass loss 92%), also point to a higher amount of protein impurities in the Tr sample (Table S6† and Fig. 3d), as an increase of active groups on the surface of CNCs can impact the thermal degradation of cellulose. All mechanoenzymatically obtained materials have higher thermal stability compared to sulfate-CNCs (onset 160 °C) and phosphate-CNCs (onset 207 °C) obtained from acid hydrolysis,73 and similar stability to enzymatically obtained CNCs (onset 205–316 °C, depending on the cellulose source, hydrolysis time and the enzyme loading).16,22,27,28 The poor dispersibility of the particles restricts the characterization of the apparent particle size by dynamic light scattering and complicates statistical particle size measurements based on transmission electron microscopy (TEM) and atomic force microscopy (AFM) imaging techniques. Long cellulose fibres are clearly retained in blank reactions (Table 1 entry 4, Fig. 3e, additional images in Fig S3†). Contrastingly, cotton hydrolysed by Tr enzymes to the levelling off DPn predominantly contains shorter elements, albeit poorly individualized on the TEM images (Table 1 entry 7, TEM on Fig. 3e, additional images in Fig S4†). The estimated length of the particles (150–350 nm, see ESI Fig. S4†) corresponds well to that reported for CNCs obtained by sulphuric acid hydrolysis from cotton (100–300 nm).75 Some larger fragments could be observed, which aligns with the observed increased dispersity of cellulose after hydrolysis.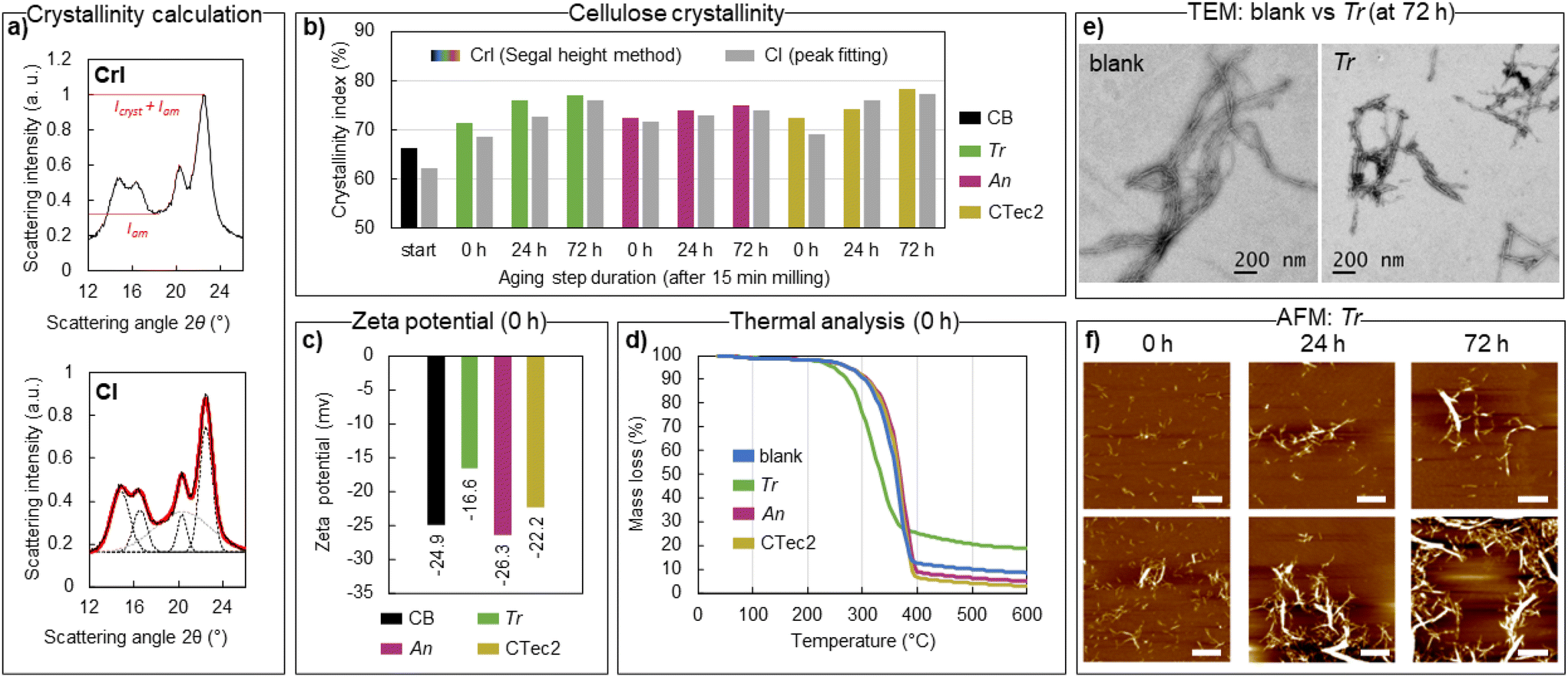 | ||
| Fig. 3 Characterization of the isolated hydrolysed cellulose by X-ray diffraction (a and b), zeta potential measurements (c), thermal analysis (d), transmission electron microscopy TEM (e) and atomic force microscopy AFM (f). Panel b compiles the CrI (in colour) and CI (in grey) for the isolated hydrolysed cellulose, right after milling, and after subsequent aging for 24 or 72 h at 55 °C, by comparing Trichoderma reesei (Tr), Aspergillus niger (An) and Cellic CTec2 enzymes to the starting material (CB start), calculated as shown in panel a, based on data shown in Fig. S2.† Zeta potentials (c) and thermal analysis (d) were used to compare the isolated materials from mechanoenzymatic treatment (milled only, Table 1 entries 5, 8 and 11) with the starting material (CB) and with the blank reaction (no enzyme, Table 1 entry 2), respectively. The TEM images (e) were used to compare the cellulose isolated from a blank reaction (no enzyme) with that hydrolysed under the same reaction conditions with Tr cellulases (Table 1 entries 4 vs. 7). The AFM images (f) were used to compare the cellulose hydrolysed by Tr cellulases: right after milling (0 h) and after subsequent aging for 24 or 72 h at 55 °C. Two images of each timepoint are shown. Scale bar is 1 μm, and all images have the height scale from −10 to 10 nm. | ||
The AFM images of Tr-hydrolyzed samples (Table 1, entries 5–7) dispersed in formic acid, one of the few solvents shown to improve the dispersion of non-charged cellulose nanocrystals,76 reveal that individual CNCs are generated. The length distribution (150–400 nm, ESI Fig. S5†) of the particles shown in Fig. 3f (at 0 h) agrees well with the TEM data (at 72 h), showing that the nanoscale particles are present in the sample right after milling. These observations, together with bulk characterization by GPC and XRD, indicate that cellulose is transformed to nanoscale crystallites within the initial 15 minutes of milling. Limited conversion of cellulose to soluble sugars at this time point ensures that a high proportion of hydrolysed cellulose is recovered (>90%, Table 1, entries 5, 8 and 11), giving process mass intensities of 2.3 (Tr), 2.1 (An) and 2.2 (CTec2). Process mass intensity (PMI)77 is a sustainability metric that evaluates the total input mass of materials (including reaction media) that is incorporated into the product (ESI, eqn (S3)†), with an optimal value of 1. The mechanoenzymatic approach reported herein shows a 50-fold improvement in PMI, compared to the reported in-solution enzymatic methods to produce cellulose nanomaterials (details in ESI, Table S7†), and a ca. 2.5-fold improvement compared to the previously reported HefCel process.24 However, due to the short reaction time and minimized reaction volume (0.25 h, 50 wt% solid loading), more than a 100-fold improvement in the space–time-yield (3.5–3.8 kg L−1 h−1) is achieved compared to the HefCel process, and more than 30000-fold improvement over the reported in-solution methods (Table S7 and eqn (S4)†). It must be noted, however, that the reported in-solution processes often investigate the co-production of glucose and CNCs for integrated biorefineries, which maximizes the glucose production and therefore a low yield of recovered cellulose (ca. 10%) is reported, affecting the space–time-yield. The mechanoenzymatic approach also provides significant economic and environmental benefits compared to acid hydrolysis. The reagent cost estimate, for transforming 1 gram of starting material (cotton), comparing the mechanoenzymatic methods developed here with conventional acid hydrolysis using sulfuric,6 hydrochloric78–80 or phosphoric acid81 (Table S8†), shows that the mechanoenzymatic process using An and CTec2 brings a 7- and 4- fold decrease in costs, respectively, compared to the cheapest sulfuric acid treatment. The process using the Tr enzyme matches the price of HCl hydrolysis and is only 2-fold more expensive than the sulfuric acid treatment, due to the higher enzyme loading. The phosphoric acid hydrolysis proved to have the highest reagent cost. Notably, this cost estimate does not account for the economic and environmental benefits of the mechanoenzymatic method, which arise from avoiding equipment corrosion, avoiding the generation of corrosive wastes, avoiding hazards to personnel and avoiding the large amount of water and/or base required for the removal of the concentrated acids. The energy consumption of the used equipment (Retsch MM400 mixer mill, loaded with two 15 mL stainless steel jars) for the reaction described in Table 1, entry 5, was found to be 0.016 kW h, which is less than the energy required for 45 minutes of sulfuric acid hydrolysis6 at 45 °C using a heating mantle with stirring (0.024 kW h). The energy consumed for hydrochloric acid78 and phosphoric acid81 hydrolysis carried out at 100 °C is 10–20× higher than that of the method reported herein (ca. 0.250 kW h and ca. 0.160 kW h for the 180- and 90-minute reactions, respectively). Notably, no energy is directly consumed during HCl-gas hydrolysis. For the details of the measurements of energy consumption, see ESI Table S8.†
Conclusions
We have examined the production of cellulose nanocrystals from cotton by solid-state mechanoenzymatic approaches using three commercially available cellulase systems from Trichoderma reesei, Aspergillus niger and a Cellic CTec2 cellulase blend. All applied cellulase systems lead to a rapid drop in the degree of polymerization near the levelling-off degree of polymerization of cotton and to a sharp increase in the crystallinity of cellulose, during a short 15 minutes mechanoenzymatic reaction. This indicates that the bulk material is quickly transformed to cellulose nanomaterials, at a space–time-yield of 3.5–3.8 kg L−1. Subsequent aging of the solid reaction mixture at 55 °C did not further decrease the DPn, but resulted in higher material losses due to the increased production of glucose. An cellulases appear particularly suitable for this process, leading to a low DPn at a significantly lower enzyme loading (0.085 wt%) compared to Tr and CTec2 cellulases (1.56 wt% and 0.3 wt%, respectively). Utilizing renewable catalysts (enzymes) and avoiding much of the waste and energy lost in heating dilute aqueous solutions, the mechanoenzymatic method for producing cellulose nanomaterials is highly efficient (PMI 2.1–2.3) and offers notable economic and environmental benefits.Experimental
The following is a concise overview of the materials and methods used. Full experimental details are included in the ESI file.†Materials
Cotton was purchased as non-sterile raw cotton balls (CB) along with Whatman type 1 filter paper (FP). Both materials were milled to powders using a Thomas Scientific Wiley Mini Mill 475-A equipped with a 30-mesh screen and thereafter used in reactions without further purification. The cellulases from Trichoderma reesei, Aspergillus niger and the CTec2® blend were purchased from Sigma-Aldrich and had a comparable activity of 1–2 U mg−1 based on the producer's specifications and in the hydrolysis assay of microcrystalline cellulose (Sigma Aldrich, average 51 μm particles). The enzymes were stored at 4 °C. Buffers were prepared using Milli-Q water with a conductivity of 1.8 μS cm−1 at 25 °C, and included 0.02 wt% of sodium azide as a bacteriostatic preservative.Methods
In a typical reaction, pre-milled cotton powder (400 mg) was weighed into a 15 mL stainless steel jar with a 4 g stainless steel ball, to which the commercial enzyme preparation (either 20 mg of Trichoderma reesei, 20 mg of Aspergillus niger or 20 mg (17.4 μL) of the CTec2® blend and buffer (420 μL)) was added, bringing the total liquid-to-solid ratio to 1.0 μL mg−1, corresponding to a solid loading of 50% w/w. The milling jars were then closed and set to mill at 25 Hz for 15 minutes. The resulting uniform solids were aliquoted into 1.5 mL Eppendorf tubes affording analysis samples for glucose assays (20–30 mg) and for recovered cellulose isolation (200 mg) at different time points: immediately after milling, after subsequent aging for 24 h at 55 °C, and after subsequent aging for 72 h at 55 °C. The commercial Glucose (HK) Assay Reagent (purchased from Sigma Aldrich) was used to quantify glucose in the reaction mixtures. An aliquot of 200–250 mg of the reaction mixture (solid content: 48–50%) was taken at the end of the reaction, suspended in 1 wt% washing buffer (50 mM citrate buffer, adjusted with NaOH to pH 10) and incubated for 1 h with gentle magnetic stirring at 100 rpm to remove the enzyme. Thereafter, the solids were collected by vacuum filtration and washed on a 0.22 μm polypropylene or nylon membrane, once with the washing buffer and three times with Milli-Q water. The solids were collected from the membrane by rinsing with Milli-Q water into a Falcon tube and freeze dried. Some loss of solids could not be avoided upon flushing of the membrane, which is reflected in the recovery% of cellulose. The weight of the dry solids was recorded, from which the recovery% was calculated as follows:
Specific reaction conditions, together with the hydrolysis yields, are compiled in Table S1.† 50 ± 5 mg of the washed and freeze-dried recovered cellulose samples were prepared for GPC analysis by the water–acetone–N,N-dimethylacetamide (DMAc) solvent exchange procedure, followed by dissolution in 90 g L−1 LiCl/DMAc, analysis at 1 mg mL−1 against Pullulan standards on a Dionex UltiMate 3000 HPLC system, and elution with 9 g−1 LiCl/DMAc at a flow rate of 0.75 mL min−1. About 15–20 mg of the dry recovered cellulose samples was used for collecting wide-angle X-ray scattering (WAXS) data using a Xeuss 3.0 C scattering device (Xenocs, France). Crystallinity changes were estimated based on CrI, calculated according to the empirical Segal peak height method,70 and CI was calculated based on peak fitting. Atomic Force Microscopy (AFM) images were recorded on a Bruker MultiMode8 instrument, from samples dispersed at 1 wt% in formic acid and spin-coated on UV-cleaned silicon wafers. Formic acid adsorbs onto the surfaces of the CNCs and disrupts the hydrogen bonds between aggregated particles, which enables the dispersion of non-charged CNCs.76 A small amount of formate ester moieties may also be formed on the surface of CNCs,76 which may also facilitate the dispersion in formic acid. Zeta potentials were measured on the Malvern Zetasizer, and thermal analysis was carried out with the Netzsch STA 449 F3 Jupiter system. Transmission electron microscopy imaging was performed on an FEI Tecnai 12 microscope at an acceleration voltage of 120 kV. A Thermo Scientific Flash Smart CHNS elemental analyzer was used to qualitatively confirm the presence of protein impurities in washed and freeze-dried recovered cellulose samples. Changes in the surface charge of recovered cellulose were probed by zeta potential measurements using a Malvern Zetasizer Nano ZS90. Further details are provided in the ESI.†
Author contributions
The authors have contributed to the following tasks: S.K.: conceptualization, investigation (data acquisition and analysis), writing the original draft and funding acquisition; I.S.-H. investigation (thermal analysis data acquisition and analysis); M.K. and E.K.: investigation (data analysis), supervision, and writing (review and editing).Data availability
The data supporting this article have been included as part of the ESI.† The ESI† file compiles detailed information on the materials, methods, and equipment used. It also contains the description of the equations used for calculations (eqn (S1)–(S4)), ESI Fig. (S1–S3) and Tables (S1–S6)† referenced in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Leena Pitkänen for her help with GPC analysis and Dr Ville Liljeström for his help in obtaining the wide-angle X-ray scattering data. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 101027061. We also acknowledge the funding from the Academy of Finland Centers of Excellence Program (2022–2029) in Life-Inspired Hybrid Materials (LIBER), project number (346110, 364201), and the provision of facilities and technical support by Aalto University Bioeconomy and Raw Materials Research Infrastructure (RAMI) facilities. We are also grateful for the support by the FinnCERES Materials Bioeconomy Ecosystem.References
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior, D. Kralisch, F. Kramer, T. Kondo, T. Lindström, S. Nietzsche, K. Petzold-Welcke and F. Rauchfuß, Mater. Today, 2018, 21, 720–748 CrossRef CAS.
- J. A. Shatkin, T. H. Wegne, E. M. T. Bilek and J. Cowie, Tappi J., 2014, 13, 9–16 CAS.
- F. A. G. S. Silva, F. Dourado, M. Gama and F. Poças, Nanomaterials, 2020, 10, 2041 CrossRef CAS PubMed.
- S. S. Ahankari, A. R. Subhedar, S. S. Bhadauria and A. Dufresne, Carbohydr. Polym., 2021, 255, 117479 CrossRef CAS PubMed.
- O. Nechyporchuk, M. N. Belgacem and J. Bras, Ind. Crops Prod., 2016, 93, 2–25 CrossRef CAS.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- G. Delepierre, O. M. Vanderfleet, E. Niinivaara, B. Zakani and E. D. Cranston, Langmuir, 2021, 37, 8393–8409 CrossRef CAS.
- M. Jonoobi, R. Oladi, Y. Davoudpour, K. Oksman, A. Dufresne, Y. Hamzeh and R. Davoodi, Cellulose, 2015, 22, 935–969 CrossRef CAS.
- O. M. Vanderfleet and E. D. Cranston, Nat. Rev. Mater., 2021, 6, 124–144 CrossRef CAS.
- P. Squinca, S. Bilatto, A. C. Badino and C. S. Farinas, ACS Sustainable Chem. Eng., 2020, 8, 2277–2286 CrossRef CAS.
- P. Squinca, S. Bilatto, A. C. Badino and C. S. Farinas, Carbohydr. Polym. Technol. Appl., 2022, 3, 100212 CAS.
- C. R. Bauli, D. B. Rocha, S. A. de Oliveira and D. S. Rosa, J. Cleaner Prod., 2019, 211, 408–416 CrossRef CAS.
- R. S. S. Teixeira, A. S. A. Da Silva, J. H. Jang, H. W. Kim, K. Ishikawa, T. Endo, S. H. Lee and E. P. S. Bon, Carbohydr. Polym., 2015, 128, 75–81 CrossRef CAS PubMed.
- C. R. Bauli, D. B. Rocha and D. D. S. Rosa, SN Appl. Sci., 2019, 1, 774 CrossRef.
- P. B. Filson, B. E. Dawson-Andoh and D. Schwegler-Berry, Green Chem., 2009, 11, 1808 RSC.
- J. De Aguiar, T. J. Bondancia, P. I. C. Claro, L. H. C. Mattoso, C. S. Farinas and J. M. Marconcini, ACS Sustainable Chem. Eng., 2020, 8, 2287–2299 CrossRef CAS.
- S. R. Anderson, D. Esposito, W. Gillette, J. Y. Zhu, U. Baxa and S. E. McNeil, Tappi J., 2014, 13, 35–42 CAS.
- G. A. Siqueira, I. K. R. Dias and V. Arantes, Int. J. Biol. Macromol., 2019, 133, 1249–1259 CrossRef CAS PubMed.
- A. A. Domingues, F. V. Pereira, M. R. Sierakowski, O. J. Rojas and D. F. S. Petri, Cellulose, 2016, 23, 2421–2437 CrossRef CAS.
- J. M. Yarbrough, R. Zhang, A. Mittal, T. Vander Wall, Y. J. Bomble, S. R. Decker, M. E. Himmel and P. N. Ciesielski, ACS Nano, 2017, 11, 3101–3109 CrossRef CAS PubMed.
- P. Satyamurthy, P. Jain, R. H. Balasubramanya and N. Vigneshwaran, Carbohydr. Polym., 2011, 83, 122–129 CrossRef CAS.
- X. Tong, W. Shen, X. Chen, M. Jia and J. Roux, J. Appl. Polym. Sci., 2020, 137, 48407 CrossRef CAS.
- X. Q. Chen, G. X. Pang, W. H. Shen, X. Tong and M. Y. Jia, Carbohydr. Polym., 2019, 207, 713–719 CrossRef CAS PubMed.
- J. Pere, T. Tammelin, P. Niemi, M. Lille, T. Virtanen, P. A. Penttilä, P. Ahvenainen and S. Grönqvist, ACS Sustainable Chem. Eng., 2020, 8, 18853–18863 CrossRef CAS.
- J. T. Xu, X. Q. Chen, W. H. Shen and Z. Li, Carbohydr. Polym., 2021, 256, 117493 CrossRef CAS PubMed.
- R. S. A. Ribeiro, N. Bojorge and N. Pereira, Biotechnol. Appl. Biochem., 2020, 67, 366–374 CAS.
- B. Alonso-Lerma, L. Barandiaran, L. Ugarte, I. Larraza, A. Reifs, R. Olmos-Juste, N. Barruetabeña, I. Amenabar, R. Hillenbrand, A. Eceiza and R. Perez-Jimenez, Commun. Mater., 2020, 1, 57 CrossRef.
- B. Alonso-Lerma, I. Larraza, L. Barandiaran, L. Ugarte, A. Saralegi, M. A. Corcuera, R. Perez-Jimenez and A. Eceiza, Carbohydr. Polym., 2021, 254, 117478 CrossRef CAS PubMed.
- F. Hammerer, L. Loots, J. Do, J. P. D. Therien, C. W. Nickels, T. Friščić and K. Auclair, Angew. Chem., Int. Ed., 2018, 57, 2621–2624 CrossRef CAS PubMed.
- F. Hammerer, S. Ostadjoo, K. Dietrich, M.-J. J. Dumont, L. F. Del Rio, T. Friščić and K. Auclair, Green Chem., 2020, 22, 3877–3884 RSC.
- S. Ostadjoo, F. Hammerer, K. Dietrich, M.-J. Dumont, T. Friščić and K. Auclair, Molecules, 2019, 24, 4206 CrossRef CAS PubMed.
- F. Hammerer, S. Ostadjoo, T. Friščić and K. Auclair, ChemSusChem, 2020, 13, 106–110 CAS.
- M. Käldström, N. Meine, C. Farès, R. Rinaldi and F. Schüth, Green Chem., 2014, 16, 2454–2462 RSC.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schüth, Energy Environ. Sci., 2013, 6, 92–96 RSC.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- M. Kessler and R. Rinaldi, Front. Chem., 2022, 9, 816553 CrossRef PubMed.
- Y. Dong, L. Schneider, T. Hu, M. Jaakkola, J. Holm, J. M. Leveque and U. Lassi, Biomass Bioenergy, 2016, 86, 36–42 CrossRef CAS.
- Y. Yu, Y. Long and H. Wu, Energy Fuels, 2016, 30, 1571–1578 CrossRef CAS.
- L. Schneider, J. Haverinen, M. Jaakkola and U. Lassi, Bioresour. Technol., 2017, 234, 1–7 CrossRef CAS PubMed.
- P. Dornath, H. J. Cho, A. Paulsen, P. Dauenhauer and W. Fan, Green Chem., 2015, 17, 769–775 RSC.
- Q. Zhang and F. Jérôme, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–447 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef.
- S. Kaabel, T. Friščić and K. Auclair, ChemBioChem, 2020, 21, 742–758 CrossRef CAS PubMed.
- C. C. Piras, S. Fernández-Prieto and W. M. De Borggraeve, Nanoscale Adv., 2019, 1, 937–947 RSC.
- J. Gröndahl, K. Karisalmi and J. Vapaavuori, Soft Matter, 2021, 17, 9842–9858 RSC.
- T. Yang, X. Li, Y. Guo, S. Peng, G. Liu and J. Zhao, Ind. Crops Prod., 2020, 155, 112755 CrossRef CAS.
- S. Cui, S. Zhang, S. Ge, L. Xiong and Q. Sun, Ind. Crops Prod., 2016, 83, 346–352 CrossRef CAS.
- M. Qian, H. Lei, E. Villota, Y. Zhao, C. Wang, E. Huo, Q. Zhang, W. Mateo and X. Lin, Chem. Eng. Process., 2021, 160, 108292 CrossRef CAS.
- Q. Zhang, Z. Lu, C. Su, Z. Feng, H. Wang, J. Yu and W. Su, Bioresour. Technol., 2021, 331, 125015 CrossRef CAS PubMed.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- O. M. Vanderfleet and E. D. Cranston, Nat. Rev. Mater., 2021, 6, 124–144 CrossRef CAS.
- M. Pérez-Venegas and E. Juaristi, ChemSusChem, 2021, 14, 2682–2688 CrossRef PubMed.
- J. Arciszewski and K. Auclair, ChemSusChem, 2022, 15, e202102084 CrossRef CAS PubMed.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712–715 CrossRef.
- L. Chen, M. Regan and J. Mack, ACS Catal., 2016, 6, 868–872 CrossRef CAS.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- S. K. Brady, S. Sreelatha, Y. Feng, S. P. S. Chundawat and M. J. Lang, Nat. Commun., 2015, 6, 10149 CrossRef CAS PubMed.
- Y. Shang, R. Su, R. Huang, Y. Yang, W. Qi, Q. Li and Z. He, Appl. Microbiol. Biotechnol., 2014, 98, 5765–5774 CrossRef CAS PubMed.
- B. Henrissat, Cellulose, 1994, 1, 169–196 CrossRef CAS.
- J. Pere, M. Siika-aho, J. Buchert and L. Viikari, Tappi J., 1995, 78, 71–78 CAS.
- J. Rahikainen, O. Mattila, T. Maloney, V. Lovikka, K. Kruus, A. Suurnäkki and S. Grönqvist, Cellulose, 2020, 27, 5311–5322 CrossRef CAS.
- S. Grönqvist, T. Kamppuri, T. Maloney, M. Vehviläinen, T. Liitiä and A. Suurnäkki, Cellulose, 2015, 22, 3981–3990 CrossRef.
- P. Spiliopoulos, S. Spirk, T. Ppaakkonen, M. Viljanen, K. Svedstrom, L. Pitkanen, M. Awais and E. Kontturi, Biomacromolecules, 2021, 22, 1399–1405 CrossRef CAS.
- K. Igarashi, M. Wada, R. Hori and M. Samejima, FEBS J., 2006, 273, 2869–2878 CrossRef CAS PubMed.
- P. Väljamäe, V. Sild, G. Pettersson and G. Johansson, Eur. J. Biochem., 1998, 253, 469–475 CrossRef.
- J. P. D. Therien, F. Hammerer, T. Friščić and K. Auclair, ChemSusChem, 2019, 12, 3481–3490 CrossRef CAS.
- S. Kaabel, J. P. D. Therien, C. E. Deschênes, D. Duncan, T. Friščić and K. Auclair, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026452118 CrossRef CAS.
- S. Kaabel, J. Arciszewski, T. H. Borchers, J. P. D. Therien, T. Friščić and K. Auclair, ChemSusChem, 2023, 16, e202201613 CrossRef CAS PubMed.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- K. S. Salem, N. K. Kasera, M. A. Rahman, H. Jameel, Y. Habibi, S. J. Eichhorn, A. D. French, L. Pal and L. A. Lucia, Chem. Soc. Rev., 2023, 52, 6417–6446 RSC.
- P. Ahvenainen, I. Kontro and K. Svedström, Cellulose, 2016, 23, 1073–1086 CrossRef CAS.
- M. Kröger, O. Badara, T. Pääkkönen, I. Schlapp-Hackl, S. Hietala and E. Kontturi, Biomacromolecules, 2023, 24, 1318–1328 CrossRef.
- T. J. Bondancia, C. Florencio, G. S. Baccarin and C. S. Farinas, Int. J. Biol. Macromol., 2022, 207, 299–307 CrossRef CAS.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS PubMed.
- O. van der Berg, J. R. Capadona and C. Weder, Biomacromolecules, 2007, 8, 1353–1357 CrossRef.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- H. Yu, Z. Qin, B. Liang, N. Liu, Z. Zhou and L. Chen, J. Mater. Chem. A, 2013, 1, 3938–3944 RSC.
- E. Kontturi, A. Meriluoto, P. A. Penttilä, N. Baccile, J. Malho, A. Potthast, T. Rosenau, J. Ruokolainen, R. Serimaa, J. Laine and H. Sixta, Angew. Chem., Int. Ed., 2016, 55, 14455–14458 CrossRef CAS PubMed.
- T. Pääkkönen, P. Spiliopoulos, A. Knuts, K. Nieminen, L.-S. Johansson, E. Enqvist and E. Kontturi, React. Chem. Eng., 2018, 3, 312–318 RSC.
- S. Camarero Espinosa, T. Kuhnt, E. J. Foster and C. Weder, Biomacromolecules, 2013, 14, 1223–1230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization protocols and data tables. See DOI: https://doi.org/10.1039/d4gc05113k |
| This journal is © The Royal Society of Chemistry 2025 |